
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Green-light photocleavable meso-methyl BODIPY building blocks for macromolecular chemistry†
Paul
Strasser
 a,
Marina
Russo
bc,
Pauline
Stadler
a,
Patrick
Breiteneder
a,
Günther
Redhammer
d,
Markus
Himmelsbach
e,
Oliver
Brüggemann
a,
Uwe
Monkowius
*f,
Petr
Klán
*bc and
Ian
Teasdale
*a
a,
Marina
Russo
bc,
Pauline
Stadler
a,
Patrick
Breiteneder
a,
Günther
Redhammer
d,
Markus
Himmelsbach
e,
Oliver
Brüggemann
a,
Uwe
Monkowius
*f,
Petr
Klán
*bc and
Ian
Teasdale
*a
aInstitute of Polymer Chemistry, Johannes Kepler University Linz, Altenberger Straße 69, A-4040 Linz, Austria. E-mail: ian.teasdale@jku.at
bDepartment of Chemistry, Faculty of Science, Masaryk University, Kamenice 5, 625 00 Brno, Czech Republic. E-mail: klan@sci.muni.cz
cRECETOX, Faculty of Science, Masaryk University, Kamenice 5, 625 00 Brno, Czech Republic
dChemie und Physik der Materialien, Abteilung für Materialwissenschaften und Mineralogie, Paris-Lodron Universität Salzburg, Jakob-Haringerstr. 2A, 5020 Salzburg, Austria
eInstitute of Analytical Chemistry, Johannes Kepler University Linz, Altenberger Straße 69, A-4040 Linz, Austria
fLinz School of Education, Johannes Kepler University Linz, Altenberger Straße 69, A-4040 Linz, Austria. E-mail: uwe.monkowius@jku.at
First published on 16th November 2021
Abstract
We report the design of easily accessible, photocleavable meso-methyl BODIPY monomers suitably functionalised for incorporation into macromolecules. Firstly a BODIPY-diol as a novel AA-type bifunctional monomer is reported. Secondly, from the same common BODIPY precursor, a clickable, azide functionalised AB-type hetero-bifunctional monomer was prepared. Photochemical studies of model compounds confirmed the ability of these compounds to undergo photocleavage in green light (λ > 500 nm). Their usefulness for photoclippable macromolecular systems is then demonstrated: firstly by incorporating the diols into polyurethane hydrogels shown to undergo photocleavage and hence dissolution under visible light irradiation and secondly, the preparation of water-soluble macromolecular photocages able to photorelease small molecules. Thus the results presented herein describe a proof-of-principle for BODIPY-based photoresponsive materials, for example, for use as degradable polymers, sacrificial materials for lithography or for the delivery of caged pharmaceuticals.
Introduction
Selective and efficient covalent bond-breaking reactions, recently coined “clip” chemistry by Shieh and Johnson,2 has become an extremely valuable tool for the present and the future of materials chemistry. Photoclip reactions, using light as the energy source to cleave bonds, are highly attractive as they enable unparalleled control in a spatial and temporal sense. Light is a non-invasive external trigger which offers qualitative and quantitative control, and even allows for differentiation between responsive entities based on different wavelengths.3,4 Applications of such photocleavable polymers range from sequence controlled polymer synthesis5 or surface reconfiguration6 through to sacrificial materials such as positive resist photolithography7,8 and degradable polymers for controlled release and drug delivery.4,9–12A significant class of photoclip compounds include those classically termed “photocages”, defined as materials that release a molecule upon irradiation due to cleavage of a specific bond.13,14 Examples of commonly investigated photocages include o-nitrobenzyl15,16 or coumarinyl17–19 groups and metal-complexes such as ruthenium8,20 among others.4,13,14,21 The vast majority of reported chromophores are predominantly receptive to light-absorption in the ultra-violet (UV) or near UV range and consequently suffer from a low material penetration depth, as well as photodamage when used in biological tissue.4 Moving from the UV range towards visible (VIS) or near-infrared (NIR) wavelengths is therefore highly desirable.22 Different methods have been suggested to avoid UV and instead use red-shifted visible or NIR light, namely two-photon absorption (TPA), upconverting nanoparticle (UCNP)-assisted photochemistry or chemical modification of the chromophores to bathochromically shift the wavelength of light necessary for photoreactivity.21,22 Whereas TPA and UCNP-assisted photochemistry either require highly sophisticated set-ups with high-intensity laser systems or the presence of additives, certain VIS or NIR-absorbing chromophores avoid such drawbacks.22
Boron-dipyrromethene (BODIPY) compounds, in particular meso-methyl BODIPY derivatives, (Fig. 1) as developed predominantly by Weinstain,1,23–25 Winter1,25,26 and Klán,1,27 are standout photocages,13 offering excellent molar absorption coefficients (ε > 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) with absorptions in the visible to near-infrared region. They are widely regarded as biologically benign and their photoreactivity can be precisely tuned by chemical design.1 For example, via halogenation at position 2 and 6, the stability of the triplet state and hence the efficiency of the cleavage reaction can be enhanced while substituents at positions 3 and 5 can increase the conjugated system and red-shift the absorption wavelength.1,26 Owing to the broad adaptability of BODIPYs, they not only show considerable promise in polymer and materials chemistry28 but were also already employed as therapeutic agents.29 Nevertheless, studies into their use as macromolecular photocages or indeed as photoclippable polymers are scarce. Only a few studies have been conducted investigating their use as polymerization initiators30,31 and even less regarding light-induced debonding.32 Herein, we report the synthesis of novel AA-type bifunctional and AB-type hetero-bifunctional BODIPY monomers and investigate their potential implementation into macromolecular systems.
000 M−1 cm−1) with absorptions in the visible to near-infrared region. They are widely regarded as biologically benign and their photoreactivity can be precisely tuned by chemical design.1 For example, via halogenation at position 2 and 6, the stability of the triplet state and hence the efficiency of the cleavage reaction can be enhanced while substituents at positions 3 and 5 can increase the conjugated system and red-shift the absorption wavelength.1,26 Owing to the broad adaptability of BODIPYs, they not only show considerable promise in polymer and materials chemistry28 but were also already employed as therapeutic agents.29 Nevertheless, studies into their use as macromolecular photocages or indeed as photoclippable polymers are scarce. Only a few studies have been conducted investigating their use as polymerization initiators30,31 and even less regarding light-induced debonding.32 Herein, we report the synthesis of novel AA-type bifunctional and AB-type hetero-bifunctional BODIPY monomers and investigate their potential implementation into macromolecular systems.
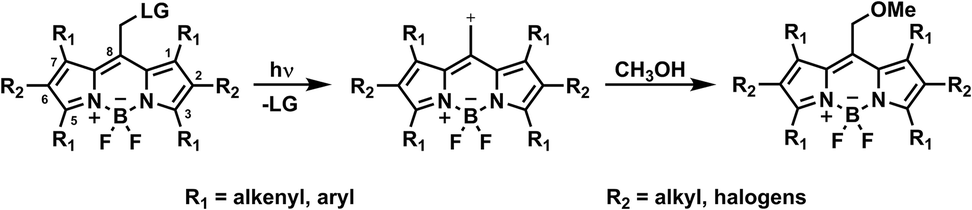 | ||
| Fig. 1 General structure of meso-methyl boron-dipyrromethene (BODIPY) derivatives and their photorelease in methanol, adapted from literature.1 | ||
Experimental
Materials and methods
Chemicals were commercially available and used as received unless otherwise stated. Boron trifluoride diethyletherate, 2-chloro-2-oxoethyl acetate (acetoxyacetyl chloride), 3-ethyl-2,4-dimethyl-1H-pyrrole (kryptopyrrole), 1,4-diazabicyclo[2.2.2]octane (DABCO), Celite® (325 Mesh powder), anhydrous tetrahydrofuran (THF) and anhydrous dichloromethane (DCM) were purchased from Alfa Aesar, acetoxyacetyl chloride and kryptopyrrole from Acros Organics and Sigma Aldrich, respectively. Phenylacetic acid, (+)-sodium-L-ascorbate, dichlorotriphenylphosphorane, 4-dimethylaminopyridine (DMAP), N,N′-dicyclohexylcarbodiimide (DCC), sodium azide, benzyl isocyanate, Jeffamine ED-2003 and poly(hexamethylene diisocyanate) were bought from Sigma Aldrich and ethanol (EtOH), ethyl acetate (EtOAc), conc. hydrochloric acid, magnesium sulfate, methanol (MeOH), N,N-dimethylformamide (DMF), n-heptane, sodium hydrogen carbonate, tetrahydrofuran (THF) and toluene from VWR. Pentyne was purchased from TCI, N-bromosuccinimide (NBS) and propargylamine from Fluorochem, copper sulfate pentahydrate and sodium hydroxide from J.T. Baker and trimethylamine and silica gel 60 (0.015–0.040 mm) from Merck. DCM was bought from Chem-Lab and chloroform-d from Eurisotop. 4,4′-Diisocyanato dicyclohexylmethane (H12-MDI, Desmodur® W/1) was provided by Covestro and Jeffamine® M-1000 was provided by Huntsman. The poly(organo)phosphazene used was synthesised in-house according to well established literature procedures33 (see ESI†). Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance III 300 MHz spectrometer in deuterated chloroform at 25 °C. IR (infra-red) spectra were measured with a PerkinElmer 100 Series FTIR spectrometer equipped with ATR using a scan number of 128. Size exclusion chromatography (SEC) in aqueous media was performed on an Agilent Technologies 1260 Infinity II system equipped with a Shodex OHpak LB-802.5 (300 mm × 8 mm, 6 μm particle size) and a Shodex OHpak LB-804 (300 mm × 8 mm, 10 μm particle size) column. Samples were filtered through 0.2 μm nylon syringe filters prior to injection, eluted with Milli-Q water with 0.1 M NaNO3 and 0.025 wt% NaN3 at a flow rate of 0.5 ml min−1 and detected via a UV-vis detector (540 nm) from Agilent, a refractive index detector RI-501 from Shodex and a TREOS II light scattering detector from Wyatt technology. Results were analysed with ASTRA 7.3.2 from Wyatt. High-resolution mass spectra were recorded on an Agilent 6520 ESI-QTOF in positive mode with methanol with 0.1% formic acid as the eluent. UV-vis and emission spectra were recorded on a SpectraMax® M2e Multi-Mode Microplate Reader with applied wavelengths of 350–780 nm and on a Jobin Yvon Fluorolog 3 emission spectrometer, depending on wavelengths and experiment. Photorheology measurements were performed on a MCR 502 Anton Paar Rheometer. The samples were investigated by oscillatory rheology, with a plate–plate geometry attached. The samples were kept at 25 °C by a temperature adjustable bottom plate. For measurements an 8 mm stamp, which was developed and provided by Anton Paar GmbH, was installed. To initiate the decomposition the light was guided from the top through the rheometer stamp onto the sample. The parameters were set to 1% deformation, a frequency of 10 rad s−1 and a gap size of 1.15 mm. As light source a Lumatec Superlite 400 portable light source with a set wavelength of 550 nm and 1.4 W was used.Absorption spectra were obtained on a UV–vis spectrometer with matched 1.0 cm quartz cuvettes. Molar absorption coefficients were determined from the absorption spectra (the average values were obtained from three independent measurements with solutions of different concentrations). The emission and excitation spectra were measured on an automated luminescence spectrometer in 1.0 cm quartz fluorescence cuvettes at 22 ± 1 °C. The corresponding optical filters were used to avoid the second harmonic excitation/emission bands induced by the grating. The samples were prepared with absorbances of ∼0.1 at the excitation wavelength. Each sample was measured five times, and the spectra were averaged. Emission and excitation spectra are normalized. Fluorescence quantum yields were determined using an integration sphere as the absolute values. For each sample, the quantum yield was measured five times and then it was averaged.
Ultrafiltration was performed with Vivaspin® 20 centrifugal concentrators with a MWCO of 3 kDa and dialysis was carried out using Spectrum™ Spectra/Por™ RC membrane tubing, EtOH used as a solvent was recycled by distillation.
Dry column vacuum chromatography was performed with silica gel 60 (0.015–0.040 mm) from Merck and EtOAc in heptane (0%–100%) as the mobile phase. The sample was absorbed on Celite® (325 mesh powder from alfa aesar) and 30 ml fractions were collected with an increasing EtOAc content of 1 ml. Thin layer chromatography was performed on pre-coated TLC sheets ALUGRAM® SIL G/UV254 with 0.20 mm silica gel 60, purchased from Macherey-Nagel.
Dry solvents have been degassed by bubbling with N2 and stored over molecular sieves (3 Å). 18 MΩ ultra pure water (Milli-Q water) was obtained from a Millipore® device with a Millipak express 40 filter (0.22 μm pore size). Dichloromethane (DCM) was purified via a solvent purification column (MBRAUN SPS compact). Water-sensitive and highly reactive chemicals were stored under an inert atmosphere in an argon filled glovebox (MBRAUN).
Suitable single crystals for X-ray diffraction for 1 and 4 were obtained by evaporation of the solvent from their solutions in n-heptane/ethyl acetate. Diffraction data were collected on a Bruker Smart Apex diffractometer operating with Mo Kα radiation (λ = 0.71073 Å). The structures were solved by direct methods (SHELXL-2014/7) and refined by full-matrix least squares on F2 (SHELXL-2014/7).34 The H atoms were calculated geometrically, and a riding model was applied in the refinement process. These data were deposited with CCDC with the following numbers: 2109136 and 2109137.† Crystal data are given in ESI Table 1.†
The conversion of the coupling reactions was determined by UV-vis spectroscopy and compound 6a as a reference. Briefly, the BODIPY concentration of the sample solution was determined via a suitable calibration curve and the corresponding conversion and content on the polymer calculated from the respective weight percent.
All BODIPY-compounds were stored in the absence of light at room temperature.
Synthetic procedures
1H-NMR (300 MHz, CDCl3, δ): 1.06 (t, 3JHH = 7.5 Hz, 6H), 2.15 (s, 3H), 2.27 (s, 6H), 2.40 (q, 3JHH = 7.5 Hz, 4H), 2.52 (s, 6H), 5.33 ppm (s, 2H); MS (ESI, positive mode) m/z calculated for C20H28BF2N2O2 377.2211 [M + H]+, found 377.2214.
1H-NMR (300 MHz, CDCl3, δ): 1.06 (t, 3JHH = 7.6 Hz, 6H), 2.40 (q, 3JHH = 7.6 Hz, 4H), 2.42 (s, 6H), 2.50 (s, 6H), 4.91 ppm (s, 2H); MS (ESI, positive mode) m/z calculated for C18H26BF2N2O [M + H]+ 335.2106, found 335.2099.
1H-NMR (300 MHz, CDCl3, δ): 1.08 (t, 3JHH = 7.5 Hz, 3H), 1.12 (t, 3JHH = 7.5 Hz, 3H), 2.43 (q, 3JHH = 7.5 Hz, 2H), 2.47 (s, 6H), 2.50 (q, 3JHH = 7.5 Hz, 2H), 2.54 (s, 3H), 4.73 (s, 2H), 4.96 ppm (s, 2H); 13C-NMR (HSQC, 75 MHz, CDCl3, δ): 12.6, 13.0, 14.5, 15.6, 17.1, 54.9, 56.1 ppm; MS (ESI, positive mode) m/z calculated for C18H25BF2N2O2Na 373.1875 [M + Na]+, found 373.1877.
1H-NMR (300 MHz, CDCl3, δ): 1.07 (t, 3JHH = 7.5 Hz, 3H), 1.13 (t, 3JHH = 7.5 Hz, 3H), 2.44 (q, 3JHH = 7.5 Hz, 2H), 2.47 (s, 6H), 2.48 (q, 3JHH = 7.6 Hz, 2H), 4.55 (s, 2H), 4.97 ppm (s, 2H); 13C-NMR(HSQC, 75 MHz, CDCl3, δ): 12.0, 12.5, 14.02, 14.5, 16.8, 44.8, 55.9 ppm; MS (ESI, positive mode) m/z calculated for C18H24BF2N5ONa 398.1940 [M + Na]+, found 398.1936.
1H-NMR (300 MHz, CDCl3, δ): 1.04 ppm (t, 3JHH = 7.6 Hz, 6H), 2.31 (s, 3H), 2.35 (s, 3H), 2.37–2.51 (m, 4H), 2.64 (s, 3H), 4.40 (s, 4H), 5.38 (s, 4H), 7.28 ppm (m, 10H); 13C-NMR (HSQC, 75 MHz, CDCl3, δ): 12.1, 13.4, 14.8, 17.1, 45.3, 57.9, 127.7 ppm.
6a: The whole reaction was carried out under argon in the dark. BODIPY-azide 4 (40.5 mg, 0.108 mmol, 1 eq.) was dissolved in 2–3 ml DCM and a mixture of copper sulfate pentahydrate (37.5 mg, 0.150 mmol, 1.5 eq.) and (+)-sodium-L-ascorbate (59.4 mg, 0.300 mmol, 3 eq.) in 2–3 ml Milli-Q water was added. After addition of pentyne (213 μl, 2.15 mmol, 20 eq.) the reaction was stirred overnight, subsequently diluted with 30 ml DCM, washed with brine and the combined aqueous phases were re-extracted with DCM. The combined organic phases were dried over MgSO4 and the solvent was evaporated. The intermediate product was purified by dry column vacuum chromatography (0%–100% EtOAc in n-heptane) to yield 6a as a shiny red solid (46.4 mg, 0.105 mmol, yield 97%).
6: To synthesise the final model compound 6, 6a (41.2 mg 0.0929 mmol, 1 eq.) was dissolved in 20 ml dry DCM and DCC (38.3 mg, 0.185 mmol, 2 eq.), DMAP (1.1 mg, 9.0 μmol, 0.1 eq.) and phenylacetic acid (18.9 mg, 0.139 mmol, 1.5 eq.) in 3 ml dry DCM were added. The reaction was stirred under argon in the dark and its progression checked via TLC. After completion (∼22 h), the solvent was evaporated and the product purified by dry column vacuum chromatography (0%–100% EtOAc in n-heptane) yielding 6 (2.6 mg, 4.6 μmol, yield 4%) as a red solid.
6a: 1H-NMR (300 MHz, CDCl3, δ): 0.76 (t, 3JHH = 7.5 Hz, 3H), 0.91 (t, 3JHH = 7.3 Hz, 3H), 1.09 (t, 3JHH = 7.6 Hz, 3H), 1.64 (q, 3JHH = 7.5 Hz, 2H), 2.42 (s, 3H), 2.43–2.48 (m, 4H), 2.49 (s, 3H), 2.58 (s, 3H), 2.6 (t, 3JHH = 7.7 Hz, 2H), 4.98 (s, 2H), 5.76 (s, 2H), 7.53 ppm (s, 1H); 13C-NMR (HSQC, 75 MHz, CDCl3, δ): 12.5, 13.0, 14.0, 16.8, 22.7, 27.8, 30.1, 44.5, 56.2, 121.5 ppm.
6: 1H-NMR (300 MHz, CDCl3, δ): 0.74 (t, 3JHH = 7.5 Hz, 3H), 0.93 (t, 3JHH = 7.5 Hz, 3H), 1.07 (t, 3JHH = 7.7 Hz, 3H), 1.59–1.71 (m, 2H), 2.09 (s, 3H), 2.16 (s, 3H), 2.34–2.49 (m, 4H), 2.59 (s, 3H), 2.64 (t, 3JHH = 7.6 Hz, 2H), 3.70 (s, 2H), 5.34 (s, 2H), 5.76 (s, 2H), 7.23–7.35 (m, 5H), 7.54 ppm (s, 1H); 13C-NMR (HSQC, 75 MHz, CDCl3, δ): 12.0, 14.0, 16.8, 22.2, 27.8, 41.2, 44.8, 58.0, 121.0, 127.3, 128.9 ppm.
1H-NMR (300 MHz, CDCl3, δ): 1.05 (t, 3JHH = 7.6 Hz, 3H), 1.10 (t, 3JHH = 7.6 Hz, 3H), 2.13 (s, 6H), 2.39 (q, 3JHH = 7.5 Hz, 2H), 2.45 (q, 3JHH = 7.5 Hz, 2H), 2.57 (s, 3H), 3.71 (s, 2H), 4.56 (s, 2H), 5.33 (s, 2H), 7.27–7.36 ppm (m, 5H); 13C-NMR (HSQC, 75 MHz, CDCl3, δ): 12.6, 13.9, 15.0, 16.6, 40.9, 45.1, 58.2, 127.2, 128.7 ppm.
:water (1:1) and subsequently dialysed (MWCO = 6–8 kDa) against ethanol. The solvent was evaporated, the product isolated as a red solid and the BODIPY content determined by UV-vis spectroscopy using compound 6a as reference. Conversion = 31%. BODIPY content = 9.6 wt%.
:water (1:1) and subsequently dialysed (MWCO = 6–8 kDa) against ethanol. After evaporation of the solvent the polymer was isolated as a red solid. The BODIPY content was determined by UV-vis spectroscopy with compound 6a as reference. Conversion = 33%, BODIPY content = 9.9 wt%.
Results
For the preparation of photoresponsive macromolecular systems based on boron dipyrromethene (BODIPY), we designed a readily accessible, modifiable and stable BODIPY platform. For this reason, compound 1 was chosen because it is relatively easily synthetically accessible,35–37 offers multiple options for modification of the framework1,38 and enables the release of caged compounds upon irradiation with green-light.23–25 After the reported synthesis of the BODIPY framework 1, hydrolysis of the ester in the meso-position resulted in the common precursor 2, which could be used for subsequent synthesis of both the AA-type bifunctional and AB-type heterobifunctional BODIPY monomers, as summarised in Scheme 1. Building on the method by Ulrich et al.,38mono-bromination in position 3 was performed, followed by substitution with a suitable nucleophile, H2O or NaN3, to give compounds 3 and 4, respectively. The prepared monomers 3 and 4 were extensively characterised by NMR-spectroscopy (1H and HSQC), UV-Vis spectroscopy and MS, as well as FTIR-spectroscopy in the case of compound 4. In addition, the molecular structures of 1 and 4 were determined by single-crystal X-ray diffraction (Scheme 1). A clear and characteristic shift in the 1H-NMR signal can be observed upon substitution at position 3 from 2.52 ppm in compound 2 to 4.71 and 4.55 ppm for 3 and 4, respectively (Fig. 3, complete spectra in ESI Fig. 2, 3 and 5†). The peak at ∼4.9 ppm, corresponding to the methylene group at the meso-position, shifts slightly downfield for compound 3, and somewhat more for 4. A splitting of the triplet and quartet of the ethyl-moiety was also observed due to the lost symmetry for both the AA-type and AB-type monomers 3 and 4 (ESI Fig. 3 and 5†). Chemical functionalization of this position does not appear to considerably change the electronic structure of the BODIPY core as all UV-vis spectra of the monomers are very similar with only slight shifts in the absorbance wavelengths (cf. ESI Fig. 19–22†). Both compounds 3 and 4 were further investigated by ESI-MS in positive mode indicating a quite labile molecule with multiple fragmentations despite the soft ionisation method. Nevertheless, the molecular ion peak or its sodium adducts are observable for each monomer (ESI Fig. 17 and 18†). For compound 4, a sharp and characteristic peak for the azide group at 2100 cm−1 can be found in the FTIR-spectrum (ESI Fig. 37†).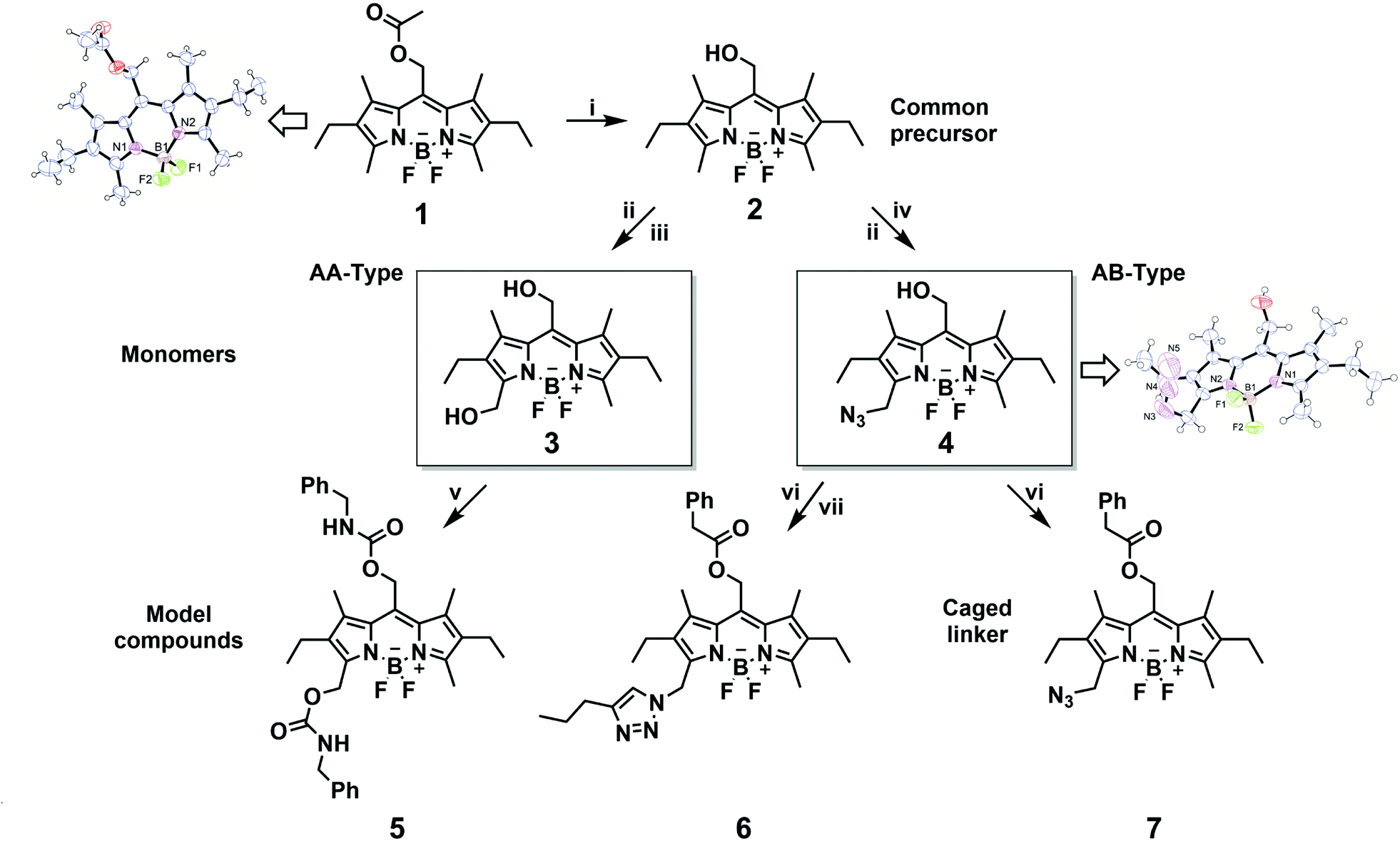 | ||
| Scheme 1 Synthesis pathway towards bifunctional and hetero-bifunctional BODIPY monomers 3 and 4 and the synthesis of their respective small-molecular model compounds 5–7. (i) NaOH, DCM/MeOH, H2O; (ii) NBS, DCM; (iii) H2O/DCM; (iv) NaN3, DCM/DMF; (v) benzyl isocyanate, toluene; (vi) phenylacetic acid, DCC, DMAP, DCM; (vii) pentyne, CuSO4·5H2O, (+)-L-Na ascorbate, DCM/H2O. | ||
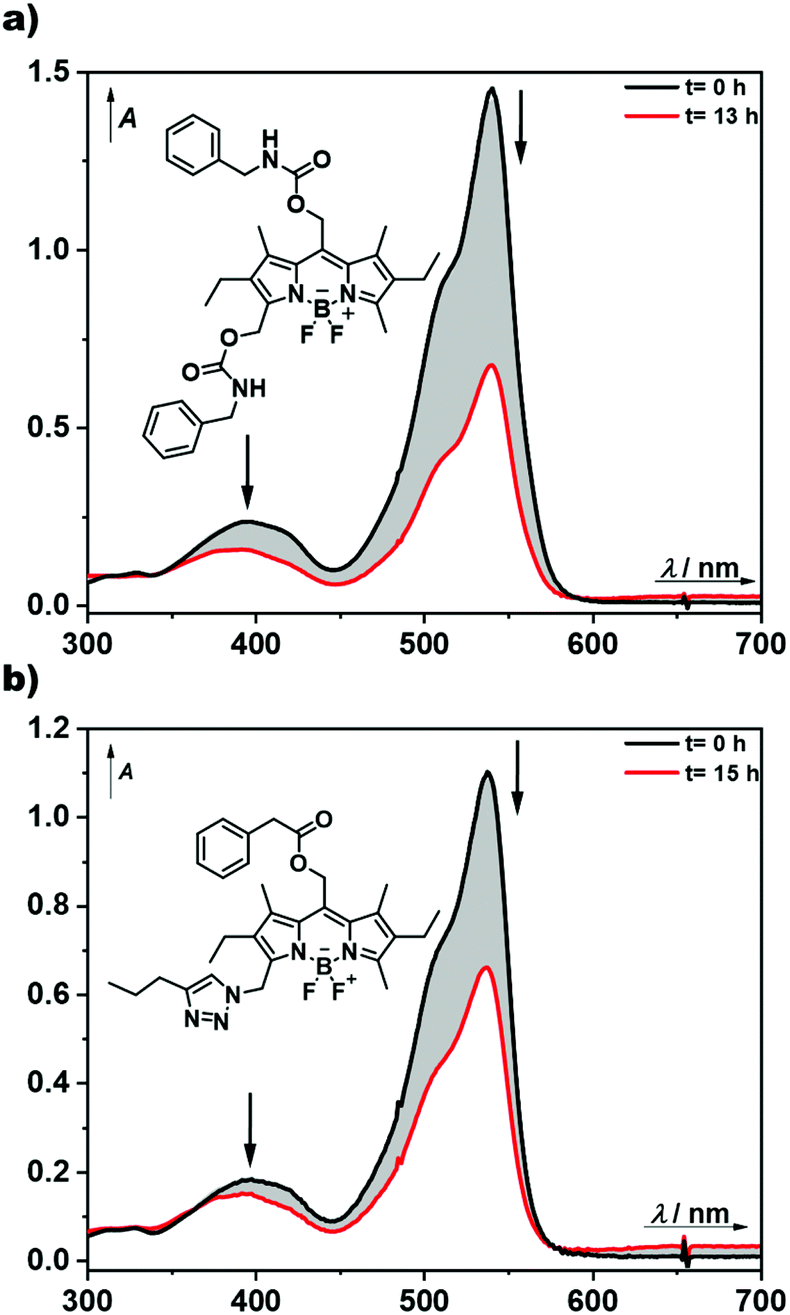 | ||
| Fig. 2 Evolution of UV-vis spectra during the photoreaction of BODIPY-model compounds 5 (a) and 6 (b) in aerated methanol solutions (5.3 × 10−5 M and 4 × 10−5 M, respectively) upon 505 nm LED irradiation for the indicated time. | ||
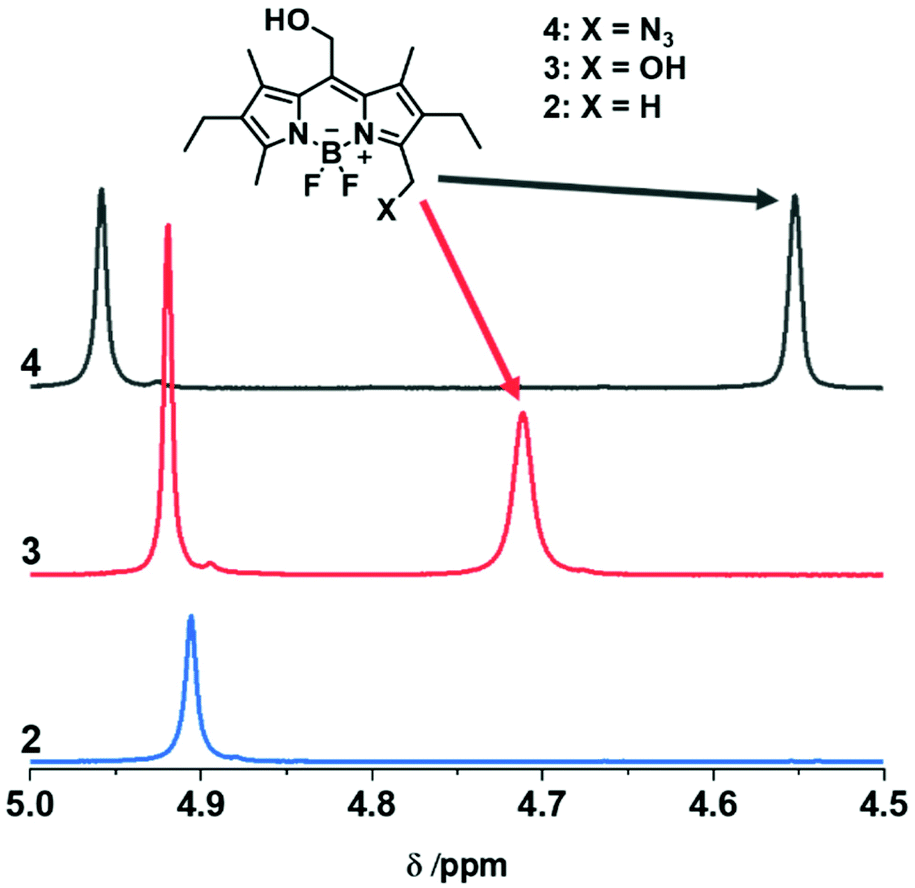 | ||
| Fig. 3 1H-NMR spectra in CDCl3 of the hydrolysed BODIPY compound 2 (blue), the BODIPY-diol compound 3 (red) and the BODIPY-azide compound 4 (black) showing the proton signals arising upon the substitution at position 3 at 4.71 ppm and 4.55 ppm for the diol and the azide, respectively. | ||
To obtain photophysical and photochemical data of the BODIPY chromophores, small-molecular models were then synthesised. For the bifunctional diol 3, isocyanate chemistry will later be used for the synthesis of hydrogels (vide infra). Therefore, the small-molecule model 5 was synthesised by reaction of both alcohol moieties in 3 with an excess of benzyl isocyanate (Scheme 1). The successful synthesis of the model compound 5 was confirmed by NMR and UV-Vis spectroscopy (ESI Fig. 7, 8 and 23†) and its photophysical properties were investigated (Fig. 2a, ESI Fig. 25a†).
To mimic the subsequent polymer-bound heterobifunctional compound 4, model compound 6 was synthesised with a cleavable ester group in the meso-position. To this end a copper-catalysed azide–alkyne cycloaddition (CuAAC) with pentyne was performed alongside a standard coupling reaction at the meso-alcohol with phenylacetic acid to act as a leaving group. The model compound 6 was characterized by NMR and FTIR-spectroscopy (ESI Fig. 9–12, 24 and 38†) and thoroughly photophysically investigated (Fig. 2b, ESI Fig. 25b†).
Compound 5 in methanol shows an absorption band at λmax = 541 nm with a shoulder at ∼500 nm, typical for BODIPY chromophores, and an emission band at λmax = 559 nm (the quantum yield of fluorescence ΦF is 0.47; Table 1, ESI Fig. 23†). The absorption and emission properties of 6 are very similar to those of 5 (Table 1, ESI Fig. 24†).
a
| Compd. | Absorptiona | Emissionb | Photoreactionc | |||
|---|---|---|---|---|---|---|
| λ absmax /nm | ε/M−1 cm−1 | λ emmax/nm | Φ f | Yieldd/% |
Φ
re |
|
| a Absorption, b emission (Φf = fluorescence quantum yield in methanol), and c photochemical properties (d chemical yields of release and e degradation quantum yields Φr in f aerated and g degassed methanol solutions; λirr = 505 nm). | ||||||
| 5 | 541 | 35700 |
559 | 0.47 ± 0.03 | 100 ± 10f | 1.0 × 10−4f |
| 124 ± 12g | 1.8 × 10−4g |
|||||
| 6 | 538 | 42100 |
559 | 0.68 ± 0.05 | 65 ± 3f | 4.6 × 10−5f |
| 68 ± 4g | 7.8 × 10−5g |
|||||
BODIPY 5 bears two carbamate substituents, one directly photocleavable from the meso-methyl position,1,13,23,39 and the second one at the 3-methyl position. Upon photolysis, the first anticipated step is the release of benzylcarbamate from the meso-methyl group, which slowly decarboxylates in dark40–42 to give the final benzylamine. The side-product of this transformation is a meso-methoxymethyl BODIPY derivative, formed by photosolvolysis of 5 with methanol, which is also photochemically active as it is composed of the same BODIPY chromophore.1 Upon exhaustive irradiation of 5 in an aerated methanol solution at 505 nm, the chemical yield (HPLC) of the released final leaving group benzylamine was found to be very high (100%). Interestingly, prolonged irradiation of a degassed solution of 5 gave more than one equivalent of benzylamine (124%). We conclude that the second carbamate group at position 3 was also released, most probably in a subsequent step, that is photodegradation of the meso-methylmethoxy BODIPY side-product. Indeed, the absorption spectra of irradiated samples in Fig. 2a demonstrate that the BODIPY chromophore is substantially consumed during the photolysis, further enhancing the degradation process. Relatively smaller release chemical yields of benzylamine in the presence of oxygen must be related to an alternative secondary degradation oxidative pathway, such as the oxidation of benzylamine by singlet oxygen43 generated by the triplet sensitization of an excited BODIPY chromophore.1 The degradation quantum yields Φr in degassed solutions were found to be larger than those in aerated solutions by a factor of 2, which means that oxygen quenches the productive triplet excited state of BODIPY. Their magnitudes match those found for analogous meso-methyl BODIPY protected carboxylates.
The photolysis of 6, which does not possess a good leaving group at position 3, under the same conditions (Fig. 2b) provided only 65 and 68% maximum chemical yields of the phenylacetate leaving group from its meso-methyl position in aerated and degassed solutions, respectively (Table 1). The chemical yield of ∼70% can thus be an upper limit of the carboxylate leaving group release from the meso-position of such pentaalkyl BODIPY derivatives. In addition, the stability of the two model compounds in the dark was tested in methanol as a model protic solvent and showed completely stable chromophores within at least 24 h (ESI Fig. 31†).
Subsequently, the synthesised BODIPY monomers were incorporated into macromolecular materials. The AA-type monomer 3 was employed as a photocleavable linker in hydrogels. The monomer was first reacted with H12-MDI in excess to prepare a diisocyanate BODIPY compound in situ. Afterwards, Jeffamine ED-2003, a hydrophilic diamine spacer, and poly(hexamethylene diisocyanate) as a crosslinker were added, and the mixture stirred vigorously until formation of the gel (Fig. 5a and b). The spacer was chosen to ensure a certain distance between the separate BODIPY molecules to reduce any quenching effects between chromophores. As can be seen in Fig. 5c, a light purple colored hydrogel was obtained, glowing bright green upon irradiation with 365 nm light. When subjected to prolonged irradiation at 365 nm for 72 h, the gel dissolved completely due to photocleavage of the BODIPY linkages. In contrast, a control gel without any BODIPY monomer (see ESI† for details) remained intact upon irradiation for the same timeframe (see Fig. 5c and d). The kinetics of the degradation process was followed by fluorescence spectroscopy and photorheology. The hydrogel was directly irradiated in the fluorescence spectrometer using the excitation light of the instrument at wavelengths of 365 and 510 nm. At regular time intervals, fluorescence spectra were recorded. A decrease in fluorescence intensity can be observed after 2 h, by 9% (@365 nm, ESI Fig. 32†) and by 23% (@510 nm, Fig. 4a). Notably, the kinetics of the degradation process at the different wavelengths are in accordance with the respective molar absorption coefficient of the BODIPY monomer. Additionally, the fluorescence upon irradiation at 510 nm was plotted against time (ESI Fig. 33†) showing a exponential fit (R2 = 0.99844) of the degradation process, slowing down over time, as already established for the model compound (ESI Fig. 29†). For photorheology, the hydrogel was irradiated at 550 nm as determined by instrument set-up. Measurements of the hydrogel showed a clear decrease in the storage modulus G′ and the loss modulus G′′ after approximately 135 min irradiation at 550 nm upon which the gel was considerably degraded (Fig. 4b). These measurements clearly show that a degradation of the gel upon irradiation is possible with green-light. A control measurement of the BODIPY-hydrogel in the dark showed no decrease in modulus over the same time period.
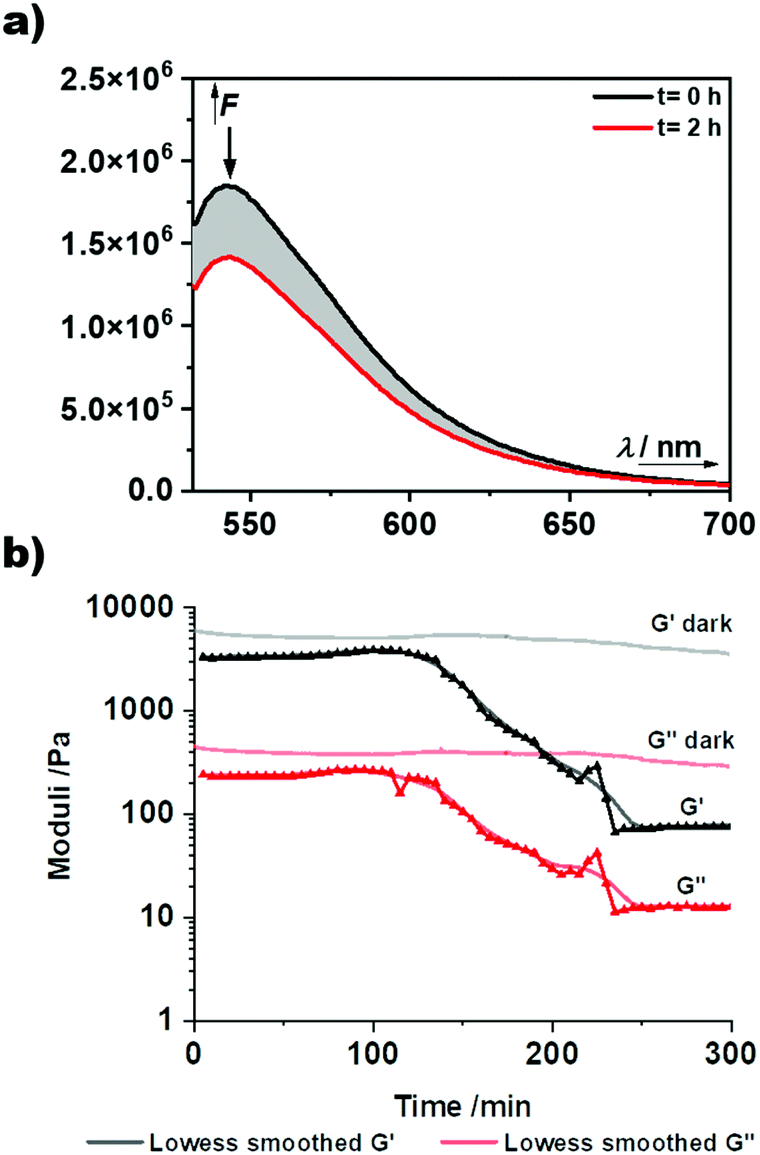 | ||
| Fig. 4 Kinetic studies of the photodegradation of the BODIPY-diol containing hydrogel. (a) Decrease of the fluorescence intensity upon irradiation of the BODIPY-hydrogel at 510 nm for 2 h. (b) Photorheology measurements of the BODIPY-hydrogel and the stability of the gel in the dark. Light on after 2 min. | ||
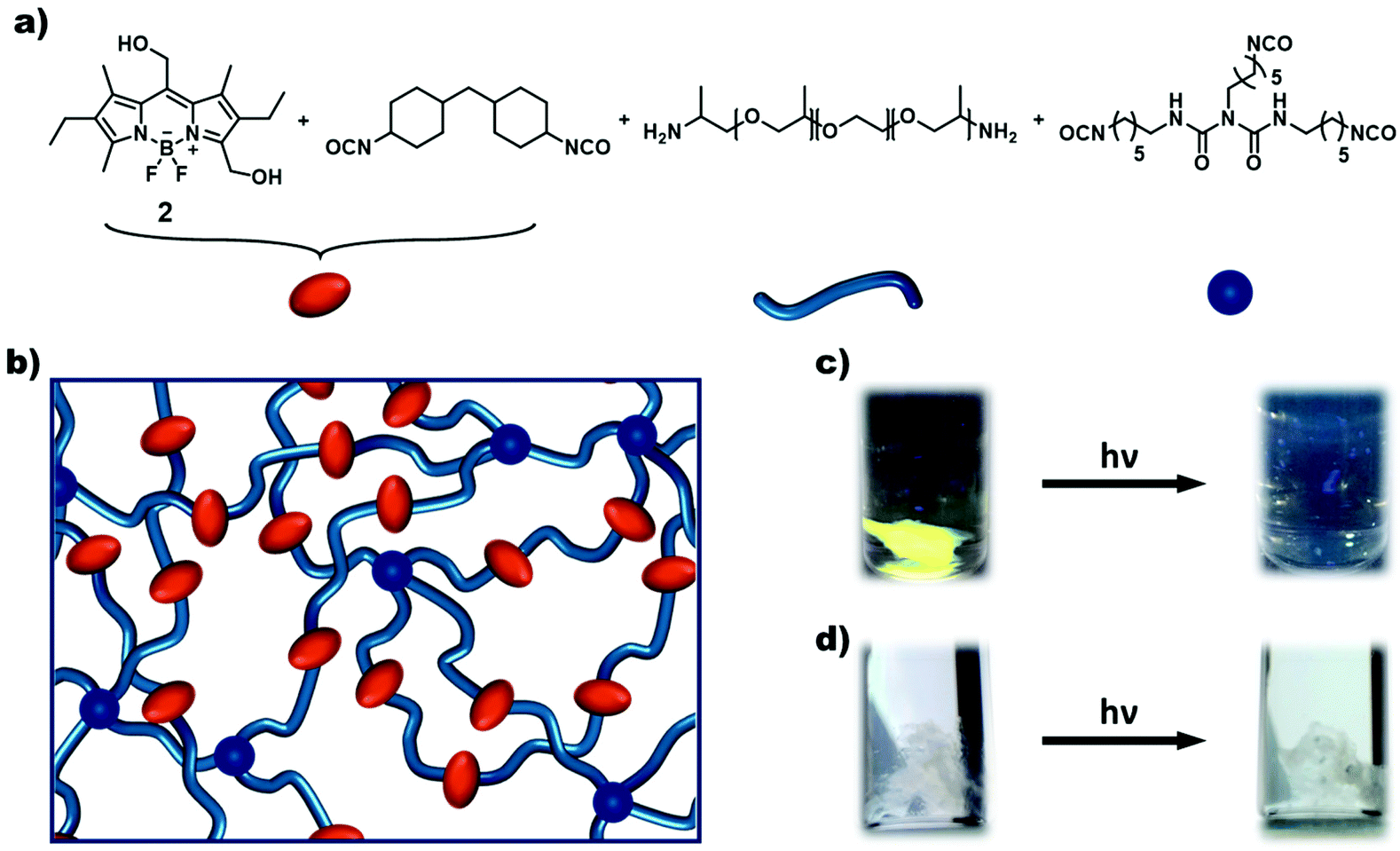 | ||
| Fig. 5 Schematic representation and photograph of the synthesised BODIPY-hydrogels. (a) Constituents of the synthesised BODIPY-hydrogel and their cartoon representation. (b) Cartoon representation of a BODIPY-hydrogel. (c) Photograph of the fluorescent BODIPY-hydrogel and the dissolution of the gel after prolonged irradiation (λexc = 365 nm). (d) Photograph of a control sample (polyurethane hydrogel without incorporated BODIPY). No fluorescence and dissolution after prolonged irradiation can be observed. | ||
The AB-type BODIPY monomer 4 was employed as a photo-labile linker for potential degradable polymeric photocages. As a first proof of principle compound 4 was coupled to a water-soluble poly(organo)phosphazene bottlebrush polymer bearing propargylamine groups (approx. one alkyne per repeat unit) accessible for the CuAAC cycloaddition (Fig. 6a). The photostability of the polymer carrier was tested via aqueous SEC measurements before and after irradiation of the sample solution at 550 nm over night (ESI Fig. 36†). Successful loading onto the polymer was demonstrated by aqueous SEC (ESI Fig. 36†). The efficiency of the coupling reaction was determined to be 31% by UV-Vis spectroscopy. Following these initial results the BODIPY-azide 7 decorated with coupled phenylacetic acid was added to the polymer. As depicted in Fig. 6b, a clear overlap of the refractive index (RI) and UV detector traces indicates a successful loading of the BODIPY onto the poly(organo)phosphazene. The incorporation of the BODIPY moiety was again determined by UV-Vis spectroscopy and was comparable to the previous experiment at 33% with a BODIPY content of around 10 wt%. Unfortunately, the solubility of the polymer was limited after loading with the BODIPY compound possibly due to cross-linking by π–π-stacking interactions. However, after acidification of the solution (resulting concentration: 0.6 M HCl) the polymer becomes completely soluble. Continuous irradiation of the poly(organo)phosphazene at 505 nm leads to a decrease of the absorbance (Fig. 7), mirroring the behavior of the small-molecular model compound 6, indicating a stimulus controlled release of the conjugated phenylacetic acid. As for the model compound, a slowing down of the degradation process can be observed, fitted exponentially (ESI Fig. 30 and 34†).
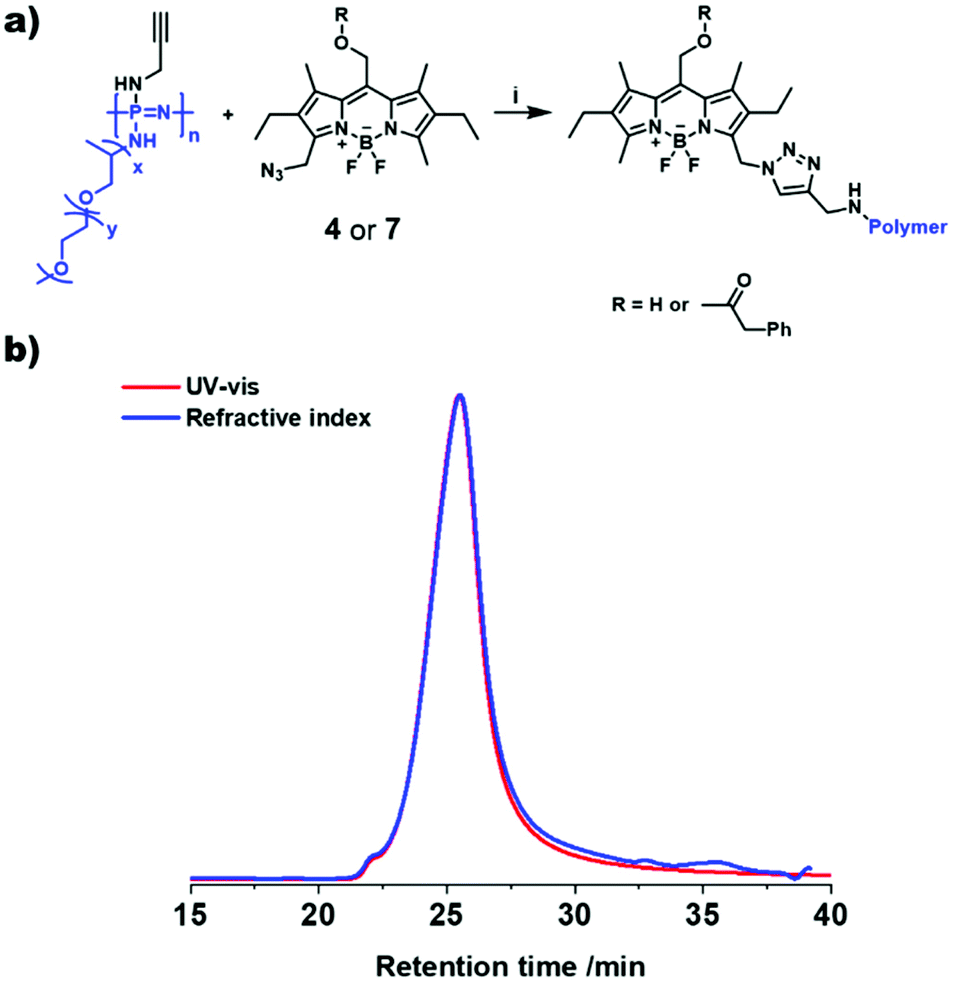 | ||
| Fig. 6 Loading of the BODIPY 4 or 7 onto the poly(organo)phosphazene. (a) Scheme of the reaction of BODIPY 4 or 7 with poly(organo)phosphazene. (b) SEC-traces of the UV–vis (540 nm) and the refractive index detectors, showing an overlap of the signals and indicating a successful loading of the BODIPY compound 7 onto the poly(organo)phosphazene carrier. (i) CuSO4·5H2O, (+)-L-Na ascorbate, DCM/H2O. | ||
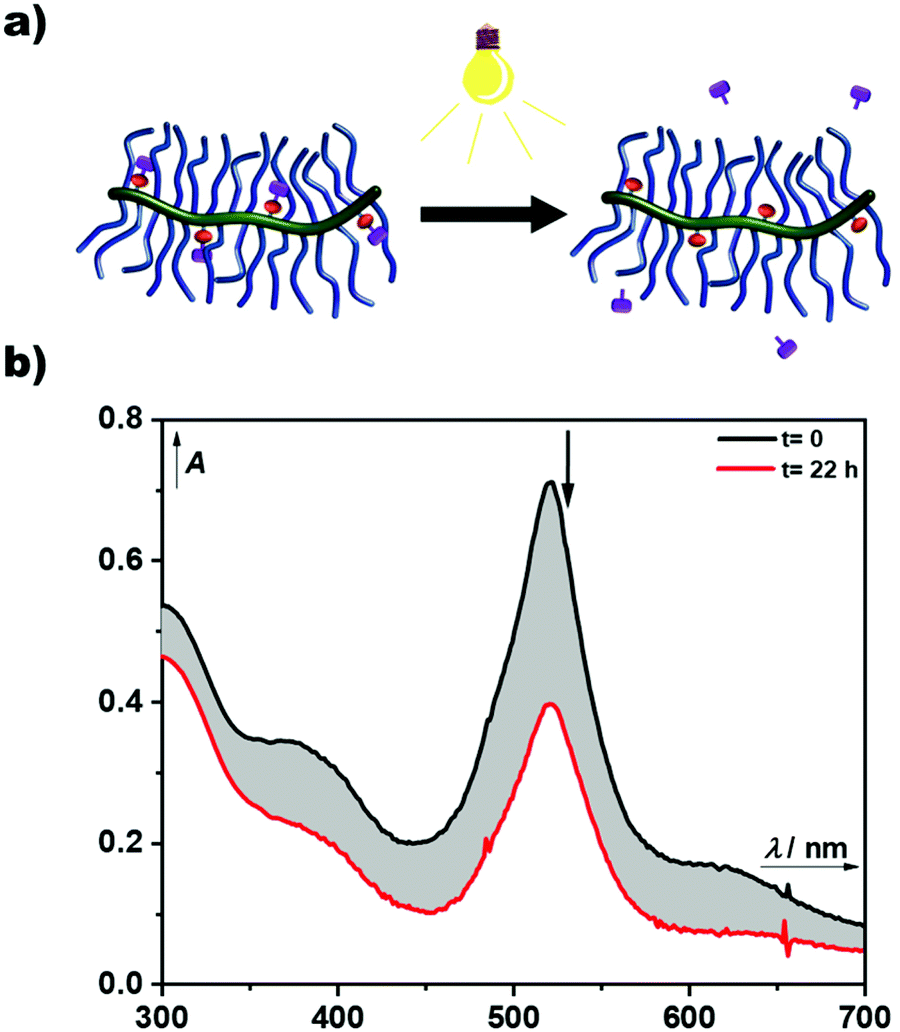 | ||
| Fig. 7 Photoreaction of the BODIPY decorated poly(organo)phosphazene. (a) Cartoon visualising the light-stimulus controlled release of conjugated phenylacetic acid form the BODIPY decorated polymer carrier. (b) Kinetic study of the photodegradation of the BODIPY decorated poly(organo)phosphazene in MeOH upon 505 nm LED irradiation for the indicated time. | ||
Conclusions
We described the synthesis of novel bifunctional AA- and heterobifunctional AB-type BODIPY monomers via an easily accessible common precursor. Suitable small-molecular model compounds were established and thoroughly characterised, showing their capability to undergo photocleavage in response to irradiation in the visible-light region. The BODIPY monomers were incorporated into macromolecular systems, namely hydrogels and a bottle-brush polyphosphazene, and demonstrated to undergo selective covalent-bond cleavage in response to green light. Although the response kinetics of both BODIPY monomers is relatively slow, rate enhancement is feasible in future work with relatively minor adjustments to the BODIPY framework.1 Thus the herein presented results offer a proof-of-principle for the design of photo-clippable meso-methyl BODIPY monomers in macromolecular systems and a solid foundation for future visible-light cleavable materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from OEAD (Scientific & Technological Cooperation Austria/Czech Republic, Project No. CZ 20/2019: “Photocleavable metallopolymer nanogels”). NMR spectrometers were acquired in collaboration with the University of South Bohemia (CZ) with financial support from the European Union through the EFRE INTERREG IV ETC-AT-CZ program (project M00146, “RERI-uasb”). Support for this work was also provided by the Czech Science Foundation (GA21-01799S), and we thank the CETOCOEN EXCELLENCE Teaming 2 project (supported by the Czech Ministry of Education, Youth and Sports: CZ.02.1.01/0.0/0.0/17_043/0009632) and the RECETOX research infrastructure (LM2018121). The authors want to thank Martin Ertl for technical support with the fluorescence spectrometer and Milan Kracalik for supporting the photorheology measurements. P. Strasser is the recipient of a DOC Fellowship of the Austrian Academy of Sciences at the Institute of Polymer Chemistry.Notes and references
- T. Slanina, P. Shrestha, E. Palao, D. Kand, J. A. Peterson, A. S. Dutton, N. Rubinstein, R. Weinstain, A. H. Winter and P. Klán, J. Am. Chem. Soc., 2017, 139, 15168–15175 CrossRef CAS PubMed.
- P. Shieh, M. R. Hill, W. Zhang, S. L. Kristufek and J. A. Johnson, Chem. Rev., 2021, 121, 7059–7121 CrossRef CAS PubMed.
- M. J. Hansen, W. A. Velema, M. M. Lerch, W. Szymanski and B. L. Feringa, Chem. Soc. Rev., 2015, 44(11), 3358–3377 RSC.
- P. Xiao, J. Zhang, J. Zhao and M. H. Stenzel, Prog. Polym. Sci., 2017, 74, 1–33 CrossRef CAS.
- F. A. Leibfarth, K. M. Mattson, B. P. Fors, H. A. Collins and C. J. Hawker, Angew. Chem., Int. Ed., 2013, 52, 199–210 CrossRef CAS PubMed.
- C. Xie, W. Sun, H. Lu, A. Kretzschmann, J. Liu, M. Wagner, H.-J. Butt, X. Deng and S. Wu, Nat. Commun., 2018, 9, 3842 CrossRef PubMed.
- M. Lunzer, L. Shi, O. G. Andriotis, P. Gruber, M. Markovic, P. J. Thurner, D. Ossipov, R. Liska and A. Ovsianikov, Angew. Chem., Int. Ed., 2018, 57, 15122–15127 CrossRef CAS PubMed.
- S. Theis, A. Iturmendi, C. Gorsche, M. Orthofer, M. Lunzer, S. Baudis, A. Ovsianikov, R. Liska, U. Monkowius and I. Teasdale, Angew. Chem., Int. Ed., 2017, 56, 15857–15860 CrossRef CAS PubMed.
- G. Pasparakis, T. Manouras, P. Argitis and M. Vamvakaki, Macromol. Rapid Commun., 2012, 33, 183–198 CrossRef CAS PubMed.
- L. Ma, R. Baumgartner, Y. Zhang, Z. Song, K. Cai and J. Cheng, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1161–1168 CrossRef CAS.
- X. Wang, C. Wang, Q. Zhang and Y. Cheng, Chem. Commun., 2016, 52, 978–981 RSC.
- H. Okada, K. Tanaka, W. Ohashi and Y. Chujo, Bioorg. Med. Chem., 2014, 22, 3435–3440 CrossRef CAS PubMed.
- R. Weinstain, T. Slanina, D. Kand and P. Klán, Chem. Rev., 2020, 120(24), 13135–13272 CrossRef CAS PubMed.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- D. Kehrloesser, P. J. Behrendt and N. Hampp, J. Photochem. Photobiol., A, 2012, 248, 8–14 CrossRef CAS.
- H. Zhao, B. Hou, Y. Tang, W. Hu, C. Yin, Y. Ji, X. Lu, Q. Fan and W. Huang, Polym. Chem., 2016, 7, 3117–3125 RSC.
- Z. Gao, P. Yuan, D. Wang, Z. Xu, Z. Li and X. Shao, Bioorg. Med. Chem. Lett., 2017, 27, 2528–2535 CrossRef CAS PubMed.
- Q. Lin, C. Bao, Y. Yang, Q. Liang, D. Zhang, S. Cheng and L. Zhu, Adv. Mater., 2013, 25, 1981–1986 CrossRef CAS PubMed.
- S. Kumar, J. F. Allard, D. Morris, Y. L. Dory, M. Lepage and Y. Zhao, J. Mater. Chem., 2012, 22, 7252–7257 RSC.
- I. Teasdale, S. Theis, A. Iturmendi, M. Strobel, S. Hild, J. Jacak, P. Mayrhofer and U. Monkowius, Chem. – Eur. J., 2019, 25, 9851–9855 CrossRef CAS PubMed.
- X. Zeng, X. Zhou and S. Wu, Macromol. Rapid Commun., 2018, 39(14), 1800034 CrossRef PubMed.
- J. T. Offenloch, M. Gernhardt, J. P. Blinco, H. Frisch, H. Mutlu and C. Barner-Kowollik, Chem. – Eur. J., 2019, 25, 3700–3709 CrossRef CAS PubMed.
- N. Rubinstein, P. Liu, E. W. Miller and R. Weinstain, Chem. Commun., 2015, 51, 6369–6372 RSC.
- D. Kand, L. Pizarro, I. Angel, A. Avni, D. Friedmann-Morvinski and R. Weinstain, Angew. Chem., Int. Ed., 2019, 58, 4659–4663 CrossRef CAS PubMed.
- D. Kand, P. Liu, M. X. Navarro, L. J. Fischer, L. Rousso-Noori, D. Friedmann-Morvinski, A. H. Winter, E. W. Miller and R. Weinstain, J. Am. Chem. Soc., 2020, 142, 4970–4974 CrossRef CAS PubMed.
- J. A. Peterson, C. Wijesooriya, E. J. Gehrmann, K. M. Mahoney, P. P. Goswami, T. R. Albright, A. Syed, A. S. Dutton, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2018, 140, 7343–7346 CrossRef CAS PubMed.
- E. Palao, T. Slanina, L. Muchová, T. Šolomek, L. Vítek and P. Klán, J. Am. Chem. Soc., 2016, 138, 126–133 CrossRef CAS PubMed.
- P. Lu, K.-Y. Chung, A. Stafford, M. Kiker, K. Kafle and Z. A. Page, Polym. Chem., 2021, 12, 327–348 RSC.
- M. Liu, J. Meng, W. Bao, S. Liu, W. Wei, G. Ma and Z. Tian, ACS Appl. Bio Mater., 2019, 2, 3068–3076 CrossRef CAS.
- E. Faggi, J. Aguilera, R. Sáez, F. Pujol, J. Marquet, J. Hernando and R. M. Sebastián, Macromolecules, 2019, 52, 2329–2339 CrossRef CAS.
- M. Li, A. P. Dove and V. X. Truong, Angew. Chem., Int. Ed., 2020, 59, 2284–2288 CrossRef CAS PubMed.
- N. G. Patil, N. B. Basutkar and A. V. Ambade, Chem. Commun., 2015, 51, 17708–17711 RSC.
- M. Kneidinger, A. Iturmendi, C. Ulbricht, T. Truglas, H. Groiss, I. Teasdale and Y. Salinas, Macromol. Rapid Commun., 2019, 40, 1900328 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- K. Sitkowska, B. L. Feringa and W. Szymański, J. Org. Chem., 2018, 83, 1819–1827 CrossRef CAS PubMed.
- K. Krumova and G. Cosa, J. Am. Chem. Soc., 2010, 132, 17560–17569 CrossRef CAS PubMed.
- M. H. R. Beh, K. I. B. Douglas, K. T. E. House, A. C. Murphy, J. S. T. Sinclair and A. Thompson, Org. Biomol. Chem., 2016, 14, 11473–11479 RSC.
- G. Ulrich, R. Ziessel and A. Haefele, J. Org. Chem., 2012, 77, 4298–4311 CrossRef CAS PubMed.
- P. P. Goswami, A. Syed, C. L. Beck, T. R. Albright, K. M. Mahoney, R. Unash, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2015, 137, 3783–3786 CrossRef CAS PubMed.
- G. Papageorgiou and J. E. T. Corrie, Tetrahedron, 1997, 53, 3917–3932 CrossRef CAS.
- L. Kammari, L. Plíštil, J. Wirz and P. Klán, Photochem. Photobiol. Sci., 2007, 6, 50–56 CrossRef CAS PubMed.
- I. Lee, C. K. Kim and B. C. Lee, J. Comput. Chem., 1987, 8, 794–800 CrossRef CAS.
- D. B. Ushakov, M. B. Plutschack, K. Gilmore and P. H. Seeberger, Chem. – Eur. J., 2015, 21, 6528–6534 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2109136 and 2109137. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1py01245b |
| This journal is © The Royal Society of Chemistry 2021 |