
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
RAFT aqueous emulsion polymerization of methyl methacrylate: observation of unexpected constraints when employing a non-ionic steric stabilizer block†
Derek H. H.
Chan
a,
Amy A.
Cockram
a,
Rebecca R.
Gibson
a,
Emily L.
Kynaston
b,
Christopher
Lindsay
b,
Philip
Taylor
b and
Steven P.
Armes
 *a
*a
aDainton Building, Department of Chemistry, University of Sheffield, Brook Hill, Sheffield, South Yorkshire S3 7HF, UK. E-mail: s.p.armes@sheffield.ac.uk
bSyngenta, Jealott's Hill International Research Centre, Bracknell, Berkshire, RG42 6EY, UK
First published on 27th September 2021
Abstract
The RAFT aqueous emulsion polymerization of methyl methacrylate (MMA) is conducted at 70 °C using poly(glycerol monomethacrylate) (PGMA) as a steric stabilizer block. This non-ionic precursor has previously proved to be highly effective for the RAFT aqueous emulsion polymerization of various vinyl monomers such as benzyl methacrylate (BzMA), 2,2,2-trifluoroethyl methacrylate (TFEMA), isopropylideneglycerol monomethacrylate (IPGMA) or glycidyl methacrylate. However, an unexpected constraint was encountered in the case of MMA. Targeting a degree of polymerization (DP) of 20 to 100 for the PMMA block led to colloidal dispersions of kinetically-trapped spherical nanoparticles ranging in size from 17 nm to 31 nm. On the other hand, targeting DPs above 100 invariably led to the formation of highly flocculated spherical nanoparticles. This rather limited DP range is in striking contrast to the much higher DPs that can be targeted without loss of colloidal stability when using more hydrophobic monomers such as BzMA, TFEMA or IPGMA. The same flocculation problem was also evident when employing a PGMA precursor containing an anionic carboxylate end-group, but a series of colloidally stable dispersions could be obtained when using an anionic poly(methacrylic acid) stabilizer. Finally, the efficient removal of RAFT end-groups from PGMA50-PMMA80 nanoparticles was achieved by visible light irradiation using a blue LED source (λ = 405 nm). UV GPC studies confirmed that up to 87% dithiobenzoate end-groups can be removed from such nanoparticles within 12 h at 80 °C. On the other hand, using excess H2O2 under the same conditions only led to 24% end-group removal. This is because this water-soluble reagent has restricted access to the hydrophobic PMMA cores.
Introduction
Reversible addition–fragmentation chain transfer (RAFT) polymerization is a well-established pseudo-living technique based on the principle of rapid reversible chain transfer that enables the convenient synthesis of a wide range of controlled-structure vinyl polymers using various organosulfur-based reagents.1–5 RAFT polymerization is exceptionally tolerant of monomer functionality and is usually conducted in either organic solvents or aqueous solution to afford soluble polymer chains.6 However, it is equally well-suited to heterogeneous conditions such as suspension,7 dispersion8–12 or emulsion13–15 polymerization. In particular, RAFT aqueous emulsion polymerization – which is an important example of polymerization-induced self-assembly (PISA)16,17 − enables the controlled polymerization of a wide range of water-immiscible vinyl monomers in aqueous media.17–19 Typically, spherical nanoparticles of tunable particle size can be readily prepared at high solids,16 although so-called ‘higher order’ copolymer morphologies (e.g. worms or vesicles) are also accessible for at least some formulations.18–20 Potential applications for such nano-objects include surface modifiers for cellulose fibres,21 viscosity modifiers,22 organic opacifiers for paints23 and additives for the reinforcement of latex films.24There are many reports of the RAFT aqueous emulsion polymerization of commodity monomers such as styrene,12,23,25–27n-butyl acrylate13,28–30 or vinyl acetate.31 Moreover, there are also various reports of the use of speciality vinyl monomers such as benzyl methacrylate,32 2,2,2-trifluoroethyl methacrylate,33 isopropylideneglycerol monomethacrylate,34 2-methoxyethyl methacrylate,35 glycidyl methacrylate,36–39 or hydroxybutyl methacrylate.40 However, given its undoubted importance as a commodity monomer, there are surprisingly few literature reports of the RAFT aqueous emulsion polymerization of methyl methacrylate (MMA).21,29,41–43 Moreover, given the moderately high aqueous solubility of this monomer (15.9 g dm−3 at 20 °C), such PISA formulations might be expected to provide convenient access to a wide range of block copolymer nano-objects (e.g. spheres, worms and vesicles).35,39,40,44
In the present study, we examine the RAFT emulsion polymerization of MMA using poly(glycerol monomethacrylate) (PGMA) as a water-soluble precursor (Scheme 1), which is known to act as an effective non-ionic steric stabilizer for various other water-immiscible vinyl monomers under such conditions.32–36 In view of the unexpected limited utility of this PISA formulation, a PGMA stabilizer containing an anionic carboxylate end-group and an anionic poly(methacrylic acid) stabilizer were also evaluated for the RAFT emulsion polymerization of MMA.
 | ||
| Scheme 1 Synthesis of PGMA50-PMMAx diblock copolymer nanoparticles by RAFT aqueous emulsion polymerization of methyl methacrylate MMA at 70 °C using a dithiobenzoate-capped PGMA50 precursor to target x = 20 to 130 at pH 7. | ||
Experimental section
Materials
Glycerol monomethacrylate (GMA) was donated by GEO Specialty Chemicals (Hythe, UK). Methyl methacrylate (MMA; 99%), 4,4′-azobis(4-cyanopentanoic acid) (ACVA; 98%), methacrylic acid (MAA; 99%), [2-(methacryloyloxy)ethyl]tri methylammonium chloride (METAC; 80% solution), 2-cyano-2-propyl benzodithioate (CPDB; 97%) and trimethylsilyl diazomethane solution (2.0 M in hexanes) were each purchased from Sigma-Aldrich (UK) and used as received. 4-Cyano-4-(2-phenylethanesulfanylthiocarbonyl) sulfanylpentanoic acid (PETTC) was prepared according to a literature protocol.45 Deionized water from an Elga Medica DV25 water purification unit was used in all the experiments.Synthesis of the PGMA50 precursor by RAFT aqueous solution polymerization
GMA monomer (30.0 g, 187 mmol), CPDB RAFT agent (0.589 g, 2.66 mmol; target PGMA DP = 70) and ACVA initiator (0.149 g, 0.53 mmol; CPDB/ACVA molar ratio = 5.0) were weighed into a 250 mL round-bottom flask. Ethanol (46.5 g, 60% w/w) was added and the flask was cooled by immersing in an ice bath while degassed with N2 gas for 30 min. The flask was then immersed in an oil bath at 70 °C for 165 min. The polymerization was quenched by exposing the reaction mixture to air and cooling the flask to 20 °C. 1H NMR spectroscopy indicated a final GMA conversion of 71%. The reaction solution was diluted with methanol (30 mL) and then precipitated into a ten-fold excess of dichloromethane (three times). End-group analysis via1H NMR spectroscopy indicated a mean degree of polymerization (DP) of 50 (the integrated aromatic proton signals at 7.4–7.8 ppm were compared to the integrated methacrylic backbone proton signals at 0.7–2.5 ppm).Synthesis of the HOOC-PGMA54 precursor by RAFT aqueous solution polymerization
GMA monomer (10.0 g, 62.4 mmol), PETTC RAFT agent (0.302 g, 0.892 mmol; target PGMA DP = 70) and ACVA initiator (0.050 g, 0.18 mmol; PETTC/ACVA molar ratio = 5.0) were weighed into a 100 mL round-bottom flask. Ethanol (15.5 g, 60% w/w) was added and the flask was cooled by immersing in an ice bath while degassed with N2 gas for 30 min. The flask was then immersed in an oil bath at 70 °C for 120 min. The polymerization was quenched by exposing the reaction mixture to air and cooling the flask to 20 °C. 1H NMR spectroscopy indicated a final GMA conversion of 77%. The solution was diluted with methanol (10 mL) and then precipitated into a ten-fold excess of dichloromethane (three times). End-group analysis via1H NMR spectroscopy indicated a mean DP of 54.Synthesis of PGMA50-PMMAx diblock copolymer nanoparticles by RAFT aqueous emulsion polymerization
A typical protocol for the synthesis of PGMA50-PMMAx diblock copolymer nanoparticles was as follows. PGMA50 precursor (0.150 g, 18.2 μmol), MMA monomer (0.146 g, 1.46 mmol, target DP = 80), ACVA initiator (1.00 mg, 3.65 μmol, PGMA50/ACVA molar ratio = 5.0) and deionized water (2.675 g, 10% w/w solution) were added to a 20 mL round-bottom flask. The mixture was adjusted to pH 7 using 1 M NaOH. The flask was then placed in an ice bath and degassed with N2 gas for 30 min, before immersing the flask in an oil bath set at 70 °C. After 3 h, the polymerization was quenched by exposing the reaction mixture to air while cooling the flask to 20 °C. Essentially the same protocol was employed for the synthesis of HOOC-PGMA54-PMMAx diblock copolymer nanoparticles but in this case the HOOC-PGMA54 precursor was used instead of the PGMA50 precursor.Synthesis of the PMAA56 precursor by RAFT aqueous solution polymerization
MAA monomer (45.0 g, 0.523 mol), PETTC RAFT agent (3.165 g, 9.34 mmol; target DP = 56) and ACVA initiator (0.523 g, 1.87 mmol; PETTC/ACVA molar ratio = 5.0) and ethanol (73.0 g, 40% w/w) were weighed into a 500 mL round-bottom flask. The solution was placed in an ice bath and degassed with N2 gas for 30 min, before the flask was immersed in an oil bath at 70 °C. The polymerization was quenched after 3 h by exposing the reaction mixture to air while cooling the flask to 20 °C. The crude polymer was then precipitated into a ten-fold excess of diethyl ether. The insoluble polymer was redissolved in ethanol prior to a second precipitation step and then freeze-dried overnight. End-group analysis by 1H NMR spectroscopy indicated a DP of 56 for this PMAA precursor.Synthesis of PMAA56-PMMAx diblock copolymer nanoparticles by RAFT aqueous emulsion polymerization
A typical protocol for the synthesis of PMAA56-PMMA100 diblock copolymer nanoparticles was conducted as follows. PMAA56 precursor (0.100 g, 19.4 μmol), MMA monomer (0.194 g, 1.94 mmol, target DP = 100), ACVA initiator (1.10 mg, 3.88 μmol, PMAA56/ACVA molar ratio = 5.0) and deionized water (1.18 g, 20% w/w solution) were added to a 20 mL round-bottom flask. The mixture was adjusted to pH 5 using 1 M HCl. The reaction mixture was then placed in an ice bath and degassed with N2 gas for 30 min, before immersing in an oil bath set at 70 °C. The polymerization was allowed to proceed at this temperature for 6 h and then quenched by exposing the reaction mixture to air while cooling the flask to 20 °C.Methylation of PMAA56-PMMAx diblock copolymer nanoparticles
Methylation of the carboxylic acid groups on the PMAA block was performed before THF GPC analysis. PMAA56-PMMAx diblock copolymer powder (20 mg) was diluted in THF (2.0 mL). Excess trimethylsilyldiazomethane was then gradually added to this solution until it turned yellow. The reaction mixture was stirred overnight, dried and analysed by THF GPC.Protocols for cleavage of RAFT end-groups
The dithiobenzoate end-groups within PGMA50-PMMA80 nanoparticles were cleaved using the following protocol. A 10% w/w copolymer dispersion (1.00 g) was placed in a water-jacketed Schlenk tube wrapped in blue LED light strips (λ = 405 nm, 0.37 mW cm−2) with the temperature of the water within the recirculating jacket set to 80 °C. Aliquots of this reaction mixture were extracted periodically and analyzed using UV GPC (with the UV detector set at λ = 308 nm). As a control experiment, the same protocol was used but without the blue LED light strips (i.e. no visible light irradiation). The same 10% w/w dispersion of PGMA50-PMMA80 nanoparticles was also treated with H2O2 (using a five-fold excess relative to the concentration of dithiobenzoate end-groups, as reported by Jesson and co-workers46) at 80 °C with no visible light irradiation. The same protocol was used for cleaving trithiocarbonate RAFT end-groups from a 10% w/w aqueous dispersion of PGMA54-PMMA80 nanoparticles. This included cleavage at 80 °C using blue LED strips, H2O2 or no visible light irradiation for 24 h.Dynamic light scattering and aqueous electrophoresis
DLS and aqueous electrophoresis studies were conducted on 0.50% w/w aqueous dispersions using a Malvern Zetasizer NanoZS instrument. The hydrodynamic z-average diameter was determined at 20 °C at a scattering angle of 173° and averaged over three measurements. Aqueous electrophoresis studies were conducted in the presence of 1 mM KCl as background electrolyte. The solution pH was adjusted using either NaOH or HCl. Zeta potentials (also averaged over three measurements) were calculated via the Henry equation using the Smoluchowski approximation.Gel permeation chromatography (GPC)
Molecular weight distributions of the PGMA50 and HOOC-PGMA54 precursors and a series of PGMA50-PMMA80 diblock copolymers were assessed using DMF eluent (containing 10 mM LiBr) at 60 °C. Two Polymer Laboratories PL gel 5 μm Mixed-C columns were connected in series to a Varian 290-LC pump injection module and a Varian 390-LC multidetector suite (refractive index detector) at a flow rate of 1.0 mL min−1. Near-monodisperse poly(methyl methacrylate) standards with Mp values ranging from 645 g mol−1 to 618![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 were used for calibration. Molecular weights distributions of methylated PMAA56-PMMAx diblock copolymers were assessed using THF eluent (containing 0.05% w/v butylhydroxytoluene and 2.0% v/v trimethylamine). Two Polymer Laboratories PL gel 5 μm Mixed-C columns were connected in series to a WellChrom K-2301 refractive index detector at a flow rate of 1.0 mL min−1. Near-monodisperse poly(methyl methacrylate) standards were used for calibration.
000 g mol−1 were used for calibration. Molecular weights distributions of methylated PMAA56-PMMAx diblock copolymers were assessed using THF eluent (containing 0.05% w/v butylhydroxytoluene and 2.0% v/v trimethylamine). Two Polymer Laboratories PL gel 5 μm Mixed-C columns were connected in series to a WellChrom K-2301 refractive index detector at a flow rate of 1.0 mL min−1. Near-monodisperse poly(methyl methacrylate) standards were used for calibration.
Transmission electron microscopy (TEM)
Copper/palladium TEM grids (Agar Scientific, UK) were coated with a thin film of amorphous carbon. Grids were treated with a plasma glow discharge for 30 s to produce a hydrophilic surface. A 10 μL droplet of a 0.10% w/w aqueous dispersion was placed on a grid and left for 1 min before blotting. The adsorbed nanoparticles were then stained using uranyl formate (9.0 μL of a 0.75% w/w aqueous solution) for 20 s followed by blotting to remove excess stain. Grids were carefully dried under vacuum and images were recorded at 100 kV using a Philips CM100 instrument equipped with a Gatan 1k CCD camera.Small-angle X-ray scattering (SAXS)
SAXS patterns were collected using a Xeuss 2.0 (Xenocs) SAXS instrument equipped with a Dectris Pilatus 1 M detector and an Excillum liquid gallium MetalJet X-ray source (λ = 1.34 Å). SAXS patterns were recorded for 1.0% w/w aqueous dispersions of PGMA50-PMMAx nanoparticles over a scattering vector q range of 0.04–0.4 Å−1 using 2.0 mm diameter glass capillary cells. The scattering of deionized water was used for absolute intensity calibration. Irena SAS macros for Igor Pro software were used to perform background subtraction, normalization and data analysis.47Results and discussion
RAFT aqueous emulsion polymerization of MMA
A PGMA precursor was prepared by RAFT solution polymerization of GMA in ethanol using a dithiobenzoate-based RAFT agent (CPDB) at 70 °C. End-group analysis by 1H NMR spectroscopy indicated had a mean DP of 50. This PGMA50 precursor was subsequently chain-extended via RAFT aqueous emulsion polymerization of MMA. A series of PGMA50-PMMAx nanoparticles were obtained by systematically varying the target DP of the PMMA core-forming block (Scheme 1). High MMA conversions were achieved within 3 h at 70 °C for all syntheses (Table 1). DMF GPC analysis indicated relatively high blocking efficiencies and unimodal molecular weight distributions in each case (Fig. 1). Increasing the target PMMA DP led to progressively higher Mn values, albeit with a gradual increase in dispersity (from Mw/Mn = 1.17 for PGMA50-PMMA20 up to Mw/Mn = 1.37 for PGMA50-PMMA130). Similar observations have been reported for various other PISA formulations in the literature.21,29,41 | ||
| Fig. 1 DMF GPC curves recorded for the PGMA50 precursor and a series of PGMA50-PMMAx nanoparticles for which the target PMMA DP (x) is 20, 40, 60, 80, 100 or 130 (see Table 1 for further details). | ||
| Target PMMA DP | Conversion (%) | DLS z-average diameter (nm) | DLS polydispersity | TEM morphology | SAXS core diameter (nm) |
|---|---|---|---|---|---|
| 20 | >99 | 18 | 0.04 | Spheres | 14 |
| 40 | >99 | 20 | 0.04 | Spheres | 15 |
| 60 | >99 | 29 | 0.08 | Spheres | 18 |
| 80 | >99 | 38 | 0.10 | Spheres | 21 |
| 100 | >99 | 66 | 0.12 | Spheres | 24 |
| 130 | 99 | 422 | 0.49 | Spheres | 30 |
TEM studies confirmed that kinetically-trapped spherical nanoparticles of increasing size were obtained when systematically varying the DP of the core-forming PMMA block (x) from 20 to 130 (Fig. 2). Furthermore, dynamic light scattering (DLS) studies revealed a systematic increase in the hydrodynamic z-average diameter while DLS polydispersities remained reasonably low up to x = 100, signifying relatively narrow particle size distributions (see Table 1 and Fig. 2). However, a substantial increase in both the apparent z-average diameter and DLS polydispersity was observed when targeting x = 130. Moreover, the TEM image recorded for this PISA synthesis indicates aggregates or clusters of nanoparticles on the grid, suggesting colloidal instability. Various PISA syntheses were conducted targeting x > 100 and similar results were invariably obtained (data not shown). This was an unexpected limitation, not least because we had previously reported that non-ionic dithiobenzoate-based PGMA precursors with similar (or lower) DPs were effective when performing RAFT aqueous emulsion polymerization syntheses with more hydrophobic methacrylic monomers. For example, Akpinar and co-workers33 were able to prepare PGMA28-PTFEMA500 and PGMA43-PTFEMA1000 spherical nanoparticles with no loss in colloidal stability. Similar results were also obtained by Jesson and co-workers when preparing PGMA39-PIPGMA1000 spheres34 and by Cunningham et al. when preparing PGMA51-PBzMA1000 spheres.32
 | ||
| Fig. 2 (a) Representative TEM images and (b) DLS particle size distributions obtained for a series of PGMA50-PMMAx nanoparticles prepared by RAFT aqueous emulsion polymerization of MMA where the target PMMA DP (x) is 20, 40, 60, 80, 100 or 130. DLS indicates a bimodal particle size distribution in the latter case owing to nanoparticle flocculation. | ||
Thus, at first sight, this unforeseen limitation appeared more likely to be associated with the choice of PMMA as the core-forming block, rather than the use of PGMA as a non-ionic stabilizer block. A few additional experiments were conducted using a longer PGMA101 block as a steric stabilizer. However, nanoparticle aggregation was still observed when targeting PMMA DPs above 100. For example, DLS studies indicated incipient flocculation for PGMA101-PMMA200 nanoparticles prepared at 70 °C when targeting 10% w/w solids using the CPDB RAFT agent (data not shown).
SAXS patterns were recorded for 1.0% w/w aqueous dispersions of the PGMA50-PMMA20–130 nanoparticles. Representative I(q) vs. q plots for six dispersions are shown in Fig. 3. The local minimum observed for each pattern is shifted to lower q as higher PMMA DPs are targeted. This indicates a systematic increase in the nanoparticle core diameter, d, according to the well-known relation q = 2π/d.48 These findings are consistent with the TEM and DLS data discussed above. Moreover, it is well-known that the low q gradient can be used to infer the predominant copolymer morphology.49 More specifically, a low q gradient of zero indicates a spherical morphology, which is indeed observed when targeting PMMA DPs of between 20 and 80. However, non-zero low q gradients are observed for PGMA50-PMMA100 and PGMA50-PMMA130, which suggests incipient nanoparticle aggregation and the formation of mass fractals for these two dispersions. Again, this is consistent with the corresponding TEM and DLS data shown in Fig. 2. Fitting the SAXS patterns using a well-established spherical micelle model50 and also a unified fit51–53 (to account for nanoparticle aggregation) enables the volume-average diameter of the PMMA cores to be determined in each case (Table 1).
 | ||
| Fig. 3 SAXS patterns recorded for a series of 1.0% w/w aqueous dispersions of PGMA50-PMMAx nanoparticles for which the target PMMA DP has been systematically varied from 20 to 130. | ||
Aqueous electrophoresis studies were performed on PGMA50-PMMA80 nanoparticles prepared using the dithiobenzoate-capped PGMA50 precursor (Fig. 4a). These nanoparticles exhibited zeta potentials close to zero (approximately −3 mV) across the whole pH range, which is consistent with the non-ionic nature of this steric stabilizer block. Given the unexpected colloidal stability problems associated with this PISA formulation, an alternative PGMA stabilizer bearing a carboxylic acid end-group54 and an anionic poly(methacrylic acid) stabilizer29 were also evaluated for the RAFT aqueous emulsion polymerization of MMA (see Scheme 2). A trithiocarbonate-based RAFT agent, PETTC, was used to prepare a carboxylic acid-capped PGMA54 precursor, which was then chain-extended via RAFT aqueous emulsion polymerization of MMA at pH 7 to produce either HOOC-PGMA54-PMMA80 or HOOC-PGMA54-PMMA150 spherical nanoparticles (Scheme 2a). The associated DLS, TEM and GPC data for these two dispersions are summarized in Fig. S1.†
 | ||
| Fig. 4 Zeta potential vs. pH curves obtained for dilute aqueous dispersions of: (a) PGMA50-PMMA80 spheres prepared using the non-ionic dithiobenzoate-based PGMA50 precursor shown in Scheme 1; (b) HOOC-PGMA54-PMMA80 spheres prepared using the carboxylic acid-functionalized trithiocarbonate-based PGMA54 precursor shown in Scheme 2a; (c) PMAA56-PMMA100 spheres prepared using the anionic PMAA precursor shown in Scheme 2b. | ||
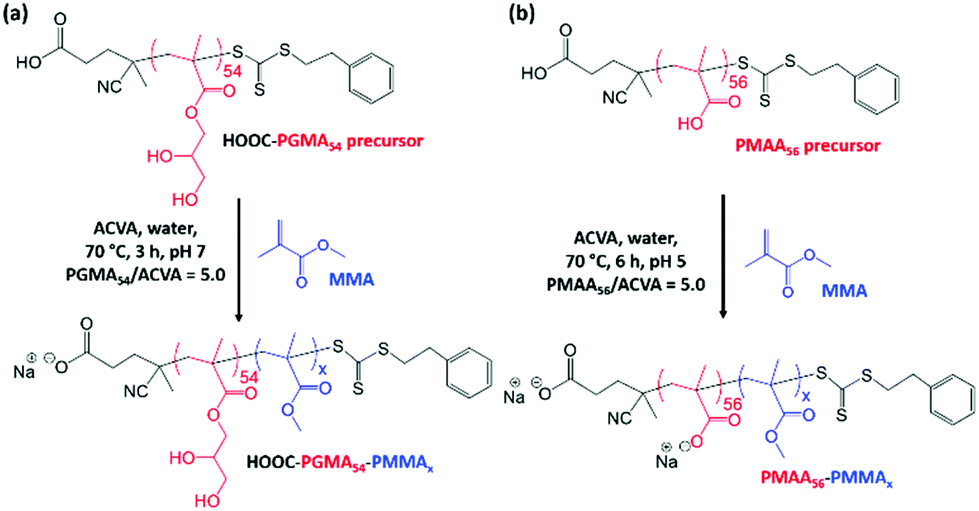 | ||
| Scheme 2 (a) Synthesis of HOOC-PGMA54-PMMAx diblock copolymer nanoparticles by RAFT aqueous emulsion polymerization of methyl methacrylate using a trithiocarbonate-based HOOC-PGMA54 precursor at 70 °C and targeting x = 80 or 150 at pH 7. (b) Synthesis of PMAA56-PMMAx diblock copolymer nanoparticles by RAFT aqueous emulsion polymerization of methyl methacrylate at 70 °C targeting x = 50 to 2000 at pH 5. | ||
The anionic charge conferred by the terminal ionized carboxylic group led to significant anionic character under the PISA synthesis conditions (pH 7) as judged by aqueous electrophoresis studies (see Fig. 4). More specifically, the nanoparticles become progressively more anionic between pH 2 and 6, before a maximum zeta potential of approximately −14 mV is observed at or above pH 6. DLS studies confirmed the formation of relatively small nanoparticles with a z-average diameter of 26 nm when targeting a PMMA DP of 80, which indicated good colloidal stability in this case (Fig. S1†). However, such anionic character was not sufficient to allow the PISA synthesis of colloidally stable spherical nanoparticles when targeting a PMMA DP of 150. In this case, DLS and TEM studies indicated extensive nanoparticle aggregation similar to that observed for the non-ionic PGMA50-PMMA130 nanoparticles.
Accordingly, a poly(methacrylic acid) (PMAA) precursor was evaluated for the RAFT aqueous emulsion polymerization of MMA.29 A series of PMAA56-PMMAx nanoparticles was prepared using this polyelectrolytic steric stabilizer by systematic variation of the target PMMA DP from 50 to 2000 (see Scheme 2b). All syntheses were performed at pH 5 targeting 20% w/w solids, as reported by Cockram and co-workers for aqueous PISA syntheses using a PMAA steric stabilizer.401H NMR spectroscopy studies confirmed that relatively high conversions (94–99%) were obtained in each case, with a modest reduction in the final conversion being observed when targeting DPs above 500 (Table 2). Mean hydrodynamic diameters determined by DLS are summarized in Table 2 and the relationship between DLS diameter and the PMMA DP is shown in Fig. 5. As expected, the particle size increases monotonically as the target DP is increased from 50 to 2000. Indeed, an approximate linear relationship is observed between DP 50 (29 nm) and DP 500 (73 nm). Above DP 500, the particle size continues to increase up to DP 2000, albeit more slowly. TEM images for selected PMAA56-PMMAx nanoparticles are shown in Fig. 6 when targeting x = 50 to 2000. As expected, a kinetically-trapped spherical morphology was obtained in each case.
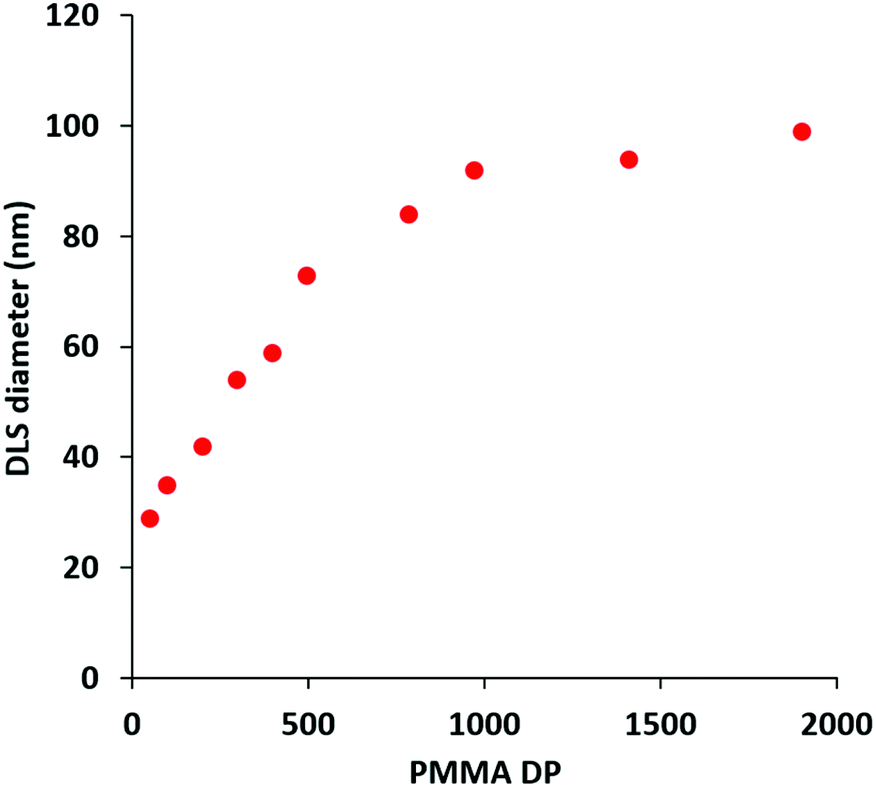 | ||
| Fig. 5 Variation in z-average diameter with PMMA DP (x; corrected for the final monomer conversion) for a series of PMAA56-PMMAx diblock copolymer nanoparticles prepared via RAFT aqueous emulsion polymerization of MMA at 70 °C targeting 20% w/w solids at pH 5. | ||
 | ||
| Fig. 6 TEM images recorded for a series of PMAA56-PMMAy diblock copolymer nanoparticles prepared by RAFT aqueous emulsion polymerization of MMA when targeting a PMMA DP (x) of 50, 100, 200, 500, 1000 or 2000 (see Table 2 for further details). | ||
| Target PMMA DP | Conversion (%) | Actual PMMA DP | DLS Diameter (nm) | DLS PDI | TEM morphology |
|---|---|---|---|---|---|
| 50 | 99 | 50 | 29 | 0.14 | Spheres |
| 100 | 99 | 99 | 35 | 0.13 | Spheres |
| 200 | 99 | 198 | 42 | 0.16 | Spheres |
| 300 | 99 | 297 | 54 | 0.14 | Spheres |
| 400 | 99 | 396 | 59 | 0.10 | Spheres |
| 500 | 99 | 495 | 73 | 0.11 | Spheres |
| 800 | 98 | 784 | 84 | 0.19 | Spheres |
| 1000 | 97 | 970 | 92 | 0.17 | Spheres |
| 1500 | 94 | 1410 | 94 | 0.20 | Spheres |
| 2000 | 95 | 1900 | 99 | 0.24 | Spheres |
Selected PMAA56-PMMAx diblock copolymers were methylated to convert such copolymers into the corresponding PMMA56+x homopolymers for THF GPC analysis using PMMA calibration standards (see Fig. 7). In each case, unimodal MWDs and high blocking efficiencies were observed and targeting higher PMMA DPs led to the expected monotonic increase in the GPC Mn. Notably, no systematic GPC error was incurred for this particular data set and the Mn values were reasonably close to the expected theoretical values. However, dispersities were relatively high (Mw/Mn = 1.51–1.76). Similar GPC data were reported by Chaduc and co-workers for the RAFT aqueous emulsion polymerization of MMA using a PMAA precursor at pH 3.5.29 Clearly, this aqueous PISA formulation does not suffer from colloidal instability problems when targeting relatively high PMMA DPs. This is perhaps not surprising, because aqueous electrophoresis studies of PMAA56-PMMA100 nanoparticles (Fig. 4c) indicated highly negative zeta potentials (approximately −40 mV at or above pH 5) owing to the strongly anionic nature of the ionized PMAA56 chains. Similar results were obtained when employing a cationic steric stabilizer comprising poly(2-(methacryloyloxy)ethyl trimethylammonium chloride) [PMETAC], see the ESI† for full synthesis details and also Fig. S2.† Thus, if relatively large PMMA-core nanoparticles are required for a given application, it is clear that polyelectrolytic stabilizers offer a decisive advantage over non-ionic stabilizers such as PGMA.
 | ||
| Fig. 7 THF GPC curves recorded for the methylated PMAA56 precursor and a series of methylated PMAA56-PMMAx diblock copolymer nanoparticles for which the target PMMA DP (x) was 100, 200, 500 or 1000. | ||
End-group removal from PGMA-PMMA nanoparticles using visible light irradiation
For many potential applications, it is desirable to remove the RAFT end-groups after the polymerization because these organosulfur groups confer color and malodour.5 For soluble copolymers, this is readily achieved using various chemistries.55–57 However, there are rather few studies of the removal of RAFT end-groups from diblock copolymer nanoparticles. In 2015, Destarac and co-workers demonstrated that xanthate groups could be efficiently cleaved from aqueous poly(n-butyl acrylate) latexes using ozone at ambient temperature.58 Subsequently, Jesson et al.46 reported that dithiobenzoate end-groups can be efficiently removed from PGMA52-PHPMA135 (where PHPMA denotes poly(2-hydroxypropyl methacrylate) diblock copolymer worms after treatment with H2O2 at 70 °C for 3 h using a H2O2/dithiobenzoate molar ratio of 5.0. However, removal of a trithiocarbonate end-group from essentially the same diblock copolymer proved to be much slower, with around 24% organosulfur groups remaining after 8 h under the same reaction conditions. Furthermore, the removal of dithiobenzoate end-groups from PGMA61-PBzMA100 (where PBzMA denotes poly(benzyl methacrylate) spheres using this H2O2 protocol was relatively ineffective, with UV GPC analysis indicating that more than 60% of the original end-groups remained intact after 8 h. This was attributed to the relatively hydrophobic nature of the core-forming PBzMA block, which is likely to impede ingress of the H2O2 into the nanoparticle cores. More recently, Gibson and co-workers59 reported that visible light irradiation (blue LED; λ = 405 nm) at 50 °C removed dithiobenzoate end-groups from aqueous dispersions of PNMEP28-PLMA87 [where PNMEP denotes poly(N-(2-methacryloyloxy)ethyl pyrrolidone) and PLMA denotes poly(lauryl methacrylate)] diblock copolymer vesicles. In principle, this approach is attractive because it should not suffer from the retarded diffusion of reagents observed by Jesson and co-workers.46 Thus, we decided to study the removal of dithiobenzoate end-groups from 10% w/w aqueous dispersions of PGMA50-PMMA80 nanoparticles using similar visible light irradiation conditions as those reported by Gibson and co-workers.59 Bearing in mind the aqueous solubilities of the respective monomers at room temperature, the hydrophobic character of the core-forming PMMA block is significantly greater than that of PHPMA but rather less than that of either PBzMA or PLMA. Moreover, it is perhaps worth emphasizing that PMMA has a significantly higher glass transition temperature (Tg) than these three alternative core-forming blocks and it is not obvious whether the glassy nature of the PMMA nanoparticle cores might impede removal of the RAFT end-groups.In our initial RAFT end-group removal studies, visible light irradiation experiments were conducted at 70 °C. However, only rather slow and incomplete end-group cleavage (86% within 24 h; data not shown) was achieved under such conditions for PGMA50-PMMA80 nanoparticles prepared using CPDB. Fortunately, we found that significantly higher rates of end-group cleavage could be achieved at 80 °C. Accordingly, the kinetics of dithiobenzoate end-group removal by visible light irradiation (blue LED source; λ = 405 nm) of a 10% w/w aqueous dispersion of PGMA50-PMMA80 nanoparticles at 80 °C was monitored by periodic sampling of the reaction mixture followed by DMF GPC analysis using a UV detector set at 309 nm. Unlike visible absorption spectrophotometry, this technique enables the RAFT end-groups that remain attached to the copolymer chains to be distinguished from those that have been cleaved to produce small molecule by-products.46 Representative UV GPC curves are shown in Fig. 8a and the fraction of remaining dithiobenzoate end-groups is plotted against time in Fig. 8b. Initially, relatively rapid cleavage occurs, with 87% of the original end-groups being removed within 12 h at 80 °C. After continuous irradiation for 24 h, 94% end-group removal can be achieved and the initial pink copolymer dispersion is converted into a colorless dispersion (see inset digital photographs). As a comparison, H2O2 was employed for oxidative end-group removal under the same conditions, as previously reported.46 However, this only led to 24% end-group removal within 12 h and 58% after 24 h (Fig. 8b and S3†). This is presumably because this water-soluble reagent cannot readily diffuse into the glassy hydrophobic PMMA cores. The extent of end-group removal was also monitored for the PGMA50-PMMA80 nanoparticles at 80 °C in the absence of either visible light irradiation or H2O2. For this control experiment, UV GPC studies (Fig. S3†) indicated that 92% end-groups remained intact after 24 h, suggesting minimal thermally-induced hydrolysis.
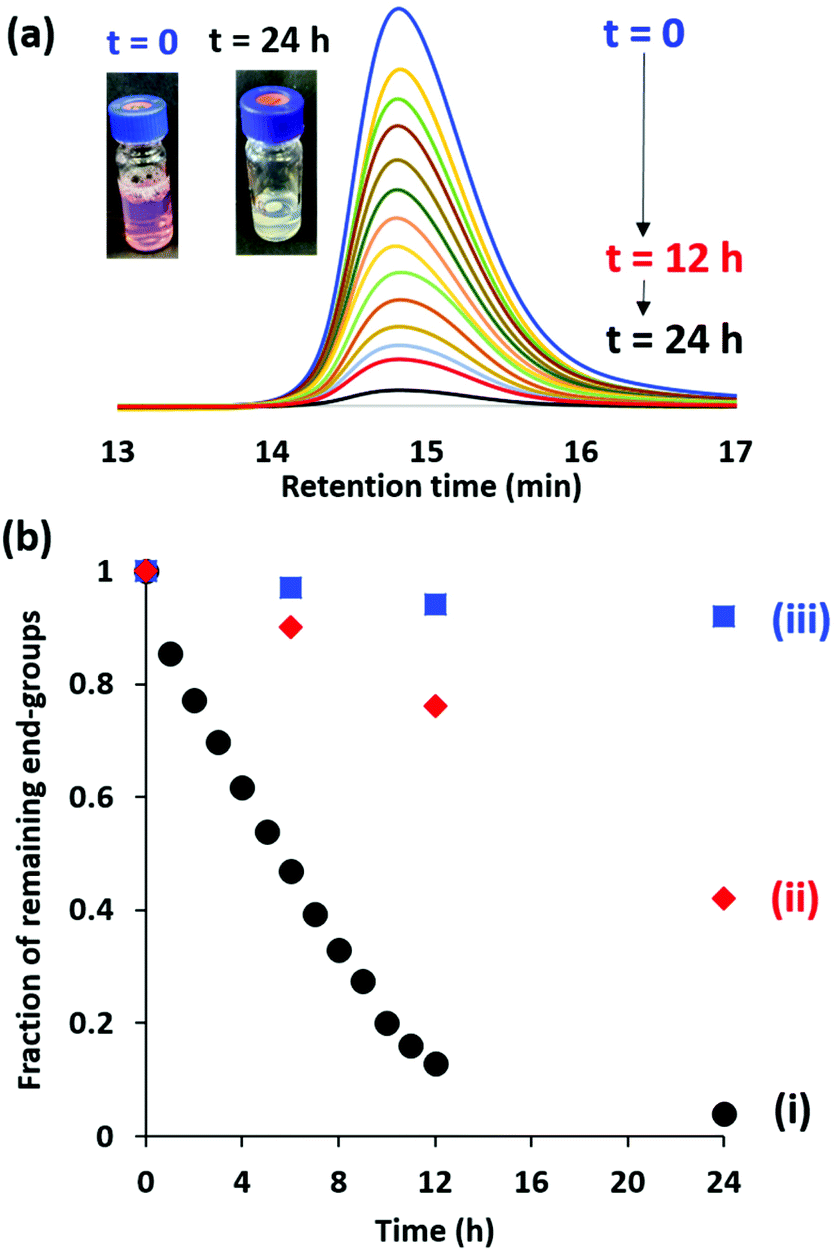 | ||
| Fig. 8 (a) Representative UV GPC curves (λ = 309 nm) recorded during kinetic studies of the cleavage of dithiobenzoate end-groups for a 10% w/w aqueous dispersion of PGMA50-PMMA80 spherical nanoparticles at 80 °C. (b) Fraction of remaining dithiobenzoate RAFT end-groups over time determined by UV GPC analysis when a 10% w/w aqueous dispersion of PGMA50-PMMA80 spherical nanoparticles at 80 °C is exposed to: (i) continuous visible light irradiation (λ = 405 nm), (ii) H2O2 (using a H2O2/dithiobenzoate molar ratio of 5.0) and (iii) neither visible light nor H2O2 (control). | ||
Precisely the same protocol was adopted when studying trithiocarbonate end-group removal from a 10% w/w (aqueous dispersion of HOOC-PGMA54-PMMA80 nanoparticles. The GPC curves and kinetics are summarized in Fig. S4.† Cleavage of trithiocarbonate end-groups was achieved, albeit at a somewhat slower rate than that found for the dithiobenzoate end-groups. More specifically, 72% of the original trithiocarbonate groups were cleaved within 12 h at 80 °C with 87% being removed after 24 h. As a control experiment, the trithiocarbonate-capped HOOC-PGMA54-PMMA80 nanoparticles were exposed to H2O2 at 80 °C using a H2O2/trithiocarbonate molar ratio of 5.0 (see Fig. S4 and S5†). However, UV GPC studies indicated that more than 80% of the original RAFT end-groups remained intact within 24 h. Similarly, a control experiment conducted in the absence of either H2O2 or visible light irradiation indicated that 96% trithiocarbonate end-groups survived intact after 24 h at 80 °C.
Clearly, visible light irradiation can be a highly effective means of removing trithiocarbonate end-groups as well as dithiobenzoate end-groups. This approach works well for both high Tg core-forming blocks such as PMMA (as demonstrated herein) and low Tg core-forming blocks such as PLMA.59 The only disadvantage appears to be the relatively long reaction time required at 80 °C but presumably this could be reduced by increasing the intensity of the visible light source.
Conclusions
Sterically-stabilized diblock copolymer nanoparticles are prepared by RAFT aqueous emulsion polymerization of MMA using PGMA as a non-ionic steric stabilizer block. For a target PMMA DP of 20 to 100, kinetically-trapped spherical nanoparticles ranging in size from 17 nm to 31 nm are obtained. However, highly flocculated spherical nanoparticles are produced when targeting DPs above 100. Similar flocculation problems are encountered when employing a PGMA stabilizer block that possesses a terminal anionic carboxylate group. Moreover, this unexpected limitation does not appear to apply to various other RAFT aqueous emulsion polymerization formulations: core-forming block DPs of up to 1000 can be targeted without any loss of colloidal stability when employing alternative (and more hydrophobic) water-immiscible monomers such as benzyl methacrylate, 2,2,2-trifluoroethyl methacrylate or isopropylideneglycerol monomethacrylate when using a non-ionic PGMA stabilizer block. This perplexing constraint appears to be related to the high Tg of the PMMA block, which exceeds the reaction temperature of 70 °C used for such PISA syntheses. However, we demonstrate that colloidally stable dispersions can be obtained when targeting PMMA DPs of up to 2000 using a highly anionic PMAA stabilizer block. Finally, visible light irradiation is used to cleave dithiobenzoate end-groups from a 10% w/w aqueous dispersion of PGMA50-PMMA80 nanoparticles. UV GPC studies indicated 87% end-group removal from such nanoparticles within 12 h at 80 °C. In contrast, using excess H2O2 only led to 24% end-group removal under the same conditions. This striking difference is attributed to the water-soluble reagent having restricted access to the hydrophobic PMMA nanoparticle cores. Furthermore, the same visible light irradiation protocol can be used to remove trithiocarbonate end-groups from HOOC-PGMA50-PMMA80 nanoparticles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. P. A. acknowledges a four-year EPSRC Established Career Particle Technology Fellowship (EP/R003009). Syngenta is thanked for an EPSRC Industrial CASE studentship for D. H. H. C. and also for permission to publish this work.References
- J. Chiefari, Y. K. B. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS.
- J. D. Biasutti, T. P. Davis, F. P. Lucien and J. P. A. Heuts, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2001–2012 CrossRef CAS.
- W. Wan and C. Pan, Polym. Chem., 2010, 1, 1475–1484 RSC.
- Y. Li and S. P. Armes, Angew. Chem., Int. Ed., 2010, 49, 4042–4046 CrossRef CAS.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- X. Zhang, J. Rieger and B. Charleux, Polym. Chem., 2012, 3, 1502–1509 RSC.
- O. J. Deane, O. M. Musa, A. Fernyhough and S. P. Armes, Macromolecules, 2020, 53, 1422–1434 CrossRef CAS.
- C. J. Ferguson, R. J. Hughes, B. T. T. Pham, B. S. Hawkett, R. G. Gilbert, A. K. Serelis and C. H. Such, Macromolecules, 2002, 35, 9243–9245 CrossRef CAS.
- C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such and B. S. Hawkett, Macromolecules, 2005, 38, 2191–2204 CrossRef CAS.
- J. Rieger, F. Stoffelbach, C. Bui, D. Alaimo, C. Jérôme and B. Charleux, Macromolecules, 2008, 41, 4065–4068 CrossRef CAS.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- X. Zhang, S. Boissé, W. Zhang, P. Beaunier, F. D'Agosto, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 4149–4158 CrossRef CAS.
- S. Boissé, J. Rieger, G. Pembouong, P. Beaunier and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3346–3354 CrossRef.
- W. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2012, 45, 4075–4084 CrossRef CAS.
- L. Carlsson, A. Fall, I. Chaduc, L. Wågberg, B. Charleux, E. Malmström, F. D'Agosto, M. Lansalot and A. Carlmark, Polym. Chem., 2014, 5, 6076–6086 RSC.
- W. Zhang, B. Charleux and P. Cassagnau, Macromolecules, 2012, 45, 5273–5280 CrossRef CAS.
- B. T. T. Pham, D. Nguyen, V. T. Huynh, E. H. Pan, B. Shirodkar-Robinson, M. Carey, A. K. Serelis, G. G. Warr, T. Davey, C. H. Such and B. S. Hawkett, Langmuir, 2018, 34, 4255–4263 CrossRef CAS PubMed.
- R. Albigès, P. Klein, S. Roi, F. Stoffelbach, C. Creton, L. Bouteiller and J. Rieger, Polym. Chem., 2017, 8, 4992–4995 RSC.
- N. P. Truong, M. V. Dussert, M. R. Whittaker, J. F. Quinn and T. P. Davis, Polym. Chem., 2015, 6, 3865–3874 RSC.
- W. Zhao, G. Gody, S. Dong, P. B. Zetterlund and S. Perrier, Polym. Chem., 2014, 5, 6990–7003 RSC.
- I. Chaduc, O. Boyron, B. Charleux, F. D. Agosto and M. Lansalot, Macromolecules, 2013, 46, 6013–6023 CrossRef CAS.
- J. Rieger, G. Osterwinter, C. Bui and B. Charleux, Macromolecules, 2009, 42, 5518–5525 CrossRef CAS.
- I. Chaduc, M. Girod, R. Antoine, B. Charleux, F. D'Agosto and M. Lansalot, Macromolecules, 2012, 45, 5881–5893 CrossRef CAS.
- M. Chenal, C. Véchambre, J. M. Chenal, L. Chazeau, V. Humblot, L. Bouteiller, C. Creton and J. Rieger, Polymer, 2017, 109, 187–196 CrossRef CAS.
- S. Binauld, L. Delafresnaye, B. Charleux, F. Dagosto and M. Lansalot, Macromolecules, 2014, 47, 3461–3472 CrossRef CAS.
- V. J. Cunningham, A. M. Alswieleh, K. L. Thompson, M. Williams, G. J. Leggett, S. P. Armes and O. M. Musa, Macromolecules, 2014, 47, 5613–5623 CrossRef CAS.
- B. Akpinar, L. A. Fielding, V. J. Cunningham, Y. Ning, O. O. Mykhaylyk, P. W. Fowler and S. P. Armes, Macromolecules, 2016, 49, 5160–5171 CrossRef CAS PubMed.
- C. P. Jesson, V. J. Cunningham, M. J. Smallridge and S. P. Armes, Macromolecules, 2018, 51, 3221–3232 CrossRef CAS PubMed.
- E. E. Brotherton, F. L. Hatton, A. A. Cockram, M. J. Derry, A. Czajka, E. J. Cornel, P. D. Topham, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2019, 141, 13664–13675 CrossRef CAS PubMed.
- F. L. Hatton, J. R. Lovett and S. P. Armes, Polym. Chem., 2017, 8, 4856–4868 RSC.
- J. Tan, D. Liu, C. Huang, X. Li, J. He, Q. Xu and L. Zhang, Macromol. Rapid Commun., 2017, 38, 1–7 Search PubMed.
- F. L. Hatton, A. M. Park, Y. Zhang, G. D. Fuchs, C. K. Ober and S. P. Armes, Polym. Chem., 2019, 10, 194–200 RSC.
- F. L. Hatton, M. J. Derry and S. P. Armes, Polym. Chem., 2020, 11, 6343–6355 RSC.
- A. A. Cockram, T. J. Neal, M. J. Derry, O. O. Mykhaylyk, N. S. J. Williams, M. W. Murray, S. N. Emmett and S. P. Armes, Macromolecules, 2017, 50, 796–802 CrossRef CAS PubMed.
- W. Zhang, F. D'Agosto, P. Y. Dugas, J. Rieger and B. Charleux, Polymer, 2013, 54, 2011–2019 CrossRef CAS.
- F. L. Hatton, M. Ruda, M. Lansalot, F. D. Agosto, E. Malmstro and A. Carlmark, Biomacromolecules, 2016, 17, 1414–1424 CrossRef CAS PubMed.
- J. Engström, H. Asem, H. Brismar, Y. Zhang, M. Malkoch and E. Malmström, Macromol. Chem. Phys., 2020, 221, 1900443 CrossRef.
- S. J. Hunter, J. R. Lovett, O. O. Mykhaylyk, E. R. Jones and S. P. Armes, Polym. Chem., 2021, 12, 3629–3639 RSC.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2013, 29, 7416–7424 CrossRef CAS PubMed.
- C. P. Jesson, C. M. Pearce, H. Simon, A. Werner, V. J. Cunningham, J. R. Lovett, M. J. Smallridge, N. J. Warren and S. P. Armes, Macromolecules, 2017, 50, 182–191 CrossRef CAS PubMed.
- J. Ilavsky and P. R. Jemian, J. Appl. Crystallogr., 2009, 42, 347–353 CrossRef CAS.
- A. Guinier and G. Fournet, Small-Angle Scattering of X-rays, John Wiley & Sons, New York, 1955 Search PubMed.
- O. Glatter and O. Kratky, Small-angle X-ray Scattering, Academic Press, London, 1982 Search PubMed.
- J. S. Pedersen, J. Appl. Crystallogr., 2000, 33, 637–640 CrossRef CAS.
- G. Beaucage and D. W. Schaefer, J. Non.-Cryst. Solids, 1994, 172–174, 797–805 CrossRef CAS.
- G. Beaucage, J. Appl. Crystallogr., 1995, 28, 717–728 CrossRef CAS.
- G. Beaucage, J. Appl. Crystallogr., 1996, 29, 134–146 CrossRef CAS.
- J. R. Lovett, N. J. Warren, L. P. D. Ratcliffe, M. K. Kocik and S. P. Armes, Angew. Chem., Int. Ed., 2015, 54, 1279–1283 CrossRef CAS PubMed.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- B. Quiclet-Sire and S. Z. Zard, Bull. Korean Chem. Soc., 2010, 31, 543–544 CrossRef.
- D. Matioszek, P. E. Dufils, J. Vinas and M. Destarac, Macromol. Rapid Commun., 2015, 36, 1354–1361 CrossRef CAS PubMed.
- R. R. Gibson, E. J. Cornel, O. M. Musa, A. Fernyhough and S. P. Armes, Polym. Chem., 2020, 11, 1785–1796 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Summary of GPC, DLS and TEM data obtained for HOOC-PGMA54-PMMA80 nanoparticles and HOOC-PGMA54-PMMA150 nanoparticles synthesised with PETTC RAFT agent; synthesis of the PMETAC46 precursor and the corresponding PMETAC46-PMMAx nanoparticles; DLS and TEM data obtained for PMETAC46-PMMAx nanoparticles; UV GPC curves (λ = 309 nm) recorded during kinetic studies of the removal of dithiobenzoate end-groups from an aqueous dispersion of PGMA50-PMMA80 nanoparticles using either excess H2O2 or no light irradiation at 80 °C. UV GPC curves (λ = 309 nm) recorded during the removal of trithiocarbonate end-groups from an aqueous dispersion of HOOC-PGMA54-PMMA80 nanoparticles using visible light irradiation, excess H2O2 or neither condition at 80 °C. See DOI: 10.1039/d1py01008e |
| This journal is © The Royal Society of Chemistry 2021 |