
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crosslinked porous polyimides: structure, properties and applications
Basiram Brahma
Narzary
 a,
Benjamin C.
Baker
a,
Neha
Yadav
b,
Valerio
D'Elia
b and
Charl F. J.
Faul
*a
a,
Benjamin C.
Baker
a,
Neha
Yadav
b,
Valerio
D'Elia
b and
Charl F. J.
Faul
*a
aSchool of Chemistry, University of Bristol, Bristol, UK. E-mail: charl.faul@bristol.ac.uk
bSchool of Molecular Science and Engineering, VISTEC, Thailand
First published on 21st September 2021
Abstract
Porous polyimides (pPIs) represent a fascinating class of porous organic polymers (POPs). Not only do they exhibit high thermal and chemical stabilities, high surface areas, and energy storage capabilities, but their formation relies upon simple polycondensation reactions. A wide library of linker (dianhydrides) and core (amines) starting materials offers a vast range of crosslinked pPIs. This review details and carefully compares the unique properties and functions of both amorphous and crystalline pPIs. Furthermore, their applications in current global challenges in the fields of gas storage and separation, electrical energy storage, catalysis, drug delivery and sensors are reported. Finally, the review highlights the progress of pPIs since 2010 and offers an outlook and suggestions for future areas for exploration and potential applications within the field.
1. Introduction
Polyimide (PI) polymers were first synthesised and reported by Borget et al. in 1908.1 PIs can be divided into two classes, aromatic and aliphatic. The first aromatic PI was developed and commercialised by DuPont™ in 1960 and an aliphatic first reported in 1971 by Hirsch et al.2–4 Initially, aromatic PIs were found to exhibit higher thermal, chemical and mechanical stabilities than aliphatic PIs, whereas, aliphatic PIs possess good solubility, low dielectric constant and high optical transparency owing to their molecular packing and polarisability.5 PIs have found general use and application in the fields of aviation, aerospace, micro-electronics, gas separation, membranes, fuel cells, batteries, electronic memory devices, shape memory devices, optical devices, biomedical applications, sensors, aerogels and polymer matrices in composites/hybrid materials (Fig. 1).6–12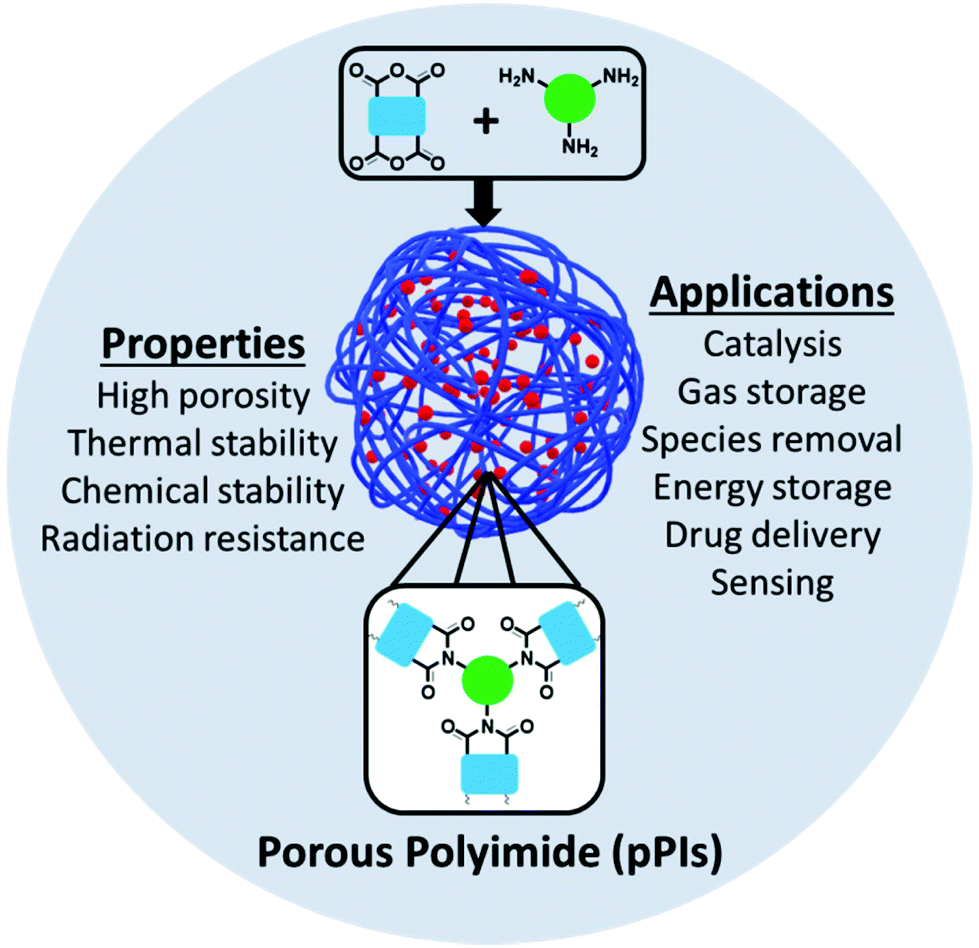 | ||
| Fig. 1 Porous polyimide properties and applications. | ||
Generally speaking, porous PIs (pPIs) are a class of porous organic polymers (POPs) synthesised by polycondensation reaction between amines and anhydrides (at high temperatures, 180–200 °C). Crosslinking is achieved either by multiple (>2) reactive sites or cross-linkable groups (e.g. alkylene) present in the starting materials. pPIs offer high flexibility of their molecular design by variation of the monomer units to yield 3D-crosslinked networks. Moreover, pPIs can also be classified according to their initial building blocks: conjugated/aromatic and non-conjugated/aliphatic. Highly crosslinked pPI networks are advantageous over other porous materials owing to the excellent physical and chemical properties such as high mechanical, chemical and thermal stabilities, radiation resistance and high surface areas. Owing to their unique properties, PIs have shown great potential applications for addressing current energy and environmental global challenges. Specifically, pPIs can be utilised in gas adsorption and separation, electrical energy storage, heterogeneous catalysis, drug delivery, sensor, and species removal from aqueous environment.
This review covers the broader field of crosslinked and porous PIs, i.e., pPIs, with a specific focus on the monomer design, synthetic advances and the exploration of function and potential applications. The content covers materials published since 2010, and considers amorphous, crystalline and gelatinous crosslinked pPIs, thus ensuring a range of applications and properties demonstrated in each are discussed. Non-crosslinked PIs are not discussed in this review, and the authors refer readers to other reviews and studies that address this topic in detail.5,6,8,13,14
2. Monomer structure
The properties of polymers are inextricably related to the choice of monomers used for their synthesis. In the case of polyimides, polytopic amines and anhydrides are reacted exploiting several strategies to produce the desired crosslinked and porous pPIs. The structures of amine monomers are shown in Fig. 2, organised according to the number of amino moieties in the monomers (and hence the overall geometry of the formed pPIs, see section 3.2.1), grouped into mono (D1), di- (A1–16), tri- (B1–14) and tetra- (C1–8) amine units; the anhydrides are provided in Fig. 3 (L1–26). To react this wide range of monomers several synthetic strategies have been applied, including solvothermal, ionothermal, interfacial synthetic approaches (exhaustively described in a recent review by Zhang et al.15 and, therefore, not discussed in detail here). The authors of the latter work described also how the choice of monomers affected the geometry of the polymeric skeleton, however they only focused on pPIs in covalent organic framework (COFs) states. In this section we will focus on those cases where the rational design and multi-step synthesis of functional monomers allowed control over the properties of the final polymers in crystalline, amorphous and gelatinous pPIs.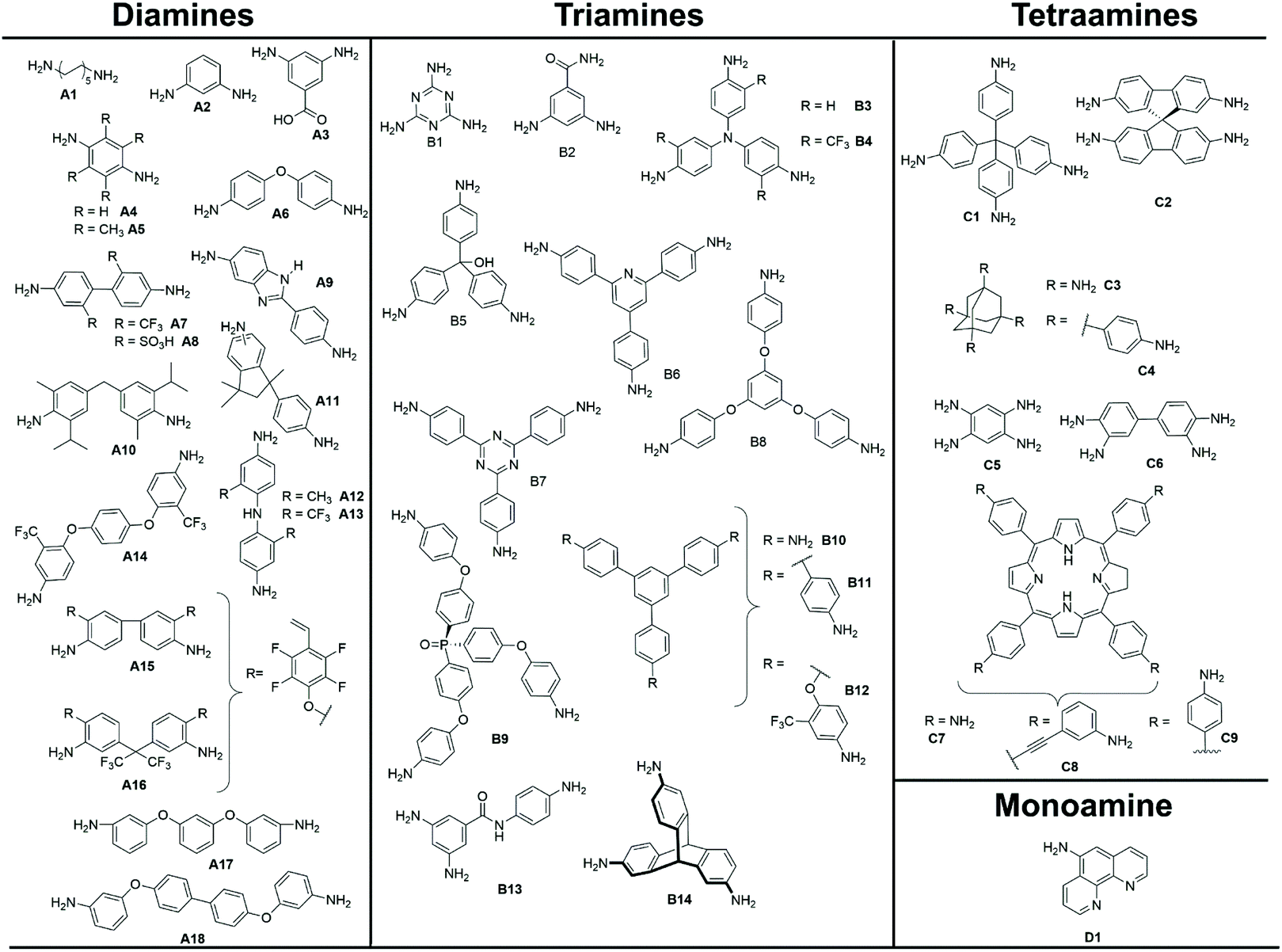 | ||
| Fig. 2 Amine monomers, grouped into different reactive sites/geometries (di-,tri- and tetra-amines), utilised in the formation of pPI networks covered within this review. | ||
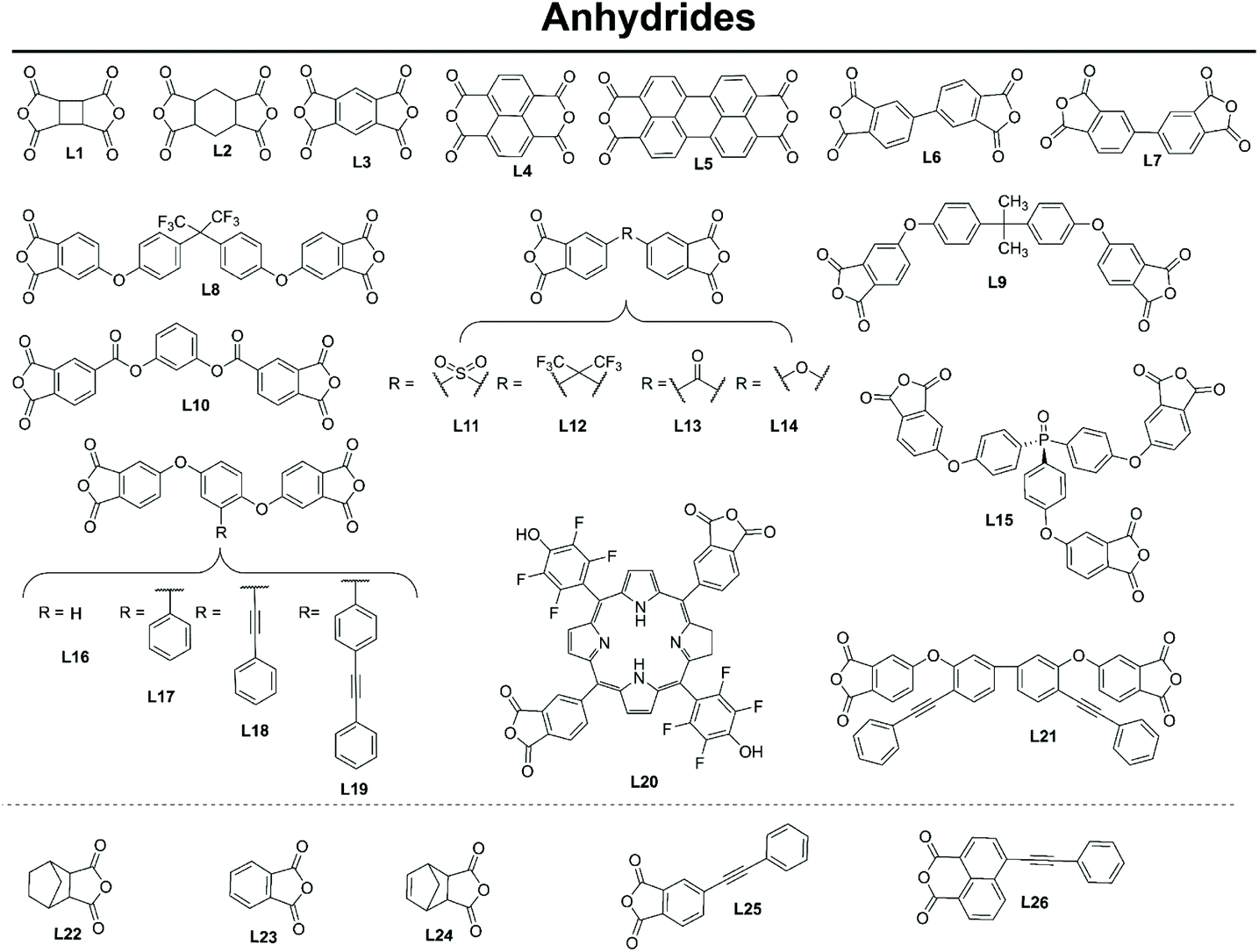 | ||
| Fig. 3 Anhydride monomers utilised in formation of pPI networks covered within this review. | ||
Most amine monomers reported in the literature for pPI syntheses are commercially available and require little to no synthetic modification. Here we focus on those monomers with rational geometric or functional design targeting specific properties (see section 4 and 5). Within the amine monomers reported there are 3 major aspects that are noteworthy:
(1) Amount of reactive amine sites. We have found that a variety of amine monomers with 2, 3 or 4 reactive amine sites are available, both commercially and synthetically, and are shown in Fig. 2. The number of reactive sites is closely related to the geometry of the amine, e.g., 2 reactive sites = linear (A1–18), 3 = trigonal pyramidal or planar (B1–14), which has the additional effect of crosslinking (with distinct effects on properties such as surface area and applications such as gas absorption, see section 4.1). For the creation of 4 reactive amine sites, many papers cite the use of tetrahedral starting monomers C1–6, or the square planar porphyrin ring (C7–8, both synthetically modified specifically for the purpose of creating pPIs).
(2) Introduction of heteroatoms and functional groups. The introduction of heteroatoms and functional groups into the imide backbone of the polymers have a variety of impacts on the properties and applications (discussed later in sections 3 and 4). In the case of such functionalized amine monomers several are worthy of discussion. With respect to di-reactive amine monomers, the use of commercially available fluorinated aromatic A7 and the synthetically modified A13 and A14 (achieved via a dehydration reaction from the fluorinated ethylene benzene and phenol diamine) are used to include novel functionality. For tri-reactive amine monomers such as B4, B12, and B13, heteroatom-containing functionalities are introduced in the form of CF3 moieties or oxygen ether/ketone linkages. For the monomer B4 multistep synthesis is required; firstly, 4-nitro-3-(trifluoromethyl)aniline and 4-chloro-1-nitro-2-(trifluoromethyl)benzene were coupled and the intermediate reduced in the presence of palladium and hydrazine hydrate by Song et al.16 In the case of monomers with additional nitrogen heteroatoms (with respect to those involved in the imide linkage), B7 was synthesised by Liebl et al.17 from a two-step synthetic procedure involving the trimerisation of 1-bromo-4-cyanobenzene in the presence of CF3SO3H, followed by a nucleophilic substitution reaction in the presence of Pd(dba)2.
(3) Introduction of cross-linkable structures or moieties in the monomers. Modifications of the monomers, to bear cross-linkable groups, lead to the formation of networks with improved control over their microporous structures (which has a direct effect on sorption and separation applications as discussed in section 4.1). Perhaps the best examples of these are found in the porphyrin-based tetraamine monomer C8, synthesised by Shi et al.18 Here C8 was functionalised with a cross-linkable ethynyl functionality in a two-step synthetic procedure (condensation and oxidation of 4-bromobenzaldehyde and pyrrole in the presence of acetic anhydride and propionic acid). Further examples can be found in the linear fluorinated diamines A15 and A16, modified with ethylene functionalities for post-polymerisation crosslinking. In the cases of non-alkene or alkyne cross-linkable moieties the triamines B2 (synthesised from an addition–elimination reaction between 3,5-dinitrobenzoylchloride followed by –NO2 reduction in the presence of Pd/C) and B13 (synthesised from an acylation reaction between 3,5-dinitro benzoylchloride and 4-nitroaniline, followed by reduction with hydrazine hydride in the presence of Pd/C by Rangel Rangel et al.19) have also been employed.
Anhydride monomers typically possess two reactive sites (especially with benzene L3, naphthalene L4 and perylene L5 linkers). A few exceptions are the use of the mono reactive anhydrides such as L22, used as endcaps by Li et al.20 to control polymerisation.
The majority of variations for this monomer are seen in the introduction of heteroatom and functional groups and cross-linkable moieties;
(1) Introduction of heteroatoms and functional groups. The commercially available di-linkers L8 and L12 have been used to introduce fluorine (useful for increasing hydrophobicity, see section 4 and 5), whereas the porphyrin-based L20 was synthesised in a multistep procedure by Shultz et al.21 (involving the formation of dimethyl 4-carboxaldehydephthalate and cyclisation with 5-pentafluorophenyl dipyrromethane in the presence of BF3·Et2O and DDQ) to introduce fluorine, porphyrin and phenol functionalities.21 Anhydrides L9, L10, L13 and L14 are commercially available and used to introduce oxygen into the polyimide backbone, an important addition to influence gas absorption (see section 4.1).
(2) Introduction of cross-linkable structures and moieties in the monomers. Shi et al.18,22 have synthesised a range of anhydride monomers containing two cross linkable pendant alkynyl functionalities, L18, L19 and L21, and crosslinked them with amine monomers bearing the alkynyl functionalities (A15 and A16).
Several preformed diimide-containing monomers are documented in the literature and shown in Fig. 4. Roy et al.23 synthesised a range of aromatic-based diimide monomers M1–3, from dianhydrides L2–4, respectively, via condensation of 5-aminoisophthalic acid. The formed diimide, functionalised with dicarboxylic acids, was then condensed with the tetra-amine monomers C5 and C6 to produce benzimidazole linked pPIs. Lu et al.24 and Zhu et al.25 condensed maleic anhydride with the appropriate diamine linker to yield the preformed monomer diimides M4–6. Lu et al.24 then used diimide M4 and crosslinkers N1 and N2 to generate pPIs via click reactions. Finally, Zhu et al.25 used preformed diimides M4–6 to generate pPIs via homo-thermal coupling.
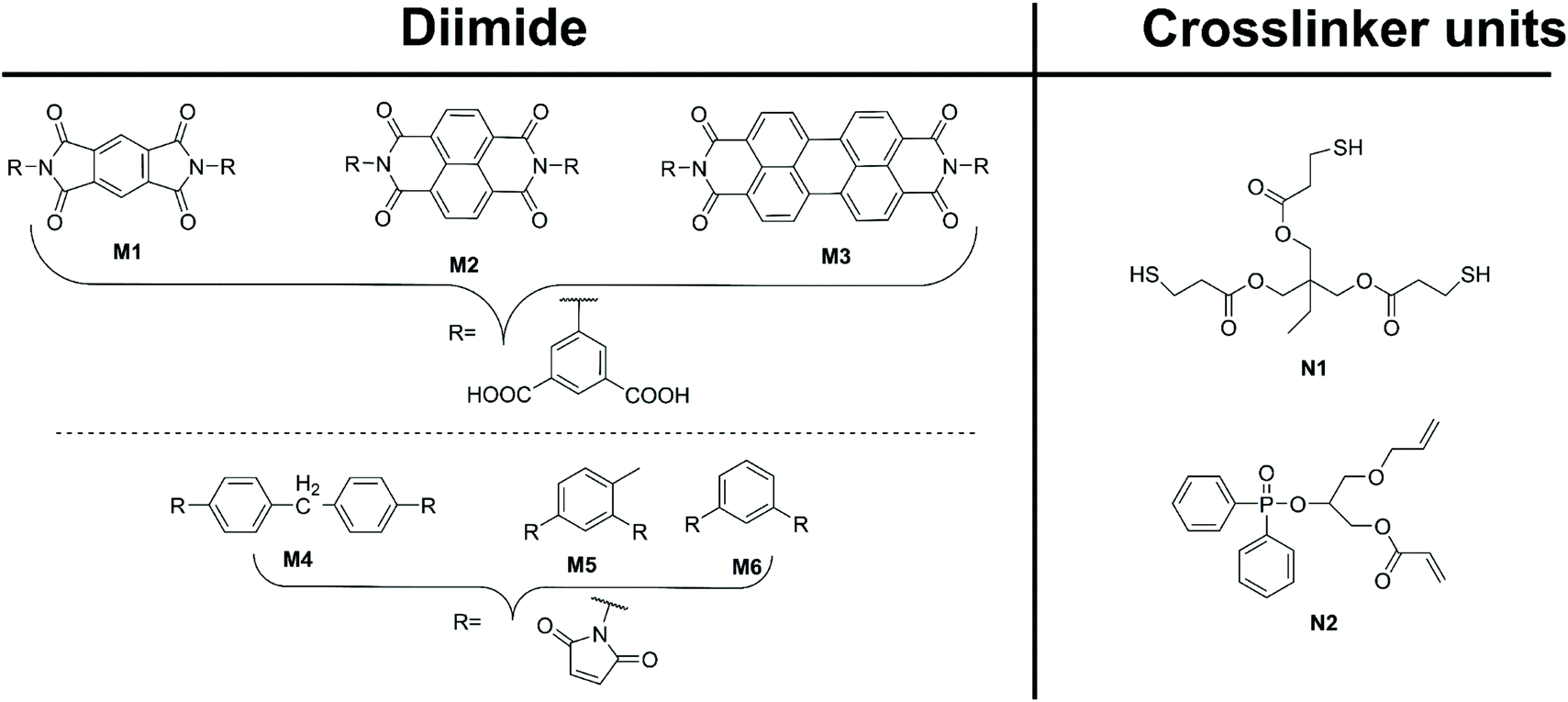 | ||
| Fig. 4 Preformed diimides M1–6 (left) and crosslinker units (right) utilised in pPI formation in ref. 23–25. | ||
3. Properties
3.1. Thermal stability
Crosslinked pPIs present excellent thermal stabilities, making them suitable for applications such as gas capture in high-temperature environments (e.g. industrial flue-gas outlets), battery and electrode environments or as protective coatings.79 Weight loss above 400 °C can often be attributed to imidisation of unreacted amic acid groups (from anhydride ring opening but failed imide formation) and water loss.45 In many cases in the literature, temperatures of degradation (Tdeg) and char yields (wt% remaining after heating to 800 °C) are not reported. In this review we have attempted to calculate those values from thermo gravimetric analysis (TGA) graphs (utilising ImageJ image analysis software) that were not calculated in the original literature and display these generated values in Table 2 alongside those reported.| Name in paper | Amine | Anhydride | Ref. | Name in paper | Amine | Anhydride | Ref. |
|---|---|---|---|---|---|---|---|
| PDI-250 | B1 | L3 | 26 and 27 | PI | B1 | L3 | 28 |
| PI0.01–0.05 | A6, B3 | L7 | 29 | PI-COF-201 | B1 | L3 | 30 |
| 3,5-DABA-TFMB | A7, B2 | L3 | 31 | PI-COF-202 | B1 | L4 | 30 |
| MPI-Phen/MS/MPI-Phe | B10, D1 | L3 | 32 | PEDA-PI | A15 | L17 | 33 |
| NP1 | C1 | L4 | 34 | PEQDA-PI | A15 | L18 | 33 |
| NP2 | B3 | L4 | 34 | ODPA PB | B5 | L14 | 35 |
| NP3 | B10 | L4 | 34 | ODPA TAPA | B3 | L14 | 35 |
| MPI-1 | C1 | L3 | 19 | ODPA TAPP | B6 | L14 | 35 |
| MPI-2 | B3 | L3 | 19 | ODPA MDA | A2 | L14 | 35 |
| MPI-3 | B10 | L3 | 19 | BPADA MDA | A2 | L9 | 35 |
| PI-1 | B1 | L4 | 36 | BPADA PB | B5 | L9 | 35 |
| PI-2 | B1 | L3 | 36 | TAPOB-HBPI-CR | B8 | L19 | 37 |
| Tr-PPI | B3 | L5 | 38 and 39 | TAPB-HBPI | B10 | L19 | 40 |
| Td-PPI | C1 | L5 | 38 and 39 | TAPA-HBPI | B3 | L19 | 40 |
| TPI-1 | B7 | L3 | 17 | TAPM-HBPI | C1 | L19 | 40 |
| TPI-2 | B7 | L4 | 17 | STPI-1 | B14 | L3 | 41 |
| TPI-3 | B7 | L5 | 17 | STPI-2 | B14 | L4 | 41 |
| TPI-4 | B7 | L13 | 17 | STPI-3 | B14 | L6 | 41 |
| TPI-5 | B7 | L14 | 17 | 6FDA-DAPI | A11, A11 | L12 | 42 |
| TPI-6 | B7 | L12 | 17 | PI (10, 30, 50 wt%) | C1 | L3 | 43 |
| TPI-7 | B7 | L11 | 17 | PI-COF-4 | C3 | L3 | 44 |
| PI-1 | B1 | L3 | 45 | PI-COF-5 | C1 | L3 | 44 |
| PI-2 | B1 | L7 | 45 | Pristine PI | A5 | L12 | 46 |
| PI-3 | B1 | L4 | 45 | MMPI | B10 | L1 | 47 |
| BIBDZ | C6 | M1 | 23 | MPI-6FA | C1 | L12 | 48 |
| NIBDZ | C6 | M2 | 23 | API-6FA | C4 | L12 | 48 |
| PIBDZ | C6 | M3 | 23 | MPI-BPA | C1 | L6 | 48 |
| BIBZ | C5 | M1 | 23 | MPI-BTA | C1 | L13 | 48 |
| NIBZ | C5 | M2 | 23 | PI & CFs | A6 | L13, L24 | 20 |
| PIBZ | C5 | M3 | 23 | FPI | A6 | L8, L12 | 49 |
| PPBPI-1-CR | C7 | L21 | 18 | FHBPAA | B12 | L12 | 49 |
| PPBPI-2-CR | C8 | L21 | 18 | BTMP-6-11 | M4 | N1 | 24 |
| PPBPI-PM-CR | C8 | L3 | 50 | BTMP-6-11-AGDP | BTMP-6-11 | N2 | 24 |
| PPBPI-BP-CR | C8 | L7 | 50 | LCP2 | A17 | L12 | 51 |
| PPBPI-NT-CR | C8 | L4 | 50 | xCP2-Am(1,2,5) | A17, B9 | L12 | 51 |
| PPBPI-PTC-CR | C8 | L5 | 50 | xCP2-An(1,2,5) | A17 | L12, L15 | 51 |
| PPBPI-x-CR | C7 | L19 | 22 | LPI-BP | A18 | L8 | 51 |
| PPBPI-PA-CR | C7 | L19, L23/L25/26 | 52 | SPI-DMDAs | A1, A9, A8 | L4 | 53 |
| FB POPP | C1 | L20 | 21 | PPI-1 | B10 | L3 | 54 |
| PI/ZIF | A6 | L7 | 55 | PPI-2 | C1 | L3 | 54 |
| PI nanosheets | B1 | L3 | 56 | PI-ADPM | C4 | L3 | 57 |
| B/P/T-0 | A4·B8 | L7 | 58 | PI-ADNT | C4 | L4 | 59 |
| PI-COF-1 | B3 | L3 | 9 | TF-PI | A15 | L19 | 60 |
| PI-COF-2 | B10 | L3 | 9 | 6FA-PI | A16 | L19 | 60 |
| PI-COF-3 | B11 | L3 | 9 | PEQDA-HBPI-CL | C1 | L18 | 61 |
| PI-1 | B1 | L5 | 62 | PEPHQDA-HBPI-CL | C1 | L19 | 61 |
| TAPB-PTCDA-COF | B10 | L5 | 63 | HBPI-TAPP-6FDA | C7 | L12 | 64 |
| TAPB-PMDA-COF | B10 | L3 | 63 | HBPI-TAPEPP-6FDA | C8 | L12 | 64 |
| TT-PMDA-COF | B7 | L3 | 63 | PI-1 | C1 | L3 | 65 |
| TAPA-PMDA-COF | B3 | L3 | 63 | PI-2 | C1 | L4 | 65 |
| PPI-1 | B13 | L3 | 19 | PIA-6FDA(0, 10, 20, 30, 40, 50) | A7, C2 | L7, L12 | 66 |
| PPI-2 | B10 | L3 | 19 | sPI-A-H | C3 | L2 | 67 |
| PPI-3 | B10 | L5 | 19 | sPI-M-H | C1 | L2 | 67 |
| CH3-PI | A12 | L18 | 68 | sPI-A-B | C3 | L1 | 67 |
| CF3-PI | A13 | L18 | 68 | sPI-M-B | C1 | L1 | 67 |
| TAPA-HBPI | B3 | L18 | 16 | MPI | C1 | L3 | 69 |
| CF3TAPA-HBPI | B4 | L18 | 16 | PBDM | — | M4 | 25 |
| PI-1 | B10 | L3 | 70 | PBMP | — | M5 | 25 |
| PI-2 | B10 | L4 | 70 | PPDM | — | M6 | 25 |
| PAF-110 | B3 | L4 | 71 | PI-3 | B10 | L5 | 70 |
| PPPP-1 | C9 | L3 | 72 | NT-COF | B3 | L4 | 73 |
| PPPP-1 | C9 | L4 | 72 | PI-COF | C7 | L5 | 74 |
| PIA | B10 | L3 | 75 | NDI-COF | B10 | L4 | 76 |
| PIB | B3 | L3 | 75 | TP-COF | B7 | L3 | 77 |
| PIC | B10 | L4 | 75 | CPI | B10 | L4 | 78 |
| PID | B3 | L4 | 75 | PI – 0%6FAPB | A6, A14, B8 | L7 | 79 |
| Name in paper | T deg (°C) | Char (wt%) | Ref. | Name in paper | T deg (°C) | Char (wt%) | Ref. |
|---|---|---|---|---|---|---|---|
| MPI-1 | 530 | 57.4 | 19 | TAPA-HBPI-CR | 500* | 60* | 37 |
| MPI-2 | 530 | 53.0 | 19 | TAPA-HBPI-GEL | 600* | 60* | 37 |
| MPI-3 | 530 | 59.3 | 19 | TAPM-HBPI | 450* | 60* | 37 |
| PI-COF-1 | 520 | 60* | 9 | TAPM-HBPI-CR | 530* | 60* | 37 |
| PI-COF-2 | 535 | 60* | 9 | TAPM-HBPI-GEL | 580* | 60* | 37 |
| PI-COF-3 | 530 | 60* | 9 | TAPOB-HBPI | 500* | 70* | 37 |
| TFMB | 537 | — | 31 | TAPOB-HBPI-CR | 550* | 75* | 37 |
| 3,5-DABA (20)TFMB (80) | 520 | — | 31 | TAPOB-HBPI-GEL | 600* | 60* | 37 |
| 3,5-DABA (50)TFMB (50) | 489 | — | 31 | STPI-1 | 610* | 5* | 41 |
| 3,5-DABA (80)TFMB (20) | 467 | — | 31 | STPI-2 | 580* | 50* | 41 |
| m-PDA (20)TFMB (80) | 527 | — | 31 | STPI-3 | 600* | 60* | 41 |
| m-PDA (50)TFMB (50) | 536 | — | 31 | PI-COF-4 | 450 | 40* | 80 |
| m-PDA (80)TFMB (20) | 532 | — | 31 | PI-COF-5 | 460 | 42* | 80 |
| PAA0.03 | 200* | 30* | 29 | BI-PEG2-xPI | 510* | — | 46 |
| PI0.03 | 550 | 50 | 29 | BI-PEG3-xPI | 510* | — | 46 |
| NPI1 | 480 | 50.0 | 34 | BI-PEG4-xPI | 500* | — | 46 |
| NPI 2 | 485 | 46.7 | 34 | BI-PEG6-xPI | 450* | — | 46 |
| NPI 3 | 490 | 64.7 | 34 | MPI-6FA | 550* | 50* | 48 |
| PI1 | 400 | 36 | MPI-BTA | 560* | 55* | 48 | |
| PI 1 | 400 | 45 | 62 | MPI-BPA | 610* | 58* | 48 |
| PI 2 | 405 | 45 | 62 | API-6FA | 550* | 53* | 48 |
| PI-1-C | 68 | 62 | PI 300 | — | — | 20 | |
| TPI1 | 419 | — | 17 | CF 600 | — | 73 | 20 |
| TPi2 | 456 | — | 17 | CF 900 | — | 65 | 20 |
| TPI3 | 342 | — | 17 | CF 1200 | — | 55 | 20 |
| TPI4 | 427 | — | 17 | CF 1500 | — | 47 | 20 |
| TPI5 | 421 | — | 17 | FPI | 542 | 60* | 49 |
| TPI6 | 396 | — | 17 | FPI/FHBPI-5% | 530 | 58* | 49 |
| TPI7 | 450 | — | 17 | FPI/FHBPI-10% | 528 | 58* | 49 |
| PI1 | 410 | 10* | 45 | FPI/FHBPI-15% | 524 | 57* | 49 |
| PI2 | 350 | 0* | 45 | FPI/FHBPI-20% | 519 | 57* | 49 |
| PI3 | 350 | 30* | 45 | FPI/FHBPI-25% | 516 | 56* | 49 |
| PI – 0%6FAPB | 591 | T g 202.1 | 79 | FHBPI | 514 | 55* | 49 |
| PI – 25%6FAPB | 589 | T g 221.6 | 79 | BTMP-6-AGDP | 328 | 30 | 24 |
| PI – 37.5%6FAPB | — | T g 234.3 | 79 | BTMP-9-AGDP | 327 | 32 | 24 |
| PI – 50%6FAPB | 589 | T g 259.2 | 79 | BTMP-11-AGDP | 326 | 34 | 24 |
| PPI1 | 560 | — | 19 | PPI-1 | 570 | 62* | 54 |
| PPI2 | 570 | — | 19 | PPI-1-NH2 | 405 | 54* | 54 |
| PPI3 | 525 | — | 19 | PPI-2 | 540 | 65* | 54 |
| Tr-PPI | 550* | 60* | 38 | PPI-2-NH2 | 410 | 35* | 54 |
| Td-PPI | 450* | 60* | 38 | PI-ADNT | 600* | — | 59 |
| BIBDZ | 100* | 70* | 23 | PI-NO2-1 | 400* | — | 59 |
| PIBDZ | 100* | 60* | 23 | PI-NO2-2 | 400* | — | 59 |
| NIBDZ | 100* | 65* | 23 | PI-NO2-3 | 400* | — | 59 |
| BIPZ | 100* | 70* | 23 | TF-PI | 500* | 45* | 60 |
| PIBZ | 100* | 60* | 23 | TF-PI-CL | 560* | 65* | 60 |
| NIBZ | 100* | 68* | 23 | 6FA-PI | 500* | 45* | 60 |
| PPBPI-H | 520* | 65* | 22 | 6FA-PI-CL | 550* | 60* | 60 |
| PPBPI-Mn | 580* | 75* | 22 | PI-1 | 588 | 72* | 81 |
| PPBPI-Fe | 518* | 66* | 22 | PI-2 | 519 | 65* | 81 |
| PPBPIR-H CR | 522* | 66* | 22 | PIA/6FDA-0 | 593 | 60* | 82 |
| PPBPI-Mn-Cr | 520* | 68* | 22 | PIA/6FDA-10 | 573 | 59* | 82 |
| PPBPI-Fe-Cr | 580* | 70* | 22 | PIA/6FDA-20 | 565 | 59* | 82 |
| PPBPI-PA | 580* | 60* | 52 | PIA/6FDA-30 | 551 | 59* | 82 |
| PPBPI-PEPA | 600* | 70* | 52 | PIA/6FDA-40 | 550 | 59* | 82 |
| PPBPI_PENA | 560* | 68* | 52 | PIA/6FDA-50 | 536 | 58* | 82 |
| PPBPI-PA-CR | 600* | 68* | 52 | sPI_A-H | 540* | 52* | 67 |
| PPBPI-PEPA-CR | 560* | 70* | 52 | sPI-M-H | 530* | 47* | 67 |
| PPBPI-PENA-CR | 380* | 60* | 52 | sPI_A-B | 430* | 48* | 67 |
| TAPA-HBPI | 510* | 54* | 16 | sPI-M-B | 420* | 48* | 67 |
| TAPA-HBPI-CL | 610* | 65* | 16 | MMPI | 520* | 60* | 83 |
| CF3TAPA-HBPI | 510* | 55* | 16 | LCP2 | 526 | — | 51 |
| CF3TAPA-HBPI-CL | 600* | 65* | 16 | xCP2-Am1 | 514 | — | 51 |
| PI-1 | 473 | — | 70 | xCP2-Am2 | 511 | — | 51 |
| PI-2 | 502 | — | 70 | xCP2-Am5 | 518 | — | 51 |
| PI-3 | 490 | — | 70 | xCP2-An1 | 519 | — | 51 |
| PI | 380* | — | 28 | xCP2-An2 | 518 | — | 51 |
| PI-COF 201 | 380* | 10* | 30 | xCP2-An5 | 518 | — | 51 |
| PI-COF 202 | 420* | 10* | 30 | LPI-BP | — | — | 51 |
| PEDA-PI | 500* | 55* | 33 | CSPI-DMDA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) :3) |
340* | 49* | 53 |
| PEDA-PI-CL | 550* | 68* | 33 | CSPI-DMDA (1:1) |
340* | 52* | 53 |
| PEQDA-PI | 480* | 50* | 33 | CSPI-DMDA (3:1) |
330* | 40* | 53 |
| PEQDA-PI-CL | 550* | 67* | 33 | PAF-110 | 530* | 5* | 71 |
| TAPB-HBPI | 520* | 75* | 40 | MPI-0-10 | 363 | 55* | 84 |
| TAPB-HBPI-CR | 570* | 75* | 40 | MPI-30-10 | 485 | 60* | 84 |
| TAPB-HBPI-GEL | 570* | 70* | 40 | MPI-40-10 | 465 | 60* | 84 |
| TAPA-HBPI | 510* | 60* | 40 | MPI-50-10 | 443 | 60* | 84 |
| PIA | 535 | 65* | 75 | MPI-60-10 | 452 | 60* | 84 |
| PIB | 525 | 60* | 75 | MPI-70-10 | 476 | 60* | 84 |
| PIC | 535 | 65* | 75 | MPI-100-10 | 391 | 61* | 84 |
| PID | 520 | 50* | 75 | MPI-60-5 | — | — | 84 |
| CPI | 275* | 55* | 78 | MPI-60-7 | 348 | 60* | 84 |
| NDI-COF | 500 | 60 | 76 | MPI-60-9 | 490 | 61* | 84 |
| PI-COF | 500 | 60 | 74 | MPI-60-11 | 492 | 62* | 84 |
| PPPP-1 | 560 | 70 | 72 | MPI-60-13 | 503 | 63* | 84 |
| PPPP-2 | 570 | 70 | 72 |
Conjugated pPIs show typical degradation temperatures between 400–600 °C (10 wt% loss), with the highest stabilities reported by Shi et al.,18,22,50,52 Song et al.,16,60,61,68 and Yao et al.37,40 (each slightly above 600 °C). Conjugated pPIs quite often exhibit a decrease in Tdeg when increasing the weight percentage of heteroatoms in the network, as seen, for example, in the studies by Wang et al.28,30 with the Tdeg increasing with increasing carbon-based dianhydride monomer linker units L3 and L4 (although the reasons behind this trend were not investigated by the authors). The introduction of post-polymerisation crosslinking units into pPI networks tends to lead to an increase in pPI thermal stability (with respect to Tdeg) after crosslinking. However, if the crosslinking moieties lead to an increased heteroatom content, a decrease in thermal stability can be observed (see the effect of the introduction of the amide crosslinker unit B2 by Hasegawa et al.31). Interestingly, very little variation in thermal stabilities of conjugated pPI networks is observed when varying the molar ratio of amine centres on the starting monomers from tri- or tetra-amines (see Rao et al.38 utilizing the tetra-armed amine C1, and Shi et al.22 using the porphyrin-based tetramines C7 and C8). Perhaps the most interesting take on control over the thermal properties of conjugated pPI networks is found in the study by Qiao et al.,79 where they investigated polyimide aerogels. Here frustrated pPI chain growth is found to leave pendant carboxylic acid groups from the anhydride monomer L8, that are then crosslinked post polymerisation (by addition of triethyl amine) under supercritical CO2 to control Tdegvia the degree of crosslinking.
The majority of conjugated pPIs reported in this review are highly crosslinked amorphous materials; thermal analysis such as dynamic scanning calorimetry (DSC) to examine thermal transitions before the Tdeg are thus largely unreported or unexplored. An exception can be found in the pPIs synthesised by Hasegawa et al.,31 where both DSC and dynamic mechanical analysis are used to demonstrate the manipulation of glass transition temperatures (Tg) by post-polymerisation crosslinking of pPIs utilizing the amide crosslinker unit B2. Further to this exception, Qiao et al.79 show manipulation of Tgvia introduction of CF3 side groups (to increase the steric hinderance and free movement of chains, hence increase in Tg), using the amine monomer A14 into their polyimide aerogels. Song et al.68 and Shi et al.22 utilised post-polymerisation cross-coupling of alkynes present in the anhydride monomers L18 and L19. In the majority of cases DSC is primarily used for verification of post-polymerisation crosslinking, with exotherms being recorded in the first heating scan but absent in the second.
The properties of pPIs beyond their Tdeg temperatures were explored by Liao et al.,62 using the charred product after heating beyond Tdeg to form ‘derived carbons’ (from pPIs of B1 and L5). They found both increased surface area and CO2 uptakes for these derived carbons with respect to the non-charred pPIs (see further discussion on gas absorption properties). This leads to the question whether higher Tdeg is desirable when trying to access char products with enhanced absorption properties efficiently.31 The mechanisms of charring and carbonisation have also been explored by Li et al.,20,49 using Raman, XPS spectroscopy and XRD diffractometry to analyse the products post carbonisation. They concluded that ‘the decomposition of imide rings and the breakage of the ether oxygen in the bridging part of polyimide’ are responsible for charring, leading to the proposition that increased heteroatom content can facilitate carbonisation (and hence a decrease in Tdeg). It is evident that carbonisation (and its beneficial effects on properties such as surface area or gas absorption, specifically CO2 uptake) are relatively unexplored in the literature to date and is an area for further future exploration and development.
Many of the reported non-conjugated pPIs show similar properties and trends to the conjugated pPIs summarized above. However, there are some aspects to be highlighted in this section. Studies for non-conjugated pPIs tend to conclude that Tdeg as well as char yields in N2 tend to be optimized when using pPIs with the least amount of heteroatoms within the network (see, for example, Li et al.19,34,36). Rangel Rangel et al.19 demonstrated that functionalization of their pPIs with nitro groups (for catalysis applications, see section 4.2) results in a decrease in thermal stability, from the 540–570 °C to the low 400 °C region. Similar results were observed by Shen et al.59 investigating a tetra-armed pPI with a high thermal stability of 621 °C that experiences a considerably drop in stability after nitration to 370 °C (as a result of C-NO2 cleavage throughout the pPI). However, in contrast to this behaviour, Wang et al.81 found that a change in anhydride linker from benzene L3 to naphthalene L4 (an increase in carbon percentage as well as conjugation) in their non-conjugated pPIs resulted in a drop in the Tdeg. Lu et al.24 also showed how increased thiol linkages in pPIs (present from linker N2) can result in increased Tdeg and increased char yields. Finally, Wu et al.82 found, counterintuitively, a decrease in Tdeg with increasing crosslinking percentage (and hence increased carbon–carbon bond formation), with no further explanation provided.
The majority of crosslinked pPIs do not demonstrate melts or glass transitions (Tg). However, Lu et al.24 showed how control over the length of thiol linkages (N2) in pPIs allow manipulation of the Tg, with a maximum of 59 °C appearing for moieties with 9 thiol linkers per linker unit. It is worth nothing that although this polymer is classified here as a pPI, the thiols are used for the formation of the final crosslinked product from the preformed diimide M2. The manipulation of the Tg of the final product is important as it has been linked to a deformation temperature. Below this temperature, the polymer will hold a deformed shape and above the polymer recovers to its original shape (i.e., displaying shape memory function) due to the high degree of crosslinking and stability. Li et al.49 use blends of both linear (from A12 and L12) and crosslinked (using B12 to crosslink) fluorinated pPIs to manipulate both Tdeg and Tg. A decrease of Tdeg is observed with increasing crosslinked content in the blend, attributed to the increase in terminal hydrides in the crosslinked structure and hence increased post-polymerisation carbonisation. Both the linear and crosslinked pPIs demonstrate Tg values of approximately 260 °C; mixing provides a route to manipulation of the Tg, with a maximum value of Tg realized at 5 wt% crosslinker (as confirmed by dynamic mechanical analysis).
3.2. Porosity
We have found several trends in the surface areas of pPIs, and discuss below the most relevant and important factors influencing this important property of this class of porous materials:| Name in paper | S BET (m2 g−1) | PV (cm3 g−1) | CO2 (wt%) | Q st (kJ mol−1) | Ref. | Name in paper | S BET (m2 g−1) | PV (cm3 g−1) | CO2 (wt%) | Q st (kJ mol−1) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PI-250 | 16.8A | — | — | — | 26 | PI-1 | — | 8ii | — | 70 | |
| PI-275 | 7.5C | — | — | — | 26 | PI-2 | — | — | — | 70 | |
| PI-300 | 6.8C | — | — | — | 26 | PI-3 | — | — | — | — | 70 |
| PI-325 | 5.1C | — | — | — | 26 | PI | 635.5A | 0.98 | — | — | 28 |
| PI-350 | 4.3C | — | — | — | 26 | PI-COF-201 | 3.9C | — | — | — | 30 |
| NPI-1 | 721A | 0.51 | 12.3ii | 33.5 | 34 | PI-COF-202 | 9.1C | — | — | — | 30 |
| NPI-2 | 291A | 0.2 | 7.3ii | 30.1 | 34 | PEDA-PI | 78A | 0.09 | — | — | 33 |
| NPI-3 | 373A | 0.29 | 8.2ii | 33.3 | 34 | PEDA-PI-CL | 399A | 0.29 | 6.32ii | 31 | 33 |
| MPI-1 | 1454A | 1.07 | 16.8ii | 34.8 | 19 | PEQDA-PI | 64A | 0.08 | — | — | 33 |
| MPI-2 | 814A | 0.59 | 13.8ii | 30.4 | 19 | PEQDA-PI-CL | 607A | 0.42 | 9.88 ii | 30.4 | 33 |
| MPI-3 | 586A | 0.31 | 9.9ii | 31.4 | 19 | TAPOB-HBPI CR | 322 | 0.33 | 5.5ii | 30.3 | 37 |
| Tr-PPI | 400A | 4.33 | 45i | — | 38 | TAPB-HBPI-CR | 385 | 0.19 | 1.23ii | 20.2–28.3 | 40 |
| Td-PPI | 2213A | 0.63 | 31i | — | 38 | TAPA-HBPI-CR | 497 | 0.24 | 1.01ii | 40 | |
| TPI-1 | 809A | 0.45 | 10.78ii | 34.4 | 17 | TAPM-HBPI-CR | 492 | 0.25 | 1.22ii | 40 | |
| TPI-2 | 796A | 0.4 | 10.78ii | 31.4 | 17 | STPI-1 | 4A | 0.16 | 7ii | 31 | 41 |
| TPI-3 | 40A | — | 2.99ii | 32.3 | 17 | STPI-2 | 541A | 0.32 | 14ii | 36 | 41 |
| TPI-4 | 245A | 0.22 | 8.1ii | 33.6 | 17 | STPI-3 | 378A | 0.34 | 10ii | 28 | 41 |
| TPI-5 | 201A | 0.18 | 6.9ii | 30 | 17 | PI-COF-4 | 2403C | — | — | — | 80 |
| TPI-6 | 510A | 0.31 | 9.6ii | 29.2 | 17 | PI-COF-5 | 187C | — | — | — | 80 |
| TPI-7 | <10A | — | 7.9ii | 32.4 | 17 | MMPI | 530A | 0.394 | — | — | 47 |
| PI-1 | 660A | 0.6 | 7.3ii | — | 45 | MPI-6FA | 781A | 0.53 | 13.5ii | — | 48 |
| PI-2 | 265A | 0.27 | 2.7ii | — | 45 | API-6FA | 752A | 0.61 | 12.4ii | — | 48 |
| PI-3 | 366A | 0.76 | 6.0ii | — | 45 | MPI-BPA | 677A | 0.53 | 11.1ii | — | 48 |
| BIBDZ | 177A | — | 11.33i | — | 23 | MPI-BTA | 490A | 0.39 | 10.4ii | — | 48 |
| NIBDZ | 118A | — | 14.56i | — | 23 | PPI-1 | 474A | — | — | — | 54 |
| PIBDZ | 75.35A | — | 11.85i | — | 23 | PPI-1-NH2 | 206A | — | — | — | 54 |
| PPBPI-1-CR | 682A | 0.429 | 8.8ii | 25.1 | 18 | PPI-2 | 604A | — | — | — | 54 |
| PPBPI-2-CR | 693A | 0.457 | 7.3ii | 30.1 | 18 | PPI-2-NH2 | 274A | — | — | — | 54 |
| PPBPI-PM-CR | 628A | 0.379 | 10.25ii | 22.2 | 50 | PI-ADPM | 868 | 0.37 | 14.6ii | 34.4 | 85 |
| PPBPI-BP-CR | 582A | 0.573 | 7.92ii | 24.7 | 50 | PI-ADNT | 774 | 0.415 | 15ii | 35.2 | 59 |
| PPBPI-NT-CR | 415A | 0.425 | 5.89ii | 19.7 | 50 | PI-NO2-1 | 286 | 0.155 | 17.7ii | 43.3 | 59 |
| PPBPI-PTC-CR | 115A | 0.838 | 2.94ii | 29.6 | 50 | PI-NO2-2 | 57 | 0.08 | 10ii | 37.9 | 59 |
| PPBPI-H-CR | 733A | 0.532 | 9.94ii | 24.5 | 22 | PI-NO2-3 | 26 | 0.03 | 8.6ii | 37.9 | 59 |
| PPBPI-Mn-CR | 144A | 0.124 | 6.33ii | 29.2 | 22 | TF-PI | 66 | 0.22 | — | 31.9–32 | 60 |
| PPBPI-Fe-CR | 172A | 0.135 | 5.63ii | 31 | 22 | TF-PI-CL | 727 | 0.44 | 10ii | — | 60 |
| PPBPI-PA-CR | 1.8A | 0.001 | 6.02ii | 34.8 | 52 | 6FA-PI | 67 | 0.29 | — | — | 60 |
| PPBPI-PEPA CR | 373A | 0.267 | 8.84ii | 27.5 | 52 | 6FA-PI-CL | 635 | 0.37 | 7.28ii | — | 60 |
| PPBPI-PENA CR | 633A | 0.858 | 10.43ii | 22.6 | 52 | TF-6FDA-PI | 154 | 0.29 | — | — | 60 |
| PI-COF-1 | 1027C | — | — | — | 9 | TF-6FDA-PI-CL | 472 | 0.33 | 6.63ii | — | 60 |
| PI-COF-2 | 1297C | — | — | — | 9 | PEQDA-HBPI | 138 | 0.79 | — | — | 61 |
| PI-COF-3 | 2346C | — | — | — | 9 | PEQDA-HBPI-CL | 339A | 0.26 | 9.82ii | 32.8 | 61 |
| PI-1 | 19A | 0.043 | 3ii | — | 62 | PEPHQDA-HBPI | 155 | 0.6 | — | — | 61 |
| PI-2 | 16A | — | 0.9ii | — | 62 | PEPHQDA-HBPI-CL | 593A | 0.35 | 10.1ii | 32.9 | 61 |
| PI-opt | — | 0.084 | 5.8ii | — | 62 | PI-1 | 1407A | 0.78 | — | 5.3 | 81 |
| PI-1-C | 13A | 0.125 | 10.3ii | — | 62 | PI-2 | 732A | 0.51 | — | 7 | 81 |
| PI-opt-C | 181A | 0.177 | 15ii | — | 62 | PIA/6FDA-50 | 607 | 4.34 | — | — | 82 |
| TAPB PTCDA COF | 460C | — | — | — | 63 | PIA/6FDA-40 | 604 | 2.9 | — | — | 82 |
| TAPB PMDA COF | 1250C | — | — | — | 63 | PIA/6FDA-30 | 594 | 2.59 | — | — | 82 |
| TT-PMDA-COF | 706C | — | — | — | 63 | PIA/6FDA-20 | 577 | 2.87 | — | — | 82 |
| TAPA-PMDA-COF | 1592C | — | — | — | 63 | PIA/6FDA-10 | 463 | 1.41 | — | — | 82 |
| PI – 0%6FAPB | 327gel | 2.69 | — | — | 79 | PIA/6FDA-0 | 441 | 1.86 | — | — | 82 |
| PI – 25%6FAPB | 346gel | 2.43 | — | — | 79 | sPI-A-H | 25A | 0.02 | 12ii | 31.7 | 67 |
| PI – 37.5%6FAPB | 368gel | 2.28 | — | — | 79 | sPI-M-H | 492A | 0.27 | 13.24ii | 34 | 67 |
| PI – 50%6FAPB | 402gel | 2.77 | — | — | 79 | sPI-A-B | 620A | 0.46 | 10.69ii | 32.5 | 67 |
| PPI-1 | 18A | 0.038 | — | — | 19 | sPI-M-B | 618A | 0.47 | 11.48ii | 34.3 | 67 |
| PPI-2 | 604A | 0.285 | — | — | 19 | MPI | 1001 | 0.68 | 12ii | 32.8 | 69 |
| PPI-3 | 707C | 1 | — | — | 19 | MPI-S | 448 | 0.31 | 6.9ii | 36.3 | 69 |
| CH3-PI | 20A | 0.09 | — | — | 68 | MPI-Ag | 103 | 0.1 | 6.4ii | 37.1 | 69 |
| CH3-PI-CL | 33A | 0.05 | 7.3ii | 31.3 | 68 | TAPB-HBPI-CR | 385 | 0.19 | 1.56ii | 28.6–30.0 | 40 |
| CF3-PI | 21A | 0.05 | — | — | 68 | TAPA-HBP-CR | 497 | 0.24 | 1.68ii | — | 40 |
| CF3-PI-CL | 575A | 0.43 | 9.4ii | 33 | 68 | TAPM-HBPI-CR | 492 | 0.25 | 2.04ii | — | 40 |
| TAPA-HBPI | 123A | 0.34 | — | — | 16 | PBDM* | 954 | 0.79 | 13 | — | 25 |
| TAPA-HBPI-CL | 43A | 0.045 | 7.47ii | 27.4 | 16 | PBMP* | 1025 | 0.87 | — | — | 25 |
| CF3TAPA-HBPI | 88A | 0.23 | — | — | 16 | PPDM* | 930 | 1.36 | — | — | 25 |
| CF3TAPA-HBPI-CL | 294A | 0.2 | 8.56ii | 32.3 | 16 | MPI-0-10 | 300 | — | — | — | 84 |
| NT-COF | 1276C | 1.23 | — | — | 73 | MPI-30-10 | 302 | — | — | — | 84 |
| PI-COF | 894C | 0.47 | — | — | 74 | MPI-40-10 | 303 | — | — | — | 84 |
| PIA | 580C | — | 10ii | — | 75 | MPI-50-10 | 276 | — | — | — | 84 |
| PIB | 760C | — | 12ii | — | 75 | MPI-60-10 | 258 | — | — | — | 84 |
| PIC | 990C | — | 11ii | — | 75 | MPI-70-10 | 250 | — | — | — | 84 |
| PID | 1430C | — | 13ii | — | 75 | MPI-100-10 | 251 | — | — | — | 84 |
| PAF-110 | 910C | 0.59 | — | — | 71 | MPI-60-5 | — | — | — | — | 84 |
| NDI-COF | 1138C | 0.77 | — | — | 76 | MPI-60-7 | 202 | — | — | — | 84 |
| PPPP-1 | 295C | — | — | — | 72 | MPI-60-9 | 226 | — | — | — | 84 |
| PPPP-2 | 301C | — | — | — | 72 | MPI-60-11 | 262 | — | — | — | 84 |
| TP-COF | 960C | — | — | — | 77 | MPI-60-13 | 281 | — | — | — | 84 |
| CPI | 65 | — | — | — | 78 |
Surface area and pore size can also be tuned by varying the length and size of the monomer linkers and cores. Rao et al.38 reported two pPIs consisting of either tetra-amine C1 (tetrahedral) or tri-amine B3 (trigonal pyramidal), but with a longer anhydride linker L5 (when compared with NPI-1–3 synthesised by Li et al.19,34). The surface areas were 2213 m2 g−1 (C1 tetrahedral) and 400 m2 g−1 (B3 trigonal pyramidal), respectively, which were higher than pPIs synthesised with the shorter dianhydride L4 linker with the same core (see Table 3).34 Analogously, Fang et al.9 showed that increases in the core size increased the surface area. The condensation of the dianhydride linker L3 with either B10 or B11 (larger core than B3) resulted in a doubling of surface area from 1297 m2 g−1 to 2346 m2 g−1 when the core length was increased to contain an extra benzene moiety. The average pore size also increased from 37 Å to 53 Å with this extended architecture (Table 3).
3.2.2.1. Carbonisation. It is important to note that uncontrolled post-polymerisation crosslinking in the form of carbonisation is reported to increase the surface area of most conjugated porous networks. Despite this general observation, the pPIs reported in this review have largely not been investigated in regard to the effects of carbonisation on surface area. In the specific case of pPIs we have found that only Liao et al.62 investigated this effect, with an increase from 19 m2 g−1 to 181 m2 g−1 after carbonisation of pPI synthesised from B1 and L5. Wu et al.55 also used carbonisation to crosslink linear PIs (from L7 and A6) to form pPIs, which were blended, in the linear form (pre-carbonisation), with zeolite imidazolate frameworks (ZIF). However, the surface area decreased significantly after carbonisation (from 200–300 m2 g−1 to 10 m2 g−1); the reasons behind this decrease were not investigated.
3.2.2.2. Post-polymerisation crosslinking. Surface areas can be further tuned via controlled post-polymerisation crosslinking. Shi et al.22 synthesised porphyrin-based pPIs (based on amine C7) with a cross-linkable alkynyl group present in the starting anhydride monomer L19. The surface area of the pPIs after crosslinking (without metal absorption) were 733 m2 g−1, significantly higher than the non-crosslinked pPI (167 m2 g−1 see Table 3). Another study by Wang et al.33 used an amine monomer A15 bearing a cross-linkable ethynyl group in combination with non-cross-linkable L17 and cross-linkable alkene-containing L18 dianhydride, respectively. The surface areas of the pPIs from the non-cross-linkable dianhydride was 78 m2 g−1 whilst that using the cross-linkable group was 64 m2 g−1. After thermal crosslinking, the surface area increased to 399 m2 g−1 and 604 m2 g−1, respectively. The increase in surface area was attributed to the crosslinking restricting movement (and hence restricting denser packing) of polymer chains.
3.2.2.3. Post polymerisation chemical functional modification. Rangel Rangel et al.54 utilised dianhydride linker L3 and triamine B10 or tetraamine C1 to synthesize PPI-1 and PPI-2, respectively. PPI-1 and PPI-1 were then functionalised with additional amino groups (via nitration and subsequent reduction), which resulted in a decrease of surface areas from 570 to 405 m2 g−1 and 540 to 410 m2 g−1, respectively. Shi et al.22 also reported that the metalloporphyrin-based pPIs (from monomers C7 and C8) have lower surface areas than the materials prepared from metal-free cores. The incorporation of metal ions increased the unit mass and blocked the micropores of the polyimide skeleton, leading to a decrease in their surface area from 733 m2 g−1 to 144 m2 g−1 for Mn and 172 m2 g−1 for Fe, respectively. It is noteworthy that these values were obtained via post-polymerisation crosslinking; the surface areas compared with the non-crosslinked pPIs were higher by a factor of 10. Yan et al.69 also reported that the post-polymerisation chemical modification of pPI MPI (formed from C1 and L3) decreased the surface area from 1001 m2 g−1 to 448 m2 g−1 after sulfonation (MPI-S), and further to 103 m2 g−1, after forming a silver-pPI ionically bound complex (MPI-Ag).
Outliers to the trend are found for the pPIs synthesised from L3 and B1 by Wang et al.,28,30 where the surface area of the amorphous analogue was significantly higher (by a factor of over 70) than that of the crystalline counterpart (i.e., 635 vs. 9 m2 g−1). Although it is not immediately obvious why higher surface areas were obtained for the amorphous pPI, one of the factors that could contribute to lower surface area for COFs could be the chosen synthesis conditions; specifically, the COF pPI was synthesised by direct heating without any solvent, resulting in potential uncontrolled polymerisation, whereas the amorphous pPI was synthesised in solution (that that might have led to the a higher degree of control in regard to surface area, but less order). A further similar example was presented by Luo et al.45 with L4 and B1 as starting materials, where the surface area of amorphous materials were higher than that of the crystalline COF analogue synthesised by Wang et al.30 However, it is noteworthy that these were the only two cases found where amorphous pPIs possessed higher surface areas than their COF counterparts.
3.3. Energy storage capabilities and related properties
Polymers with an extended π-conjugated backbone and high surface area with excellent microporosity (with an average pore size of <2 nm) are advantageous for electrical energy storage applications.87 Conjugation is beneficial with respect to both electrochemical stability and electronic conductivity.36 It is to be noted that we are only considering conjugated pPI networks in this section owing to their attractive electronic properties. The electronic properties of non-conjugated pPIs are explored in two recent publications by Tian et al.,64 and Gao et al.,43 highlighting attempts to increase conductivity focusing on increasing pPI conjugation, or via addition of conjugated additives.Batteries and capacitors, as the most common energy storage devices, operate via electric double layer capacitance and pseudo-capacitance mechanisms. In electric double layer capacitance (EDLC), energy is stored and release by a physical ion adsorption–desorption mechanism at the electrode–electrolyte interface.88 The EDLC depends on the pore size and surface area of the materials, as well as the electrical conductivity of the electrode.89 Therefore, high surface areas are crucial for larger numbers of ions or charge accumulation at the electrode–electrolyte interface, i.e., the higher the specific surface area or the micropore volume of the materials the higher the capacitance value (although this relationship is not actively explored in the literature).90 Pseudo-capacitive mechanisms depend on charge storage involving fast surface redox reactions, which consist mainly of surface electron transfer by the intercalation or adsorption of charge-compensating ions.91,92 Therefore, high surface areas as well as redox-active moieties (e.g., carbonyl compounds) are desirable to benefit pseudo-capacitive mechanisms.
The key advantages of porous pPIs are their tuneable molecular structure, e.g. redox-active groups can be introduced within the backbone, allowing manipulation of beneficial properties for electrochemical applications.87,93 Highly crosslinked porous pPI networks possess the above properties and create actives sites for charge or ions to be stored within the porous surface. In addition, pPIs (as part of the broader class of porous organic polymers) are presented as environmentally friendly, low safety risk (with respect to toxicity) and low cost electrodes when compared with metal-containing electrode counterparts.70 Specifically, the major advantage is the presence of carbonyl groups in the polyimide framework structure, which provides redox stability and multi-electron transfer capabilities (see Fig. 5).36,70
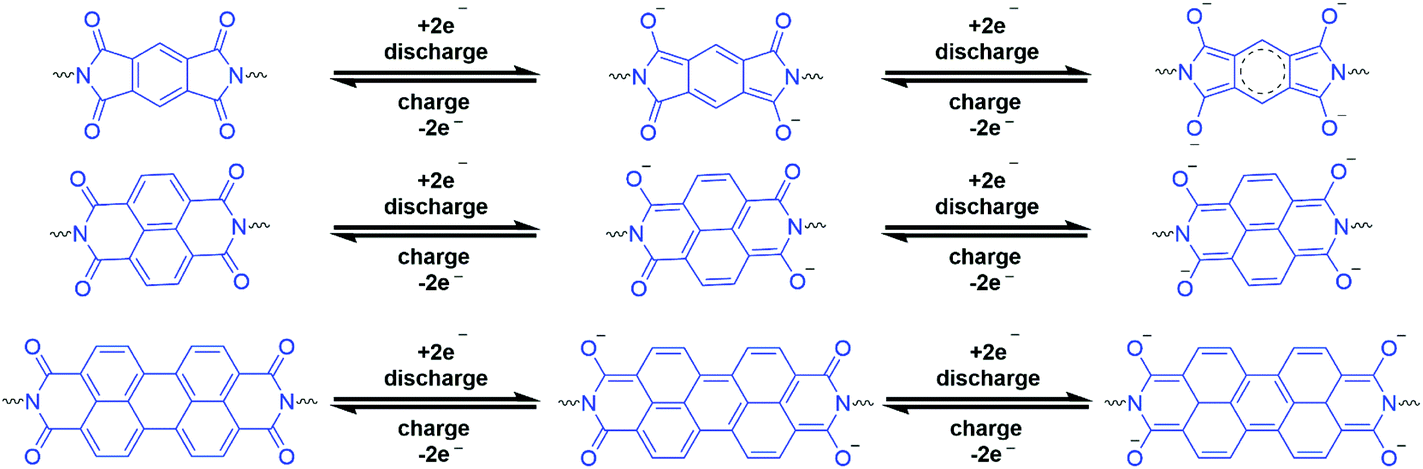 | ||
| Fig. 5 Various oxidation states of common anhydride linkers L3–5.70 | ||
One major drawback for using porous organic polymers (POPs) as electrode materials is poor bulk conductivity, limiting their effectiveness in electrochemical applications.90 To overcome this challenge, materials can be carbonised, or conductive additives incorporated into the bulk (e.g. carbon nanotubes, graphene or reduced graphene oxide).43,77,90,94 pPIs containing aromatic linkers L3–5 have demonstrated semiconductive properties without the need for additives; however, they can only transfer two electrons in the reversible charge/discharge process as shown in Fig. 5.70 In an initial study, Song et al. employed non-crosslinked linear PIs for redox-active systems (from anhydrides L3–4 and various diamines). However, dissolution in the electrolyte during cycling resulted in inefficiency as well as degradation of the PI.95,96 To overcome this practical problem, Tian et al.70 utilised triamine B10 with anhydrides L3–5 to form crosslinked pPIs as electrode materials (cathodes) for Li-ions batteries (LIBs). The highly crosslinked networks were able to sustain harsh chemical and thermal environments without degradation/dissolution. These naphthalene- and perylene-based pPIs utilise carbonyl groups within the structure to effectively conjugate with the aromatic rings as well as ionically bonding to Li ions. The aromatic carbonyl-derived pPIs show high capacity and high cycling stabilities, with those incorporating the perylene linker L5 giving the best electrochemical performance, around 74% retention of discharge capacity (57.9 mA h g−1) after 65 cycles. A further approach is detailed by Lv et al.73 utilizing B3 and L4 to form cathodes that are able to exhibit intramolecular charge transfer with lithium anodes to form solar-to-electrochemical energy storage and conversion devices. Finally, Zhao et al.77 reported atomic-layer modification of pPI COFs used as a cathode material for LIBs. They used a mechanical exfoliation method to create atomic-layered or nano-sheet pPIs (thickness ca. 2.6 nm) to improve the electrochemical performance (with respect to the bulk material). The dual active site modified atomic-layered pPIs shows initial capacity of 110 mA h g−1 (unmodified pPI 25 mA h g−1) with 87.3% retention after 500 cycles.
Li et al.36 reported polycondensation of melamine (B1) with the benzene (L3) and naphthalene (L4) linkers to generate pPIs for anode materials for Na ion batteries. When compared to graphitic carbon (used as anodic materials in both Na and Li-ion batteries),97 these pPI networks facilitate higher mobility of the larger sized Na ions (owing to increased pore size relative to graphitic carbon). These materials thus performed more effectively as anodes, possessing more reactive sites for electrode–electrolyte interactions and ion transport during the reversible sodiation/desodiation processes. Furthermore, the randomly arranged conjugated systems and larger conjugated units provided higher stability and conductivity than the graphitic carbon. The discharge capacity of PI-1 (using linker L4) and PI-2 (using linker L3) are 330.8 and 137.02 mA h g−1, respectively, at 100 mA g−1 after 20 cycles, when compared with the discharge capacity of graphitic carbon at 31 mA h g−1 (see Fig. 6). The higher redox activity and conductivity of PI-1 can be attributed to the higher electron affinity values and the ionisation potential and lower HOMO–LUMO gap as compared to PI-2 (L3 linker). Effectively, PI-1 is more conjugated than PI-2. The long-term stability of PI-1 and PI-2 are shown in Fig. 6, with minimal capacity degeneration (ca. 10% for PI-1) even after 1000 cycles.
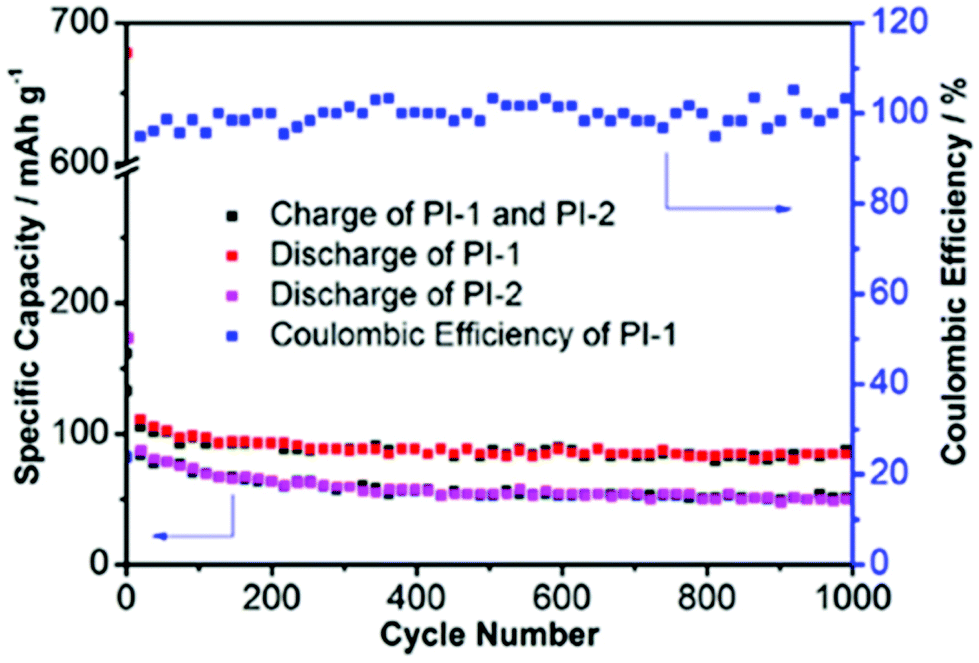 | ||
| Fig. 6 Long-term cycling performance of PI-1 (B1 and L4) and PI-2 (B1 and L3) at 5 A g−1, image reproduced with permission.36 Copyright Elsevier 2016. | ||
Roy et al.23 condensed dicarboxylic acids M1–3 (containing diimides) with tetra-amines C5–6 to form polybenzimidazole rings within pPI networks for use as electrode materials for energy storage applications. The incorporation of the polybenzimidazole units (present in M1–3) in the pPIs provides dynamic dipolar interactions between the electrolyte cations (1 M H3PO4) and the pPI pore walls, facilitating proton storage on the pore walls during the charge–discharge process. In addition, the extended π-conjugation and presence of heteroatoms throughout the pPI networks provide conductivity and enhance the redox activity of the network. The specific capacitance of networks formed from the tetraamine core C4 and linker L3 (BIBDZ), L4 (NIBDZ) and L5 (PIBDZ) were 88.4, 66.56 and 5.65 F g−1, respectively, at 5 A g−1. The higher capacitance value of L3 was attributed to the higher surface area (accommodating larger number of interactions between the electrode–electrolyte interface, assisting EDLC) when compared to L4 and L5, overriding the increased units available for π-conjugation of the other pPIs. Most recently, Royuela et al.76 reported a crystalline pPI NDI-COF (synthesised from L4 and B10), as an electrocatalyst in the oxygen reduction reaction (ORR). The majority of known ORR catalysts rely on containing metals within their structure, or pyrolysis or addition of conductive materials to achieve conductivity.98–100 The NDI-COF for ORR represents one of the first metal-free catalysts in this field that doesn't rely upon pyrolysis or additives. Owing to the increased electroactive area of the NDI-COF vs. the bare glassy carbon electrode, the capacitance current increases upon addition of the pPI, as shown by cyclic voltammetry. It is noteworthy that pPI networks are largely unexplored in the field of electrocatalysis and electrocatalytic behaviour.
4. Applications
4.1. Gas storage and separation
The high surface areas, high stabilities preventing degradation (as discussed in the previous section), alongside their synthetic simplicity and tunability (including starting materials, chemical make-up, porosity), make pPIs attractive candidates for gas capture in harsh environments. The literature focuses on CO2, H2, C6H6, CH4 and CH3OH (water is considered in a separate section in this review, see section 4.1.2), and are discussed below:The major factors responsible for high CO2 uptakes in pPIs are not (solely) reliant on high specific surface areas; for example, the pPIs synthesised by Rao et al.38 possess a surface area of 2213 m2 g−1 (Td-PPI, synthesised from C1 and L5) and CO2 uptake of 31 wt%, while Tr-PPI, synthesised from B3 and L5, has as surface are of 400 m2 g−1 and uptake of 45 wt% (both measured at 195 K and 1 bar). Direct comparison of the reported pPIs surface areas with CO2 uptake efficiency is challenging, as the conditions for CO2 uptake vary greatly between each experiment with little standardisation evident across the studies surveyed in this review. For example, the pPIs synthesised by Roy et al.23 (NIBDZ from C6 and the dianhydride M2) has a low surface area (118 m2 g−1) but fair CO2 uptake (14.56 wt%). In contrast, the pPIs synthesised by Li et al.19 has far higher surface areas, by a factor of up to 10 (e.g. MPI-1, 1454 m2 g−1 synthesised from C1 and L3) but this significant increase is not reflected in an increase in CO2 uptake, with similar values of 10–16 wt% CO2 uptake. However NIBDZ measurements were recorded at a far lower temperature (195 K) than that of MPI-1 (273 K), highlighting the discrepancies mentioned previously.19
More important than high surface areas are pPIs with high specific surface areas that contain high concentrations of micropores (<2 nm) or ultramicropores (<0.7 nm), and ideally without high mesopore (2–50 nm) volumes that will absorb competitive gas species and potentially block the pores.101 The smaller volumes are more compatible with the kinetic diameter of CO2. As an example, Luo et al.45 synthesised PI1–3 with mesoporous volumes and poorer CO2 uptakes (7.3 wt% for 660 m2 g−1 surface area), whereas pPIs with similar surface areas (e.g. Shi et al.,52 PPBPI-PENA-CR, 633 m2 g−1) but with higher overall pore volume and microporous volume exhibited higher CO2 uptake (10.43 wt%). A further example is found in the linear based PIs from L7 and A6 by Wu et al.55 Once formed and carbonised with ZIF aerogels the composites formed structures with pore sizes matching that of the kinetic diameter of CO2, giving an increased CO2 capture capacity of 9.81 wt% at 298 K when compared to non-carbonised, though overall still low (however these linear polymers and the effect of ZIF on CO2 uptakes are not investigated individually in this paper). It is important to note that if the pPIs have only ultramicropores, the CO2 present may block the pores and so stop further uptake of CO2.101
The isosteric heat of adsorption (Qst) provides a general measure of the interactions of gases and porous structures, measuring the heat produced when a gaseous molecule is physiosorbed onto the polymeric surface. Higher Qst means better interactions and higher absorbance values, though values over 50 kJ mol−1 typically indicate chemisorption.102,103 Specifically, the interaction of CO2 and the pore walls in CO2-pPI gas uptake studies is of interest here (see Table 4). The strength of the interactions can be tuned in pPIs via chemical modification of the starting monomers. As an example, NPI-3 (B10 and L4) synthesised by Li et al.34 has similar CO2 uptake to NPI-1 (C1 and L4) as it has similar Qst, despite having far lower surface area (see Table 3). A method of affecting the heat of absorption, as a gauge of the strength of CO2 interactions with the pore walls, is found by the incorporation of heteroatoms or polar groups into the pPIs. Incorporation of heteroatoms and polar groups affects the interactions between the CO2 and the pore wall via dipole–quadruple interactions, H-bonding or Lewis acid–base interactions. As an example, the pPI Tr-PPI (B3 triamine) synthesised by Rao et al.38 has a higher N content per repeat unit than the Td-PPI (C1 tetraamine) and therefore, despite having a far lower surface area, has a higher CO2 uptake of 45 wt% vs. 31 wt% (please note: both these measurements were performed at 195 K and 1 bar).
| Name in paper | H2 (wt%) | H2O (wt%) | CH4 (wt%) | C6H6 (wt%) | C6H12 (wt%) | Ref. |
|---|---|---|---|---|---|---|
| NPI-1 | — | 14.1iv | 1.35iii | 90.5iv | 58.1iv | 34 |
| NPI-2 | — | 9.5iv | 0.78iii | 41.5iv | 17.9iv | 34 |
| NPI-3 | — | 10.2iv | 1.07iii | 59.9iv | 37.4iv | 34 |
| MPI-1 | — | 16.7iv | — | 119.8iv | 54.1iv | 19 |
| MPI-2 | — | 9.9iv | — | 76.6iv | 44.8iv | 19 |
| MPI-3 | — | 9.6iv | — | 54.9iv | 41.5iv | 19 |
| PI-1 | 0.66i | — | — | — | — | 45 |
| PI-2 | 0.23 | — | — | — | — | 45 |
| PI-3 | 0.58 | — | — | — | — | 45 |
| BIBDZ | 0.75i* | — | 0.79iv* | — | — | 23 |
| PPBPI-1-Cr | — | — | 1iii* | — | — | 18 |
| PPBPI-2-CR | — | — | 1iii* | — | — | 18 |
| PPBPI-H-CR | — | — | 0.62iii* | — | — | 22 |
| PPBPI_Fe-CR | — | — | 0.24 | — | — | 22 |
| PPBPI-Mn-CR | — | — | 0.37 | — | — | 22 |
| PPBPI-PA-CR | — | NA | 0.6iii* | — | — | 52 |
| PPBPI-PEPA-CR | — | NA | 1.1 | — | — | 52 |
| PPBPI-PENA-CR | — | 19.27iv* | 1.1 | — | — | 52 |
| PI-1 | 0.7i* | — | 0.5iii* | — | — | 70 |
| PI-0%6FAPB | — | 383.8 | — | — | — | 79 |
| PI-25%6FAPB | — | 12.5 | — | — | — | 79 |
| PI-37.5%6FAPB | — | 9.8 | — | — | — | 79 |
| PI-50%6FAPB | — | 9.5 | — | — | — | 79 |
| MPI-6FA | — | 8.5iv | 1.42iv* | 80.9iv | 44iv | 48 |
| API-6FA | — | — | 1.07 | — | — | 48 |
| MPI-BTA | — | 10.4 | 1.07 | 72.8 | 40.4 | 48 |
| MPI-BPA | — | 13.3 | 0.78 | 104.9 | 60.2 | 48 |
| PI | 1.217i | 30iv | — | 99.2iv | 59.7iv | 85 |
| PI-1 | 3.2i | — | — | — | — | 81 |
| PI-2 | 2.6 | — | — | — | — | 81 |
| sPI-A-H | — | — | 0.62 | — | — | 67 |
| sPI_M-H | — | — | 0.6 | — | — | 67 |
| sPI-A-B | — | — | 0.56 | — | — | 67 |
| sPI-m-B | — | — | 0.49 | — | — | 67 |
| MPI | — | — | 0.92iii | — | — | 69 |
| MPI-S | — | — | 0.59 | — | — | 69 |
| MPI-Ag | — | — | 0.53 | — | — | 69 |
It is noted that pPIs synthesised by Hossain et al.46 from A5 and L12 and crosslinked with PEG units are not examined in regard to surface area or CO2 uptake, but showed promise in CO2-selective permeability.
| Name in paper | C2H6 (wt%) | C2H4 (wt%) | C3H8 (wt%) | C3H6 (wt%) | C4H10 (wt%) | C4H8 (wt%) | C4H6 (wt%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| sPI-A-H | 4.77iv | — | 8.8iv | 11.25iv | 18.27iv | 19.82iv | 25.05iv | 67 |
| sPI_M-H | 4.47iv | — | 8.44iv | 10.50iv | 17.80iv | 19.60iv | 25.86iv | 67 |
| sPI-A-B | 3.99iv | — | 8.05iv | 9.61iv | 14.50iv | 14.95iv | 19.65iv | 67 |
| sPI-m-B | 3.72iv | — | 7.61iv | 9.70iv | 12.52iv | 12.99iv | 18.30iv | 67 |
| MPI | 4.02iii | 4.35iii | — | — | — | — | — | 69 |
| MPI-S | 4.16iii | 4.68iii | — | — | — | — | — | 69 |
| MPI-Ag | 4.17iii | 4.96iii | — | — | — | — | — | 69 |
| PAF-110 | — | 4.83iii | — | — | — | — | — | 71 |
The efficient absorbance and storage of hydrogen gas is important for future fuel applications and a move to net zero emissions.104–106 The pPIs reported in Table 4 show hydrogen gas uptake of up to 3 wt% at 77 K and 1 bar, which is low when compared with the published data from high performing MOFs (e.g. Furukawa et al.'s MOF-210, 17.6 wt% at 77 K and 80 bar, although these frameworks rely on potentially scarce metal centres, e.g. zinc).107–109 With respect to pPIs, the best uptake was demonstrated by Wang et al.81 with the tetrahedral core C1 and dianhydrides L3 (PI-1) and L4 (PI-2). These two pPIs possess large surface areas (1407 m2 g−1 and 732 m2 g−1, respectively) with a higher ultramicroporous pore volume. These ultramicropores are compatible in size with the kinetic diameter of hydrogen (of 0.29 nm), which facilitates increased hydrogen uptake. The authors found that increased π-electron delocalisation groups present in the aromatic linker L4 (when compared to L3) aided Qst values at a range of hydrogen wt% uptakes (0.5–1 wt%) and hence overall hydrogen uptake. Despite the importance of H2 uptake and storage, this remains a largely unexplored area of application for pPIs.
Water sorption and harvesting will be an important future application for future proofing water resources, especially in arid environments. The crosslinked conjugated pPIs synthesised by Qiao et al.79 demonstrate the highest water sorption at 383.8 wt% (Table 4). This high uptake is attributed to the synthetic conditions employed, forming aerogels from A6, A14 and L7 owing to the slow reactions at ambient conditions, and slow introduction of crosslinking agent B8. However, the exact mechanism of water uptake was not fully explored in this study, leading to interesting avenues for research into future water-harvesting applications of pPIs, potentially fully tuneable by controlling aerogel formation (in addition to controlling chemical content).
High water uptake in other studies are attributed to either the introduction of heteroatoms, especially oxygen-containing linkers such as ester A6 and ketone L13 (see Qiao et al.79 and Li et al.48), or a decrease in (hydrophobic) aromaticity of the formed pPI by Shen et al.85 Those pPIs synthesised from the hydrophobic monomers L4 and L5 show low water uptakes.34,48,52 In the case of the materials with the largest water uptake,79 the introduction of the hydrophobic CF3-containing monomer A14 into the formed pPI aerogels leads to dramatically decreased water absorption capabilities (from 383.8 to 9.5 wt% when the CF3-containing monomer A14 content is increased to 50 mol%).
Volatile organic compound (VOC) sorption by pPIs can be separated into two specific focus areas: cyclic (benzene and cyclohexane) and aliphatic hydrocarbon adsorption. In the case of the cyclic hydrocarbons, Li et al.34,48,86 synthesised several pPIs based on naphthalene L4 and pyromellitic (a single benzene ring linker) L3 anhydrides for successful absorption of cyclic VOCs. With respect to benzene absorption, it was found that the more aromatic the skeleton the higher the benzene absorption (owing to π-interactions). Differences between the individual pPIs in benzene uptake were attributed to differences in surface areas, with lower surface areas leading to lower benzene uptake.85 The same pPIs were also used for cyclohexane sorption, though absorbing significantly less (ca. 40% lower uptake when compared with benzene) due to the lack of π-interactions.34,85,86 The increased absorption of cyclohexane by the pPI synthesised by Shen et al.85 from C4 and L3 was due to the cycloaliphatic core used – maximizing aliphatic–aliphatic interactions.
For the case of aliphatic hydrocarbon uptake, methane absorption was reported for a large number of papers (Table 5); however, the recorded absorbed weight percentages were never higher than 1.42 wt% (Li et al.48). This low absorption is theorised to be a result of poor interactions with the pPIs and incompatible kinetic diameters of the CH4 (0.38 nm) and pPI pore volumes.18,22,23,34,48,50,67,69,70 In regards to absorbance of other aliphatic VOCs (alkanes and alkenes), this review only found two papers by Yan et al.69,110 that addressed this specific application: a Ag-doped pPI MPI-Ag (from C1 and L3) and its four non-doped pPI counterparts sPI-A-H (C1 and L1), sPI-M-H (C4 and L1), sPI-A-B (C1 and L2) and sPI-M-B (C4 and L2). Interestingly, the pPIs reported were predominantly non-conjugated, which aided interaction with the aliphatic VOCs and their sorption. In parallel to cyclic VOC (benzene and cyclohexane) sorption studies, the pPIs MPI, MPI-S, and MPI-Ag (from C1 and L3) were able to adsorb unsaturated aliphatic VOCs when Ag was present in the pPI owing to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and Ag+ d-orbital overlap interactions (π-Ag+). In contrast, the pPI without Ag+ units were better suited to adsorb saturated aliphatic VOCs, although the reasons behind this observation were not explored in the study. Further studies by Jiang et al.71 show selective adsorption of acetylene from ethylene/acetylene mixtures due to Qst values clearly demonstrating a greater affinity of the material for acetylene (up to 9.0 wt% uptake).
C bond and Ag+ d-orbital overlap interactions (π-Ag+). In contrast, the pPI without Ag+ units were better suited to adsorb saturated aliphatic VOCs, although the reasons behind this observation were not explored in the study. Further studies by Jiang et al.71 show selective adsorption of acetylene from ethylene/acetylene mixtures due to Qst values clearly demonstrating a greater affinity of the material for acetylene (up to 9.0 wt% uptake).
We found several reports of nitrogen sorption, rather than just related to surface area calculations. However, in the majority of cases the level of N2 gas absorbed was extremely low: in the cases where these values are calculated and reported, or from our own calculations taken from isotherms provided, the values never exceeded 1 wt%, with the majority below 0.2 wt%.
4.2. Heterogeneous catalysis
Porous organic polymers play an important role in modern catalytic applications.111,112 In this context, pPI networks can be applied either by incorporating the active site within the polymeric structure (as pendant group or incorporated into one of the monomers), or by directly exploiting the activity and properties of conjugated pPIs. Nevertheless, application of pPI networks in catalysis has been, thus far, limited to a few examples as discussed below.In a first application of polyimides in catalysis, Shultz et al.21 prepared a metalloporphyrin ring functionalised with aromatic moieties (L20), two of which bore anhydride functionalities that could be used for the condensation reaction with tetramine C1. This reaction generated a polyimide network that was subsequently metalated at the porphyrin rings with iron(II) or manganese(II). The synthesised polyimide performed as recyclable catalyst for the epoxidation of styrene using iodosylbenzene as the oxidant; however, the recyclability was only partially effective as most of the catalytic activity was lost after three reaction cycles. Considering the excellent catalytic activity of Lewis acidic centres in metalloporphyrins for the cycloaddition of CO2 to epoxides,113,114 it is expected that appropriately designed and constructed pPI catalysts would perform well in this reaction;115,116 to the best of our knowledge, these promising materials have not yet been explored for this specific application.
A different catalytic application was reported by Chu et al.27 in the search of organic photocatalysts for hydrogen generation. To this aim, a crystalline pPI network was fabricated from the thermal reaction of melamine (B1) and pyromellitic dianhydride (L3) at 325 °C. The good crystallinity of the polymer was attributed to the solvent-free, high-temperature synthesis that led to the initial precipitation of crystals of oligomeric pPI. Whereas the individual monomers were colourless, the produced pPI network displayed a yellow color due to the extended π-conjugation. The pPI displayed attractive optoelectronic properties with a bandgap of 2.7 eV and a spatially separated HOMO (on the melamine moiety) and LUMO (on the pyromellitic moiety), potentially favouring charge separation during photocatalytic processes. The performance of the catalyst in the H2 evolution reaction (HER) was tested in the presence of methanol as sacrificial reagent and using Pt as co-catalyst under visible light irradiation; 70 mmol of H2 was produced after 10 h of irradiation. The authors compared the photocatalytic activity of the pPI with that of graphitic carbon nitride (g-C3N4) finding that, the two materials displayed similar photocatalytic performance despite the pPI possessing half the surface area of g-C3N4. Interestingly, the crystallinity of the pPI was found to play an important role: in a control experiment with a non-crystalline analogue of the pPI no photocatalytic activity was detected. In a subsequent study Chu et al.26 attempted to achieve bandgap modulation and optimize catalytic activity for the pPIs by varying the degree of polymerisation by using different temperatures (in the range from 250–350 °C) during syntheses using the same starting monomers B1 and L3 as in the previous example. The increase of the polymerisation temperature above 250 °C had a clear positive effect on crystallinity and on the degree of polymerisation; the UV-Vis optical absorption edge was progressively bathochromically shifted by increasing the polymerisation temperature, with the bandgaps decreasing from 3.39 eV (polymer prepared at 250 °C) to a more application-relevant value of 2.56 eV for the polymer prepared at 350 °C. The latter effect was attributed to achieving stronger orbital overlap by increasing the degree of polymerisation. DFT calculations supported this observation as the bandgap of oligomeric pPIs was found to decrease with chain length.117 When pPIs produced at different temperatures were tested in hydrogen generation, it was found that the materials produced at intermediate temperatures performed better. Specifically, the material produced at 300 °C was the best catalyst under full arc light whereas that produced at 325 °C was the best photocatalyst under visible light irradiation. This effect was attributed to the balance of several factors contributing to or affecting the photocatalytic activity. Indeed, despite a higher degree of polymerisation, the pPI prepared at the highest temperatures suffered from strong particle aggregation (with consequent decrease of surface area) and decreased redox power. Overall, this investigation highlighted a promising route worthy of further exploration, showing that the reaction conditions, and thus the degree of polymerisation of pPIs, can be used to engineer and modulate the bandgap of polymeric photocatalysts.
Kim et al.32 synthesised pPI networks from the triamine B10, anhydride L3 and the mono amine D1, (yielding MPI-Phen), showing catalytic activity in the Suzuki reactions. These pPI networks were synthesised both on and without a melamine sponge support, allowing ease of handling under catalytic conditions, placing in or removal from reaction vessels, whilst still allowing access to high surface areas (723 m2 g−1 without the sponge and 524 m2 g−1 on the sponge support). The catalytic activity of the networks was observed upon coordination of Pd(II) ions to the mono amine D1, and activity evaluated for the Suzuki coupling reaction between bromobenzene and phenylboronic acid in toluene (and other solvents) at 95 °C, to give a maximum yield of 83.3%. The recycling results showed a slight decrease in yields, which could be attributed to the leaching of Pd(II) ions from the network. Zhu et al.72 reported porphyrin-based pPI networks synthesised from dianhydride L3 and L4 with tetraamine porphyrin C9 monomer (PPPP-1 and PPPP-2, respectively). The major advantage of porphyrin-based pPIs in heterogeneous catalysis applications is found in the large size of the porphyrin linker (∼2 nm), creating large pores inside the framework. Furthermore, catalytically active sites can be modified by binding various metal ions to the porphyrin linker (strong π-metal orbital coupling also enhances the heterogeneous binding of the ions). The authors also loaded the porphyrin-based pPI networks with Pd nanoparticles (to form Pd@PPPP-1 and Pd@PPPP-2 from PPPP-1 and PPPP-2 respectively) to catalyse the Suzuki–Miyaura coupling reaction. Pd@PPPP-1 and Pd@PPPP-2 were utilised to catalyse substituted phenylboronic acid and aryl halides to form C–C bonds with high catalytic activity and product yield greater than 90%. Furthermore, the pPI catalytic networks were recovered and reused 5 times without any loss of catalytic activity of Pd@PPPP-1 and Pd@PPPP-2.
Rangel Rangel et al.54 synthesised 2 different pPI networks with triamine B10 and tetraamine C1 with dianhydride L3, respectively. The networks were nitrated with HNO3/H2SO4 and subsequently reduced with SnCl2 to yield amine-decorated aromatic cores (B10 or C1). These modified networks were then reacted with picolinaldehyde to yield iminopyridine ligands for coordination with palladium (Fig. 7). It is noteworthy that these modifications resulted in a significant decrease in thermal stability of the final network structures (as discussed in section 3.1). The catalytic activity was monitored using the Suzuki coupling reaction of halobenzenes with various aryl boronic acids; when compared to the palladium ligand only (i.e., not bound to the pPI networks) the pPI was found to yield on average twice the efficiency (calculated from GC and GCMS of the Suzuki coupling reaction). The increase in efficiency is related to the hydrophobicity of the pPI and the products from the coupling, as well as increased surface area for contact. Furthermore, the pPIs’ excellent catalytic activity in aqueous media provide very useful greener alternatives to other heterogeneous palladium catalyst systems.
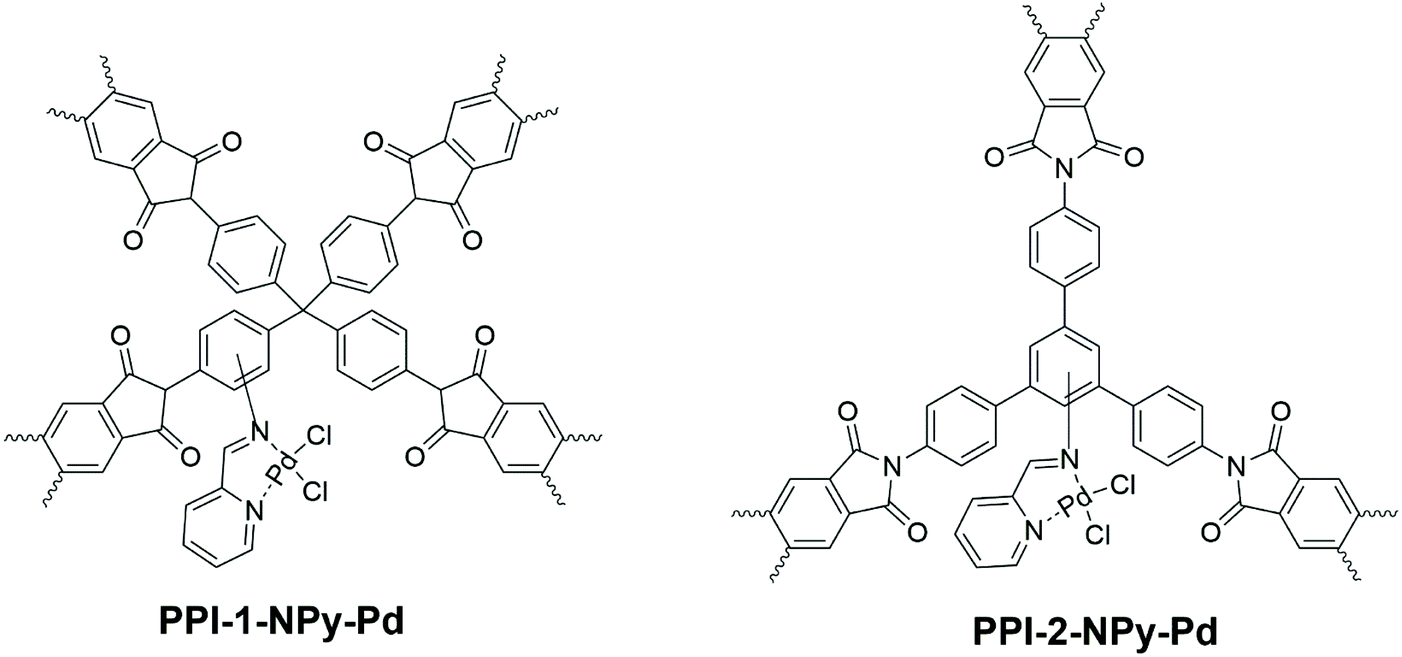 | ||
| Fig. 7 Schematic representation of heterogeneous Pd catalysts based on porous polyimide networks from Rangel Rangel et al.54 | ||
4.3. Other applications
Four further broad areas of application were found besides those listed above and are detailed below:Wang et al.28 used a pPI synthesised from B1 and L3 to investigate the removal of organic pollutants (the dye methyl orange and the antibiotic tetracycline) from aqueous solutions. Removal was monitored by UV/Vis spectroscopy and further investigated with IR spectroscopy to show how the amino groups of the pPI could interact ionically with the sulfonic groups of methyl orange, leading to adsorption. These pPIs absorbed methyl orange and tetracycline at 555.6 and 122.2 mg g−1, respectively, at 298 K from aqueous solutions.
A further good example of the potential of crosslinked pPIs for the removal of undesired species from aqueous systems is found in the study by Simón-Herrero et al.29 Here linear polyimides from L7 and A6 with hydrophobic properties were used to separate oil from water (please note: the crosslinked pPI formed via addition of B3 was not studied in this investigation, despite the potential for separation enhancement). In contrast, Qiao et al.79 reported on high surface area CF3-functionalised pPI-based aerogels with high water repulsion for the adsorption of organic species from aqueous systems. The pPI aerogels removed both chloroform and toluene from aqueous mixtures. However, the investigation only reported empirical observations, with no quantification of the performance of the materials presented. Although a range of other CF3-containing water-repellent pPIs have been synthesised,16,18,22,50,52,60,61 they have not been exploited for application in the area of solvent removal from water.
With respect to metal ion removal from aqueous systems, mention is only made of one study related to the porphyrin-containing materials prepared by Shi et al.18,22,50,52 Here iron and manganese binding capabilities were demonstrated but not explored in regards to species removal. Binding studies focused on either blocking or increasing pore sizes and surface areas rather than the removal of metal ions from aqueous systems, leaving this aspect wide open for further exploration.
Further sensing of heavy metal ions by fluorescence quenching was demonstrated by Xiao et al.,56 specifically sensing Cr3+ ions. It was theorised that the strong binding of Cr3+ ions to the triazine nitrogen atoms (B1), as well as oxygen atoms from the imide bonds within the pPI framework, led to a reduction in antenna efficiency and hence an increase in quenching of the luminescence.
Beyond heavy metal sensing, Zhang et al.74 demonstrated a porphyrin (C7) and perylenetetracarboxylic dianhydride (L5) derived pPI for 2,4,6-trinitrophenol (TNP) detection, an explosive with higher efficacy than its counterpart 2,4,6-trinitrotoluene (TNT), via fluorescence dampening. Interestingly, the pPI shows selectivity for TNP detection over other nitroaromatics, which was tentatively attributed to a reduction in π–π stacking (although not explored in any detail).
Further applications of pPIs as coatings can be found in the work by Qiao et al.,79 who prepared fluorinated pPIs as water-repellent coatings. They found that increasing the content of the CF3-functionalised monomers of the pPI from 0 to 50 wt% increased the water contact angle from 87.7° to 112.1°, allowing optimisation of water repulsion.
Finally, it is noted that the majority of publications do not investigate the chemical stability of pPIs in great detail. One example of stability investigations can be found in van der Jagt et al.,75 submerging pPIs PIC and PID (see Table 1) for 14 days in organic (1 M NaClO4 in ethylene carbonate (EC)/dimethylcarbonate (DMC)) and aqueous (1 M Na2SO4 in neutral water) electrolytes, both of which are representative electrolytes used in energy storage application. Both crystalline pPIs (PIC and PID) were found to have identical powder XRD spectra before and after, used as a proof of stability of these highly crosslinked materials in these application-relevant environments.
5. Future outlook
One common theme throughout the studies cited in this review was centred around the ability and opportunities available to control the properties and the function of the prepared pPIs. We have endeavoured to highlight aspects of deliberate design where encountered, and attempted to show the influence of careful choice of starting materials and their functionality, and synthesis conditions on the chosen areas of applications. There are however a number of challenges that the broader field of porous functional materials, and certainly also pPIs, face that are connected to fine control of properties and function.Although pPIs can be synthesised solvent free to avoid issues with insolubility and the incompatibility of the growing and crosslinked polymer chains with reaction solvents, Chen et al.120,121 have recently explored tuning Hansen solubility parameters (HSPs) in the preparation of CMPs through the addition of simple inorganic salts to reaction mixture. They showed that this approach, the so-called Bristol–Xi'an Jiaotong (BXJ) approach, provides a novel route to accurately match the HSPs of the growing polymer construct with that of the reaction solvent, thus exerting fine control over the properties and functionality (as expressed through increased CO2 uptake) in the obtained porous materials. Significant opportunities therefore exist, especially when keeping Tables 1 and 3 in mind in terms of scope of materials, to apply the BXJ approach for the production optimized and fully tuned functional pPI networks.
In terms of application areas as potential future growth areas, especially those with an environmental application is of global interest. Given the current world shortages in water (for safe human consumption, but also for wider agricultural use), the use of pPIs as water harvesting motifs presents a significant opportunity. Although pPIs synthesised by Wu et al.82 demonstrate large water uptake (over 300 wt%), and while many other systems were reported as having the capability (Table 4), water harvesting remains largely unexplored in the majority of papers where gas absorption is presented as an application (Table 3). It is expected that significant increases in performance would be possible with focused efforts in this area of importance, thus leading to significant global socio-economic impact on local communities where water is a precious commodity.
To the best of our knowledge biocompatibility of pPIs has not been reported in the literature. This omission is especially worthy of attention when one considers the potential of these networks as controlled drug delivery or drug removal motifs (as discussed in sections 5.1 and 5.2), as potential scaffolds for cell culture and growth (for biocompatible implants), as well as the possibility for in vitro drug detection, section 5.3.
Finally, the excellent surface areas reported, high wt% of CO2 capture, and tuneable conductivity of the reported pPIs make them excellent candidates for electro- and photo-catalytic CO2 capture and conversion. Other POFs have been utilised successfully in this field.101,122–127 Recent advances show that the photocatalytic CO2 conversion can be carried out in porous polymers even in the absence of metal atoms;112,128,129 yet this area has not been explored at all with pPIs to date, and presents a significant opportunity to address global challenges presented by continued and growing human activity and influence on the environment. In particular, recent focus has been placed on the direct conversion of CO2 to useful chemicals (rather than storage).130,131 For instance, robust and moisture-insensitive pPIs incorporating functional groups such as hydrogen-bond donors,132,133 nucleophilic species such as pyridines,134,135 or Lewis acidic metal centres136,137 could serve as components of catalytic systems for the direct conversion of CO2 and epoxides to cyclic carbonates. Further opportunities in energy conversion and storage (as touched on in section 4.2 focusing on photocatalysis) are also available for further significant effort and exploitation.
In conclusion, we believe that pPIs, as a class of materials, present a range of exciting possibilities to contribute to solutions for a range of global challenges. The hope is that this review will provide readers and researchers with a range of opportunities for future exploration, thus contributing to solutions for the significant problems that society currently faces.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
B.B.N acknowledges support from the National Overseas Scholarship for ST Students, Government of India. N. Y. and V. D'E. acknowledge support from Vidyasirimedhi Institute of Science and Technology (VISTEC), Thailand. B. C. B. and C. F. J. F. acknowledge the EPSRC (EP/T020792/1) for support. The authors thank Marcos Villeda Hernandez for graphic design and images.Notes and references
- M. T. Borget and R. R. Renshaw, J. Am. Chem. Soc., 1908, 30, 1135–1144 CrossRef.
- C. E. Sroog, Prog. Polym. Sci., 1991, 16, 561–694 CrossRef CAS.
- W. M. Edwards and I. M. Robinson, United States Patent Office, 14 June 1955, 2710853.
- L. T. C. Lee, E. M. Pearce and S. S. Hirsch, J. Polym. Sci., 1971, 9, 3169–3174 CrossRef CAS.
- Y. Zhuang, J. G. Seong and Y. M. Lee, Prog. Polym. Sci., 2019, 92, 35–88 CrossRef CAS.
- D.-J. Liaw, K.-L. Wang, Y.-C. Huang, K.-R. Lee, J.-Y. Lai and C.-S. Ha, Prog. Polym. Sci., 2012, 37, 907–974 CrossRef CAS.
- M. Hasegawa and K. Horie, Prog. Polym. Sci., 2001, 26, 259–335 CrossRef CAS.
- M. Ding, Prog. Polym. Sci., 2007, 32, 623–668 CrossRef CAS.
- Q. Fang, Z. Zhuang, S. Gu, R. B. Kaspar, J. Zheng, J. Wang, S. Qiu and Y. Yan, Nat. Commun., 2014, 5, 4503 CrossRef PubMed.
- M.-C. Fu, T. Higashihara and M. Ueda, Polym. J., 2017, 50, 57–76 CrossRef.
- M. Hasegawa, Polymers, 2017, 9, 520 CrossRef PubMed.
- L. Y. Jiang, Y. Wang, T.-S. Chung, X. Y. Qiao and J.-Y. Lai, Prog. Polym. Sci., 2009, 34, 1135–1160 CrossRef CAS.
- K. Vanherck, G. Koeckelberghs and I. F. J. Vankelecom, Prog. Polym. Sci., 2013, 38, 874–896 CrossRef CAS.
- E. P. Favvas, F. K. Katsaros, S. K. Papageorgiou, A. A. Sapalidis and A. C. Mitropoulos, React. Funct. Polym., 2017, 120, 104–130 CrossRef CAS.
- Y. Zhang, Z. Huang, B. Ruan, X. Zhang, T. Jiang, N. Ma and F. C. Tsai, Macromol. Rapid Commun., 2020, 41, 2000402 CrossRef CAS PubMed.
- N. Song, T. Ma, T. Wang, K. Shi, Y. Tian, H. Yao, Y. Zhang and S. Guan, Microporous Mesoporous Mater., 2020, 293, 109809 CrossRef CAS.
- M. R. Liebl and J. Senker, Chem. Mater., 2013, 25, 970–980 CrossRef CAS.
- K. Shi, N. Song, Y. Zou, S. Zhu, H. Tan, Y. Tian, B. Zhang, H. Yao and S. Guan, Polymer, 2019, 169, 160–166 CrossRef CAS.
- E. Rangel Rangel, E. M. Maya, F. Sánchez, J. de Abajo and J. G. de la Campa, J. Membr. Sci., 2013, 447, 403–412 CrossRef CAS.
- J. Li, Y. Ding, N. Yu, Q. Gao, X. Fan, X. Wei, G. Zhang, Z. Ma and X. He, Carbon, 2020, 158, 45–54 CrossRef CAS.
- A. M. Shultz, O. K. Farha, J. T. Hupp and S. T. Nguyen, Chem. Sci., 2011, 2, 686–689 RSC.
- K. Shi, H. Yao, S. Zhang, Y. Wei, W. Xu, N. Song, S. Zhu, Y. Tian, Y. Zou and S. Guan, Ind. Eng. Chem. Res., 2019, 58, 14146–14153 CrossRef CAS.
- A. Roy, S. Mondal, A. Halder, A. Banerjee, D. Ghoshal, A. Paul and S. Malik, Eur. Polym. J., 2017, 93, 448–457 CrossRef CAS.
- Y. F. Lu, Y. M. Wang, X. D. Chen, M. H. Miao and D. H. Zhang, eXPRESS Polym. Lett., 2020, 14, 192–204 CrossRef CAS.
- T. Zhu, Q. Yu, L. Ding, T. Di, T. Zhao, T. Li and L. Li, Green Chem., 2019, 21, 2326–2333 RSC.
- S. Chu, Y. Wang, C. Wang, J. Yang and Z. Zou, Int. J. Hydrogen Energy, 2013, 38, 10768–10772 CrossRef CAS.
- S. Chu, Y. Wang, Y. Guo, P. Zhou, H. Yu, L. Luo, F. Kong and Z. Zou, J. Mater. Chem., 2012, 22, 15519–15521 RSC.
- Y. Wang, Q. Gao, Q. You, G. Liao, H. Xia and D. Wang, React. Funct. Polym., 2016, 103, 9–16 CrossRef CAS.
- C. Simón-Herrero, X.-Y. Chen, M. L. Ortiz, A. Romero, J. L. Valverde and L. Sánchez-Silva, J. Mater. Res. Technol., 2019, 8, 2638–2648 CrossRef.
- T. Wang, R. Xue, H. Chen, P. Shi, X. Lei, Y. Wei, H. Guo and W. Yang, New J. Chem., 2017, 41, 14272–14278 RSC.
- M. Hasegawa, R. Tokunaga, K. Hashimoto and J. Ishii, React. Funct. Polym., 2019, 139, 181–188 CrossRef CAS.
- J. G. Kim, J. Lee, J. Lee, S. I. Jo and J. Y. Chang, Macromol. Res., 2017, 25, 629–634 CrossRef CAS.
- T. Wang, H. Yao, N. Song, Y. Yang, K. Shi and S. Guan, Ind. Eng. Chem. Res., 2020, 59, 2953–2959 CrossRef CAS.
- G. Li and Z. Wang, J. Phys. Chem. C, 2013, 117, 24428–24437 CrossRef CAS.
- J. Wu, L. Xia, S. Zhang and X. Liu, Mater. Today Commun., 2020, 24, 101030 CrossRef CAS.
- Z. Li, J. Zhou, R. Xu, S. Liu, Y. Wang, P. Li, W. Wu and M. Wu, Chem. Eng. J., 2016, 287, 516–522 CrossRef CAS.
- H. Yao, N. Zhang, K. Shen, N. Song, K. Shi, S. Zhu, Y. Zhang and S. Guan, Polymer, 2017, 115, 176–183 CrossRef CAS.
- K. V. Rao, R. Haldar, C. Kulkarni, T. K. Maji and S. J. George, Chem. Mater., 2012, 24, 969–971 CrossRef CAS.
- K. V. Rao, R. Haldar, T. K. Maji and S. J. George, Polymer, 2014, 55, 1452–1458 CrossRef CAS.
- H. Yao, N. Zhang, N. Song, K. Shen, P. Huo, S. Zhu, Y. Zhang and S. Guan, Polym. Chem., 2017, 8, 1298–1305 RSC.
- C. Zhang, T.-L. Zhai, J.-J. Wang, Z. Wang, J.-M. Liu, B. Tan, X.-L. Yang and H.-B. Xu, Polymer, 2014, 55, 3642–3647 CrossRef CAS.
- M. E. Dose, M. Chwatko, I. Hubacek, N. A. Lynd, D. R. Paul and B. D. Freeman, Polymer, 2019, 161, 16–26 CrossRef CAS.
- H. Gao, B. Tian, H. Yang, A. R. Neale, M. A. Little, R. S. Sprick, L. J. Hardwick and A. I. Cooper, ChemSusChem, 2020, 13, 5571–5579 CrossRef CAS PubMed.
- Q. Fang, J. Wang, S. Gu, R. B. Kaspar, Z. Zhuang, J. Zheng, H. Guo, S. Qiu and Y. Yan, J. Am. Chem. Soc., 2015, 137, 8352–8355 CrossRef CAS PubMed.
- Y. Luo, B. Li, L. Liang and B. Tan, Chem. Commun., 2011, 47, 7704–7706 RSC.
- I. Hossain, A. Z. Al Munsur, O. Choi and T.-H. Kim, Sep. Purif. Technol., 2019, 224, 180–188 CrossRef CAS.
- J. Lee and J. Y. Chang, Chem. Commun., 2016, 52, 10419–10422 RSC.
- G. Li, B. Zhang, J. Yan and Z. Wang, J. Mater. Chem. A, 2016, 4, 11453–11461 RSC.
- Q. Li, R. Chen, Y. Guo, F. Lei, Z. Xu, H. Zhao and G. Liao, Polymers, 2020, 12, 88 CrossRef CAS PubMed.
- K. Shi, H. Yao, Y. Zou, Y. Wei, N. Song, S. Zhang, Y. Tian, S. Zhu, B. Zhang and S. Guan, Microporous Mesoporous Mater., 2019, 287, 246–253 CrossRef CAS.
- D. H. Wang and L.-S. Tan, ACS Macro Lett., 2019, 8, 546–552 CrossRef CAS.
- K. Shi, H. Yao, Y. Zou, Y. Wei, S. Zhang, Y. Song, N. Song, T. Wang, Y. Tian, S. Zhu and S. Guan, Microporous Mesoporous Mater., 2020, 292, 109739 CrossRef.
- L. Yu, L. Wang, L. Yu, D. Mu, L. Wang and J. Xi, J. Membr. Sci., 2019, 572, 119–127 CrossRef CAS.
- E. Rangel Rangel, E. M. Maya, F. Sánchez, J. G. de la Campa and M. Iglesias, Green Chem., 2015, 17, 466–473 RSC.
- T. Wu, J. Dong, K. De France, P. Zhang, X. Zhao and Q. Zhang, Chem. Eng. J., 2020, 395 CrossRef CAS.
- J.-D. Xiao, L.-G. Qiu, Y.-P. Yuan, X. Jiang, A.-J. Xie and Y.-H. Shen, Inorg. Chem. Commun., 2013, 29, 128–130 CrossRef CAS.
- C. Shen, Y. Bao and Z. Wang, Chem. Commun., 2013, 49, 3321 RSC.
- H. Zhou, H. Lei, J. Wang, S. Qi, G. Tian and D. Wu, Polymer, 2019, 162, 116–120 CrossRef CAS.
- C. Shen and Z. Wang, J. Phys. Chem. C, 2014, 118, 17585–17593 CrossRef CAS.
- N. Song, T. Wang, H. Yao, T. Ma, K. Shi, Y. Tian, Y. Zou, S. Zhu, Y. Zhang and S. Guan, Polym. Chem., 2019, 10, 4611–4620 RSC.
- N. Song, H. Yao, T. Ma, T. Wang, K. Shi, Y. Tian, Y. Zou, S. Zhu, Y. Zhang and S. Guan, Chem. Eng. J., 2019, 368, 618–626 CrossRef CAS.
- Y. Liao, J. Weber and C. F. J. Faul, Macromolecules, 2015, 48, 2064–2073 CrossRef CAS.
- J. Maschita, T. Banerjee, G. Savasci, F. Haase, C. Ochsenfeld and B. V. Lotsch, Angew. Chem., 2020, 59, 15750–15758 CrossRef CAS PubMed.
- Y. Tian, K. Shi, H. Yao, T. Wang, H. Liu, Y. Song, Y. Zhang and S. Guan, J. Phys. Chem. C, 2020, 124, 2872–2878 CrossRef CAS.
- Z. Wang, B. Zhang, H. Yu, L. Sun, C. Jiao and W. Liu, Chem. Commun., 2010, 46, 7730 RSC.
- Z. Wu, B. Han, C. Zhang, D. Zhu and Z. Yang, Polymer, 2019, 179, 121605 CrossRef CAS.
- J. Yan, B. Zhang and Z. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 26618–26627 CrossRef CAS PubMed.
- N. Song, T. Ma, T. Wang, Z. Li, H. Yao and S. Guan, J. Colloid Interface Sci., 2020, 573, 328–335 CrossRef CAS PubMed.
- J. Yan, B. Zhang, L. Guo and Z. Wang, J. Phys. Chem. C, 2019, 123, 575–583 CrossRef CAS.
- D. Tian, H.-Z. Zhang, D.-S. Zhang, Z. Chang, J. Han, X.-P. Gao and X.-H. Bu, RSC Adv., 2014, 4, 7506–7510 RSC.
- L. Jiang, Y. Tian, T. Sun, Y. Zhu, H. Ren, X. Zou, Y. Ma, K. R. Meihaus, J. R. Long and G. Zhu, J. Am. Chem. Soc., 2018, 140, 15724–15730 CrossRef CAS PubMed.
- W. Zhu, X. Wang, T. Li, R. Shen, S.-J. Hao, Y. Li, Q. Wang, Z. Li and Z.-G. Gu, Polym. Chem., 2018, 9, 1430–1438 RSC.
- J. Lv, Y. X. Tan, J. Xie, R. Yang, M. Yu, S. Sun, M. D. Li, D. Yuan and Y. Wang, Angew. Chem., Int. Ed., 2018, 57, 12716–12720 CrossRef CAS PubMed.
- C. Zhang, S. Zhang, Y. Yan, F. Xia, A. Huang and Y. Xian, ACS Appl. Mater. Interfaces, 2017, 9, 13415–13421 CrossRef CAS PubMed.
- R. van der Jagt, A. Vasileiadis, H. Veldhuizen, P. Shao, X. Feng, S. Ganapathy, N. C. Habisreutinger, M. A. van der Veen, C. Wang, M. Wagemaker, S. van der Zwaag and A. Nagai, Chem. Mater., 2021, 33, 818–833 CrossRef CAS PubMed.
- S. Royuela, E. Martinez-Perinan, M. P. Arrieta, J. I. Martinez, M. M. Ramos, F. Zamora, E. Lorenzo and J. L. Segura, Chem. Commun., 2020, 56, 1267–1270 RSC.
- G. Zhao, H. Li, Z. Gao, L. Xu, Z. Mei, S. Cai, T. Liu, X. Yang, H. Guo and X. Sun, Adv. Funct. Mater., 2021, 31, 2101019 CrossRef CAS.
- X. Zhang, X. Cui, C.-H. Lu, H. Li, Q. Zhang, C. He and Y. Yang, Chem. Eng. J., 2020, 401, 126031 CrossRef CAS.
- S. Qiao, S. Kang, Z. Hu, J. Yu, Y. Wang and J. Zhu, J. Porous Mater., 2019, 27, 237–247 CrossRef.
- Q. Fang, J. Wang, S. Gu, R. B. Kaspar, Z. Zhuang, J. Zheng, H. Guo, S. Qiu and Y. Yan, J. Am. Chem. Soc., 2015, 137, 8352–8355 CrossRef CAS PubMed.
- Z. Wang, B. Zhang, H. Yu, L. Sun, C. Jiao and W. Liu, Chem. Commun., 2010, 46, 7730–7732 RSC.
- Z. Wu, B. Han, C. Zhang, D. Zhu and Z. Yang, Polymer, 2019, 179, 121605 CrossRef CAS.
- J. Lee and J. Y. Chang, Chem. Commun., 2016, 52, 10419–10422 RSC.
- S. Liu, W. Chen and X. Zhou, J. Porous Mater., 2021, 28, 1155–1165 CrossRef CAS.
- C. Shen, Y. Bao and Z. Wang, Chem. Commun., 2013, 49, 3321–3323 RSC.
- G. Li and Z. Wang, Macromolecules, 2013, 46, 3058–3066 CrossRef CAS.
- J.-S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- F. Wang, X. Wu, X. Yuan, Z. Liu, Y. Zhang, L. Fu, Y. Zhu, Q. Zhou, Y. Wu and W. Huang, Chem. Soc. Rev., 2017, 46, 6816–6854 RSC.
- R. Heimbockel, F. Hoffmann and M. Froba, Phys. Chem. Chem. Phys., 2019, 21, 3122–3133 RSC.
- J.-S. M. Lee, T.-H. Wu, B. M. Alston, M. E. Briggs, T. Hasell, C.-C. Hu and A. I. Cooper, J. Mater. Chem. A, 2016, 4, 7665–7673 RSC.
- Y. Wang, Y. Song and Y. Xia, Chem. Soc. Rev., 2016, 45, 5925–5950 RSC.
- Y. Jiang and J. Liu, Energy Environ. Mater., 2019, 2, 30–37 CrossRef.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- H. Wu, Q. Meng, Q. Yang, M. Zhang, K. Lu and Z. Wei, Adv. Mater., 2015, 27, 6504–6510 CrossRef CAS PubMed.
- Z. Song, H. Zhan and Y. Zhou, Angew. Chem., Int. Ed., 2010, 49, 8444–8448 CrossRef CAS PubMed.
- X. Han, C. Chang, L. Yuan, T. Sun and J. Sun, Adv. Mater., 2007, 19, 1616–1621 CrossRef CAS.
- W. Luo, C. Bommier, Z. Jian, X. Li, R. Carter, S. Vail, Y. Lu, J. J. Lee and X. Ji, ACS Appl. Mater. Interfaces, 2015, 7, 2626–2631 CrossRef CAS PubMed.
- J. Guo, C. Y. Lin, Z. Xia and Z. Xiang, Angew. Chem., Int. Ed., 2018, 57, 12567–12572 CrossRef CAS PubMed.
- Q. Xu, Y. Tang, X. Zhang, Y. Oshima, Q. Chen and D. Jiang, Adv. Mater., 2018, 30, 1706330 CrossRef PubMed.
- D. Wu, Q. Xu, J. Qian, X. Li and Y. Sun, Chemistry, 2019, 25, 3105–3111 CAS.
- G. Singh, J. Lee, A. Karakoti, R. Bahadur, J. Yi, D. Zhao, K. AlBahily and A. Vinu, Chem. Soc. Rev., 2020, 49, 4360–4404 RSC.
- A. Nuhnen and C. Janiak, Dalton Trans., 2020, 49, 10295–10307 RSC.
- P. Atkins, J. de Paula and J. Keeler, Physical Chemistry, Oxford University Press, 2018 Search PubMed.
- P. P. Edwards, V. L. Kuznetsov, W. I. F. David and N. P. Brandon, Energy Policy, 2008, 36, 4356–4362 CrossRef.
- S. Sharma and S. K. Ghoshal, Renewable Sustainable Energy Rev., 2015, 43, 1151–1158 CrossRef CAS.
- I. P. Jain, Int. J. Hydrogen Energy, 2009, 34, 7368–7378 CrossRef CAS.
- M. P. Suh, H. J. Park, T. K. Prasad and D. W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed.
- S. S. Han, H. Furukawa, O. M. Yaghi and W. A. Goddard, J. Am. Chem. Soc., 2007, 130, 11580–11581 CrossRef PubMed.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- J. Yan, B. Zhang and Z. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 26618–26627 CrossRef CAS PubMed.
- P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS.
- T.-X. Wang, H.-P. Liang, D. A. Anito, X. Ding and B.-H. Han, J. Mater. Chem. A, 2020, 8, 7003–7034 RSC.
- Y. Chen, R. Luo, Q. Xu, W. Zhang, X. Zhou and H. Ji, ChemCatChem, 2017, 9, 767–773 CrossRef CAS.
- R. R. Shaikh, S. Pornpraprom and V. D’Elia, ACS Catal., 2018, 8, 419–450 CrossRef CAS.
- F. Della Monica and A. W. Kleij, Catal. Sci. Technol., 2020, 10, 3483–3501 RSC.
- V. Aomchad, À. Cristòfol, F. Della Monica, B. Limburg, V. D'Elia and A. W. Kleij, Green Chem., 2021, 23, 1077–1113 RSC.
- Z. Wei and C. F. J. Faul, Macromol. Rapid Commun., 2008, 29, 280–292 CrossRef CAS.
- F. Qu, G. Zhu, H. Lin, W. Zhang, J. Sun, S. Li and S. Qiu, J. Solid State Chem., 2006, 179, 2027–2035 CrossRef CAS.
- P. Horcajada, C. Serre, M. Vallet-Regí, M. Sebban, F. Taulelle and G. Férey, Angew. Chem., 2006, 118, 6120–6124 CrossRef.
- J. Chen, W. Yan, E. J. Townsend, J. Feng, L. Pan, V. Del Angel Hernandez and C. F. J. Faul, Angew. Chem., Int. Ed., 2019, 58, 11715–11719 CrossRef CAS PubMed.
- J. Chen, T. Qiu, W. Yan and C. F. J. Faul, J. Mater. Chem. A, 2020, 8, 22657–22665 RSC.
- P. Bhanja, A. Modak and A. Bhaumik, ChemCatChem, 2018, 11, 244–257 CrossRef.
- H. Liu, J. Chu, Z. Yin, X. Cai, L. Zhuang and H. Deng, Chem, 2018, 4, 1696–1709 CAS.
- M. Lu, M. Zhang, C. G. Liu, J. Liu, L. J. Shang, M. Wang, J. N. Chang, S. L. Li and Y. Q. Lan, Angew. Chem., 2021, 60, 4864–4871 CrossRef CAS PubMed.
- L. Majidi, A. Ahmadiparidari, N. Shan, S. N. Misal, K. Kumar, Z. Huang, S. Rastegar, Z. Hemmat, X. Zou, P. Zapol, J. Cabana, L. A. Curtiss and A. Salehi-Khojin, Adv. Mater., 2021, 33, 2004393 CrossRef CAS PubMed.
- R. Sharma, A. Bansal, C. N. Ramachandran and P. Mohanty, Chem. Commun., 2019, 55, 11607–11610 RSC.
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang, J. Zhang and J. Huang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS PubMed.
- Y. Fu, X. Zhu, L. Huang, X. Zhang, F. Zhang and W. Zhu, Appl. Catal., B, 2018, 239, 46–51 CrossRef CAS.
- K. Lei, D. Wang, L. Ye, M. Kou, Y. Deng, Z. Ma, L. Wang and Y. Kong, ChemSusChem, 2020, 13, 1725–1729 CrossRef CAS PubMed.
- J. W. Maina, J. M. Pringle, J. M. Razal, S. Nunes, L. Vega, F. Gallucci and L. F. Dumee, ChemSusChem, 2021, 14, 1805–1820 CrossRef CAS PubMed.
- O. Sodpiban, C. Phungpanya, S. Del Gobbo, S. Arayachukiat, T. Piromchart and V. D’Elia, Chem. Eng. J., 2021, 422, 129930 CrossRef CAS.
- P. Yingcharoen, W. Natongchai, A. Poater and V. D'Elia, Catal. Sci. Technol., 2020, 10, 5544–5558 RSC.
- C. J. Whiteoak, A. Nova, F. Maseras and A. W. Kleij, ChemSusChem, 2012, 5, 2032–2038 CrossRef CAS PubMed.
- S. Subramanian, J. Park, J. Byun, Y. Jung and C. T. Yavuz, ACS Appl. Mater. Interfaces, 2018, 10, 9478–9484 CrossRef CAS PubMed.
- W. Natongchai, J. A. Luque-Urrutia, C. Phungpanya, M. Solà, V. D'Elia, A. Poater and H. Zipse, Org. Chem. Front., 2021, 8, 613–627 RSC.
- J. Meléndez, M. North and R. Pasquale, Eur. J. Inorg. Chem., 2007, 2007, 3323–3326 CrossRef.
- O. Sodpiban, S. Del Gobbo, S. Barman, V. Aomchad, P. Kidkhunthod, S. Ould-Chikh, A. Poater, V. D'Elia and J.-M. Basset, Catal. Sci. Technol., 2019, 9, 6152–6165 RSC.
| This journal is © The Royal Society of Chemistry 2021 |