
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoengineering with RAFT polymers: from nanocomposite design to applications
Wentao
Peng
 ,
Yingying
Cai
,
Luise
Fanslau
and
Philipp
Vana
*
,
Yingying
Cai
,
Luise
Fanslau
and
Philipp
Vana
*
Institut für Physikalische Chemie, Georg-August-Universität Göttingen, Tammannstrasse 6, 37077 Göttingen, Germany. E-mail: pvana@uni-goettingen.de
First published on 20th October 2021
Abstract
Reversible addition–fragmentation chain-transfer (RAFT) polymerization is a powerful tool for the precise formation of macromolecular building blocks that can be used for the construction of well-defined nanocomposites. Especially when combining RAFT polymers with uniform inorganic/metallic nanoparticles, a vast variety of complex nanocomposites become available that may find applications especially in the field of life sciences, e.g., bio-imaging, drug delivery or cancer-therapy, but also in areas such as energy conversion or catalysis. RAFT polymerization not only provides the possibility to control the size and the macromolecular architecture of the polymer building blocks, but inherently delivers highly functional end-groups that can often directly be employed as linker-sites for installing the polymeric components into the final nanocomposites. This review describes recent advances in this vivid field and concentrates on innovations in the fabrication method and design strategies for polymer/inorganic nanohybrids. The methodology of synthesizing RAFT polymer with the aim of surface functionalization and the design options for anchoring RAFT polymer on surfaces are covered. A series of core–shell nanostructures with the focus on novel functionalities brought by RAFT polymer brushes will be reviewed with focus on the detailed macromolecular design for each specific application's scenario. Examples of ordered nano-assemblies using RAFT polymer linkers will be reviewed in order to demonstrate the advantages of RAFT polymer for introducing different morphologies, interactions and chirality to the final functional nanostructures.
 Wentao Peng | Wentao Peng received his B. Sc. in Chemistry from the University of Göttingen in Germany in 2013, and obtained his Ph. D. in Chemistry under the supervision of Prof. Philipp Vana in 2020. He is currently a Postdoctoral Associate in Prof. Vanás group with a focus on the self-assembly of nanomaterials (metal and silica-based nanoparticles) using functional macromolecular linkers. |
 Yingying Cai | Yingying Cai received her B. Sc. in Chemistry in 2015 before completing her M. Sc. in 2017 from the University of Göttingen. Currently, she is a doctoral student in the group of Prof. Philipp Vana at the University of Göttingen. Her research interest focuses on developing new polymer-inorganic nanodevices for photon luminary application using silica coating and surface-initiated RAFT polymerization. She is also developing new approaches for colloidal self-assembly of silica and noble metal nanoparticles using functional polymer. |
 Luise Fanslau | Luise Fanslau received her B. Sc. degree in Molecular Biotechnology from Heidelberg University, Germany, where she was working on photoswitchable DNA in the group of Prof. Dr Andres Jäschke. After working one year at GlaxoSmithKline Biologicals in Dresden, Germany, she pursued an MPhil Biotechnology degree at the University of Cambridge, UK, researching on novel DNA vaccine delivery nanoparticles in the group of Dr Ljiljana Fruk and graduated with distinction. Currently, she is a doctoral student in the group of Prof. Philipp Vana at the University of Göttingen, working on RAFT polymer nanocomposites. She receives a Kekulé scholarship of the Fonds der Chemischen Industrie. |
 Philipp Vana | Philipp Vana studied Chemistry at the University of Vienna, Austria, where he obtained a Master of Natural Sciences in 1996 and a Doctor of Natural Sciences in 1999 after investigating chain-length dependent termination kinetics in radical polymerisation in Professor Oskar-Friedrich Olaj's group. Parallel to his PhD, he studied economics and law and obtained a diploma from the Technical University Vienna and a Master of Business Administration from the Danube-University Krems, Austria. Between 2001 and 2003 he was a Schrödinger-Fellow of the Austrian Science Fund at the Centre for Advanced Macromolecular Design (CAMD) at the University of New South Wales, Australia, where he work in the group of Professor Tom Davis in the field of RAFT polymerisation kinetics. In 2003, he established an independent research group at the University of Göttingen, Germany, specifically focusing on macromolecular design and functional polymer materials. In 2005 he became a Fellow of the Japan Society for the Promotion of Science at Kyoto University in Professor Takeshi Fukudas group, where he moved into the field of polymer brushes and surface modifications. In 2008 he was granted the prestigious Heisenberg-Professorship of the German Research Foundation (DFG) at the University of Göttingen, where he finally became a Full Professor for Macromolecular Chemistry in 2010 after declining offers from Leipzig University and Duisburg-Essen University. Philipp has published more than 140 original research papers in addition to several book chapters, reviews and patents. |
1. Introduction
Since its debut in 1998,1 reversible addition–fragmentation chain-transfer (RAFT) polymerization has quickly become a fantastic tool for designing and fabricating polymers with special functions. The precision engineering using RAFT polymerization includes well-defined polymer size,2 versatile end-group design,3,4 convenient access for block copolymer (BCP) synthesis,5–7 and flexible topological design.8–12 These options can be combined simultaneously to give RAFT polymers the ability for a wide range of applications including e.g., stimuli-responsiveness,9,13–15 therapeutic-delivery,16–20 bioimaging and diagnosis,17,21,22 optoelectronics,23 polymerization induced self-assembly,24–27 interface engineering,28 smart nanoreactors,29 structured templating,30 bioseparation,31 and photo-curing 3D printing.32,33The rise of RAFT polymers has revolutionarily changed the game of surface engineering of nanoparticles (NPs). A great variety of anchoring strategies was developed to immobilize RAFT polymer brushes onto a plethora of nanosurfaces.28,34 The polymer-functionalized inorganic nanomaterials carrying a combined talent from both functional polymers and inorganic nanomaterials have received extensive product proliferation to encompass an extraordinary array of applications especially in the biomedical field.35–37
The colloidal self-assembly of nanomaterials has stimulated enormous research interest in the last decade.38–43 In many cases, the construction of these well-defined nanostructures all share the same goal: creating synergic interaction between nanocomponents. On this topic, RAFT polymer has the auspicious talent as a linker for guiding the colloidal self-assembly of nanomaterials using its topological and anchoring design. We will see how RAFT polymer engineering can create the synergy between nanocomponents and dynamically changes the colloidal arrangement of each nanocomponent for modern applications.
The current review focus on the recent advances in both fabrication methods and design strategies for polymer/inorganic nanohybrids for multifunctional applications (illustrated in Fig. 1). We will start with the methodology of synthesizing RAFT polymers with the aim of surface functionalization. We will go through the important design options for RAFT polymers including a detailed discussion on anchoring strategies for diverse nanomaterials. Then we will review a series of classic core–shell nanostructures with the focus on the novel functionalities brought by the polymer brush. These functional nanostructures cover a wide range of applications including bioimaging, drug-delivery, cancer treatment, fluorescence energy transfer based nanosensors and hybrid responsiveness. We will focus on the detailed macromolecular design for each specific application scenario. Thereafter, examples of ordered nanoassemblies using the RAFT polymer linker will be reviewed to showcase the advantages brought by the design flexibility of RAFT polymers. This part also highlights the possibility to introduce novel morphologies, interactions and chirality to the nanostructure using the sophisticated design of the RAFT polymer shells. The review will be concluded by discussing the future challenges and opportunities for surface-bound RAFT polymers.
 | ||
| Fig. 1 Schematic illustration of design options for the surface modification of inorganic NPs with RAFT polymer and the fields of applications for the hybrid nanomaterial. | ||
It is worth mentioning here that the offering of RAFT polymerization in nanoengineering cannot be fully covered within the scope of this review. Especially polymerization-induced self-assembly (PISA) has been a surge of interest in the fabrication of block copolymer nano-objects with controlled morphology, e.g., nano-sphere, -worm, and -vesicles. In this research area, a great number of the literatures are based on RAFT technique. The readers interested in this topic can refer to more elaborate publications24,44–50 on PISA technique.
2. Construction options for RAFT polymer towards nanosurface
RAFT polymerization is a type of reversible deactivation radical polymerization (RDRP) developed by the Australian Commonwealth Scientific and Industrial Research Organization (CSIRO).1 This polymerization technique confers the favored living characteristics to radical polymerization. It combines many useful features of radical polymerization, such as ease and cost-effectiveness of synthesis, compatibility with many different monomers, and broad reaction conditions.23,51 The control of the polymerization kinetic enables precise design and engineering of functional polymers which induced an explosion of innovation in polymer science over the last decades. Compared with other RDRP techniques, the major competitor for RAFT is the atom transfer radical polymerization (ATRP). Both of them offer a very high degree of design and control on polymer architecture and flexible end-group modification. The major advantage of RAFT polymerization is the straight-forward and insensitive reaction condition: in ATRP, a copper-based complex must be usually added for the polymerization and removed from the product. However, compared with other RDRP techniques, the color of RAFT moieties in the polymer product could be an issue for many applications unless they are removed by one extra reaction step. Notably, for the long-chain polymer (n > 500), ATRP often offers better control than the RAFT counterpart. A selection of recent work and reviews is included here on the comparison of RDRP polymerization techniques.52–54A series of grafting strategies have been developed to immobilize RAFT polymers onto the surface of nanomaterials. Understanding the surface chemistry between RAFT polymer and the specific surface of NPs is crucial for the effective polymer functionalization.
2.1. RAFT polymerization
The mechanism of RAFT polymerization is illustrated in Fig. 2A. After a radical initiation event, the propagating radical chains react with the RAFT agent, which usually consists of a thiocarbonylthio group. This leads to a reversible degenerate chain transfer, rendering the former radical chain inactive (“dormant”) and releasing the previously dormant chain as radical species able to propagate. The RAFT equilibrium offers a rapid exchange between the active propagating radical and the dormant radicals, while the majority of the chains are kept in dormant form. As result, all polymer chains have equal time and possibility to propagate. This controlled kinetic gives the polymer product a narrow molecular weight distribution, i.e., a similar degree of polymerization. Practically, the amount of radical initiator must be carefully considered to maintain the favored RAFT kinetic, since the concentration of active species in the reaction mixture depends on the concentration of the initiator.2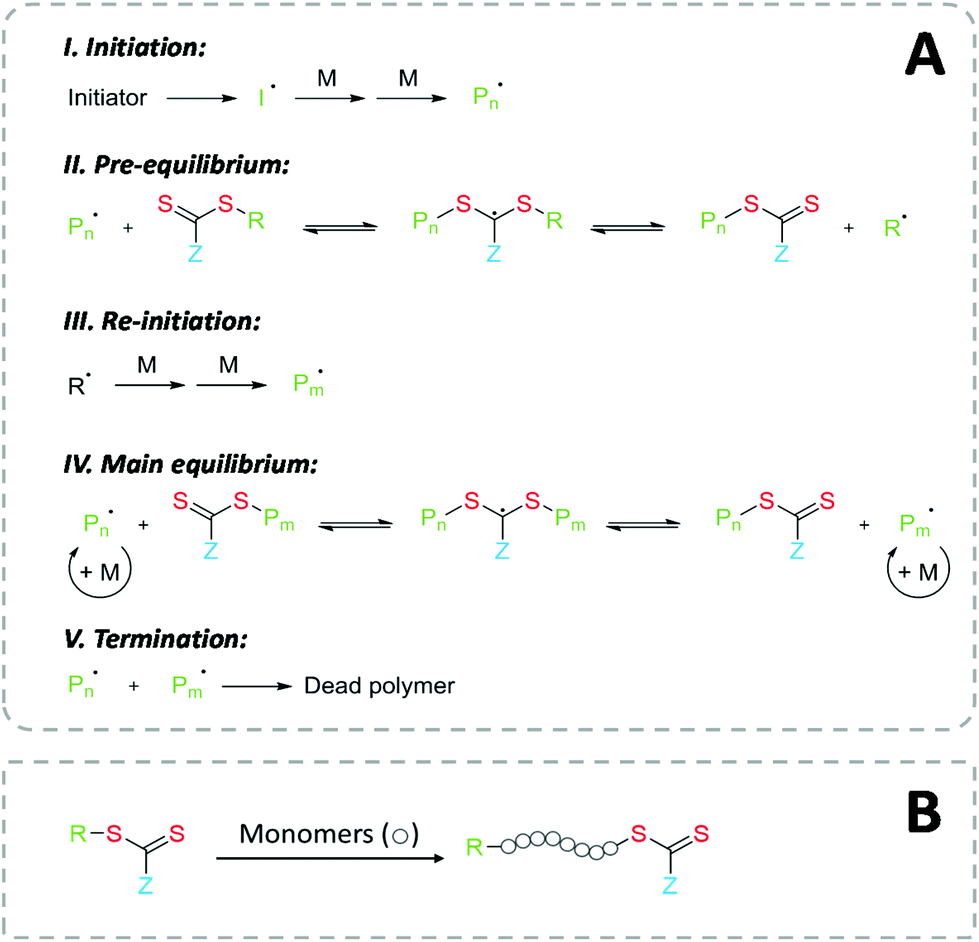 | ||
| Fig. 2 (A) Proposed Mechanisms of RAFT polymerization. (B) Overall RAFT polymerization process. | ||
Owing to the clear mechanism of polymer growth in the controlled radical polymerization, RAFT technique allows for a high degree of control over resulting molecular weight distribution, copolymer composition, and macromolecular architecture.23 Due to the living character of the polymerization, BCPs can be easily synthesized. Also, the end-group functionality is preserved, providing a reactive handle for further modification or serving directly as anchoring moieties for surface functionalization.23
Since the invention of RAFT polymerization, extensive research efforts were initiated towards developing versatile RAFT agents which are suitable for a great number of monomer sand reaction conditions, including aerobic, biological, or continuous-flow conditions.23,55,56 Boosted by growing industry demands, the range of reported RAFT agents as well as their commercial availability is rapidly expanding.23,57,58 Development of the initiation process for RAFT polymerization also has been developed rapidly, amongst others via light, enzymes, electric fields, or ultra-sonication.56
Over the years, the precision, versatility, and simplicity in both design and synthesis provided by RAFT polymerization quickly found application in various fields.23 The offering of RAFT polymers has quickly gone beyond the conventional branches for polymers. For further information on RAFT polymerization, e.g., the match of RAFT polymer with its suitable monomer classes, the recent development of new RAFT agents, the process of polymerization, and more studies on the mechanism and kinetics, the reader is referred to more elaborate publications on those specific topics.2,23,51,56,57,59
2.2. End group modification
Introducing functional groups at specific positions in the polymer chain is the most basic and practical approach for extending the functionality of polymers. RAFT polymerization offers flexible options for positioning the RAFT group after polymerization: both in-chain and at the chain ends. The chain-end design has the advantage of further chemical modification without considering the degeneration of the polymer chain. Fig. 2B shows the position of the RAFT agent after polymerization: the RAFT moiety part (Z group) retains at their ω-end, while the R-group functionality stays at the α-end.60 It is worth mentioning that besides the favored RAFT polymer with end-group functionalities, polymer species not possessing both of these end groups always exist in the product. This “dead” polymer originates from irreversible termination of propagating polymer radicals, which also border the molecular weight distribution since the terminating product has twice the molecular weight of the propagating radical and this process occurs during the entire polymerization period. The formation of the termination product can be controlled by reducing the amount of initiator.59 A low initiator concentration keeps the ratio of active/dormant species healthy, i.e., maintains the living character of the reaction.51We have seen that using RAFT polymerization allows a simultaneous end-group design for both chain ends of the polymer. However, one needs to consider that both Z- and R-group of the chain transfer agent (CTA) must be designed not only in terms of required end group functionality, but also the reactivity of the CTA during polymerization. R-Groups need to be a good leaving group and effective at reinitiating polymerization as free radicals, whereas Z-groups modulate the rate of addition and fragmentation and need to provide stability to the intermediate radical species.3,59 On this topic, a great library of RAFT agents with distinct end group functionalities, such as carboxyls, hydroxyls, protected amines, clickable groups, and fluorophores, their syntheses, and compatibility with monomers has been reported in the literature.51,61–64
Another widely used approach bypassing the limitation of the design option for RAFT agents is the modification of the ω-group after polymerization: the thiocarbonylthio moiety provides a suitable and chemically versatile handle for introducing desired functional moieties, as shown in Fig. 3. On this topic, the chemical modification options for the RAFT group are reviewed in detail in the literature.3,4,60
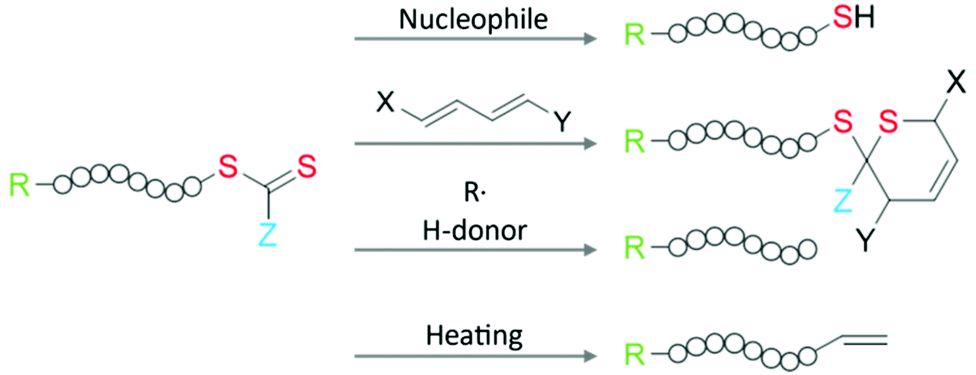 | ||
| Fig. 3 Schematic illustration of commonly used chemical pathways for RAFT group modification. | ||
Employing a reaction with nucleophiles or ionic reducing agents, the thiocarbonylthio moiety can be converted into a thiol group. Prominent examples include the reaction with borohydrides,65,66 aminolysis,67,68 or hydroxyls.60 Owing to the specific color of RAFT moieties, this process can be optically monitored. The resulting thiol end group can be directly used for binding to various nanosurfaces (see section 2.4) or further reactions including nucleophilic substitution at carbons or sulfurs, nucleophilic addition to activated double bonds, radical addition with alkenes and alkynes, or oxidation to disulfides.69,70 It is worth mentioning here that the oxidation of thiols will yield a disulfide bond that links two thiolated polymers together and doubles the molecular weight. For multi-thiolated polymers, the oxidation will cause a covalently cross-linked polymer network. Special attention must be given to the synthesis and storage of thiolated polymer to avoid oxidation (inert atmosphere or low pH condition).67 Alternatively, the thiocarbonylthio moiety can be used as a dienophile in hetero Diels–Alder reactions with dienes.60 The reaction of the thiocarbonylthio group with oxidizing agents such as hydrogen peroxide, air, or ozone results in e.g., hydroxyl, hydroperoxide, or thiolocarbonate end groups.4
Furthermore, the RAFT moieties can be easily and completely removed from the polymer, if necessary, especially when it comes to the color, chemical reactivity, and biosafety of the product.4 To completely remove the thiocarbonylthio moiety, the RAFT polymer can be reduced with radicals and hydrogen atom donors without a significant side reaction.60,71 Interestingly, by choosing an appropriate radical species, it is possible to even retrieve the RAFT agent.72 Lastly, thermolysis can be used to completely remove the thiocarbonylthio moiety and involves heating of the polymer to 120–200 °C, resulting in an unsaturated end group functionality.60,73
Bioconjugation with RAFT polymers has always been a highlight for diverse hybrid applications in the biomedical field. Especially when it comes to the surface-bound RAFT polymer brush, converting RAFT end groups to specific bio-binding moieties quickly provides the direct conjugation for biomolecules. For example, well-established site-specific bioconjugation methods targeting cysteines (in native proteins)74 can be used on the thiol group. Maleimide,75–77 vinyl sulfone,78 and activated disulfide79,80 moieties can be built in the RAFT polymer or converted from RAFT moieties for bioconjugation. On this topic, large efforts were made to optimize the detailed bioconjugation from RAFT polymers. The recent advances in this area are extensively reviewed elsewhere.54,81–88
2.3. Grafting strategies
The power of functional RAFT polymers is rarely applied in their bulk form. Since their discovery, RAFT polymers were used to enhance the performance of diverse materials and quickly found applications in various fields of research and industry.2 RAFT polymers are particularly useful for surface modification on nanoscale materials, such as NPs or membranes. The enormous surface area and comparable size of nanomaterials and single polymer chains maximize the effect brought by the polymer functionalization.The binding of RAFT polymers onto the surface of nanomaterials requires specific chemistry towards each type of surface. A number of different anchoring strategies are available to link RAFT polymers to a surface, which often come with their own merits and shortcomings. Researchers should carefully consider suitable anchoring options depending on the properties of their system, including the type of polymer, the surface character of the NP, the properties of the protecting ligand and the required capping density of the polymer on the surface.
In general, there are two most commonly employed grafting strategies: grafting-to and grafting-from approaches. In the grafting-to strategy, the pre-synthesized polymer is introduced to the surface, whereas in grafting-from, the polymer synthesis is initiated from the NP surface (Fig. 4). Grafting-to is considered a more straightforward method,89 since the grafting step can often be facilitated by a simple ligand exchange reaction: polymers carrying a functional group with a stronger affinity towards the NP surface tends to replace the weaker binding protecting ligand. Also, polymer and NPs are individually prepared to allow for separate syntheses conditions and hence tight control over polymer characteristics.88,90,91 In contrast, grafting from surfaces demands the attachment of the RAFT agent before RAFT polymerization. In this stage, the surface-bound RAFT agent might alter the colloidal stability of the system since the protecting effect of these small molecules is very limited. Special care on the storage and purification steps must be given to avoid irreversible aggregation.65,92 Moreover, the polymerization conditions must be optimized for the NPs as well to ensure the complete dispersion of NPs in the polymerization mixture. For the modification of biosurfaces or bio-containing surfaces such as proteins, the grafting-from approach can struggle to retain the biological functionality after polymerization.91 Furthermore, the characterization of the surface-bound polymer is not as straightforward as for grafting-to approaches.65,93 Oftentimes, the free polymers formed in solution are characterized as a substitute for the surface-anchored ones, as they are quite similar with respect to molecular weight and dispersity.94,95
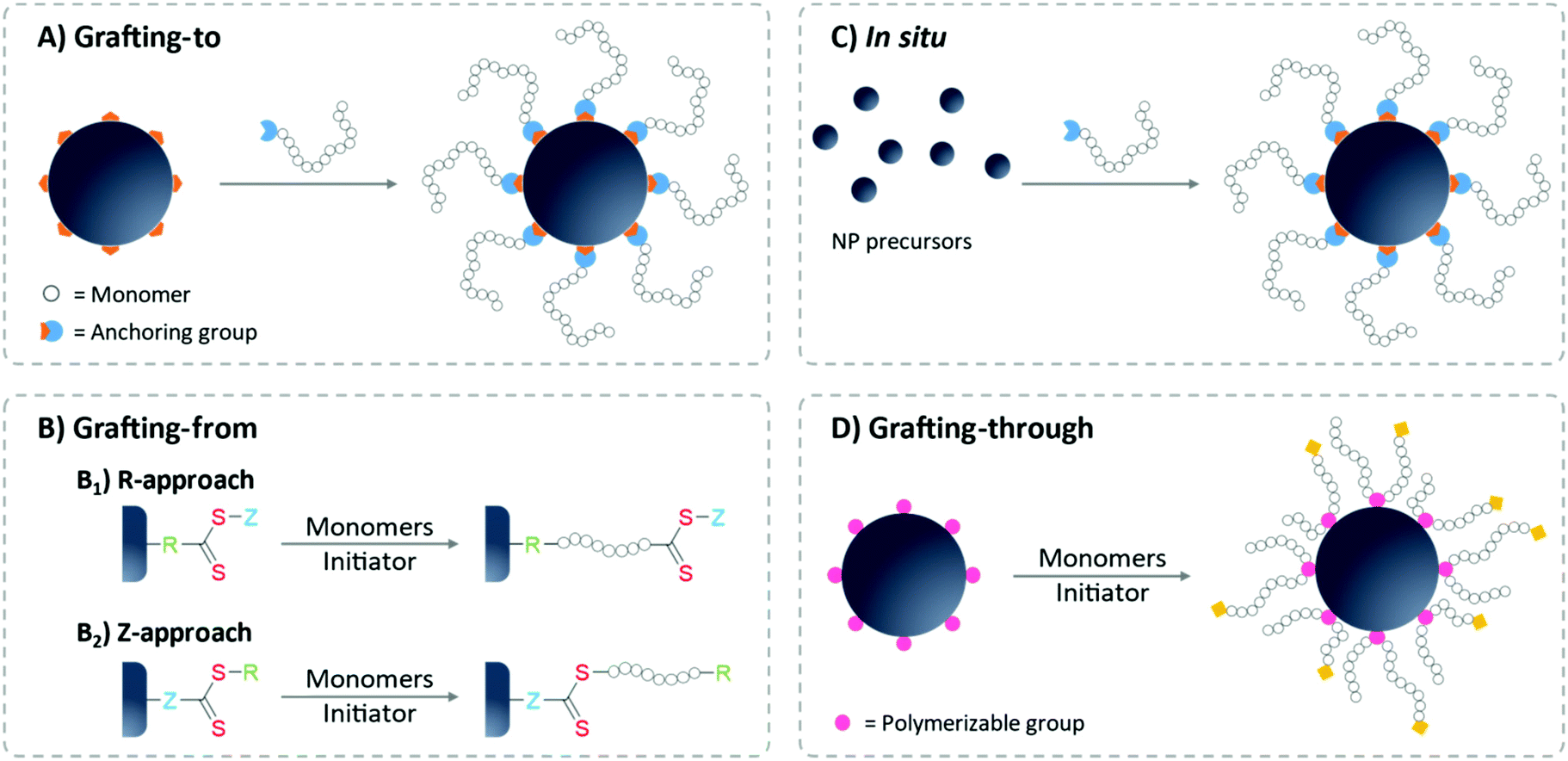 | ||
| Fig. 4 Illustration of the most commonly used strategies to immobilize polymers onto the surface of NPs. (A) Grafting-to, (B) grafting-from with B1 for R-approach and B2 for Z-approach, (C) in situ synthesis and (D) grafting-through approach. | ||
However, in many cases, grafting-from outperform grafting-to approaches in terms of obtained grafting density.35,90,91,96,97 This difference can be explained by the steric hindrance effect during the grafting-to process: the approaching polymer chain is sterically blocked by the grafted polymer chains as well as by binding site masking via random coil formation of the approaching polymers.90,91 The grafting-to approach often shows chain length dependence and performs better with shorter polymer chains.89,98 Other challenges for grafting-to approaches are the ligand exchange process and the efficiency of the anchoring chemistry. Those issues are highly specific and will be discussed in section 2.4. The reader should keep in mind that the surface properties of NPs differ strongly from their bulk counterpart, mainly due to the protection ligand from the synthesis. Oftentimes, choosing NPs with suitable ligand and solvent combination is the key to a successful polymer-ligand exchange.
Notably, the molecular design of the attached RAFT agent plays a critical role when it comes to polymerization characteristics of the grafting-from approach. If the RAFT agent is coupled to the surface via its R-group (R-group approach), the polymer chains grow while being attached to the surface. The propagating radical is located at the solvent-facing polymer terminus, thereby able to readily engage with monomers and RAFT agents in the solution during the whole polymerization process, even at high molecular weights (Fig. 4B). Consequently, high monomer conversion and narrow polymer dispersity of the grafted polymer can be achieved.88,95,99 Concerns could arise due to the termination between surface-bound radicals (both inter- and intra-particle termination). However, since an excess of free RAFT agent is often added to the polymerization mixture, the termination reaction is then dominated between the solution radicals.100 Similar to the solution polymerization without NPs, the amount of initiator must be controlled to avoid the formation of termination product on the surface-grafted polymer shell,101 which precludes further polymerization to longer polymers or to BCPs. An important advantage brought by R-approach is that the RAFT moieties will stay on the outward-facing chain termini of the surface-grafted polymer brushes which can be utilized for further modifications.
In contrast, if the RAFT agent is anchored via the Z-group (also called transfer-to), the thiocarbonylthio groups are permanently attached to the surface during and after polymerization. This means that radical chain propagation and termination only occur in solution. This approach only allows active radicals to recombine back onto the surface-grafted Z-groups, giving the advantage of avoiding the anchoring of any terminated product (Fig. 4B).100 However, since the recombination of polymer radicals with the surface RAFT Z-groups has a similar steric issue as the grafting-to approach, in practical, R-group approaches are often favored over Z-group approaches, especially for longer polymers.88,90,101,102 Furthermore, for the first step of anchoring RAFT agents onto the surface, the R-group offers easier access to introducing anchoring moieties over the Z-group.100 Also, the Z-group approach locates the thiocarbonylthio group directly on the surface of the NPs, which comes with a potential risk of chain stability loss due to hydrolysis or aminolysis of the RAFT agent.103
Another approach for introducing polymers onto nanosurfaces is the grafting-through method, in which the monomer is anchored on the surface instead of the RAFT agent.104,105 During polymerization, this group is incorporated into the growing polymer chain (Fig. 4D). This often leads to intermolecular cross-coupling as one polymer chain can incorporate polymerizable groups from more than one NP surface.106 This characteristic can be beneficial for the fabrication of gels or matrices, but is rather undesired for colloidal systems.
In all aforementioned grafting approaches, the particles are synthesized prior to polymer functionalization. Another important strategy to fabricate polymer-capped NPs is using the polymer micelle as a template during NP synthesis. This approach can often be designed in a one-pot and in situ experiment, performing synthesis and polymer capping simultaneously (Fig. 4C). This direct synthesis approach brings several advantages, for one the polymer usually acts as a stabilizer or surfactant during the synthesis, circumventing the need for additional chemicals, which might require further clean-up/ligand exchange.35,107 On this topic, the design of the BCP offers a very intriguing architecture of the polymer micelle morphology which can further be used to create nanomaterials according to the shape of the polymer template. Readers can refer to a recent review article on this topic.108
In summary, understanding the mechanism and character of each approach is necessary for the successful design of the experiment. The approach of introducing polymer shells on the surface of NPs must be chosen according to the specific type of NP including its surface ligands and solvent conditions. More specific strategies to introduce RAFT polymers on distinct types of NPs are discussed in section 2.4.
2.4. Surface chemistry for anchoring polymers
One of the most crucial chemical engineering tasks for the fabrication of polymer-NP structures is the junction point between the polymer and NPs. The linkage between the synthetic polymer and surface of NPs requires strong interaction, or even covalent bonds, to prevent the structure from disassembling. The anchoring itself is a dynamic process and must be approached using an effective chemical reaction or strong adsorption. For this task, customized to the property of the surface, functional groups bearing such anchoring competence are introduced to the polymer chain. The position for these anchoring moieties in the polymer architecture is highly flexible, they can be present as part of the RAFT agent (end group),109,110 integrated as polymer side chains,111,112 or introduced after polymerization.113,114 Here, we demonstrate strategies for polymer functionalization of a selection of the most popular NPs. Table 1 and Fig. 5 give an overview of the content in this section.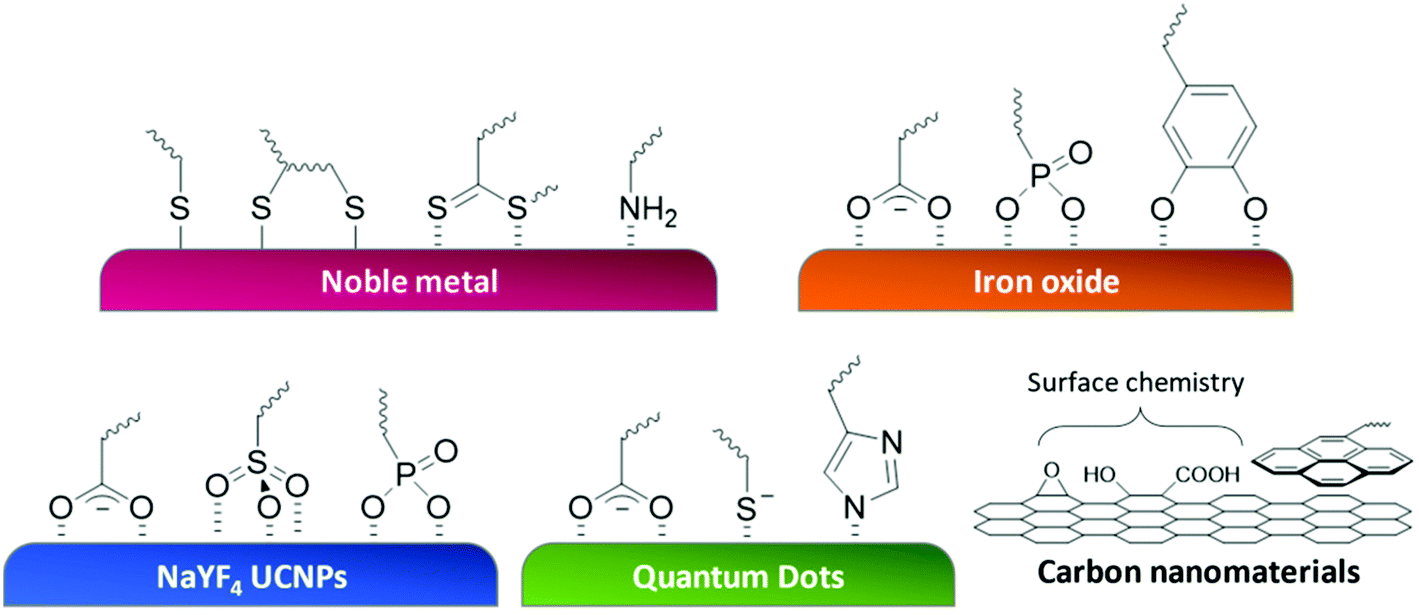 | ||
| Fig. 5 Schematic illustration of commonly used anchoring strategies to immobilize polymers on different nanosurfaces. | ||
| Gold | Silver | Iron oxide | NaYF4 UCNPs | QDs | Carbon nano-materials | |
|---|---|---|---|---|---|---|
| Carboxylic groups | 141–143 | 144 and 145 | 146 and 147 | 148–153 | ||
| Thiol groups | 65, 68, 89 and 154–162 | 163 | 164 | |||
| Dithiol groups | 161 | 138 and 165 | ||||
| Dithiobenzoate groups | 136 and 166 | 138 | ||||
| Trithiocarbonate groups | 109, 136, 139, 156 and 167–174 | 140 and 175 | ||||
| Thiocarbamate groups | 157 | |||||
| Sulfo group | 144 | |||||
| Amine groups | 155 and 176–179 | 105 | ||||
| Phosphate groups | 111 and 180–182 | 144 and 183 | ||||
| Catechol groups | 184 | 185–187 | ||||
| Imidazole groups | 112, 188 and 189 | |||||
| Pyrene moieties | 110, 190 and 191 |
In general, sulfur-containing groups, i.e., thiols,134 disulfides135 and RAFT groups136,137 have strong interaction with noble metal NPs. The inherently thiocarbonylthio-terminated RAFT polymer offers a well-known and straightforward approach to form dense polymer shells on the surface of Au and Ag NPs.138–140
Owing to the strong interaction between the RAFT group and the surface of noble metal NPs, the grafting-to approach works well here: the RAFT polymer can be synthesized separately without the presence of NPs avoiding the risk of causing any aggregation of NPs during the polymerization step. The self-assembly of RAFT polymers with noble metal NPs often has simple protocols by typically mixing them in a good solvent for both polymer and particles. However, a sufficient NP surface activity is required: strongly bound surfactants on NPs often jeopardize the ligand exchange process, especially for macromolecules. The detailed ligand exchange of each type of nanocomponent must be studied specifically. In some cases, the partial removal of original protecting ligand is necessary.167,175,192
To discuss the performance of RAFT polymer shells on noble metal NPs, the interaction between polymer and particle as well as the grafting density are the key factors. Here, thiol and RAFT groups can be both applied as anchoring moiety. The thiol group is covalently bound to the surface of gold NPs through a redox reaction. The strong RS–Au bonds134,193 provide rapid and robust binding with a binding strength of approximately 170 kJ mol−1 (ref. 194) and total net stabilization energy of roughly −20 kJ mol−1 considering the break of the S–H bond and formation of the H–H bond.195 The RAFT group, e.g., dithiocarbonate, has a bonding energy of −16 kJ mol−1 (ref. 136) and free energy of adsorption of −36 kJ mol−1 (ref. 137) towards the gold surface. The thiol group seems to be a better choice due to its stronger and irreversible bonding despite extra reaction and purification steps from the RAFT polymer. Under this consideration, RAFT groups are often reduced to thiol groups prior to particle functionalization.196–199 Slavin et al. demonstrated that the capping density of the polymer brush on the gold surface with diverse sulfur-containing groups has the following trend: disulfide > dithiocarbonate ∼ trithiocarbonate > free thiol for diverse polymer chain lengths.200 Wang et al. showed that the end group fidelity of thiol-functionalized polymer has a significant impact on the capping density of polymers on AuNPs: the unfunctionalized polymer (from radical termination processes) has a jeopardizing effect on the grafting-to process.201
For RAFT groups, Blakey et al. also confirmed that the RAFT group does not dissociate upon adsorption to the AuNPs.137 In fact, a number of high-density RAFT polymer-capped noble metal NP hybrids have been fabricated by successful ligand exchange with e.g., citrate,139,202,203 oleylamine,140 cetyltrimethylammonium bromide (CTAB)167,192,204 and tetraoctylammonium bromide (TOAB).140 Our group also confirmed the high long-term thermal stability of the RAFT–Au interaction up to 90 °C on both AuNPs and AgNPs covered with RAFT-terminated polyethylene shells.140 Thiol and RAFT moieties both have numerous successful results in fabricating well-defined polymer brush shells on noble metal NPs. The binding efficiency of RAFT and thiol groups on palladium NPs was compared by Ruiu et al.205 As result, no significant difference was found. In the author's opinion, both RAFT moieties and thiols work great as anchoring functionality for noble metal NPs.
The grafting-to approach serves as the dominating synthesis strategy for modification of noble metal NPs with polymers. As the sulfur-containing groups of the RAFT agent act as the reactive center during chain transfer reactions, surface anchoring via RAFT groups is less suitable for grafting-from syntheses. Other functional groups like amines,176 alkynes,206,207 catechols208 or phosphonates209 can be used for attachment to gold surfaces. However, the stronger aurophilicity and inherent presence of the RAFT group make the sulfur-containing anchoring approaches much more popular.
Similar to gold, silver NPs possess an affinity towards sulfur-containing moieties (thiols,163 disulfides,165 and RAFT moieties.138,175 The Ag–S bound has a binding energy of 217 kJ mol−1 (ref. 210) and a colloid formation free energy of−25 kJ mol−1 (ref. 211) (ethanethiol) for NP surface. The thermodynamic values of thiol-binding on AgNPs and AuNPs are very similar. The strong anchoring performance of thiocarbonate group on AgNPs is also confirmed.175 For further noble metal NPs, thiol also provide a well-known solid anchoring performance, e.g., on Pt and Rh NPs.197,212 Notably, the situation of Pd–S interaction is much complicated since the thiol can react with Pd forming PdSx layer. Readers should refer to the detailed works213,214 on this specific topic if the surface properties of the NPs are crucial for the applications.
Besides AuNPs and PtNPs, metal NPs are notorious for their oxidation tendency. Depending on the synthesis route, oxygen-free conditions could be required for maintaining the surface of metal NPs active for surface modification. In fact, anchoring strategies targeted on the oxidized surface also have been reported e.g. for ZnNPs,215 AgNPs,184 and gallium–indium (liquid metals) based NPs216 typically using catechol or carboxylic anchoring groups.
Two other surface affinity groups found in the literature for attachment of polymers to iron oxide NPs are phosphonates and carboxyls. Out of the two, phosphonates create more stable linkages with higher grafting densities;223,231 however, the use of phosphonate moieties often requires protection prior to polymerization.180,232 Carboxyls can also be used for surface modification,142 but it has been reported that this anchoring is temperature-sensitive and replaceable with other ligands.223
The surface modification of QD often brings an impact on its fluorescence performance since the photon luminance properties of QD are strongly dependent on the surface properties and surface–ligand interaction. First, a strong ligand–surface interaction and dense ligand shell are required to prevent QD surface oxidation since the oxidation layer destroys the perfection of the QD surface causing the change in the energy level and luminance.235,236 Also, the type of surface ligand can influence the electronic properties of the QD.237
Although small molecules, e.g., thiol derivates are oftentimes used for QD surface modification.236,238 The nature of the thiol–QD interaction is very distinct from the simple Au–S interaction: on CdTe QD, a thin layer of CdS is formed and the anchoring originates from the interaction between CdS (or ZnS shell) and thiolate rather than with thiol moieties.239,240 The colloidal stability provided by small molecules is generally limited. One way of circumventing this issue is to use macromolecular ligands, which can be easily prepared via RAFT polymerization. Here, a multiply-binding copolymer design has been proven to be very effective: one type of monomer contains an anchoring group for QD binding, such as thiol,164 carboxyl,146,147 imidazole112,188,189 and 8-hydroxyquinoline241 moieties. Another part of the polymer adjusts dispersibility and provides additional properties.
An alternative option for surface modification and QD stabilization is the use of amphiphilic BCPs. QD synthesis often employs hydrophobic surfactant molecules, rendering them water-insoluble.233 The hydrophobic part of the amphiphilic BCP can interact with this outer QD layer, whilst the hydrophilic block confers hydrophilicity and added functionalities such as pH-responsiveness as demonstrated by Liu et al.242
One of the most applied approach to effectively introduce polymers or biomolecules to UCNPs is using a silica coating. With a nano-thin silica-shell around UCNPs, surface modification with well-established silane chemistry can be applied without any special modification. RAFT polymerization can also proceed from the silica surface (see section 2.4.6 for more information).248–250
The choice of oxidation depends on the desired anchoring chemistry. An esterification reaction between the hydroxyl group on carbon nanomaterials and carboxyl groups on the RAFT moieties is often used to introduce RAFT agents to the surface for grafting-from polymerizations.150–153 Alternatively, one can perform a nucleophilic ring-opening reaction of epoxide groups using amines, which has been demonstrated by Mardani et al. to introduce a polymerizable methacrylate on graphene oxide (GO) for a subsequent grafting-through RAFT polymerization.105 Moreover, it would be possible to couple the surface carboxyl groups with primary amine moieties using N-hydroxysuccinimide (NHS) click chemistry.252 A detailed comparison between grafting strategies for RAFT polymers and click chemistry is reported by Hwang and coworkers.253
Another useful approach to immobilize polymers to the surface of GO is using pyrene moieties as anchoring groups due to a strong π–π stacking interaction.110,190,191 Owing to this strong binding energy (comparable with covalent attachment), the straightforward grafting-to protocols have been proven to be very effective.110,190
The classic Stöber process for silica coating requires the dispersion of NPs in ethanol (or mixture with water). With the addition of ammonia, tetraethylorthosilicate (TEOS) is hydrolyzed into silica precursors and subsequently condensates, forming silica on the surface of NPs layer by layer.256,257 The silica shell thickness can be tuned by varying e.g., the amount of ammonia and TEOS or the reaction time.258–261 For a successful silica coating, perfect dispersion of the nanomaterial during the reaction is crucial to avoid aggregation. The perfect dispersion of originally hydrophilic nanomaterials in ethanol requires surface modification. For this task, polyvinylpyrrolidone (PVP) provides generally good adsorption on diverse nanomaterials, making them suitable for the silica coating condition.255
Another approach based on the water-in-oil (reverse) microemulsion technique has the special ability to perform one-on-one silica coating if the dosing of particles roughly matches the number of micelles. In these approaches, NPs must be dispersed in nonpolar media, e.g., cyclohexane.258 Silica coating takes place on the NP surface isolated in water micelles. The resulting particles are highly spherical and possess a narrow size distribution. Generally, microemulsion systems are able to work at higher NP concentrations compared to the Stöber coating with much lower risk of aggregation.262 However, microemulsion-based approaches possess an upper limit for the size of the used NP cores as well as the resulting silica-shell thickness, which is amongst other influenced by the size of the microemulsion droplets and the type of used surfactant.263 For instance, Han et al. reported an upper size limit for the resulting silica-coated particles of approximately 90 nm.262
Grafting strategies for polymer attachment onto silica surfaces has been extensively studied. By far the most efficient and widely used strategy to introduce RAFT polymers on silica is the surface-initiated grafting-from approach. The RAFT moieties are often first introduced to the surface of silica via a two-step reaction. First, the surface is functionalized with amine groups using aminosilanes followed by RAFT agent anchoring via activated carboxyl groups.99,168,248–250,264,265 The choice of silane group has strong impact on the result since the number of alkoxysilyl groups on each silane has very different performance: the tri- and di-alkoxysilanes have the risk of formation of interparticle crosslinking causing the formation of aggregated material.266 Although the monoalkoxysilane is sensitive to moisture during the reaction, the crosslinking can be completely suppressed.99 In a similar way, carboxyl-containing polymers can be linked to the aminosilane-functionalized particles in a grafting-to approach using the same coupling reaction.267,268 The large selection of silanes also allows for other approaches to anchor RAFT groups on the surface of silica, such as via chlorosilanes269 or alkyne-containing silanes.154 Furthermore, the silica core can be removed by etching with HF or NaOH.270 This allows the formation of hollow polymer shell by cross-linking the grafted polymer before the etching process.271–274
2.5. Topological design
RAFT polymerization also provides a vast selection of topological design options. To this end, star,8 branched, hyperbranched, network,10,11,275 comb-like, bottlebrush,12 and in-chain polyfunctional139,172,173 RAFT polymers are extensively studied. Readers interested in the design, synthesis, and selection of the RAFT agents for those polymer structures are referred to the abovementioned literature. With the knowledge of RAFT mechanisms and suitable R- and Z-approaches, the transfer of topology from RAFT agent to the RAFT polymer can be executed in a very precise manner with the control of the positioning of the RAFT moieties in the polymer.Using linear polymer with a single anchoring end group can only form a simple core–shell nanostructure. However, RAFT polymers with a higher order of topological design can provide intriguing competence for further functional extension, e.g., linking further NPs onto the existing core–shell structure. As an example, our group reported the formation of spherical AuNPs superstructures crosslinked with multiblock RAFT polymers containing several in-chain RAFT groups along their backbone. The interparticle distance inside these superstructures can be controlled with the chain length of the linking polymer.172,173
The topological design of the polymers has a key impact on the self-assembly of nanomaterials. In section 3.2.3, more examples of hierarchical nanoassemblies fabricated by multi-arm and hyperbranched polymer linkers are reviewed in detail. It is worth mentioning that a change in the topology could affect the stimuli-responsive parameter of the polymer, the polymer content should be adjusted according to the application scenario by e.g., adding another monomer to the polymer.9
3. Design with RAFT polymers in nanoengineering
The innovation in molecular engineering for a RAFT polymer can be applied to every point of the polymer chain. The development of new functional monomers combined with sophisticated structural design makes RAFT polymers the perfect key building block for fabricating a wide range of nanostructures. In this chapter, we will demonstrate the recent advances in the development of surface-bound RAFT polymers in the realm of nano application.The “smartness” of RAFT polymers has been rapidly growing during the last decade. The possibility to build multiple intriguing abilities into one (block) copolymer makes RAFT polymers a versatile tool for the surface modification of nanomaterials. With a wide selection of well-established anchoring strategies, the RAFT polymers can be applied as smart linkers for connecting distinct nanomaterials. We will demonstrate how the structural coding from the polymer design can be used to introduce and dynamically control the self-assembly, interparticle-interaction, energy transfer and stimuli-responsiveness of the hybrid nanostructures.
3.1. Creating functions with RAFT polymer shells for NPs
The most straightforward application of polymers for NP modification is using them as capping ligands, forming core–shell nanostructures. Once the NP is covered with the polymer shell, these nanostructures often carry the solubility (dispersibility) and functionality of the polymer shell. In recent years, with the rapid innovation of both NP synthesis and functional polymer design, new types of polymer-capped NPs with diverse hybrid functions are emerging. Especially the combination of orthogonal functions in RAFT polymer brushes using copolymer design strategies opens up towards the next level of smart NPs.To introduce multiple functions to one nanostructure, the (block) copolymer brush design provides the most straightforward and reliable approach. The synergy between different polymer units gives rise to many advantages. This section will review the recent development of RAFT polymer-functionalized core–shell nanomaterials and showcase the novel design options and collaborative properties between the polymer and nanocore. We will focus diverse research topics covering enhancement of the dispersion behavior, hybrid stimuli responsiveness and smart linkers for fluorescence energy transfer-based nanosensors. Table 2 summarizes our selection of publications on the novel functions brought by modern RAFT polymer shells.
| Nanomaterial | Grafted polymer | Applications | Ref. |
|---|---|---|---|
| AgNP | Branched PEG-star-poly((2-hydroxypropyl methacrylate)-co-(dithio-di-2,1-ethanediyl bismethacrylate)-co-(lipoic acid methacrylate)-co-(2-(4-nitro-3-benzyl carbonate camptothecin)phenoxyethyl methacrylate)) | Water dispersibility; biocompatibility; photo-responsive drug-release tracked by fluorescence (NSET) | 165 |
| AgNP | Fluorescent dye functionalized PAA | pH responsive metal-enhanced fluorescence behavior | 276 |
| AgNP | Poly(2-(2-hydroxyethoxy)ethyl methacrylate-co-methacryloyloxy-3-thiahexanoyl-camptothecin) | pH-sensitive drug-delivery with tracking of drug-release progress via NSET controlled fluorescence | 138 |
| AgNP | Poly(methyl methacrylate-co-(2-hydroxypropyl-9-anthroate methacrylate)) | Surface plasmon resonance enhanced upconversion of fluorescence | 163 |
| Au nanocage | PNIPAM-co-poly(acrylamide) | High-intensity focused ultrasound induced drug release | 277 |
| AuNP | Poly(PEG methacrylate) | Controlled aggregation upon addition of 2-phenyl-2-(phenylcarbonothioylthio) acetate, yielding nanoaggregation with tunable optical properties via surface plasmonic coupling | 278 |
| AuNP | ω-Carboxyl-terminated PNIPAM | Thermo- and pH-controlled reversible phase transfer between water and chloroform | 279 |
| AuNP | Azobenzene-containing methacrylic polymer | UV-light triggered aggregation of AuNPs | 166 |
| AuNP | PNIPAM with pillararene end-group | Introduces hexafluorophosphate-pillararene host–guest interaction alone with thermoresponsiveness from PNIPAM | 280 |
| AuNP | Poly(di-(ethylene glycol) methyl ether methacrylate)-b-(poly(6-O-vinylazelaioyl-D-glucose)) | Water dispersibility; biocompatibility; lectin recognition | 66 |
| AuNP | Polystyrene-b-PEG with trithiocarbonate group as the junction between two blocks | Amphiphilic colloid properties | 281 |
| AuNP | Poly(hydroxyethyl acrylamide) with amino-benzylguanine or tris-NTA amine end-group | Bioconjugation with anti-freeze proteins | 198 |
| AuNP, AgNP, CdSe QD | Thiol-terminated meta-triphenylamine polymer | Electron conductivity | 282 |
| AuNR | Poly(MPC-co-HEMA-DHLA) | Water dispersibility; high resistance against cyanide ion etching; antifouling; cytocompatibility | 161 |
| AuNR | PNIPAM | NIR light triggered photothermal drug-release | 199 |
| AuNR | PEG-b-poly(N-vinylcaprolactam) | NIR triggered drug-release | 283 |
| AuNR | 3-Arm PNIPAM | NIR-light triggered photothermal aggregation of AuNRs in simulated blood fluids | 167 |
| Carbon NP | Copolymer of NIPAM and spiropyran containing units | Reversible thermo- and light-responsive fluorescent behaviors | 284 |
| CdSe/CdS/ZnS QD | Random copolymer or BCP of PEG methacrylate and N-methacrylol succinimide | Water dispersibility; stable interaction with QDs against L-glutathione | 189 |
| CdSe/CdS/ZnS QD | Copolymer consist of three monomers: PEG side chain monomer; imidazole groups containing unit; primary amines or biotin groups | Water dispersibility; antifouling; high stability; conjugation with dye or protein | 188 |
| CdSe/CdS/ZnS QD | Poly((methacrylamidosulfobetaine-co-N-(3-aminopropyl)methacrylamide hydrochloride)-b-(4-vinylimidazole)) | Extracellular stability; antifouling; bioconjugation | 112 |
| CdSe/ZnS and CdTe/ZnS QDs | Polymers consist of styrene, NIPAM, vinyl carbazoleand and 8-hydroxyquinoline derivative monomers | Multi-emission hybrid micelles | 241 |
| CdSe/ZnS QD | Poly(2-(N,N-diethylamino) ethyl methacrylate-b-poly(2-methacryloyloxyethyl phosphorylcholine-co-(n-Butyl methacrylate)-co-(p-Nitrophenyloxycarbonyl oligo(ethylene glycol) methacrylate)) conjugated with fluorescent dye | Water dispersibility; cytocompatibility; pH-sensitive FRET between QD and dye | 242 |
| CdSe/ZnS QD, Au | Poly(St) on AuNPs, poly(4-vinylpyridine) on QDs | By forming doubly alternating arrays of Au NPs and QDs within onion-like poly(St-b-(4-vinylpyridine)) BCP particle, the hybrid micropheres has a solvent-responsive fluorescence switching ability | 285 |
| CdSe/ZnS QD | PNIPM with end-functinalized fluorescent dye | Thermoresponsive dual photoemission nanosensor using LCST of PNIPAM and FRET between QD and dye | 286 |
| CdTe QD | Dye-labeled poly(N-(2-thiolethyl methacrylamide) | Water dispersibility; pH-controlled release of the DOX; FRET between donor QDs and acceptor dye on the polymer | 164 |
| CdTe QD | Poly(PEG methacrylate) with adenosine attached to the end of the PEG branches | Water dispersibility; antifouling; bioconjugation | 238 |
| GO | Poly(NIPAM-co-butyl methacrylate), poly(NIPAM-co-dimethylaminopropylacrylamide) and PNIPAM, all with addition fluorescent block | Precise detection of temperature between 25–45 °C within micronized domains using fluorescence color | 110 |
| GO and CdSe/ZnS QD | PAA and poly(2-vinylpyridine) | Efficient colorimetric pH sensor based on FRET between GO and QDs | 287 |
| GQD | Poly(7-(4-(acryloyloxy)butoxy)coumarin)-b-PNIPAM | pH, temperature and metal ion responsive multicolor fluorescence behavior based on FRET | 288 |
| IONP | Homopolymer or BCP of POEGA and PDMAEA | IONP siRNA nano-carriers with antifouling-shell; cytocompatibility | 181 |
| IONP | Thiabendazole imprinted PNIPAM | Reusable and specific recognition of benzimidazole | 269 |
| IONP | PNIPAM | Hyperthermia and heat-triggered drug-delivery | 187 |
| IONP | Poly(oligo(2-ethyl-2-oxazine) methacrylate) | Water dispersibility; negligible cytotoxicity; thermoresponsive behavior with tunable LCST | 180 |
| IONP | DBM-terminated poly(OEGA)-b-(poly(phosphonate acrylate)) | Water dispersibility; antifouling; bioconjugation; cell tracking via fluorescence labelling | 111 |
| IONP | PNIPAM-b-PAA | Thermal and pH dual responsive drug-delivery | 268 |
| Mesoporous silica NP | Poly(PEG acrylate) | Thermoresponsive drug-delivery with large cargo capacity | 289 |
| Montmorillonite | Poly(methyl acrylate) | Enhanced mechanical properties of poly(methyl acrylate) | 104 |
| MoS2 | P7AC-b-PNIPAM | Real-time photothermal heating and imaging using LCST of polymer and FRET between MoS2 and fluorescent polymer block | 114 |
| NaYF4 based UCNP | PAA-b-PEG with terminated RGD peptide | Cell imaging and labeling with drug-delivery functions | 183 |
| NaYF4:Yb/Er UCNP | POEGA-b-PAA, POEGA-b-poly(monoacryloxyethyl phosphate) and POEGA-b-poly(2-acrylamido-2-methyl-1-propanesulphonic acid) | Colloidal stability in water | 144 |
| PbS/CdS QD | Copolymer bearing carboxylic acid anchoring group and PEG chains | Long-term colloidal stability in isotonic saline; in vivo NIR imaging | 146 |
| Silica coated Fe3O4 NP | PNIPAM | Inhibition of magnetic interaction between Fe3O4 NPs cores | 264 |
| Silica coated Fe3O4 NP | Poly(NIPAM-co-GMA) with hydrazine moieties in GMA units | Temperature and pH-controlled drug release | 265 |
| Silica NP | Polyisoprene | Incorporation into a polyisoprene matrix | 290 |
| Silica NP | PAA-b-poly(St) | Proton conductive membrane | 291 and 292 |
| Silica NP | Poly(NIPAM-b-spirooxazine acryloyl-b-DMAEA) | Multi-responsiveness towards UV-vis light, pH, temperature | 293 |
The requirement of surface modification for nanomaterials regarding this topic is especially challenging when it comes to biomedical applications. Since numerous types of NPs must be synthesized using hydrophobic ligands, e.g., QDs, iron oxide NPs and UCNPs, these NPs are rendered non-dispersible in aqueous medium without modification.
Besides good water-dispersibility and long-term colloidal stability, the biomedical application further requires the nanomaterials to have high biocompatibility and minimal non-specific interaction in biological environment (antifouling). A very wide choice of capping polymers have been established for this purpose, including PEG,296,297 poly(2-hydroxyethyl methacrylate) (PHEMA),298 PVP,299,300 poly(2-oxazoline)180 and zwitterionic polymers.301,302 For general biological applications, PEG and its derivatives are the most widely used ones for their well-known character and commercial availability. Notably, the capping density is crucially important for the performance of PEG capped nanoparticles in biological environment.303,304 To create high capping density of grafted polymer, efficient and reliable anchoring strategies are required. Again, RAFT polymerization provides a very straightforward synthesis of functional polymers containing PEG derivatives with suitable anchoring groups for diverse nanomaterials.111,146,181,188,189,238
Zwitterionic polymers possess equal cationic and anionic moieties on the chain: the polymer maintains charge neutrality while carrying high ion density. This unique property makes them highly resistant to nonspecific protein adsorption due to their strong hydration ability resulting from the high charge density.302 RAFT polymerization can also polymerize carboxy betaine, sulfobetaine, and phosphorylcholine-based zwitterionic monomers and anchor these polymers onto various NPs.68,112,161,305
In many cases, the limiting factor for stability is the anchoring point between the polymer and surface. To avoid any detachment of the functional polymer, in addition, multiple anchoring groups can be introduced as anchoring blocks in the polymer design. Combing both hydrophilic block and anchoring blocks provides long-term colloidal stability across a wide pH-range and against contaminations.112,161,188,189
Lequeux and co-workers fabricated zwitterionic vinylimidazole BCPs using RAFT polymerization to decorate the surface of QDs (Fig. 6A). To immobilize the polymer onto the surface of QDs, they used a multidentate anchoring approach via the imidazole block. The sulfobetaine-zwitterion polymer provides robust and long-term stability. The inserted primary amines in the zwitterionic block allow for oriented bioconjugation with IgG antibodies and carry remarkable targeting performance towards specific proteins with a very low level of unspecific binding to live cells. Combining high stability provided by the polymer shell and the robust luminance performance of QDs, the observation time can exceed the 48 h window which is rarely feasible with conventional approaches.112
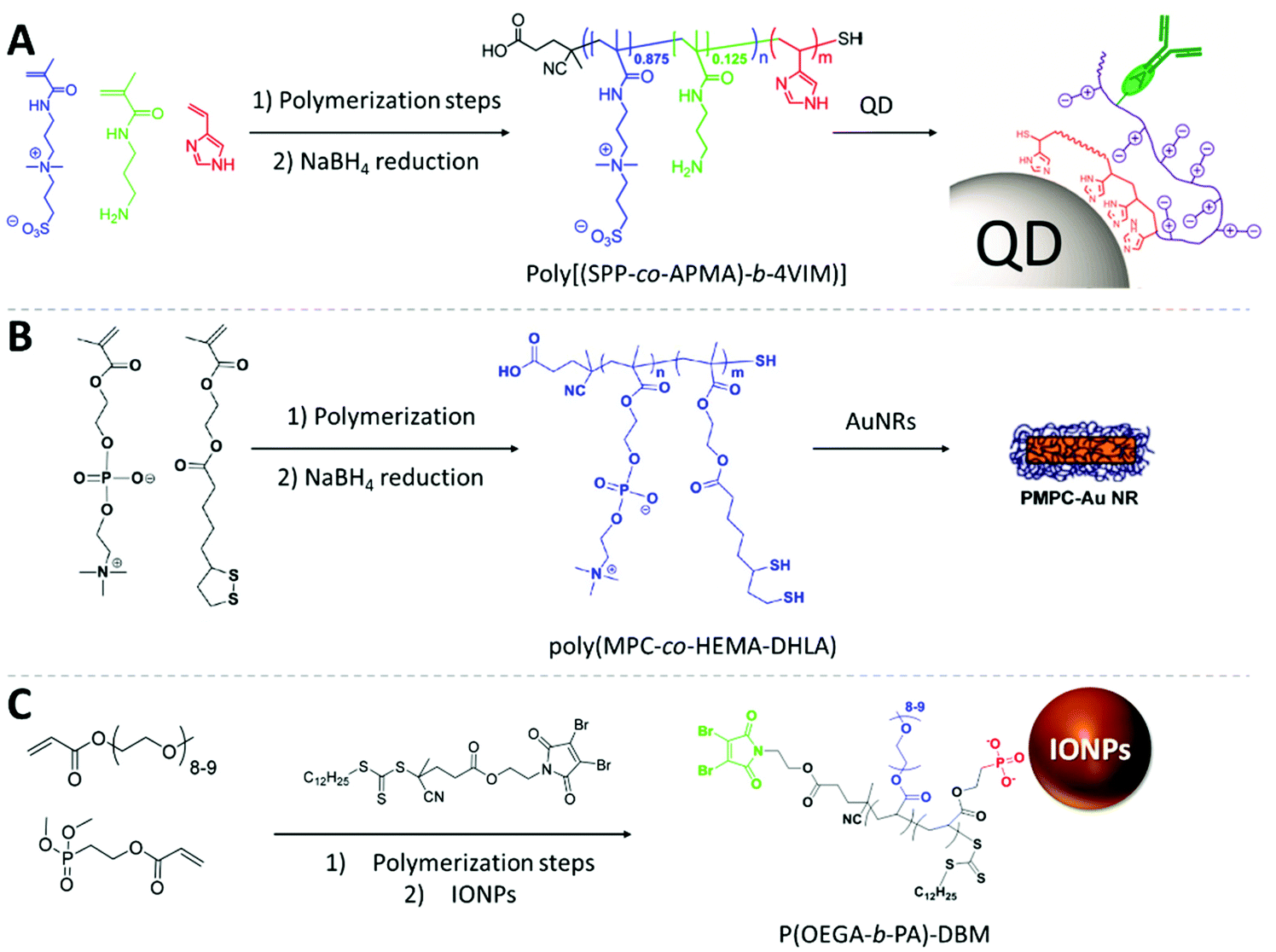 | ||
| Fig. 6 Fabrication strategies of RAFT copolymers and the illustrated core–shell polymer-capped NPs. (A) The 3-[3-methacrylamidopropyl-(dimethyl)-ammonio]propane-1-sulfonate-co-(N-(3-aminopropyl)methacrylamide hydrochloride) block serves as hydrophilic and bio-binding block. The 4-vinylimidazole block is used as a multidentate anchoring block for the QD surface. Adapted with permission from ref. 112. Copyright 2015 American Chemical Society. (B) The anchoring of the copolymers on AuNRs is enabled by the thiol groups on the HEMA-DHLA unit, and MPC zwitterionic units provide the antifouling property. Adapted with permission from ref. 161. Copyright 2013 American Chemical Society. (C) The phosphonate groups from the PA block anchor on the surface of IONPs, the POEGA block provides biocompatibility, the DBM end groups serves as binding site for further functionalization with fluorophores. Adapted with permission from ref. 111. Copyright 2018 American Chemical Society. | ||
Emrick and co-workers coated AuNRs with zwitterionic polymers with multi-thiol anchoring monomer units (Fig. 6B). A random copolymer was prepared by lipoic acid-substituted hydroxyethyl methacrylate (LA-HEMA) and methacrylamide phosphorylcholine (MPC) monomers. The polymer is then reduced with NaBH4 to convert RAFT and disulfide moieties of LA into thiols. They used a grafting-to approach to immobilize poly(MPC-co-HEMA-DHLA) onto AuNRs. Notably, the ligand exchange of the polymer with CTAB on AuNRs is challenging due to the dense protection provided by the original ligand: the CTAB excess must be removed carefully. Here, the multiply-anchoring moieties could have promoted the ligand exchange process. The poly(MPC-co-HEMA-DHLA)-capped AuNRs demonstrated exceptionally high stability against challenging conditions. The polymer shell even offers the resistance against cyanide ion etching for AuNRs. This structure was shown to be non-cytotoxic and demonstrated antifouling properties.161
Davis and co-workers have reported the synthesis of superparamagnetic iron oxide NPs coated with dibromomaleimide (DBM)-terminated BCPs consisting of PEG acrylate and phosphonate acrylate (PA) monomers (Fig. 6C). The PA block serves as a multiply-binding anchor for the iron oxide surface providing a stable grafting performance. The poly(oligoethylene glycol) methyl ether acrylate (POEGA) block offers biocompatibility. The DBM group was integrated in the R-group of the RAFT agent and remains at the brush termini after polymerization which allows for covalent conjugation with biomolecules via chemoselective reactions with both thiol and amino-terminal molecules. Using DBM moieties, they demonstrated the conjugation with an NIR dye and peptides. This work impressively demonstrated the possibility to introduce the highly favored bio-targeting and fluorescence tracking function to iron oxide NPs including high colloidal stability and anti-fouling character with only one type of RAFT polymer. The combination of all these features created an all-around nanostructure for cancer treatment.111
We have seen that both random and BCP design with hydrophilic monomers are used for enhancement of colloidal stability. An interesting study was published recently by Dunlap et al., who compared the relative stability between block and random copolymers consisting of PEG and imidazole-containing (QD-anchoring) monomers. They found that BCPs have a more stable interaction with QDs than random copolymers against L-glutathione. Also, higher molecular weights contributed to higher colloidal stability of QDs for both types of polymers.189
By anchoring the thermoresponsive polymer onto the surface of NPs, the cloud transition or aggregation can be effectively transferred to the nanostructure.309 This approach was employed successfully for a wide range of applications especially in the biomedical field including drug delivery, tissue engineering, bioimaging and hyperthermia.289,310–313 The convenient temperature-dependent reversible dispersibility also finds its applications in catalysis314,315 and sensing.316–318 Furthermore, combining the thermoresponsiveness with other types of stimuli-responsive polymers can create multi-responsive properties.319,320
Although the temperature-triggered aggregation of thermoresponsive polymer-capped NPs is a very useful function, the origin of these transitions is the release of solvent molecules attached to the polymer chain. As an example, the widely used poly(N-isopropylacrylamide) (PNIPAM) collapses above its lower critical solution temperature (LCST) due to a loss of most of its attached water molecules in the fully hydrated chain.321 Jones et al. have recently demonstrated the importance of residual free PNIPAM in the LCST-triggered aggregation of PNIPAM capped AuNPs. They have used five centrifugation cycles to completely remove unbound PNIPAM. Interestingly, only with the absence of free polymer, the polymer-capped AuNPs do not aggregate above the LCST.322 If the LCST-induced aggregation can be avoided, the LCST transition of the polymer can be applied as colloidal actuator to switch the length of the polymer in the hierarchical core–satellite nanostructures.154,168,171,174 Furthermore, the switch of the surface property can also trigger colloidal self-assembly of NPs into ordered structures.323,324 Polymer possessing upper critical solution temperature (UCST) behavior306,325 is also a topic of interest for nano-applications.326 Especially polymer simultaneously possesses both LCST and UCST (e.g., using block-copolymer approach) offers very interesting dual-responsiveness: (I) LCST < UCST (insoluable temperature window).327 (II) LCST > UCST (soluble window).113,328 Section 3.2.3 will give some detailed examples for the stimuli-responsive hybrid nanocomposites.
The synergies of thermoresponsive polymer brushes with its nanocore opened up possibilities to induce temperature change not only by simple heating, but also through other stimuli. Recent works have used nanocores to produce heat from e.g., NIR light,114,160,167,283 magnetic fields,187 and ultrasound.277 The highlight in these works includes the simultaneous drug release from the polymer shell: upon application of these external stimuli, the thermal therapy and drug therapy can be induced simultaneously.
RAFT polymerization technique also offers effective designs for creating pH-responsive polymer-functionalized core–shell nanostructures. Pourjavadi et al. demonstrated a very good example of the combination of pH and thermal dual responsive magnetic drug-delivery nanohybrids. They fabricated a copolymer of NIPAM and glycidyl methacrylate (GMA) monomers on the surface of silica coated Fe3O4 NPs via surface-initiated RAFT polymerization and introduced acid-labile hydrazine moieties on the epoxide ring of the GMA monomer. Doxorubicin (DOX) is then bound onto the hydrazine moieties which will be released at lower pH values (<5.4). The advantage of the double responsiveness is that the LCST transition of PNIPAM at higher temperatures helps to further “squeeze out” the remaining DOX on the GMA units and accomplishes a high drug release in a short time.265
Sahoo et al. also introduced pH and thermal dual responsive polymer shells onto Fe3O4 NPs. They first synthesized a BCP of PNIPAM and polyacrylic acid (PAA) block. The polymer is immobilized on the surface of amine-functionalized Fe3O4 NPs using carboxyl groups from the PAA block. DOX is then introduced onto the polymer shell using the charge interaction between positively charged DOX and negatively charged PAA. At a lower pH value (<5.0), the PAA block gets protonated causing the release of DOX and the LCST transition of the PNIPAM block at higher temperatures further induces a more complete drug release performance.268
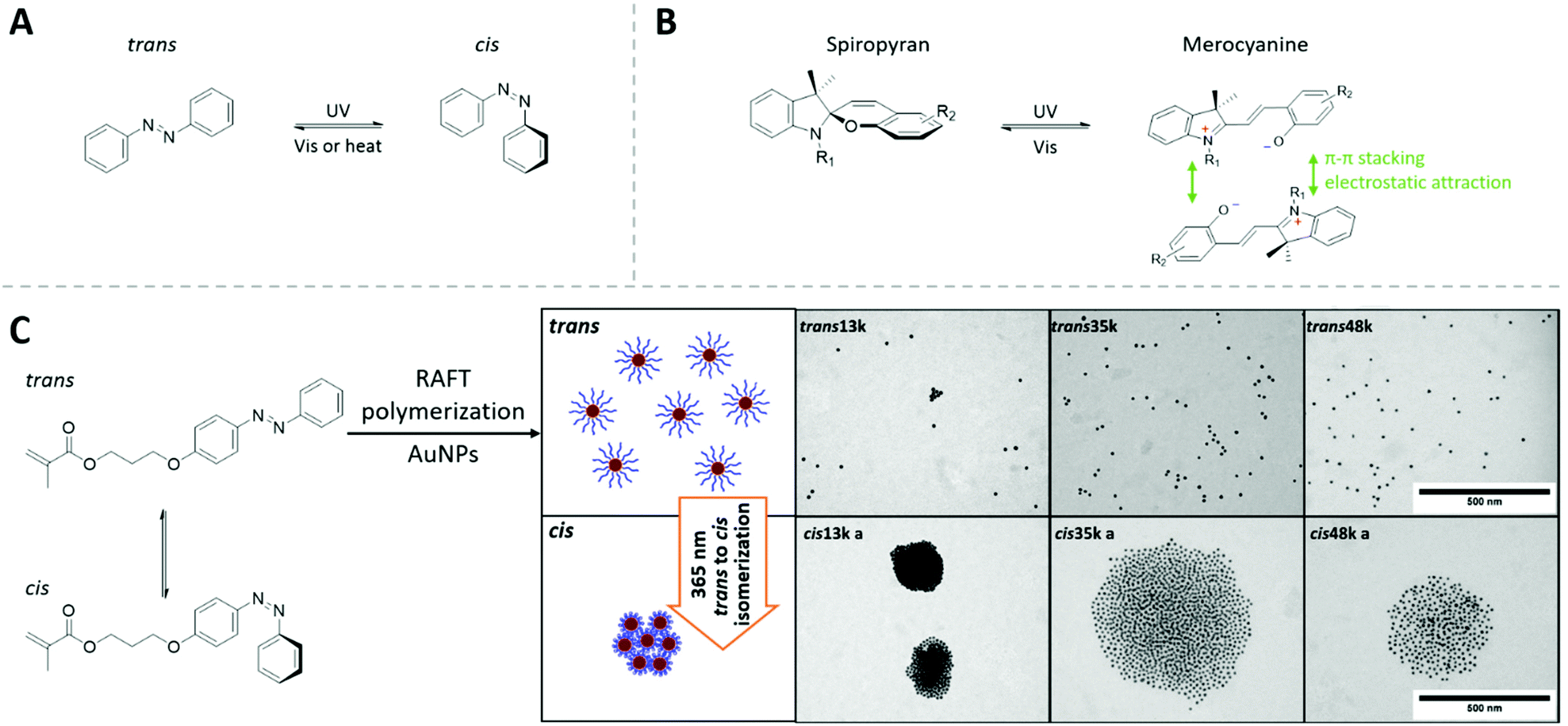 | ||
| Fig. 7 (A) The light-switchable cis-to-trans transition of azobenzene molecules. (B) The photon-switching mechanism of spiropyran derivatives between its spiropyran form and merocyanine form, as well as the attractive interaction between two merocyanine units. (C) Cis and trans azobenzene monomer and the light-switchable aggregation of AuNPs mediated by azobenzene-containing RAFT polymer. Adapted with permission from ref. 166. Copyright 2016 Elsevier Ltd. | ||
The light-switchable solubility and interaction can be introduced onto the surface of the NPs via small molecules containing these light-switchable units. Upon irradiation, these NPs tend to self-assembly into aggregates with controlled size (“supraspheres”) in suitable solvent conditions. The disassembly of these aggregates can be achieved via the switch-back process with the NPs returning to their dispersed state.335,336
The photoswitching property can be introduced to polymers, e.g., our group has demonstrated the fabrication of AuNPs covered with photoresponsive polymer shells made by azeobenzene-contanining methacrylic monomers using RAFT polymerization. In toluene, the light-induced cis-to-trans transition varied the interaction between polymer-capped AuNPs and induces the controlled aggregation of AuNPs. The tunable chain length of grafted polymer brush brought the advantage to control the interparticle-distance in a very straightforward manner (Fig. 7C).166
Zhang et al. synthesized AuNPs covered with spiropyran-containing polymers by surface-initiated ATRP polymerization. These nanostructures also self-assemble into AuNP oligomers upon light irradiation. Since these AuNPs are brought in very close range, their surface plasmonic coupling provides a strong surface-enhanced Raman (SER) signal in the oligomer state.337
The first requirement here is the overlap between donor emission and acceptor absorption. In general, materials with tunable and broad absorption and emission spectra are good candidates for energy transfer-based applications. Nobel metal nanocrystals, e.g., gold and silver nanocrystals, have broad plasmonic bands which are tunable by changing their size and aspect ratio, and thus can be used as super quenching unit.347 The unique electronic properties of graphene and MoS2 also make them perfect quenching materials.338,348–350 The fluorescent NPs including semiconductor QDs, graphene QDs (GQDs) and UCNPs have strong, broad and tunable absorbance and luminance, which make them good candidates as both donors338,351 and acceptors.352,353
The energy transfers process between the donor and acceptor is strongly dependent on their distance. In case of FRET, an efficient energy transfer only takes place when the distance between donor and acceptor is smaller than 10 nm.342 This makes nanomaterial-induced energy transfer perfect candidates as sensors to indicate near-field changes.
Smart polymers have the ability to change their size towards certain stimuli, for example, PNIPAM has a significantly shrunken size above its LCST. These stimuli-responsive polymers can be built between the donor and acceptor molecules, using their transition to switch on and off the fluorescence. Kim and coworkers have demonstrated a series of smart fluorescent nanosensors using diverse nanomaterials based on fluorescence energy transfer. These nanosensors are capable to detect thermal,110,288,354 photothermal,114 pH,287,288and metal ion288 changes due to a stimuli-responsive RAFT polymer linker.
In their recent publication, they used MoS2 as acceptor (strong FRET quencher) and a RAFT BCP containing a thermoresponsive PNIPAM block and a fluorescent poly(7-(4-(acryloyloxy)butoxy)coumarin) block (P7AC, emits blue light at ∼392 nm) as donor (Fig. 8). After RAFT polymerization, dithiolsulfide moieties were introduced to the BCPs on their chain ends (R-group), which provide strong anchoring on the surface of ultrathin MoS2 nanosheets. The BCP brush-capped MoS2 was then fabricated by a simple grafting-to experiment with the PNIPAM block positioned between the MoS2 nanosheet and the fluorescent block. MoS2 has very high absorption coefficient for both the visible and NIR region. Upon NIR irradiation, the photothermal ability of MoS2 induces the LCST transition of the PNIPAM block. After LCST transition, the PNIPAM block contracts and pulls the P7AC block close to MoS2 causing FRET quenching (reduced fluorescence intensity). They then dispersed BCP-MoS2 nanohybrids in a hydrogel and demonstrated the high-resolution (sub-micrometer) fluorescence thermal mapping during photothermal treatment.114
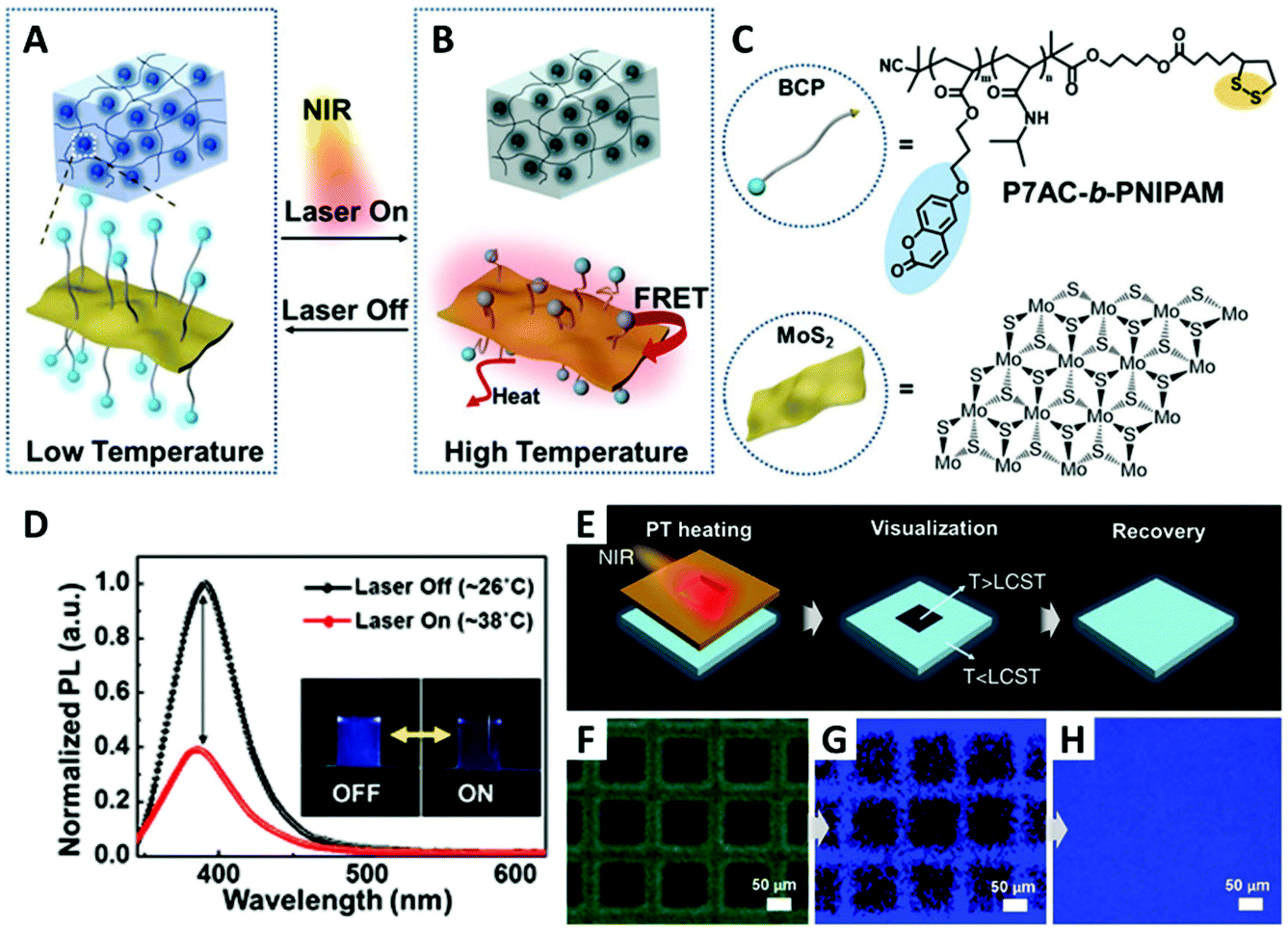 | ||
| Fig. 8 (A and B) Schematic illustration of the BCP-functionalized MoS2 nanosheets in hydrogel and the on/off transition controlled by NIR laser irradiation. Upon NIR irradiation, the PNIPAM block collapses due to the rise of local temperature, and the fluorescence of the P7AC block is quenched by MoS2. (C) Structure of P7AC-b-PNIPAM and MoS2. (D) The change of photoluminescence spectra of BCP-MoS2 nanocomposites by NIR light-induced heating. (E) Illustration of BCP-MoS2-film upon NIR irradiation through a photomask. (F) Optical microscope image of the BCP-MoS2-film visualized using the photomask. (G) Fluorescence image of selective local heating by NIR irradiation. (H) Recovered fluorescence image at 2 min after laser shutdown. Adapted with permission from ref. 114. Copyright 2017 WILEY-VCH Verlag GmbH & Co. KGaA. | ||
In another example, they have introduced three types of BCPs with blue, green, or red fluorescent polymer blocks (donor) onto the surface of graphene (acceptor/quencher) via pyrene anchoring groups (by a strong π–π stacking interaction) (Fig. 9). The three different fluorophore blocks are copolymerized with three thermal responsive polymer blocks with different LCSTs, respectively. The LCST of the thermoresponsive block was tuned to be 27, 32 and 39 °C for blue, green, red, respectively. The precise adjustment of LCST for PNIPAM is achieved by copolymerizing PNIPAM with different monomers. After the BCP is anchored to the surface of graphene, the PNIPAM block acts as thermoresponsive spacer between fluorophore donor and acceptor: by increasing temperature, the LCST is surpassed, resulting in PNIPAM block shrinkage, bringing the fluorescent block close to graphene and causing fluorescence quenching. In this way, the intensity of the three fluorescent colors represents three different LCSTs and offers sensing within a wide range of temperature between 25 to 45 °C.110
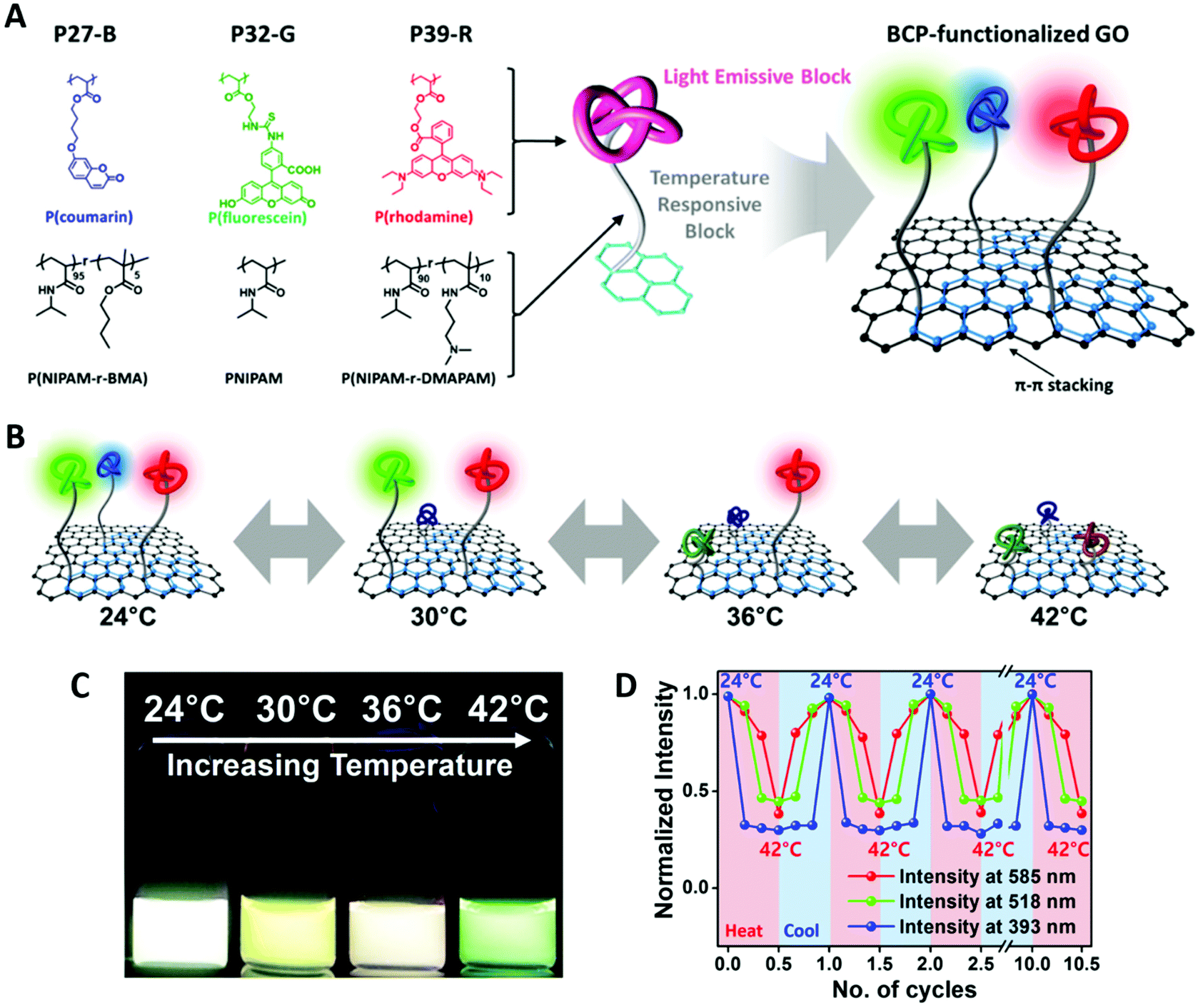 | ||
| Fig. 9 (A) Structures of pyrene-functionalized RAFT BCP with different thermoresponsive and fluorescent blocks (27 °C-blue, 32 °C-green, 39 °C-red). Schematic illustration of the BCP-GO nanohybrid. (B and C) Illustration and the actual color change of BCP-GO with increasing temperature. (D) Reversibility of the thermoresponsive photoluminescent behavior at three different wavelengths of the BCP-GO nanostructure. Adapted with permission from ref. 110. Copyright 2016 American Chemical Society. | ||
Furthermore, they exchanged the quenching GO nanosheet with green emitting GQDs. They used surface-initiated RAFT polymerization and synthesized PNIPAM-b-P7AC copolymers on GQDs. The GQD serves as the acceptor for the blue emitting P7AC block. If the temperature is above the LCST, the PNIPAM block collapses and the P7AC blocks enter suitable FRET ranges. As the result, the blue intensity drops distinctly while the green emission increases.288
RAFT polymers with pH-responsive conformation change, i.e., PAA and poly(2-vinylpyridine), can also be used as a responsive spacer in energy transfer systems. Kim and coworkers demonstrated multiple colorimetric pH sensors based on QDs (donor) and GO (quencher) with two types of QDs linked to the surface of GO using RAFT polymer linkers with distinct pH-responsiveness.287
Plasmonic noble metal NPs can also be used as quencher in energy transfer processes like NSET. Li et al.165 and Qiu et al.138 have both fabricated AgNPs decorated with RAFT copolymers carrying a block binding drug molecules via a UV or pH-cleavable bond. Furthermore, these drugs are fluorescent and both interact with AgNPs via NSET when they are attached to the surface-bound polymer, causing fluorescence quenching. After drug release upon UV or pH stimuli, the fluorescent drug recovers its fluorescent emission, enabling real-time monitoring of the drug release process.
In summary, we can clearly see the flexible role of stimuli-responsive RAFT polymers as tunable soft nanospacers and molecule carriers for energy transfer-related applications. The precise structural design and anchoring group positioning make RAFT polymers a perfect tool for connecting and adjusting the interaction between diverse energy donors and acceptors in a smart and dynamic manner.
3.2. Creating precise nanoassemblies using RAFT polymers
Recent advances are being made in the development of novel polymer-functionalized nanomaterials with the aim beyond the conventional definition of functions. The introduction of asymmetry, chirality, and interaction between each nanocomponent has triggered a very intriguing self-assembly process according to the corresponding properties. Transferring these symmetrical and directional codes from functional polymers (molecular level) to the assembly of NPs (nanoscale) requires innovations in the polymer design and a deep understanding of the colloidal interactions between polymers, solvent and nanosurfaces.Using RAFT polymers as structured linkers, well-defined hierarchical core–satellite nanoassemblies can also be created with tunable interparticle distance and interaction. The flexible polymerization technique and topological design enable the fabrication of polymer-capped core NPs with a strong ability to capture colloidal nanosatellite particles. By building stimuli-responsiveness into the polymer linker, the core–satellite NPs can smartly switch their structure and properties.
The challenge for manipulating the chirality of nanoassemblies is to create the chiral interaction between NPs. On this point, Cheng et al. impressively demonstrated a method to endow AuNRs with chirality utilizing the RAFT polymers poly(methacrylate hydroxyethyl-3-indole propionate) (PIPEMA) and PHEMA (Fig. 10A).192 Both polymers exhibit preferred-handed helical conformation, which could induce Au NRs to self-assemble into a left-handed rotating side-by-side structure (Fig. 10B and C) at high pH conditions. The chiral nanoassembly of AuNRs showed strong circular dichroism signals in the Vis-NIR region. Notably, both monomers are achiral and no chiral reagents are involved in the polymerization process, the chirality is originated from the preferred-handed helical conformation of the polymer main chain caused by their high syndiotacticity and their bulky pendant side groups as demonstrated by MD simulation.
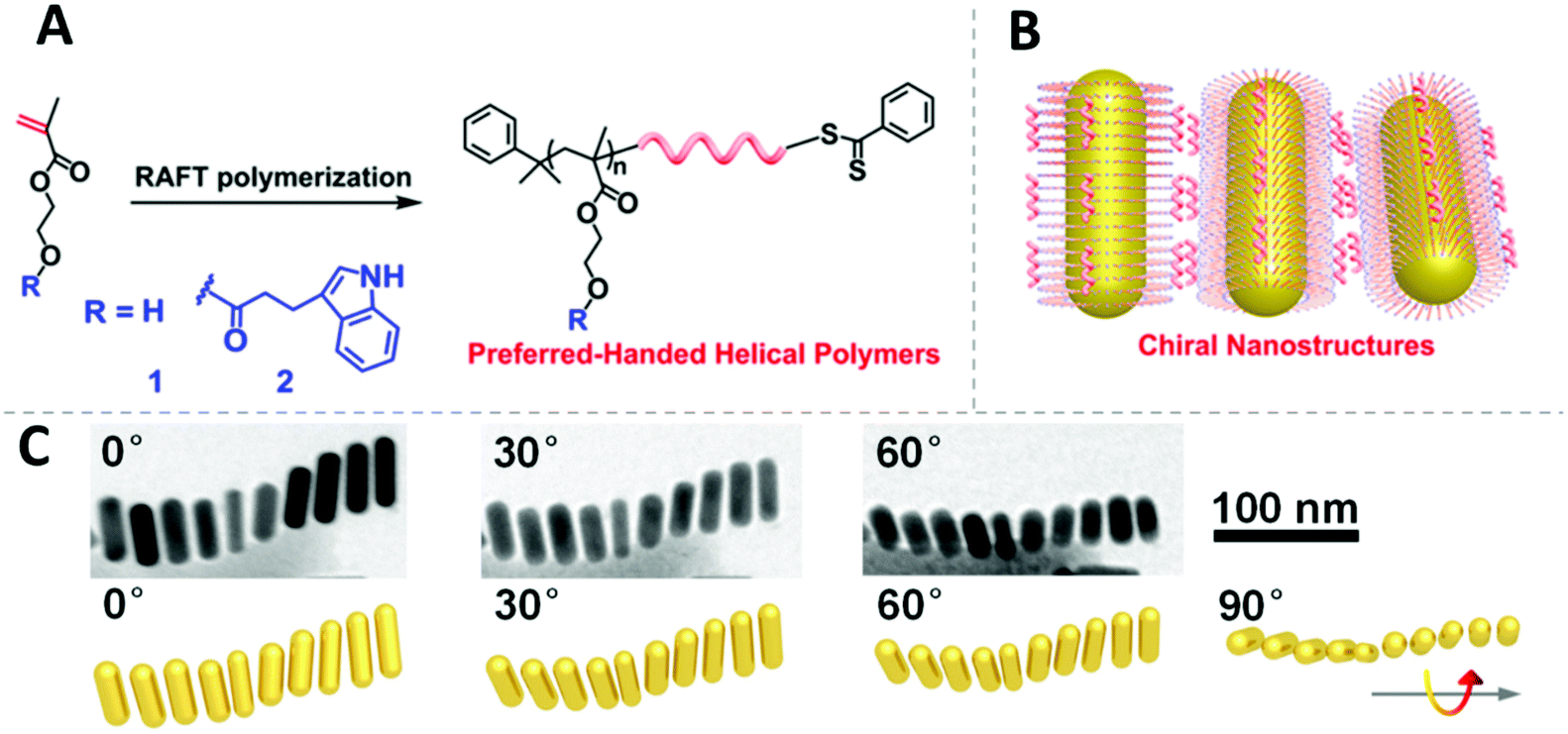 | ||
| Fig. 10 (A) Design and synthesis of the chiral RAFT polymer. (B) Illustration of chiral polymer capped AuNRs and the self-assembly of them. (C) The chiral arrangement of polymer capped AuNRs. Adapted with permission from ref. 192. Copyright 2019 American Chemical Society. | ||
On this topic, Rossner et al. reported an approach for fabricating patchy polymer shell-capped AuNPs using unfavorable solvent interaction (1st stimulus) and subsequent self-assembly of these asymmetrical NPs via their functional polymer patches upon application of a 2nd stimulus ([Cu2(OAc)4] complex). The two-step orthogonal stimuli-responsiveness requires sophisticated design of the BCP (Fig. 11A). AuNPs were capped with RAFT poly(styrene-block-(4-vinylbenzoic acid)) [poly(St-b-4VBA)], with poly(St) as NP-adjacent block and poly(4VBA) as NP-remote block. Upon reducing solvent quality, the Poly(St) block segregated to patches on the AuNP surface to minimize unfavorable polymer-solvent interaction, while the solvated Poly(4VBA) block stabilized the patchy NPs against aggregation. The subsequent self-assembly of these NPs is triggered by adding Cu2+ precursor complexes to the colloids: junction of patchy NPs was mediated via coordination of Cu24+ dimetal-ions by Poly(4VBA) blocks.368 Moreover, the size and shape of the self-assembled nanocluster are highly dependent on the BCP design: the short Poly(4VBA) block leads to a small, well-defined cluster containing 3–4 NPs (Fig. 11C(a)); while longer Poly(4VBA) blocks form rather large irregularly shaped assemblies (Fig. 11C(c)).169
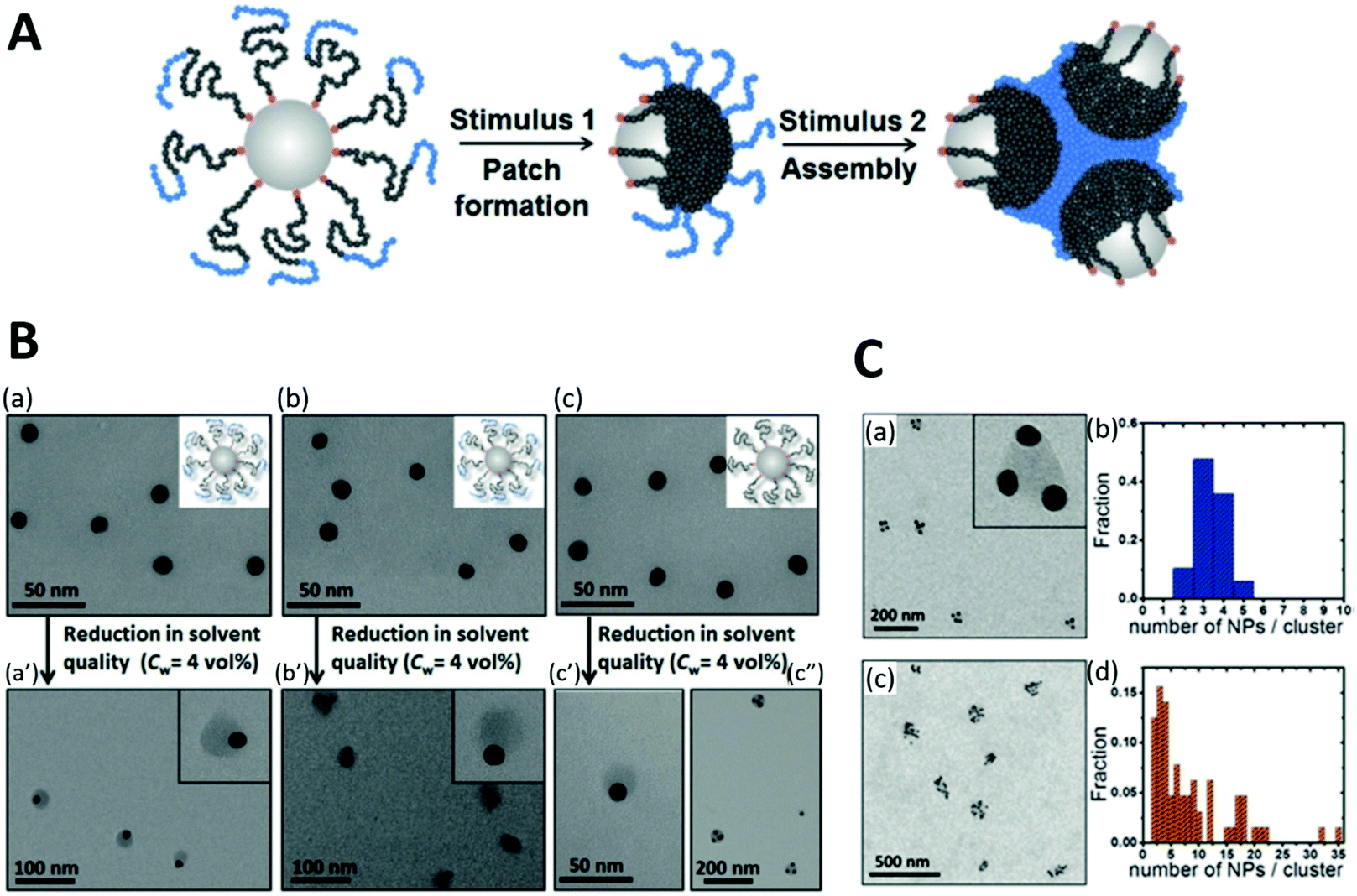 | ||
| Fig. 11 (A) Schematic illustration of the design principle for the stimuli-triggered surface patterning and self-assembly of AuNPs with block-copolymer ligands. (B) TEM images of AuNPs capped with BCP (a, b, distinct chain length) and poly(St) (c). a′, b′ and c′ are the corresponding patchy NPs yielded from the poorer solvent condition. (C) Self-assembly of patchy NPs after 2nd stimulus with a statistic of the number of NPs in each cluster. Adapted with permission from ref. 169. Copyright 2019 WILEY-VCH Verlag GmbH & Co. KGaA. | ||
Yi et al. have impressively demonstrated the fabrication of a series of directional core–satellite-like NP assemblies utilizing the charge interaction on the grafted BCP.196 In their work, gold, silver, and magnetite (Fe3O4) were used as building blocks for diverse nanoassemblies. Both RAFT and ATRP polymerization were applied to fabricate the BCPs used to form dense polymer shells on the NPs: thiol-terminated BCPs synthesized from RAFT polymerization were grafted onto gold and silver surfaces via simply mixing them under sonication. For anchoring polymers onto Fe3O4, phosphonate-terminated BCP was synthesized from ATRP polymerization.
To simultaneously introduce complementary reactive bonding between core and satellite NPs and the repulsive interaction between the charged polymer patches bonds, an acid–base neutralization reaction between two types of NPs carrying polymer brushes with opposite acid–base groups was applied. Those acid/base functionalities are tuned with the build-in moieties in the BCP using acrylic acid (AA) as acid and N,N-dimethylaminoethyl methacrylate (DMAEMA) as the base monomer. The molecular design of these acid/base polymers is illustrated in Fig. 12A, which is comprised of two blocks: the first polymer block consists of a statistic copolymer of acid/base and styrene monomers, the ratio of both monomers in this block was kept constant (∼30%) while the chain length is varied to adjust the content of the acid/base moieties in the polymer shell. The second PEO block is acting as a steric stabilizer of the NPs.
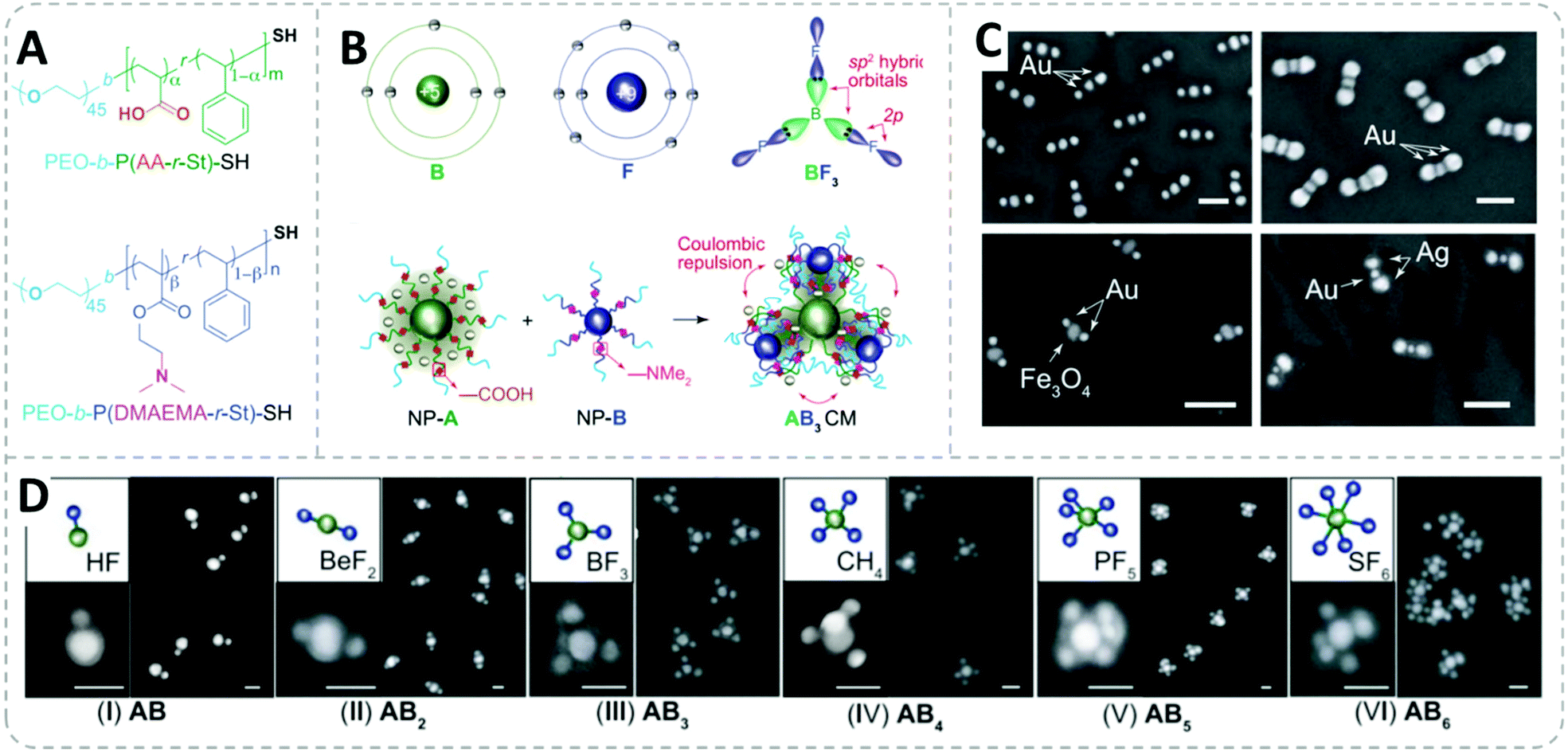 | ||
| Fig. 12 (A) Molecular structure of the BCPs used for surface modification of AuNPs and AgNPs. (B) Schematic illustration of an sp2-hybridized BF3 molecule. Illustration of directional bonding of NP-A and NP-B and the formation of an AB3 structure by the stoichiometric reaction between the complementary reactive polymer shells. (C) SEM images of AB2 nanoclusters formed from diverse types of NPs, scale bar = 100 nm. (D) SEM images of ABx structures (x = 1 to 6) assembled from 36 nm AuNPs (A) and 20 nm AuNPs (B). The change in the number and geometry of the satellite NPs is controlled by increasing the chain length of the poly(DMAEMA-r-St) block. Adapted with permission from ref. 196. Copyright 2020 American Association for the Advancement of Science. | ||
Once the acid copolymer-capped NPs are mixed with the base copolymer-capped NPs, the interparticle neutralization reaction takes place. The neutralization yields a strong ionic attractive interaction which provides a powerful binding effect between the two types of NPs, resulting in “colloidal molecules” nanoassemblies (Fig. 12B). The charged polymer regimes also confer a repulsing interaction between the polymer brushes of adjacent satellite NPs, thus leading to a highly ordered geometry (Fig. 12C and D). Here, the Coulomb repulsion can be analogously compared with the valence electron pair model which can predict the geometry of simple molecules in ABx form. Now the same principles and rules can be applied to the “colloidal molecules” nanoassemblies. To adjust the number of satellite NPs in each nanostructure, they varied the chain length of the polymer shell of the satellite. Since the ratio of the DMAEMA is kept the same in the polymer chain, a shorter polymer brush chain contains fewer base groups which resulted in a smaller neutralization capacity of the satellites, which then allowed the acid core NP to catch more satellite NPs. As a result of the interaction brought by the reactive polymer shell, the 3D structure of the nano “colloidal molecules” including the arrangement and the number of the satellite particles can be precisely tuned by only engineering the chain length of the surface BCP brush.
Choosing appropriately designed synthetic polymers as a linker brings multiple advantages such as enforced colloidal stability owing to the steric shielding of a polymer shell (as we have seen in section 3.1), the tunable interparticle distance between planet–satellite,109,168,175 and last but not least, its high efficiency and low cost to produce. As a linker for nanomaterials, the design flexibility in the macromolecular structure including topology, end-group functionality and the choice of monomers/BCPs offers very open access for creating hierarchical nanostructures with desired functions.
In general, the number of free anchoring end groups for satellite particles on the polymer-capped core NP surface is the key for the self-assembly process. For the case that both core and satellite are noble metal NPs, the multi-arm polymer with anchoring groups at each chain termini is a widely used approach. Dey et al. demonstrated a practical example to increase the free end groups using hyperbranched polymers carrying multiple anchoring RAFT and alkyne end groups (Fig. 13).207 The nanostructure made of AuNPs presents a well-defined structural arrangement with a tunable number of satellite NPs. Due to both the structural nature of the hyperbranched polymer and its low molecular weight (∼10 kg mol−1), the interparticle distance between core and satellites was very short. This close distance between the NPs induced a strong SER effect which can be easily tuned by the number of attached satellite AuNPs (Fig. 13D).
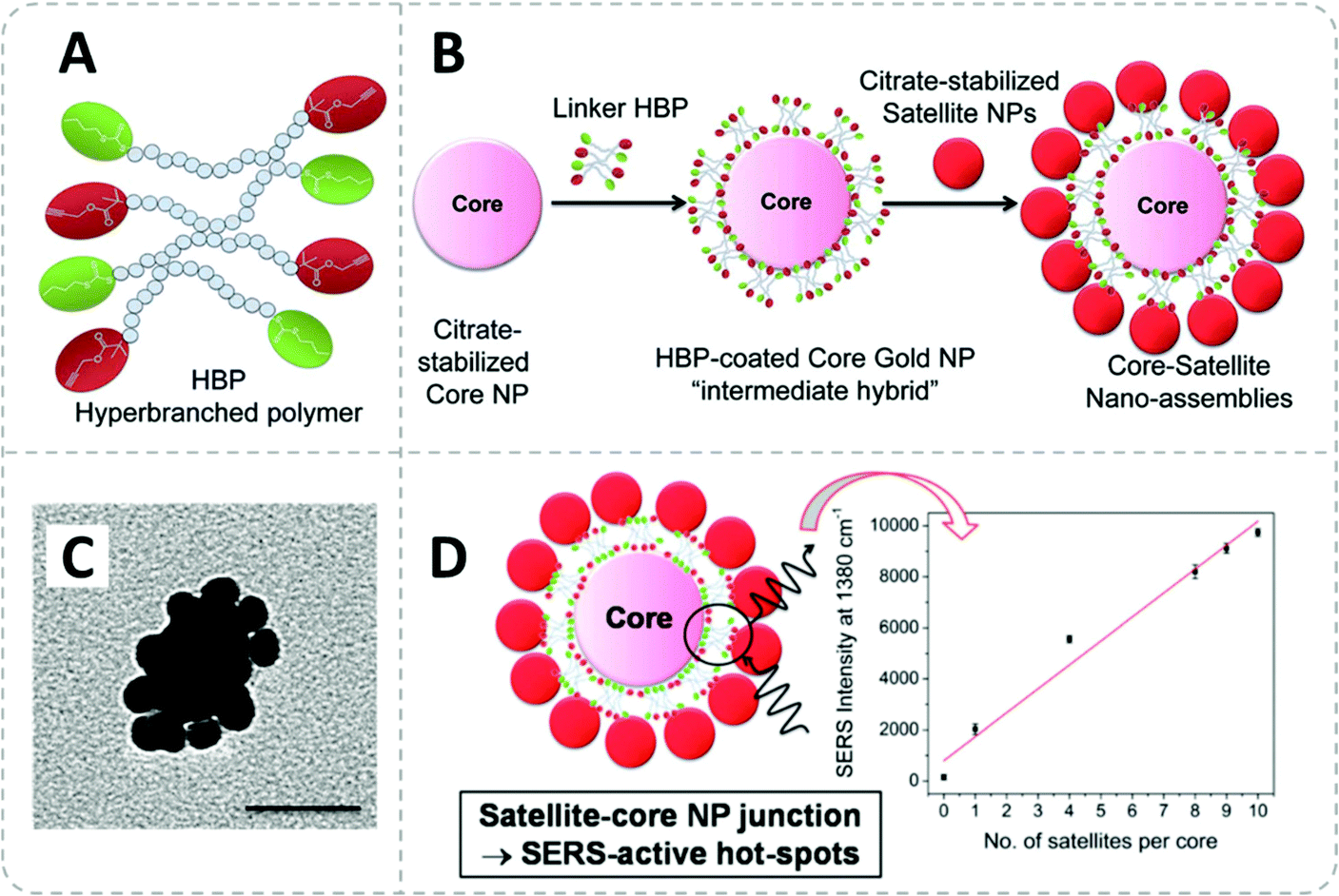 | ||
| Fig. 13 (A) Structural design of hyperbranched polymer. (B) Schematic illustration of the fabrication strategy of core–satellite nanostructures using hyperbranched polymer linkers. (C) TEMicrograph of core–satellite nanostructure with a satellite/core ratio of 10. Scale bar = 50 nm. (D) Increasing SER scattering intensity with growing number of satellites per core. Adapted with permission from ref. 207. Copyright 2014 The Royal Society of Chemistry. | ||
To form a connection between two NPs, at least two end groups are required. However, if both end groups have attached to the surface of the core NP, the linking effect becomes unavailable: linear bi-end-functionalized polymers have shown diminished performance of forming core–satellite nanostructures compared with star polymers containing more than three arms.379 Gooding and coworkers have reported a method to first saturate the planet AuNPs with thiol end-functionalized polymers carrying a carboxylic acid group on the other terminus.113,156 An additional step was applied to convert the outer carboxylic acid group into thiol groups which allows for self-assembly of additional satellite AuNPs.
Our group has reported a method to prepare noble metal core–satellite nanostructures based on multi-arm star PNIPAM with RAFT end groups as linkers (Fig. 14).109,175 The planet is constructed by self-assembly of star RAFT polymers onto citrate-capped AuNPs using the grafting-to approach. The free (not surface-bound) polymer arms of the grafted polymer can capture satellite NPs.379 The same planet nanostructure is able to capture both gold (Fig. 14A)109 and silver (Fig. 14B)175 satellite NPs. Notably, the well-defined structure of the star polymer offers full control over the arrangement of the satellite nanocomponents: the average distance between planet satellites NPs can be precisely tuned between approximately 3 to 20 nm by varying the molecular weight of the linker.
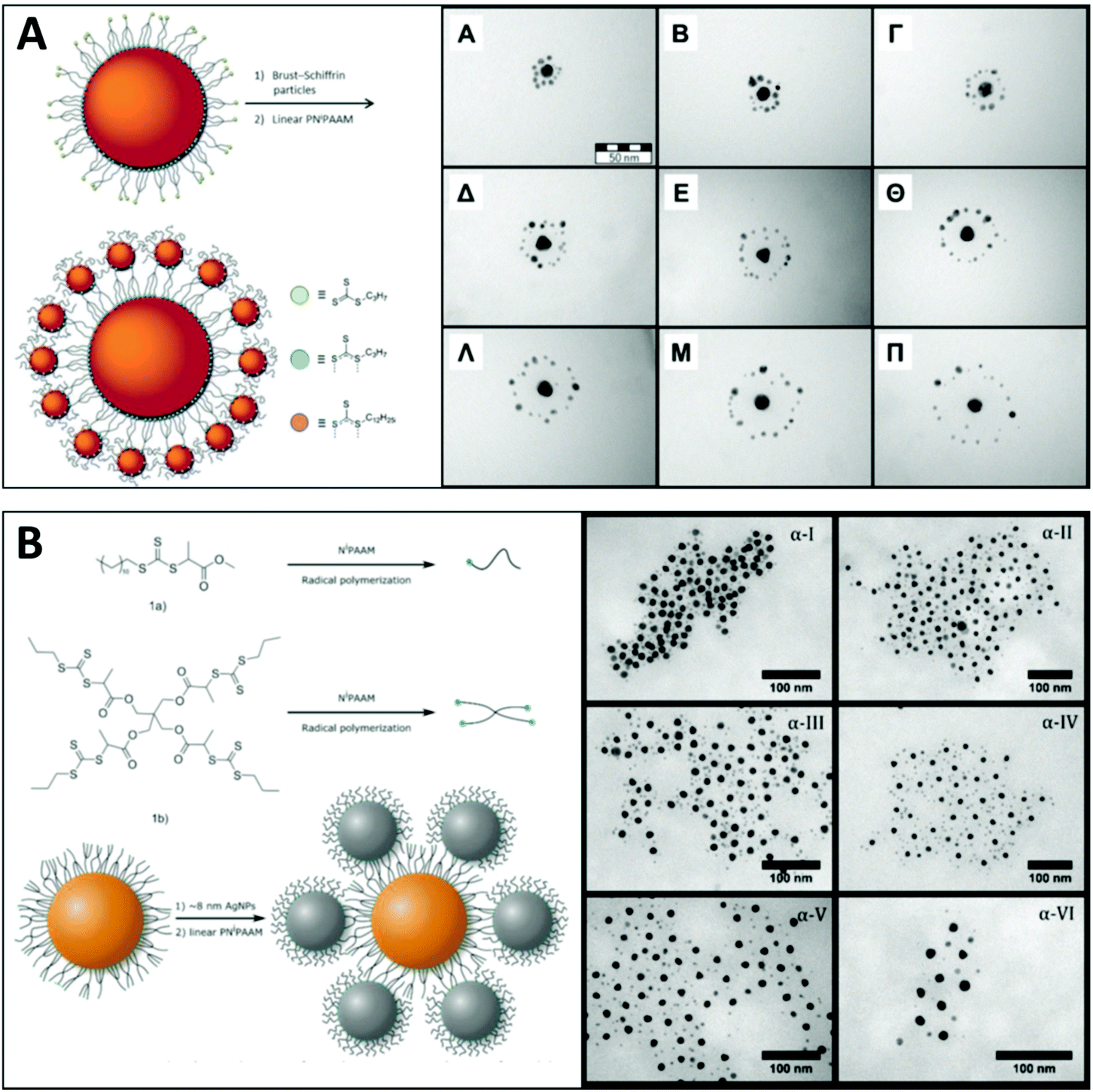 | ||
| Fig. 14 (A) Synthetic strategy (left) and TEMicrograph of gold-planet–gold-satellite nanostructures. Adapted with permission from ref. 109. Copyright 2014 WILEY-VCH Verlag GmbH & Co. KGaA. (B) Structure of star and linear RAFT agents, synthetic scheme, and TEMicrograph of gold-planet–silver-satellite nanostructures. Adapted with permission from ref. 175. Copyright 2016 American Chemical Society. | ||
Sulfur-terminated polymers with suitable topological designs have been proven to be a powerful platform for the self-assembly of hierarchical nanostructures made from noble-metal nanomaterials. However, for constructing structures with non-metal NPs, other approaches are inevitably required. As building material for hierarchical nanostructures, silica brings very intriguing potential owing to the intensively studied silica coating methods which allow almost every nanomaterial to be introduced as a planet. To bring polymer brushes onto the silica surface, the well-established silane chemistry offers a great variety of protocols.380,381 However, creating well-defined silica planets with a dense polymer shell requires an effective route and careful treatment of silica NPs for their tendency to form aggregation. For this task, the surface-initiated RAFT polymerization via grafting-from has been proven to be suitable.95,99,264 By designing the surface-initiated RAFT polymerization using an R-approach, the RAFT moieties will be positioned at the outer termini of the polymer shell which are capable for attaching to satellite NPs. Based on this principle, Wu et al. and Tian et al. have both reported protocols for fabricating AuNPs decorated silica-RAFT PNIPAM core–satellite nanostructures (Fig. 15).154,168 The nanoassemblies showed a well-defined structure with full control over the number of satellite NPs154 and interparticle distance.168 Tian et al. also demonstrated the access of introducing further Fe3O4 NPs in the core position using the silica coating technique.168 These complicated nanostructures can be fabricated using a simple synthetic method and a very efficient route.
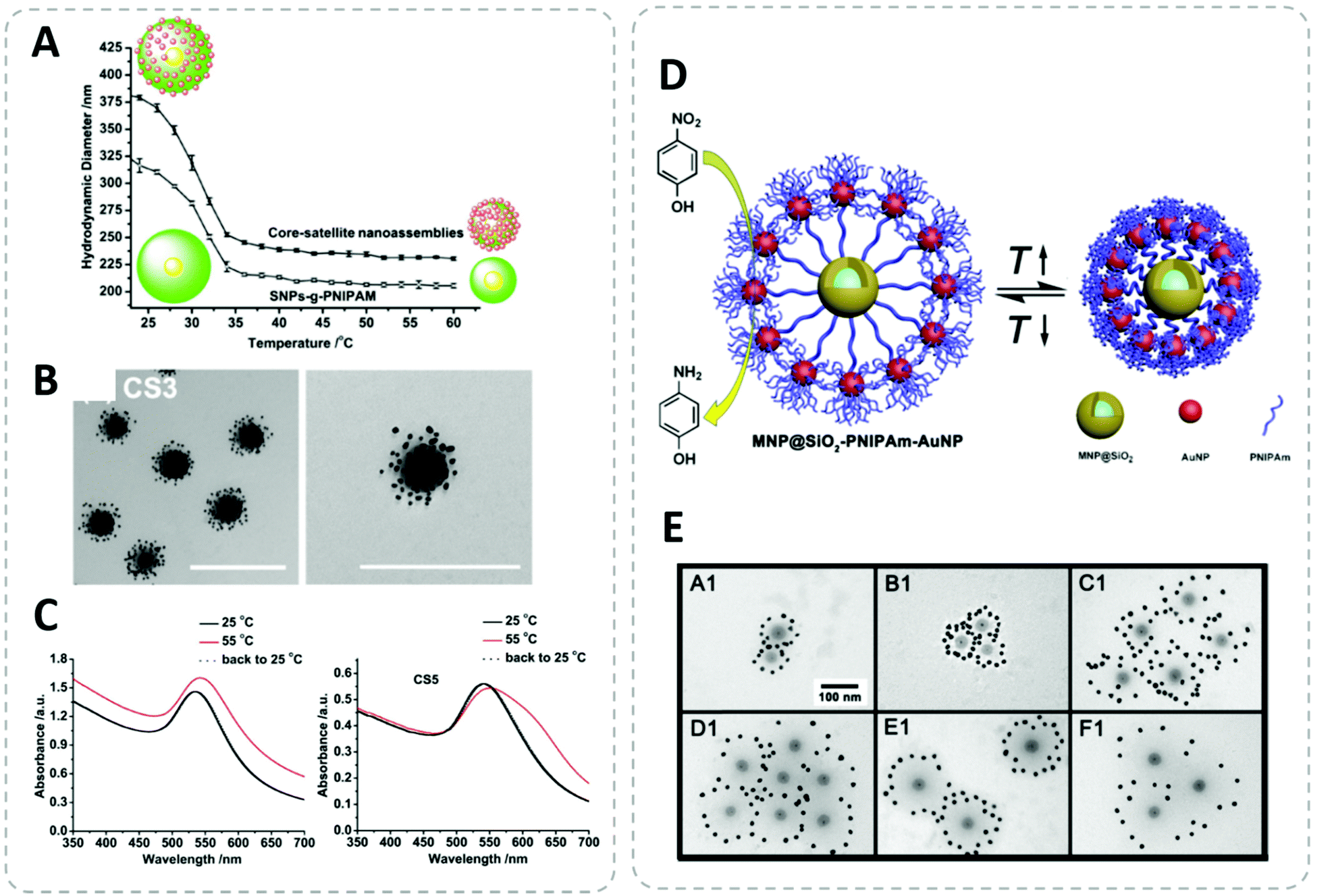 | ||
| Fig. 15 (A) Temperature dependence of the hydrodynamic diameter of PNIPAM-functionalized silica-core–gold-satellite nanoassemblies and PNIPAM-capped silica NPs (core). (B) TEMicrograph of silica-core–gold-satellite nanoassemblies. (Scale bar = 500 nm) (C) UV-vis spectra of silica-core–gold-satellite nanostructures at different temperatures. The structure for the right spectrum has a higher gold/silica ratio. The additional absorption originates from the surface plasmonic coupling in the shrunken state. A–C: Adapted with permission from ref. 154. Copyright 2017 WILEY-VCH Verlag GmbH & Co. KGaA. (D) Schematic illustration of the fabrication strategy of Fe3O4NP@silica–PNIPAM–AuNPs nanostructures. (E) TEMicrograh of Fe3O4NP@silica–PNIPAM–AuNPs nanostructure with increasing molecular weight of the PNIPAM linker. D–E: Adapted with permission from ref. 168. Copyright 2019 American Chemical Society. | ||
Due to the close distance between the NPs brought by the polymer linker, the nanostructure often shows a strong surface plasmonic coupling between noble metal NPs, which has the potential to be used for SER applications.113,207 Moreover, a stimuli-responsive polymer linker, e.g., PNIPAM, also brings its ability to these core–satellite nanostructures as a thermoresponsive colloidal actuator reversibly switching the structure between hydrated and shrunken state.113,154,171 The temperature-triggered switch of the interparticle distance in the nanostructure also induces a shift in the optical properties of the colloid, which may render them as flexible candidates for nanosensors.
Notably, the versatile silica-coating technique enables a wide choice of nanomaterials for the role of the core and makes this strategy a general protocol to fabricate core–satellite nanostructures with tunable distance and interactions.
4. Conclusions and outlook
It was demonstrated that RAFT polymers can nicely be used to decorate and/or connect nanoparticles of different nature in order to arrive at complex and functional nanocomposites. The success of RAFT polymerization in this field is clearly due to the great versatility in terms of polymerization reaction conditions as well as in terms of polymer-to-surface grafting strategies. It has for instance been shown that RAFT polymer with its thiocarbonylthio-moeties as reactive end-groups can often directly be used in grafting-to approaches without preceding end-group transformation. This clearly simplifies nanoengineering with these reactive building blocks. Another great advantage of RAFT is its good tolerance against water-based systems, which is key for bio-related applications.While we observe that RAFT polymers and the nanocomposites formed from them are constantly finding new fields of application, the question arises as to what the next big goals are in order to achieve a higher level in quality and function. While we can't see into the future, we think the following goals will be relevant and hopefully solved in the near future:
1. The combination of different types of polymers and different types of particles within one nanocomposite will become important. Some first initial work into this direction has been performed, but increasing the control over different types of nanocomponents will enhance the functionality. By tuning the interplay of components, e.g., via different stimulus sensitivity and or via orthogonal (surface) chemistry, the functionality of such nanohybrids can be greatly enhanced and more than one function, e.g., imaging and disease treatment at the same time, may become possible.
2. In the early days of this scientific field, polymer was mainly applied to the surface of inorganic materials using grafting-from methods. However, progress in surface chemistry using polymers has shown that similarly good end results can be achieved with grafting-to methods, while the synthetic effort is greatly reduced. With this synthetic simplification, however, the complexity of the overall system can be increased at the same time while the overall expenditure remains the same. These functional nanohybrids, after all, need to remain accessible in short times and with an acceptable spending of resources. Efforts must therefore be made to further simplify the grafting-to methods for a large number of surfaces, including those that are less reactive. An effective grafting-to method for silica surfaces would, e.g., be a huge step forward.
3. Most of the studies so far have addressed the action of these functional nanocomposites in solution, where they have been formed. Little is known so far about the behavior of these nanoarchitectures when cast to a surface or how their function can be exploited in a dry bulk material state. Ordering behavior and regularity control in the dry state will be future challenges to exploit these nanohybrids in the design of novel functional materials.
It is fascinating to see, how RAFT polymerization evolved during the last years from a chain-transfer effect that is capable of controlling radical polymerization to a powerful tool for constructing functional nanocomposites. It is very likely, that this is not the end of the story and that many exciting new developments using RAFT polymers will emerge.
Abbreviations
| AA | Acrylic acid |
| ATRP | Atom transfer radical polymerization |
| BCP | Block copolymer |
| CNT | Carbon nanotube |
| CSIRO | Commonwealth scientific and industrial research organization |
| CTA | Chain transfer agent |
| CTAB | Cetyltrimethylammonium bromide |
| DBM | Dibromomaleimide |
| DMAEMA | N,N-Dimethylaminoethyl methacrylate |
| DOX | Doxorubicin |
| FRET | Förster resonance energy transfer |
| GMA | Glycidyl methacrylate |
| GO | Graphene oxide |
| GQD | Graphene quantum dot |
| IONP | Iron oxid nanoparticle |
| LA-HEMA | Lipoic acid-substituted hydroxyethyl methacrylate |
| LCST | Lower critical solution temperature |
| MPC | Methacrylamide phosphorylcholine |
| NHS | N-Hydroxysuccinimide |
| NIR | Near-infrared |
| NP | Nanoparticle |
| NR | Nanorod |
| NSET | Nanometal surface energy transfe |
| P7AC | Poly(7-(4-(acryloyloxy)butoxy)coumarin) |
| PA | Phosphonate acrylate |
| PAA | Polyacrylic acid |
| PEG | Polyethylene glycol |
| PHEMA | Poly(2-hydroxyethyl methacrylate) |
| PIPEMA | Poly(methacrylate hydroxyethyl-3-indole propionate) |
| PNIPAM | Poly(N-isopropylacrylamide) |
| POEGA | Poly(oligoethylene glycol) methyl ether acrylate |
| Poly(4VBA) | Poly(4-vinylbenzoic acid) |
| Poly(St) | Poly(styrene) |
| PRET | Plasmon resonance energy transfer |
| PVP | Polyvinylpyrrolidone |
| QD | Quantum dot |
| RAFT | Reversible addition–fragmentation chain-transfer |
| RDRP | Reversible deactivation radical polymerization |
| SER | Surface-enhanced Raman |
| TEOS | Tetraethylorthosilicate |
| TOAB | Tetraoctylammonium bromide |
| UCNP | Upconverting nanoparticle |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of our work by the Fonds der Chemischen Industrie is gratefully acknowledged.References
- J. Chiefari, Y. K. B. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polym. Int., 2011, 60, 9–25 CrossRef CAS.
- A. Skandalis, T. Sentoukas, D. Giaouzi, M. Kafetzi and S. Pispas, Polymers, 2021, 13, 1698 CrossRef CAS PubMed.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC.
- M. Semsarilar and V. Abetz, Macromol. Chem. Phys., 2021, 222, 2000311 CrossRef CAS.
- J. M. Ren, T. G. McKenzie, Q. Fu, E. H. H. Wong, J. Xu, Z. An, S. Shanmugam, T. P. Davis, C. Boyer and G. G. Qiao, Chem. Rev., 2016, 116, 6743–6836 CrossRef CAS PubMed.
- D. Kuckling and A. Wycisk, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2980–2994 CrossRef CAS.
- Y. Zheng, S. Li, Z. Weng and C. Gao, Chem. Soc. Rev., 2015, 44, 4091–4130 RSC.
- S. I. Bhat, Y. Ahmadi and S. Ahmad, Ind. Eng. Chem. Res., 2018, 57, 10754–10785 CrossRef CAS.
- Z. Li, M. Tang, S. Liang, M. Zhang, G. M. Biesold, Y. He, S.-M. Hao, W. Choi, Y. Liu, J. Peng and Z. Lin, Prog. Polym. Sci., 2021, 101387 CrossRef CAS.
- G. Moad, Polym. Chem., 2016, 8, 177–219 RSC.
- C. Lu and M. W. Urban, Prog. Polym. Sci., 2018, 78, 24–46 CrossRef CAS.
- C. Zhao, Z. Ma and X. X. Zhu, Prog. Polym. Sci., 2019, 90, 269–291 CrossRef CAS.
- N. Deirram, C. Zhang, S. S. Kermaniyan, A. P. R. Johnston and G. K. Such, Macromol. Rapid Commun., 2019, 40, 1800917 CrossRef PubMed.
- B. D. Fairbanks, P. A. Gunatillake and L. Meagher, Adv. Drug Delivery Rev., 2015, 91, 141–152 CrossRef CAS PubMed.
- A. Zhang, K. Jung, A. Li, J. Liu and C. Boyer, Prog. Polym. Sci., 2019, 99, 101164 CrossRef CAS.
- M. Ahmed and R. Narain, Prog. Polym. Sci., 2013, 38, 767–790 CrossRef CAS.
- P. Gurnani and S. Perrier, Prog. Polym. Sci., 2020, 102, 101209 CrossRef CAS.
- X. Jin, P. Sun, G. Tong and X. Zhu, Biomaterials, 2018, 178, 738–750 CrossRef CAS PubMed.
- R. Yang, X. Wang, S. Yan, A. Dong, S. Luan and J. Yin, Prog. Polym. Sci., 2021, 118, 101409 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Chem. – Asian J., 2013, 8, 1634–1644 CrossRef CAS PubMed.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 CrossRef PubMed.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- J. Yeow and C. Boyer, Adv. Sci., 2017, 4, 1700137 CrossRef PubMed.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- J. O. Zoppe, N. C. Ataman, P. Mocny, J. Wang, J. Moraes and H.-A. Klok, Chem. Rev., 2017, 117, 1105–1318 CrossRef CAS PubMed.
- J. Gaitzsch, X. Huang and B. Voit, Chem. Rev., 2016, 116, 1053–1093 CrossRef CAS PubMed.
- C. K. Wong, X. Qiang, A. H. E. Müller and A. H. Gröschel, Prog. Polym. Sci., 2020, 102, 101211 CrossRef CAS.
- K. Nagase, T. Okano and H. Kanazawa, Nano-Struct. Nano-Objects, 2018, 16, 9–23 CrossRef CAS.
- K. Jung, N. Corrigan, M. Ciftci, J. Xu, S. E. Seo, C. J. Hawker and C. Boyer, Adv. Mater., 2020, 32, 1903850 CrossRef CAS PubMed.
- Z. Zhang, N. Corrigan, A. Bagheri, J. Jin and C. Boyer, Angew. Chem., Int. Ed., 2019, 58, 17954–17963 CrossRef CAS PubMed.
- G. Palui, F. Aldeek, W. Wang and H. Mattoussi, Chem. Soc. Rev., 2015, 44, 193–227 RSC.
- X. Huang, J. Hu, Y. Li, F. Xin, R. Qiao and T. P. Davis, Biomacromolecules, 2019, 20, 4243–4257 CrossRef CAS PubMed.
- N. Zhao, L. Yan, X. Zhao, X. Chen, A. Li, D. Zheng, X. Zhou, X. Dai and F.-J. Xu, Chem. Rev., 2019, 119, 1666–1762 CrossRef CAS PubMed.
- S. O. Pereira, A. Barros-Timmons and T. Trindade, Polymers, 2018, 10, 189 CrossRef PubMed.
- X. Bouju, É. Duguet, F. Gauffre, C. R. Henry, M. L. Kahn, P. Mélinon and S. Ravaine, Adv. Mater., 2018, 30, 1706558 CrossRef PubMed.
- M. A. Boles, M. Engel and D. V. Talapin, Chem. Rev., 2016, 116, 11220–11289 CrossRef CAS PubMed.
- M. Grzelczak, L. M. Liz-Marzán and R. Klajn, Chem. Soc. Rev., 2019, 48, 1342–1361 RSC.
- K. Deng, Z. Luo, L. Tan and Z. Quan, Chem. Soc. Rev., 2020, 49, 6002–6038 RSC.
- W. B. Rogers, W. M. Shih and V. N. Manoharan, Nat. Rev. Mater., 2016, 1, 1–14 Search PubMed.
- C. Yi, S. Zhang, K. T. Webb and Z. Nie, Acc. Chem. Res., 2017, 50, 12–21 CrossRef CAS PubMed.
- S. Samanta, S. L. Banerjee, K. Bhattacharya and N. K. Singha, ACS Appl. Mater. Interfaces, 2021, 13, 36307–36319 CrossRef CAS PubMed.
- N. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS.
- C. Liu, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2020, 11, 3673–3689 RSC.
- X. Wang and Z. An, Macromol. Rapid Commun., 2019, 40, 1800325 CrossRef PubMed.
- S. Y. Khor, J. F. Quinn, M. R. Whittaker, N. P. Truong and T. P. Davis, Macromol. Rapid Commun., 2019, 40, 1800438 CrossRef PubMed.
- J. Cao, Y. Tan, Y. Chen, L. Zhang and J. Tan, Macromol. Rapid Commun., 2021, 2100498 CrossRef PubMed.
- E. J. Cornel, J. Jiang, S. Chen and J. Du, CCS Chem., 2021, 3, 2104–2125 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- J. Cuthbert, S. V. Wanasinghe, K. Matyjaszewski and D. Konkolewicz, Macromolecules, 2021, 54, 8331–8340 CrossRef CAS.
- U. C. Palmiero, M. Sponchioni, N. Manfredini, M. Maraldi and D. Moscatelli, Polym. Chem., 2018, 9, 4084–4099 RSC.
- M. S. Messina, K. M. M. Messina, A. Bhattacharya, H. R. Montgomery and H. D. Maynard, Prog. Polym. Sci., 2020, 100, 101186 CrossRef CAS PubMed.
- F. L. Hatton, Polym. Chem., 2020, 11, 220–229 RSC.
- M. D. Nothling, Q. Fu, A. Reyhani, S. Allison-Logan, K. Jung, J. Zhu, M. Kamigaito, C. Boyer and G. G. Qiao, Adv. Sci., 2020, 7, 2001656 CrossRef CAS PubMed.
- G. Moad and E. Rizzardo, Polym. Int., 2020, 69, 658–661 CrossRef CAS.
- M. Destarac, Polym. Chem., 2018, 9, 4947–4967 RSC.
- C. Barner-Kowollik, Handbook of RAFT Polymerization, John Wiley & Sons, 2008 Search PubMed.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- M. Mazloomi-Rezvani, M. Salami-Kalajahi and H. Roghani-Mamaqani, J. Phys. Chem. Solids, 2018, 123, 183–190 CrossRef CAS.
- F.-W. Shen, K.-C. Zhou, H. Cai, Y.-N. Zhang, Y.-L. Zheng and J. Quan, Colloids Surf., B, 2019, 173, 504–511 CrossRef CAS PubMed.
- W. Shen, Q. Qiu, Y. Wang, M. Miao, B. Li, T. Zhang, A. Cao and Z. An, Macromol. Rapid Commun., 2010, 31, 1444–1448 CrossRef CAS PubMed.
- C. Wu, Y. Zhou, H. Wang, J. Hu and X. Wang, J. Saudi Chem. Soc., 2019, 23, 1080–1089 CrossRef CAS.
- P. J. Roth, C. Boyer, A. B. Lowe and T. P. Davis, Macromol. Rapid Commun., 2011, 32, 1123–1143 CrossRef CAS PubMed.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2007, 40, 4446–4455 CrossRef CAS.
- S. Perrier, P. Takolpuckdee and C. A. Mars, Macromolecules, 2005, 38, 2033–2036 CrossRef CAS.
- A. Postma, T. P. Davis, G. Moad and M. S. O'Shea, Macromolecules, 2005, 38, 5371–5374 CrossRef CAS.
- M. H. Stenzel, ACS Macro Lett., 2013, 2, 14–18 CrossRef CAS.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288–11289 CrossRef CAS PubMed.
- L. Tao, C. S. Kaddis, R. R. O. Loo, G. N. Grover, J. A. Loo and H. D. Maynard, Macromolecules, 2009, 42, 8028–8033 CrossRef CAS PubMed.
- K. L. Heredia, G. N. Grover, L. Tao and H. D. Maynard, Macromolecules, 2009, 42, 2360–2367 CrossRef CAS PubMed.
- G. N. Grover, S. N. S. Alconcel, N. M. Matsumoto and H. D. Maynard, Macromolecules, 2009, 42, 7657–7663 CrossRef CAS PubMed.
- L. Tao, J. Liu and T. P. Davis, Biomacromolecules, 2009, 10, 2847–2851 CrossRef CAS PubMed.
- C. Boyer, J. Liu, V. Bulmus and T. P. Davis, Aust. J. Chem., 2009, 62, 830–847 CrossRef CAS.
- V. Bulmus, Polym. Chem., 2011, 2, 1463–1472 RSC.
- M. Scherger, H. J. Räder and L. Nuhn, Macromol. Rapid Commun., 2021, 42, 2000752 CrossRef CAS PubMed.
- K. L. Heredia and H. D. Maynard, Org. Biomol. Chem., 2006, 5, 45–53 RSC.
- C. Chen, D. Y. W. Ng and T. Weil, Prog. Polym. Sci., 2020, 105, 101241 CrossRef CAS.
- Y. Wang and C. Wu, Biomacromolecules, 2018, 19, 1804–1825 CrossRef CAS PubMed.
- X. Liu and W. Gao, Angew. Chem., Int. Ed., 2021, 60, 11024–11035 CrossRef CAS PubMed.
- E. M. Pelegri-O'Day and H. D. Maynard, Acc. Chem. Res., 2016, 49, 1777–1785 CrossRef PubMed.
- R. A. Olson, A. B. Korpusik and B. S. Sumerlin, Chem. Sci., 2020, 11, 5142–5156 RSC.
- C. I. Biggs, M. Walker and M. I. Gibson, Biomacromolecules, 2016, 17, 2626–2633 CrossRef CAS PubMed.
- J. L. M. Gonçalves, E. J. Castanheira, S. P. C. Alves, C. Baleizão and J. P. Farinha, Polymers, 2020, 12, 2175 CrossRef PubMed.
- R. Falatach, C. McGlone, M. S. Al-Abdul-Wahid, S. Averick, R. C. Page, J. A. Berberich and D. Konkolewicz, Chem. Commun., 2015, 51, 5343–5346 RSC.
- M. Mazloomi-Rezvani, M. Salami-Kalajahi, H. Roghani-Mamaqani and A. Pirayesh, Appl. Organomet. Chem., 2018, 32, e4079 CrossRef.
- J. E. Gargari, A. Shakeri, H. S. Kalal, A. Khanchi and H. Rashedi, Polym. Adv. Technol., 2017, 28, 1884–1891 CrossRef CAS.
- A. J. Chancellor, B. T. Seymour and B. Zhao, Anal. Chem., 2019, 91, 6391–6402 CrossRef CAS PubMed.
- K. Ohno, Y. Ma, Y. Huang, C. Mori, Y. Yahata, Y. Tsujii, T. Maschmeyer, J. Moraes and S. Perrier, Macromolecules, 2011, 44, 8944–8953 CrossRef CAS.
- E. Roeven, A. R. Kuzmyn, L. Scheres, J. Baggerman, M. M. Smulders and H. Zuilhof, Langmuir, 2020, 36, 10187–10199 CrossRef CAS PubMed.
- S. Ghasemi and S. Karim, Mater. Chem. Phys., 2018, 205, 347–358 CrossRef CAS.
- G. Conzatti, S. Cavalie, C. Combes, J. Torrisani, N. Carrere and A. Tourrette, Colloids Surf., B, 2017, 151, 143–155 CrossRef CAS PubMed.
- C. Li, J. Han, C. Y. Ryu and B. C. Benicewicz, Macromolecules, 2006, 39, 3175–3183 CrossRef CAS.
- J. C. Foster, S. C. Radzinski and J. B. Matson, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2865–2876 CrossRef CAS.
- J. Moehrke and P. Vana, Macromol. Chem. Phys., 2017, 218, 1600506 CrossRef.
- P. Eskandari, Z. Abousalman-Rezvani, H. Roghani-Mamaqani, M. Salami-Kalajahi and H. Mardani, Adv. Colloid Interface Sci., 2019, 273, 102021 CrossRef CAS PubMed.
- C. Boyer, M. H. Stenzel and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 551–595 CrossRef CAS.
- J. E. Rauschendorfer, K. M. Thien, M. Denz, S. Köster, F. Ehlers and P. Vana, Macromol. Mater. Eng., 2021, 306, 2000595 CrossRef CAS.
- H. Mardani, H. Roghani-Mamaqani, K. Khezri and M. Salami-Kalajahi, Appl. Phys. A, 2020, 126, 251 CrossRef CAS.
- M. A. Macchione, C. Biglione and M. Strumia, Polymers, 2018, 10, 527 CrossRef PubMed.
- H. Liu, M. Ding, Z. Ding, C. Gao and W. Zhang, Polym. Chem., 2017, 8, 3203–3210 RSC.
- X. Li, J. Iocozzia, Y. Chen, S. Zhao, X. Cui, W. Wang, H. Yu, S. Lin and Z. Lin, Angew. Chem., Int. Ed., 2018, 57, 2046–2070 CrossRef CAS PubMed.
- C. Rossner and P. Vana, Angew. Chem., Int. Ed., 2014, 53, 12639–12642 CAS.
- J. Lee, H. Yang, C. H. Park, H.-H. Cho, H. Yun and B. J. Kim, Chem. Mater., 2016, 28, 3446–3453 CrossRef CAS.
- R. Qiao, L. Esser, C. Fu, C. Zhang, J. Hu, P. Ramírez-arcía, Y. Li, J. F. Quinn, M. R. Whittaker, A. K. Whittaker and T. P. Davis, Biomacromolecules, 2018, 19, 4423–4429 CrossRef CAS PubMed.
- M. Tasso, E. Giovanelli, D. Zala, S. Bouccara, A. Fragola, M. Hanafi, Z. Lenkei, T. Pons and N. Lequeux, ACS Nano, 2015, 9, 11479–11489 CrossRef CAS PubMed.
- F. Han, A. H. Soeriyadi and J. J. Gooding, Macromol. Rapid Commun., 2018, 39, 1800451 CrossRef PubMed.
- C. H. Park, H. Yun, H. Yang, J. Lee and B. J. Kim, Adv. Funct. Mater., 2017, 27, 1604403 CrossRef.
- M. Brust, D. Bethell, C. J. Kiely and D. J. Schiffrin, Langmuir, 1998, 14, 5425–5429 CrossRef CAS.
- G. Schmid and B. Corain, Eur. J. Inorg. Chem., 2003, 2003, 3081–3098 CrossRef.
- A. N. Shipway, E. Katz and I. Willner, ChemPhysChem, 2000, 1, 18–52 CrossRef CAS PubMed.
- C. E. Talley, J. B. Jackson, C. Oubre, N. K. Grady, C. W. Hollars, S. M. Lane, T. R. Huser, P. Nordlander and N. J. Halas, Nano Lett., 2005, 5, 1569–1574 CrossRef CAS PubMed.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, in Nanoscience And Technology: A Collection of Reviews from Nature Journals, World Scientific, 2010, pp. 308–319 Search PubMed.
- J. Sun, Y. Lu, L. He, J. Pang, F. Yang and Y. Liu, TrAC, Trends Anal. Chem., 2020, 122, 115754 CrossRef CAS.
- N. Lopez, T. V. W. Janssens, B. S. Clausen, Y. Xu, M. Mavrikakis, T. Bligaard and J. K. Nørskov, J. Catal., 2004, 223, 232–235 CrossRef CAS.
- A. Bej, K. Ghosh, A. Sarkar and D. W. Knight, RSC Adv., 2016, 6, 11446–11453 RSC.
- I. Favier, D. Pla and M. Gómez, Chem. Rev., 2020, 120, 1146–1183 CrossRef CAS PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911 CrossRef CAS PubMed.
- K. L. McGilvray, M. R. Decan, D. Wang and J. C. Scaiano, J. Am. Chem. Soc., 2006, 128, 15980–15981 CrossRef CAS PubMed.
- S. Kruss, T. Wolfram, R. Martin, S. Neubauer, H. Kessler and J. P. Spatz, Adv. Mater., 2010, 22, 5499–5506 CrossRef CAS PubMed.
- J. F. Hainfeld, D. N. Slatkin, T. M. Focella and H. M. Smilowitz, Br. J. Radiol., 2006, 79, 248–253 CrossRef CAS PubMed.
- S. A. Bansal, V. Kumar, J. Karimi, A. P. Singh and S. Kumar, Nanoscale Adv., 2020, 2, 3764–3787 RSC.
- P. Ghosh, G. Han, M. De, C. K. Kim and V. M. Rotello, Adv. Drug Delivery Rev., 2008, 60, 1307–1315 CrossRef CAS PubMed.
- M. Sharifi, F. Attar, A. A. Saboury, K. Akhtari, N. Hooshmand, A. Hasan, M. A. El-Sayed and M. Falahati, J. Controlled Release, 2019, 311–312, 170–189 CrossRef CAS PubMed.
- R. R. Arvizo, S. Bhattacharyya, R. A. Kudgus, K. Giri, R. Bhattacharya and P. Mukherjee, Chem. Soc. Rev., 2012, 41, 2943–2970 RSC.
- B. Klębowski, J. Depciuch, M. Parlińska-Wojtan and J. Baran, Int. J. Mol. Sci., 2018, 19, 4031 CrossRef PubMed.
- Y. Vlamidis and V. Voliani, Front. Bioeng. Biotechnol., 2018, 6, 143 CrossRef PubMed.
- Y. Xue, X. Li, H. Li and W. Zhang, Nat. Commun., 2014, 5, 4348 CrossRef CAS PubMed.
- H. Grönbeck, A. Curioni and W. Andreoni, J. Am. Chem. Soc., 2000, 122, 3839–3842 CrossRef.
- A.-S. Duwez, P. Guillet, C. Colard, J.-F. Gohy and C.-A. Fustin, Macromolecules, 2006, 39, 2729–2731 CrossRef CAS.
- I. Blakey, T. L. Schiller, Z. Merican and P. M. Fredericks, Langmuir, 2010, 26, 692–701 CrossRef CAS PubMed.
- L. Qiu, J.-W. Li, C.-Y. Hong and C.-Y. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 40887–40897 CrossRef CAS PubMed.
- B. Ebeling and P. Vana, Macromolecules, 2013, 46, 4862–4871 CrossRef CAS.
- J. Wagner, W. Peng and P. Vana, Polymers, 2018, 10, 407 CrossRef PubMed.
- M. I. Majeed, J. Guo, W. Yan and B. Tan, Polymers, 2016, 8, 392 CrossRef PubMed.
- A. Zengin, U. Tamer and T. Caykara, Anal. Chim. Acta, 2014, 817, 33–41 CrossRef CAS PubMed.
- X. He, X. Wu, X. Cai, S. Lin, M. Xie, X. Zhu and D. Yan, Langmuir, 2012, 28, 11929–11938 CrossRef CAS PubMed.
- H. T. T. Duong, Y. Chen, S. A. Tawfik, S. Wen, M. Parviz, O. Shimoni and D. Jin, RSC Adv., 2018, 8, 4842–4849 RSC.
- W. Zhang, B. Peng, F. Tian, W. Qin and X. Qian, Anal. Chem., 2014, 86, 482–489 CrossRef CAS PubMed.
- D. M. Montana, M. Nasilowski, W. R. Hess, M. Saif, J. A. Carr, L. Nienhaus and M. G. Bawendi, ACS Appl. Mater. Interfaces, 2020, 12, 35845–35855 CrossRef CAS PubMed.
- G. Marcelo, F. Burns, T. Ribeiro, J. M. G. Martinho, M. P. Tarazona, E. Saiz, M. G. Moffitt and J. P. S. Farinha, Langmuir, 2017, 33, 8201–8212 CrossRef CAS PubMed.
- M. S. Amini-Fazl, M. Barzegarzadeh and R. Mohammadi, J. Inorg. Organomet. Polym. Mater., 2021, 31, 2959–2970 CrossRef CAS.
- H. Roghani-Mamaqani and K. Khezri, Appl. Surf. Sci., 2016, 360, 373–382 CrossRef CAS.
- J. Wang, W. Zhang and J. Wei, J. Mater. Chem. A, 2019, 7, 2055–2065 RSC.
- P. Ding, J. Zhang, N. Song, S. Tang, Y. Liu and L. Shi, Composites, Part A, 2015, 69, 186–194 CrossRef CAS.
- H. Hosseinzadeh, S. Pashaei, S. Hosseinzadeh, Z. Khodaparast, S. Ramin and Y. Saadat, Sci. Total Environ., 2018, 640–641, 303–314 CrossRef CAS PubMed.
- B. Zhang, Y. Chen, L. Xu, L. Zeng, Y. He, E.-T. Kang and J. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2043–2050 CrossRef CAS.
- L. Wu, U. Glebe and A. Böker, Adv. Mater. Interfaces, 2017, 4, 1700092 CrossRef.
- M. S. Strozyk, M. Chanana, I. Pastoriza-Santos, J. Pérez-Juste and L. M. Liz-Marzán, Adv. Funct. Mater., 2012, 22, 1436–1444 CrossRef CAS.
- F. Han, S. R. C. Vivekchand, A. H. Soeriyadi, Y. Zheng and J. J. Gooding, Nanoscale, 2018, 10, 4284–4290 RSC.
- C. K. Adokoh, S. Quan, M. Hitt, J. Darkwa, P. Kumar and R. Narain, Biomacromolecules, 2014, 15, 3802–3810 CrossRef CAS PubMed.
- K. Matsuura, K. Ohno, S. Kagaya and H. Kitano, Macromol. Chem. Phys., 2007, 208, 862–873 CrossRef CAS.
- H. Yun, Y. J. Lee, M. Xu, D. C. Lee, G. E. Stein and B. J. Kim, ACS Nano, 2020, 14, 9644–9651 CrossRef CAS PubMed.
- M. S. Yavuz, Y. Cheng, J. Chen, C. M. Cobley, Q. Zhang, M. Rycenga, J. Xie, C. Kim, K. H. Song, A. G. Schwartz, L. V. Wang and Y. Xia, Nat. Mater., 2009, 8, 935–939 CrossRef CAS PubMed.
- X. Chen, J. Lawrence, S. Parelkar and T. Emrick, Macromolecules, 2013, 46, 119–127 CrossRef CAS.
- K. Ohno, K.-m. Koh, Y. Tsujii and T. Fukuda, Macromolecules, 2002, 35, 8989–8993 CrossRef CAS.
- E. G. Westbrook and P. Zhang, Dalton Trans., 2018, 47, 8638–8645 RSC.
- S. Nasri, G. R. Bardajee and M. Bayat, Colloids Surf., B, 2018, 171, 544–552 CrossRef CAS PubMed.
- J.-Y. Li, L. Qiu, X.-F. Xu, C.-Y. Pan, C.-Y. Hong and W.-J. Zhang, J. Mater. Chem. B, 2018, 6, 1678–1687 RSC.
- D. Huebner, C. Rossner and P. Vana, Polymer, 2016, 107, 503–508 CrossRef CAS.
- D. Rohleder and P. Vana, Biomacromolecules, 2021, 22, 1614–1624 CrossRef CAS PubMed.
- J. Tian, B. Huang and W. Zhang, Langmuir, 2019, 35, 266–275 CrossRef CAS PubMed.
- C. Rossner, E. B. Zhulina and E. Kumacheva, Angew. Chem., Int. Ed., 2019, 58, 9269–9274 CrossRef CAS PubMed.
- K. Hendrich, W. Peng and P. Vana, Polymers, 2020, 12, 1214 CrossRef CAS PubMed.
- C. Rossner, O. Glatter and P. Vana, Macromolecules, 2017, 50, 7344–7350 CrossRef CAS.
- C. Rossner, B. Ebeling and P. Vana, ACS Macro Lett., 2013, 2, 1073–1076 CrossRef CAS.
- C. Rossner, O. Glatter, O. Saldanha, S. Köster and P. Vana, Langmuir, 2015, 31, 10573–10582 CrossRef CAS PubMed.
- F. Han, T. Armstrong, A. Andres-Arroyo, D. Bennett, A. Soeriyadi, A. A. Chamazketi, P. Bakthavathsalam, R. D. Tilley, J. J. Gooding and P. J. Reece, Nanoscale, 2020, 12, 1680–1687 RSC.
- W. Peng, C. Rossner, V. Roddatis and P. Vana, ACS Macro Lett., 2016, 5, 1227–1231 CrossRef CAS.
- D. Miyamoto, M. Oishi, K. Kojima, K. Yoshimoto and Y. Nagasaki, Langmuir, 2008, 24, 5010–5017 CrossRef CAS PubMed.
- Y. Nagasaki, Chem. Lett., 2008, 37, 564–569 CrossRef CAS.
- J. Huang, D. Li, H. Liang and J. Lu, Macromol. Rapid Commun., 2017, 38, 1700202 CrossRef PubMed.
- R. Poupart, A. Benlahoues, B. Le Droumaguet and D. Grande, ACS Appl. Mater. Interfaces, 2017, 9, 31279–31290 CrossRef CAS PubMed.
- T. Klein, J. Parkin, P. A. J. M. de Jongh, L. Esser, T. Sepehrizadeh, G. Zheng, M. De Veer, K. Alt, C. E. Hagemeyer, D. M. Haddleton, T. P. Davis, M. Thelakkat and K. Kempe, Macromol. Rapid Commun., 2019, 40, e1800911 CrossRef PubMed.
- C. Boyer, P. Priyanto, T. P. Davis, D. Pissuwan, V. Bulmus, M. Kavallaris, W. Y. Teoh, R. Amal, M. Carroll, R. Woodward and T. St Pierre, J. Mater. Chem., 2010, 20, 255–265 RSC.
- G. Kahraman, D.-Y. Wang, J. von Irmer, M. Gallei, E. Hey-Hawkins and T. Eren, Polymers, 2019, 11, 613 CrossRef PubMed.
- Z. Xie, X. Deng, B. Liu, S. Huang, P. Ma, Z. Hou, Z. Cheng, J. Lin and S. Luan, ACS Appl. Mater. Interfaces, 2017, 9, 30414–30425 CrossRef CAS PubMed.
- K. W. Fan and A. M. Granville, Polymers, 2016, 8, 81 CrossRef PubMed.
- H. Kakwere, M. E. Materia, A. Curcio, M. Prato, A. Sathya, S. Nitti and T. Pellegrino, Nanoscale, 2018, 10, 3930–3944 RSC.
- J. Yu, R. J. Nap, I. Szleifer and J. Y. Wong, Langmuir, 2019, 35, 15864–15871 CrossRef CAS PubMed.
- H. Kakwere, M. P. Leal, M. E. Materia, A. Curcio, P. Guardia, D. Niculaes, R. Marotta, A. Falqui and T. Pellegrino, ACS Appl. Mater. Interfaces, 2015, 7, 10132–10145 CrossRef CAS PubMed.
- W. Liu, A. B. Greytak, J. Lee, C. R. Wong, J. Park, L. F. Marshall, W. Jiang, P. N. Curtin, A. Y. Ting, D. G. Nocera, D. Fukumura, R. K. Jain and M. G. Bawendi, J. Am. Chem. Soc., 2010, 132, 472–483 CrossRef CAS PubMed.
- J. H. Dunlap, A. F. Loszko, R. A. Flake, Y. Huang, B. C. Benicewicz and A. B. Greytak, J. Phys. Chem. C, 2018, 122, 26756–26763 CrossRef CAS.
- J. Liu, W. Yang, L. Tao, D. Li, C. Boyer and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 425–433 CrossRef CAS.
- Y. Xu, H. Bai, G. Lu, C. Li and G. Shi, J. Am. Chem. Soc., 2008, 130, 5856–5857 CrossRef CAS PubMed.
- G. Cheng, D. Xu, Z. Lu and K. Liu, ACS Nano, 2019, 13, 1479–1489 CAS.
- E. Pensa, E. Cortés, G. Corthey, P. Carro, C. Vericat, M. H. Fonticelli, G. Benítez, A. A. Rubert and R. C. Salvarezza, Acc. Chem. Res., 2012, 45, 1183–1192 CrossRef CAS PubMed.
- R. G. Nuzzo, L. H. Dubois and D. L. Allara, J. Am. Chem. Soc., 1990, 112, 558–569 CrossRef CAS.
- L. Kankate, A. Turchanin and A. Gölzhäuser, Langmuir, 2009, 25, 10435–10438 CrossRef CAS PubMed.
- C. Yi, H. Liu, S. Zhang, Y. Yang, Y. Zhang, Z. Lu, E. Kumacheva and Z. Nie, Science, 2020, 369, 1369–1374 CrossRef CAS PubMed.
- W. Yu, L.-L. Lou, S. Li, T. Ma, L. Ouyang, L. Feng and S. Liu, RSC Adv., 2017, 7, 751–757 RSC.
- L. E. Wilkins, M. Hasan, A. E. R. Fayter, C. Biggs, M. Walker and M. I. Gibson, Polym. Chem., 2019, 10, 2986–2990 RSC.
- Y. Kwon, Y. Choi, J. Jang, S. Yoon and J. Choi, Pharmaceutics, 2020, 12, 204 CrossRef CAS PubMed.
- S. Slavin, A. H. Soeriyadi, L. Voorhaar, M. R. Whittaker, C. R. Becer, C. Boyer, T. P. Davis and D. M. Haddleton, Soft Matter, 2012, 8, 118–128 RSC.
- Y.-X. Wang, Y. Li, S.-H. Qiao, J. Kang, Z.-L. Shen, N.-N. Zhang, Z. An, X. Wang and K. Liu, J. Phys. Chem. Lett., 2021, 12, 4713–4721 CrossRef CAS PubMed.
- H.-J. Li, P.-Y. Li, L.-Y. Li, A. Haleem and W.-D. He, Molecules, 2018, 23, 921 CrossRef PubMed.
- P. Dey, K. J. Thurecht, P. M. Fredericks and I. Blakey, Macromolecules, 2020, 53, 7469–7478 CrossRef CAS.
- B. Huang, J. Tian, D. Jiang, Y. Gao and W. Zhang, Biomacromolecules, 2019, 20, 3873–3883 CrossRef CAS PubMed.
- A. Ruiu, B. Bauer-Siebenlist, M. Senila, T. Jänisch, D. Foix, K. Seaudeau-Pirouley and P. Lacroix-Desmazes, J. CO2 Util., 2020, 41, 101232 CrossRef CAS.
- S. Zhang, K. L. Chandra and C. B. Gorman, J. Am. Chem. Soc., 2007, 129, 4876–4877 CrossRef CAS PubMed.
- P. Dey, S. Zhu, K. J. Thurecht, P. M. Fredericks and I. Blakey, J. Mater. Chem. B, 2014, 2, 2827–2837 RSC.
- S. Moulay, Polym. Rev., 2014, 54, 436–513 CrossRef CAS.
- M. Monteil, H. Moustaoui, G. Picardi, F. Aouidat, N. Djaker, M. L. de La Chapelle, M. Lecouvey and J. Spadavecchia, J. Colloid Interface Sci., 2018, 513, 205–213 CrossRef CAS PubMed.
- S. W. Han, S. J. Lee and K. Kim, Langmuir, 2001, 17, 6981–6987 CrossRef CAS.
- W. Gan, B. Xu and H.-L. Dai, Angew. Chem., Int. Ed., 2011, 50, 6622–6625 CrossRef CAS PubMed.
- A. B. Lowe, B. S. Sumerlin, M. S. Donovan and C. L. McCormick, J. Am. Chem. Soc., 2002, 124, 11562–11563 CrossRef CAS PubMed.
- G. Corthey, A. A. Rubert, A. L. Picone, G. Casillas, L. J. Giovanetti, J. M. Ramallo-López, E. Zelaya, G. A. Benitez, F. G. Requejo, M. José-Yacamán, R. C. Salvarezza and M. H. Fonticelli, J. Phys. Chem. C, 2012, 116, 9830–9837 CrossRef CAS.
- P. Carro, G. Corthey, A. A. Rubert, G. A. Benitez, M. H. Fonticelli and R. C. Salvarezza, Langmuir, 2010, 26, 14655–14662 CrossRef CAS PubMed.
- A. Isakova, P. D. Topham and A. J. Sutherland, Macromolecules, 2014, 47, 2561–2568 CrossRef CAS.
- B. Fan, J. Wan, Y. Liu, W. W. Tian and S. H. Thang, Polym. Chem., 2021, 12, 3015–3025 RSC.
- L. Xiao, J. Li, D. F. Brougham, E. K. Fox, N. Feliu, A. Bushmelev, A. Schmidt, N. Mertens, F. Kiessling, M. Valldor, B. Fadeel and S. Mathur, ACS Nano, 2011, 5, 6315–6324 CrossRef CAS PubMed.
- J. Park, N. R. Kadasala, S. A. Abouelmagd, M. A. Castanares, D. S. Collins, A. Wei and Y. Yeo, Biomaterials, 2016, 101, 285–295 CrossRef CAS PubMed.
- K. Ulbrich, K. Holá, V. Šubr, A. Bakandritsos, J. Tuček and R. Zbořil, Chem. Rev., 2016, 116, 5338–5431 CrossRef CAS PubMed.
- I. Andreu, E. Natividad, L. Solozábal and O. Roubeau, ACS Nano, 2015, 9, 1408–1419 CrossRef CAS PubMed.
- R. Bazak, M. Houri, S. El Achy, S. Kamel and T. Refaat, J. Cancer Res. Clin. Oncol., 2015, 141, 769–784 CrossRef CAS PubMed.
- M. Lévy, C. Wilhelm, J.-M. Siaugue, O. Horner, J.-C. Bacri and F. Gazeau, J. Phys.: Condens. Matter, 2008, 20, 204133 CrossRef PubMed.
- K. Turcheniuk, A. V. Tarasevych, V. P. Kukhar, R. Boukherroub and S. Szunerits, Nanoscale, 2013, 5, 10729–10752 RSC.
- C. Xu, K. Xu, H. Gu, R. Zheng, H. Liu, X. Zhang, Z. Guo and B. Xu, J. Am. Chem. Soc., 2004, 126, 9938–9939 CrossRef CAS PubMed.
- E. Amstad, A. U. Gehring, H. Fischer, V. V. Nagaiyanallur, G. Hähner, M. Textor and E. Reimhult, J. Phys. Chem. C, 2011, 115, 683–691 CrossRef CAS.
- E. Amstad, T. Gillich, I. Bilecka, M. Textor and E. Reimhult, Nano Lett., 2009, 9, 4042–4048 CrossRef CAS PubMed.
- J. Xie, C. Xu, Z. Xu, Y. Hou, K. L. Young, S. X. Wang, N. Pourmand and S. Sun, Chem. Mater., 2006, 18, 5401–5403 CrossRef CAS PubMed.
- D. Burnand, C. A. Monnier, A. Redjem, M. Schaefer, B. Rothen-Rutishauser, A. Kilbinger and A. Petri-Fink, J. Magn. Magn. Mater., 2015, 380, 157–162 CrossRef CAS.
- K. Ohno, C. Mori, T. Akashi, S. Yoshida, Y. Tago, Y. Tsujii and Y. Tabata, Biomacromolecules, 2013, 14, 3453–3462 CrossRef CAS PubMed.
- Y. Huang, K. Ohno and R. Ishige, Langmuir, 2014, 31, 1172–1179 CrossRef PubMed.
- L. Sandiford, A. Phinikaridou, A. Protti, L. K. Meszaros, X. Cui, Y. Yan, G. Frodsham, P. A. Williamson, N. Gaddum, R. M. Botnar, P. J. Blower, M. A. Green and R. T. M. de Rosales, ACS Nano, 2013, 7, 500–512 CrossRef CAS PubMed.
- A. J. McGrath, C. Dolan, S. Cheong, D. A. J. Herman, B. Naysmith, F. Zong, P. Galvosas, K. J. Farrand, I. F. Hermans, M. Brimble, D. E. Williams, J. Jin and R. D. Tilley, J. Magn. Magn. Mater., 2017, 439, 251–258 CrossRef CAS.
- R. Bilan, F. Fleury, I. Nabiev and A. Sukhanova, Bioconjugate Chem., 2015, 26, 609–624 CrossRef CAS PubMed.
- O. T. Bruns, T. S. Bischof, D. K. Harris, D. Franke, Y. Shi, L. Riedemann, A. Bartelt, F. B. Jaworski, J. A. Carr, C. J. Rowlands, M. W. B. Wilson, O. Chen, H. Wei, G. W. Hwang, D. M. Montana, I. Coropceanu, O. B. Achorn, J. Kloepper, J. Heeren, P. T. C. So, D. Fukumura, K. F. Jensen, R. K. Jain and M. G. Bawendi, Nat. Biomed. Eng., 2017, 1, 1–11 CrossRef.
- T. M. Inerbaev, A. E. Masunov, S. I. Khondaker, A. Dobrinescu, A.-V. Plamadă and Y. Kawazoe, J. Chem. Phys., 2009, 131, 044106 CrossRef PubMed.
- H. R. Chandan, J. D. Schiffman and R. G. Balakrishna, Sens. Actuators, B, 2018, 258, 1191–1214 CrossRef CAS.
- P. N. Goswami, D. Mandal and A. K. Rath, Nanoscale, 2018, 10, 1072–1080 RSC.
- T. D. Nguyen, H. Vu-Quang, T. S. Vo, D. C. Nguyen, D.-V. N. Vo, D. H. Nguyen, K. T. Lim, D. L. Tran and L. G. Bach, Polymers, 2019, 11, 77 CrossRef PubMed.
- S. Jeong, M. Achermann, J. Nanda, S. Ivanov, V. I. Klimov and J. A. Hollingsworth, J. Am. Chem. Soc., 2005, 127, 10126–10127 CrossRef CAS PubMed.
- M. Green, J. Mater. Chem., 2010, 20, 5797–5809 RSC.
- Y. Zheng, Y. Tang, J. Yu, L. Xie, H. Dong, R. Deng, F. Jia, B. Liu, L. Gao and J. Duan, Materials, 2020, 13, 440 CrossRef CAS PubMed.
- Y. Liu, Y. Inoue and K. Ishihara, Colloids Surf., B, 2015, 135, 490–496 CrossRef CAS PubMed.
- L. Zhang, D. Jin and M. H. Stenzel, Biomacromolecules, 2021, 22, 3168–3201 CrossRef CAS PubMed.
- D. Yang, P. Ma, Z. Hou, Z. Cheng, C. Li and J. Lin, Chem. Soc. Rev., 2015, 44, 1416–1448 RSC.
- V. Muhr, S. Wilhelm, T. Hirsch and O. S. Wolfbeis, Acc. Chem. Res., 2014, 47, 3481–3493 CrossRef CAS PubMed.
- A. Bagheri, H. Arandiyan, C. Boyer and M. Lim, Adv. Sci., 2016, 3, 1500437 CrossRef PubMed.
- A. Sedlmeier and H. H. Gorris, Chem. Soc. Rev., 2015, 44, 1526–1560 RSC.
- A. Bagheri, H. Arandiyan, N. N. M. Adnan, C. Boyer and M. Lim, Macromolecules, 2017, 50, 7137–7147 CrossRef CAS.
- A. Kavand, C. Blanck, F. Przybilla, Y. Mély, N. Anton, T. Vandamme, C. A. Serra and D. Chan-Seng, Polym. Chem., 2020, 11, 4313–4325 RSC.
- A. Bagheri, Z. Sadrearhami, N. N. M. Adnan, C. Boyer and M. Lim, Polymer, 2018, 151, 6–14 CrossRef CAS.
- J. Chen, Q. Chen and Q. Ma, J. Colloid Interface Sci., 2012, 370, 32–38 CrossRef CAS PubMed.
- W. Zhao, Z. Hou, Z. Yao, X. Zhuang, F. Zhang and X. Feng, Polym. Chem., 2015, 6, 7171–7178 RSC.
- Y.-S. Ye, Y.-N. Chen, J.-S. Wang, J. Rick, Y.-J. Huang, F.-C. Chang and B.-J. Hwang, Chem. Mater., 2012, 24, 2987–2997 CrossRef CAS.
- A. Guerrero-Martínez, J. Pérez-Juste and L. M. Liz-Marzán, Adv. Mater., 2010, 22, 1182–1195 CrossRef PubMed.
- C. Graf, D. L. J. Vossen, A. Imhof and A. van Blaaderen, Langmuir, 2003, 19, 6693–6700 CrossRef CAS.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- Y. Han, Z. Lu, Z. Teng, J. Liang, Z. Guo, D. Wang, M.-Y. Han and W. Yang, Langmuir, 2017, 33, 5879–5890 CrossRef CAS PubMed.
- H. L. Ding, Y. X. Zhang, S. Wang, J. M. Xu, S. C. Xu and G. H. Li, Chem. Mater., 2012, 24, 4572–4580 CrossRef CAS.
- A. M. El-Toni, S. Yin and T. Sato, J. Colloid Interface Sci., 2006, 300, 123–130 CrossRef CAS PubMed.
- K. Nozawa, H. Gailhanou, L. Raison, P. Panizza, H. Ushiki, E. Sellier, J. P. Delville and M. H. Delville, Langmuir, 2005, 21, 1516–1523 CrossRef CAS PubMed.
- Y. Kobayashi, H. Inose, T. Nakagawa, K. Gonda, M. Takeda, N. Ohuchi and A. Kasuya, J. Colloid Interface Sci., 2011, 358, 329–333 CrossRef CAS PubMed.
- Y. Han, J. Jiang, S. S. Lee and J. Y. Ying, Langmuir, 2008, 24, 5842–5848 CrossRef CAS PubMed.
- M. A. López-Quintela, C. Tojo, M. C. Blanco, L. G. Rio and J. R. Leis, Curr. Opin. Colloid Interface Sci., 2004, 9, 264–278 CrossRef.
- Y. Cai, W. Peng, S. Demeshko, J. Tian and P. Vana, Macromol. Rapid Commun., 2018, 39, 1800226 CrossRef PubMed.
- A. Pourjavadi, M. Kohestanian and C. Streb, Mater. Sci. Eng., C, 2020, 108, 110418 CrossRef CAS PubMed.
- D. Huebner, V. Koch, B. Ebeling, J. Mechau, J. Steinhoff and P. Vana, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 103–113 CrossRef CAS.
- S. Ghasemi and S. Karim, Colloid Polym. Sci., 2018, 296, 1323–1332 CrossRef CAS.
- B. Sahoo, K. S. P. Devi, R. Banerjee, T. K. Maiti, P. Pramanik and D. Dhara, ACS Appl. Mater. Interfaces, 2013, 5, 3884–3893 CrossRef CAS PubMed.
- F. Chen, J. Wang, H. Chen, R. Lu and X. Xie, Appl. Surf. Sci., 2018, 435, 247–255 CrossRef CAS.
- A. Liberman, N. Mendez, W. C. Trogler and A. C. Kummel, Surf. Sci. Rep., 2014, 69, 132–158 CrossRef CAS PubMed.
- Y. Zheng, Y. Huang, Z. M. Abbas and B. C. Benicewicz, Polym. Chem., 2016, 7, 5347–5350 RSC.
- K. C. Bentz and D. A. Savin, Polym. Chem., 2018, 9, 2059–2081 RSC.
- X. Huang, D. Appelhans, P. Formanek, F. Simon and B. Voit, Macromolecules, 2011, 44, 8351–8360 CrossRef CAS.
- M. Dhara, S. Rudra, N. Mukherjee and T. Jana, Polym. Chem., 2021, 12, 3976–3991 RSC.
- G. Moad, Polym. Int., 2015, 64, 15–24 CrossRef CAS.
- H. Ma, A. Li, Y. Xu, W. Zhang and J. Liu, Eur. Polym. J., 2015, 72, 212–221 CrossRef CAS.
- W. Li, X. Cai, C. Kim, G. Sun, Y. Zhang, R. Deng, M. Yang, J. Chen, S. Achilefu, L. V. Wang and Y. Xia, Nanoscale, 2011, 3, 1724–1730 RSC.
- I. Blakey, Z. Merican and K. J. Thurecht, Langmuir, 2013, 29, 8266–8274 CrossRef CAS PubMed.
- T. Honold, D. Skrybeck, K. G. Wagner and M. Karg, Langmuir, 2017, 33, 253–261 CrossRef CAS PubMed.
- X. Liao, L. Guo, J. Chang, S. Liu, M. Xie and G. Chen, Macromol. Rapid Commun., 2015, 36, 1492–1497 CrossRef CAS PubMed.
- Z.-L. Wang, J.-T. Xu, B.-Y. Du and Z.-Q. Fan, J. Colloid Interface Sci., 2011, 360, 350–354 CrossRef CAS PubMed.
- P. E. Williams, S. T. Jones, Z. Walsh, E. A. Appel, E. K. Abo-Hamed and O. A. Scherman, ACS Macro Lett., 2015, 4, 255–259 CrossRef CAS.
- J. Liu, C. Detrembleur, M.-C. De Pauw-Gillet, S. Mornet, E. Duguet and C. Jérôme, Polym. Chem., 2014, 5, 799–813 RSC.
- B. Liao, X. Liu, S. Liao, W. Liu, S. Yi, Q. Liu and B. He, Polym. J., 2020, 52, 1289–1298 CrossRef CAS.
- T. Kim, M. Xu, Y. J. Lee, K. H. Ku, D. J. Shin, D. C. Lee, S. G. Jang, H. Yun and B. J. Kim, Small, 2021, 17, 2101222 CrossRef CAS PubMed.
- J. Zhou, K. Mishra, V. Bhagat, A. Joy and M. L. Becker, Polym. Chem., 2015, 6, 2813–2816 RSC.
- K. Paek, H. Yang, J. Lee, J. Park and B. J. Kim, ACS Nano, 2014, 8, 2848–2856 CrossRef CAS PubMed.
- C. H. Park, H. Yang, J. Lee, H.-H. Cho, D. Kim, D. C. Lee and B. J. Kim, Chem. Mater., 2015, 27, 5288–5294 CrossRef CAS.
- T. Ribeiro, E. Coutinho, A. S. Rodrigues, C. Baleizão and J. P. S. Farinha, Nanoscale, 2017, 9, 13485–13494 RSC.
- M. M. Khani, Z. M. Abbas and B. C. Benicewicz, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1493–1501 CrossRef CAS.
- T. Nohara, K. Koseki, K. Tabata, R. Shimada, Y. Suzuki, K. Umemoto, M. Takeda, R. Sato, S. Rodbuntum, T. Arita and A. Masuhara, ACS Sustainable Chem. Eng., 2020, 8, 14674–14678 CrossRef CAS.
- K. Shito, J. Matsui, Y. Takahashi, A. Masuhara and T. Arita, Chem. Lett., 2018, 47, 9–12 CrossRef CAS.
- A. Pourjavadi, S. Rahemipoor and M. Kohestanian, Compos. Sci. Technol., 2020, 188, 107951 CrossRef CAS.
- C. Durand-Gasselin, R. Koerin, J. Rieger, N. Lequeux and N. Sanson, J. Colloid Interface Sci., 2014, 434, 188–194 CrossRef CAS PubMed.
- Z. Cao, N. N. M. Adnan, G. Wang, A. Rawal, B. Shi, R. Liu, K. Liang, L. Zhao, J. J. Gooding, C. Boyer and Z. Gu, J. Colloid Interface Sci., 2018, 521, 242–251 CrossRef CAS PubMed.
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2012, 64, 116–127 CrossRef.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- S. Lowe, N. M. O'Brien-Simpson and L. A. Connal, Polym. Chem., 2015, 6, 198–212 RSC.
- Â. Serrano, O. Sterner, S. Mieszkin, S. Zürcher, S. Tosatti, M. E. Callow, J. A. Callow and N. D. Spencer, Adv. Funct. Mater., 2013, 23, 5706–5718 CrossRef.
- Z. Wu, H. Chen, X. Liu, Y. Zhang, D. Li and H. Huang, Langmuir, 2009, 25, 2900–2906 CrossRef CAS PubMed.
- Z. G. Estephan, P. S. Schlenoff and J. B. Schlenoff, Langmuir, 2011, 27, 6794–6800 CrossRef CAS PubMed.
- Q. Shao and S. Jiang, Adv. Mater., 2015, 27, 15–26 CrossRef CAS PubMed.
- J. Manson, D. Kumar, B. J. Meenan and D. Dixon, Gold Bull., 2011, 44, 99–105 CrossRef CAS.
- J. Tournebize, A. Boudier, A. Sapin-Minet, P. Maincent, P. Leroy and R. Schneider, ACS Appl. Mater. Interfaces, 2012, 4, 5790–5799 CrossRef CAS PubMed.
- E. Giovanelli, E. Muro, G. Sitbon, M. Hanafi, T. Pons, B. Dubertret and N. Lequeux, Langmuir, 2012, 28, 15177–15184 CrossRef CAS PubMed.
- D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214 RSC.
- K. J. Hogan and A. G. Mikos, Polymer, 2020, 211, 123063 CrossRef CAS.
- A. Bordat, T. Boissenot, J. Nicolas and N. Tsapis, Adv. Drug Delivery Rev., 2019, 138, 167–192 CrossRef CAS PubMed.
- E. Reimhult, M. Schroffenegger and A. Lassenberger, Langmuir, 2019, 35, 7092–7104 CrossRef CAS PubMed.
- O. Veiseh, J. W. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 284–304 CrossRef CAS PubMed.
- S. Shen, B. Ding, S. Zhang, X. Qi, K. Wang, J. Tian, Y. Yan, Y. Ge and L. Wu, Nanomedicine, 2017, 13, 1607–1616 CrossRef CAS PubMed.
- M. A. Ward and T. K. Georgiou, Polymers, 2011, 3, 1215–1242 CrossRef CAS.
- I. Yildiz and B. S. Yildiz, J. Nanomater., 2016, 16, 357 Search PubMed.
- J. Lü, Y. Yang, J. Gao, H. Duan and C. Lü, Langmuir, 2018, 34, 8205–8214 CrossRef PubMed.
- G. Liu, D. Wang, F. Zhou and W. Liu, Small, 2015, 11, 2807–2816 CrossRef CAS PubMed.
- Q. Xiao, Y. Li, F. Li, M. Zhang, Z. Zhang and H. Lin, Nanoscale, 2014, 6, 10179–10186 RSC.
- Y. Zheng, A. H. Soeriyadi, L. Rosa, S. H. Ng, U. Bach and J. J. Gooding, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- S. Maji, B. Cesur, Z. Zhang, B. G. De Geest and R. Hoogenboom, Polym. Chem., 2016, 7, 1705–1710 RSC.
- R. Cheng, F. Meng, C. Deng, H.-A. Klok and Z. Zhong, Biomaterials, 2013, 34, 3647–3657 CrossRef CAS PubMed.
- X. Liu, Y. Yang and M. W. Urban, Macromol. Rapid Commun., 2017, 38, 1700030 CrossRef PubMed.
- R. Pelton, J. Colloid Interface Sci., 2010, 348, 673–674 CrossRef CAS PubMed.
- S. T. Jones, Z. Walsh-Korb, S. J. Barrow, S. L. Henderson, J. del Barrio and O. A. Scherman, ACS Nano, 2016, 10, 3158–3165 CrossRef CAS PubMed.
- D. Fava, M. A. Winnik and E. Kumacheva, Chem. Commun., 2009, 2571–2573 RSC.
- H. Han, J. Y. Lee and X. Lu, Chem. Commun., 2013, 49, 6122–6124 RSC.
- J. Niskanen and H. Tenhu, Polym. Chem., 2017, 8, 220–232 RSC.
- F. Liu and S. Agarwal, Macromol. Chem. Phys., 2015, 216, 460–465 CrossRef CAS.
- X. Cai, L. Zhong, Y. Su, S. Lin and X. He, Polym. Chem., 2015, 6, 3875–3884 RSC.
- Z. Dong, J. Mao, D. Wang, M. Yang, W. Wang, S. Bo and X. Ji, Macromol. Chem. Phys., 2014, 215, 111–120 CrossRef CAS.
- G. K. Such, Y. Yan, A. P. R. Johnston, S. T. Gunawan and F. Caruso, Adv. Mater., 2015, 27, 2278–2297 CrossRef CAS PubMed.
- A.-H. Ranneh, H. Takemoto, S. Sakuma, A. Awaad, T. Nomoto, Y. Mochida, M. Matsui, K. Tomoda, M. Naito and N. Nishiyama, Angew. Chem., Int. Ed., 2018, 57, 5057–5061 CrossRef CAS PubMed.
- H. Tang, W. Zhao, J. Yu, Y. Li and C. Zhao, Molecules, 2018, 24, 4 CrossRef PubMed.
- X. Lv, J. Zhang, D. Yang, J. Shao, W. Wang, Q. Zhang and X. Dong, J. Mater. Chem. B, 2020, 8, 10700–10711 RSC.
- H. Akiyama, K. Tamada, J. Nagasawa, K. Abe and T. Tamaki, J. Phys. Chem. B, 2003, 107, 130–135 CrossRef CAS.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- P. K. Kundu, D. Samanta, R. Leizrowice, B. Margulis, H. Zhao, M. Börner, T. Udayabhaskararao, D. Manna and R. Klajn, Nat. Chem., 2015, 7, 646–652 CrossRef CAS PubMed.
- T. Bian, Z. Chu and R. Klajn, Adv. Mater., 2020, 32, 1905866 CrossRef CAS PubMed.
- L. Zhang, L. Dai, Y. Rong, Z. Liu, D. Tong, Y. Huang and T. Chen, Langmuir, 2015, 31, 1164–1171 CrossRef CAS PubMed.
- J. Shi, F. Tian, J. Lyu and M. Yang, J. Mater. Chem. B, 2015, 3, 6989–7005 RSC.
- Z. Gueroui and A. Libchaber, Phys. Rev. Lett., 2004, 93, 166108 CrossRef PubMed.
- S. Jiang and Y. Zhang, Langmuir, 2010, 26, 6689–6694 CrossRef CAS PubMed.
- C. Chen and N. Hildebrandt, TrAC, Trends Anal. Chem., 2020, 123, 115748 CrossRef CAS.
- C. S. Yun, A. Javier, T. Jennings, M. Fisher, S. Hira, S. Peterson, B. Hopkins, N. O. Reich and G. F. Strouse, J. Am. Chem. Soc., 2005, 127, 3115–3119 CrossRef CAS PubMed.
- P. F. Gao, Y. F. Li and C. Z. Huang, TrAC, Trends Anal. Chem., 2020, 124, 115805 CrossRef CAS.
- G. L. Liu, Y.-T. Long, Y. Choi, T. Kang and L. P. Lee, Nat. Methods, 2007, 4, 1015–1017 CrossRef CAS PubMed.
- Y. Choi, T. Kang and L. P. Lee, Nano Lett., 2009, 9, 85–90 CrossRef CAS PubMed.
- C. Chen and N. Hildebrandt, TrAC, Trends Anal. Chem., 2020, 123, 115748 CrossRef CAS.
- E. Morales-Narváez, B. Pérez-López, L. B. Pires and A. Merkoçi, Carbon, 2012, 50, 2987–2993 CrossRef.
- C. Zhu, Z. Zeng, H. Li, F. Li, C. Fan and H. Zhang, J. Am. Chem. Soc., 2013, 135, 5998–6001 CrossRef CAS PubMed.
- F. Prins, A. J. Goodman and W. A. Tisdale, Nano Lett., 2014, 14, 6087–6091 CrossRef CAS PubMed.
- A. Kasry, A. A. Ardakani, G. S. Tulevski, B. Menges, M. Copel and L. Vyklicky, J. Phys. Chem. C, 2012, 116, 2858–2862 CrossRef CAS.
- J. Saha, A. D. Roy, D. Dey, D. Bhattacharjee and S. A. Hussain, Mater. Today: Proc., 2018, 5, 2306–2313 CAS.
- K. Paek, S. Chung, C.-H. Cho and B. J. Kim, Chem. Commun., 2011, 47, 10272–10274 RSC.
- M. Dadsetan, K. E. Taylor, C. Yong, Ž. Bajzer, L. Lu and M. J. Yaszemski, Acta Biomater., 2013, 9, 5438–5446 CrossRef CAS PubMed.
- H. Yang, K. Paek and B. J. Kim, Nanoscale, 2013, 5, 5720–5724 RSC.
- M. Hwang and B. Yeom, Chem. Mater., 2021, 33, 807–817 CrossRef CAS.
- W. Ma, H. Kuang, L. Xu, L. Ding, C. Xu, L. Wang and N. A. Kotov, Nat. Commun., 2013, 4, 1–8 Search PubMed.
- X. Wu, L. Xu, L. Liu, W. Ma, H. Yin, H. Kuang, L. Wang, C. Xu and N. A. Kotov, J. Am. Chem. Soc., 2013, 135, 18629–18636 CrossRef CAS PubMed.
- Y. Zhou, R. L. Marson, G. Van Anders, J. Zhu, G. Ma, P. Ercius, K. Sun, B. Yeom, S. C. Glotzer and N. A. Kotov, ACS Nano, 2016, 10, 3248–3256 CrossRef CAS PubMed.
- A. D. Merg, J. C. Boatz, A. Mandal, G. Zhao, S. Mokashi-Punekar, C. Liu, X. Wang, P. Zhang, P. C. van der Wel and N. L. Rosi, J. Am. Chem. Soc., 2016, 138, 13655–13663 CrossRef CAS PubMed.
- W. Yan, L. Xu, C. Xu, W. Ma, H. Kuang, L. Wang and N. A. Kotov, J. Am. Chem. Soc., 2012, 134, 15114–15121 CrossRef CAS PubMed.
- Q. Jiang, Q. Liu, Y. Shi, Z.-G. Wang, P. Zhan, J. Liu, C. Liu, H. Wang, X. Shi, L. Zhang, J. Sun, B. Ding and M. Liu, Nano Lett., 2017, 17, 7125–7130 CrossRef CAS PubMed.
- X. Lan, X. Lu, C. Shen, Y. Ke, W. Ni and Q. Wang, J. Am. Chem. Soc., 2015, 137, 457–462 CrossRef CAS PubMed.
- C. Li, Q. Li, Y. V. Kaneti, D. Hou, Y. Yamauchi and Y. Mai, Chem. Soc. Rev., 2020, 49, 4681–4736 RSC.
- T. N. Hoheisel, K. Hur and U. B. Wiesner, Prog. Polym. Sci., 2015, 40, 3–32 CrossRef CAS.
- A. H. Gröschel, A. Walther, T. I. Löbling, F. H. Schacher, H. Schmalz and A. H. E. Müller, Nature, 2013, 503, 247–251 CrossRef PubMed.
- R. M. Choueiri, E. Galati, H. Thérien-Aubin, A. Klinkova, E. M. Larin, A. Querejeta-Fernández, L. Han, H. L. Xin, O. Gang, E. B. Zhulina, M. Rubinstein and E. Kumacheva, Nature, 2016, 538, 79–83 CrossRef CAS PubMed.
- J.-M. Rabanel, V. Adibnia, S. F. Tehrani, S. Sanche, P. Hildgen, X. Banquy and C. Ramassamy, Nanoscale, 2019, 11, 383–406 RSC.
- N. D. Knöfel, H. Rothfuss, C. Barner-Kowollik and P. W. Roesky, Polym. Chem., 2019, 10, 86–93 RSC.
- M. Sun, L. Xu, W. Ma, X. Wu, H. Kuang, L. Wang and C. Xu, Adv. Mater., 2016, 28, 898–904 CrossRef CAS PubMed.
- A. Qu, L. Xu, M. Sun, L. Liu, H. Kuang and C. Xu, Adv. Funct. Mater., 2017, 27, 1703408 CrossRef.
- L. He, M. Brasino, C. Mao, S. Cho, W. Park, A. P. Goodwin and J. N. Cha, Small, 2017, 13, 1700504 CrossRef PubMed.
- Y. Zheng, T. Thai, P. Reineck, L. Qiu, Y. Guo and U. Bach, Adv. Funct. Mater., 2013, 23, 1519–1526 CrossRef CAS.
- D. Sun, Y. Tian, Y. Zhang, Z. Xu, M. Y. Sfeir, M. Cotlet and O. Gang, ACS Nano, 2015, 9, 5657–5665 CrossRef CAS PubMed.
- N. Gandra, A. Abbas, L. Tian and S. Singamaneni, Nano Lett., 2012, 12, 2645–2651 CrossRef CAS PubMed.
- J. H. Yoon, J. Lim and S. Yoon, ACS Nano, 2012, 6, 7199–7208 CrossRef CAS PubMed.
- Y. Yang, J. Zhu, J. Zhao, G.-J. Weng, J.-J. Li and J.-W. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 3617–3626 CrossRef CAS PubMed.
- R. Schreiber, J. Do, E.-M. Roller, T. Zhang, V. J. Schüller, P. C. Nickels, J. Feldmann and T. Liedl, Nat. Nanotechnol., 2014, 9, 74–78 CrossRef CAS PubMed.
- L. Y. T. Chou, K. Zagorovsky and W. C. W. Chan, Nat. Nanotechnol., 2014, 9, 148–155 CrossRef CAS PubMed.
- C. Rossner, Q. Tang, O. Glatter, M. Müller and P. Vana, Langmuir, 2017, 33, 2017–2026 CrossRef CAS PubMed.
- J. Moraes, K. Ohno, T. Maschmeyer and S. Perrier, Chem. Commun., 2013, 49, 9077–9088 RSC.
- H. Zou, S. Wu and J. Shen, Chem. Rev., 2008, 108, 3893–3957 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |