
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Slightly congested amino terminal dendrimers. The synthesis of amide-based stable structures on a large scale†
Anjara
Morgado
 ab,
Francisco
Najera
ab,
Anna
Lagunas
cd,
Josep
Samitier
cde,
Yolanda
Vida
*ab and
Ezequiel
Perez-Inestrosa
*ab
ab,
Francisco
Najera
ab,
Anna
Lagunas
cd,
Josep
Samitier
cde,
Yolanda
Vida
*ab and
Ezequiel
Perez-Inestrosa
*ab
aUniversidad de Málaga - IBIMA, Dpto. Química Orgánica, Campus de Teatinos s/n, 29071 Málaga, Spain. E-mail: inestrosa@uma.es; yolvida@uma.es
bCentro Andaluz de Nanomedicina y Biotecnología-BIONAND. Parque Tecnológico de Andalucía, c/Severo Ochoa, 35, 29590 Campanillas, Málaga, Spain
cInstitute for Bioengineering of Catalonia (IBEC), Barcelona Institute of Science and Technology (BIST), 08028 Barcelona, Spain
dBiomedical Research Networking Center in Bioengineering, Biomaterials, and Nanomedicine (CIBER-BBN), 28029 Madrid, Spain
eDepartment of Electronics and Biomedical Engineering, University of Barcelona (UB), 08028 Barcelona, Spain
First published on 17th August 2021
Abstract
Nowadays, amino terminal dendrimers are appealing materials for biological applications due to their multivalence and the versatile conjugation of the amino groups. However, the high reactivity of these terminal groups can be decreased by steric hindrance, limiting their possible bioapplications. Herein, we report the divergent synthesis of slightly sterically hindered amino terminal polyamide dendrimers. A simple and unique AB2 scaffold has been chosen to build the dendritic structures, where only amide bonds have been used as the connecting unit. The 1–7 relative positions of the amino groups in the AB2 monomers avoid the steric congestion of the macromolecules, allowing the construction of robust dendrimers up to the fifth generation. The construction of the dendrimers is based on two well-established reactions, using simple and cheap reactants, with yields above 90% on a gram scale and easy purification procedures. This synthetic methodology constitutes an easy and efficient way for the preparation of stable and aqueous soluble dendrimers on a gram scale, representing a substantial improvement over the synthesis of this kind of aliphatic polyamide amino terminal dendrimer. The prepared structures were completely characterized and evaluated by size exclusion chromatography, diffusion ordered spectroscopy and atomic force microscopy to determine their size. Molecular dynamics simulations were also carried out and the values obtained were consistent with the experimentally determined values.
Introduction
Nowadays, dendrimers are considered one of the best appealing materials for biological applications. The interesting properties of dendritic structures arise from their scaling size and the number of terminal functional groups with generations. Their hyperbranched and well-defined structure allows dendrimers to confine a large number of functional groups in a relatively small space, and this controlled multivalence makes dendrimers unique for certain bioapplications.1 Among all the dendrimers established, those with amino-terminal aqueous solubility are an excellent alternative for biological applications. Out of these, the most suitable and available are polypropylenimine (PPI),2 poly(L-lysine) (PLL),3 polyamidoamine (PAMAM)4 and a modification of polyester (2,2-bis(methylol)propionic acid, bis-MPA).5 Among all, PAMAM is probably the most extensively used.6 Nonetheless, limitations regarding the synthesis, quality or stability of these dendrimers have been previously reported. While PLL is different from the others in its asymmetry, retro-Michael reactions and intramolecular cyclization may cause defects in the structures of PAMAM and PPI; it is reported for the former that only 29% is defect-free up to generation five.7–9 Polyester (bis-MPA) dendrimers avoid such kinds of drawbacks; however, their lack of stability due to hydrolysis reactions may be a problem for certain applications.5In recent years, the chemistry of dendrimers has proliferated with the introduction of different methodologies, with the aim to optimize and accelerate the construction of these complex macromolecules. The first divergent and convergent approaches, proposed by Vögtle,2 Tomalia,10 Newkome,11 and Fréchet,12 laid the basis for the synthesis of dendrimers, independently or in combination.4,13–15 These are now complemented with new procedures, such us accelerated strategies, which focus on reducing the number of reaction steps.16 To achieve this, orthogonal and chemoselective reactions are used, where click reactions play the most important role.17 The one-pot reaction strategy has also been used in the synthesis of dendrimers,18,19 and has recently been applied to obtain up to four generation dendrimers using four chemoselective reactions.20 However, even though the synthesis time is reduced, the preparation of up to four different monomers with different functionalities is required.
Previously, we reported new classes of amino-terminal dendrimers by applying different methodologies. Bisaminoalkylpolyamide dendrimers (BAPAD) were synthesized using 3,3′-diaminopivalic acid as a scaffold, following a divergent approach.21 However, the inherent structure of the dendrimers, together with the synthetic methodology proposed, allows the construction of the dendrimers only until the third generation.21 This could probably be caused by a back-folding of the terminal groups, which decreases dramatically their reactivity. This setback can partially be solved by changing the core of the dendrimer, obtaining thus the dendrimer until generation four.22 Using the same scaffold, but changing the synthetic strategy completely, we develop a new class of amino terminal dendrimers taking advantage of click reactions. Thus, dendrons up to generation 3 were prepared and used to couple with different multifunctional groups. In this convergent approach, large quantities of dendrons were obtained with more than 85% yield. Different dendrimers were also prepared using cores of different multiplicities, demonstrating the potential of this methodology for the assembly of dendrimers with a controlled shape and number of amino terminal groups.23
These amide-based dendritic structures are completely stable, aqueous soluble and versatile enough to be used in different bioapplications. Thus, dendrons based on this structure were used as platforms in diagnosis tests of allergic reactions to drugs.24 The dendritic moieties have also been used as scaffolds for the covalent immobilization of bioactive peptides in titanium disks, increasing the biocompatibility of oral titanium implants.25 Furthermore, the insertion of these dendritic scaffolds into Pt(II) complexes endows them with unique properties such as aqueous solubility, shielding from quenching by dioxygen, and binding to bacterial surfaces, opening the way for the development of new phosphorescent biomarkers with promising properties.26
Despite the versatility of the system, only relatively low generations of dendrimers can be prepared on a large scale. Another aspect to improve is the proximity of the terminal amino groups. The use of 3,3′-diaminopivalic acid as a scaffold promotes a 1–3 relative position between the terminal amino groups in the dendrimers. The proximity of the terminal groups may promote an extremely dense surface packing that, together with their back-folding, can be responsible for a steric hindrance inhibited reaction rate.22,27 Above this critical point, the lower reactivity of the terminal amino groups can be responsible not only for the limited success in obtaining higher-generation dendrimers, but may also have contributed towards incomplete functionalization of the amino terminal groups necessary for certain bioapplications.
Herein, we report the divergent synthesis of a less sterically hindered amino terminal polyamide dendrimer. Based on the excellent properties of our previous developments, only amide bonds were used in the construction of these new dendritic architectures. 5-Amino-2-(3-aminopropyl)pentanoic acid has been chosen as a single and simple AB2 scaffold to build the dendrimer. The presence of two amino groups in 1–7 relative positions resulted in the synthesis of slightly congested structures, while the carboxylic acid acted as the connecting unit. Using ethylenediamine (EDA) as a core, two steps are involved in increasing the dendrimer generation: amide formation and amine deprotection. Both are well-established procedures, using simple and cheap reactants, with yields above 90%, on a gram scale and easy purification procedures. The use of a unique scaffold in the synthesis also avoids the tedious work to construct a family of different monomers, while simplifying the structure of the final dendrimer. This synthetic methodology constitutes an easy and efficient way for the preparation of stable and aqueous soluble dendrimers on a gram scale in higher generations. The prepared structures were completely characterized and evaluated by size exclusion chromatography (SEC), diffusion-ordered spectroscopy (DOSY) and atomic force microscopy (AFM) to determine their size. Molecular dynamics simulations (MDS) were also carried out, with the values consistent with the experimentally determined values.
Experimental
All reactions were performed using commercially available reagents and solvents from the manufacturer without further purification. Unless otherwise stated, all reactions were performed in air. Column chromatography and TLC were performed on silica gel 60 (0.040–0.063 mm) using UV light and/or stains to visualize the products. SephadexTM G-10 column chromatography (100 g sephadex in a 5 cm diameter column) and SephadexTM G-10 pre-packed columns were used to purify the final dendrimers. Chemicals were purchased from Sigma-Aldrich (di-tert-butyl-dicarbonate, N-methylmorpholine, 1-hydroxybenzotriazole), Alfa Aesar (Amberlyst A-26, Celite, sodium hydride, di-tert-butyl malonate, ethylenediamine, 4 M hydrochloric acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dioxane) or panreac (triethylamine, acetic acid, hydrochloric acid, sodium hydroxide) and used without further purification, unless otherwise indicated. Water was purified with a Mili-Q purification system from Millipore.
:dioxane) or panreac (triethylamine, acetic acid, hydrochloric acid, sodium hydroxide) and used without further purification, unless otherwise indicated. Water was purified with a Mili-Q purification system from Millipore.
1H-NMR and 13C-NMR experiments were performed in the indicated deuterated solvent at 25 °C on a Bruker Ascend 400 MHz spectrometer. Proton chemical shifts (δ) are reported with the solvent resonance employed as the internal standard CDCl3δ 7.26, DMSO-d6δ 2.50 and D2O δ 4.79. Carbon chemical shifts are reported in ppm with the solvent resonance as the internal standard CDCl3δ 77.16 and DMSO-d6δ 39.52. 1H-RMN data are reported as follows: chemical shift, multiplicity, coupling constants (Hz) and integration. The HRMS (Electrospray Ionization Time of Flight, ESI-TOF mass spectrometry (MS)) was performed on a high resolution mass spectrometer Orbitrap, Q-Exactive (Thermo Fisher Scientific, Waltham, MA, USA), in either positive or negative ion mode. Matrix-Assisted Laser Desorption Ionization Time of Flight Mass Spectrometry (MALDI-TOF MS) was used to measure the mass of the dendritic materials, and it was conducted on a Bruker MicroFlex MALDI-TOF MS. The obtained spectra were analyzed with FlexAnalysis Bruker Daltonics versión. The detector mass range was set from 100 to 5000 Da, depending on the size of the dendritic structure analysed. A solution of 10 mg mL−1 of dithranol (DIT) or 2,5-dihydroxybenzoic acid (DHB) was used as the matrix.
HPLC chromatograms were obtained using a JASCO HPLC system equipped with a refractive index (RI) detector and a size exclusion column (SEC) Styragel® HR 4E DMF at room temperature using DMF LiCl 10 mM as the mobile phase. The concentration of the samples was 1 mg mL−1 and the flow rate was 0.7 mL min−1.
General procedure for deprotection of terminal amine groups
Gx-NHBoc (1 eq.) was dissolved in THF (10 mL) and the solution was cooled in an ice-water bath. 4 M HCl in dioxane (10 mL) was added dropwise and the mixture was stirred until the reaction was completed. Afterwards, the solvent was evaporated under vacuum to obtain the products in a quantitative way. The compounds were purified by size-exclusion chromatography.General procedure for dendrimer growth reactions
Amino terminal dendrimer Gx (1 eq.) was dissolved in the minimum volume of methanol. Amberlyst A-26(OH) ion exchange resin (250 mg mmol−1 of the amine group) was added and left in a shaker for one hour. Afterwards, the resin was removed by filtration through MeOH-pre-wetted Celite and the solvent was removed under vacuum. The amine-free terminal dendrimer was dissolved in dry DMF under a nitrogen atmosphere, 1 (1.1 eq. per amine group) and N-methylmorpholine (1.5 eq. per amine group) were added and the mixture was stirred at room temperature. Once finished, the solvent was removed. The crude was dissolved in dichloromethane (60 mL) and washed with water (2 × 30 mL) and saturated NaHCO3 solution (2 × 30 mL). The organic layer was dried over MgSO4 and the solvent was removed. The product was purified by precipitation in hexane.DOSY nuclear magnetic resonance (NMR) experiments
The samples were prepared in deuterium oxide at a concentration between 0.5 and 2 mM (within the infinite dilution range for similar samples at 0.1–2.1 mM).28 For all samples, DOSY experiments were performed with decreasing concentrations until no differences between the diffusion coefficients were observed. The experiments have been performed on a Bruker AscendTM 400 MHz spectrometer equipped with a 5 mm BBFOPLUS probe with a 2H “lock” channel and Z gradient. The spectrometer is also equipped with a control temperature unit prepared to work at temperatures ranging from 0 °C to +50 °C. The gradient strength was calibrated by measuring the diffusion rate of pure water of residual protons in D2O. All experiments were conducted at 300 K. The samples were allowed to equilibrate for no less than 15 min. To determine the diffusion rates, a 2D sequence using double stimulated echo for convection compensation and a LED using bipolar gradient pulses for diffusion were used. The diffusion coefficients (D) were determined from the slope of the Stejskal–Tanner plot, which relates it to the signal intensity through the following equation:where I is the integral of the peak area at a given value of G, I0 is the integral of the peak area at G = 0, G is the gradient field strength, γ is the gyromagnetic ratio, δ is the gradient duration and Δ is the time between the gradient pulses.29 The diffusion coefficients determined were used to calculate the hydrodynamic radius via the Stokes–Einstein equation:
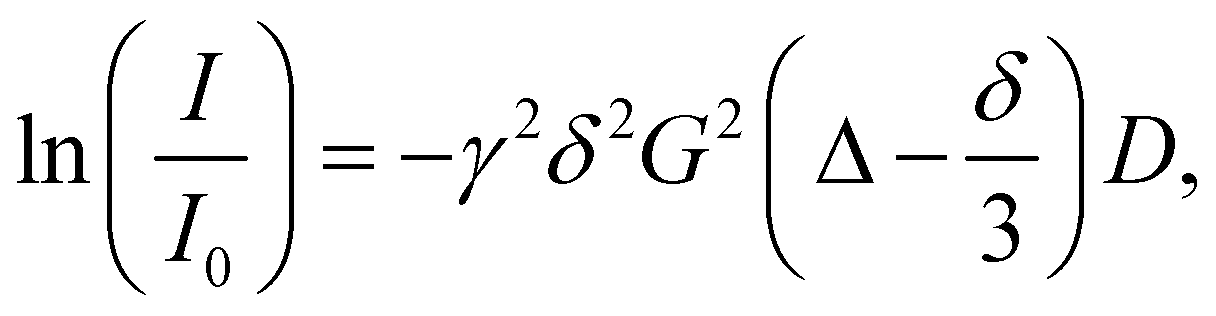
| Rh = KBT/6πηD, |
Dendrimer deposition and AFM analysis
Atomically flat Au(111) single-crystal disks of 10 mm diameter and 1 mm thickness (MaTecK) were flame annealed prior to use and incubated with 150 μL of an aqueous solution of the dendrimer at 1.5 × 10−7, 1.1 × 10−7 and 7.2 × 10−8 w/w for G3, G4 and G5, respectively. All solutions were prepared in pure water (18 MΩ cm−1 <4 ppb TOC Milli-Q, Millipore) and sonicated for 10 min before use. Dendrimer stock solutions were used within 6 months of preparation.Substrates were imaged by AFM using a Dimension 3100 AFM instrument (Veeco Instruments) operated in tapping mode in air. Silicon AFM probes (Budget Sensors) with a spring constant k = 40 N m−1 and a resonant frequency ν = 300 kHz were used. At least 4 areas in two independent substrates were analyzed. AFM images were processed with WSxM software.30
Molecular dynamics simulations
Simulations were performed in water as an explicit solvent using the AMBER12 MD software package.31 Briefly, AMBER force field (parm99) parameters have been used and the General Atom Force Field (GAFF) parameters to include parameters when needed.32 Initial dendrimer conformations were made using the Dendrimer Building Tool (DBT).33 The systems were minimized and then heated to 300 K over 40 ps. Non-bonded interactions were cutoff at 9 Å. Time steps of 2 fs were carried out employing the SHAKE routine.34 Simulations were run at physiological pH (7.4) in the NPT ensemble (300 K and 1 atm). Dendrimers were equilibrated for 2 ns and starting from these configurations were performed production runs of 20 ns trajectories. Trajectory analyses were performed using the Amber modules ptraj and cpptraj. Snapshots from the trajectories within this paper were produced with VMD software.35 The details of the molecular dynamics simulations are thoroughly detailed in the ESI.†Results and discussion
In general, amino terminal dendrimers are excellent scaffolds for a series of bioapplications where the conjugation of such groups with bioactive molecules plays a crucial role. These terminal groups impart aqueous solubility to the dendrimers, while can also become positively charged, essential aspects for some request. One more important feature to take into account is stability, a critical aspect in some applications where the role of the dendrimer is a carrier vehicle. In our dendrimer design, an exclusive amide based scaffold has been selected to avoid stability problems or protonation of internal amino groups that could alter the size or the charge. A unique monomer was carefully chosen to simplify not only the synthesis, but also the structure of the resulted dendrimer. A divergent approach was designed to build the dendritic structures, with just a couple of well-known, effective, easy and cheap reaction steps.Synthesis
For the synthesis of these amide based structures, a simple AB2 type monomer was designed. In order to achieve a higher separation between amine terminal groups, 5-amino-2-(3-aminopropyl)pentanoic acid was selected as a platform to build the dendrimers. This will arrange the terminal amino groups in a 1,7 relative position, achieving less steric hindrance than in previously prepared BAPAD21 or clicked dendrimers,23 where the amino groups were in a 1,3 relative position.The presence of a carboxylic acid and two amino groups in the selected AB2 monomer will ensure that all connections will be exclusively amide bonds. 5-Amino-2-(3-aminopropyl)pentanoic acid was prepared according to procedures previously described (detailed synthetic protocols are described in the ESI†).36 For the growth of the dendrimers, the amine groups of 5-amino-2-(3-aminopropyl)pentanoic acid were protected with tert-butoxycarbonyl (Boc) derivatives and the carboxylic group was activated using 1-hydroxybenzotriazole (HOBt), obtaining 1 (Scheme 1) in >90% yield. A simple nucleus (EDA) has been selected to build the dendrimers in a divergent way. Two iterative steps of amidation/deprotection reactions produced consistently high generations of dendrimers in up to 90% yield in all cases on a multigram scale.
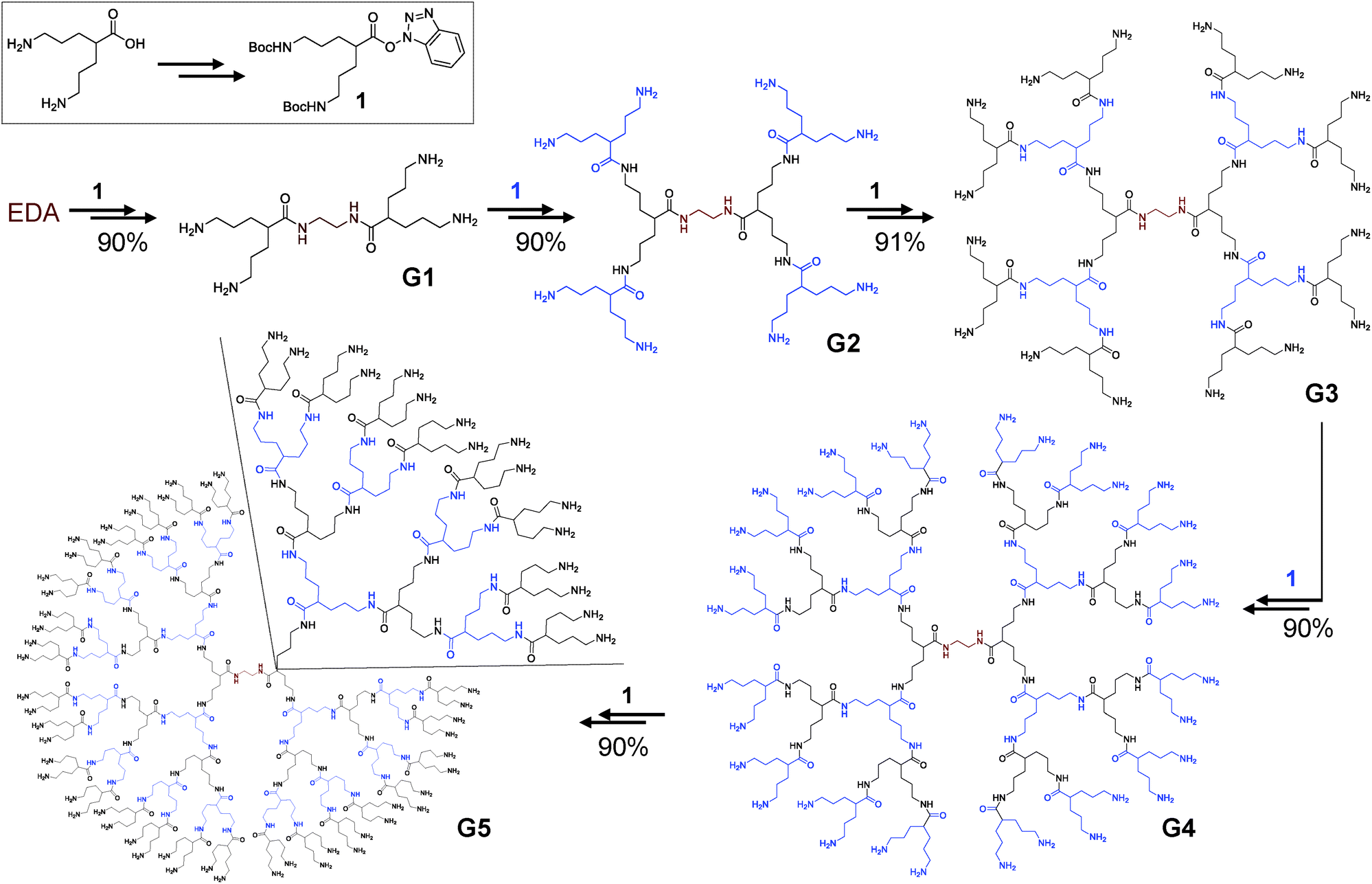 | ||
| Scheme 1 Synthesis of all-amide dendrimers up to the 5th generation. Growth steps consist of an amidation reaction (N-methylmorpholine, DMF, r.t.; yield indicated for each reaction) followed by the deprotection of the terminal amino groups (4 M HCl in dioxane, THF; quantitative). | ||
Amidation reactions were carried out with a slight excess of 1 for lower generations; nevertheless, to ensure the reaction of all branches of the dendrimers, refreshing with different amounts of 1 was needed when increasing generation. For an effective coupling, a pre-treatment with an Amberlyst A-26(OH) ion exchange resin has been made to the dendrimers to guarantee that all terminal amino groups were in a non-protonated form. The amidation reactions afforded Boc-protected dendrimers (Gx-NHBoc) in excellent yields. It is noteworthy that no further purifications were needed for G1-NHBoc and G2-NHBoc. For higher generations, precipitation in hexane yielded pure compounds in excellent yields (Scheme 1). No chromatography was needed for the purification of Gx-NHBoc, avoiding tedious and time consuming procedures. The deprotection of the terminal amino groups was carried out under acidic conditions, and all amino terminal dendrimers were filtered by size-exclusion chromatography. G1–5 dendrimers were obtained in an almost quantitative way in all cases.
Characterization
The structures of G1-5 dendrimers were analyzed by nuclear magnetic resonance (NMR) spectroscopy. 1H-NMR spectra in DMSO-d6 are shown in Fig. 1. For G1, protons corresponding to the ethylene core (EDA in Fig. 1) can be clearly distinguished as a singlet at 3.13 ppm. Regarding the branching moieties, protons corresponding to the adjacent methylenes to the amino terminal groups (d1 in Fig. 1) appear as a multiplet centered at 2.75 ppm. The broad signal at 2.38 ppm corresponds to the proton adjacent to the carbonyl group (a1 in Fig. 1). Finally, protons corresponding to the rest of methylenes (b1 and c1 in Fig. 1) appear as multiplets. Methylene b1 protons are constitutionally non-equivalent and present different chemical shifts, appearing at both 1.33 ppm and 1.51 ppm (together with c1 protons). The same behavior was also observed in BAPAD type dendrimers.21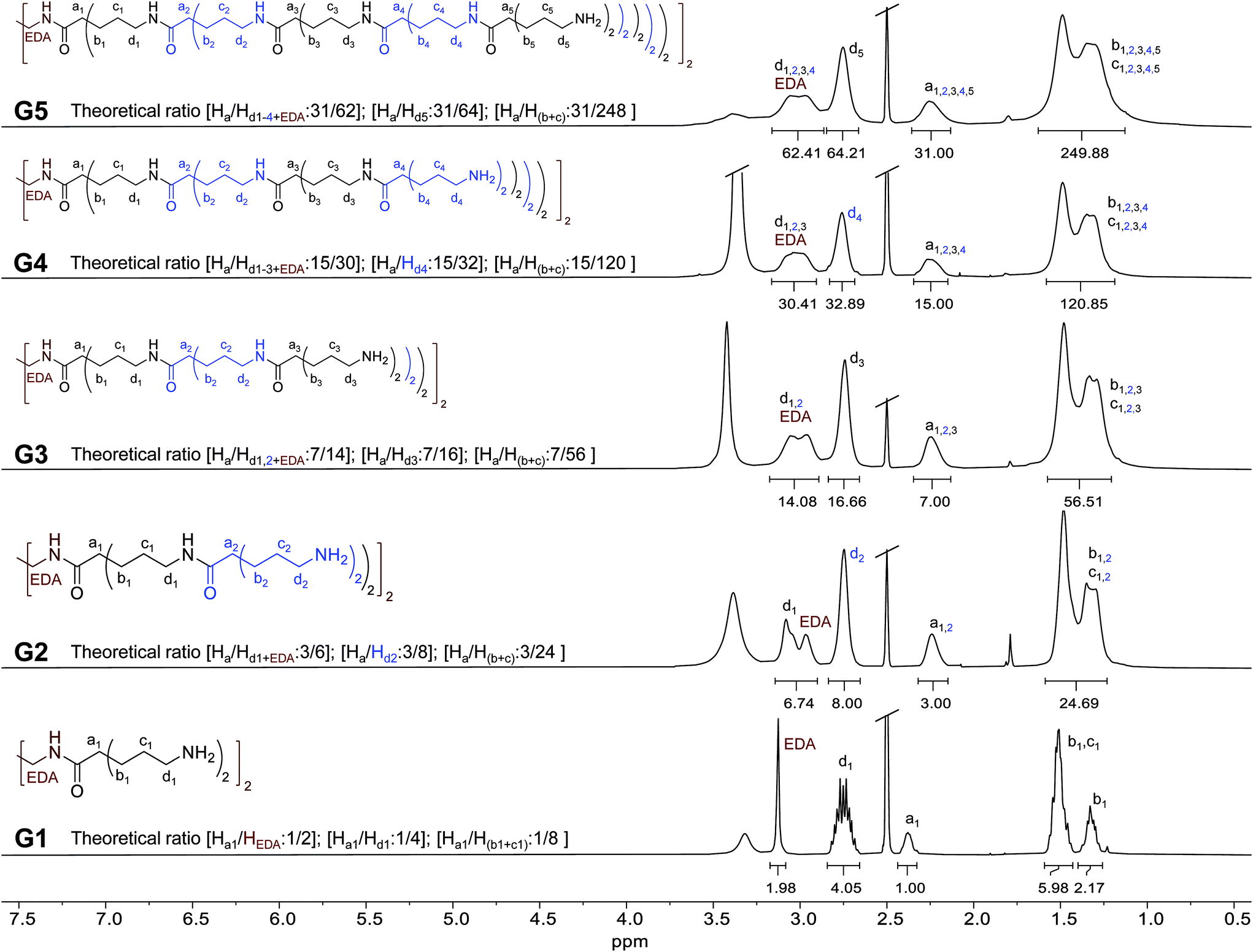 | ||
| Fig. 1 1H-NMR spectra in DMSO-d6 of G1–5 dendrimers. Integral values refer to the numbers of Ha protons in half dendrimers. The theoretical relationship between the numbers of Ha protons with respect to each type of signal is indicated for each dendrimer. Integral values refer to half dendrimers for clarity. | ||
Growth in a generation can be clearly observed in the 1H-NMR spectra of G2. The signal corresponding to d1 shifted to 3.08 ppm, indicating the reaction of the amino groups of G1, forming amide bonds in G2. New signals appear overlapping those corresponding to G1, causing a widening of the peaks. The most relevant is the new signal at 2.74 ppm, corresponding to the new methylenes adjacent to the new terminal amino groups (d2 in Fig. 1). The same trend is observed in the 1H-NMR spectra for higher generation dendrimers. A new layer involves the overlapping of internal d signals (as the formation of amide bonds involved an upper field shift of the signals of the previous generation d-terminal). The broadening of these signals also implies an overlap with the signal of the ethylene core (EDA) protons, while the new generation d protons (d-terminal) appear at around 2.75 ppm. In all cases, the peaks corresponding to a, b and c protons overlap, promoting a broadening of the signals in each generation.
To evaluate the integral values, the signal corresponding to Ha protons has been used as a reference. This value was adjusted to the number of Ha in each dendrimer (we used half dendrimers for clarity, since the number of protons increases significantly with generations). The theoretical ratio between the integral values is indicated in Fig. 1 for each compound. As can be observed, these values correlate very well with the values of the integral in each spectrum (experimental values are indicated under the corresponding signal in Fig. 1). This seems to indicate a complete growth reaction in each case. More details about the NMR characterization of G1–5 as well as the Boc-protected derivatives can be found in the ESI (Fig. S1 to S56, ESI†). 1H-NMR spectra of an aqueous solution of G5 kept at 37 °C for one week were also recorded. No differences between both the spectra of G5 (freshly prepared or after one week) were observed (Fig. S57, ESI†), as may be expected given the stability of these amide-based dendrimers.
MALDI-TOF obtained values for the smaller generation dendrimers correlated well with the theoretical values, verifying their structures (Fig. S58 to S62, ESI†). However, as previously reported, characterization of dendrimers with high molecular weights and charges by MALDI-TOF is extremely difficult.23,37,38 For G4-5 no MALDI spectrum could be obtained, probably due to the high lability of the Boc protecting groups (in Gx-NHBoc) and the high degree of positive charges (in G4-5) during measurements.
Protected Gx-NHBoc dendrimers were analyzed by size exclusion chromatography (SEC). SEC chromatograms corroborate the conversions to higher generations, showing bands shifted to lower elution times as generations increased (Fig. 2). For generations up to four, SEC bands show a shoulder at lower elution times (higher molar mass) probably due to the presence of potential aggregates, as has been reported in other cases for generation-4 dendrimers.20
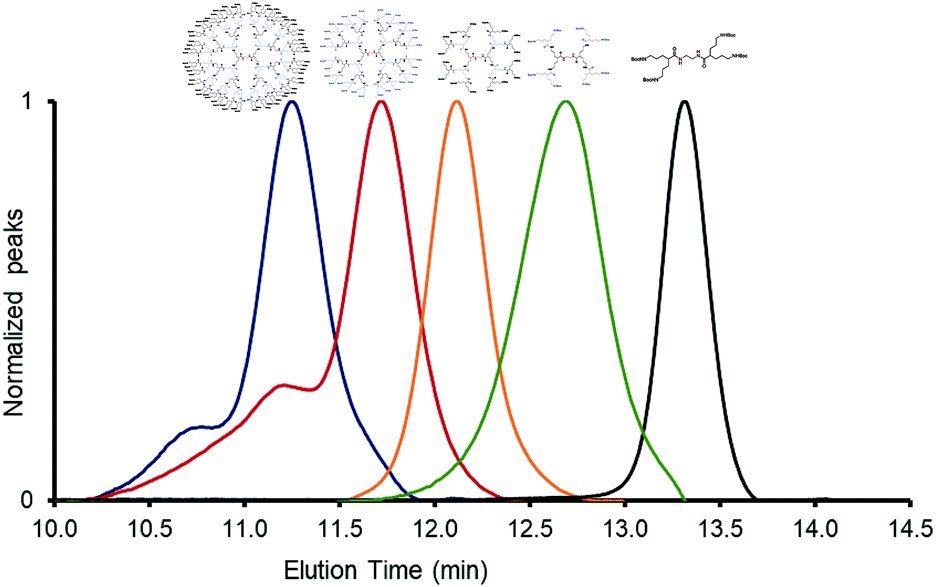 | ||
| Fig. 2 SEC chromatograms of Gx-NHBoc dendrimers, using DMF/LiCl as an eluent and a (SEC) Styragel® HR 4E DMF column at 0.7 mL min−1 flow rate. Black (generation 1), green (generation 2), orange (generation 3), red (generation 4) and blue (generation 5). | ||
Amino terminal obtained dendrimers were examined by diffusion NMR techniques. DOSY (diffusion-ordered spectroscopy) experiments were carried out in D2O solutions. Decays of the signals were monoexponential in all cases, which is translated into linear Stejskal–Tanner plots (Fig. 3a and Fig. S63 to S67, ESI†).39Fig. 3a shows the amplitude decays of dendrimers, where the logarithms of the normalized signal intensities are plotted against the gradient squared. The linear relationship obtained for all samples indicates that they are pure and monodisperse macromolecules. Diffusion coefficients (D) were also determined and used to estimate the size of all dendrimers in solution, by calculating the hydrodynamic radius (Rh) using the Stokes–Einstein equation (Table 1).28,29
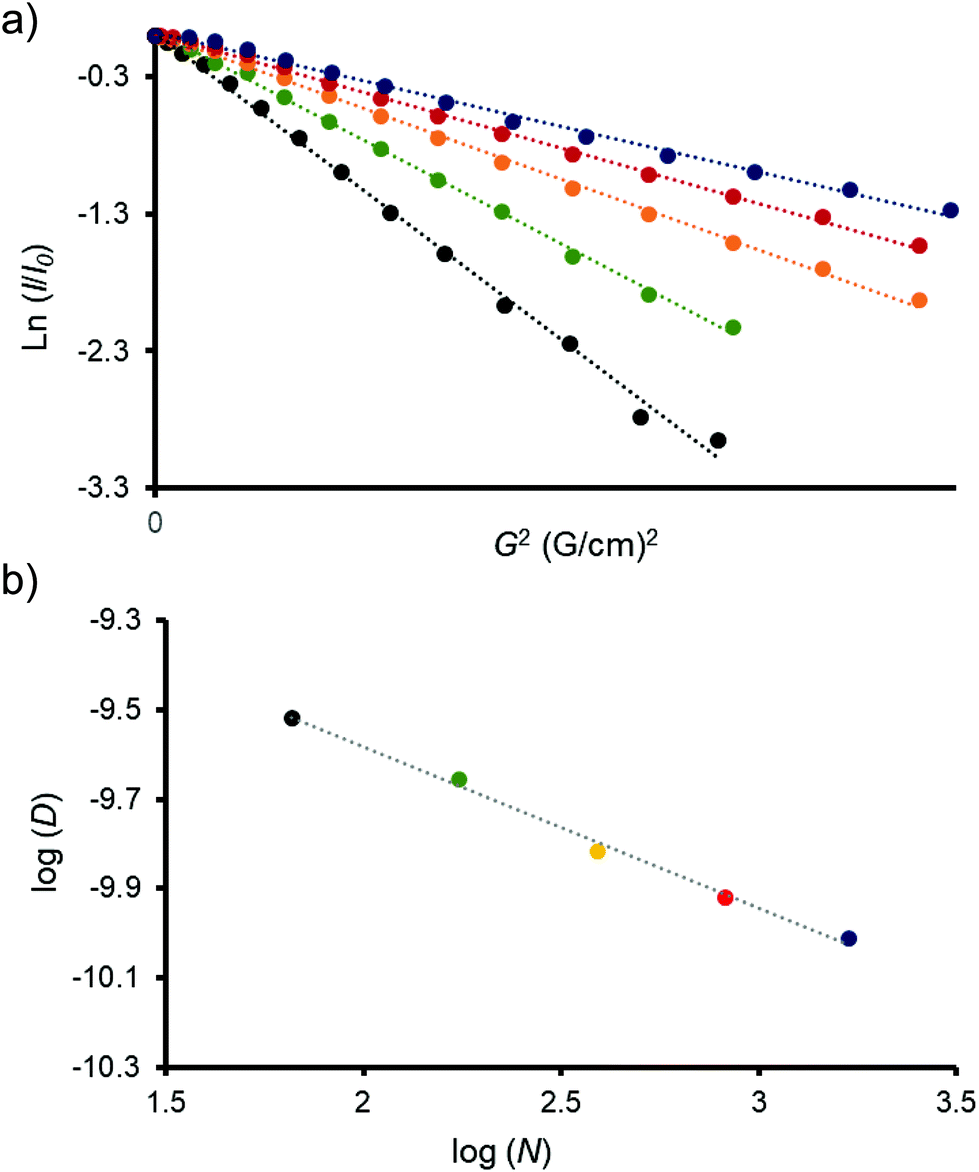 | ||
| Fig. 3 (a) Stejskal–Tanner plots of the synthesized dendrimers in D2O. (b) Log–log representation of the diffusion coefficients versus the approximate number of atoms for each dendrimer. Values of D were taken from the slopes of the Stejskal–Tanner plots in (a). In both cases black (generation 1), green (generation 2), orange (generation 3), red (generation 4) and blue (generation 5). | ||
| Dendrimer | G1 | G2 | G3 | G4 | G5 |
|---|---|---|---|---|---|
| a Values averaged over the last 1 ns simulation run taking each snapshot after 1 ps. | |||||
| Formula | C18H40N6O2 | C50H104N14O6 | C114H232N30O14 | C242H488N62O30 | C498H1000N126O62 |
| Terminal-NH2 | 4 | 8 | 16 | 32 | 64 |
| M.W. (gmol−1) | 372.56 | 997.47 | 2247.31 | 4746.97 | 9746.30 |
| D (m2 s−1) | 3.039 × 10−10 | 2.2135 × 10−10 | 1.5275 × 10−10 | 1.2035 × 10−10 | 9.7121 × 10−11 |
| R h (Å) | 6.59 | 9.05 | 13.12 | 16.65 | 20.63 |
| R g (Å) | 5.26 ± 0.26 | 8.06 ± 0.19 | 11.02 ± 0.24 | 14.87 ± 0.23 | 18.81 ± 0.28 |
| I x /Iy | 1.19 ± 0.10 | 1.53 ± 0.10 | 1.37 ± 0.09 | 1.08 ± 0.04 | 1.26 ± 0.08 |
| I x /Iz | 3.08 ± 0.49 | 2.05 ± 0.21 | 1.68 ± 0.12 | 1.57 ± 0.11 | 1.58 ± 0.09 |
| δ | 0.080 ± 0.016 | 0.045 ± 0.008 | 0.024 ± 0.006 | 0.017 ± 0.005 | 0.018 ± 0.003 |
As expected, both the diffusion constants and hydrodynamic radius scale with dendrimer generation. G1 diffused the fastest, at almost three times the rate of G5 (Table 1), corresponding to the smallest observed Rh. It has been previously reported that the diffusion coefficients of PAMAM dendrimers (up to the fifth generation) scale exponentially with the number of atoms in the dendrimer structure, following the relation D ≈ Nα (where N is the number of atoms and α is a scaling constant).28,29,39Fig. 3b shows the log–log relation between D and N in the prepared dendrimers. As can be clearly observed, dendrimers from generations 1 to 5 adjust perfectly in a linear dependence. From the slope, we assume a scale factor of α = −0.36. This value is practically identical to the observed value for the aqueous solutions of PAMAM dendrimers (α = −0.35).28,29
Amino terminal dendrimers were also analyzed by atomic force microscopy (AFM) by depositing diluted solutions of the dendrimers on (111) gold single-crystal substrates (Fig. 4).40,41 Isolated dendrimers could mostly be found in the G3 and G4 samples, together with some two-dimensional aggregates. Conversely, in G5 aggregation was predominant, leading to both chains of dendrimers and three-dimensional aggregates (Fig. 4a and Fig. S68 in the ESI†). Analysis of single dendrimers in the zoomed-in sections of the AFM images (Fig. 4b) shows the measured heights of approximately 25, 30 and 45 Å for G3, G4 and G5 respectively. These values are in concordance with the results observed from DOSY experiments (Table 1).
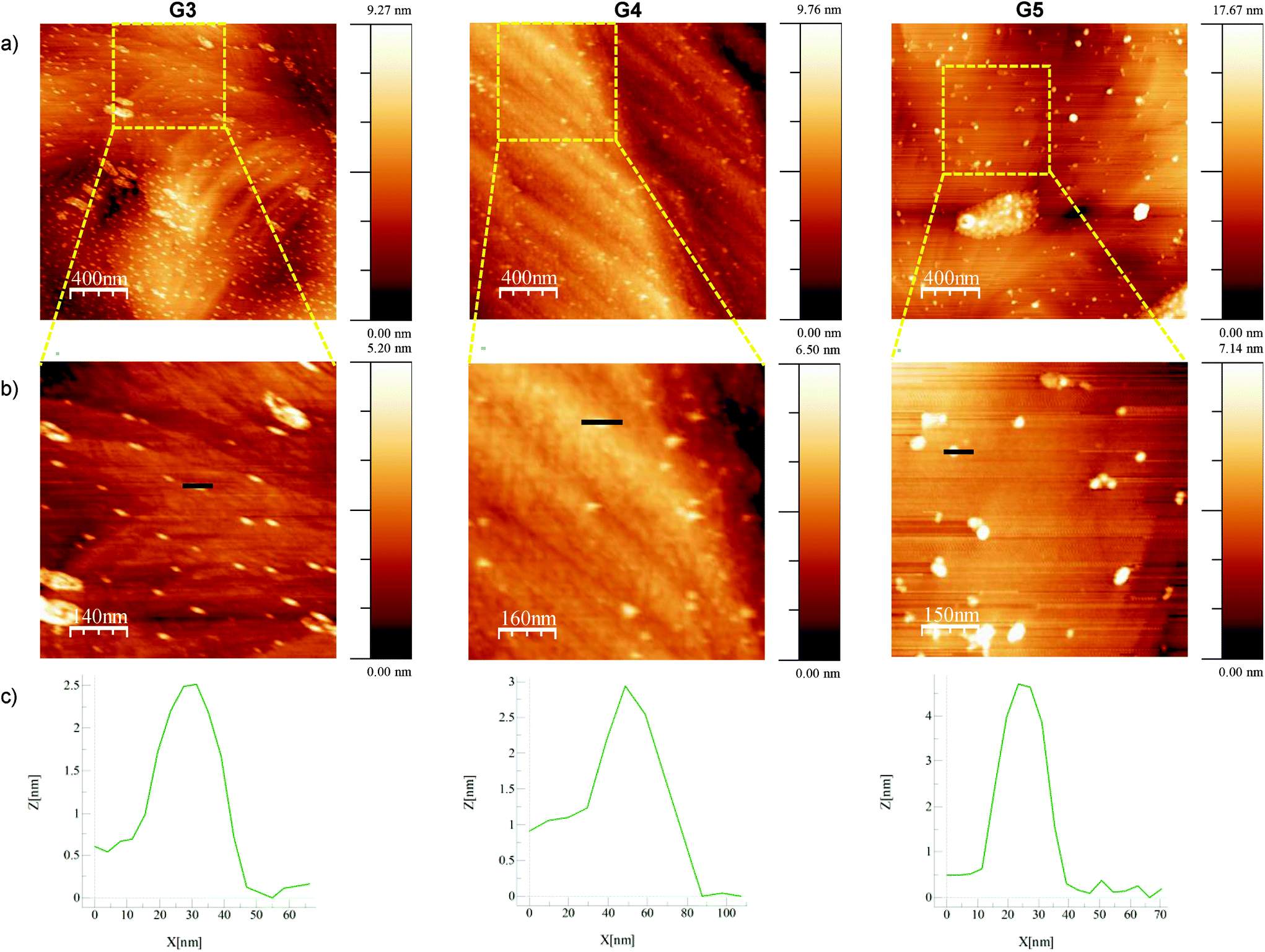 | ||
| Fig. 4 (a and b) AFM images obtained for G3, G4 and G5 deposited on (111) gold substrates. Images in (b) correspond to the zoomed-in sections of the dashed areas depicted in (a). (c) High-distance profiles obtained for the representative features selected in (b) (bold black lines). | ||
Molecular dynamics simulations
Molecular models of these dendrimers were built and subjected to molecular dynamic simulations in water as an explicit solvent (Fig. 5). Three different residues were used to define these compounds: the core (COR), the repeating fragment (REP), and the terminal ones (TAM) (Fig. S69, ESI†). The structures obtained from MDS have been analyzed and properties such as the radius of gyration (Rg), the aspect ratio and the asphericities have been calculated (Table 1 and Fig. S76 and S77 in the ESI†).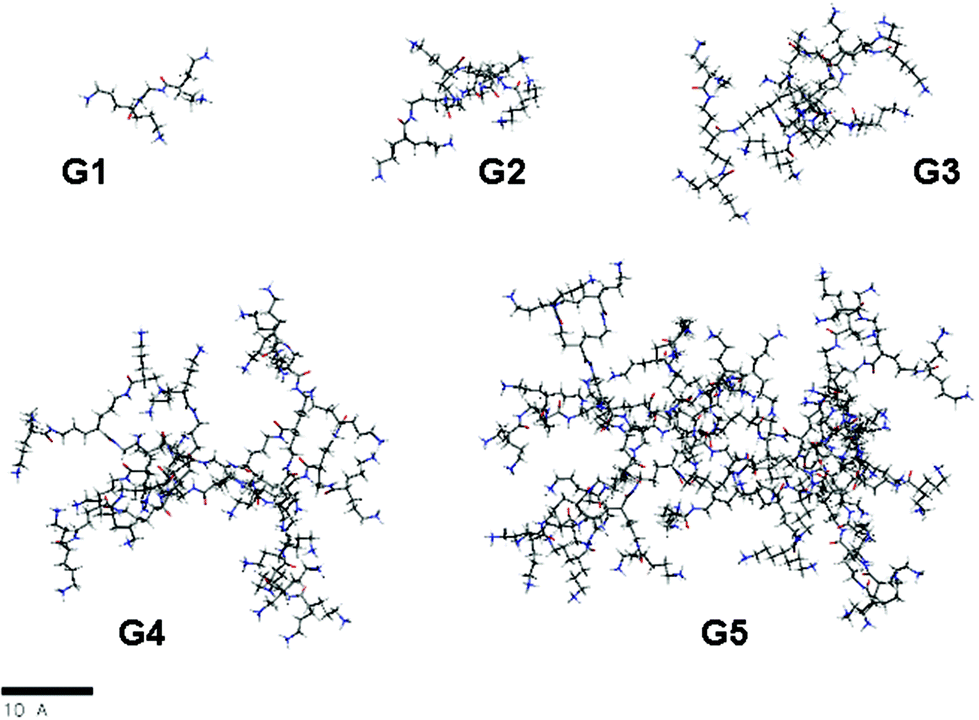 | ||
| Fig. 5 Snapshots from molecular dynamics simulations of G1–5; (to simplify, carbon atoms are depicted in gray, oxygen atoms in red, nitrogen atoms in blue and hydrogens in white). | ||
The radius of gyration obtained from the MDS calculations (Table 1) is in good agreement with the values quantified from the DOSY experiments and shows an increase in the size of these dendrimers from generation 1 to 5.
The aspect ratio of these dendrimers can be inferred from the average of the three principal moments of inertia (Ix, Iy, and Iz in decreasing order). The ratios (Ix/Iy) and (Ix/Iz) are the measures of the eccentricity (minor-major axes ratio) of the ellipsoid shape of the dendrimers. As the generation of dendrimers increases from G1 to G5, the relationship between the Ix/Iy and Ix/Iz values becomes smaller, indicating a change in the shape from ellipsoid to globular. This is also corroborated by the decrease in the asphericity values (Table 1 and Fig. S77†).
The logarithmic linear relationship between the number of atoms (logN) and the radius of gyration (logRg) points to a space-filling structure and the correlated fractal dimension (df) gives an idea of the compactness of the dendrimer series. The size scale found for this dendrimer series is N ≈ Rg2.51 (df = 2.51, Fig. S78, ESI†). This implies that these dendrimers have a more polymeric-like structure than a perfect compact sphere where df is 3.42
The radial density profiles can describe the atom distribution within the dendrimers. Those corresponding to G1–5 dendrimer generations are shown in Fig. S79, ESI.† All compounds have the maximum density close to the core of the dendrimers and it decreases towards the edge of the molecule. In G3 to G5 dendrimers appears a plateau, corresponding to the REP unit distribution, along the entire molecule decaying slowly at the end. This suggests a region with high localization and low atom mobility, showing a dense dendrimer shell pattern. With each generation, the number of TAM units doubles and the terminal amino groups spread over a larger part of the molecule. This points to a high degree of back-folding (Fig. S79, ESI†), especially for G4 and G5 dendrimers where an appreciable distribution of the amino terminal groups can be found close to the core, although with increasing density toward the outer region of the dendrimer (Fig. 6).
 | ||
| Fig. 6 Radial distribution function of the terminal amino groups in Gn-NH3term using the dendrimer center of mass as a reference. The unit value for ρ(r) is expressed in atoms per Å3. | ||
The design of these dendrimers present some advantages relative to our previously published BAPAD dendrimers decorated with amino terminal groups.21 Although both dendrimers present compact structures (similar df), those described in this work are slightly larger and more globular (better aspect ratios and less asphericities). Finally, they present less back-folding relative to BAPAD, allowing access to higher generations.
Conclusions
The presented methodology allows the preparation of a new family of stable and aqueous soluble high-generation dendrimers on a gram scale, representing a substantial improvement in the synthesis of aliphatic polyamide amino terminal dendrimers. Our design resulted in the achievement of a high amount of robust and stable dendrimers using a cheap, easy and efficient synthesis method together with high yields and the avoidance of tedious purification procedures. NMR, SEC, DOSY and AFM techniques were applied to evaluate G1–5, and all of them concur with the estimated size of these dendrimers, observing an exponential increase in size with generations. Molecular dynamics simulations (MDS) were also carried out, with the values consistent with the experimentally determined values. G1–5 are promising scaffolds for applications where the combination of stability, multivalence and efficient conjugation is required.Author contributions
The manuscript has been written through contributions of all authors. E. Perez-Inestrosa and Y. Vida conceived and designed the experiments. A. Morgado performed the chemical synthesis and analysis of the compounds helped by Y. Vida. F. Najera performed molecular dynamics simulation studies. A. Lagunas and J. Samitier performed the AFM studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ministerio de Ciencia y Educación (PID2019-104293GB-I00), Ministerio de Ciencia e Innovación (Proyectos de I+D+I “Programación Conjunta Internacional”), EuroNanoMed 2019 (PCI2019-111825-2), Instituto de Salud Carlos III (ISCIII; RETIC ARADYAL RD16/0006/0012) and Junta de Andalucía and Universidad de Málaga (UMA18-FEDERJA-007). We gratefully acknowledge the computer resources provided by the SCBI (Supercomputing and Bioinformatics Center) of the University of Malaga. NMR experiments have been performed in the ICTS “NANBIOSIS”, in the U28 Unit at the Andalusian Centre for Nanomedicine and Biotechnology (BIONAND).Notes and references
- R. M. Kannan, E. Nance, S. Kannan and D. A. Tomalia, J. Intern. Med., 2014, 276, 579–617 CrossRef CAS.
- E. Buhleier, W. Wehner and F. Vögtle, Synth., 1978, 1978, 155–158 CrossRef.
- R. G. Denkewalter, J. F. Kolc, W. J. Lukasavage, K. E. Stroup, R. C. Stewart and A. M. Doernberg, Macromolecular highly branched homogeneous compound, US Pat., 4410688, 1983 Search PubMed.
- D. A. Tomalia and J. M. J. Fréchet, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2719–2728 CrossRef CAS.
- P. Stenström, E. Hjorth, Y. Zhang, O. C. J. Andrén, S. Guette-Marquet, M. Schultzberg and M. Malkoch, Biomacromolecules, 2017, 18, 4323–4330 CrossRef PubMed.
- Z. Lyu, L. Ding, A. Y. T. Huang, C. L. Kao and L. Peng, Mater. Today Chem., 2019, 13, 34–48 CrossRef CAS.
- M. A. Mintzer, M. W. Grinstaff, I. Paetsch, P. Hunold, M. Mahler, K. Shamsi, E. Nagel, C. F. Price, L. J. Clark, J. R. A. Paull, C. K. Fairley, X. Lou and E. W. Meijer, Chem. Soc. Rev., 2011, 40, 173–190 RSC.
- J. R. Lloyd, P. S. Jayasekara and K. A. Jacobson, Anal. Methods, 2016, 8, 263–269 RSC.
- S. Falkovich, D. Markelov, I. Neelov and A. Darinskii, J. Chem. Phys., 2013, 139, 064903 CrossRef CAS PubMed.
- D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Polym. J., 1985, 17, 117–132 CrossRef CAS.
- G. R. Newkome, Z. Yao, G. R. Baker and V. K. Gupta, J. Org. Chem., 1985, 50, 2003–2004 CrossRef CAS.
- C. J. Hawker and J. M. J. Frechet, J. Am. Chem. Soc., 1990, 112, 7638–7647 CrossRef CAS.
- F. Vögtle, G. Richardt and N. Werner, Dendrimer chemistry: Concepts, Synthesis, Properties, Applications, Wiley-VCH, 2009 Search PubMed.
- D. A. Tomalia, J. B. Christensen and U. Boas, Dendrimers, Dendrons, and Dendritic Polymers. Discovery, Applications, and the Future, Cambridge University Press The Edinburgh Building, Cambridge CB2 8RU, UK, 2012 Search PubMed.
- K. L. Wooley, C. J. Hawker and J. M. J. Frechet, J. Am. Chem. Soc., 1991, 113, 4252–4261 CrossRef CAS.
- M. V. Walter, M. Malkoch, E. Soderlind, K. Ohshimizu, J. N. Hunt, K. Sivanandan, M. I. Montanez, M. Malkoch, M. Ueda, C. J. Hawker and J. M. DeSimone, Chem. Soc. Rev., 2012, 41, 4593 RSC.
- G. Franc and A. K. Kakkar, Chem. Soc. Rev., 2010, 39, 1536–1544 RSC.
- C. Ornelas, J. R. Aranzaes, E. Cloutet and D. Astruc, Org. Lett., 2006, 8, 2751–2753 CrossRef CAS PubMed.
- L. Brauge, G. Magro, A. M. Caminade and J. P. Majoral, J. Am. Chem. Soc., 2001, 123, 6698–6699 CrossRef CAS PubMed.
- S. García-Gallego, O. C. J. Andrén and M. Malkoch, J. Am. Chem. Soc., 2020, 142, 1501–1509 CrossRef.
- A. J. Ruiz-Sanchez, P. Mesa-Antunez, N. Barbero, D. Collado, Y. Vida, F. Najera and E. Perez-Inestrosa, Polym. Chem., 2015, 6, 3031–3038 RSC.
- D. Jishkariani, C. M. MacDermaid, Y. N. Timsina, S. Grama, S. S. Gillani, M. Divar, S. S. Yadavalli, R.-O. Moussodia, P. Leowanawat, A. M. Berrios Camacho, R. Walter, M. Goulian, M. L. Klein and V. Percec, Proc. Natl. Acad. Sci. U.S.A., 2017, 114, E2275–E2284 CrossRef CAS PubMed.
- N. Molina, F. Nájera, J. A. Guadix, J. M. Perez-Pomares, Y. Vida and E. Perez-Inestrosa, J. Org. Chem., 2019, 84, 10197–10208 CrossRef CAS.
- P. Mesa-Antunez, D. Collado, Y. Vida, F. Najera, T. Fernandez, M. Torres and E. Perez-Inestrosa, Polymers, 2016, 8, 111 CrossRef PubMed.
- N. Molina, A. González, D. Monopoli, B. Mentado, J. Becerra, L. Santos-Ruiz, Y. Vida and E. Perez-Inestrosa, Polymers, 2020, 12, 770 CrossRef CAS.
- N. Molina, M. Cnudde, J. A. Guadix, J. M. Perez-Pomares, C. A. Strassert, Y. Vida and E. Perez-Inestrosa, ACS Omega, 2019, 4, 13027–13033 CrossRef CAS PubMed.
- P. G. de Gennes and H. Hervet, J. Phys., Lett., 1983, 44, 351–360 CrossRef CAS.
- V. A. Jiménez, J. A. Gavín and J. B. Alderete, Struct. Chem., 2012, 23, 123–128 CrossRef.
- M. A. van Dongen, B. G. Orr and M. M. Banaszak Holl, J. Phys. Chem. B, 2014, 118, 7195–7202 CrossRef CAS PubMed.
- I. Horcas, R. Fernández, J. M. Gómez-Rodríguez, J. Colchero, J. Gómez-Herrero and A. M. Baro, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef CAS PubMed.
- D. A. Case, T. A. Darden, T. E. Cheatham, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. P. Roberts, S. Hayik, A. E. Roitberg, G. Seabra, J. M. Swails, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wo, A. Kovalenko and P. A. Kollman, AMBER 12, University of California, San Francisco (Computer program), 2012 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- V. Maingi, V. Jain, P. V. Bharatam and P. K. Maiti, J. Comput. Chem., 2012, 33, 1997–2011 CrossRef CAS PubMed.
- J.-P. Ryckaert, G. Ciccotti and H. J. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- K. T. Nguyen, S. Syed, S. Urwyler, S. Bertrand, D. Bertrand and J.-L. Reymond, ChemMedChem, 2008, 3, 1520–1524 CrossRef CAS PubMed.
- S. P. Amaral, M. H. Tawara, M. Fernandez-Villamarin, E. Borrajo, J. Martínez-Costas, A. Vidal, R. Riguera and E. Fernandez-Megia, Angew. Chem., Int. Ed., 2018, 57, 5273–5277 CrossRef CAS PubMed.
- J. W. Lee, H. J. Kim, S. C. Han, J. H. Kim and S.-H. Jin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1083–1097 CrossRef CAS.
- B. Fritzinger and U. Scheler, Macromol. Chem. Phys., 2005, 206, 1288–1291 CrossRef CAS.
- A. Lagunas, A. G. Castaño, J. M. Artés, Y. Vida, D. Collado, E. Pérez-Inestrosa, P. Gorostiza, S. Claros, J. A. Andrades and J. Samitier, Nano Res., 2014, 7, 399–409 CrossRef CAS.
- A. Lagunas, I. Tsintzou, Y. Vida, D. Collado, E. Pérez-Inestrosa, C. Rodríguez Pereira, J. Magalhaes, J. A. Andrades and J. Samitier, Nano Res., 2017, 10, 1959–1971 CrossRef CAS.
- P. K. Maiti, T. Çaǧin, G. Wang and W. A. Goddard, Macromolecules, 2004, 37, 6236–6254 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C, COSY and HSQC spectra of all of the described compounds, DOSY NMR experiments, AFM experiments and theoretical calculations. See DOI: 10.1039/d1py00667c |
| This journal is © The Royal Society of Chemistry 2021 |