
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurfactant-free emulsion polymerization of vinylidene fluoride mediated by RAFT/MADIX reactive poly(ethylene glycol) polymer chains†
Mathieu
Fuentes-Exposito
 a,
Sébastien
Norsic
a,
Thibaut
Février
a,
Pierre-Yves
Dugas
a,
Salima
Boutti
b,
Samuel
Devisme
c,
Anthony
Bonnet
b,
Franck
D'Agosto
*a and
Muriel
Lansalot
*a
a,
Sébastien
Norsic
a,
Thibaut
Février
a,
Pierre-Yves
Dugas
a,
Salima
Boutti
b,
Samuel
Devisme
c,
Anthony
Bonnet
b,
Franck
D'Agosto
*a and
Muriel
Lansalot
*a
aUniv Lyon, Université Claude Bernard Lyon 1, CPE Lyon, CNRS, UMR 5128, Catalysis, Polymerization, Processes and Materials (CP2M), 43 Bd du 11 Novembre 1918, 69616 Villeurbanne, France. E-mail: franck.dagosto@univ-lyon1.fr; muriel.lansalot@univ-lyon1.fr
bARKEMA, Centre de Recherche Rhône-Alpes (CRRA), Rue Henri Moissan - CS 42063, 69491 Pierre-Bénite Cedex, France
cARKEMA, Centre de recherche, développement, applications et technique de l'ouest (Cerdato), 13 route de Launay, 27470 Serquigny, France
First published on 13th September 2021
Abstract
A robust and straightforward synthesis of surfactant-free poly(vinylidene fluoride) (PVDF) latexes is presented using RAFT/MADIX-mediated emulsion polymerization. VDF emulsion polymerizations were conducted in the presence of commercial poly(ethylene glycol) chains (PEG-OH) and in the presence of the same chains carrying a dithiocarbonate (xanthate) chain end (PEG-X). The identification of the stabilization mode in both cases is carefully examined, considering the irreversible chain transfer reactions well known in VDF free radical polymerization. The particle size is significantly smaller when PEG-X was used, demonstrating the positive effect on particle stabilization of the involvement of xanthate chain end in the free radical process.
Introduction
Poly(vinylidene fluoride) is a semicrystalline fluoropolymer with unique thermal and mechanical properties combined with excellent chemical, UV and abrasion resistance, and electrochemical and oxidation stability. It thus finds applications in various fields, such as outdoor coatings, lithium ion batteries, photovoltaics, porous membranes, cables and wires, just to name a few.1–3 PVDF is industrially produced by aqueous free-radical emulsion and suspension polymerization. Nevertheless, VDF emulsion polymerization is poorly documented in the open literature in terms of kinetics and processes.4–7 This probably comes from the fact that it has to combine the use of high pressure reactors to work with gaseous VDF, the high reactivity of the propagating radicals and the resulting complex physical chemistry of this free radical polymerization when conducted in water. Besides, VDF emulsion polymerization requires a surfactant to stabilize the PVDF particles. However, low molar mass surfactants can have negative effects on the final properties of the material as they can migrate with time.8 A very attractive way of avoiding the use of such molecules is to employ reactive hydrophilic macromolecules, which can be involved in the emulsion polymerization.9 Poly(ethylene glycol) (PEG)-based molecules are commonly used as stabilizers in the latex industry. Indeed, steric stabilization provided by PEG chains can strongly increase the particle stability against freeze–thaw, shear or in the presence of polyelectrolytes.10 In that respect, PEG-based macromolecules incorporating reactive groups such as macromonomers,11–13 macroinitiators,14–16 and macromolecular chain transfer agents (macroCTA)10,17–20 have been reported for polymerization in dispersed media.To address the constraints related to the use of fluorinated and low molar surfactants, various poly(ethylene glycol) (PEG) based-molecules have been used and depicted in the patent literature related to VDF emulsion polymerization, however mainly for the synthesis of poly(vinylidene fluoride-co-hexafluoropropylene).21–25 Surfactant-free PVDF-based latexes were thus obtained using reactive PEG (meth)acrylate macromonomers. Surprisingly, unreactive hydrosoluble PEG-OH chains alone can also act as precursor of stabilizer during the emulsion polymerization process by providing stable latexes. The patent literature mentioned above does however not provide any mechanistic details on how commercial PEG-OH can bring particle stability. Nevertheless, VDF free radical polymerization is known to be prone to chain transfer reactions to the monomer, to the polymer (via inter- (long chain branching) or intramolecular (short chain branching) transfer reaction) and to chain transfer agents such as e.g. ethyl acetate.4,5,7,26 Indeed, labile hydrogen atoms carried by these species could be abstracted by the very reactive PVDF propagating macroradicals leading to new radicals, which should initiate new PVDF polymer chains. In that context, chain transfer reactions occurring along PEG-OH chains during the emulsion (co)polymerization of VDF could thus form short PVDF grafts along the PEG chains and result in the in situ formation of an amphiphilic stabilizer.
The advances achieved in polymer synthesis by reversible-deactivation radical polymerization (RDRP) techniques over the last 20 years have pushed forward the use of living hydrophilic polymers for the synthesis of polymer particles in emulsion. Indeed, these well-defined and reactive macromolecules can be chain-extended with a hydrophobic monomer directly in water leading to the in situ formation of amphiphilic block copolymers that simultaneously self-assemble into polymer nanoparticles. The process, coined polymerization-induced self-assembly (PISA), can be applied to different polymerization techniques,27–30 but the most studied one undoubtedly remains reversible addition–fragmentation chain transfer (RAFT)/macromolecular design by interchange of xanthates (MADIX).31,32 PISA is also applicable in dispersion polymerization (where the core forming monomer is soluble in the continuous phase), both in aqueous and organic media.31,33–35 Hence, water-soluble chains obtained either by the RAFT polymerization of hydrophilic monomers or via the chemical modification of preformed polymers can act as chain transfer agent (and referred to as macroCTA) generating stabilizers during an emulsion polymerization by forming amphiphilic block copolymers in situ. PEG-based macroCTA have thus been reported for the PISA synthesis of various kinds of particles,36–51 but to the best of our knowledge, never for PVDF latexes.
Using very low amounts of such macroCTA in an emulsion polymerization seems to be an economically viable approach to access surfactant-free latexes. In that case, the initial amount of the macroCTA is such that enough amphiphilic block copolymers are formed in situ to ensure stabilization of the particles produced simultaneously by emulsion polymerization. This approach is actually quite interesting for the synthesis of industrially relevant polymer latexes, for instance in paint technology, where the fraction of hydrophilic species in the final coating must remain low. Using this strategy, we indeed prepared high solids content (ca. 40 wt%) polymer latexes incorporating less than 2 wt% of hydrophilic species with poly(vinylidene chloride)52,53 and polyacrylics.54–57 The stabilizer being strongly anchored to the particle surface at the end of the emulsion polymerization, the obtained latexes led to polymer films with enhanced water barrier properties. The strategy was also successful for the synthesis of poly(vinyl acetate-co-ethylene).58
In the present paper, emulsion polymerization of VDF is conducted for the first time in the presence of hydrophilic RAFT/MADIX macroCTA. In order to accurately comprehend the role of the thiothiocarbonylated chain end in the process, PEG-OH chains and their analogues equipped with a dithiocarbonate (xanthate) moiety (PEG-X) have been comparatively evaluated in the synthesis of surfactant-free synthesis of PVDF latexes. Several parameters are varied such as the PEG molar mass and the macroCTA structure (mono versus bifunctional xanthate) (Scheme 1).
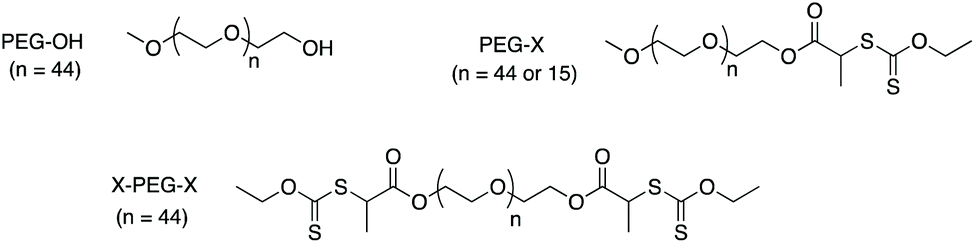 | ||
| Scheme 1 PEG-based macroCTAs used for VDF emulsion polymerization. | ||
Experimental
Materials
Poly(ethylene glycol) methyl ether (PEG-OH, Mn ≈ 2000 and 750 g mol−1, Aldrich), α,ω-dihydroxy poly(ethylene glycol) (HO-PEG-OH, Mn ≈ 2050 g mol−1, Aldrich), poly(ethylene glycol) methyl ether thiol (PEG-SH, Mn ≈ 2000 g mol−1, Aldrich), 1,4-dioxane (Alfa Aesar, 99.8%), triethylamine (Aldrich, 99.5%), 2-bromopropionyl bromide (Aldrich, 97%), sodium hydrogen carbonate (NaHCO3, Aldrich, 99.7%), ammonium chloride (NH4Cl, Aldrich, 99.5%), dichloromethane (Aldrich, 99.8%), magnesium sulfate (Aldrich, >99.9%), O-ethyl xanthic acid (Aldrich, 96%), potassium persulfate (KPS, Aldrich, 99%), sodium acetate (Aldrich, 99%) were used as received. Vinylidene fluoride (VDF) was kindly provided by Arkema (Pierre Bénite, France) and used as received. Water was deionized with a PureLab system (Purelab Classic UV, Elga LabWater). Tetrahydrofuran (THF, HPLC, stabilized/BHT, Sigma Aldrich) was used for SEC analyses.Methods
1H NMR (400 MHz, CDCl3, δ): 4.6 (q, 2H, O–CH2–CH3); 4.4 (q, 1H, CH–S); 4.3 (t, 2H, CH2–CH2–O); 3.75–3.5 (s, 180H, (CH2–CH2–O)n); 3.35 (s, 3H, CH3–O); 1.6 (d, 3H, CHCH3); 1.4 (t, 3H, CH2–CH3).
The same procedure was followed with another commercial PEG-OH (Mn = 750 g mol−1), leading to PEG-X with Mn,SEC = 1300 g mol−1 and Đ = 1.10. The 1H NMR spectrum of the final product is shown in Fig. S3,† while Fig. S4† displays the chromatogram obtained by SEC analyses in THF using PS standards.
1H NMR (400 MHz, (CD3)2CO, δ): 4.6 (q, 2H, O–CH2–CH3); 4.4 (q, 1H, CH–S); 4.3 (t, 2H, CH2–CH2–O); 3.8–3.5 (s, 70H, (CH2–CH2–O)n); 3.3 (s, 3H, CH3–O); 1.55 (d, 3H, CH–CH3); 1.4 (t, 3H, CH2–CH3).
Synthesis of a di-functional poly(ethylene glycol)-xanthate (X-PEG-X) macroCTA agent
α,ω-Dihydroxy poly(ethylene glycol) (Mn = 2050 g mol−1) (20 g; 0.01 mol) was dissolved in dichloromethane (80 mL) in a round bottom flask and triethylamine (5.46 g; 0.054 mol) was then added. The mixture was added dropwise in 2-bromopropionyl bromide (9.94 g; 0.046 mol) putting an ice bath. The flask was removed from the ice bath after the mixture addition and the reaction mixture stirred for 16 h. After filtration of the residual salts, the organic phase was washed with saturated aqueous solution of NH4Cl (1 × 15 mL), NaHCO3 (1 × 15 mL) and water (1 × 15 mL). The washed organic phase was then dried with magnesium sulfate and the solvent evaporated under vacuum. The product obtained (16.20 g; 0.0079 mol) was dissolved in dichloromethane (55 mL). Then, O-ethyl xanthic acid (7.60 g; 0.0474 mol) was added by small amounts under stirring. The reaction mixture was stirred overnight. KBr salts were removed by filtration. The mixture was washed with saturated aqueous solution of NH4Cl (2 × 15 mL) and of NaHCO3 (2 × 15 mL) then water (1 × 15 mL). The washed organic phase was then dried with magnesium sulfate and the solvent evaporated under vacuum. The polymer was then precipitated in cold petroleum ether. Finally, the product was dried under vacuum. The 1H NMR spectrum of the final product is shown in Fig. S5,† while Fig. S6† displays the chromatogram obtained by SEC analyses in THF (Mn,SEC = 3420 g mol−1; Đ = 1.10/PS standards).1H NMR (400 MHz, CDCl3, δ): 4.6 (q, 2H, O–CH2–CH3); 4.4 (q, 1H, CH–S); 4.3 (t, 2H, CH2–CH2–O); 3.75–3.5 (s, 220H, (CH2–CH2–O)n); 1.6 (d, 3H, CHCH3); 1.4 (t, 3H, CH2–CH3).
Emulsion polymerization of vinylidene fluoride
VDF emulsion polymerizations were all carried out in a 50 mL stainless steel autoclave equipped with a nitrogen inlet, a thermometer, a mechanical stirrer and a pressure sensor. In a typical polymerization procedure, KPS, PEG-OH (or PEG-X or X-PEG-X), sodium acetate used as buffer and deionized water (25 mL) were introduced in the reactor. The medium was deoxygenated under nitrogen for 30 min. VDF gas was fed into the reactor until the targeted pressure (30 bar). Immediately after, the injection port was closed and the medium heated at a set point temperature of 80 °C. At the end of the experiment, the reactor was cooled with iced water. When the temperature inside the reactor dropped below 25 °C, the remaining pressure was carefully released, and the obtained latex was collected, and the particle size was measured. A fraction of the obtained latex was dried for solids content (SC) measurement, which after subtraction of nonpolymeric species gave the polymer content (PC, %). The dried polymer was then used for polymer characterization by DSC.For a scale-up experiment, VDF emulsion polymerization was carried out in a 4 L high pressure stainless steel autoclave equipped with a nitrogen inlet, a thermometer, a mechanical stirrer and a pressure sensor. The reactor temperature was measured by a thermocouple J Atex, protected by a metal tube and placed inside the reactor. Oil (Ultra 350, Lauda) circulating in the jacket was used to control the reactor temperature (no oil circulated in the cover neither the bottom). The inlet and outlet temperature of the circulating oil in the reactor jacket was measured with a platinum resistance Pt100. The reactor pressure was monitored with a pressure sensor Atex (type PA-23EB, Keller). In a typical polymerization procedure, KPS, PEG-OH (or PEG-X), buffer (sodium acetate) and deionized water (2 L) were introduced in the reactor. The medium was deoxygenated under nitrogen for 30 min. VDF gas was fed into the reactor until the targeted pressure (30 bar). Immediately after, the injection port was closed and the medium heated at a set point temperature of 80 °C. When the temperature inside the reactor dropped below 25 °C, the remaining pressure was carefully released, and the obtained latex was collected, and the particle size was measured. A fraction of the obtained latex was dried for SC measurement, which after subtraction of nonpolymeric species gave the PC. The dried polymer was then used for polymer characterization by DSC.
Characterization
where Np is the number of particles (L−1), PC the polymer content (g L−1), Dz the average particle size (cm) and dp the polymer density (g cm−3). Here, dp was set at 1.78 g cm−3.
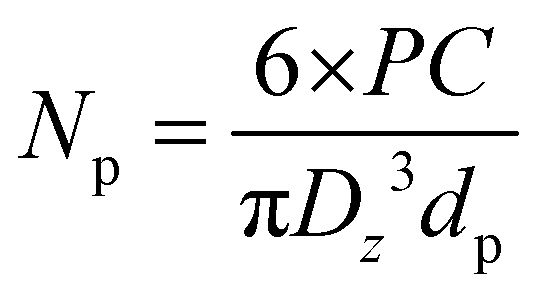
| Xc(%) = (ΔHf,measured/ΔHf,∞) × 100 |
Results and discussion
As mentioned in the Introduction, examples on the use of PEG-OH chains as stabilizer precursors in emulsion (co)polymerization of VDF has only been depicted in the patent literature without any indication on the stabilization mode of the particles. In the following, we first investigated the polymerization of VDF under 30 bar pressure in 25 mL of water in absence of molecular surfactant and using potassium persulfate (KPS, 50 mg) as initiator at 80 °C. Several attempts were first performed in the presence of various quantities of PEG-OH (Mn = 2000 g mol−1) with always a KPS/PEG-OH weight ratio lower than 1, i.e. PEG-OH amounts higher than 50 mg. These first experiments were indeed designed with the view of employing the minimum amount of PEG-OH (and further on of PEG-X) while still being able to characterize structural modifications on the PEG backbone during the emulsion polymerization. However, none of these experiments led to polymerization. Indeed, with 50 mg of KPS in the recipe, the weight ratio KPS/PEG-OH had to be adjusted to 2.5 to form a stable PVDF latex and measure appreciable conversions of VDF (Table 1 – L01, 11.2 wt% solids after 4 h).| Exp.a | Stabilizer | KPS/stabilizerb | Time (h) | SCc (%) | PEG fractione (wt%) |
D
z
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) f (nm) f (nm) |
PdIf | N p (×1016 L−1latex) | |
|---|---|---|---|---|---|---|---|---|---|
| Name | n | ||||||||
| a T = 80 °C; P = 30 bar. Fixed amount of KPS (7.3 mmol L−1) and sodium acetate – Variable PEG nature and weight. n = number of ethylene glycol units. b Weight ratio. c Solids content taking into account all the non-volatile species (including stabilizer and PVDF). d Performed in a 4 L reactor in the presence of 2 L of water, 4 g of KPS, 2.5 g of sodium acetate and 1.6 g of PEG-X or PEG-OH. e Weight fraction of PEG in the final latex considering the initial amount of PEG species and the final amount of PVDF. f Determined by DLS. g The PdI of this sample was high (0.7) and the Dz value was thus not considered as relevant. | |||||||||
| L01 | PEG-OH | 44 | 2.5 | 4 | 11.2 | 0.7 | 234 | 0.01 | 0.9 |
| L02 | — | — | — | 1 | 7.8 | — | 378 | 0.31 | — |
| L03 | PEG-X | 44 | 2.5 | 4 | 10.4 | 0.8 | 72 | 0.06 | 30.3 |
| L04 | PEG-SH | 44 | 2.5 | 3.25 | 8.1 | 1.0 | 95 | 0.04 | 10.1 |
| L05 | PEG-X | 44 | 1.25 | 8 | 2.4 | 6.7 | —g | —g | — |
| L06 | 5 | 4 | 14.4 | 0.3 | 101 | 0.05 | 15.2 | ||
| L07 | PEG-X | 15 | 2.5 | 4 | 10.4 | 0.8 | 62 | 0.06 | 47.5 |
| L08 | 6.7 | 4 | 15.9 | 0.2 | 99 | 0.03 | 17.9 | ||
| L09 | X-PEG-X | 44 | 2.5 | 4 | 8.5 | 0.9 | 72 | 0.06 | 24.1 |
| L10d | PEG-OH | 44 | 2.5 | 4.25 | 7.6 | 1.1 | 225 | 0.04 | 0.7 |
| L11d | PEG-X | 44 | 2.5 | 4.25 | 6.8 | 1.2 | 72 | 0.06 | 19.5 |
This result was explained by the peculiar reactivity of the propagating radicals in VDF polymerization. Indeed, as mentioned in the introduction and depicted in the literature, irreversible transfer reactions are particularly pronounced in the presence of hydrogenated species. All the hydrogens atoms of PEG-OH are adjacent to the oxygen atoms and thus labile. They are probably responsible for the inhibition observed. These first unsuccessful experiments give an additional information. The inhibition observed is indeed consistent with a degradative chain transfer, i.e. without subsequent efficient reinitiation of the VDF polymerization (Scheme 2A-a). Decreasing the amount of PEG versus KPS would allow, once enough reactive transfer sites on the PEG chains (hydrogen atoms adjacent to the oxygen ones) have been neutralized by forming an amphiphilic structure after coupling (Scheme 2A-b), to produce enough radicals to efficiently start the polymerization. Indeed, L01 provided a stable latex with particle size of 234 nm. The stability of the latex obtained cannot be explained only by the charges provided by the initiator KPS. The latter contribution is indeed real, as attested by the formation of a stable PVDF latex in the sole presence of KPS under the same conditions (Table 1, L02, 7.8 wt% solids after 1 h). However, the large particle size obtained in that case (378 nm) compared to the latex formed in the presence of PEG-OH (L01, 234 nm) indicates a beneficial contribution of this macromolecule on the particle stability. As mentioned above, in L01, the amount of PEG used (20 mg, for a final weight fraction of 0.7 wt% with respect to PVDF, Table 1) makes the isolation and the characterization of the stabilizer formed almost impossible. Taking into account the observed inhibition when the KPS/PEG-OH weight ratio is too low, these structures would not be the result of a reinitiation of the polymerization of VDF on the PEG after a transfer reaction, although this cannot be completely rule out.
Indeed, reinitiation of VDF polymerization by –CH2–CH˙(OMe) has been reported in the literature.59 In this work, the successful synthesis of block copolymers based on poly(methyl vinyl ether) (PMVE) and PVDF segments was achieved by a combination of cationic RAFT and RAFT polymerizations. PMVE chains carrying a dithiocarbamate chain end (PMVE-DTC) were used to control RAFT polymerization of VDF. This however takes place in dimethylcarbonate, a good solvent for VDF, whereas VDF is only slightly soluble in water. In addition, VDF affinity for a hydrophilic polymer such as PEO dissolved in water is low compared to the affinity of VDF for PMVE in DMC. Besides, oligomers of PVDF formed in water in an emulsion process (+K,−OSO3-PVDF˙) will carry an anionic charge (coming from the KPS initiator) and their affinity with hydrosoluble species will be increased favoring both transfer reaction but also coupling reactions with PEG chains. In that case, the bimolecular termination between a growing hydrosoluble +K,−OSO3-PVDF˙ oligoradical and the radical generated by transfer between another growing hydrosoluble +K,−OSO3-PVDF˙ oligoradical and PEG (Scheme 2A-b) will likely be favored. The negative charges carried by−OSO3-PVDF˙ oligomers or by the grafted structure, would favorably contribute to stabilization of the particles.
As mentioned above, the presence of additional PVDF grafts resulting from reinitiation after transfer (thus not carrying sulfate groups) cannot completely be ruled out (Scheme 2B, red PVDF grafts). Eventually, transfer reactions on the hydrogen of the tertiary carbon atom formed by the previous assumed reactions (coupling or reinitiation) are also possible although, considering steric effects, the formation of two grafts on the same carbon may be disfavored (Scheme 2B, green PVDF grafts). Scheme 2B represents a structure in which all the above possibilities are compiled. For the sake of clarity, this structure only accounts for the normal head-to-tail additions of propagating radicals (–CH2CF2–CH2CF2–) onto VDF and not for the well-known and more complicated microstructure of PVDF produced by free radical polymerization, which additionally includes head-to-head (–CH2CF2–CF2CH2–) and tail-to-tail (–CF2CH2–CH2CF2–) additions.1,60
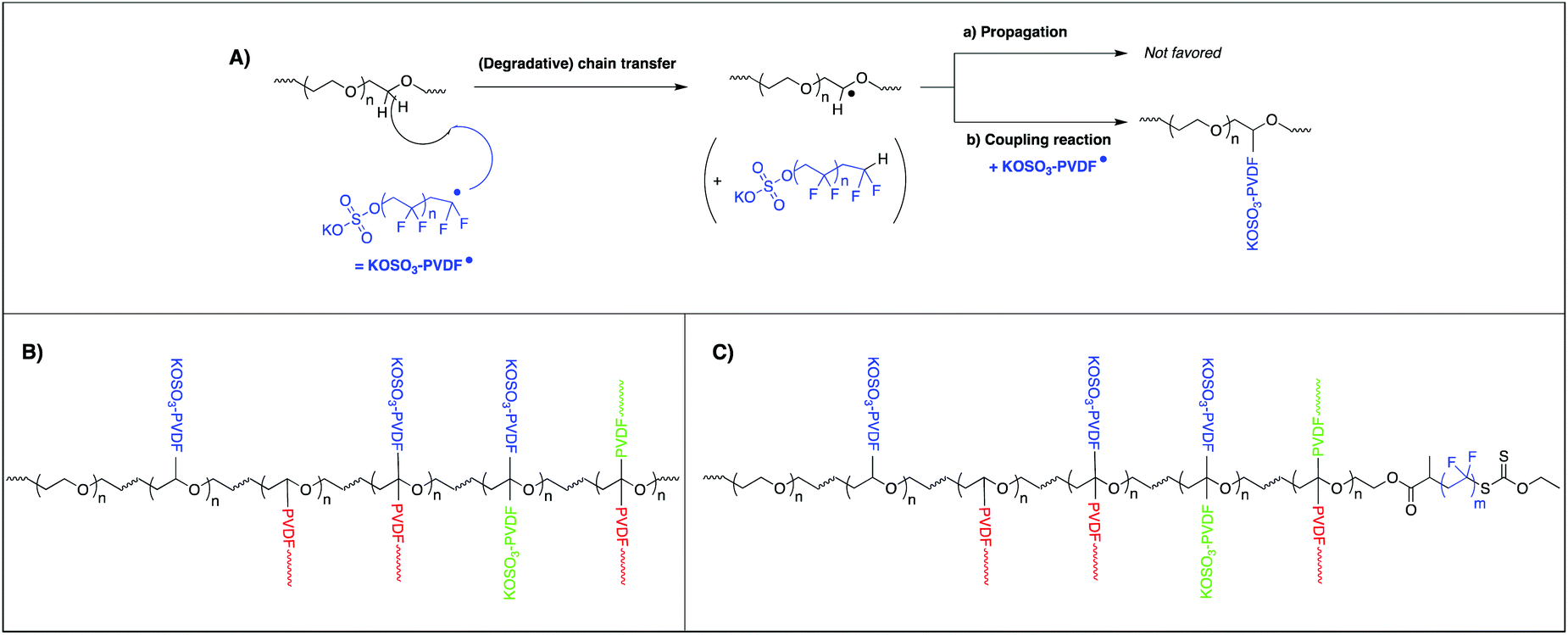 | ||
| Scheme 2 (A) Degradative chain transfer onto PEG chains followed by coupling reaction with PVDF oligoradicals, and possible chemical modifications occurring during emulsion polymerization of VDF onto (B) PEG-OH chains and (C) PEG-X chains. Please note that in the structures presented in (B) and (C), the two carbon atoms of the PEG repetitive units may carry a branch but for the sake of simplicity only one carbon atom has been considered to illustrate the chain transfer events. | ||
Strong of these first understandings on the potential stabilization mechanisms and on the role of PEG-OH as precursor of stabilizer in VDF emulsion polymerization, we then investigated the use of PEG-X (Mn = 2300 g mol−1 and Đ = 1.03) as stabilizer precursor. The choice of the installation of a xanthate moiety at the end of the PEG chains was dictated by the good control observed when RAFT polymerization of VDF was conducted in organic solvent61–68 and by the successful block copolymer syntheses reported in organic solvent by chain extension using polymers functionalized with xanthate chain ends,69–72 including an attempt to perform VDF PISA in dimethyl carbonate from a poly(vinyl acetate) macroCTA.70 These results were a strong indication that a chain extension of PEG-X with VDF could take place in our systems although it does not exclude the occurrence of the grafting reactions very specific from the emulsion polymerization process and identified above. The commercial PEG-OH used above was chemically modified to introduce the xanthate functionality. The xanthate is meant to promote reversible transfer reactions at the end of the chain and minimize degradative chain transfer reactions along the PEG chain as observed when RDRP of VDF was performed.61,63,73–75 A stable PVDF latex is indeed obtained in presence of PEG-X (Table 1 – L03). After 4 hours of polymerization the solids content (10.4%) is very similar to the one obtained with PEG-OH (11.2%, L01) showing that the presence of the xanthate chain ends does not significantly affect the yield of polymer produced. It is worth mentioning that a comparative kinetic study presented in Fig. S7† shows that the snapshot given after 4 hours of polymerization does not hide a completely different kinetic behaviour between PEG-OH and PEG-X. Indeed, rather similar kinetic profiles were observed, with a different but steady polymerization rate in both cases.
Besides, isometric particles with a size of 72 nm are obtained compared to 234 nm with PEG-OH. This result shows the strong and beneficial impact of favoring chain transfer reaction via the xanthate group on particle stabilization. This goes along with a higher number of formed particles when using PEG-X (almost 30 times higher, see L01 and L03 in Table 1). This is further attested by cryo-TEM pictures of the corresponding PVDF latexes (Fig. 1). The difference in sizes observed with DLS is confirmed by cryo-TEM. Besides, brighter areas corresponding to less electron dense parts are visible by cryo-TEM inside the PVDF particles synthesized with PEG-OH (Fig. 1A). This phenomenon has already been reported for particles synthesized with PEG-based macroCTA,19,76 PEG-based stabilizers77,78 or initiators,79 or even KPS alone.80 These parts probably correspond to the formation of water pockets during the emulsion polymerization, resulting from buried PEG-based species inside the particles and leading to the brighter area observed during cryo-TEM analysis. This corroborates the fact that the structure shown in Scheme 2B is probably not the optimum one to act as stabilizer and to stabilize water/PVDF interfaces. The much smaller size and the absence of less electron dense parts inside the particles when PEG-X is used (Fig. 1C) confirm the superior ability of PEG-X to act as a (precursor of) stabilizer in this case. The formation of a PVDF segment from the chain end of PEG-X, PVDF segment probably longer than the side PVDF grafts as resulting from a reversible transfer from the xanthate chain end, would then drive the anchorage of the overall structure as depicted in Scheme 2C, at the surface of the particles optimizing the stabilization.
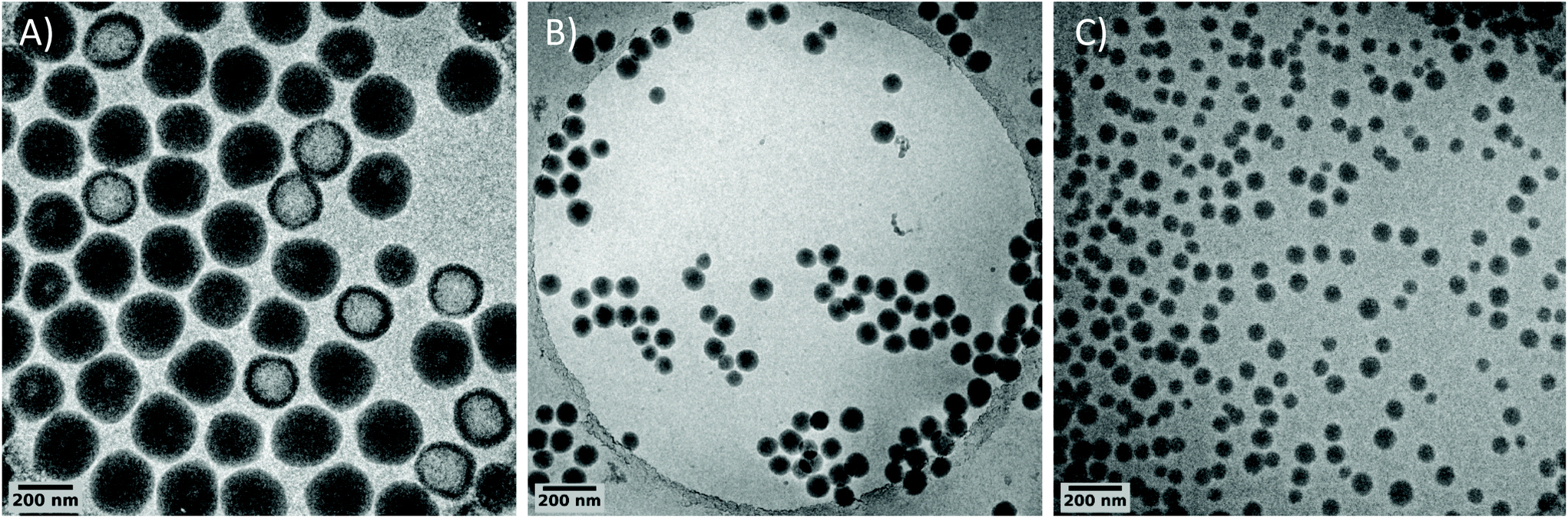 | ||
| Fig. 1 Cryo-TEM images of PVDF particles prepared with (A) PEG-OH (L01), (B) PEG-SH (L04) and (C) PEG-X (L03). | ||
The beneficial effect of the reversibility of the transfer reaction induced by the xanthate moiety can be supported by experiment L04 in which PEG chains carrying a thiol chain end (PEG-SH – Mn = 2000 g mol−1) were used instead of PEG-X. In this case, a lower yield (solids content 8.1%) was obtained after 3.25 h while the particle size was already larger (95 nm, Fig. 1B) than the one obtained with PEG-X. PEG-SH has been previously used in emulsion polymerization of styrene to generate block copolymers surfactant during the process.14 Besides, thiols are known to induce irreversible chain transfer reactions in VDF polymerization.81 As a result, the small size particles obtained with PEG-SH confirm the formation of short PVDF segments from PEG via irreversible chain transfer and thus in situ formation of stabilizers. It further shows the superiority of the reversible chain transfer reaction induced by the xanthate for the stabilization of PVDF latex.
The above results showed that the presence of xanthate chain end favors reversible chain transfer at the end of PEG chains over the degradative chain transfer reactions along the PEG backbone. In that respect, we hoped to be able to slightly increase the amount of PEG-X in the formulation without lowering too much the final solids content for the same polymerization time. This could have also eased the identification of the chemical modifications occurring on the PEG during the emulsion process. However, when increasing the amount of PEG-X in the recipe (KPS/PEG-X weight ratio of 1.25 in L05, Table 1), the solids content greatly decreased to only 2.4% even after a prolonged polymerization time (8 hours). This phenomenon is directly linked to the increase in the content of hydrogenated species. Indeed, decreasing on the other hand PEG-X content to reach a KPS/PEG-X ratio of 5 (L06) allowed the formation of 100 nm PVDF particles, with a solids content of 14.4% in 4 hours.
As mentioned above, the initial amount of PEG-X cannot allow a classical structural characterization of the PEG chains after polymerization. Indeed, the final weight fraction of PEG (with respect to the formed PVDF) in these latexes is typically lower than 1 wt% (except for the low SC latex L05, Table 1). Nevertheless, the strategy remains very interesting as it minimizes the amount of stabilizer precursor and in the end the fraction of hydrophilic species in the final latexes. Indirect information on the structure of the stabilizer can however be gathered by additional physico-chemical characterizations of the final latex. Surface tension analyses were thus performed on L01 (PVDF latex from PEG-OH) and L03 (PVDF latex from PEG-X) in order to quantify in both systems, the amount of free PEG chains, i.e., not participating to particle stabilization. Calibration curves were first established by measuring the surface tension of aqueous solutions of various concentrations of PEG-OH and PEG-X (Fig. S8†). According to the calibration curve, 82.5 wt% of the initial amount of PEG-OH is present as free chains in the final latex L01, whereas only 1.0 wt% of the initial amount of PEG-X is present as free polymer chains in the latex L03. These very contrasted results are fully consistent with the superiority of PEG-X compared to PEG-OH to stabilize PVDF latex obtained by emulsion polymerization. As a comparison, using the same surface tension measurements, 23 wt% of the initial PEG-SH was present as free chains, results consistent with a lower efficacity of stabilization in this case, as discussed above. All these data should however be considered with care as the surface tension measured may not be only the reflect of the contribution of free PEG chains in the water phase. Indeed, the aqueous phase may also contain grafted fluorinated PEG species (see Scheme 2), the contribution of which will not be accurately reflected by the calibration curves obtained from the initial PEG species (i.e., PEG-OH, PEG-X or PEG-SH).
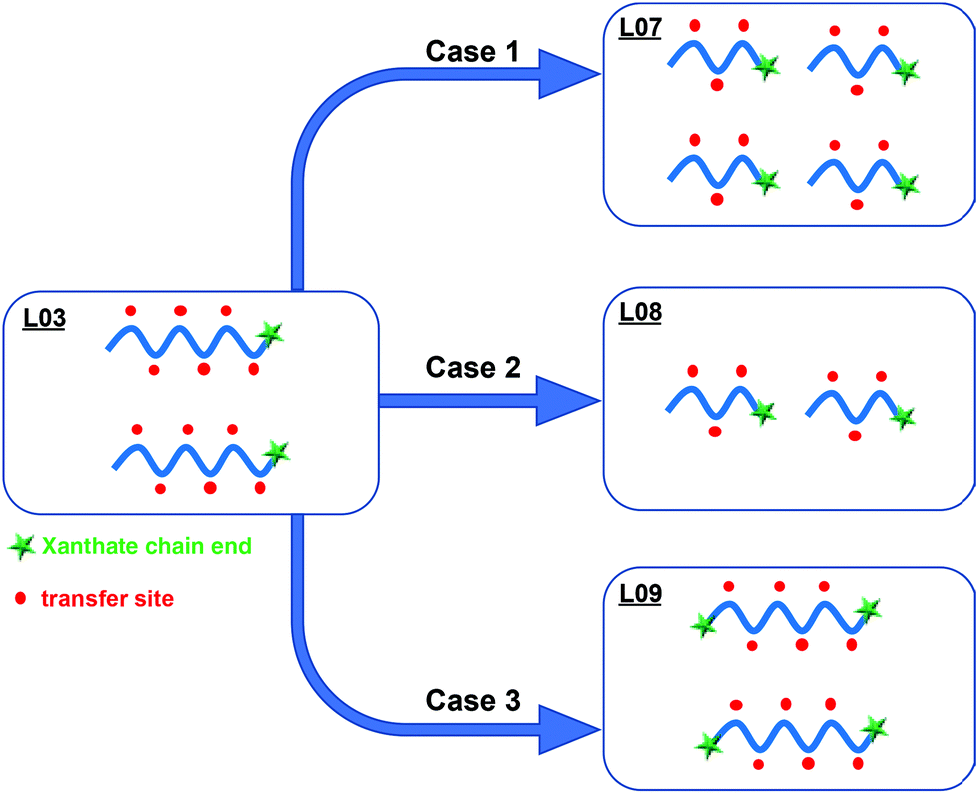
The VDF emulsion polymerizations conducted with PEG-X showed that the presence of the xanthate chain end on PEG was relevant, but also that the solids content that can be reached for a given polymerization time of the polymerization was linked to the number of hydrogenated species (in other words, the amount of PEG-X initially introduced). To emphasize the positive contribution of the reactive xanthate moiety on the formation of PVDF latex, experiments were designed with a lower molar mass PEG-X (Mn = 1300 g mol−1) or by introducing xanthate moieties on both chain ends (X-PEG-X, Mn = 3420 g mol−1) (Scheme 1). Experiment L07 in Table 1 was thus conducted with PEG-X of 1300 g mol−1 and the same KPS/PEG-X weight ratio as in L03. The number of xanthates moieties is thus doubled compared to L03 with a number of potential sites along the PEG chains for transfer reaction remaining the same (Scheme 3, case 1). The particle size obtained is smaller in L07 (62 nm) compared to L03 (72 nm) while the solids content remained unchanged (10.4%), leading to the formation of more particles (47.5 × 1016vs. 30.3 × 1016). As anticipated, this result shows the beneficial effect of favoring reversible chain transfer reaction on the xanthate chain ends over chain transfer along PEG backbone. In the same vein, decreasing the initial amount of PEG-X for a similar number of xanthate chain ends with respect to L03 (L08, KPS/PEG-X = 6.7, Scheme 3, case 2) effectively decreased the occurrence of irreversible chain transfer reactions leading to a 99 nm PVDF latex with a higher solids content (15.9%), in good agreement of the results obtained in experiment L06.
 | ||
| Scheme 3 Experiments performed with various amounts of PEG-X with different Mn (1300 or 2300 g mol−1). | ||
The positive impact of the reactive xanthate group on the particle stabilization was also demonstrated in a last experiment performed with X-PEG-X, keeping the KPS/X-PEG-X ratio to 2.5 (L09). This means that the number of xanthate moieties involved in this polymerization is twice the number involved in L03, for the same number of potential irreversible chain transfer sites along the PEG backbone (Scheme 3, case 3). This also means that if both xanthate moieties are efficiently reacting, the PEG chains will form loops on the particle surface. The particle size is similar for the two latexes (72 nm). However, the SC is lower in the case of L09 (8.5%) compared to L03 (10.5%). Larger particle size is thus expected for L09 for similar solids contents. The result is not against the formation of the expected two outer PVDF blocks at both PEG chain ends. However, the anchorage of the resulting amphiphilic structure onto the forming PVDF particles may disfavor the deployment of the hydrophilic part (PEG backbone with side PVDF grafts) of the corresponding stabilizer at the surface. The lower number of particles obtained for L09 (24.1 × 1016) compared to L03 (30.3 × 1016) might reflect a slightly lower stabilization efficiency.
The thermal properties of the PVDF polymers obtained using the different above-mentioned PEG-based species as stabilizer were investigated using DSC (Table S1†). The analyses showed that semi-crystalline polymers were formed, with Tm, Tc and Xc values in the range of what is commonly observed for PVDF polymers formed by emulsion polymerization.2,82
Eventually, experiment L01 and L03 in Table 1, performed with PEG-OH and PEG-X respectively, were scaled up in a 4 L reactor in experiments L10 and L11, respectively (Table 1). The solids content obtained after 4 h15 (7.6 wt% and 6.8 wt%) are similar to those for L01 and L03 (11.2 wt% and 10.4 wt%) although slightly lower. This probably accounts for the very different stirring and volume over surface ratios in the two sets of conditions. Nevertheless, it shows this technology is robust and that its transposition to a larger scale is possible.83
Conclusions
This work describes the synthesis of self-stabilized PVDF particles by combining the advantages of emulsion polymerization with those of controlled radical polymerization using the RAFT process. First, a commercially available methoxy poly(ethylene glycol) carrying a hydroxyl function (PEG-OH, Mn = 2000 g mol−1) was used for the stabilization of PVDF particles. Stable PVDF particles were obtained with a diameter of 234 nm. The stabilization is provided by irreversible transfer reactions occurring along the PEG-OH chains leading to the formation of a grafted copolymer stabilizer in situ. The same PEG-OH was then chain-end functionalized to introduce a xanthate group (PEG-X). The experiments carried out in the presence of PEG-X demonstrated the beneficial implication of the reactive xanthate chain-end in the VDF emulsion polymerization process. Indeed, without significant impact on the solids content (ca. 10 wt% obtained after 4 h for PEG-OH- or PEG-X-mediated emulsion polymerization), the particle size was strongly reduced (72 nm) in the presence of PEG-X. Additional experiments conducted in the presence of PEG-X with a lower molar mass (Mn = 1300 g mol−1) or carrying two xanthate chain ends (X-PEG-X, Mn = 3420 g mol−1) confirmed the previous results and the positive contribution of the installation of a xanthate chain end on the PEG chains. In all cases, the fraction of PEG species in the final PVDF remains low (typically ≤1 wt%). The robustness of this technology was confirmed by scale-up emulsion polymerizations of VDF in a 4 L reactor that resulted in a surfactant-free stabilized PVDF latex obtained in the sole presence of PEG-X as precursor of stabilizer and with the same characteristics as the one obtained in smaller volumes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Timothy F. L. McKenna (CP2M), Andrew Kahn, Béatrice Allard-Breton, Stéphanie Bruel and Manuel Hidalgo (Arkema) for fruitful discussions.Notes and references
- B. Ameduri, Chem. Rev., 2009, 109, 6632–6686 Search PubMed.
- J. T. Goldbach, R. Amin-Sanayei, W. He, J. Henry, W. Kosar, A. Lefebvre, G. O'Brien, D. Vaessen, K. Wood and S. Zerafati, in Fluorinated Polymers: Volume 2: Applications, The Royal Society of Chemistry, 2017, vol. 2, pp. 127–157 Search PubMed.
- B. Améduri, Macromol. Chem. Phys., 2020, 221, 1900573 Search PubMed.
- A. C. Mendez Ecoscia, N. Sheibat-Othman and T. F. L. McKenna, Can. J. Chem. Eng., 2019, 97, 207–216 Search PubMed.
- M. Apostolo, V. Arcella, G. Storti and M. Morbidelli, Macromolecules, 1999, 32, 989–1003 Search PubMed.
- P. Pladis, A. H. Alexopoulos, J. Bousquet and C. Kiparissides, in Computer Aided Chemical Engineering, ed. L. Puigjaner and A. Espuña, Elsevier, 2005, vol. 20, pp. 319–324 Search PubMed.
- P. Pladis, A. H. Alexopoulos and C. Kiparissides, Ind. Eng. Chem. Res., 2014, 53, 7352–7364 CrossRef CAS.
- J. L. Keddie and A. F. Routh, Fundamentals of Latex Film Formation: Processes and Properties, Springer Netherlands, Dordrecht, 2010, pp. 185–212 Search PubMed.
- A. Guyot and K. Tauer, in Reactions and Synthesis in Surfactants Systems, ed. J. Texter, Marcel Dekker, New York, 2001, vol. 100(XXVIII), pp. 547–576 Search PubMed.
- A. Martins dos Santos, J. Pohn, M. Lansalot and F. D'Agosto, Macromol. Rapid Commun., 2007, 28, 1325–1332 Search PubMed.
- S. Kawaguchi, K. Tano, M. Maniruzzaman and K. Ito, Macromol. Symp., 2000, 150, 101–108 Search PubMed.
- A. Tuncel and E. Serpen, Colloid Polym. Sci., 2001, 279, 240–251 CrossRef CAS.
- C. K. Hong, M.-J. Hwang, D.-W. Ryu and H. Moon, Colloids Surf., A, 2008, 331, 250–256 CrossRef CAS.
- F. Vidal, J. Guillot and A. Guyot, Polym. Adv. Technol., 1995, 6, 473–479 CrossRef CAS.
- K. Tauer, M. Antonietti, L. Rosengarten and H. Müller, Macromol. Chem. Phys., 1998, 199, 897–908 CrossRef CAS.
- M. Kimoto, K. Yamamoto, A. Hioki and Y. Inoue, J. Appl. Polym. Sci., 2008, 110, 1469–1476 CrossRef CAS.
- F. Vidal, J. Guillot and A. Guyot, Colloid Polym. Sci., 1995, 273, 999–1007 Search PubMed.
- F. Vidal and R. G. Gilbert, Macromol. Chem. Phys., 1996, 197, 1835–1840 CrossRef CAS.
- A. Martins dos Santos, B. T. Le, C. Graillat, F. D'Agosto and M. Lansalot, Macromolecules, 2009, 42, 946–956 CrossRef CAS.
- T. Boursier, I. Chaduc, J. Rieger, F. D'Agosto, M. Lansalot and B. Charleux, Polym. Chem., 2011, 2, 355–362 RSC.
- R. Amin-Sanayei, M. Durali, P. Kappler and G. Burch, US8080621B2, 2011.
- R. Amin-Sanayei, M. Durali, P. Kappler and G. Burch, US8765890B2, 2014.
- R. Amin-Sanayei and C. B. Olmstead, WO2006135543A2, 2006.
- R. Amin-Sanayei and C. B. Olmstead, WO2008073685A1, 2008.
- R. Amin-Sanayei and C. B. Olmstead, US8338518B2, 2012.
- I. Stefanichen Monteiro, A. C. Mendez Ecoscia and T. F. L. McKenna, Ind. Eng. Chem. Res., 2019, 58, 20976–20986 Search PubMed.
- P. B. Zetterlund, S. C. Thickett, S. Perrier, E. Bourgeat-Lami and M. Lansalot, Chem. Rev., 2015, 115, 9745–9800 Search PubMed.
- J. Jennings, G. He, S. M. Howdle and P. B. Zetterlund, Chem. Soc. Rev., 2016, 45, 5055–5084 Search PubMed.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS.
- D. Le, D. Keller and G. Delaittre, Macromol. Rapid Commun., 2019, 40, 1800551 Search PubMed.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., 2020, 59, 8368–8392 Search PubMed.
- J. Zhou, H. Yao and J. Ma, Polym. Chem., 2018, 9, 2532–2561 RSC.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 Search PubMed.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- X. Wang, L. Shen and Z. An, Prog. Polym. Sci., 2018, 83, 1–27 CrossRef CAS.
- J. Rieger, F. Stoffelbach, C. Bui, D. Alaimo, C. Jérôme and B. Charleux, Macromolecules, 2008, 41, 4065–4068 CrossRef CAS.
- J. Rieger, G. Osterwinter, C. Bui, F. Stoffelbach and B. Charleux, Macromolecules, 2009, 42, 5518–5525 CrossRef CAS.
- I. Chaduc, W. Zhang, J. Rieger, M. Lansalot, F. D'Agosto and B. Charleux, Macromol. Rapid Commun., 2011, 32, 1270–1276 CrossRef CAS PubMed.
- E. Velasquez, J. Rieger, F. Stoffelbach, B. Charleux, F. D'Agosto, M. Lansalot, P.-E. Dufils and J. Vinas, Polymer, 2013, 54, 6547–6554 Search PubMed.
- M. Chenal, J. Rieger, A. Philippe and L. Bouteiller, Polymer, 2014, 55, 3516–3524 Search PubMed.
- S. Binauld, L. Delafresnaye, B. Charleux, F. D'Agosto and M. Lansalot, Macromolecules, 2014, 47, 3461–3472 CrossRef CAS.
- J. Lesage de la Haye, X. Zhang, I. Chaduc, F. Brunel, M. Lansalot and F. D'Agosto, Angew. Chem., Int. Ed., 2016, 55, 3739–3743 Search PubMed.
- P. Galanopoulo, P.-Y. Dugas, M. Lansalot and F. D'Agosto, Polym. Chem., 2020, 11, 3922–3930 Search PubMed.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 1023–1033 Search PubMed.
- J. Tan, Y. Bai, X. Zhang and L. Zhang, Polym. Chem., 2016, 7, 2372–2380 RSC.
- X. Wang, C. A. Figg, X. Lv, Y. Yang, B. S. Sumerlin and Z. An, ACS Macro Lett., 2017, 6, 337–342 Search PubMed.
- S. Piogé, T. N. Tran, T. G. McKenzie, S. Pascual, M. Ashokkumar, L. Fontaine and G. Qiao, Macromolecules, 2018, 51, 8862–8869 CrossRef.
- L. Shen, H. Guo, J. Zheng, X. Wang, Y. Yang and Z. An, ACS Macro Lett., 2018, 7, 287–292 Search PubMed.
- B. Couturaud, P. G. Georgiou, S. Varlas, J. R. Jones, M. C. Arno, J. C. Foster and R. K. O'Reilly, Macromol. Rapid Commun., 2019, 40, 1800460 Search PubMed.
- N. Busatto, V. Stolojan, M. Shaw, J. L. Keddie and P. J. Roth, Macromol. Rapid Commun., 2019, 40, 1800346 Search PubMed.
- R. A. E. Richardson, T. R. Guimarães, M. Khan, G. Moad, P. B. Zetterlund and S. Perrier, Macromolecules, 2020, 53, 7672–7683 CrossRef CAS.
- J. Vinas, E. Velasquez, P.-E. Dufils, J. Rieger, F. Stoffelbach, M. Lansalot, F. D'Agosto, B. Charleux and Y. Vanderveken, WO2013092587Al, 2013.
- E. Velasquez, J. Rieger, F. Stoffelbach, F. D'Agosto, M. Lansalot, P.-E. Dufils and J. Vinas, Polymer, 2016, 106, 275–284 CrossRef CAS.
- J. Lesage de la Haye, I. Martin-Fabiani, M. Schulz, J. L. Keddie, F. D'Agosto and M. Lansalot, Macromolecules, 2017, 50, 9315–9328 Search PubMed.
- V. Dehan, E. Bourgeat-Lami, F. D'Agosto, B. Duffy, A. Fortini, S. Hilton, K. Krassa, J. L. Keddie, M. L. Koh, M. Lansalot, M. Lee, J. Lesage de la Haye, I. Martin-Fabiani, C. Mantzaridis, D. P. Mazeffa, R. P. Sear, M. Schulz, M. Sibbald, B. Skerry and B. Thomas, Steel Constr., 2017, 10, 254–259 Search PubMed.
- I. Martín-Fabiani, J. Lesage de la Haye, M. Schulz, Y. Liu, M. Lee, B. Duffy, F. D'Agosto, M. Lansalot and J. L. Keddie, ACS Appl. Mater. Interfaces, 2018, 10, 11221–11232 CrossRef PubMed.
- I. Martín-Fabiani, D. K. Makepeace, P. G. Richardson, J. Lesage de la Haye, D. A. Venero, S. E. Rogers, F. D'Agosto, M. Lansalot and J. L. Keddie, Langmuir, 2019, 35, 3822–3831 Search PubMed.
- J. Delorme, O. Boyron, P.-Y. Dugas, P.-E. Dufils, D. J. Wilson, V. Monteil, F. D'Agosto and M. Lansalot, Polym. Chem., 2020, 11, 7410–7420 RSC.
- M. Guerre, M. Uchiyama, E. Folgado, M. Semsarilar, B. Améduri, K. Satoh, M. Kamigaito and V. Ladmiral, ACS Macro Lett., 2017, 6, 393–398 CrossRef CAS.
- N. Eid, B. Améduri, O. Gimello, A. Bonnet and S. Devisme, Polym. Chem., 2021, 12, 926–938 Search PubMed.
- M. Guerre, B. Campagne, O. Gimello, K. Parra, B. Ameduri and V. Ladmiral, Macromolecules, 2015, 48, 7810–7822 CrossRef CAS.
- M. Guerre, G. Lopez, T. Soulestin, C. Totée, B. Améduri, G. Silly and V. Ladmiral, Macromol. Chem. Phys., 2016, 217, 2275–2285 Search PubMed.
- M. Guerre, S. M. W. Rahaman, B. Améduri, R. Poli and V. Ladmiral, Macromolecules, 2016, 49, 5386–5396 CrossRef CAS.
- E. Folgado, M. Guerre, C. Bijani, V. Ladmiral, A.-M. Caminade, B. Ameduri and A. Ouali, Polym. Chem., 2016, 7, 5625–5629 Search PubMed.
- M. Guerre, M. Semsarilar, C. Totee, G. Silly, B. Ameduri and V. Ladmiral, Polym. Chem., 2017, 8, 5203–5211 RSC.
- D. E. Apostolides, C. S. Patrickios, T. Sakai, M. Guerre, G. Lopez, B. Améduri, V. Ladmiral, M. Simon, M. Gradzielski, D. Clemens, C. Krumm, J. C. Tiller, B. Ernould and J.-F. Gohy, Macromolecules, 2018, 51, 2476–2488 CrossRef CAS.
- Q. Yang, V. Ladmiral and B. Améduri, ChemPhotoChem, 2019, 3, 1095–1099 CrossRef CAS.
- E. Folgado, M. Guerre, A. Da Costa, A. Ferri, A. Addad, V. Ladmiral and M. Semsarilar, Polym. Chem., 2020, 11, 401–410 Search PubMed.
- M. Guerre, S. M. Wahidur Rahaman, B. Ameduri, R. Poli and V. Ladmiral, Polym. Chem., 2016, 7, 6918–6933 Search PubMed.
- M. Guerre, M. Semsarilar, F. Godiard, B. Ameduri and V. Ladmiral, Polym. Chem., 2017, 8, 1477–1487 RSC.
- M. Guerre, J. Schmidt, Y. Talmon, B. Améduri and V. Ladmiral, Polym. Chem., 2017, 8, 1125–1128 RSC.
- E. Folgado, M. Mayor, D. Cot, M. Ramonda, F. Godiard, V. Ladmiral and M. Semsarilar, Polym. Chem., 2021, 12, 1465–1475 Search PubMed.
- C. Boyer, D. Valade, L. Sauguet, B. Ameduri and B. Boutevin, Macromolecules, 2005, 38, 10353–10362 CrossRef CAS.
- A. D. Asandei, O. I. Adebolu and C. P. Simpson, J. Am. Chem. Soc., 2012, 134, 6080–6083 CrossRef CAS PubMed.
- S. Banerjee, V. Ladmiral, A. Debuigne, C. Detrembleur, R. Poli and B. Améduri, Angew. Chem., Int. Ed., 2018, 57, 2934–2937 Search PubMed.
- W. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 7584–7593 CrossRef CAS.
- M. Okubo, H. Kobayashi, T. Matoba and Y. Oshima, Langmuir, 2006, 22, 8727–8731 CrossRef CAS PubMed.
- H. Kobayashi, E. Miyanaga and M. Okubo, Langmuir, 2007, 23, 8703–8708 CrossRef CAS PubMed.
- K. Tauer, M. Antonietti, L. Rosengarten and H. Müller, Macromol. Chem. Phys., 1998, 199, 897–908 CrossRef CAS.
- K. Tauer, R. Deckwer, I. Kühn and C. Schellenberg, Colloid Polym. Sci., 1999, 277, 607–626 CrossRef CAS.
- G. Haran and D. W. A. Sharp, J. Chem. Soc., Perkin Trans. 1, 1972, 34–38 RSC.
- P. Martins, A. C. Lopes and S. Lanceros-Mendez, Prog. Polym. Sci., 2014, 39, 683–706 Search PubMed.
- S. Devisme, A. Kahn, M. Fuentes-Exposito, T. McKenna, F. D'Agosto, M. Lansalot and A. Bonnet, WO2019063445A1, 2019.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00728a |
| This journal is © The Royal Society of Chemistry 2021 |