
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of polyampholytic diblock copolymers via RAFT aqueous solution polymerization†
S. M.
North
and
S. P.
Armes
 *
*
Department of Chemistry, The University of Sheffield, Brook Hill, Sheffield, South Yorkshire S3 7HF, UK. E-mail: s.p.armes@shef.ac.uk
First published on 13th August 2021
Abstract
We report the synthesis of two new classes of polyampholytic diblock copolymers by RAFT aqueous solution polymerization. In each case, poly(methacrylic acid) (PMAA) is the anionic block while the cationic block comprises either poly(2-N-(morpholino)ethyl methacrylate) (PMEMA) or poly(2-(methacryloyloxy)ethyl trimethylammonium chloride) (PMETAC). Empirically, we found that polymerizing methacrylic acid as the second block afforded more well-defined diblock copolymers. Using this protocol, a series of copolymers of varying diblock composition is prepared for both classes. Robust derivatization protocols are developed to aid the characterization of such diblock copolymers via gel permeation chromatography (GPC). Thus the carboxylic acid groups within the PMAA block of the PMEMA–PMAA diblock copolymers are selectively methylated without quaternization of the tertiary amine groups on the PMEMA chains. In contrast, PMETAC–PMAA diblock copolymers are subjected to forced hydrolysis of the PMETAC ester groups to produce a PMAA homopolymer, which is then methylated to produce poly(methyl methacrylate) samples for GPC analysis. The aqueous solution properties of such polyampholytic diblock copolymers are explored using dynamic light scattering (DLS) and aqueous electrophoresis. These techniques enable identification of the isoelectric point. Unlike most other polyampholytic diblock copolymers reported in the literature, the PMEMA–PMAA diblock copolymers exhibit minimal variation in their isoelectric point when adjusting the diblock copolymer composition. This is because the pKa of the acidic PMAA block is close to the pKa of the conjugate acid form of the basic PMEMA block. For the PMETAC–PMAA system, no IEP is observed for PMETAC-rich copolymers because there is insufficient anionic charge to compensate for the cationic charge even if the PMAA chains are fully ionized.
Introduction
Polyzwitterions are an interesting class of water-soluble polymers that contain both cationic and anionic charge.1–5 There are two main classes of polyzwitterions. Polybetaines contain both cationic and anionic charge within each monomer repeat unit, whereas polyampholytes contain a binary mixture of cationic and anionic comonomers within every copolymer chain.6–9 In the particular case of polyampholytic diblock copolymers, one block contains all the cationic comonomer units and the other contains all the anionic comonomer units.10,11 The latter copolymers usually precipitate from aqueous solution at a specific pH when the number of cationic charges is equal to the number of anionic charges, since this results in no overall net charge.12 This is known as the isoelectric point (IEP) and such behavior is analogous to that observed for many proteins.13 For polyampholytic diblock copolymers that comprise a weak polyacid block and a weak polybase block, the IEP depends on the pKa of the former block, the pKa of the conjugate acid form of the latter block, and the diblock copolymer composition.14 For example, this is well known in the case for poly(2-(dimethylamino)ethyl methacrylate)–poly(methacrylic acid) (PDMA–PMAA) diblock copolymers.3,15–18 Potential applications suggested for polyampholytic diblock copolymers include protein purification,19,20 ion exchange,21 trace metal chelation,22 and pigment dispersion.23Traditionally, polyampholytic diblock copolymers have been prepared using anionic polymerization combined with protecting group chemistry for the polyacid block.23,24 However, with the development of various pseudo-living polymerization techniques over the past twenty-five years,25–27 such copolymers can now be prepared using radical chemistry without recourse to protecting groups.11,28 Indeed, we have recently reported the direct synthesis of well-defined PDMA–PMAA diblock copolymers in aqueous solution using reversible addition–fragmentation chain transfer (RAFT) polymerization.29 Such polyampholytes proved to be useful aqueous dispersants for a nano-sized iron oxide pigment.
Herein we report the synthesis of two new classes of well-defined polyampholytic diblock copolymers by RAFT aqueous solution polymerization. In each case, poly(methacrylic acid) (PMAA) is the anionic block while the cationic block comprises either poly(2-N-(morpholino)ethyl methacrylate) (PMEMA) or poly(2-(methacryloyloxy)ethyl trimethylammonium chloride) (PMETAC). PMEMA is a significantly weaker polybase than PDMA, while PMETAC remains permanently cationic at all solution pH (unlike PDMA or PMEMA). We examine whether it is better to prepare the anionic or the cationic block first for such aqueous syntheses and develop robust new derivatization protocols to aid the characterization of such diblock copolymers via gel permeation chromatography (GPC). The aqueous solution properties of such copolymers are briefly explored using dynamic light scattering (DLS), 1H NMR spectroscopy and aqueous electrophoresis. These techniques enable identification of the isoelectric point and reveal atypical behavior for the PMEMA–PMAA system compared to that of other polyampholytic diblock copolymers reported in the literature.2,5,16 Similarly, the PMETAC–PMAA system also exhibits unusual aqueous solution behaviour: colloidal complexes can be formed in alkaline solution and macroscopic precipitation can occur even when no IEP is observed.
Experimental
Materials
[2-(Methacryloyloxy)ethyl] trimethylammonium chloride (METAC), 2-(N-morpholino)ethyl methacrylate (MEMA) and trimethylsilyldiazomethane (supplied as a 2.0 M solution in diethyl ether) were purchased from Sigma-Aldrich (Dorset, UK) and were used as received. Methacrylic acid (MAA) was purchased from Merck (Germany) and was used as received. 2,2′-Azobis(2-(2-imidazolin-2-yl)propane) dihydrochloride (VA-044) was purchased from Wako Pure Chemical Industries (Japan). 4,4′-Azobis(4-cyanovaleric acid) (ACVA; 98%) was purchased from Alfa Aesar (Heysham, UK) and was used as received. 4-Cyano-4-(2-phenylethanesulfanylthiocarbonyl)-sulfanylpentanoic acid (PETTC) was synthesized as previously reported.30 4-((((2-Carboxyethyl)thio)carbonothioyl)thio)-4-cyanopentanoic acid (CECPA) was purchased from Boron Molecular (Australia). CD3OD and CD2Cl2 were purchased from Goss Scientific Instruments Ltd (Cheshire, UK). CDCl3, D2O, sodium deuteroxide (NaOD) and deuterium chloride (DCl) were purchased from Sigma-Aldrich (Dorset, UK). All other solvents were purchased from Fisher Scientific (Loughborough, UK) and were used as received. Deionized water was used for all experiments and the solution pH was adjusted using either HCl or NaOH.One-pot synthesis of poly(methacrylic acid)–poly(2-(methacryloyloxy)ethyl trimethylammonium chloride) (PMAA–PMETAC) diblock copolymer
The wholly aqueous one-pot synthesis of a PMAA–PMETAC diblock copolymer was conducted as follows. MAA (1.0 g, 11.6 mmol) and CECPA (59.4 mg, 0.19 mmol), were stirred thoroughly in a 50 ml round-bottomed flask prior to the addition of ACVA (10.8 mg, 38.7 μmol) and deionized water (6.07 g). The resulting 15% w/w aqueous acidic solution (pH 2) was then purged for 30 min with nitrogen and heated to 70 °C. Approximately 99% MAA conversion was achieved within 3 h as determined by 1H NMR spectroscopy. In a separate vial, METAC (2.41 g, 11.6 mmol), ACVA (20.8 mg, 48.4 μmol) and deionized water (7.89 g, target solids concentration = 20% w/w) were purged with nitrogen for 30 min. This degassed aqueous acidic solution (pH 3) was then added under a nitrogen atmosphere and the second-stage polymerization was allowed to continue for 6 h at 70 °C to yield a viscous yellow solution. A final METAC conversion of 96% was determined by 1H NMR spectroscopy.One-pot synthesis of poly(2-(methacryloyloxy)ethyl trimethylammonium chloride)–poly(methacrylic acid) (PMETAC–PMAA) diblock copolymer
The wholly aqueous one-pot synthesis of a PMETAC–PMAA diblock copolymer was conducted as follows. METAC (5.00 g of 80% w/w aqueous solution, 19.3 mmol) and CECPA (98.6 mg, 0.32 mmol) were stirred thoroughly to ensure full solubility for the RAFT agent at 30% w/w solids in a 100 ml round-bottomed flask prior to the addition of VA-044 initiator (20.7 mg, 0.064 mmol) and deionized water (6.18 g). The solution pH was adjusted to pH 6 using 0.5 M NaOH. This aqueous solution was then purged for 30 min with nitrogen and heated to 44 °C. After 5 h, the METAC polymerization had reached more than 99% conversion as determined by 1H NMR spectroscopy. In a separate vial, MAA (2.50 g, 29.0 mmol), VA-044 initiator (19.6 mg, 0.06 mmol) and deionized water (15.2 g, target solids concentration = 20% w/w) were purged with nitrogen for 30 min. This degassed aqueous acidic solution (pH 2) was added under a nitrogen atmosphere and the second-stage polymerization was allowed to continue for 6 h at 44 °C to yield a yellow solution. A final MAA conversion of more than 99% was achieved as determined by 1H NMR spectroscopy.One-pot synthesis of poly(methacrylic acid)–poly(2-(N-morpholino)ethyl methacrylate) (PMAA–PMEMA) diblock copolymer
A typical protocol for the one-pot wholly aqueous synthesis of a PMAA–PMEMA diblock copolymer was conducted as follows. MAA (1.50 g, 17.4 mmol) and CPDB (0.064 g, 0.29 mmol) were stirred thoroughly to ensure full solubility of the RAFT agent at 60% w/w solids in a 50 ml two-necked round-bottomed flask prior to the addition of ACVA initiator (16.3 mg, 0.058 mmol) and deionized water (1.05 g). This aqueous acidic solution (pH 2) was then purged for 30 min with nitrogen and heated to 70 °C. In a second vial, deionized water (7.90 g) was degassed and then added to the reaction solution after 45 min to offset the progressively increasing viscosity of the polymerizing solution. After 3 h, the MAA polymerization had reached more than 99% conversion as determined by 1H NMR spectroscopy. In a separate vial, MEMA (3.47 g, 17.4 mmol), ACVA (23.4 mg, 0.073 mmol), NaOH (0.697 g, 17.4 mmol) and deionized water (11.28 g, target solids concentration = 20% w/w, solution pH 8.5) were purged with nitrogen for 30 min. This degassed aqueous solution was added under a nitrogen atmosphere and the second-stage polymerization was allowed to continue for 16 h at 70 °C to yield a viscous reddish-pink solution. A final MEMA conversion of more than 99% was achieved as determined by 1H NMR spectroscopy.One-pot synthesis of poly(2-(N-morpholino)ethyl methacrylate)–poly(methacrylic acid) (PMEMA–PMAA) diblock copolymer
The wholly aqueous one-pot synthesis of a PMEMA–PMAA diblock copolymer was conducted as follows. MEMA (1.00 g, 5.02 mmol), CPDB (18.5 mg, 0.084 mmol) and 12 M HCl (0.43 g, 5.02 mmol) were stirred thoroughly to ensure full solubility of the RAFT agent at 30% w/w solids in a 50 ml round-bottomed flask prior to the addition of ACVA (5.86 mg, 0.021 mmol) and deionized water (2.39 g). This aqueous acidic solution (pH 4) was then purged for 30 min with nitrogen and heated to 70 °C. After 3 h, the MEMA polymerization had reached more than 99% conversion as determined by 1H NMR spectroscopy. In a separate vial, MAA (0.38 g, 4.41 mmol), ACVA (3.87 mg, 0.014 mmol) and deionized water (2.73 g, target solids concentration = 20% w/w) were purged with nitrogen for 30 min. This degassed aqueous acidic solution (pH 2) was then added under a nitrogen atmosphere and the second-stage polymerization was allowed to continue for 16 h at 70 °C to yield a pink solution. A final MAA conversion of more than 99% was achieved as determined by 1H NMR spectroscopy.Chemical derivatization of homopolymers and diblock copolymers for GPC analysis
PMETAC homopolymers, PMAA–PMETAC diblock copolymers and PMETAC–PMAA diblock copolymers were modified for GPC analysis as follows: 1.0 g of a 20% w/w aqueous copolymer dispersion was diluted with 1.0 g ethylene glycol (2.1 g total volume, 10% copolymer solids) containing 0.18 g KOH (3 M). This reaction solution was then heated to 120 °C for 6 h to convert this PMETAC, PMAA–PMETAC or PMETAC–PMAA precursor into the corresponding PMAA homopolymer via forced ester hydrolysis. The resulting dark brown liquid was acidified using 2 M HCl (2.5 mL) and purified by precipitation (twice) into excess diethyl ether. After filtration, this PMAA homopolymer was methylated a ten-fold excess of trimethylsilyldiazomethane to afford off-white poly(methyl methacrylate) (PMMA), which was analyzed by THF GPC (see below for further details). Essentially the same methylation protocol was used to selectively esterify all the MAA repeat units within the PMAA–PMEMA and PMEMA–PMAA diblock copolymers to produce PMMA–PMEMA and PMEMA–PMMA diblock copolymers, respectively.Gel permeation chromatography (GPC)
The THF GPC set-up comprised two 5 μm (30 cm) Mixed C columns and a WellChrom K-2301 refractive index detector operating at a wavelength of 950 ± 30 nm. The mobile phase contained 2.0% v/v triethylamine and 0.05% w/v butylhydroxytoluene and the flow rate was 1.0 mL min−1. A series of ten near-monodisperse poly(methyl methacrylate) standards (Mp values ranging from 645 to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 480000 g mol−1) were used for calibration. Chromatograms were analyzed using Agilent GPC/SEC software.
480000 g mol−1) were used for calibration. Chromatograms were analyzed using Agilent GPC/SEC software.
Dynamic light scattering
Dilute (0.10% w/w) aqueous copolymer dispersions were analyzed at 25 °C using a Malvern NanoZS instrument. Scattered light was detected at 173° and hydrodynamic diameters were calculated using the Stokes–Einstein equation, which assumes dilute non-interacting spheres. Data were averaged over three consecutive measurements comprising ten runs per measurement.Aqueous electrophoresis
Zeta potentials were calculated from electrophoretic mobilities using the same Malvern NanoZS instrument. Measurements were performed as a function of pH on dilute aqueous dispersions (0.05–0.10% w/w) in the presence of 1 mM KCl as background salt and averaged over 20 runs. In each case the solution pH was first increased by addition of 1.0 M NaOH and then gradually lowered by adding 0.1 M HCl.1H NMR spectroscopy
All 1H NMR spectra were recorded using a 400 MHz Bruker Avance-400 spectrometer. The NMR solvent was CD3OD, D2O or CDCl3 and typically 64 scans were averaged per spectrum.Results and discussion
The synthesis of poly(2-(methacryloyloxy)ethyl trimethylammonium chloride)–poly(methacrylic acid) (PMETAC–PMAA) diblock copolymers was optimized as described in Scheme 1. The nomenclature used in this manuscript is as follows: the first named block was that synthesized first and the subscripts refer to the target mean degrees of polymerization for each block. As noted in our prior study involving similar aqueous syntheses,29 it was found empirically that the order of monomer addition, i.e. which block is prepared first, is important for obtaining the highest possible monomer conversions and hence well-defined diblock copolymers. These parameters were also dependent on the choice of RAFT agent, with the water-soluble trithiocarbonate CECPA being required for optimal results, see Fig. S1.† | ||
| Scheme 1 Wholly aqueous one-pot synthetic route to PMETAC–PMAA diblock copolymers via (i) RAFT aqueous solution polymerization of [2-(methacryloyloxy)ethyl]trimethylammonium chloride (METAC) at pH 6 followed by (ii) RAFT aqueous solution polymerization of methacrylic acid (MAA) at pH 2. | ||
A PMETAC60 precursor was prepared at 30% w/w solids via RAFT aqueous solution polymerization of METAC at 44 °C using a CECPA RAFT agent and VA-044 initiator at pH 6, see Scheme 1. In this case, the solution pH had little effect on the polymerization, since the METAC monomer possesses no basic character – it retains its permanent cationic charge regardless of the solution pH. This polymerization reached essentially full conversion within 5 h as judged by 1H NMR studies. The subsequent MAA polymerization was conducted at 20% w/w solids and the solution pH was adjusted to pH 2, where the PMAA block exhibits neutral character. In each case, this second-stage polymerization proceeded to approximately 99% conversion.
In principle, ampholytic diblock copolymers can be characterized by aqueous GPC.3,5,29,31 In practice, this technique suffers from a lack of suitable calibration standards. For example, using a series of poly(ethylene oxide) standards to characterise the ampholytic diblock copolymers would most likely incur significant systematic errors. In view of this well-known problem, we undertook chemical derivatization of the two series of ampholytic diblock copolymers reported herein.
Accordingly, PMETAC–PMAA diblock copolymers and PMETAC homopolymers (and also PMAA–PMETAC diblock copolymers; see Table S1†) were subjected to a two-step chemical derivatization protocol prior to GPC studies, as summarized in Fig. 1. First, PMETAC chains (or blocks) are subjected to forced ester hydrolysis using 3 M NaOH at 120 °C for 6 h. An aqueous acidic work-up (to remove choline chloride, cleaved RAFT end-groups and NaCl) affords PMAA homopolymer, as confirmed by the complete disappearance of the NMR signal c at 3.4 ppm corresponding to the nine trimethylammonium protons.
 | ||
| Fig. 1
1H NMR spectra recorded for the PMETAC60–PMAA60 precursor in CD3OD (top); the same sample after being subjected to forced hydrolysis (3 M NaOH, 120 °C, 6 h in ethylene glycol) to afford PMAA120 (middle) and finally the same sample after exhaustive methylation a ten-fold excess of trimethylsilyldiazomethane in a 3:2 toluene/methanol mixture to afford PMMA120 (bottom). | ||
This precursor was then fully methylated using a ten-fold excess of trimethylsilyldiazomethane in a 3:2 toluene/methanol mixture for 72 h at 20 °C to generate the desired PMMA homopolymer. Comparison of the integrated methoxy signal h at 3.6 ppm with that of the methacrylic backbone signals indicates a mean degree of methylation of more than 99%. This derivative is fully soluble in THF and enables GPC analysis to be conducted using PMMA calibration standards, i.e. without incurring any systematic errors (see Fig. 2). In principle, such chemical modification should not result in any C–C bond scission nor lead to any crosslinking. In practice, this seems to be case, because GPC data indicate a series of well-defined PMETAC60–PMAAx diblock copolymers (Mw/Mn = 1.13 to 1.36) and a relatively high blocking efficiency relative to the PMETAC60 precursor (see Fig. 2 and Table 1). Moreover, the PMMA molecular weights are in reasonably good agreement with the expected values. For example, chemical modification of PMETAC60–PMAA120 (see entry 4 in Table 1) should yield a PMMA180 with a theoretical Mn of 18000 g mol−1, which compares rather well with the experimental Mn of 18400 g mol−1. Furthermore, this PMMA180 has the lowest dispersity (Mw/Mn = 1.13) of the four entries shown in Table 1. Interestingly, its parent PMETAC60–PMAA120 diblock copolymer contains the longest PMAA block. Bearing in mind the relatively high dispersities observed for each of the four PMETAC precursors (see Table 1), this suggests that the second-stage MAA polymerization is more well-controlled than the initial METAC polymerization. Indeed, this accounts for the systematic reduction in dispersity that is observed as higher PMAA DPs are targeted when using a fixed PMETAC DP of 60. Moreover, there is clearly minimal difference between the Mn and Mw/Mn values observed for the four ‘identical’ PMETAC60 precursors. This indicates remarkably good reproducibility for both this synthetic protocol and the subsequent GPC analysis. As far as we are aware, this is the first time that such a derivatization route has been used to assess the molecular weight distributions of ampholytic diblock copolymers.
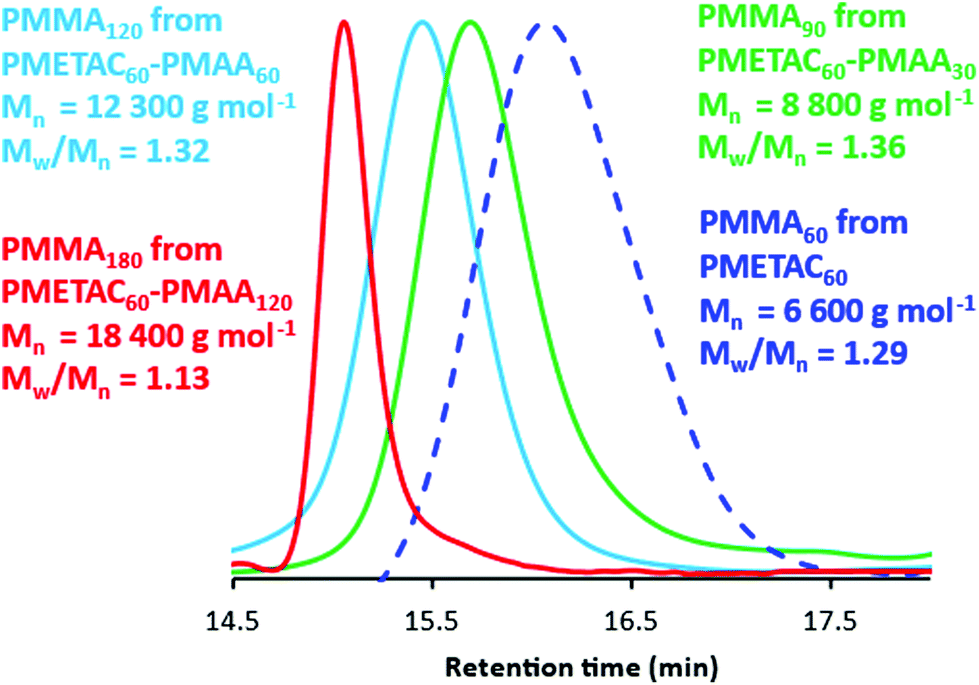 | ||
| Fig. 2 THF GPC curves recorded after the chemical modification of a PMETAC60 homopolymer and three PMETAC60–PMAAx diblock copolymers prepared at 20% w/w solids via one-pot RAFT aqueous dispersion polymerization of PMAA at pH 2. In each case, this two-step derivatization (see Fig. 1) was required to produce the corresponding PMMAy homopolymer prior to GPC analysis. Mn values are expressed relative to a series of near-monodisperse poly(methyl methacrylate) calibration standards (see also Table 1). | ||
| Target diblock copolymer composition | GPC analysis of PMETAC precursor | 1H NMR conversion (%) | GPC analysis of diblock copolymer | 1H NMR conversion (%) | IEP | ||
|---|---|---|---|---|---|---|---|
| M n (g mol−1) | M w/Mn | M n (g mol−1) | M w/Mn | ||||
| PMETAC60–PMAA30 | 6600 | 1.29 | >99 | 8800 | 1.36 | >99 | No IEP |
| PMETAC60–PMAA60 | 6600 | 1.28 | >99 | 12300 |
1.32 | >99 | No IEP |
| PMETAC60–PMAA90 | 6700 | 1.31 | >99 | 16000 |
1.23 | >99 | pH 6.0 |
| PMETAC60–PMAA120 | 6600 | 1.29 | >99 | 18400 |
1.13 | >99 | pH 5.4 |
The same diblock copolymer synthesis was also attempted by reversing the monomer sequence, see Scheme S1.† Thus, MAA was polymerized via RAFT aqueous solution polymerization at pH 2 and 20% w/w solids using ACVA initiator at 70 °C. This polymerization reaches full conversion within 3 h and the resulting PMAA precursor can then be chain-extended with METAC at 70 °C to give a PMAA60–PMETAC60 diblock copolymer at pH 3. In each case, the difference in the solution pH has minimal effect but the lower temperature required for the former synthesis in Scheme 1 is considered more favorable.32
The final monomer conversions (calculated by 1H NMR spectroscopy) and molecular weight data (determined by THF GPC analysis after their forced hydrolysis and exhaustive methylation) for PMETAC–PMAA diblock copolymers are summarized in Table 1. The latter data lie close to the theoretical molecular weights expected for these diblock copolymers after their conversion into the corresponding PMMA homopolymers, which is a strong testament to the validity of this two-step derivatization protocol. The corresponding data for the PMAA–PMETAC diblock copolymers are shown in Table S1.† Comparing Tables 1 and S1,† it is notable that the METAC-first syntheses enable more than 99% monomer conversions to be achieved in all cases. The synthesis of the PMETAC precursor is also very consistent, with comparable Mn and Mw/Mn data being obtained for these one-pot protocols. Moreover, the incomplete MAA conversions obtained for the PMAA–PMETAC syntheses (Table S1†) lead to inaccurate diblock compositions. Thus the optimum reaction sequence is to polymerize METAC first followed by MAA; this synthetic route yields well-defined PMETAC–PMAA diblock copolymers. Further synthesis optimization may well lead to better control over the molecular weight distribution.
We also explored the synthesis of PMEMA–PMAA diblock copolymers using a similar wholly aqueous one-pot protocol, see Scheme 2. In this case, the dithiobenzoate-based RAFT agent CPDB was used. This CTA resulted in consistently higher monomer conversions and narrower polydispersities by GPC compared to trithiocarbonate-based CTAs. First, a PMEMA60 precursor was prepared at 30% w/w solids using ACVA at pH 4 within 3 h at 70 °C, as judged by 1H NMR studies. This precursor was then used to polymerize MAA at pH 2, which is the optimum pH for this second-stage polymerization, to form a series of well-defined PMEMA60–PMAAx diblock copolymers as judged by THF GPC (Fig. 3).
 | ||
| Scheme 2 Wholly aqueous one-pot synthetic route to PMEMA–PMAA diblock copolymers via (i) RAFT aqueous solution polymerization of (2-(N-morpholino)ethyl methacrylate) (MEMA) at pH 4 followed by (ii) RAFT aqueous solution polymerization of methacrylic acid (MAA) at pH 2. | ||
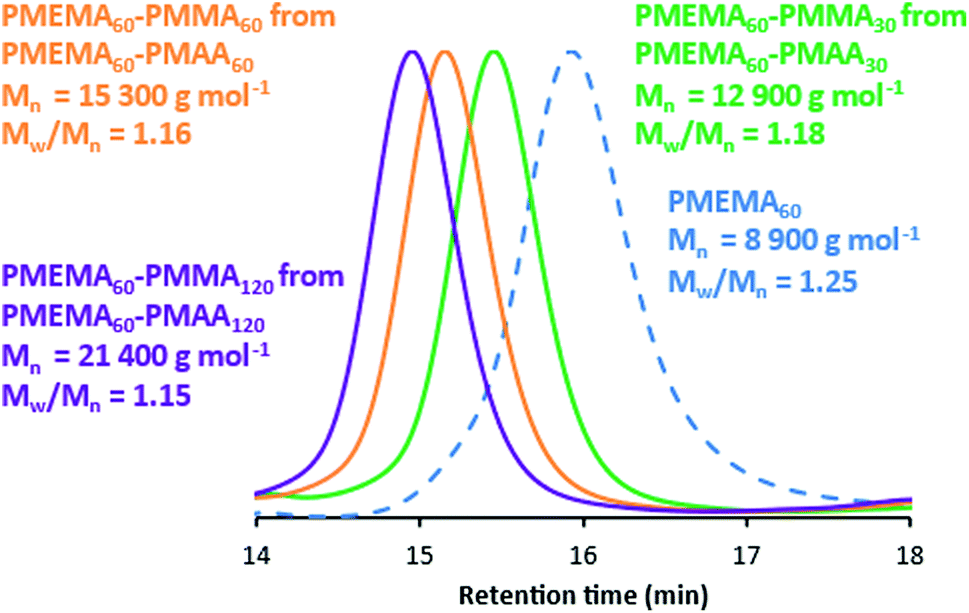 | ||
| Fig. 3 THF GPC curves recorded for a PMEMA60 homopolymer and three PMEMA60–PMAAx diblock copolymers prepared at 20% w/w solids via one-pot RAFT aqueous dispersion polymerisation of PMAA at pH 2. Mn values are expressed relative to a series of near-monodisperse poly(methyl methacrylate) calibration standards. | ||
As noted above, the reverse monomer sequence is also feasible. Thus, a PMAA precursor is prepared first via RAFT aqueous solution polymerization, then subsequently chain-extended with MEMA at a solution pH of 8.5 using ACVA at 70 °C, see Scheme S2.† This pH is required to ensure solubility of the PMEMA block during the second-stage polymerization. However, this MAA-first monomer sequence may be sub-optimal because this relatively high pH could lead to premature hydrolysis of the RAFT chain-ends, which results in reduced Mn control and broader molecular weight distributions.33,34
In order to assess the molecular weight distributions of these PMEMA–PMAA and PMAA–PMEMA diblock copolymers, chemical modification was again essential to ensure solubility in a suitable GPC eluent, as well as minimizing any unwanted interactions with the GPC columns. Fortunately, the carboxylic acid residues of such diblock copolymers can be selectively methylated using a ten-fold excess of trimethylsilyldiazomethane in a 3:2 toluene/methanol mixture for 72 h at 20 °C. To ensure that these relatively mild conditions did not result in unwanted methylation of the PMEMA block,35 a control experiment was performed in which a PMEMA60 homopolymer was subjected to the same methylation conditions.
The 1H NMR spectrum of the product isolated from this attempted reaction is more or less identical to that recorded for the PMEMA precursor (see Fig. S2 in the ESI†). Moreover, the corresponding GPC curves recorded before and after attempted methylation also remain essentially unchanged (see Fig. S2†). Such control experiments indicate that the degree of methylation of the PMEMA block is negligible when performing the esterification of the PMAA block to afford the desired PMEMA–PMMA diblock copolymer derivatives for GPC analysis. 1H NMR spectroscopy was used to determine the extent of methylation for such chemical derivatization (Fig. 4). As expected, a new methyl ester proton signal can be observed at approximately 4.4 ppm. Comparison of this integrated signal with that of the methacrylic backbone indicated a mean degree of methylation of 99%.
 | ||
| Fig. 4 1H NMR spectra recorded for a PMEMA60–PMAA60 diblock copolymer precursor in CD3OD (blue, top) and after selective methylation with excess trimethylsilyldiazomethane in d5-pyridine (red, bottom) confirming the appearance of a new methyl ester signal, e, at approximately 4.4 ppm. | ||
These THF-soluble PMEMA–PMMA diblock copolymers are amenable to GPC analysis to assess their molecular weight distributions (Table 2). The molecular weight data for the corresponding PMAA–PMEMA diblock copolymers are provided in Table S1.† Again, it is notable that the MEMA-first syntheses are more efficient, with relatively high monomer conversions (>98%) being obtained for each block in all cases as judged by 1H NMR. GPC dispersities are also significantly lower for the MEMA-first syntheses (Mw/Mn < 1.13), indicating narrow molecular weight distributions. Moreover, the Mn and Mw/Mn data obtained for the PMEMA precursor indicate good reproducibility for this one-pot protocol. This may be related to the lower solution pH used for this MEMA polymerization. In contrast, the second-stage MEMA polymerization was conducted at pH 8.5 (Scheme S2†), which is known to be sub-optimal for well-controlled RAFT syntheses.33,34
| Target diblock copolymer composition | GPC analysis of PMEMA precursor | 1H NMR conversion (%) | GPC analysis of diblock copolymer | 1H NMR conversion (%) | IEP | ||
|---|---|---|---|---|---|---|---|
| M n (g mol−1) | M w/Mn | M n (g mol−1) | M w/Mn | ||||
| PMEMA60–PMAA30 | 8900 | 1.26 | >99 | 12900 |
1.18 | >99 | pH 5.9 |
| PMEMA60–PMAA60 | 8800 | 1.25 | >99 | 15300 |
1.16 | 98 | pH 5.7 |
| PMEMA60–PMAA120 | 8900 | 1.23 | >99 | 21400 |
1.15 | >99 | pH 5.6 |
The IEPs of these ampholytic diblock copolymers can be determined from aqueous electrophoresis studies. Representative data obtained for the three PMEMA60–PMAAx copolymers are shown in Fig. 5. Unlike other ampholytic diblock copolymers reported in the literature, the IEP at which reversible macroscopic precipitation (see the shaded regions within Fig. 5) occurs is remarkably insensitive to the diblock copolymer composition: only a rather modest reduction from 5.9 to 5.3 is observed with increasing MAA content. This is simply because the pKa for the acidic PMAA block (pKa ∼ 5.5)36,37 is almost the same as the pKa for the conjugate acid form of the PMEMA block (pKa ∼ 4.9).38,39 We are not aware of any other weak polyacid – weak polybase diblock copolymer system that exhibits such behavior.
 | ||
| Fig. 5 Zeta potential vs. pH curves constructed for (a) PMEMA60–PMAA30, (b) PMEMA60–PMAA60 and (c) PMEMA60–PMAA120 in the presence of 1 mM KCl background salt. The shaded regions indicate colloidal instability, with the mid-point of these regions corresponding to the isoelectric point (IEP). | ||
1H NMR spectroscopy can be used to assess changes in the proton environment of each block for these ampholytic diblock copolymers. For the PMEMA60–PMAA60 diblock copolymer (Fig. 6), a significant change is observed in the proton signals nearest to the protonated nitrogen atom. Signals b and c are shifted from approximately 2.5 and 2.6 ppm to 3.4 and 3.7 ppm, respectively. Similarly, signals a and d are shifted from 3.7 and 4.1 ppm to 3.9 and 4.2 ppm, respectively. Integration of the methacrylic backbone signals indicates partial desolvation of the PMAA block when it is in its neutral form at pH 2. At the IEP (pH 5.7), all the diblock copolymer signals are substantially attenuated owing to its insolubility under such conditions.
 | ||
| Fig. 6 1H NMR spectra recorded for a PMEMA60–PMAA60 diblock copolymer in DCl/D2O at pH 2 (where the PMAA block is in its neutral form and the PMEMA block is in its cationic protonated form), at its IEP of pH 5.7 (where the diblock copolymer is insoluble so all signals are substantially attenuated), and in NaOD/D2O at pH 10 (where the PMAA block is ionized and the PMEMA block is present in its neutral form). | ||
For the series of PMETAC–PMAA diblock copolymers, aqueous electrophoresis studies revealed a range of interesting behavior. For example, IEPs could only be observed for PMAA-rich compositions, whereby a higher MAA content leads to a lower IEP as expected (compare Fig. 7c and d). In contrast, no IEP is observed for the symmetric PMETAC60–PMAA60 diblock copolymer with zeta potentials remaining positive regardless of the solution pH. This was rather unexpected behavior, not least because the two carboxylic acid end-groups conferred by the RAFT CTA should lead to weakly anionic character when all the methacrylic acid groups are ionized. 1H NMR studies suggested that the DP of the PMETAC block might be slightly higher than that targeted but the lack of unique protons for the PMAA block makes this difficult to confirm with certainty. Despite not exhibiting an IEP, the PMETAC60–PMAA60 diblock copolymer nevertheless undergoes macroscopic precipitation above pH 6. Finally, for the PMETAC-rich PMETAC60–PMAA30 diblock copolymer, zeta potentials remain positive across the whole pH range but colloidal complexes are formed above pH 8 with a z-average diameter of around 130 nm. Presumably, such multimolecular complexes comprise charge-compensated cores containing the relatively short anionic PMAA30 block and around half of the permanently cationic METAC repeat units, leaving the remaining uncomplexed portion of the PMETAC block to form a cationic corona.
 | ||
| Fig. 7 Zeta potential vs. pH curves constructed for (a) PMETAC60–PMAA30, (b) PMETAC60–PMAA60, (c) PMETAC60–PMAA90, and (d) PMETAC60–PMAA120 in the presence of 1 mM KCl background salt. The shaded regions indicate colloidal instability, with the mid-point of these regions corresponding to the isoelectric point (IEP). | ||
Conclusions
In summary, we report the wholly aqueous one-pot synthesis of PMEMA–PMAA and PMETAC–PMAA ampholytic diblock copolymers via RAFT solution polymerization. Under the stated reaction conditions, these syntheses are highly efficient and do not require protecting group chemistry. A two-step method is developed for the chemical derivatization of PMETAC–PMAA diblock copolymers via forced ester hydrolysis and exhaustive methylation: 1H NMR spectroscopy studies confirm that this robust protocol produces a series of PMMA homopolymers, which enables GPC analysis to be conducted without incurring any systematic error when using a series of near-monodisperse PMMA calibration standards. Similarly, the methacrylic acid repeat units within PMEMA–PMAA diblock copolymers can be selectively methylated to generate PMEMA–PMMA diblock copolymers. In each case, GPC analysis indicates relatively narrow molecular weight distributions for both PMEMA–PMAA (Mw/Mn < 1.13) and PMETAC–PMAA (Mw/Mn < 1.36) diblock copolymers. Aqueous solubility behavior was assessed by 1H NMR studies in D2O: this indicates that macroscopic precipitation occurs at around the IEP for selected PMEMA–PMAA diblock copolymers. Aqueous electrophoresis and dynamic light scattering experiments confirm that systematic variation of the diblock copolymer composition enables the isoelectric point to be tuned in the case of the PMETAC–PMAA diblock copolymer series. However, in the case of the PMEMA–PMAA series, the isoelectric point remains relatively constant. This unusual observation is simply because the pKa for the acidic PMAA block is comparable to that of the conjugate acid form of the basic PMEMA block. Finally, aqueous electrophoresis and dynamic light scattering experiments indicate the formation of colloidal complexes by a PMETAC-rich PMETAC–PMAA diblock copolymer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
EPSRC is thanked for funding a CDT PhD studentship for the first author (EP/L016281). Lubrizol (Blackley, UK) is thanked for partial financial support of this PhD project and for permission to publish this work. S. P. A. also thanks EPSRC for an Established Career Particle Technology Fellowship (EP/R003009).Notes and references
- X. Xin, Y. Wang and W. Liu, Eur. Polym. J., 2005, 41, 1539 CrossRef CAS.
- V. Bütün, S. Liu, J. V. M. Weaver, X. Bories-Azeau, Y. Cai and S. P. Armes, React. Funct. Polym., 2006, 66, 157 CrossRef.
- C. S. Patrickios, W. R. Hertler, N. L. Abbott and T. A. Hatton, Macromolecules, 1994, 27, 930 CrossRef CAS.
- S. Liu and S. P. Armes, Angew. Chem., Int. Ed., 2002, 41, 1413 CrossRef CAS PubMed.
- A. B. Lowe and C. L. McCormick, Chem. Rev., 2002, 102, 4177 CrossRef CAS PubMed.
- A. V. Dobrynin, R. H. Colby and M. Rubinstein, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 3513 CrossRef CAS.
- A. Laschewsky, Polymers, 2014, 6, 1544 CrossRef.
- K. E. B. Doncom, N. J. Warren and S. P. Armes, Polym. Chem., 2015, 6, 7264 RSC.
- L. D. Blackman, P. A. Gunatillake, P. Cass and K. E. S. Locock, Chem. Soc. Rev., 2019, 48, 757 RSC.
- C. L. McCormick and A. B. Lowe, Acc. Chem. Res., 2004, 37, 312 CrossRef CAS PubMed.
- X. Bories-Azeau, S. P. Armes and H. J. W. van den Haak, Macromolecules, 2004, 37, 2348 CrossRef CAS.
- L. I. Gabaston, S. A. Furlong, R. A. Jackson and S. P. Armes, Polymer, 1999, 40, 4505 CrossRef CAS.
- A. Hernández-Hernández, A. B. Rodríguez-Navarro, J. Gómez-Morales, C. Jiménez-López, Y. Nys and J. M. García-Ruiz, Cryst. Growth Des., 2008, 8, 1495 CrossRef.
- T. Alfrey, R. M. Fuoss, H. Morawetz and H. Pinner, J. Am. Chem. Soc., 1952, 74, 438 CrossRef CAS.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435 RSC.
- J. F. Gohy, S. Creutz, M. Garcia, B. Mahltig, M. Stamm and R. Jérôme, Macromolecules, 2000, 33, 6378 CrossRef CAS.
- S. Creutz and R. Jérôme, Langmuir, 1999, 15, 7145 CrossRef CAS.
- F. L. Baines, S. P. Armes, N. C. Billingham and Z. Tuzar, Macromolecules, 1996, 29, 8151 CrossRef CAS.
- T. Chakrabarty, M. Kumar and V. K. Shahi, Ind. Eng. Chem. Res., 2012, 51, 3015 CrossRef CAS.
- R. K. Scopes, Protein Purification, Springer, New York, 1982, p. 39 Search PubMed.
- J. Liu, Y. Ma, T. Xu and G. Shao, J. Hazard. Mater., 2010, 178, 1021 CrossRef CAS PubMed.
- P. Liu, A. J. Boyle, Y. Lu, J. Adams, Y. Chi, R. M. Reilly and M. A. Winnk, Biomacromolecules, 2015, 16, 3613 CrossRef CAS PubMed.
- S. Creutz, R. Jérôme, G. M. P. Kaptijn, A. W. Van Der Werf and J. M. Akkerman, J. Coat. Technol., 1998, 70, 41 CrossRef CAS.
- A. B. Lowe, N. C. Billingham and S. P. Armes, Macromolecules, 1998, 31, 5991 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133 CrossRef CAS PubMed.
- G. Moad and D. H. Solomon, Aust. J. Chem., 2005, 58, 379 CrossRef CAS.
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614 CrossRef CAS.
- C. D. Vo, S. P. Armes, D. P. Randall, K. Sakai and S. Biggs, Macromolecules, 2007, 40, 157 CrossRef CAS.
- S. M. North and S. P. Armes, Green Chem., 2021, 23, 1248 RSC.
- N. J. W. Penfold, J. R. Whatley and S. P. Armes, Macromolecules, 2019, 52, 1653 CrossRef CAS.
- T. C. Krasia and C. S. Patrickios, Polymer, 2002, 43, 2917 CrossRef CAS.
- Following a query by one of the reviewers of this manuscript, we attempted the synthesis of PMAA60–PMETAC120 and also PMEMA60–PMAA120 using the VA-044 initiator at 44 °C (instead of using the ACVA initiator at 70 °C). For the synthesis of a PMAA60–PMETAC120 diblock copolymer using VA-044, 1H NMR studies confirmed that 99% conversion was achieved after 16 h at 44 °C, while GPC analysis indicated an Mn of 13400 and an Mw/Mn of 1.28. These results are marginally better than those reported in Table S1† (see first entry), which were obtained when targeting the same diblock copolymer using ACVA at 70 °C. Thus there is no decisive advantage in performing this synthesis at a lower temperature. For the synthesis of a PMEMA60–PMAA120 diblock copolymer using the VA-044 initiator, 1H NMR studies confirmed 97% conversion after 16 h at 44 °C, while GPC analysis indicated an Mn of 49500 and an Mw/Mn of 1.57. Both values are significantly higher than the Mn and Mw/Mn data obtained when targeting the same diblock copolymer using ACVA at 70 °C (see Table 2), suggesting inferior control for the VA-044 polymerization.
- D. B. Thomas, A. J. Convertine, R. D. Hester, A. B. Lowe and C. L. McCormick, Macromolecules, 2004, 37, 1735 CrossRef CAS.
- S. Garnier and A. Laschewsky, Macromolecules, 2005, 38, 7580 CrossRef CAS.
- V. Bütün, S. P. Armes and N. C. Billingham, Macromolecules, 2001, 34, 1148 CrossRef.
- S. Guragain, B. P. Bastakoti and K. Nakashima, J. Colloid Interface Sci., 2010, 350, 63 CrossRef CAS PubMed.
- J. Zhang and N. A. Peppas, Macromolecules, 2000, 33, 102 CrossRef CAS.
- X. Bories-Azeau, S. P. Armes and H. J. W. Van Den Haak, Macromolecules, 2004, 37, 2348 CrossRef CAS.
- V. Bütün, S. P. Armes, N. C. Billingham, Z. Tuzar, A. Rankin, J. Eastoe and R. K. Heenan, Macromolecules, 2001, 34, 1503 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Schemes to describe the one-pot PMAA-first syntheses of PMAA–PMETAC and PMAA–PMEMA and a summary table for the copolymer molecular weight data, 1H NMR conversions and IEP data for these PMAA-first syntheses, GPC and NMR data for the attempted methylation of a PMEMA homopolymer; aqueous electrophoresis and DLS data for PMAA-first diblock copolymers. See DOI: 10.1039/d1py01020d |
| This journal is © The Royal Society of Chemistry 2021 |