
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermoresponsive polymers as macromolecular coordination ligands: complexation-dependence of thermally induced aggregation in aqueous solution†
Maximilian Felix Toni
Meier
 a,
Franck
Thetiot
b,
Narsimhulu
Pittala
b,
Ingo
Lieberwirth
c,
Cleiton
Kunzler
a,
Smail
Triki
b and
Ulrich
Jonas
*a
a,
Franck
Thetiot
b,
Narsimhulu
Pittala
b,
Ingo
Lieberwirth
c,
Cleiton
Kunzler
a,
Smail
Triki
b and
Ulrich
Jonas
*a
aMacromolecular Chemistry, Department Chemistry and Biology, University of Siegen, Adolf-Reichwein-Strasse 2, 57076 Siegen, Germany. E-mail: jonas@chemie.uni-siegen.de
bUniversité de Brest (UBO), UMR CNRS 6521, 6 Avenue Le Gorgeu, C.S., 93837-29238 Brest CEDEX 3, France
cMax-Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: lieberw@mpip-mainz.mpg.de
First published on 13th August 2021
Abstract
We have designed novel macromolecular coordination ligands (MCLs) by conjugation of thermoresponsive polymers based on poly(N-isopropylacrylamide) (![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n around 3 to 25 kg mol−1) with 1,2,4-triazole coordination sites. These triazole units were integrated into two fundamentally different MCL architectures via reversible addition–fragmentation chain transfer polymerization following two synthetic strategies: (I) The customized chain transfer agent 1-{[3-(4H-1,2,4-triazol-4-yl)propyl]amino}-2-methyl-1-oxopropan-2-yl dodecyl carbonotrithioate (DMP-APTRZ) was employed for hemi-telechelic MCLs with a single triazole end group. (II) A tailored comonomer N-[3-(4H-1,2,4-triazol-4-yl)propyl]methacrylamide (APTRZMAAm) provides access to multidentate MCLs with a controllable number of triazole side groups along the polymer backbone. The thermally controlled variation of the MCL volume demand in aqueous solution was exploited for reversible aggregate formation upon Fe2+ complexation. Thermal response was studied via UV/Vis turbidity measurements, aggregate dimensions were determined via DLS, while the aggregate morphology was analyzed via customized TEM.
n around 3 to 25 kg mol−1) with 1,2,4-triazole coordination sites. These triazole units were integrated into two fundamentally different MCL architectures via reversible addition–fragmentation chain transfer polymerization following two synthetic strategies: (I) The customized chain transfer agent 1-{[3-(4H-1,2,4-triazol-4-yl)propyl]amino}-2-methyl-1-oxopropan-2-yl dodecyl carbonotrithioate (DMP-APTRZ) was employed for hemi-telechelic MCLs with a single triazole end group. (II) A tailored comonomer N-[3-(4H-1,2,4-triazol-4-yl)propyl]methacrylamide (APTRZMAAm) provides access to multidentate MCLs with a controllable number of triazole side groups along the polymer backbone. The thermally controlled variation of the MCL volume demand in aqueous solution was exploited for reversible aggregate formation upon Fe2+ complexation. Thermal response was studied via UV/Vis turbidity measurements, aggregate dimensions were determined via DLS, while the aggregate morphology was analyzed via customized TEM.
1. Introduction
Reversible structure formation at the molecular and supramolecular level is of fundamental relevance for vital processes in living organisms, as for example showcased in the current research highlights about liquid–liquid phase transitions of dissolved proteins in membrane-less organelle formation.1–4 Such concepts also play an increasing role for the bio-inspired design of modern materials and their implementation in advanced technical applications. The underlying reversible transitions are driven by variation of the intermolecular interactions depending on the system conditions, such as temperature. This is exemplified by the aggregation behaviour of thermoresponsive polymers or by the bonding strength between ligand and central ion in coordination complexes.5,6 The combination of these two structure-forming classes is expected to yield a synergistic behaviour that goes beyond the characteristic properties of the individual building blocks, which is the motivation for the present work.The fundamental bonding motif in the class of metal complexes is typically provided by an electron–donor–acceptor arrangement (Lewis acid–base pair) between the central ion and an appropriate ligand.7,8 If the inorganic or organometallic coordination motifs occur repetitively as backbone-forming elements in an extended macromolecular scaffold, such structure is labelled coordination polymer;6,9–11 in contrast, the term metallopolymer is defined as macromolecular structures with an organic backbone, that integrates metal complexes in the molecular framework.12–15 If a single coordination site is localized at one end of the polymer chain, the particular architecture is referred to as hemi-telechelic. This structural concept was the basis for one of the two main strategies that have been pursued in the present work to engineer responsive polymer-metal complexes. The second architectural strategy is targeting macromolecular structures with multiple coordination sites distributed along the polymer backbone, termed here as multidentate ligands. Upon coordination with a metal centre, the formed complexes act as chemical network junctions, resulting in molecular scaffolds known as metal–organic coordination-networks.16
Functionalized triazole derivatives offer strong coordinative interactions with numerous metal ions such as Fe2+, Cu2+, Zn2+, Ag+ and surfaces of nanoparticles.17–19 They can be utilized as antibacterial-, antitumoral-, and anti-fouling agents, pesticides, herbicides, dyes, analytical reagents, and lubricants, beyond some unique spin crossover (SCO) properties.6,20–24 In particular, 1,2,4-triazoles exhibit three main coordination modes with Fe2+ ions, comprising mono- and bidentate bindings, as well as the more stable triple N1N2-bridging mode.6 In this latter geometry, the octahedrally coordinated iron ions are bridged via triazole rings and typically form an extended 1D-coordination structure (usually insoluble in water). This reduces the electrostatic repulsion of the central ions (Fig. 1i) and has led to far unprecedented SCO properties depending on the specific details of counterions and the ligand structure.6 Synthetic modification of the ligand in its 4-position furthermore allows convenient tuning of the coordination properties, as documented for 1,2,4-triazole-1-yl-propylamine (APTRZ).25
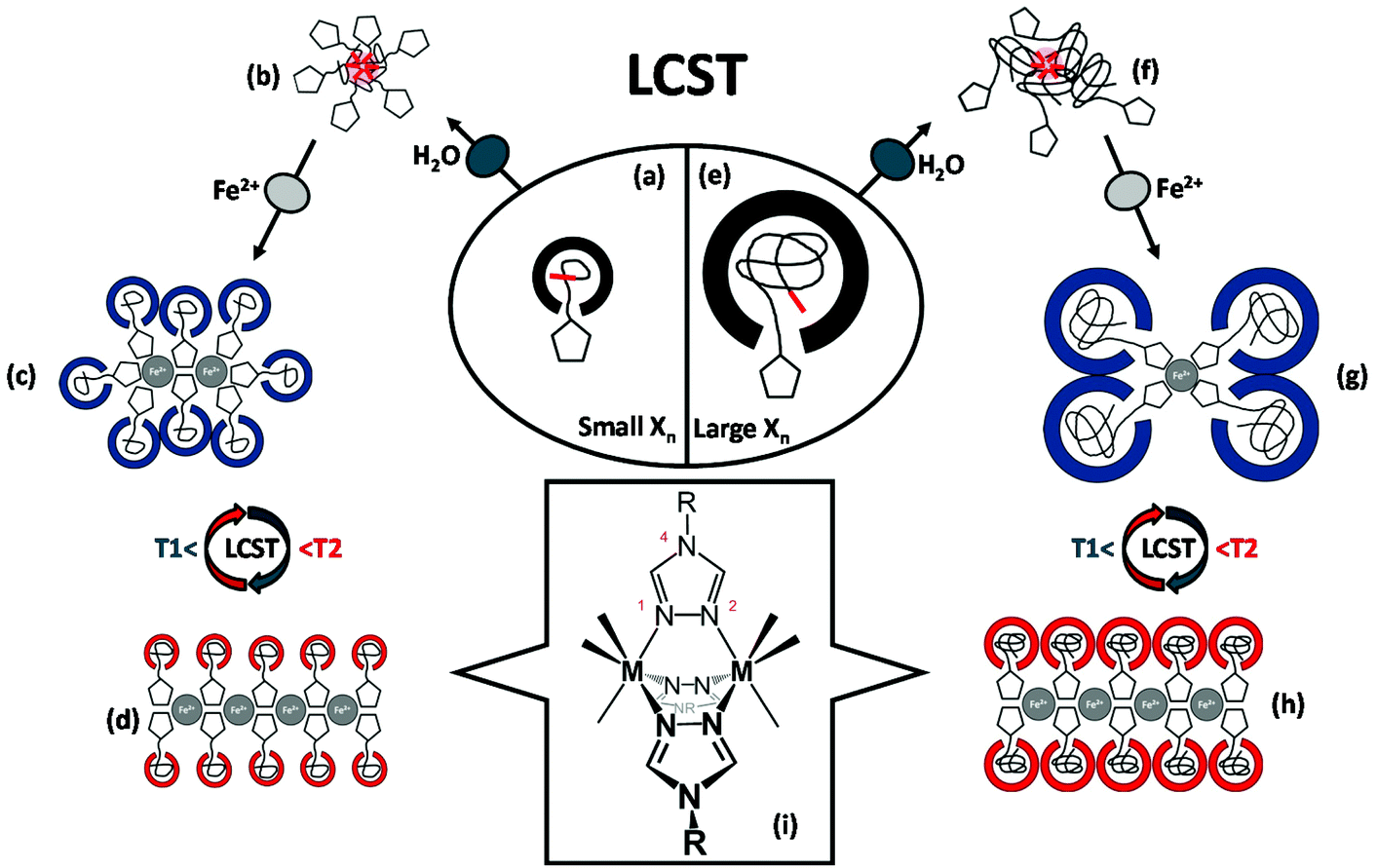 | ||
| Fig. 1 Schematics of the anticipated structural organization for short (a–d) and long (e–h) hemi-telechelic macromolecular coordination ligands P3-25k (with triazole end groups) in dependence of iron(II) ion coordination and thermal variation of the solvation state. A one-dimensional arrangement by N1,N2-bridging6 of the 1,2,4-triazole units is illustrated for the divalent iron-polymer complexes (i). The triazole units are represented by pentagons and the C12 alkyl chains by short red lines. | ||
The introduction of polymeric building blocks for the afore-mentioned coordination structures provides an elegant possibility to tune the architectural details via the design of the macromolecular framework (such as the chain lengths determined during synthesis),26 while tuning also their intrinsic properties e.g. solubility. Few examples demonstrating the versatility of polymeric ligands as building blocks to enable complex functions are reported in the recent literature: (i) mechanochromic sensors utilizing reversible metal–ligand dissociation of telechelic coordination polymers,27 (ii) self-healing polymers based on reversible triazole coordination,28 (iii) metallocopolymer networks with designable multicoloured photoluminescence.29 Beyond these examples, going one step further and endowing the polymer with thermoresponsive behaviour enables additional dynamic control over the structural dimensions by an external temperature stimulus. The solvation state of the responsive chain in aqueous solution can be reversibly switched by the external stimulus of temperature, inducing a coil-to-globule transition.30 For example, such thermoresponsive switching of the swelling behaviour was previously demonstrated for a polymeric host–guest system via a reversible complexation/decomplexation process of a cyclodextrin copolymer through molecular recognition.31 Various systems based on macromolecular structures that combine metal complexation and responsiveness have been reported in the literature.11,26,32,33 Inspired by this body of research, we have chosen to study dynamic structure formation of novel metal–polymer complexes based on metal-coordination motifs with thermoresponsive polymer ligands.
For this purpose, a 1,2,4-triazole unit was conjugated with thermoresponsive poly(N-isopropylacrylamide) (PNIPAAm) to yield a LCST-type macromolecular coordination ligand (MCL) for Fe2+ complexation. As one of the most studied examples for thermoresponsive polymers, PNIPAAm in aqueous solution shows a robust LCST transition near body temperature (32 °C) and is well-accessible via various synthetic routes.34 Amongst these, reversible addition–fragmentation chain transfer polymerization (RAFT)34,35 represents a powerful method to fabricate very defined polymers with controlled molar mass and desired end group.36 The 1,2,4-triazole end group was introduced by the chain transfer agent (CTA) via RAFT polymerization in presence of 1-{[3-(4H-1,2,4-triazol-4-yl)propyl]amino}-2-methyl-1-oxopropan-2-yl dodecyl carbonotrithioate (DMP-APTRZ, section 3.1.1).
For these thermoresponsive macromolecular coordination ligands, the thermally-dependent solution- and coordination behaviours were studied in presence of iron(II) ions via UV–Vis spectroscopy, DLS and cryo-TEM. Particularly, a structure–property relationship was established for the two distinct architectures: the hemi-telechelic and multidentate design.
2. Experimental
2.1. Materials
The used solvents (ethanol, ethyl acetate, hexane, methanol (98.5%, VWR), chloroform (99%, VWR), dichloromethane (99.9%, Fisher Scientific)) were of technical purity unless specified otherwise. The distilled water was obtained from MilliQ apparatus (resistivity σ = 18.2 MΩ cm). N-Isopropylacrylamide (99%, Acros Organics, recrystallized from hexane/toluene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4)), carbon disulfide (99%, Sigma-Aldrich), 2,2′-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (98%, Wako Chemicals), 2-methacrylic acid (99%, Sigma-Aldrich), formic hydrazide (98%, Alfa Aesar), triethyl orthoformate (98%, Alfa Aesar), 3-aminopropan-1-ol (99%, Alfa Aesar), triphenylphosphine flakes (99%, Alfa Aesar), diisopropyl azodicarboxylate (94%, Alfa Aesar) and hydrazine monohydrate (>98%) were purchased from Alfa Aesar and stored under inert atmosphere and in a dark place at 0–4 °C. Phthalimide (99%, Alfa Aesar), L-ascorbic acid (99%, Alfa Aesar), azobisisobutyronitile (AIBN) (99%, Acros Organics), 4-(dimethylamino)-pyridine (99.9%, Acros Organics), aqueous ammonia (25%, Roth Chemicals), N,N′-dicyclohexylcarbodiimide (99%, Sigma-Aldrich), 2-bromo-2-methylpropionic acid (>98%, Alfa Aesar), 3-(ethyliminomethyleneamino)-N,N-dimethylpropane-1-amine (99%, Roth), THF (99.9%, Fisher Scientific), triethylamine (99%, Alfa Aesar), 1-dodecanethiol (>98%, Sigma-Aldrich), N-hydroxysuccinimide (98%, Alfa Aesar) were used as received. Dioxane (98%, Roth Chemicals) was freshly distilled. Absolute ethanol (99.9%, Sigma Aldrich), Fe(BF4)2·6H20 (99%, Sigma Aldrich) were stored under inert conditions. Other dried solvents were prepared through refluxing for one day under nitrogen over the appropriate drying agents (sodium-naphthalene and benzophenone for THF, sodium-benzophenone and calcium hydride for diethyl ether, calcium hydride for acetonitrile, magnesium and iodine for methanol, sodium for ethanol) and degassed before use. Solvents were stored in glass ampoules under argon. 2{[(dodecylthio)carbothioyl]thio}-2-methylpropanoic acid (DMP) and 2-{[(ethylthio)carbothioyl]thio}-2-methylpropanoic acid (EMP) were synthesized and checked via TLC, 1H- and 13C NMR.
:4)), carbon disulfide (99%, Sigma-Aldrich), 2,2′-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (98%, Wako Chemicals), 2-methacrylic acid (99%, Sigma-Aldrich), formic hydrazide (98%, Alfa Aesar), triethyl orthoformate (98%, Alfa Aesar), 3-aminopropan-1-ol (99%, Alfa Aesar), triphenylphosphine flakes (99%, Alfa Aesar), diisopropyl azodicarboxylate (94%, Alfa Aesar) and hydrazine monohydrate (>98%) were purchased from Alfa Aesar and stored under inert atmosphere and in a dark place at 0–4 °C. Phthalimide (99%, Alfa Aesar), L-ascorbic acid (99%, Alfa Aesar), azobisisobutyronitile (AIBN) (99%, Acros Organics), 4-(dimethylamino)-pyridine (99.9%, Acros Organics), aqueous ammonia (25%, Roth Chemicals), N,N′-dicyclohexylcarbodiimide (99%, Sigma-Aldrich), 2-bromo-2-methylpropionic acid (>98%, Alfa Aesar), 3-(ethyliminomethyleneamino)-N,N-dimethylpropane-1-amine (99%, Roth), THF (99.9%, Fisher Scientific), triethylamine (99%, Alfa Aesar), 1-dodecanethiol (>98%, Sigma-Aldrich), N-hydroxysuccinimide (98%, Alfa Aesar) were used as received. Dioxane (98%, Roth Chemicals) was freshly distilled. Absolute ethanol (99.9%, Sigma Aldrich), Fe(BF4)2·6H20 (99%, Sigma Aldrich) were stored under inert conditions. Other dried solvents were prepared through refluxing for one day under nitrogen over the appropriate drying agents (sodium-naphthalene and benzophenone for THF, sodium-benzophenone and calcium hydride for diethyl ether, calcium hydride for acetonitrile, magnesium and iodine for methanol, sodium for ethanol) and degassed before use. Solvents were stored in glass ampoules under argon. 2{[(dodecylthio)carbothioyl]thio}-2-methylpropanoic acid (DMP) and 2-{[(ethylthio)carbothioyl]thio}-2-methylpropanoic acid (EMP) were synthesized and checked via TLC, 1H- and 13C NMR.
2.2. Analytical techniques
:5) with 0.1 vol% TFA was used as eluent. A sample volume of 5 μL was injected at temperature of T = 20 °C. A flow rate of 0.4 mL min−1 was adjusted. The editing and fitting of the raw data were carried out using the software ChemStation and Origin.
2.3. Synthesis
Step 1. 3-(4H-1,2,4-Triazol-4-yl)propan-1-ol (HYPTRZ). A mixture of triethyl orthoformate (18 mL, 108 mmol) and formyl hydrazine (5.4 g, 90 mmol) in dry methanol (100 mL) is refluxed for 6 hours under nitrogen atmosphere. Then 3-aminopropanol (6.9 mL, 90 mmol) is added and the resulting solution is refluxed for additional 24 h. Subsequently, the mixture is allowed to cool down to room temperature, and the resulting crude product is recrystallized from dry ethanol and diethyl ether mixture at low temperature (−32 °C). Finally, the moisture sensitive white crystalline product is filtered off, washed with cold diethyl ether, and dried under vacuum. Yield: 8.25 g (72%). 1H NMR (300 MHz, MeOD), δ ppm: 8.56 (s, 2H, –N–CH
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 4.27 (t, 2H, TRZ–C
N), 4.27 (t, 2H, TRZ–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2–), 3.56 (t, 2H, –C2–OH), 2.04 (m, 2H, C2–CH2–OH); 13C NMR (300 MHz, MeOD), δ ppm: 144.8 (–N–CHN), 58.9 (TRZ–
2–), 3.56 (t, 2H, –C2–OH), 2.04 (m, 2H, C2–CH2–OH); 13C NMR (300 MHz, MeOD), δ ppm: 144.8 (–N–CHN), 58.9 (TRZ–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2–), 43.3 (–H2–OH), 33.0 (–H2–CH2–OH).
H2–), 43.3 (–H2–OH), 33.0 (–H2–CH2–OH).
Step 2. 2-(3-(4H-1,2,4-Triazol-4-yl)propyl)isoindoline-1,3-dione (PHTPTRZ). Diisopropyl azodicarboxylate (9.7 mL, 49.45 mmol, 1.23 equivalent) is added with stirring in small portions to a solution of triphenylphosphine (PPh3) (12.98 g, 49.45 mmol, 1.23 equiv.) in dry THF (60 mL) at 0 °C under argon, and the mixture is stirred for 10 min. A suspension of phthalimide (5.89 g, 40 mmol, 1 equiv.) and 4-(3-hydroxypropyl)-1,2,4-triazole (HYPTRZ) (5.08 g, 40 mmol, 1 equivalent) in dry acetonitrile (60 mL) is added to the reaction mixture at 0 °C, and the resulting mixture is stirred overnight at room temperature. The white precipitate formed was collected using sintered funnel, washed with cold THF and acetonitrile to obtain a white crystalline powder. Yield: 8.45 g (82%). 1H NMR (500 MHz, DMF-D7), δ ppm: 8.62 (s, 2H, –N–CH
N–), 7.91–7.89 (m, 4H, HAr), 4.27 (t, 2H, TRZ–C2–), 3.69 (t, 2H, –C2–Pth), 2.23 (qi, 2H, –C2–CH2–Pth), 13C NMR (500 MHz, DMF-D7), δ ppm: 168.9 (–CO), 143.9 (–N–CHN–), 134.9 (–CAr), 132.8 (–CAr), 123.5 (–CAr), 42.8 (TRZ–H2–), 35.4 (–H2–Pth), 30.5 (–H2–CH2–Pth).
Step 3. 3-(4H-1,2,4-Triazol-4-yl)propan-1-amine (APTRZ). Triazole derivative 2-(3-(4H-1,2,4-triazol-4-yl)propyl)isoindoline-1,3-dione (PHTPTRZ) (5 g, 19.51 mmol) and NH2NH2·H2O (5 mL, 103.17 mmol) in 100 mL of ethanol were heated to reflux for 4 hours. After removal of the phthalhydrazide by filtration at room temperature, the solvent was removed under reduced pressure to give pale-yellow oil. To ensure that all phthalhydrazide was removed, 50 mL of acetonitrile was added and kept at −32 °C overnight, then the resulting precipitate was filtered and the filtrate concentrated to afford the pure oil. Yield: 1.98 g (81%). 1H NMR (300 MHz, CDCl3, shown in Fig. S1†), δ ppm: 8.17 (s, 2H, –N–CH
N–), 4.15 (t, 2H, TRZ–C2–), 2.71 (t, 2H, –C2–NH2), 1.89 (m, 2H, –C2–CH2–NH2), 1.26 (br s, 2H, N2), 13C NMR (300 MHz, CDCl3), δ ppm: 143.0 (–N–CHN–), 42.5 (TRZ–H2–), 38.3 (–H2–NH2), 33.7 (–H2–CH2–NH2).
:CHCl3:MeOH:NH3 = 80:13:6:1, Rf(DMP-NHS) = 0.3). The reaction mixture was concentrated and the crude product was purified by column chromatography (silica gel, EtOAc) to obtain the DMP-NHS as a yellowish solid after drying. Yield: 1535 mg (60%). 1H NMR (400 MHz, CDCl3, shown in Fig. S3a†), δ ppm: 0.88 (t, 3H, –CH2–C3), 1.25–1.39 (m, 18H, –(C2)9–CH3), 1.68 (p, 2H, –CH2–C2–(CH2)9–), 1.87 (s, 6H, –C–(C3)2), 2.81 (br. s, 4H, –NHS) and 3.30 (t, 2H, –CS3–C2–). – 13C NMR (100 MHz, CDCl3, shown in Fig. S3b†), δ ppm: 14.1 (–CH2–H3), 22.7 (–H2–CH3), 25.6 (–NHS), 27.8 (–(H2)8–), 29.0 (–(H2)8–), 29.1 (–(H2)8–), 29.4 (–(H2)8–), 29.5 (–(H2)8–), 29.6 (–(H2)8–), 29.6 (–(H2)8–), 31.9 (–H2–CH2–CH3), 37.2 (–CS3–H2–), 54.2 (––(CH3)2), 168.6 (–OO–, –ONH–) and 218.7 (–S3–). – IR (shown in Fig. S4†): 1734 cm−1 and 1777 cm−1 (5-membered imide ring).
Via active ester. DMP-NHS (520 mg, 1.13 mmol) and APTRZ (142 mg, 1.13 mmol) were dissolved in dry dichloromethane (50 ml) in a two-neck flask under argon atmosphere. TEA (114 mg, 1.13 mmol, 156 μL) was added to the above solution and the reaction mixture was continued to stir for 24 hours. The TEA-salts were removed by extraction with water (50 mL) and the organic phase was concentrated under reduced pressure. The impurities of DMP were removed by column chromatography (silica gel, EtOAc
:hexane = 3:1) and the pure product was obtained after a change of eluent (silica gel, CH2Cl2:CHCl3:MeOH:NH3 = 80:13:6:1, iodine indicator, Rf(DMP-APTRZ) = 0.44). Since DMP-APTRZ interacts strongly with the silica gel during column chromatography, the basic eluent mixture CHCl3/MeOH/NH3 (13:6:1) was required to isolate the product. The product phase was concentrated under vacuum to dryness. Yield: 78–93%. 1H NMR (400 MHz, CDCl3, shown Fig. S5a†), δ ppm: 0.88 (t, 3H, –CH2–C3), 1.26–1.38 (m, 18H, –(C2)9–CH3), 1.65 (p, 2H, –CH2–C2–(CH2)9–), 1.70 (s, 6H, –C–(C3)2), 1.97 (m, 2H, –C2–CH2–TRZ), 3.27 (m, 4H, –C2–CS3–, –C2–(CH2)2–TRZ), 4.07 (t, 2H, –C2–TRZ), 6.71 (br.s, 1H, –CON–) and 8.23 (s, 2H, –TRZ). 13C NMR (100 MHz, CDCl3, shown in Fig. S5b†), δ ppm: 14.3 (–CH2–H3), 22.9 (–H2–CH3), 26.0 (–C–(H3)2), 27.9 (–(H2)8–), 29.1 (–(H2)8–), 29.2 (–(H2)8–), 29.5 (–(H2)8–), 29.6 (–(H2)8–), 29.7 (–(H2)8–), 29.8 (–(H2)8–), 29.8 (–(H2)8–), 32.0 ((–H2–CH2–CH3, –H2–CH2–TRZ), 37.3 (–H2–CS3–, –H2–(CH2)2–TRZ), 43.2 (–H2–TRZ), 57.3 (––(CH3)2), 143.0 (–TRZ), 173.4 (–ONH–) and 221.3 (–S3–). TGA: Tdec = 389.4 K – DSC: Tg = 286.75 K (1.41 J g−1), Tm = 330.85 K (86.15 J g−1).
Via direct coupling. The triazole derivative APTRZ (174.5 mg, 1.39 mmol) was added to the dichloromethane (150 mL) solution containing excesses of 66% EDC (358.2 mg, 3.08 mmol) and 10% DMP (560 mg, 1.54 mmol) in a two-neck flask under argon flux. The 4-(dimethylamino)-pyridine (186.4 mg, 1.54 mmol) was then added to the reaction mixture with continuous stirring for two days at room temperature. The salts of 4-(dimethylamino)-pyridine were removed by threefold extraction from 1 M HCl (150 mL), water (150 mL) and 1 M NH3 (150 mL), and the organic phase was concentrated under reduced pressure. The excess of pyridine salts was removed by short-column chromatography (silica gel, EtOAc
:hexane = 3:1) and the dark yellowish solid was obtained by an eluent change (CHCl3:MeOH:NH3 = 13:6:1, iodine indicator, Rf, (DMP-APTRZ) = 0.26). The product phase was concentrated to dryness. Yield: 695 mg (95%).
:[AIBN]:[NIPAAm] indicated in Table 1. The reaction mixtures were degassed by a fivefold cycle of vacuum and argon flux to eliminate the dissolved oxygen. The reactions were performed in a preheated sample holder at a temperature of 70 °C. The polymerizations were quenched after 24 hours by air and cooling with ice. The polymers were precipitated from ice cold diethyl ether or hexane.
| [NIPAAm]0:[DMP-APTRZ]0:[AIBN]0 |
AIBN/mg (μmol) | DMP-APTRZ/mg (μmol) | NIPAAm/mg (mmol) |
|---|---|---|---|
| a Polymers were precipitated in hexane instead of diethyl ether. | |||
| 15.0:1:0.1 |
4.3 (26.4) | 250 (529) | 896 (7.98) |
| 25.0:1:0.1a |
1.3 (7.9) | 75 (159) | 448 (3.97) |
| 25.0:1:0.1 |
2.6 (15.9) | 150 (317) | 896 (7.98) |
| 50.0:1:0.1a |
0.4 (2.6) | 25 (53) | 298 (2.64) |
| 100.0:1:0.1a |
0.4 (2.6) | 25 (53) | 600 (5.28) |
| 150.0:1:0.1a |
0.4 (2.6) | 25 (53) | 900 (7.92) |
| 150.0:1:0.1 |
0.9 (5.3) | 50 (106) | 1793 (15.95) |
The solid products were collected using centrifugation and dried under reduced pressure. LCSTs and molar mass distribution of the polymers were determined (Table 3). The presence of the end groups was confirmed by 1H NMR measurements. Yield: 87–94%. 1H NMR (400 MHz, CDCl3, shown in Fig. S10†), δ ppm: 0.87 (t, 3H, C3–DMP), 1.13–4.16 (backbone), 6.40 (amide of NIPAAm), 8.48 (br. s, 2H, –TRZ).
| [APTRZMAAm]feed/mol% | AIPC/mg (μmol) | EMP/mg (μmol) | APTRZMAAm/mg (mmol) | NIPAAm/mg (mmol) |
|---|---|---|---|---|
| 5.0 | 2.38 (7.4) | 16.47 (73.5) | 16.8 (0.09) | 196.6 (1.74) |
| 10.0 | 2.38 (7.4) | 16.47 (73.5) | 33.6 (0.17) | 188.3 (1.66) |
| 15.0 | 2.38 (7.4) | 16.47 (73.5) | 50.4 (0.26) | 177.4 (1.57) |
n), degree of polymerization (Xn), cloud point (Tc), dispersity (Đ) and sample code of the hemi-telechelic macromolecular coordination ligands with triazole end groups
| [NIPAAm]0:[CTA]0:[I]0 |
n,theo/kg mol−1 (Xn) |
n,exp
/kg mol−1 (Xn) |
T c /°C | Đ | Yield/% | Sample code |
|---|---|---|---|---|---|---|
| a Values determined by GPC measurement. b Values determined by turbidity measurements. c Values not determined. | ||||||
| 15:1:0.1 |
2.2 (15) | 3.1 (21) | 34.7 | 1.38 | 68 | P3k |
| 25:1:0.1 |
3.3 (25) | 3.9 (30) | 33.4 | 1.27 | 94 | P4k |
| 50:1:0.1 |
6.1 (50) | 6.9 (57) | 32.9 | 1.38 | 92 | P7k |
| 100:1:0.1 |
11.8 (100) | 12.3 (104) | 34.1 | 1.34 | 87 | P12k |
| 150:1:0.1 |
17.5 (150) | 23.3 (199) | 34.8 | 1.12 | 86 | P23k |
| 150:1:0.1 |
17.5 (150) | 24.8 (212) | —c | 1.19 | 60 | P25k |
:5), shown in Fig. S6c and d†): 4.76 min, M = 195.1 g mol−1, 1H NMR (500 MHz, CDCl3, shown in Fig. S6a†) δ ppm: 1.97 (s, 3H, H2CCR–C3), 2.08 (quin, J = 6.7 Hz, 2H, –CH2–CH2–C2–TRZ), 3.37 (q, J = 6.2 Hz, 2H, –CONH–C2–CH2–), 4.11 (t, J = 6.9 Hz, 2H, –CH2–C2–TRZ), 5.35 (s, 1H, 2CCR–CH3), 5.74 (s, 1H, 2CCR–CH3), 6.80 (br. s., 1H, H2CC–CH3–CON–CH2–), 8.30 (s, 2H, –CH2–![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif)
![[R with combining low line]](https://www.rsc.org/images/entities/char_0052_0332.gif)
![[Z with combining low line]](https://www.rsc.org/images/entities/char_005a_0332.gif) ), 13C NMR (126 MHz, CDCl3, shown in Fig. S6b†), δ ppm: 18.7 (H2CCR–H3), 31.2 (–CH2–H2–CH2–TRZ), 36.4 (–CONH–H2–CH2–), 43.0 (–CH2–H2–TRZ), 120.0 (H2CR–CH3), 139.6 (H2CR–CH3), 142.9 (–CH2–), 169.1 (H2CC–CH3–ONH–CH2–).
:1) (1 mL) and placed in a Schlenk-tube with varying molar composition with ratio of [monomers]0:[EMP]0:[AIPC]0 = 25:1:0.1 (Table 2). The methanol solution (1.5 mL) of APTRZMAAm was added to the reaction mixtures, which then were degassed by a fivefold freeze thaw cycle. The reactions were performed in a preheated sample holder at a temperature of 55 °C. The polymerizations were quenched after 24 hours by air und ice cooling. The polymers were precipitated twice in ice cold EtOAc. The solid products were collected using centrifugation, dissolved in distilled water and freeze-dried. The LCSTs and molar mass distributions were determined (Table 4). The quantification of the APTRZ units was determined by 1H NMR measurements. Yield: 58–63%. 1H NMR (500 MHz, CDCl3, shown in Fig. S12†), δ ppm: 0.88–3.55 (CTA, backbone), 3.99 (br. s, isopropyl of NIPAAm –C–(CH3)2), 4.19 (br. s, –C2–TRZ), 5.75–7.25 (amide of NIPAAm) 8.55 (br. s, –APTRZMAAm).
n), degree of polymerization (Xn), cloud point (Tc), dispersity (Đ) and sample codes for the multidentate copolymer ligands (9) with varying numbers of triazole side chains
), 13C NMR (126 MHz, CDCl3, shown in Fig. S6b†), δ ppm: 18.7 (H2CCR–H3), 31.2 (–CH2–H2–CH2–TRZ), 36.4 (–CONH–H2–CH2–), 43.0 (–CH2–H2–TRZ), 120.0 (H2CR–CH3), 139.6 (H2CR–CH3), 142.9 (–CH2–), 169.1 (H2CC–CH3–ONH–CH2–).
:1) (1 mL) and placed in a Schlenk-tube with varying molar composition with ratio of [monomers]0:[EMP]0:[AIPC]0 = 25:1:0.1 (Table 2). The methanol solution (1.5 mL) of APTRZMAAm was added to the reaction mixtures, which then were degassed by a fivefold freeze thaw cycle. The reactions were performed in a preheated sample holder at a temperature of 55 °C. The polymerizations were quenched after 24 hours by air und ice cooling. The polymers were precipitated twice in ice cold EtOAc. The solid products were collected using centrifugation, dissolved in distilled water and freeze-dried. The LCSTs and molar mass distributions were determined (Table 4). The quantification of the APTRZ units was determined by 1H NMR measurements. Yield: 58–63%. 1H NMR (500 MHz, CDCl3, shown in Fig. S12†), δ ppm: 0.88–3.55 (CTA, backbone), 3.99 (br. s, isopropyl of NIPAAm –C–(CH3)2), 4.19 (br. s, –C2–TRZ), 5.75–7.25 (amide of NIPAAm) 8.55 (br. s, –APTRZMAAm).
n), degree of polymerization (Xn), cloud point (Tc), dispersity (Đ) and sample codes for the multidentate copolymer ligands (9) with varying numbers of triazole side chains
3. Results and discussion
3.1. Synthesis
 | ||
| Scheme 1 Functionalization of the chain-transfer agent DMP (1) by APTRZ (3). | ||
The molecular structure of the target compound 1-{[3-(4H-1,2,4-triazol-4-yl)propyl]amino}-2-methyl-1-oxopropan-2-yldodecyl carbonotrithioate (DMP-APTRZ) was confirmed by 1H and 13C NMR measurements (ESI, Fig. S5a/b†) with the presence of the characteristic signals for the 1,2,4-triazole group (1H: 2 H, s, 8.23 ppm, 13C: 143.0 ppm) and the amide proton (1H: 1H, t, 6.71 ppm). Direct EDC coupling was preferred over the NHS route based on the good yield of product (4) and the convenience of that procedure.
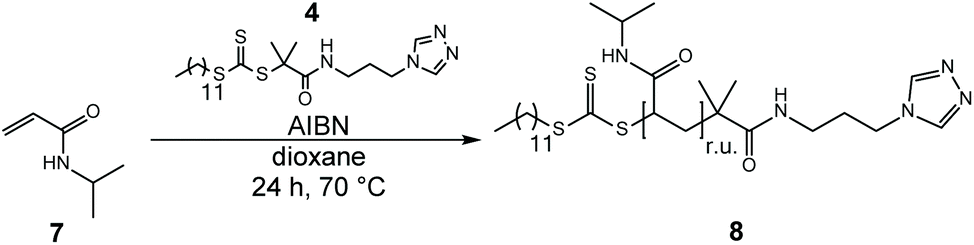 | ||
| Scheme 2 RAFT homopolymerization of NIPAAm (7) with the triazole chain transfer agent DMP-APTRZ (4). | ||
The 1H NMR spectrum (shown in ESI, Fig. S10†) confirms the presence of the terminal CTA-moiety at the polymer chain end (characteristic triazole signal at ca. 8.5 ppm). The isolated hemi-telechelic polymers (8) are characterized by narrow molar mass distributions (Đ = 1.12–1.34) and possess the targeted chain-lengths (n = 3.1–24.8 kg mol−1), corroborating full control over the RAFT polymerization process with DMP-APTRZ (4). The corresponding physical data, like monomer:CTA:initiator feed ratios, experimental (n) and theoretical molar mass (n,theo), degrees of polymerization (Xn), cloud points (Tc), dispersities (Đ), and yields are summarized in Table 3. All pNIPAAm samples (P3k–P25k) show the typical thermoresponsive behavior in aqueous solution with the transition temperatures being only slightly affected by the triazole end group (32.9–34.8 °C, see Fig. 2a and b).
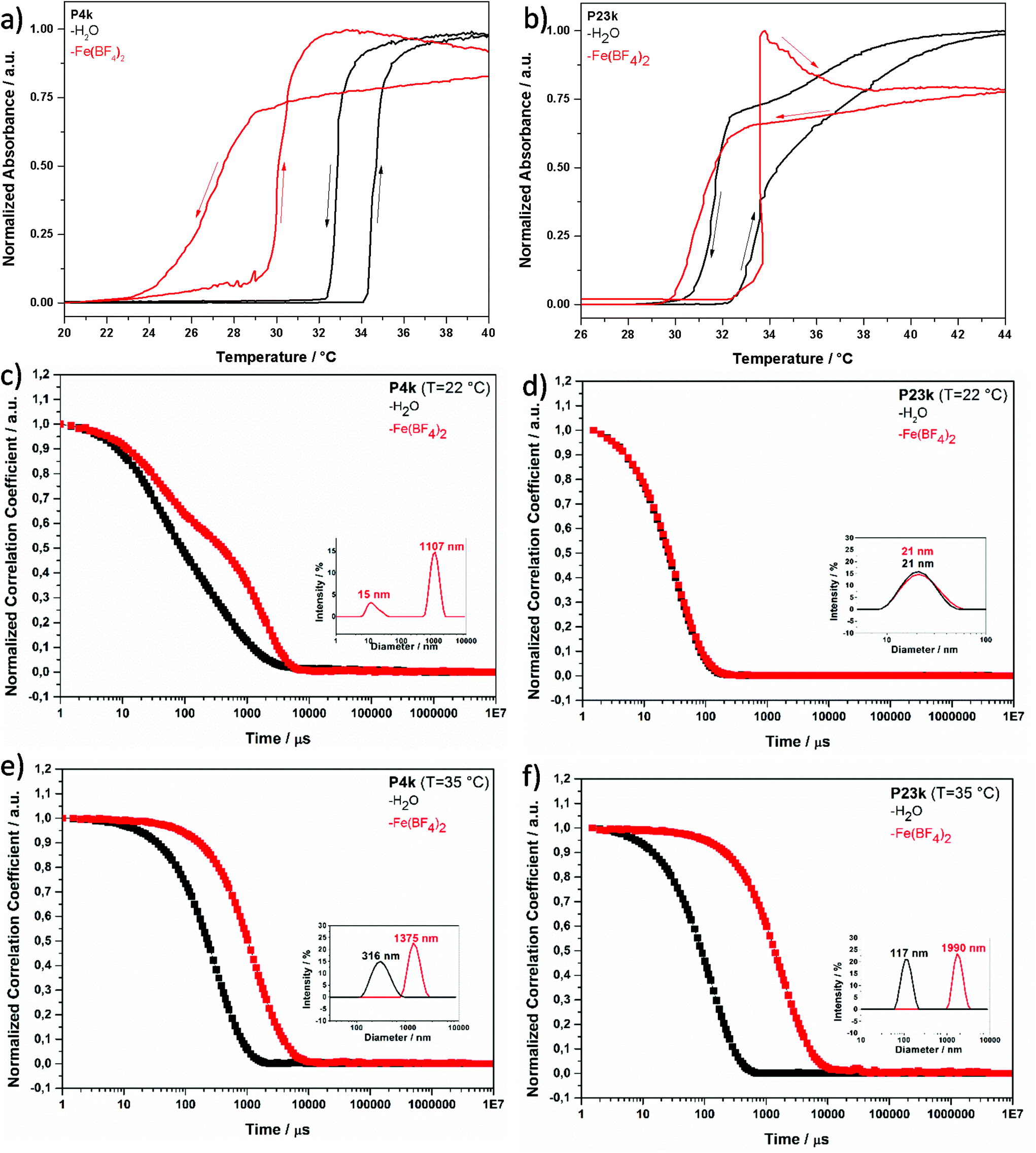 | ||
| Fig. 2 Temperature-dependent turbidity measurements by UV/VIS-spectroscopy for the cloud point determination of (a) the hemi-telechelic MCLs with short chain P4k and (b) with long chain P23k as 1 wt% aqueous polymer solution (transmission measured at ν = 550 nm) and after the addition of tenfold molar excess of Fe(BF4)2 with respect to triazole units (transmission measured at ν = 740 nm). Corresponding DLS measurements for the determination of the iron ion-sensitive aggregation behaviours in pure aqueous solution (black) and in presence of iron(II) ions (red) with 0.1 wt% hemi-telechelic polymer solutions of P4k (c–e) and P23k (d–f) below (c and d) and above (e and f) the Tc. | ||
 | ||
| Fig. 3 TEM-images of the short macromolecular coordination ligand P3k dried from a 0.1 wt% aqueous solution for varying the temperature from 22 °C (a and c) to 40 °C (b and d) and without (a–b)/with (c–d) presence of Fe2+. | ||
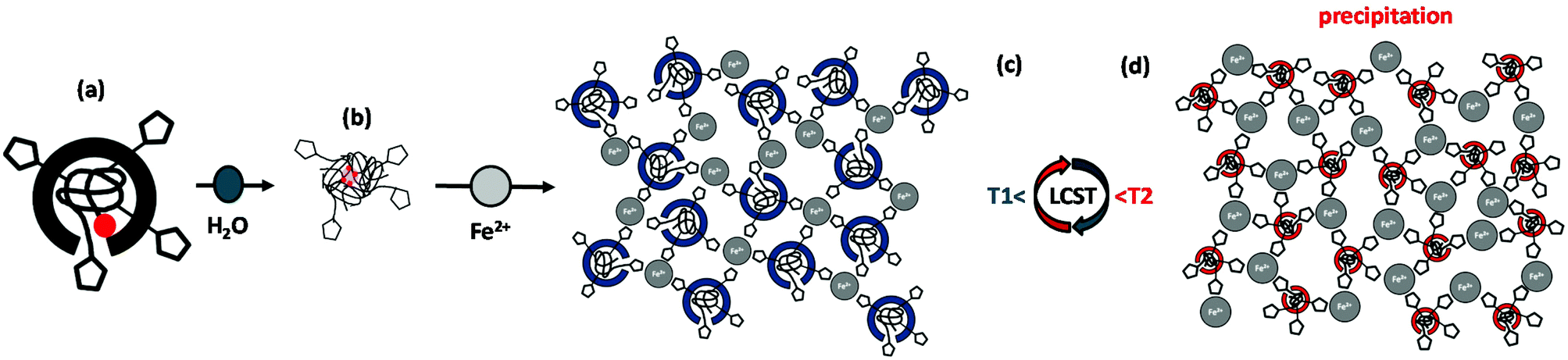 | ||
| Fig. 4 Proposed structural organization of the multidentate macromolecular coordination ligand poly(NIPAAm-co-APTRZMAAm)-EMP (C4.8k5%–4.9k5-15%) with respect to iron(II) ion coordination and thermal variation of the solvation state. These architectures represent the elemental structural units, which form the basis for the larger scattering objects notably observed in DLS. The triazole units are schematically represented by pentagons and the C2 alkyl chains as red spots. | ||
Unfortunately, all coupling attempts of APTRZ to acrylic acid via various synthetic strategies were unsuccessful, which is described in details in the ESI chapters 1.5.1–1.5.3.† This is assumingly caused by a Michael addition as side reaction. Instead, methacrylic acid was selected as monomer fragment since the Michael addition is expected to be supressed by steric hindrance of the methyl group adjacent to the double bond. The corresponding APTRZ monomer N-[3-(4H-1,2,4-triazol-4-yl)propyl]methacrylamide (APTRZMAAm) (6) was obtained by direct coupling of methacrylic acid to the primary amine group of APTRZ with N,N′-dicyclohexylcarbodiimide (DCC) in the absence of an auxiliary base. The yield was 43% after workup (Scheme 2). DCC was preferred over EDC or CDI as condensation agent since workup is facilitated by the insolubility of the formed N,N′-dicyclohexylurea in water, while the other reagents and products were readily soluble under such conditions (Scheme 3).
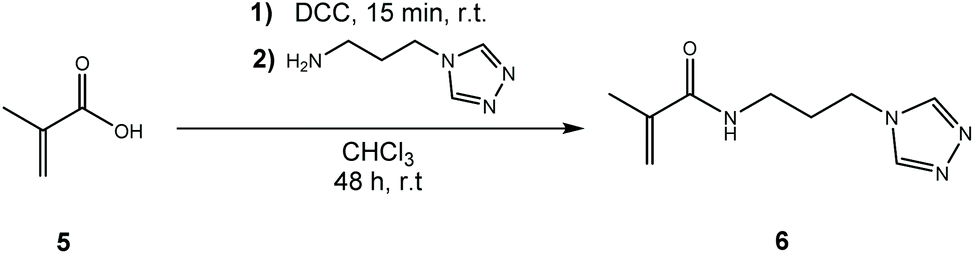 | ||
| Scheme 3 Synthesis of the coordination monomer APTRZMAAm (6). | ||
It is worthwhile to note that the neat triazole monomer (6) after drying becomes insoluble in water, suggesting strong hydrogen bonding. The brownish resin was only very slowly soluble in a solvent mixture of H2O and MeOH (1:1). Thus, the aqueous product phase from the workup was only partially concentrated by incomplete freeze drying and used as stock solution. The purity of the product was confirmed with a chiefly dried sample by TLC measurements, HPLC-MS (shown in ESI, Fig. S6c and d†), 1H and 13C NMR measurements (shown in Fig. S6a and b†). The molecular structure of the target compound (6) was notably corroborated by the NMR signals of the chemically equivalent methylene groups from the 1,2,4-triazole moiety (1H: 8.26 ppm, s, 2H, 13C: 142.9 ppm in ESI, Fig. S6a and b†), the amide (1H: 6.76 ppm, br. s, 1H, 13C: 169.1 ppm in ESI, Fig. S6a and b) and the methacrylate double bond (1H: 5.36 ppm, 1H, 5.74 ppm, 1H; 13C: 120.0 ppm and 139.9 ppm).
 | ||
| Scheme 4 RAFT copolymerization of APTRZMAAm (6) and NIPAAm (7) with the chain transfer agent EMP. | ||
The reaction was performed in a mixture of water/MeOH (1:1) to allow homogeneous dissolution of all reaction partners, including the very polar APTRZMAAm and the less polar CTA EMP. Here, EMP was chosen over DMP due to the shorter alkyl chain, which provides solubility in the polar reaction medium. Yields for all copolymerizations with EMP were in the range of 55–69%, similar to the homopolymerization of NIPAAm with DMP-APTRZ as CTA. Analysis of the multidentate ligands by gel permeation chromatography indicated successful copolymerization via the RAFT mechanism by a unimodal, narrow molar mass distribution (Đ = 1.09–1.10, shown in Table 4).
The presence of the characteristic signals for the triazole group in the 1H NMR spectra of (9) (ESI, Fig. S12†) confirmed successful copolymerization of APTRZMAAm (8.55 ppm for triazole and 6.52 ppm for amide) with NIPAAm (3.98 ppm for tertiary isopropyl proton) in the desired ratios, which are within the acceptable range of the theoretically anticipated values. As expected, the cloud temperature rises systematically with increasing amount of hydrophilic APTRZMAAm repeat units for aqueous copolymer solutions (Fig. 5a) in comparison to a PNIPAAm-DMP homopolymer reference (n = 10 kg mol−1) without triazole units.
 | ||
| Fig. 5 Cloud point determination by turbidity measurements of multidentate MCL solutions (1 wt%) with varied triazole monomer contents C4.8k5%, C4.7k10%, and C4.2k15% (a) in pure water (transmission measured at ν = 550 nm) and (b) in presence of tenfold molar excess of Fe2+ ions with respect to triazole units (transmission measured at ν = 740 nm). DLS measurements of the iron-ion-dependent change of scattering object dimensions for C4.6k15% solutions (1 wt%) with and without iron ions (c) below and (d) above Tc. | ||
3.2. Systematic study of the aggregation behaviours upon Fe2+ complexation
n = 3.1–24.8 kg mol−1).
Before discussing the experimental details of the complexation studies, we may first introduce the proposed model carefully established by considering the potential interaction motives between the respective molecular components. These interactions encompass the following substantiated components:
(1) The pivotal motive for structure formation emerges from the attraction mediated through the coordinative bond between the Fe2+-center and the triazole nitrogen, as sketched in Fig. 1i.6
(2) Secondly, the repulsion by steric demand of the polymer chain is known to be a function of the degree of polymerization Xn (Fig. 1a/e) and of the temperature-dependent swelling state (LCST volume transition), as represented in the schemes of Fig. 1c/d and g/h, respectively.
(3) In addition, the steric repulsion of the hydrophilic polymer chains in the swollen state (below the LCST) can be thermally switched to an attraction between now hydrophobic chain segments in the collapsed state (above the LCST). This is illustrated in Fig. 1c/d and Fig. 1g/h with the accompanying coil-to-globule transition.5,42
(4) Lastly, van der Waals attraction emanates from the hydrophobic dodecyl substituents from the CTA units at the polymer chain ends (Fig. 1b/f).
In essence, the solution behaviour of the macromolecular coordination ligands is governed by the temperature-dependent balance of the various afore-mentioned types of interactions. In the following section, the proposed structure models and morphological changes are discussed in the context of thermal and coordinational dependencies for the small and large MCLs (Fig. 1). The elemental structural units are sketched as building blocks for the larger scattering objects. This hypothetical representation is markedly corroborated by the experimental data (DLS, UV–Vis, TEM) as discussed hereafter.
The small polymer ligand (n ≤4 kg mol−1, Fig. 1a) with a low number of repeat units (Xn < 25) possess a pronounced amphiphilic character due to the hydrophobic dodecyl group of the CTA on one end, and the hydrophilic triazole head group on the other chain end. In Fig. 1b, the micellar aggregation of the small MCL in aqueous solution below the cloud point at 22 °C is depicted, with the dodecyl chains forming the hydrophobic core due to their van der Waals attraction and the hydrophilic triazole head groups residing at the surface of the micelles in contact with the water phase (supported by DLS, Fig. 2c, black curve). After addition of iron(II) tetrafluoroborate to the polymer ligand below the cloud point (22 °C), larger aggregates are formed by the iron-polymer complexes, as illustrated in Fig. 1c. The increase in aggregation size is driven by attractive forces due to complex formation between the iron ions and multiple triazole head groups, uniting multiple chain ends (evidenced by Fig. 2, red curve). The exact structure of the coordination sphere is currently not known for our MCLs due to the intrinsic difficulties in the characterization of such coordination process in aqueous solution. Based on the known complexation motives of 1,2,4-triazoles, an extended 1D-coordination structure is yet conceivable via a triple N1N2-bridging mode with octahedrally coordinated iron ions (Fig. 1i) as subunits inside the larger scattering objects.6 Such a postulated 1D association motive is depicted in Fig. 1d for the collapsed polymer ligand at 35 °C with a reduced steric demand, which is further enhanced by the hydrophobic attraction between the polymer chain segments above the Tc. This hydrophobic attraction may result in further aggregation of the 1D elements into larger scattering objects, as suggested by the DLS data (red curve) in Fig. 2, which eventually leads to precipitation of the short chain samples.
A comparable picture can be drawn for the larger MCLs (n >4 up to 23 kg mol−1) in Fig. 1e. Below the turbidity point at 22 °C, the longer polymer chains are well hydrated and thus reduce the relative influence of the hydrophobic dodecyl end groups, yielding an overall weaker amphiphilic character. Yet, the DLS trace in Fig. 2 (black curve) points to a micellar aggregation of these chains. Upon addition of iron(II) salt, the triazole head groups are expected to coordinate with the metal centers (sketched in Fig. 1g), but only few chains can effectively associate via complex formation in the core due to the higher steric demand of the longer polymer ligands (compare Fig. 2). Like in the case of shorter chains above the Tc at 35 °C, an extended 1D coordination structure can be realized for the large polymer ligands (Fig. 1h). A binding motive via the triple N1N2-bridging mode (Fig. 1i) may become possible by the reduced volume demand of the collapsed chains with their hydrophobic attraction above the phase transition temperature (see Fig. 2, red trace).
In the following section, the obtained experimental data are systematically correlated to the above anticipated structure models.
Turbidity measurements. Determination of the cloud point by turbidity measurements (confer to Fig. 2a and b) delivers information about the temperature-dependent aggregation process of the thermoresponsive macromolecular coordination ligands by increasing turbidity due to intra- and intermolecular chain aggregations caused by the coil-to-globule transition.5 The cloud point temperature is determined by the inflection point in the turbidity curve (in ESI, Table S1†) and reveals the influence of the APTRZ triazole end group. This moiety is hydrophilic and leads to a slightly raised cloud point in pure water by stronger polymer-solvent-interactions (cf.Fig. 2a) in comparison to the parent carboxylic acid end group.43 The complex shape of the turbidity curves above the transition is attributed to kinetic effects during the reorganization process of the colloidal structures upon polymer aggregation.
Upon addition of the divalent iron salt Fe(BF4)2, the shorter polymer ligands (n < 3 kg mol−1, Xn < 15) precipitate abruptly at all investigated temperatures. The larger polymer ligands form homogeneous, clear solutions in the presence of Fe2+ below the cloud point (Tc). Above Tc, polymer phase separation causes clouding of the solution, attributable to an attractive coordination force that leads to a substantial growth of the scattering objects. Generally, a decrease of the Tc (Fig. 2a and b) is observed upon coordination of the APTRZ moieties with Fe2+ ions. The difference in Tc between the liquid media with and without iron ions becomes larger with a decrease in the molar mass of the polymers (depicted in ESI, Table S1,† ΔTH2O/Fe(II) = 0.9–4.5 K). These results suggest that the shorter chains can more efficiently participate in an extended coordination structure with the Fe2+ ions in the hydrated state at lower temperatures, which also leads to their preferential precipitation.
Dynamic light scattering measurements. Dynamic light scattering (DLS) of 0.1 wt% hemi-telechelic polymer solutions was employed to follow the effect of complex formation by variation of the aggregation state upon addition of Fe2+ ions. For that purpose, a tenfold molar excess of Fe(BF4)2·6H2O with respect to triazole end groups was added with traces of ascorbic acid to the aqueous medium. The results for the short ligand P4k and the long ligand P23k are depicted in Fig. 2, while the complete data set for all polymer systems is provided in the ESI, Fig. S13† (including samples with intermediate chain lengths). The size distribution analysis is based on a mathematical fit assuming a spherical aggregate shape, which may not be realistic for the complex aggregate morphologies observed via TEM measurements (Fig. 3) for some samples. Since the focus lies on the relative changes in aggregation behaviour under different experimental conditions, the exact aggregate geometry is not relevant here.
In Fig. 2, the DLS curves are shown for the short MCL P4k (n < 4 kg mol−1, Xn < 25) at a temperature (22 °C) below Tc in pure water (black curve) and in Fe2+ solution (red curve). In pure water, the broad decay of the autocorrelation curve reflects the presence of several coexisting aggregation species with substantially different dimensions (thus, for this case, no size distribution analysis is included in the inset – refer to ESI, Fig. S14† for related details). Even for the smallest of these scattering species, the diameter typically lies between Dh = 15–21 nm (ESI, Fig. S13 and S14†). This suggests the presence of micellar aggregates composed of several polymer chains for the given experimental degrees of polymerization Xn = 20–200. Micellization is facilitated by the asymmetric chain structure, with one end carrying a hydrophobic dodecyl chain located in the micelle core, while the other end features a hydrophilic triazole unit at the micelle periphery in contact with water, as discussed above in section 3.2 (Fig. 1b). Addition of Fe2+ ions evokes in the DLS trace (Fig. 2, red curve) an additional slow mode at higher correlation times corresponding to larger scattering objects. At the same time, an insoluble precipitate is forming (in particular for the shortest MCL). Under these conditions, the system is out of thermodynamic equilibrium, and thus the DLS methodology may not be appropriate to determine absolute object dimensions. Nevertheless, such DLS measurements can still be consulted as indicator for the aggregation tendency of the polymer systems. These observations suggest that upon iron ion complexation the attractive forces between the triazole units lead to a reorganization of the aggregate architecture, in which the complex units are localized in the core cluster (sketched in Fig. 1b and c). Such complex clusters may serve as building blocks for the larger aggregates formed over time. Precipitation is commonly observed for Fe2+ complexation with triazole derivatives that carry no or only small substituents.44 The shorter polymer chains (P3k and P4k) below the Tc seem to have a sufficiently small space requirement to allow an unconstrained N1,N2-bridging coordination mode (Fig. 1i) even in their swollen state.
In Fig. 2, the DLS curves for the largest polymer ligand P23k below the Tc show no significant difference in water (black) or Fe2+ solution (red). Both are characterized by a fast diffusion process with a single-exponential decay. The data suggest presence of well-defined micellar aggregates with unimodal size distribution of Dh, 22 °C ∼20 nm that consist of few polymer chains (Fig. 1f). This behaviour is in stark contrast to the observations for the short MCLs P3k and P4k, that show a change of aggregation size upon iron addition. For the larger ligands P7k and beyond, no effect of Fe2+ ions on aggregate size is found in the DLS measurements (Fig. S13b†). In pure water below the cloud point, the large ligand P23k possesses a strong hydrophilic character that induces steric repulsion between neighbouring polymer chains. In comparison to the small hydrophobic dodecyl end group localized in the micelle core, the larger polymer coil volume prevents the formation of voluminous spherical micelles with high aggregation numbers. This interference is schematically presented in Fig. 1f and supported by detailed DLS analysis in ESI, Fig. S14.† Addition of iron(II) ions assumedly leads to a reorganization of the aggregates induced by the complexation with the triazole end groups without changing the overall dimensions of the scattering objects. As multiple triazole units coordinate several iron ions and the resulting iron-triazole complexes usually are insoluble in aqueous media, we reasonably assume that the binding sites are localized in the core of the micelles (Fig. 1g and h). Again, the steric demand of the large polymer coils would be responsible for the smaller number of chains per micelle indicated by the DLS data. Thus, such volume demand of the ligands prevents the formation of extended 1D coordination structures associated with N1,N2-bridging mode.
When heating the polymer solutions above the transition temperature Tc, the chains undergo a volume transition and contract as solvent molecules are expelled from the polymer coil. The DLS curve in Fig. 2 shows a single-exponential decay of the shorter polymer P4k in pure water above the cloud point temperature Tc. The thermally induced coil-to-globule transition leads to a narrower size distribution of the scattering objects, corroborating their well-defined dimensions. At T = 35 °C in presence of Fe2+ ions, the monoexponential autocorrelation curve shifts to longer correlation times and indicates an increase in the dimensions of the scattering objects from Dh,H2O = 315 nm in pure water to Dh,Fe = 1375 nm for P4k. Apparently, the attractive forces between the collapsed chains are further enhanced by the coordination processes.
The monoexponential DLS correlation curves of the larger MCL P23k above Tc are presented in Fig. 2. In pure water, the aggregate dimensions substantially increase from Dh,22 °C ∼20 nm upon heating to Dh,35 °C = 117 nm as a consequence of the stronger attraction of the collapsed, hydrophobic polymer chains. Interestingly, the longer polymer ligand displays only about half the aggregate diameter compared to the shorter MCL P4k under identical conditions (Dh,H2O = 315 nm in pure water above Tc). This effect is attributed to the increased coil volume of the longer P23k polymer chains, which arrange with their triazole end groups at the micelle periphery, while the hydrophobic dodecyl groups of the opposite chain ends are localized inside the micelles. Steric crowding with increasing polymer chain length leads to smaller aggregate numbers (Fig. S13c†) with reduced scattering object dimensions. The addition of iron(II) salt above Tc shifts the DLS curve (Fig. 2) to even longer correlation times, demonstrating a coordination-induced growth of the scattering object dimensions to an apparent diameter of Dh = 1375–2555 nm. This temperature-dependent aggregation process is reversible upon thermal cycling (Fig. S15b†), which further supports the schematic switching between the coordination structures in Fig. 1g/h.
TEM measurements. The effect of the coordination processes (Fig. 1c/d and g/h) on the aggregation structure was additionally investigated with a customized TEM method, utilizing trehalose as immobilization matrix and staining with uranyl acetate.38 This particular method allows to image polymer structures isolated from aqueous solution, in analogy to cryo-transmission electron microscopy, to corroborate and further validate the results from DLS (Fig. 2 and Fig. S13†) and UV–Vis-spectroscopy (Fig. 2a and b). The shortest and longest hemi-telechelic MCLs (P3k in Fig. 3 and P23k in ESI, Fig. S16†) were chosen for the TEM investigations, as they provided the most distinct thermoresponsive coordination behaviours (in accordance to Fig. 2).
The short polymer P3k (Fig. 3a) isolated from pure water (T = 22 °C) below the Tc showed spherical to elliptical aggregates with dimensions of 20–30 nm, in overall agreement with the DLS data (Dh = 15 nm, refer to P4k in Fig. 2). The aggregates are randomly distributed throughout the hydrophilic trehalose matrix, with a slight preference to localize at the hydrophobic carbon surface of the TEM grid at higher temperature of T = 40 °C above Tc (Fig. 3b). This indicates their increased hydrophobic character. A noteworthy peculiarity under these conditions is the internal lamellar structure of the aggregates with a width of 30 to 90 nm and an average periodicity of 5–10 nm (Fig. 3b). Again, the data are in accordance with the DLS observations (overall scattering object dimensions: TEM: 150–400 nm, DLS: 316 nm).
Addition of Fe2+ to the aqueous medium at low temperature (T = 22 °C) results in an increase of the P3k aggregate dimensions to 50–100 nm with irregular shapes. The enhanced tendency to aggregate in presence of Fe2+ ions is again in agreement with the multiple scattering species observed in the DLS measurements (compared to Fig. 2c and S13b†), which implicitly hints to the formation of triazole-iron complexes. When the temperature of the P3k-Fe2+ system is raised above Tc to 40 °C (Fig. 3d), yet another substantial reorganization becomes evident in the TEM images with a combined formation of extended, interconnected globular and fibrillar structures. The observations are congruent with the DLS data (Fig. 2) and support the hypothesis of extended 1D coordination structures.
For comparison, the TEM images with the long MCL P23k are provided and discussed in detail in the ESI (Fig. S16†). The visualized aggregate structures under the different experimental conditions are again in good agreement with the results from the DLS measurements and fit the theoretical models outlined in Fig. 1.
Turbidity measurements. The effect of the triazole comonomer APTRZMAAm in the multidentate MCL architecture on the thermal response was studied for copolymer solutions by turbidity measurements. As an interesting side note, the as-synthesized copolymer in water had to be initially heated above the phase transition and cooled down in order to obtain a clear solution. This suggests a kinetic hindrance of the solvation process by strongly interacting chains in the dry state. With increasing incorporation of APTRZMAAm (5–15 mol%) in the copolymers C5k5%–C4k15%, the cloud point shifts to higher temperatures from 36.6 to 44.7 °C (Fig. 5a). This shift reflects the polar character of the APTRZMAAm comonomer, which increases the overall hydrophilicity of the copolymer and the corresponding transition temperature Tc.
After addition of the iron salt Fe(BF4)2·6H2O, Tc drops in relation to the triazole content of the copolymer (ΔTc = 0.6–12.7 °C, Table 5 and Fig. 5a, b). While soluble in pure water above their Tc, all copolymers precipitated upon addition of the Fe2+ salt. For C4.2k15% with the highest APTRZAAm content of 15%, precipitation occurred even below the cloud point (20 °C). The ill-defined turbidity curves for 10% and 15% APTRZMAAm contents are attributed to larger flakes of polymer precipitates that circulated through the light beam in the stirred cuvette. The observed behaviours clearly indicate the increased attraction between the copolymer chains with higher content of triazole coordination sites in the presence of Fe2+ ions.
n, cloud point (Tc), and average of the apparent hydrodynamic diameter at 20 and 60 °C
| Sample code |
n/kg mol−1 |
H2O | Fe2+aq | ||||
|---|---|---|---|---|---|---|---|
| T c, heat/°C | D h, 20 °C/nm | D h, 60 °C/nm | T c, heat/°C | D h, 20 °C/nm | D h, 60 °C/nm | ||
| a Precipitation occurred. b Two scattering species present. | |||||||
| C4.8k5% | 4.8 | 36.6 | 459 106b | 3100 | 35.9 | 1400 | 9500a |
| C4.9k10% | 4.9 | 39.1 | 255 | 2700 | 33.1 | 1500 | 5600a |
| C4.6k15% | 4.6 | 44.7 | 220 | 2000 | 32.4 | 1700a | 4800a |
DLS measurements. DLS measurements were performed with aqueous solutions of the multidentate MCLs at a concentration of 1.0 wt% before and after addition of Fe(BF4)2·6H2O salt at different temperatures, in analogy to the experiments with the hemi-telechelic MCLs. The here reported average dimensions serve predominantly as indicator for structural changes of the scattering objects, since exact values depend on the specific experimental conditions and may be subject of variation due to kinetic effects. Fig. 5c and d show the autocorrelation curves for the triazole-rich copolymer ligand C4.2k15% (complete data set for all copolymers in the ESI, Fig. S17†). At low temperature of 20 °C (below Tc) in pure water, the DLS trace (black curve) in Fig. 5c with a monoexponential decay is characterized by slower scattering objects with well-defined dimensions (Dh = 220–460 nm, Table 5). This indicates micellar aggregation, where the triazole groups are expected to be in contact with the water phase (Fig. 4b). Under these conditions, all copolymer MCLs responded sensitively to the addition of iron(II) ions by an increase of the scattering object dimensions in dependence of the triazole content (red curve, Dh = 1400–1700 nm; Fig. 5c and d). This significant size increase suggests a strong Fe2+-induced attraction between the chains and the formation of coordination networks, as illustrated in Fig. 4c. Raising the temperature of the aqueous copolymer solution above Tc to 60 °C results in larger scattering object dimensions of Dh = 2000–3100 nm (black curve in Fig. 5d, data summarized in Table 5). Under these conditions, the copolymers turn hydrophobic and assemble into colloidal scattering objects. These tend to smaller diameters with larger numbers of hydrophilic triazole units per polymer chain (cf. ESI, Fig. S17†). In presence of Fe2+ salt (red curve), the colloidal scattering objects further grow in size (Dh = 4800–9500 nm, Table 5), and an insoluble precipitate is formed for all ligands (refer to Fig. 4d).
The obtained results for the multidentate MCL poly(NIPAAm-co-APTRZMAAm)-EMP show a marked variability in their colloidal behaviour and thermal response upon the addition of Fe2+ salt. This extreme change in properties is associated with the formation of a coordination network in presence of Fe2+ ions according to the hypothetical picture in Fig. 4. As the copolymers yield very large scattering objects upon Fe2+ salt addition and partially precipitate, no TEM investigations were attempted.
4. Conclusion
We successfully demonstrated the novel concept of endowing thermoresponsive polymers with an Fe2+-coordinating 1,2,4-triazole unit to thermally switch the complex structure of these macromolecular coordination ligands (MCL) in aqueous solution. Such smart building blocks are inspired by two macromolecular architectures based on a (I) hemi-telechelic MCL with triazole end group and (II) a multidentate MCL with multiple triazole substituents along the polymer backbone (ESI, Fig. S18†). The thermoresponsive polymer segments can change their solvation state and interactions in solution upon temperature variation, which consequently alters the overall chain conformation and volume demand. This thermal trigger presents the basis to dynamically influence the complexation behaviour via the switchable coil dimensions of the polymer.A convenient access to these MCLs is provided by the RAFT polymerization method, which allows introduction of the triazole end group through modification of the chain transfer agent, or as side groups by copolymerization with the appropriate triazole comonomer. These two complementary synthetic strategies provide the advantage (a) to fine tune the position and number of the coordination units in the polymer backbone, and (b) to control the MCL chain length and tailor their intrinsic thermoresponsive properties such as solvation and aggregation behaviour.
Both types of architectures feature the following fundamentally different aggregation properties depending on temperature and Fe2+-coordination.
The hemi-telechelic ligands poly(NIPAAm)-DMP-APTRZ below their LCST phase transition allow a clear distinction of the iron-induced aggregation behaviour with respect to the molecular mass. The MCLs with shorter polymer chains (n < 6.9 kg mol−1) showed an increase of the scattering object dimension in DLS upon addition of iron salt, which is attributed to an extended complex formation. In contrast, the larger MCLs did not reveal such effect in presence of Fe2+ ions. This suggests that the bulkiness of the solvated chains prevents association into larger complex structures. Notably, the coil collapse above the phase transition temperature in pure water leads to larger aggregates independent of the MCL chain lengths. Under these conditions, addition of Fe2+ causes a further significant increase of the aggregate dimensions for all MCLs. This corroborates the particular steric impact on the coordination of the iron centres by the collapsed MCLs with reduced volume demand.
In contrast, the multidentate ligands poly(NIPAAm-co-APTRZMAAm)-EMP show a growth in aggregate dimension for both a temperature increase and iron salt addition. The assumed network formation upon iron complexation is supported by the fact that the systems precipitate above the phase transition. Interestingly, the multidentate ligand with the highest triazole content (15 mol%) precipitates even at lower temperatures, evidencing a denser network.
All these results further consolidate the large potential of thermoresponsive MCLs as versatile building blocks in the development of novel multifunctional hybrid materials and as sophisticated scaffolds for reversible assembly processes. The observed dual responsiveness to variations in temperature and presence of metal ions may be exploited in thermoresponsive ion catch-release systems, for sensing, and for catalytic applications in aqueous media.11,45,46
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to kindly thank Dr Arden-Jacob, ATTO-TEC GmbH, for measuring HPLC-MS. While Petra Frank is acknowledged for the general technical assistance in the laboratories, we would like to additionally thank Niklas Jung and Thorben Jaik for the fruitful discussions concerning potential coupling methods. Lastly, we acknowledge the CNRS, the University of Brest, and the European community (FP7 Marie-Curie project: PCIG-GA-2011-304193 NANOCOORD).Notes and references
- H. Zhang, S. Elbaum-Garfinkle, E. Langdon, N. Taylor, P. Occhipinti, A. Bridges, C. P. Brangwynne and A. S. Gladfelter, Mol. Cell, 2015, 60, 220–230 CrossRef CAS PubMed.
- S. Alberti, Curr. Biol., 2017, 27, R1097–R1102 CrossRef CAS PubMed.
- E. W. Martin and T. Mittag, Biochemistry, 2018, 57, 2478–2487 CrossRef CAS PubMed.
- A. K. Rai, J.-X. Chen, M. Selbach and L. Pelkmans, Nature, 2018, 559, 211–216 CrossRef CAS.
- T. E. de Oliveira, C. M. Marques and P. A. Netz, Phys. Chem. Chem. Phys., 2018, 20, 10100–10107 RSC.
- O. Roubeau, Chem. – Eur. J., 2012, 18, 15230–15244 CrossRef CAS PubMed.
- L. H. Gade, Koordinationschemie, Wiley, Weinheim, 2010 Search PubMed.
- T. R. Cook, V. Vajpayee, M. H. Lee, P. J. Stang and K.-W. Chi, Acc. Chem. Res., 2013, 46, 2464–2474 CrossRef CAS PubMed.
- A. Y. Robin and K. M. Fromm, Coord. Chem. Rev., 2006, 250, 2127–2157 CrossRef CAS.
- W. L. Leong and J. J. Vittal, Chem. Rev., 2011, 111, 688–764 CrossRef CAS PubMed.
- W. Li, Y. Kim, J. Li and M. Lee, Soft Matter, 2014, 10, 5231–5242 RSC.
- C. Rüttiger, H. Hübner, S. Schöttner, T. Winter, G. Cherkashinin, B. Kuttich, B. Stühn and M. Gallei, ACS Appl. Mater. Interfaces, 2018, 10, 4018–4030 CrossRef PubMed.
- K. Zhang, S. Liu, Q. Zhao and W. Huang, Coord. Chem. Rev., 2016, 319, 180–195 CrossRef CAS.
- G. R. Whittell and I. Manners, Adv. Mater., 2007, 19, 3439–3468 CrossRef CAS.
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 CrossRef CAS PubMed.
- C. Janiak, Dalton Trans., 2003, 2781–2804 RSC.
- M. Freund, Ber. Dtsch. Chem. Ges., 1896, 29, 2483–2490 CrossRef CAS.
- J. G. Haasnoot, Coord. Chem. Rev., 2000, 200–202, 131–185 CrossRef CAS.
- B. Pergolese, M. Muniz-Miranda and A. Bigotto, J. Phys. Chem. B, 2004, 108, 5698–5702 CrossRef CAS.
- W. Ye, Q. Yao, S. Yu, P. Gong and M. Qin, Molecules, 2017, 22, 1759 CrossRef.
- D. Gupta and D. K. Jain, J. Adv. Pharm. Technol. Res., 2015, 6, 141–146 CrossRef CAS PubMed.
- S. R. Singer and C. N. McDaniel, Plant Physiol., 1982, 69, 1382–1386 CrossRef CAS PubMed.
- M. Erfantalab and H. Khanmohammadi, Spectrochim. Acta, Part A, 2014, 125, 345–352 CrossRef CAS PubMed.
- M. A. Trojer, A. Movahedi, H. Blanck and M. Nydén, J. Chem., 2013, 2013, 946739 Search PubMed.
- Y. Boland, P. Hertsens, J. Marchand-Brynaert and Y. Garcia, Synthesis, 2006, 1504–1512 CAS.
- A. O. Moughton and R. K. O'Reilly, Macromol. Rapid Commun., 2010, 31, 37–52 CrossRef CAS PubMed.
- D. W. R. Balkenende, S. Coulibaly, S. Balog, Y. C. Simon, G. L. Fiore and C. Weder, J. Am. Chem. Soc., 2014, 136, 10493–10498 CrossRef CAS.
- B. Sandmann, B. Happ, S. Kupfer, F. H. Schacher, M. D. Hager and U. S. Schubert, Macromol. Rapid Commun., 2015, 36, 604–609 CrossRef CAS PubMed.
- W.-X. Feng, S.-Y. Yin, M. Pan, H.-P. Wang, Y.-N. Fan, X.-Q. Lü and C.-Y. Su, J. Mater. Chem. C, 2017, 5, 1742–1750 RSC.
- Y. Ono and T. Shikata, J. Am. Chem. Soc., 2006, 128, 10030–10031 CrossRef CAS PubMed.
- H. Ohashi, Y. Hiraoka and T. Yamaguchi, Macromolecules, 2006, 39, 2614–2620 CrossRef CAS.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- M. Wei, Y. Wan and X. Zhang, J. Compos. Sci., 2021, 5, 101 CrossRef CAS.
- A. J. Convertine, N. Ayres, C. W. Scales, A. B. Lowe and C. L. McCormick, Biomacromolecules, 2004, 5, 1177–1180 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- G. Zhou and I. I. Harruna, Macromolecules, 2005, 38, 4114–4123 CrossRef CAS.
- M. Kokkinopoulou, J. Simon, K. Landfester, V. Mailänder and I. Lieberwirth, Nanoscale, 2017, 9, 8858–8870 RSC.
- P. Renz, M. Kokkinopoulou, K. Landfester and I. Lieberwirth, Macromol. Chem. Phys., 2016, 217, 1879–1885 CrossRef CAS.
- N. Pittala, F. Thétiot, S. Triki, K. Boukheddaden, G. Chastanet and M. Marchivie, Chem. Mater., 2017, 29, 490–494 CrossRef CAS.
- H. O. Bayer, R. S. Cook and W. C. Von Mayer, US Patent, 3821376, 1974 Search PubMed.
- A. Postma, T. P. Davis, G. Li, G. Moad and M. S. O'Shea, Macromolecules, 2006, 39, 5307–5318 CrossRef CAS.
- X. Wang, X. Qiu and C. Wu, Macromolecules, 1998, 31, 2972–2976 CrossRef CAS.
- S. Furyk, Y. Zhang, D. Ortiz-Acosta, P. S. Cremer and D. E. Bergbreiter, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1492–1501 CrossRef CAS.
- J. G. Haasnoot, G. Vos and W. L. Groeneveld, Z. Naturforsch., B: J. Chem. Sci., 2014, 32, 1421–1430 CrossRef.
- J.-J. Li, Y.-N. Zhou, Z.-H. Luo and S. Zhu, Polym. Chem., 2018, 10, 260–266 RSC.
- O. Roubeau, A. Colin, V. Schmitt and R. Clérac, Angew. Chem., Int. Ed., 2004, 43, 3283–3286 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures, procedures, and characterization data. See DOI: 10.1039/d1py00847a |
| This journal is © The Royal Society of Chemistry 2021 |