
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of chiral end groups on the assembly of supramolecular polyurethanes†
Daniel
Hermida-Merino‡
*a,
Lewis R.
Hart
 a,
Peter J.
Harris
b,
Andrew T.
Slark§
c,
Ian W.
Hamley
a and
Wayne
Hayes
*a
a,
Peter J.
Harris
b,
Andrew T.
Slark§
c,
Ian W.
Hamley
a and
Wayne
Hayes
*a
aDepartment of Chemistry, University of Reading, Whiteknights, Reading, RG6 6AD, UK. E-mail: w.c.hayes@reading.ac.uk
bElectron Microscopy Laboratory, University of Reading, Whiteknights, Reading, RG6 6AD, UK
cHenkel Adhesive Technologies, Wood Lane End, Hemel Hempstead, HP2 4RQ, UK
First published on 21st July 2021
Abstract
The ability to influence the physical properties of supramolecular polymers has been the subject of numerous synthetic studies in recent years. Tuning properties by adjustment of the polymer composition to modify the degree of phase separation of polar (hard) and apolar (soft) segments in addition to variation of the capability of the polar end groups to self-assemble efficiently via non-covalent interactions (such as hydrogen bonding, aromatic π–π stacking or metal–ligand binding) has yielded materials with attractive thermo-reversible characteristics. Previously, we have studied supramolecular polyurethanes (SPUs) to understand in more detail the interplay and importance of phase separation and the binding affinity of the polar end group on the physical properties of these interesting thermo-responsive materials. In this paper, we report the positive effect that chirality has upon the self-assembly and physical properties of SPUs. The synthesis of a series of novel SPUs that feature chiral polar end-groups is described in addition to how these chiral moieties improve the order of the relatively weak hydrogen bonding units in this type of supramolecular polymer. The introduction of chiral moieties in the polymer end group induces cooperative arrangements of the components of the hard segments (urethane, urea and aromatic units), leading to SPUs with increased ordered and stronger supramolecular hard segments. The supramolecular interactions of the chiral hard segments were first evaluated by CD and FTIR spectroscopies. In addition, the thermo-responsive nano-segregated structure formed by the columnar aggregates of the SPUs generated was probed by variable temperature CD and FTIR spectroscopies in addition to simultaneous SAXS/WAXS analysis. These studies confirmed the microphase separation morphology also revealed by AFM analysis. Additionally, rheological analysis of the chiral SPUs highlighted the enhancement of the rubbery plateau of chiral SPUs derivatives in comparison to analogous racemic SPUs as a result of the chiral self-assembly motifs. Finally, to explore the mechanical properties of the afforded polymers, electrospinning experiments were undertaken which revealed that defined microfibers were formed at lower concentrations in the case of the homochiral SPUs (ca. 15%) when compared to the racemic analogue, thus confirming the improvement in the supramolecular interactions afforded by the chiral groups.
Introduction
Supramolecular polymers1–3 represent a rich and diverse field of polymer research, developing the design and manipulation of molecular interactions and information through highly specific and directional recognition processes to generate self-assembled arrays in a controlled and spontaneous fashion.4,5 Supramolecular polymers can have a wide variety of structures, ranging from simple guest–host systems to complex self-assembled supramolecular objects, crystalline networks, and monolayers.5–7 These materials are comprised typically of low-molecular weight species which are able to form a pseudo-high molecular weight polymer network with attractive physical properties.8,9 The switchable nature of supramolecular polymers has been investigated for a wide range of potential applications3 including adhesives,10–12 surface-coatings,13 biomedical materials,14,15 healable materials,9,16–19 inkjet and 3D printing.20–22 As a consequence of the highly addressable assembly motifs, supramolecular polymers23 offer a route to novel materials and properties which can be easily processed at relatively low temperatures.24 Narrow processing windows are afforded through dissociation and subsequent reassociation of recognition motifs when subjected to external stimulus such as heat, light or pressure.11,25,26 However, supramolecular polymers currently only have limited industrial use, especially in real-world structural applications, as their mechanical properties require further refinement before they can truly be useful.A number of chiral supramolecular polymers have been reported,27–29 predominantly through hydrogen bonded small molecules,30–34 to yield helical assemblies although other macromolecular assemblies35–39 and peptidic arrays40–44 are known. Chiral supramolecular polymers continue to be the subject of research efforts as a result of their biomimetic nature and well-defined order, resulting in advanced functional materials.28,45,46 Furthermore, chiral supramolecular polymers may allow the development of new materials for applications in sensing, electronics,47 biomedical48 and catalysis.49 The assembly of the backbone,48,50 or pendant motifs51,52 are usually dictated by the molecular chirality of either monomers, oligomeric sequences, terminal groups or the external guest molecules that interact with these motifs by way of non-covalent or dynamic covalent interactions.33 Successful strategies in preparing chiral supramolecular polymers frequently rely on the use of templates, end cappers, or selective solvent techniques to yield chiral materials. These self-assembled materials are often found to include stacked helicoidal aggregates53 which control the morphology, size, and stability of the supramolecular polymer networks. By harnessing the intrinsic chirality of these molecules, polymer architectures and physical characteristics can be amplified or tuned to yield potentially attractive functional materials.
Previously we have reported54 the use of chiral end-groups in bis-urea super-hydrogelators which are able to form gels at extremely low concentrations (0.2 mM) as a result of enhanced self-assembly. The expression of chirality in the hydrogen bonding motif was observed to be translated from the molecular level to the macromolecular domain, resulting in homochiral assemblies. Here we report the use of this chiral recognition motif as an end-group in supramolecular polyurethanes (SPUs) and examine how the highly directional stereochemistry of the self-assembly motif enhances the phase separated morphology and improves the mechanical properties of the resulting materials. Chiral bis-urea low molecular compounds were prepared in bulk in addition to a polyurethane terminated with the analogous racemic end-group as references to enable a greater understanding of the structure property relationship. Finally, the molecular weight of the soft segment of the phase-separated polymer was increased to examine the effect of diluting the concentration of the self-assembling end-group motifs on the material properties.
Results and discussion
In order to understand the effect of chiral centres located in the recognition motifs of supramolecular polyurethanes on their self-assembly polymers, (SPUs 1–3) were first synthesised using a one-pot, two-step procedure (see Scheme 1).55 To allow focus on the physical properties arising as a consequence of the chirality of the end-groups which assemble within the hard segment phase, a polyol and diisocyanate were selected with predictable contributions to the self-assembly process. A pre-polymer 4 was first prepared by reacting the apolar polybutadiene 5 (Krasol LBH-P 3000, MW = 3000 g mol−1) with 4,4′-methylenebis(phenyl isocyanate) [4,4′-MDI] 6. The prepolymer 4 was then terminated with the appropriate amino-diol (7–9) to yield the desired polymer with homochiral (1 or 2) or racemic (3) terminal groups. Spectroscopic data for polymers 1–3 can be found in the ESI (Fig. S1–6†).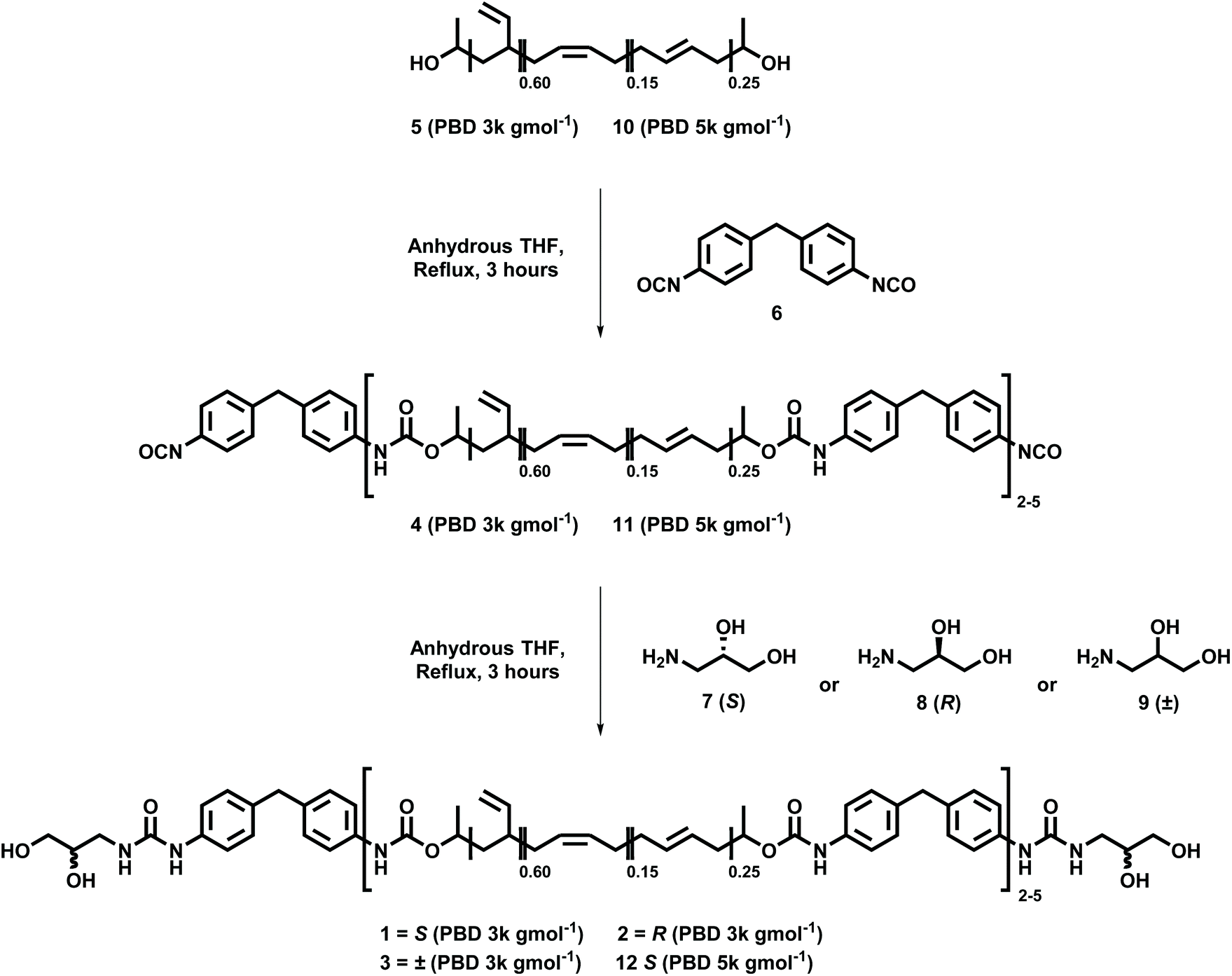 | ||
| Scheme 1 Synthesis of supramolecular polyurethanes 1–3 and 12 from a poly(butadiene) (PBD) telechelic polyol of 3000 g mol−15 or 5000 g mol−110 terminated with homochiral (7 and 8) or racemic (9) self-assembly end-groups. | ||
The successful synthesis of the SPUs was first confirmed by 1H NMR spectroscopy which revealed resonances between 4.01–4.20 ppm, characteristic of methylene residue adjacent to newly formed urethanes from the polymer core. Furthermore, the installation of the hydrogen bonding motif (7, 8 or 9) to the pre-polymer 4 was confirmed by the appearance of signals between 3.21 ppm and 3.38 ppm, assigned to the methylene adjacent to the urea produced through reaction of the primary amine of the end-group and the isocyanate terminal of the pre-polymer. 13C NMR spectroscopy was also used to confirm the establishment of urea (ca. 158 ppm) and urethane (ca. 153 ppm) linkages, respectively, in the polymer backbone. As an additional conformation of the formation of urethane/urea motifs in the supramolecular polyurethanes, infra-red spectroscopy revealed the complete consumption of isocyanates functionalities as observed by the disappearance of the vibration at 2270 cm−1. Furthermore, new absorbances at 1632–1634 cm−1 and ca. 1703–1710 cm−1 were attributed to the carbonyl stretches in the newly formed urethane/urea moieties, respectively. Finally, GPC analysis was employed to confirm the extent of chain extension in the polyurethane, with an average of 2.5 hydrogenated poly(butadiene) residues per supramolecular polymer (Mn = 9100 g mol−1).
To further explore the effect of chirality on the self-assembly and physical properties of the SPUs, the molecular weight of the linear, telechelic polymers was also altered. By increasing the length of the central polymer chain, it was proposed that the effects of self-assembly would be diluted. To investigate this, an additional SPU was prepared as previously described using polybutadiene (MW = 5000 g mol−1) 10 and 4,4′-MDI 6 to synthesise a pre-polymer 11 which was subsequently terminated with S-3-amino-1,2-propanediol 7 to yield a higher molecular weight polyurethane 12 with a homochiral end-group (Scheme 1). Furthermore, the hydrogen bonding content of the SPU 12 at room temperature was calculated to be 65% when compared to that of SPU 1 (76%) and SPU 3 (69%). Spectroscopic data for polymer 12 can be found in the ESI (Fig. S7 and S8†).
A structurally related SPU (13) that featured an achiral diol end-group (Fig. S9†) was also selected to help assess the effects of chiral end groups on the structure–property relationship of this type of SPU system. In an effort to further probe the effects of end-group chirality on supramolecular self-assembly in bulk, model compounds were synthesised54 by the reaction of 4,4′-MDI (6) with homochiral (7) or racemic (9) terminal groups to produce 14 and 15 (Fig. S9†). Interestingly, and in contrast to our previous studies56 on achiral low molecular weight analogues with hydrogen bonding recognition motifs, these model compounds were insoluble in common organic solvents and instead required a much more polar solvent, namely DMSO to solubilise them. This observation suggests that the order imparted through the inclusion of chirality in the recognition motifs was due to an enhanced association constant when compared to structurally related model compounds. Similar solubility characteristics of supramolecular materials has been reported57 by Meijer and co-workers whereby UPy dimers with high binding constants were achieved through enhanced quadruple hydrogen bonding moieties.
The stereochemical information stored within chiral centres induce macromolecules to adopt arrangements as a result the unequivocal self-recognition which may impart chirality on the self-assembled array.27,58–60 The effects of the chiral centres found in the end-groups of SPUs on the inter-macromolecular self-assembly were explored by circular dichroism (CD) analysis both in the solid and solution states (Fig. 1 and Fig. S10†), revealing that both enantiomeric SPUs 1 (S-end groups) and 2 (R-end groups) afforded CD bands of opposite ellipticity in equal intensity. The bands observed at 215 nm and 265 nm were assigned to the aromatic MDI motifs within the SPUs, in good agreement with the previously reported chiral bis-urea model compounds.54 The active chiral signals suggest that the stereogenic centre in the end-group imparts chirality to the adjacent aromatic moieties through their self-assembly, similar to the effect of stereogenic transcription observed61 by Ajayaghosh et al. in self-assembled oligo(p-phenylenevinylene)s. Similar to analysis of the model chiral bisureas,54 concentration dependent studies in solution of the analogous chiral SPUs by both UV and circular dichroism (CD) spectroscopy were performed to probe the potential cooperative effect of the non-covalent interactions triggered by the chirality of the end-group (Fig. S10†). A bathochromic shift in the maximum of the absorption band corroborates the enhancement of the degree of hydrogen bonding interactions. Additionally, the amplification of chirality with respect to increased polymer concentration, as observed in the CD analysis, confirmed the reinforcement of the supramolecular network as a result of self-assembled chiral structures. The absence of ellipticity at low concentrations below 0.155 mg mL−1 suggest sufficient distance between the chiral centres of the end groups to permit self-assembly. However, an inflection point in the sign of the CD signal was observed at 215 nm above 0.312 mg mL−1 which arose from the Cotton effect related to the self-assembly of the chiral excitons. In addition, the single elliptical activity of a chromophore with a band centered at 255 nm in the CD spectra at the minimum concentration of aggregation indicated that the aggregation of the hard segments of the polymers (i.e. a non-linear increase of the ellipticity signal with respect to concentration) arose in close proximity to an aromatic environment (Fig. S10†). The ellipticity changes with increasing the polymer concentration reflect the conformational changes of the self-assembled end-groups. Furthermore, the relative conformations of the chromophores (urea and phenyl moieties) provoked a dramatic decrease of the Cotton effect as well as a shifting of the aromatic band from 255 nm to 265 nm, highlighting the participation of the chromophore groups in supramolecular interactions. The solutions of the SPUs 1 and 2 were evaporated in the cuvette to yield thin films to assess the influence on the supramolecular interactions in the solid state (Fig. 1). The ellipticity of the bulk SPU revealed that the urea adopted only a single configuration which indicated the alignment during the self-assembly process of the end-groups. The enhancement of the directionality of interactions among the end-groups promoted the hard segment alignment, entailing the strengthening of multiple non-covalent bonds and the physical properties of the SPU material (vide infra). The enhancement of peaks in the CD spectra confirm the development of supramolecular chirality as concentration increases.61,62
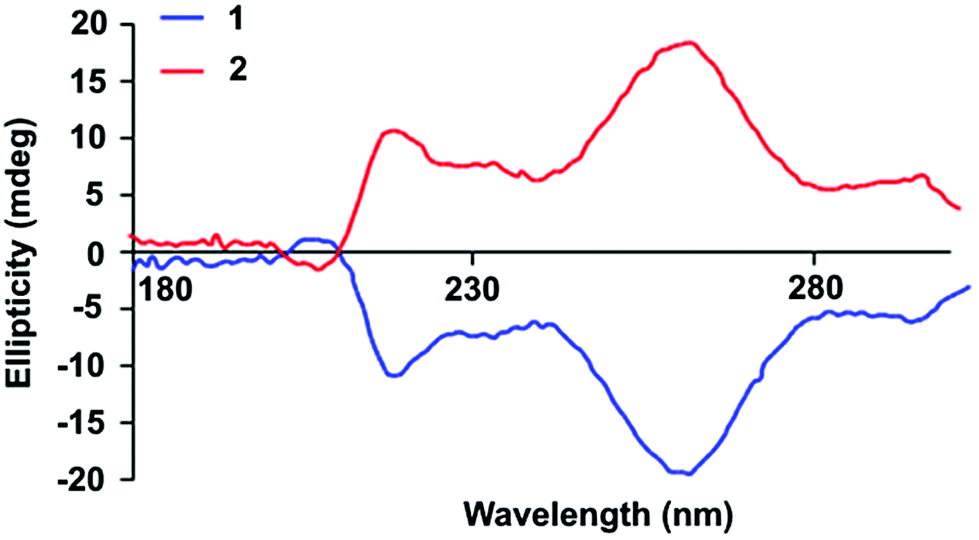 | ||
| Fig. 1 CD spectra of films formed by the chiral supramolecular SPU derivatives 1 (S-end groups) and 2 (R-end groups). | ||
For the purposes of the remainder of this study, the S-terminated polymer was used to investigate the properties of the homochiral polymer on account of facile and viable access to the desired homochiral end-group. The influence of stereogenic centres on the self-assembly mechanism was established by temperature dependent CD experiments to probe the thermo-reversible nature of the SPU interactions. The ellipticity due to the chiral end-groups was measured in 10 °C increments after heating at 5 °C min−1 and holding at a set temperature for a further 5 minutes to allow the polymer to reach thermal equilibrium. Interestingly, a disengagement of the supramolecular interactions occurs in the temperature range between 80 °C and 90 °C, as indicated by the reduction in CD signal (Fig. S11†), potentially as a result of the disruption of supramolecular network formed mainly by multiple weak hydrogen bonds as well as relatively weaker aromatic π–π stacking interactions. The intensity of the CD signal is restored on cooling through self-assembly, confirming the thermo-reversible nature of the supramolecular interactions between the SPUs under investigation that form the helical aggregates. The chirality activity detected by CD spectroscopy correlated well with the efficiency of the hydrogen bonding interactions of the end groups observed by variable temperature FTIR spectroscopy and the related mechanical properties measured by rheology experiments (vide infra).
The hydrogen bonding network in films of SPUs 1 and 3 was also probed by variable temperature FTIR spectroscopy to exemplify the difference in the specific interactions between the homochiral recognition motifs when compared to the racemic end-groups (Fig. 2). As expected, thermo-responsive behaviour analogous to that observed in variable temperature CD experiments was observed. The vibration bands for both the free (1740 cm−1) and hydrogen-bonded (1710 cm−1) urethane groups were identified in both the homochiral 1 and racemic 3 SPUs at room temperature, arising from the urethane linkers bonds from the polymer core. In addition, the ordered hydrogen bonded urea band at ca. 1640 cm−1 was predominant among urea vibration modes for both SPU derivatives. However, the urea vibration band of the racemic derivative 3 features a sharp band superimposed at slightly higher wavenumbers (Fig. 2), implying weaker association and diminished urea stacking whilst the relative intensity of the ordered urea vibration mode is greater in 1, suggesting that the homochiral centres within the end-groups improve the ordering of the urea assembly motifs. The intensity of both the urethane and urea vibrational bands diminished at elevated temperatures in both classes of SPU (1 and 3), whilst simultaneously the magnitude of the free urethane absorbance was observed to increase, attributed to the dissociation of the hydrogen bonding motifs. Upon cooling, the hydrogen bonding urea and urethane vibrational band were seen to increase in intensity as a result of the thermally reversible assembly process (Fig. S12†).
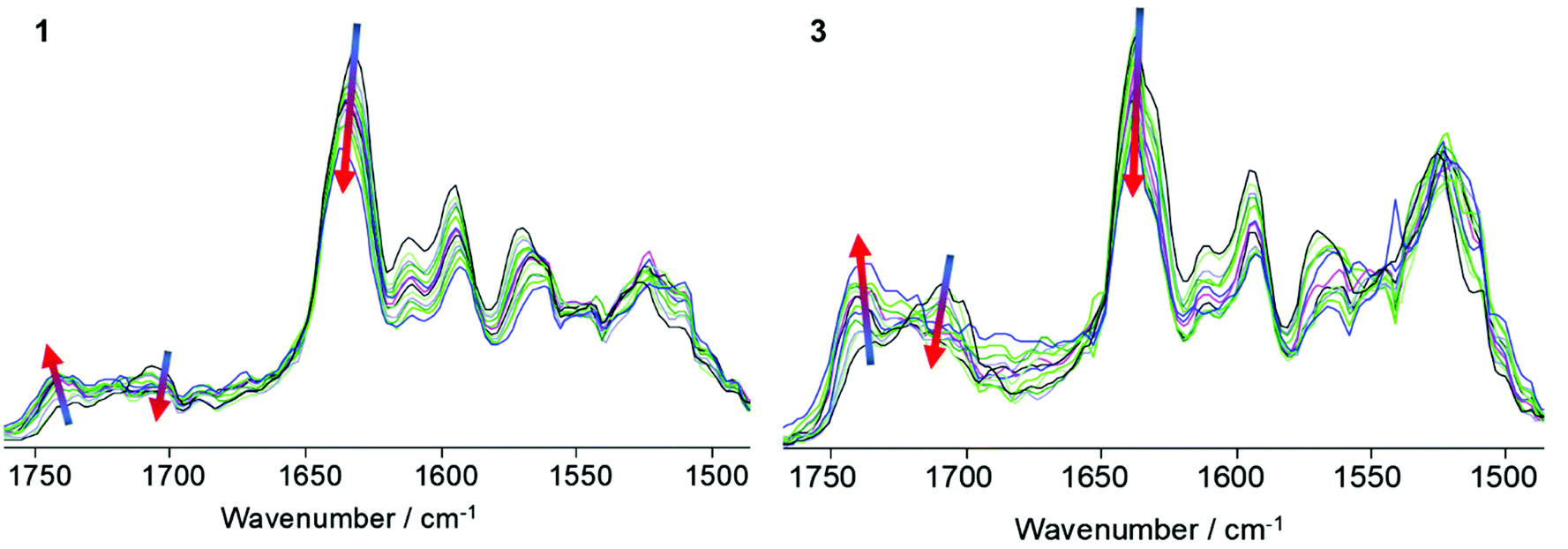 | ||
| Fig. 2 VT-FTIR spectroscopic analysis (25 °C–150 °C) for the homochiral SPU 1 and racemic SPU 3 terminated supramolecular polymers in the bulk. Trend arrows demonstrate the change in absorbance profiles with respect to temperature from low (blue arrow terminus) to high (red arrow terminus) temperature. | ||
Quantitative analysis of the urea and urethane vibration mode absorbances was then used to calculate the extent of hydrogen bonding (Fig. 3) within the supramolecular polymer networks by deconvoluting and fitting the absorptions from VT-FTIR spectroscopic data. Interestingly, the SPU with homochiral terminal groups (1) exhibited a 12% increase in hydrogen bonded ureas at room temperature when compared to the racemic analogue 3 which was attributed to enhanced ordering of the end groups. Furthermore, a 11% increase in the extent of hydrogen bonding in urethane motifs was noted in 1 when compared to 3. Upon heating, the percentage of bound urethane in both the chiral 1 and racemic 3 networks diminished rapidly although still present above 50% in both cases. Furthermore, the onset temperature for the dissociation of urea groups occurred between 40 °C and 60 °C in both SPUs, reaching a steady plateau above 80 °C before diminishing further at 130 °C for 1 (S derivative) and 110 °C for 3 (racemate). The increase in final dissociation temperature was attributed to more ordered assembly in the homochiral terminated polymer 1. Additionally, the extent of hydrogen bonding of urea and urethane groups remained greater in the homochiral polymer network 1 when compared with previously reported63–67 SPUs featuring similar end-groups, remaining stable even at 150 °C, indicating a more stable assembly formed in 1 through the enhanced order in the packing of the hard domain. After thermal equilibration of the SPU 1 film for approximately 30 minutes at room temperature, full recovery of the relative percentage of hydrogen bonding was observed within the SPUs. The recovery of the relative percentage of hydrogen bonding over time demonstrates the thermoreversible nature of the SPUs and is in good agreement63–67 with our previous studies for analogous SPUs. The homochiral end-groups enable the efficient packing within the hard domains whilst retaining the hydrogen bonding characteristics of the urethane/urea functional groups.
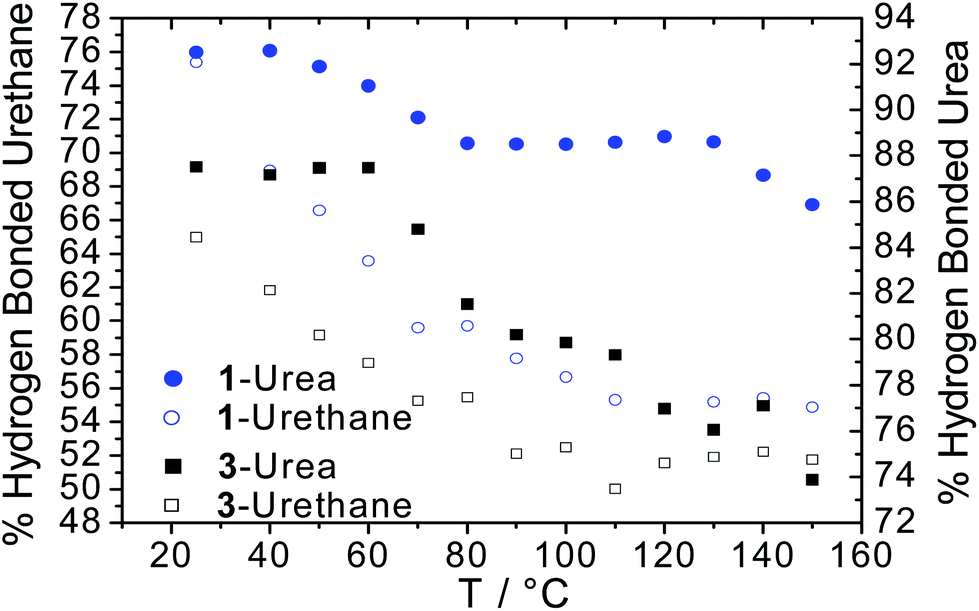 | ||
| Fig. 3 Hydrogen bonding extent of both urethane and urea moieties of homo-chiral 1 (S) enantiomer and racemic 3 (±) supramolecular derivatives. Left-hand y-axis corresponds to percentage urethane hydrogen bonding and right-hand y-axis corresponds to percentage urea hydrogen bonding. | ||
To further understand the effects of soft segment molecular weight on hydrogen bonding interactions within the polyurethane network, VT-IR spectroscopic analysis was also conducted on SPU 12 derived from a higher molecular weight polyol (Fig. S13a and c†). Although the behaviour of both urethane and urea moieties followed the equivalent trends described for SPUs 1 and 3 (Fig. 3), qualitative analysis of the extent of hydrogen bonding interactions revealed a decrease in their prevalence by approximately 10% (Fig. S13b†) at room temperature when compared to both SPU 1 and 3. The dissociation of the hydrogen bonding network in 12 as temperature increased occurred to essentially the same degree as SPU 3, despite the lower concentration of end-group.
The thermal evolution of the nanostructure of segmented SPUs (1 and 3) was explored by variable temperature SAXS experiments (Fig. 4). Initially, a broad reflection centred at ca. 0.62 nm−1 was observed, this correlates to a nanophase separation morphology. This peak shifted to ca. 1 nm−1 upon heating (80 °C), yielding a narrow reflection in agreement with the reorganisation of the hard domains resulting from the annealing above the hard domain Tg. The average domain spacing obtained (ca. 10 nm at room temperature and then 6.9 nm at high temperature) corresponded to the extended conformation of the hard domains for all the SPU derivatives with a polybutadiene (3000 g mol−1) midblock.68 The SPU 12 derived from the higher molecular weight (5000 g mol−1) polyol (11) showed a greater domain spacing as a result of the larger midblock (Fig. S14 and S15†). The development of the nanophase separated structure followed a thermo-reversible response and remained phase separated within the temperature range studied.
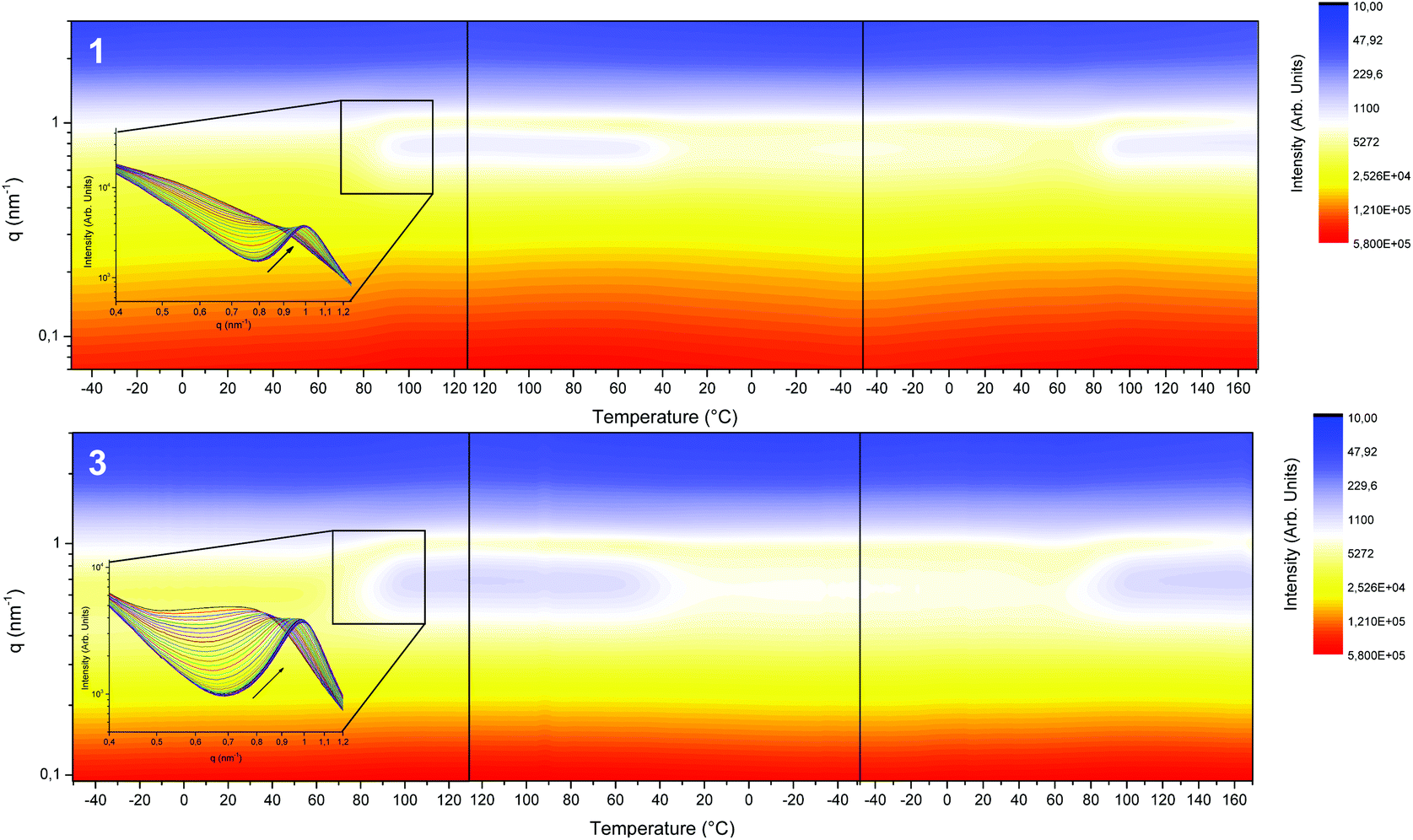 | ||
| Fig. 4 Variable temperature SAXS profiles of SPUs 1 and 3 between −50 °C and 120 °C, showing the thermoreversible nature of the morphologies observed. | ||
The internal structure of the hard domains of the SPUs was monitored by WAXS analysis. Interestingly, the local packing of the generated SPUs featured a crystalline structure (Fig. 5 and S16†) when compared to previously reported55,63,64 SPUs that feature end-groups capable hydrogen bond formation and hard segment generation. The chiral self-assembly of the end-groups promotes the packing of the hard-segments in a fashion analogous to that of end groups featuring multiple urea/urethane moieties or flexible DBDI linkers that permitted enhanced the stacking of the urethane/urea motifs.65,69 In addition, the structure of the SPU derivatives were the same as that of the analogous model compounds (Fig. S15†), indicating the assembly of hard segments to yield helical columnar stacks.54 The chiral 1 and racemic 3 SPU derivatives as well as the corresponding model compounds (14 and 15) are characterised by slightly different crystalline structures whilst generating reflections typical of staggered ureas (ca. 4.7 Å) as well as aromatic π–π stacking assemblies (ca. 3.5 Å). Furthermore, the homochiral derivatives (both model compounds54 in bulk and SPU 1) feature a reflection centred at 7 nm−1 (ca. 8.98 Å) that is comparable to the reflection observed for the xerogel of the model compound in an extended configuration. However, upon heating the SPU derivatives, a decrease of the crystallinity and shift to lower angles was triggered around 50 °C as well as the disappearance of the reflection at 7 nm−1 related to partial melting of the SPU hard segments. The less complex WAXS profile of the homochiral SPUs 1 and 12 (Fig. S15 and S16†) indicate that the hydrogen bonding interactions define a more ordered configuration when compared to the racemic SPU 3 as a result of the inherent recognition imparted by the chiral centre in the end groups.
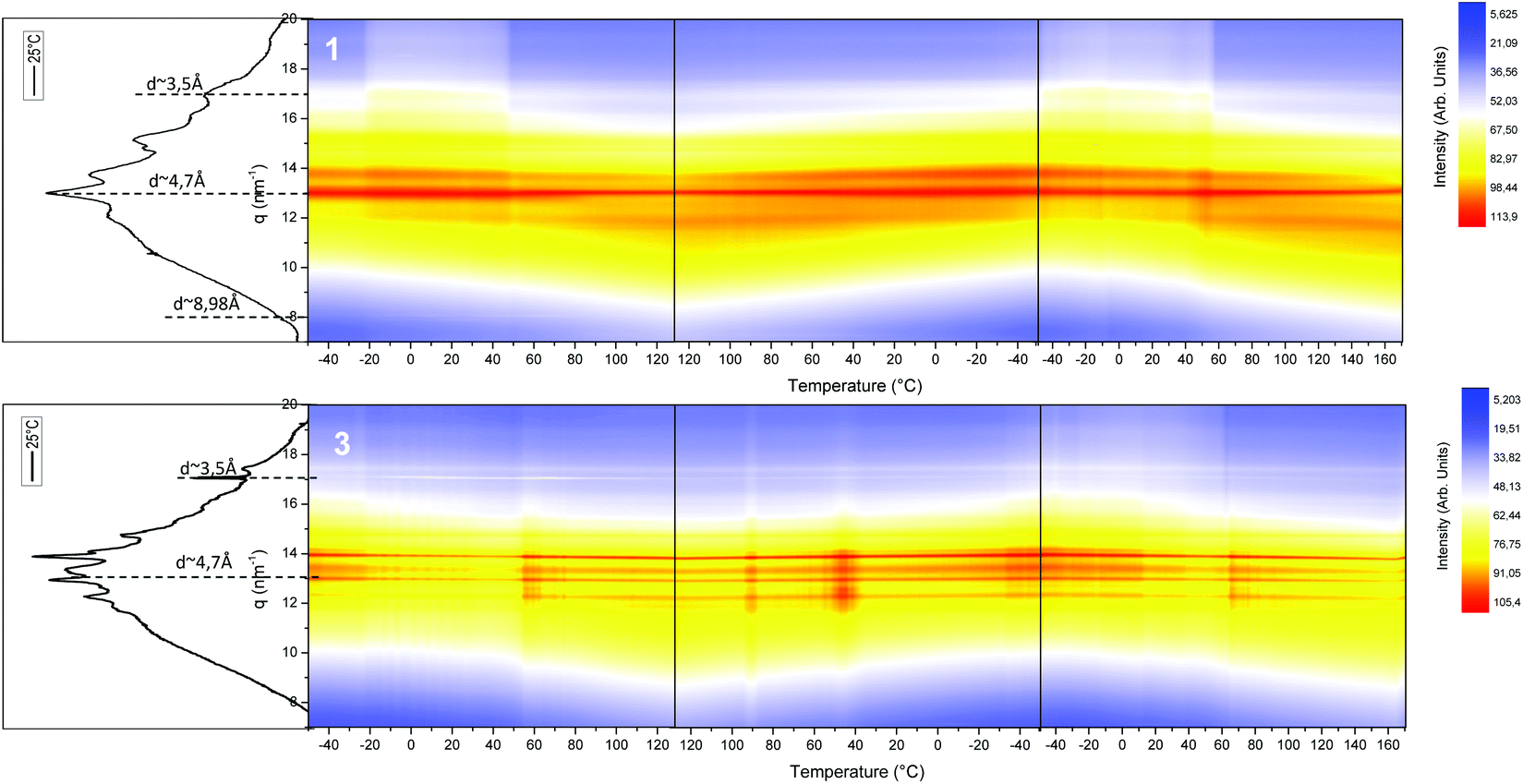 | ||
| Fig. 5 Variable temperature WAXS profiles of homochiral SPU 1 and racemic SPU 3. | ||
Alignment of the adjacent urea/urethane moieties in supramolecular polyurethanes with stereogenic end groups, could lead to the formation of fibrillar or other higher order aggregates. Therefore, the morphology of the segregated segmented SPUs was probed by AFM to assess the ability of the SPUs with chiral 1 or racemic end-groups 3 to form aggregates. Polymeric solutions were spin coated prior to analysis to yield homogeneous films. Microphase separation was observed in the SPUs because of the immiscibility of block components (consistent with the SAXS analysis discussed above) and ordered stacking of the urea/urethane moieties within the polymer backbone and association of the diol end-groups. In an attempt to allow the polymer to reach a thermodynamic equilibrium, samples were annealed for 4 hours at 80 °C.68,69 Subsequent morphological analysis by AFM (Fig. 6) on the annealed polymer films revealed fiber-like aggregates, with more defined phase separation observed when compared to the initial spin coated films. Additionally, a modest improvement in the phase separation of SPUs with the homochiral end-groups was observed when compared to the racemic SPU 3. The morphological observations from AFM imaging were also in good agreement with the assembly determined by SAXS wherein nano-phase separated regions featuring a domain spacing of ca. 10 nm were observed for the SPUs 1–3. Furthermore, AFM investigations of the SPU 12 (Fig. S17†) confirmed, as expected, that increasing the molecular weight of the soft segment resulted in smaller aggregates generated by the hard segments within the soft domain (lower hard segment content [7.49 wt%] for 12 with respect to 1 and 3 [17.49 wt%]) and was in good agreement with SAXS measurements. The ordered aggregates of the hard segment formed by 12 reflect the capability of the chiral end group to interact specifically even when diluted within the large soft segment matrix.
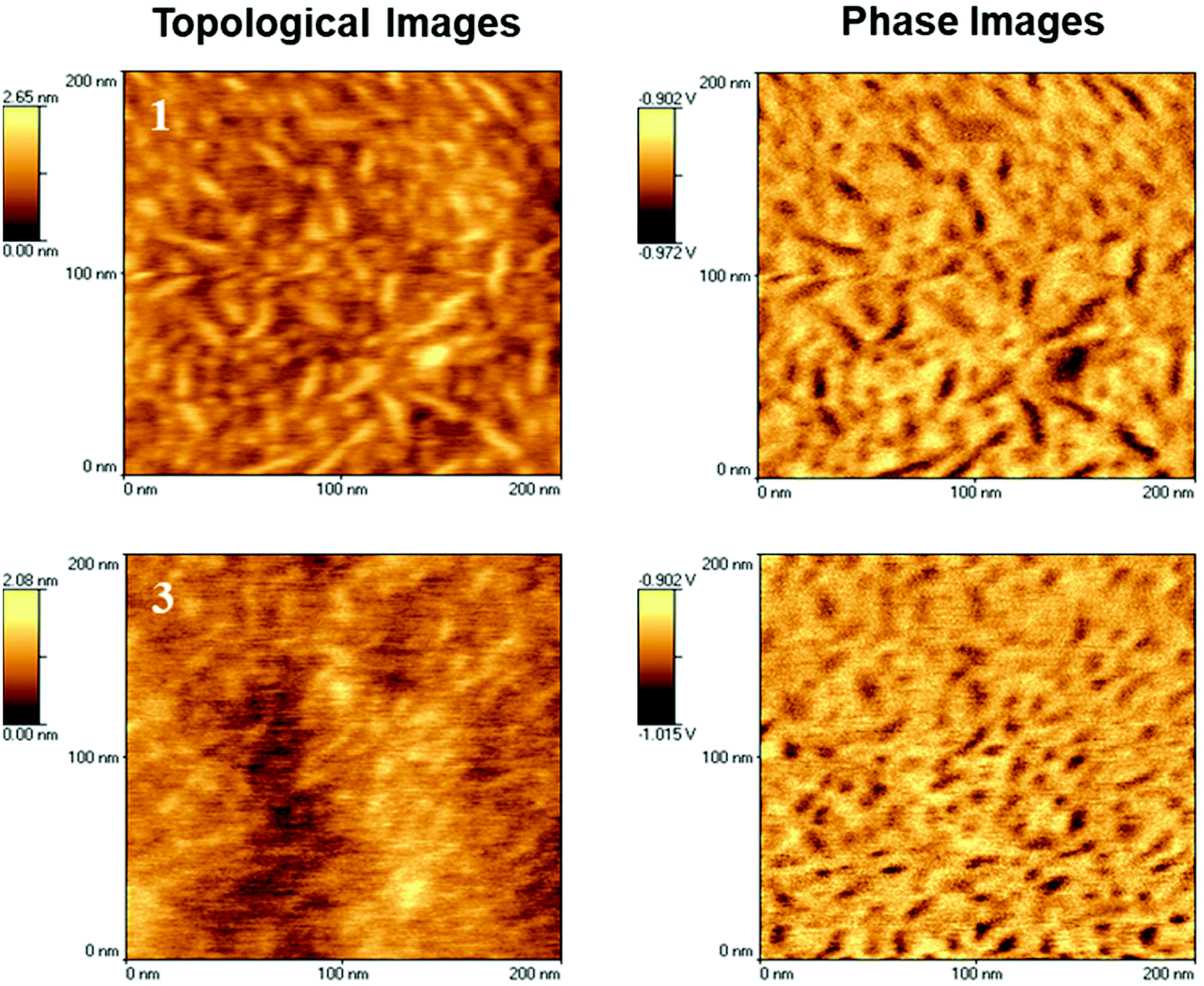 | ||
| Fig. 6 AFM phase micrographs of homochiral SPU 1 and racemic SPU 3. | ||
The ability of stereogenic transcription to strengthen the polymeric network was assessed by rheological experiments (Fig. 7a and b). The maximum storage modulus (G′) for all SPUs observed at ca. 107 Pa was attributed to the glass phase region related to the soft segment of the SPUs in agreement with comparable SPUs previously reported.12,55,63–66,68–70 The dramatic drop of the mechanical properties at higher temperatures was consistent with the disruption of the physical crosslinks between discrete supramolecular oligomers above the hard segment transition. Overall, the modifications of the hard domain69 dramatically alter the viscoelastic properties and thus the mechanical properties which are governed largely by the degree of immiscibility between the domains.
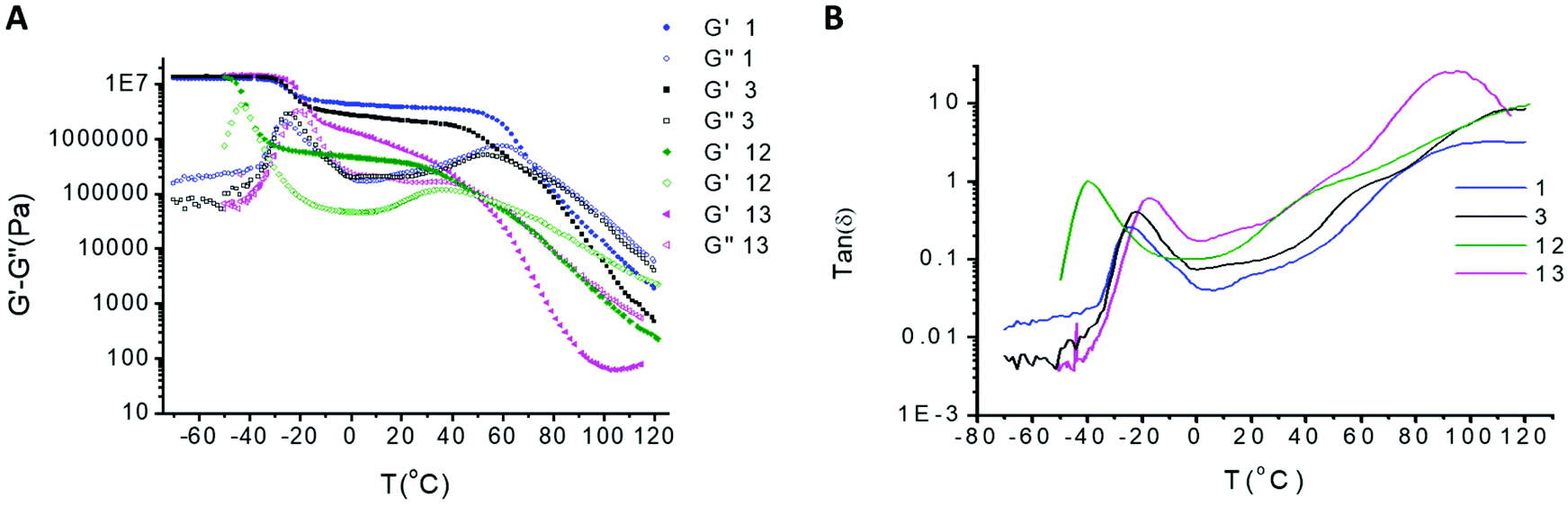 | ||
Fig. 7 Temperature sweep analysis of homochiral SPUs 1 and 12, racemic SPU 3 and achiral SPU tetrol 13 over the temperature regime of −70 °C to 120 °C (A) storage and loss modulus and (B) tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ. δ. | ||
The storage moduli of SPU 1 containing homochiral end-groups was substantially improved when compared to the racemic analogue 3 (the storage modulus of 3 is only 55% of 1 at 40 °C) and the plateau modulus for 1 was more consistent over a broader temperature range. Racemic SPU 3 possessed a characteristic phase mixing behaviour revealing a lower degree of microphase separation achieved by the hard/soft domains when compared to the homochiral SPU 1. The high modulus in the rubber plateau for 1 provided excellent evidence that the efficiency of packing within the hard segment afforded stronger phase segregation resulting from the enhancement of the supramolecular interactions between the hard segments. It is important to highlight that the rubber plateau of SPU 1 is observed at a higher modulus when compared to other analogous SPUs.63–67 The rheology of SPU 1 (Fig. 7) comprising the homochiral end groups was very different to the structurally related achiral SPU tetrol6313 (Fig. S9†). The latter does not provide a plateau modulus, likely as a result of the phase mixing generated from the end-group with lower self-recognition capability.
The increased molecular weight of the soft segment of SPU 12 revealed a reduction in the storage modulus when compared to SPU 1 (Fig. 7a and b) related to the lower hard segment content (17.49 wt% of end-group for SPUs 1 and 3 and 7.49% for SPU 12). Although both SPUs initially exhibited similar rheological characteristics at low temperatures (−70 °C), an earlier and more dramatic transition of the storage modulus plateau was observed for the higher molecular weight SPU 12 as a consequence of the lower Tg of the polymer midblock. Furthermore, the rubbery plateau regime for SPU 1 is more consistent and occurs over a broader temperature range, suggesting a more ordered assembly of the hard segments. The second transition in the rheology profile for SPU 12, which was attributed to dissociation of the hydrogen bonding network of the urea stacking related to lower hard segment content, reached an onset at a lower temperature and was observed to take place over a less well-defined temperature regime. Therefore, it was proposed that that the hard segments were hydrogen bonded but at a lower concentration than for SPU 1 which resulted in a lowering of the temperature of the viscoelastic transition.
DSC analysis also revealed a complex structural transition upon thermal treatment. This was related to the Tg of the corresponding soft and hard segment as well to the melting or crystallization of different components of the hard segment (see Fig. S18–S20†).
Electrospinning of the SPUs was conducted to explore the contribution of the chiral hydrogen bonding motifs in the formation of fibrillar aggregates during the electrospinning process64 and to further confirm the enhancement of the supramolecular interactions of the hard segments by the self-recognition of the chiral groups. Solutions of the polymers were prepared in dichloromethane at different percentages (10–38% w/v). By varying the weight percentage of the polymer, the entanglement concentration and thus the strength of the hydrogen bonding motifs required to overcome the superficial tension and the stretching forces during the evaporation process of the polymeric jet was determined by morphological SEM analysis (Fig. 8). Near spherical beads with a diameter of 80 μm were obtained when 1 was electrospun at a concentration of 10% w/v as a result of the low entanglement of the polymer chains resulted by the lower interaction of the end-groups, showing evidence of electrospraying. Increasing the concentration to 15% w/v was sufficient to form short (less than 3 mm) beaded fibres with a cross-sectional diameter of 8–32 μm as a consequence of capillary instability created in the thin fibre sections between beads. Increasing the concentration further to 20% w/v gave rise to regular wavy fibres of diameter 11–55 μm which revealed jet instabilities during the stretching of the polymer jet. Regular and continuous fibres of diameter 17 μm were electrospun with polymer concentration of 25% w/v in dichloromethane. Fibres with incompletely circular cross section and porous walls were also found which could indicate that the fibres were partially dried when they impacted the collection surface. However, the wavy fibres afforded confirmed that fibres were constantly generated and thus, the polymer jet was able to overcome the jet instabilities. Rounded fibres of a reproducible morphology were achieved successfully with a concentration of 38% w/v which indicated a homogenous evaporation of the polymer jet. The fibres featured higher porosity compared to those obtained at lower concentration which confirmed improved evaporation of the solvent. Furthermore, the disposition of the fibres in a zigzag like manner instead of coiled patterns indicated that the fibres were produced by bending instabilities.
 | ||
| Fig. 8 SEM images and tabulated data of different morphologies produced by electrospinning SPU 1 at a variety of concentrations and SPU 3 at a concentration close to the critical entanglement concentration. | ||
To explore the effect of the stereochemistry of the end-groups, SPU 3 with racemic end group was also electrospun at 15% w/v. The concentration regime where polymer chains entanglements determine the morphology development was probed by viscosity studies (Fig. S21†). For linear polymer that do not possess higher organization in solution the power law exponent in the entangled state ranges from 3.7 to 4.8.71,72 The higher power law exponent value for SPU 1 (5.52) is remarkable given the relative low molecular weight (Mn ∼ 6400) of this polymer. Furthermore, the notably lower entanglement concentration (Ce) value is indicative of stronger interactions of the homochiral end-group with respect to structurally related end-groups as found in SPU 3. When spun near the Ce value at which, in principle, the morphology will be most affected as a result of the transition from the semi-dilute unentangled regime to the semi-dilute entangled regime, SPU 3 produced non-continuous fibres with a thinner diameter of 4–24 μm (Fig. 8) in contrast to the short-beaded fibres afforded for SPU 1. Evidence of electrospraying were also found as well as a reduced density of fibres of SPU 3 at comparable humidity. The lack of stereospecificity of the end-group interactions in SPU 3 may lead to a reduction of the strength of the supramolecular entanglements when compared to the homo-chiral hydrogen bonding motifs in SPU 1 which increased the organisation of the interactions within the hard domain. Moreover, the hydrogen bonding interactions of SPU 1 were observed to enhance the fibre morphology compared to SPU 3. Notably, a consistent morphology development was determined as the polymer concentration was increased (see Fig. S21†).
The temperature at which the SPUs were electrospun also affected the morphology of the fibres afforded. Fibres were again prepared close to the critical entanglement concentration and an increase of the temperature to 30 °C led to a reduction in both the proliferation and the control of the fibre morphology. Regular fibres with diameter of 18.5 μm were obtained (Fig. S22†) which are equivalent to those previously formed with 25% w/v polymer at 25 °C. Fibres obtained at 15 °C and 20 °C (Fig. S23†) revealed a lack of fibre junctions, most probably on account of the solvent evaporation before impacting the collector.
Conclusions
The chirality of end groups in supramolecular polyurethanes has been shown to have a substantial influence on the self-assembly and physical properties of the resulting polymers. A series of novel SPUs that feature chiral polar end-groups were synthesised and chemical and physical analysis ascertained how these chiral moieties improve the order of the weak hydrogen bonding units of this type of supramolecular polymer. They induce cooperative ordering of the components of the hard segments (urethane, urea and aromatic units), ultimately affording SPUs with enhanced physical properties in comparison to racemic analogues. SAXS/WAXS analysis revealed thermo-responsive nano-segregated assemblies featuring helical columnar aggregates. Temperature sweep rheological analysis of the chiral SPUs revealed an enhancement of the rubbery plateau when compared to analogous achiral SPUs to further highlight that the enhancement of the self-assembly by the chiral end-groups to reinforce the polymeric network. Finally, electrospinning studies revealed that defined microfibers were formed at lower concentrations in the case of the homochiral SPUs (ca. 15%) when compared to the racemic analogue, thus confirming the improvement in the supramolecular interactions afforded by homochiral end groups.Experimental
Materials
The reagents used were commercially available from either Acros Chimica, Aldrich Chemical Company or Alfa Aesar and were used without further purification. Dry solvents were purchased and used as supplied or distilled with appropriate dehydration agents, i.e. tetrahydrofuran (THF) which was distilled from benzophenone and sodium. All other reagents such as polybutadiene LBH-P 3000, MW = 3000 g mol−1 and isomer content; (1,2- = 60 wt%, 1,4-cis = 15 wt%, and 1,4-trans = 25 wt%), and 4,4′-methylenebis(phenylisocyanate) (MDI) were supplied by Henkel Adhesive Technologies, Slough, UK. Thin-layer chromatography (TLC) was performed on aluminium sheets coated with Merck silica gel 60 F254. Column chromatography was performed using either SI60 sorbent silica (40–63 μm) supplied from VWR international or Brockmann grade 1, standard grade, neutral activated aluminium oxide (ca. 150 mesh) supplied from the Aldrich Chemical Company.Instrumentation
1H Nuclear Magnetic Resonance spectroscopy (1H-NMR) was conducted on either a Bruker DPX250 (250 MHz) or a Bruker AMX400 (400 MHz) spectrometer using the deuterated solvent as the lock. In the same manner, 13C Nuclear Magnetic Resonance spectroscopy (13C-NMR) was performed on a Bruker DPX250 (62.5 MHz) or a Bruker AMX400 (100 MHz) spectrometers. FT Infrared spectroscopy was performed using a PerkinElmer 1420-X spectrometer with the samples analysed as either neat films or in solution between two potassium bromide or sodium chloride disks. A Bruker MicroToF LC-MS instrument employing ionisation via electrospray was utilised to obtain mass spectrometric data and samples were injected by direct insertion via a syringe pump. Circular dichroism (CD) spectra were recorded on a Chirascan spectropolarimeter (Applied Photophysics, UK) with a 0.5 nm step, 1 nm bandwidth and 1s collection time per step at 20 °C. Samples were loaded into quartz cover slip cuvettes 0.01 or 0.1 mm thick. An electrothermal digital melting point instrument or a TA Instruments DSC 2920 differential scanning calorimeter were used to determine the melting point of the SPUs. The average molecular weight of the polymers generated was determined via Gel Permeation Chromatography (GPC) using a Polymer Laboratories PL-GPC 220 high temperature chromatograph fitted with PL Mixed Gel columns at 40 °C. The samples were dissolved in analytical grade chloroform (5 mg mL−1), and analysed against PL Easy-Cal polystyrene as the calibrants. Samples for AFM analysis were prepared by spin-coating onto a silicon wafer from a solution of the respective compound (1% w/v) in THF. AFM micrographs were collected using a Veeco Explorer fitted with a tube-type 2 μm scanner and silicon cantilever tips (spring constant 1–5 N m−1; Veeco). For all measurements, the set-point amplitude (free oscillating amplitude of the cantilever divided by the actual oscillating amplitude) was maintained between 60 and 100 kHz in a tapping mode. Time resoled SAXS/WAXS experiments during the polymer crystallization experiments were conducted on the Dutch-Belgian Beamline (DUBBLE) BM26B of the European Synchrotron Radiation Facility (ESRF) in Grenoble (France). The samples were placed in pierced DSC pans and loaded in a DSC 600 heating stage (Linkam). The windows of the DSC pans were made of mica and the DSC pans were sealed to avoid sample loss. The distance to the SAXS detector was ca. 2.848 musing a wavelength of 0.1 nm and collecting the SAXS patterns in a Pilatus 1 M detector (981 × 1043 pixels of 172 μm × 172 μm) whilst a Pilatus 300 K detector (1472 × 195 pixels of 172 μm × 172 μm was placed at a distance of ca. 0.28 m to record the WAXS patterns. The scattering data was corrected from background contributions and sample absorption as well as normalized upon incident beam fluctuations prior to the data reduction. The scattering vector q-scale was calibrated by using the scattering of AgBe (silver behenate) for the SAXS scattering angular range and α-Al2O3 (alumina) for the WAXS scattering angles. The SAXS and WAXS patterns were reduced to 1D intensity profiles using bubble72 as a function of the scattering vector (q = 4πsinθ/λ) and reported in arbitrary units for both SAXS and WAXS profiles. Thin films of the polyurethanes were produced for mechanical testing and scattering experiments via a solution casting procedure. All SPUs under study were dissolved in THF and the solution was poured into flat PTFE moulds and subsequently placed in a vacuum oven at 70 °C with a pressure of approximately 0.6 to 0.8 bar for a duration of 24 hours. SPUs films of uniform thickness between 200 and 500 μm were obtained at the end of this procedure without residual solvent. Rheological analysis was performed on circular samples (25 mm diameter) that were obtained from moulded films using a steel punch cutter. Rheology was performed using a TA instruments AR2000 rheometer. Temperature sweeps were performed at a single frequency of 10 Hz with a strain amplitude of 0.1%. Initially, the samples were placed on the rheometer, heated to 115 °C and left to stabilise before the start of the experiment. The temperature was then decreased to −70 °C at a rate of 3 °C per minute whilst data were collected. The sample was again heated to 115 °C at 3 °C per minute. Three cycles of tests were measured, and the results show that the repeating heating does not change the properties significantly. Shear rate sweeps were conducted from 1 to 1000 s−1 to characterize the specific viscosity (ηsp) of each polymer solution and understand the correlation with the solution concentration. The specific viscosity is defined as:where ηs is the solvent viscosity and η0 is the zero shear rate viscosity of the solution.
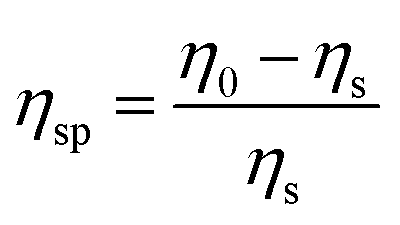
Electrospun fibres were produced successfully using a glass syringe mounted in a syringe pump fitted with a needle of length 2.5 cm and with an internal diameter of 0.71 mm (22 gauge). A flat aluminium electrode was placed normal to the needle at distances varying from 10 to 70 cm with a Glassman high voltage power supply used which allowed defined voltages over the range 7.5–20 kV.
General synthetic procedure for supramolecular polyurethanes 1–3 and 12
Hydroxy terminated poly(butadiene) 5 (molecular weight as supplied = 3000 g mol−1) or 10 (molecular weight as supplied = 5000 g mol−1) was dried under vacuum in oven at 120 °C for 1 hour. The pre-polymers 4 or 11 were first prepared using an established procedure50 by reacting the apolar polybutadienes 5 or 10 (1.68 mmol) with 4,4′-methylenebis(phenyl isocyanate) [4,4′-MDI] 6 (3.37 mmol) in the bulk under N2. The colourless pre-polymer 4 or 11 thus obtained was then dissolved in dry THF (100 mL) and allowed to cool to room temperature. The appropriate 3-amino-1,2-propanediol end-group 7–9 (0.31 g, 3.37 mmol) was then added to the solution and this was maintained under reflux for 3 hours. The solvent was then removed in vacuo and the supramolecular polymer 1–3, or 12 purified by dissolving in chloroform (30 mL) and repeated precipitation from methanol (×3) to afford a white solid elastomer 87% yield (3.92 g). Full spectroscopic data for polyurethanes 1–3 and 12 can be found in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Henkel Adhesive Technologies (post-graduate studentship for DHM), EPSRC (EP/N024818/1, post-doctoral research fellowship in support of LRH) and the University of Reading for financial support of this research. The authors would also like to acknowledge the University of Reading for access to analytical instrumentation within the Chemical Analysis Facility.References
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons Ltd, Chichester, 2nd edn, 2009 Search PubMed.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS PubMed.
- Y. Huo, Z. He, C. Wang, L. Zhang, Q. Xuan, S. Wei, Y. Wang, D. Pan, B. Dong, R. Wen, N. Naik and Z. Guo, Chem. Commun., 2020, 57, 1413–1429 RSC.
- R. F. M. Lange, M. Van Gurp and E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 3657–3670 CrossRef CAS.
- F. Huang and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 5879–5880 RSC.
- A. Ciferri, Macromol. Rapid Commun., 2002, 23, 511–529 CrossRef CAS.
- A. Harada, Supramolecular Polymer Chemistry, Wiley-VCH Verlag GmbH & Co, Weinheim, 2012 Search PubMed.
- S. Burattini, B. W. Greenland, D. Chappell, H. M. Colquhoun and W. Hayes, Chem. Soc. Rev., 2010, 39, 1973–1985 RSC.
- L. R. Hart, J. L. Harries, B. W. Greenland, H. M. Colquhoun and W. Hayes, Polym. Chem., 2013, 4, 4860–4870 RSC.
- J. Courtois, I. Baroudi, N. Nouvel, E. Degrandi, S. Pensec, G. Ducouret, C. Chanéac, L. Bouteiller and C. Creton, Adv. Funct. Mater., 2010, 20, 1803–1811 CrossRef CAS.
- C. Heinzmann, S. Coulibaly, A. Roulin, G. L. Fiore and C. Weder, ACS Appl. Mater. Interfaces, 2014, 6, 4713–4719 CrossRef CAS PubMed.
- A. Feula, X. Tang, I. Giannakopoulos, A. M. Chippindale, I. Hamley, F. Greco, C. P. Buckley, C. R. Siviour and W. Hayes, Chem. Sci., 2016, 7, 4291–4300 RSC.
- B. Folmer, R. P. Sijbesma, R. M. Versteegen, J. A. J. van der Rijt and E. W. Meijer, Adv. Mater., 2000, 12, 874–878 CrossRef CAS.
- P. Shokrollahi, H. Mirzadeh, O. A. Scherman and W. T. S. Huck, J. Biomed. Mater. Res., Part A, 2010, 95, 209–221 CrossRef PubMed.
- G. Di Palma, A. M. Kotowska, L. R. Hart, D. J. Scurr, F. J. Rawson, S. Tommasone and P. M. Mendes, ACS Appl. Mater. Interfaces, 2019, 11, 8937–8944 CrossRef CAS PubMed.
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS PubMed.
- L. R. Hart, J. H. Hunter, N. A. Nguyen, J. L. Harries, B. W. Greenland, M. E. Mackay, H. M. Colquhoun and W. Hayes, Polym. Chem., 2014, 5, 3680–3688 RSC.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- L. R. Hart, S. Li, C. Sturgess, R. Wildman, J. R. Jones and W. Hayes, ACS Appl. Mater. Interfaces, 2016, 8, 3115–3122 CrossRef CAS PubMed.
- L. R. Hart, J. L. Harries, B. W. Greenland, H. M. Colquhoun and W. Hayes, ACS Appl. Mater. Interfaces, 2015, 7, 8906–8914 CrossRef CAS PubMed.
- S. Salimi, Y. Wu, M. I. E. Barreiros, A. A. Natfji, S. Khaled, R. Wildman, L. R. Hart, F. Greco, E. A. Clark, C. J. Roberts and W. Hayes, Polym. Chem., 2020, 11, 3453–3464 RSC.
- K. A. Houton and A. J. Wilson, Polym. Int., 2015, 64, 165–173 CrossRef CAS.
- J. Li, C. L. Lewis, D. L. Chen and M. Anthamatten, Macromolecules, 2011, 44, 5336–5343 CrossRef CAS.
- M. W. Urban, Prog. Polym. Sci., 2009, 34, 679–687 CrossRef CAS.
- J. Li, J. A. Viveros, M. H. Wrue and M. Anthamatten, Adv. Mater., 2007, 19, 2851–2855 CrossRef CAS.
- D. K. Smith, Chem. Soc. Rev., 2009, 38, 684–694 RSC.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
- W. R. Henderson and R. K. Castellano, Polym. Int., 2021, 70, 897–910 CrossRef CAS.
- A. K. Mondal, M. D. Preuss, M. L. Ślȩczkowski, T. K. Das, G. Vantomme, E. W. Meijer and R. Naaman, J. Am. Chem. Soc., 2021, 143, 7189–7195 CrossRef CAS PubMed.
- K. Salikolimi, V. K. Praveen, A. A. Sudhakar, K. Yamada, N. N. Horimoto and Y. Ishida, Nat. Commun., 2020, 11, 1–12 Search PubMed.
- M. Kumar, P. Brocorens, C. Tonnelé, D. Beljonne, M. Surin and S. J. George, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- P. Besenius, G. Portale, P. H. H. Bomans, H. M. Janssen, A. R. A. Palmans and E. W. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17888–17893 CrossRef CAS PubMed.
- O. Shyshov, S. V. Haridas, L. Pesce, H. Qi, A. Gardin, D. Bochicchio, U. Kaiser, G. M. Pavan and M. von Delius, Nat. Commun., 2021, 12, 1–11 CrossRef PubMed.
- M. Miyauchi, Y. Takashima, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2005, 127, 2984–2989 CrossRef CAS PubMed.
- N. H. Evans and P. D. Beer, in Chirality in Supramolecular Assemblies: Causes and Consequences, ed. F. R. Keene, Wiley, 2017, pp. 142–158 Search PubMed.
- S. A. Ikbal, S. Brahma and S. P. Rath, Chem. Commun., 2014, 50, 14037–14040 RSC.
- S. G. Chen, Y. Yu, X. Zhao, Y. Ma, X. K. Jiang and Z. T. Li, J. Am. Chem. Soc., 2011, 133, 11124–11127 CrossRef CAS PubMed.
- X. X. Chen, X. Y. Lin, X. Wu, P. A. Gale, E. V. Anslyn and Y. B. Jiang, J. Org. Chem., 2019, 84, 14587–14592 CrossRef CAS PubMed.
- F. Paquin, J. Rivnay, A. Salleo, N. Stingelin and C. Silva, Chem. Commun., 2015, 51, 12470–12473 RSC.
- K. Sato, W. Ji, Z. Álvarez, L. C. Palmer and S. I. Stupp, ACS Biomater. Sci. Eng., 2019, 5, 2786–2792 CrossRef CAS PubMed.
- R. Garifullin and M. O. Guler, Chem. Commun., 2015, 51, 12470–12473 RSC.
- A. Sánchez-Ferrer, J. Adamcik, S. Handschin, S. H. Hiew, A. Miserez and R. Mezzenga, ACS Nano, 2018, 12, 9152–9161 CrossRef PubMed.
- A. M. Garcia, D. Iglesias, E. Parisi, K. E. Styan, L. J. Waddington, C. Deganutti, R. De Zorzi, M. Grassi, M. Melchionna, A. V. Vargiu and S. Marchesan, Chem, 2018, 4, 1862–1876 CAS.
- A. M. Castilla, W. J. Ramsay and J. R. Nitschke, Acc. Chem. Res., 2014, 47, 2063–2073 CrossRef CAS PubMed.
- S. Cantekin, T. F. A. de Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125–6137 RSC.
- H. Goto and E. Yashima, J. Am. Chem. Soc., 2002, 124, 7943–7949 CrossRef CAS PubMed.
- X. Dou, N. Mehwish, C. Zhao, J. Liu, C. Xing and C. Feng, Acc. Chem. Res., 2020, 53, 852–862 CrossRef CAS PubMed.
- Y. Wang, Y. Sun, P. Shi, M. M. Sartin, X. Lin, P. Zhang, H. Fang, P. Peng, Z. Tian and X. Cao, Chem. Sci., 2019, 10, 8076–8082 RSC.
- A. R. A. Palmans, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Angew. Chem., Int. Ed. Engl., 1997, 36, 2648–2651 CrossRef CAS.
- J. H. K. Ky Hirschberg, L. Brunsveld, A. Ramzi, J. A. J. M. Vekemans, R. P. Sijbesma and E. W. Meijer, Nature, 2000, 407, 167–170 CrossRef PubMed.
- M. M. Green, N. C. Peterson, T. Sato, A. Teramoto, R. Cook and S. Lifson, Science, 1995, 268, 1860–1866 CrossRef CAS PubMed.
- M. B. Baker, L. Albertazzi, I. K. Voets, C. M. A. Leenders, A. R. A. Palmans, G. M. Pavan and E. W. Meijer, Nat. Commun., 2015, 6, 6234 CrossRef PubMed.
- F. Rodríguez-Llansola, D. Hermida-Merino, B. Nieto-Ortega, F. J. Ramírez, J. T. López Navarrete, J. Casado, I. W. Hamley, B. Escuder, W. Hayes, J. F. Miravet and F. J. Ramirez, Chem. – Eur. J., 2012, 18, 14725–14731 CrossRef PubMed.
- P. J. Woodward, D. H. Merino, B. W. Greenland, I. W. Hamley, Z. Light, A. T. Slark and W. Hayes, Macromolecules, 2010, 43, 2512–2517 CrossRef CAS.
- K. Melia, B. W. Greenland, D. Hermida-Merino, L. R. Hart, I. W. Hamley, H. M. Colquhoun, A. T. Slark and W. Hayes, React. Funct. Polym., 2018, 124, 156–161 CrossRef CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. Ky Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- J. Kumaki, S. I. Sakurai and E. Yashima, Chem. Soc. Rev., 2009, 38, 737–746 RSC.
- C. C. Lee, C. Grenier, E. W. Meijer and A. P. H. J. Schenning, Chem. Soc. Rev., 2009, 38, 671–683 RSC.
- F. Vera, J. L. Serrano and T. Sierra, Chem. Soc. Rev., 2009, 38, 781–796 RSC.
- A. Ajayaghosh, R. Varghese, S. J. George and C. Vijayakumar, Angew. Chem., Int. Ed., 2006, 45, 1141–1144 CrossRef CAS PubMed.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- D. Hermida-Merino, A. T. Slark, H. M. Colquhoun, W. Hayes and I. W. Hamley, Polym. Chem., 2010, 1, 1263–1271 RSC.
- D. Hermida-Merino, M. Belal, B. W. Greenland, P. Woodward, A. T. Slark, F. J. Davis, G. R. Mitchell, I. W. Hamley and W. Hayes, Eur. Polym. J., 2012, 48, 1249–1255 CrossRef CAS.
- D. Hermida-Merino, A. Feula, K. Melia, A. T. Slark, I. Giannakopoulos, C. R. Siviour, C. P. Buckley, B. W. Greenland, D. Liu, Y. Gan, P. J. F. Harris, A. M. Chippindale, I. W. Hamley and W. Hayes, Polymer, 2016, 107, 368–378 CrossRef.
- S. Salimi, L. R. Hart, A. Feula, D. Hermida-Merino, A. B. R. Touré, E. A. Kabova, L. Ruiz-Cantu, D. J. Irvine, R. Wildman, K. Shankland and W. Hayes, Eur. Polym. J., 2019, 118, 88–96 CrossRef CAS.
- A. Feula, X. Tang, I. Giannakopoulos, A. M. Chippindale, I. W. Hamley, F. Greco, C. P. Buckley, C. R. Siviour, W. Hayes, C. P. Buckley, C. R. Siviour and W. Hayes, Chem. Sci., 2016, 7, 4291–4300 RSC.
- X. Tang, A. Feula, B. C. Baker, K. Melia, D. Hermida-Merino, I. W. Hamley, C. P. Buckley, W. Hayes and C. R. Siviour, Polymer, 2017, 133, 143–150 CrossRef CAS.
- D. Hermida-Merino, B. O'Driscoll, L. R. Hart, P. J. F. Harris, H. M. Colquhoun, A. T. Slark, C. Prisacariu, I. W. Hamley and W. Hayes, Polym. Chem., 2018, 9, 3406–3414 RSC.
- A. Feula, A. Pethybridge, I. Giannakopoulos, X. Tang, A. Chippindale, C. R. Siviour, C. P. Buckley, I. W. Hamley and W. Hayes, Macromolecules, 2015, 48, 6132–6141 CrossRef CAS.
- M. G. McKee, C. L. Elkins and T. E. Long, Polymer, 2004, 45, 8705–8715 CrossRef CAS.
- V. Dyadkin, P. Pattison, V. Dmitriev and D. Chernyshov, J. Synchrotron Radiat., 2016, 23, 825–829 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00714a |
| ‡ Present address: DUBBLE CRG, BM26, ESRF-The European Synchrotron, Netherlands Organization for Scientific Research, 6 rue Jules Horowitz, 38043, Grenoble, France. |
| § Present address: Department of Chemistry, The University of Sheffield, Brook Hill, Sheffield, S3 7HF, UK. |
| This journal is © The Royal Society of Chemistry 2021 |