
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoinitiating systems and kinetics of frontal photopolymerization processes – the prospects for efficient preparation of composites and thick 3D structures
Filip
Petko
 ab,
Andrzej
Świeży
ab and
Joanna
Ortyl
*ab
ab,
Andrzej
Świeży
ab and
Joanna
Ortyl
*ab
aPhoto HiTech Ltd, Bobrzyńskiego 14, 30-348 Cracow, Poland. E-mail: jortyl@pk.edu.pl
bDepartment of Biotechnology and Physical Chemistry, Faculty of Chemical Engineering and Technology, Cracow University of Technology, Warszawska 24, 30-155 Kraków, Poland
First published on 2nd July 2021
Abstract
In recent years, frontal polymerisation has found a growing interest in many applications. Thus, its ability to cure thick samples with different fillers up to 50% by local application of a reasonable portion of energy makes this polymerisation approach very promising. It can find many applications in materials science and industry as well as in the preparation of 3D structures. Herein, we present recent achievements in the field of frontal photopolymerisation and phototriggered frontal thermal polymerisation. A light application allows full control of the curing process. The combination of frontal polymerisation and light curing provides a convenient and versatile method to trigger frontal polymerisation, especially in heat sensitive materials or dentistry applications. Moreover, in this review, we focused on the photoinitiating systems in frontal polymerisation, their mechanism of action, components, and applications, dividing them according to the type of polymerisation: free-radical polymerisation, ring-opening cationic polymerisation and other minor types.
Introduction
The idea of the autocatalytic propagating thermal front was used for the first time in 1967 by Merzhanov and Borovinskaya. They called it self-propagating high-temperature synthesis and used it to prepare ceramics and intermetallic compounds.1 In the polymerisation area, this phenomenon was first used in 1972 by the Chechilo team to obtain poly-methyl methacrylate.2Nowadays, the definition created by Pojman is employed to describe frontal polymerisation. He states that frontal polymerisation (FP) is a process that occurs directionally in a localised reaction zone.3
The initial approach to FP was the thermal approach (TFP). Generally, in TFP, the process's driving force is the heat generated during a polymer chain propagating after a thermal initiator's initial cleavage. From a theoretical point of view, TFP can be started by any process that increases the initiation volume temperature.4 Therefore, TFP is the most commonly studied method. To conduct TFP, the system, where the reaction is exothermic, is needed. The system must have a low rate at the initial temperature (no reaction or very slowly reaction at room temperature) and have a significant heat release and high energy activation.3 Despite its many advantages, it quickly became apparent that TFP also had many disadvantages. The initial heat must provide enough energy to create the appropriate number of radicals, which can continue the process because of the process's exothermic character and the autocatalytic effects (Trommsdorff effect and Arrhenius dependence); the control of the TFP process is complex.5
Drawbacks of TFP have forced the search for new approaches. Potentially controllable in time and conversion rate method is frontal photopolymerisation (FPP).3 By swapping a thermal initiator with a photoinitiator system ignited with UV light is obtained.6 Consequently, light absorption leads the monomer to polymer conversion, and the rate and degree of conversion depend on the given light intensity. FPP method also allows for greater depth of conversion. The other advantage is the possibility to use fluorescent probes which not only allow to monitor systems7,8 but also sometimes accelerate the reaction.9,10 On the other hand, it has many disadvantages too.11 First of all, comparing to TFP, where after process ignition it is autocatalytic, in FPP the continuous exposure of the reaction mixture to light radiation is needed.11 Moreover, to obtain a good curing depth, photoinitiators with characteristic “bleaching” properties have to be used.12
A new approach was created by combining the above method, where both thermal and photoinitiators are entered into the system. The cleavage of photoinitiators under UV radiation leads to chain propagation which exothermic effect contributes to thermal initiator cleavage, and the process is auto-accelerated and controllable in some aspects.4
The method described above has different advantages and has individual applications. FPP, driven by continuous exposure of energy and potentially controllable parameters, has found a place in areas where it is necessary to obtain, e.g., thin layers.3
Radical frontal polymerisation
The radical approach to frontal polymerisation has many advantages. The main driving forces behind such polymerisation are two phenomena found in the use of free-radical systems. They are the Trommsdorff effect and thermal autocatalysis.13 The first of them says that the polymerisation rate increases with the degree of monomer conversion, resulting from the increase in the system's density during the process itself, and thus the possibility of contact between two macroradicals and completion of the reaction is difficult.14 The second effect is the spontaneous fuelling of the polymerisation process resulting from chain propagation's exothermic nature based on the Arrhenius dependence of the initiator decomposition.13In the case of conventional polymerisation processes, these phenomena usually do not bring any benefits and may even lead to changes in the final product's properties. However, the situation is different in the case of frontal polymerisation, where with local process initiation, it is auto-accelerated into unreacted regions due to a combination of these two effects.13
An additional advantage of radical frontal photopolymerisation (FPP) is the possibility of using UV-initiated multifunctional monomers. Due to this approach, not only the amount of energy used in the process are reduced, but also spatially and time-controllable systems are obtained.15 Free-radical systems used for FPP will be described later in this paper.
The main mechanism of FPP is analogous to other types of polymerisation and can be divided into three main steps (Fig. 1).3 In the initiation stage, the photoinitiator decomposes under UV radiation, generating radicals that react with the monomer to form a polymer network. Then the chain propagates. The solid front formed closest to the light source begins move toward to the uncross linked medium.16
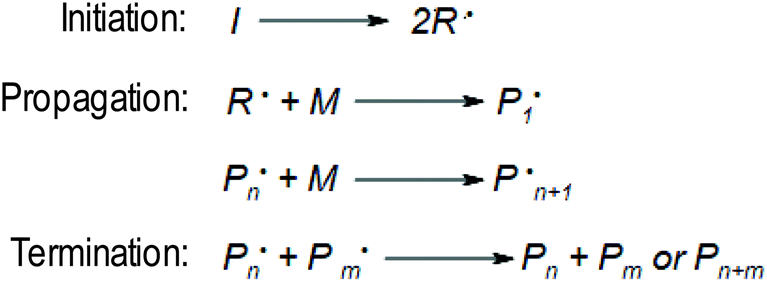 | ||
| Fig. 1 Mechanism of free radical frontal photopolymerisation. | ||
Subsequent monomer molecules are connected by a covalent bond. A characteristic thin interfacial area is created between the monomer-rich liquid phase and the polymer-rich solid phase, which separates these two phases unequivocally. As the solid front moves from the illuminated surface into the bulk, the unreacted monomer can diffuse into the polymer network. In that case, the polymer swells which can lead to instability of the whole system (Fig. 2).17 During further polymerisation, the volume of the solution decreases, and the volume of solid phase is shrinking. The layers above them press the newly formed layers, and the next ones are formed slower and slower due to the hindered flow of light radiation until the polymerisation and recombination of radicals are extinguished, which is the end of the process.18
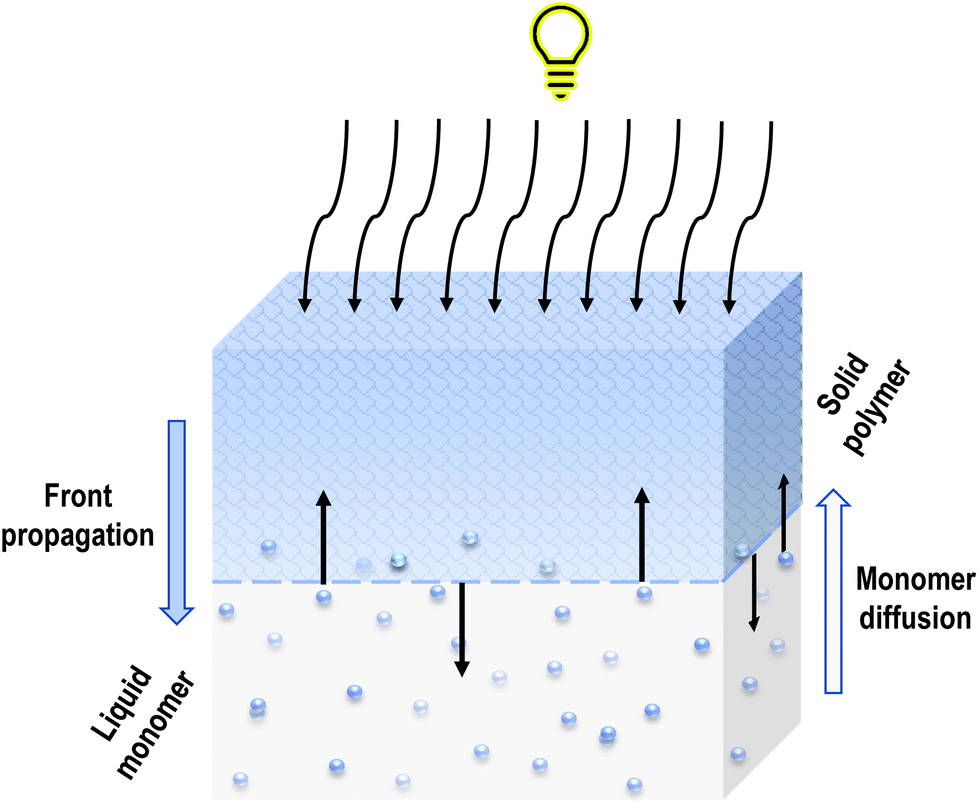 | ||
| Fig. 2 Polymer swelling during front propagation. | ||
To counteract this phenomenon, it is necessary to use photobleaching initiators (Fig. 3). The absorption maximum of such initiators is different from the absorption maximum of their decay products, so light penetrates deeper, and a higher depth of cure is obtainable. The most important thing is maintaining the process's parameters, such as photoinitiator type, concentration, and solvent type, to obtain the most efficient system.12 On the one hand, using a high concentration of photoinitiator will increase the radical formation, theoretically, the polymerisation rate. However, on the other hand, the system's absorption will increase which will translate into worse penetration into the solution. Selecting the photoinitiators concentration in such a way as to maintain deep penetration into the depth while simultaneously generating enough radicals to create a homogeneous material is the key in the frontal polymerisation approach.19
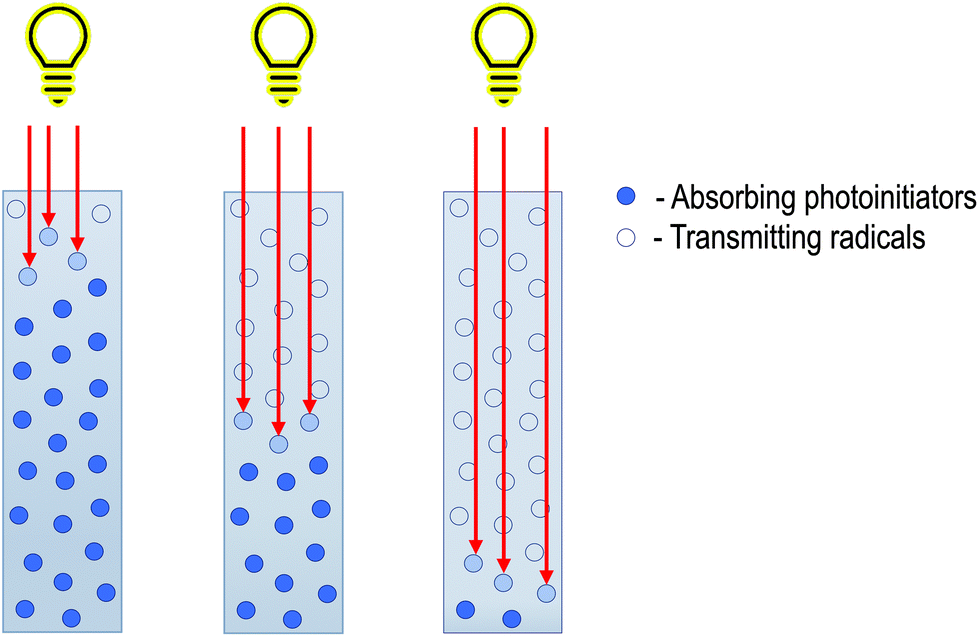 | ||
| Fig. 3 Photobleaching of initiator during FPP. | ||
The FPP operation principle is a bit different from the traditional thermally driven frontal polymerisation (TPF). In the classical approach, the heated system undergoes autocatalytic polymerisation which is the sum of the system's thermal effects.3,20 In contrast, FPP is not fully autocatalytic. The solid front is created after photocleavage of the photoinitiator, and it is moved by continuous exposure to UV rays. Further polymerisation can be stopped simply by turning off the light, but nothing prevents reactivating when the light source is restarted.11,15 Due to these properties, nowadays, FPP is the preferred method used in the fabrication process.15,16
It is important that the chain propagation process itself is exothermic, so thermal effects can also occur in FPP. It has been reported that the faster the initiation and polymerisation process, the greater the temperature rise during FPP. This translates into an even greater acceleration of the front to move deeper into the solution. This effect is strongly active in the first stages of the process, and it disappears when the front crosses a certain critical point at which UV radiation reaches, so the whole process slows down. However, it should be remembered that while this effect may be beneficial, in a controlled FPP, it may lead to a situation in which, after switching off the radiation source, the polymerisation process itself continues for a certain period, which is called “heat rush”.15
There is also an approach that combines the techniques of thermal and photo radical frontal polymerisation.21–25 In thermal frontal polymerisation (TFP), after the initiation of the system, there is almost no option to control the process.22 Additionally, the thermal propagating front temperature can rise to above 200 °C, leading to bubble creation or even a “burnout” of the initiator.26 The bubbles appearing in the reaction mixture can lead to a breakdown of the propagating front and “burnout” of the initiator due to very low monomer conversion.22 These drawbacks can be overcome with photo-induced thermal frontal polymerisation systems. Two types of initiators are introduced into the system – photo and thermal initiator. Upon exposure of the sample to light, the photoinduced initiator cleaves and starts the polymerisation process. The heat created during photo-induced polymerisation leads to the thermal initiator's cleavage, radicals are created, and the process accelerates. When the light source is turned off, the polymerisation process stops. The rate of polymerisation can also be controlled by changes in light intensity.27
There are publications in the literature focusing on approximating the kinetic basis of the phenomena occurring during the FPP techniques described above.19,28–34
Initiators of radical frontal photopolymerisation
When choosing an initiator for FPP, it is necessary to pay attention to several important aspects. First, the appropriate type of initiator for the initiating system is needed. The initiator must absorb to the extent that the light from the source is emitted while exhibiting photobleaching ability in order not to cover the light rays at the depth so that the solid front can propagate further.The selection of the photoinitiator concentration also plays a key role. The chosen concentration must allow deep penetration of the light radiation; simultaneously, it must function efficiently, creating a sufficient number of radicals.12 Based on the above properties, mainly five groups of initiators are used in FPP (Fig. 4). This group contains azo initiators,35 peroxide initiators,36,37 sulphide initiators,38 redox initiators,36 and phosphonium based initiators.37
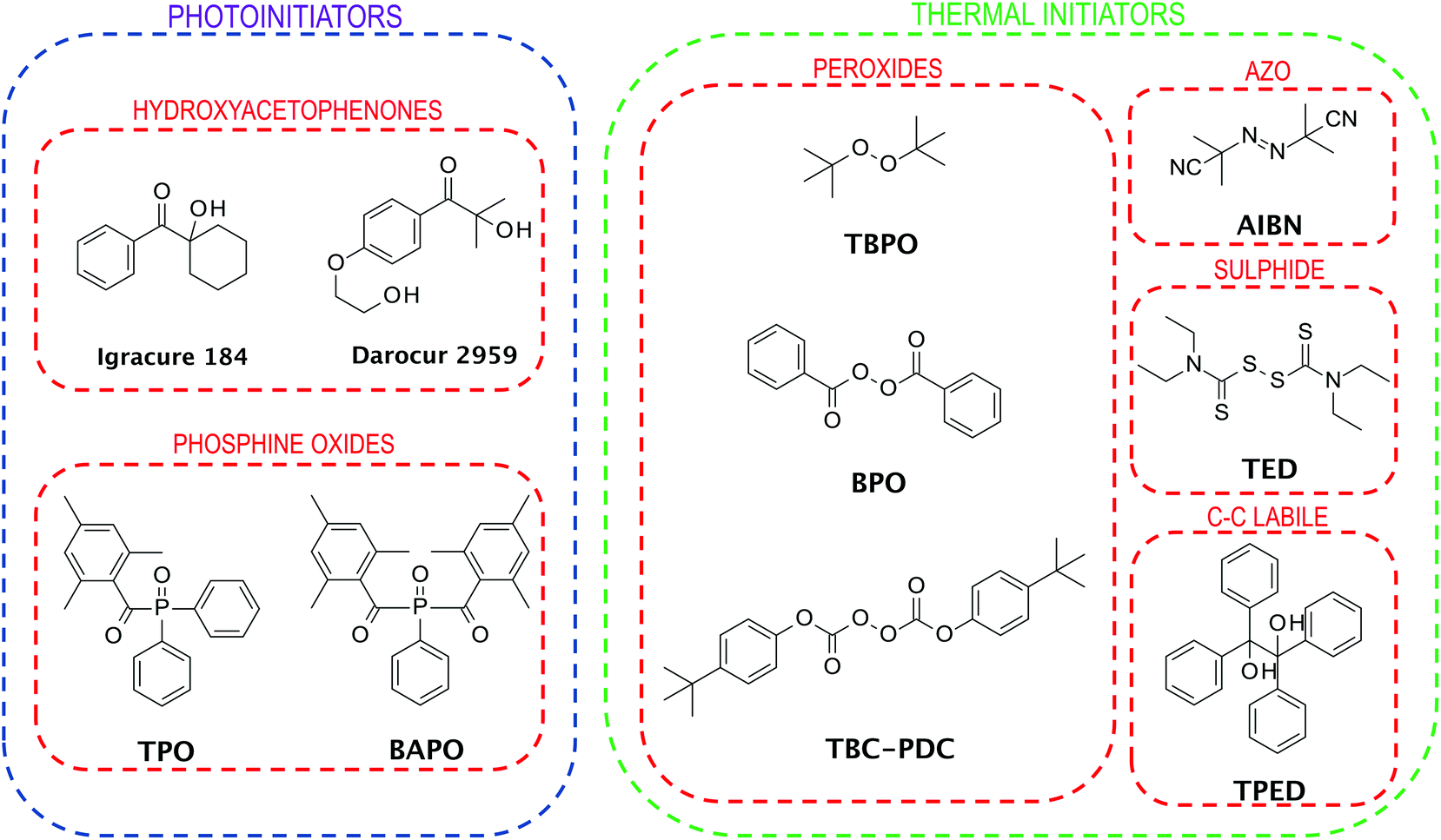 | ||
| Fig. 4 Examples of initiators used in FP. | ||
Of the above, redox initiators are characterised by the lowest activation energy, allowing for the creation of radicals in a reasonable time.39 Use of redox initiators, compared to others, allows achieving a faster induction time and lower front temperature, but their applications are limited.22 Commonly used in practical applications are initiators of peroxide and azo groups. Their main disadvantage is that during their cleavage, gaseous products are created. In open and stirred reactors, they do not show any adverse effects caused by the appearance of bubbles or the polymerisation front's recession, thus not affecting the physical and chemical properties of the obtained product. However, there are many FPP systems where the reactive monomer mixture cannot be stirred. In this case, too many bubbles deteriorate the mechanical resistance of the product. Therefore, the use of phosphine-based initiators has allowed this barrier to be overcome using such methods.37
Acrylate systems in radical frontal photopolymerisation
Acrylate systems are deeply investigated because of their potential use as hydrogels. The first paper describing frontal polymerisation was based on the polymerisation of poly(methyl methacrylate).2 Washington and Steinbock proposed a mechanism to prepare poly(N-isopropylacrylamide).40 Investigation of the mechanism of frontal polymerisation of hydrogels from a poly(acrylic acid),41,42 polyacrylamide,43 and others4,5,26,44–49 was also presented.Due to its chemical structure, monomers from acrylate species (Fig. 5) have found an application, especially in creating thick layers. The conversion of acrylate monomers into polymers is exothermic. It induces a self-propagating process of a solid polymer network, which penetrates deeply into the reaction mixture. At the same time, the polymer and liquid monomer are visibly separated.42
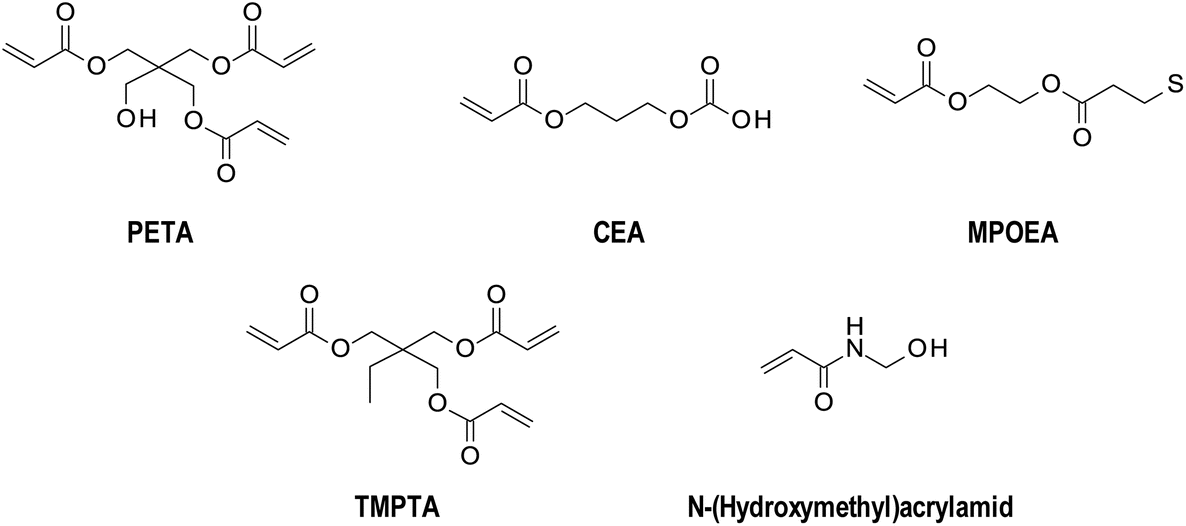 | ||
| Fig. 5 Examples of acrylate monomers used in FP. | ||
In the FPP of acrylate, the main problem is bubbling. Acrylates usually have a high boiling point that helps prevent bubble formation, but they are sensitive to oxygen which quenches the induced photoinitiator radical and deactivates it. Peroxyl radicals may also be created by the reaction of oxygen and free radicals. These new radicals are less reactive and cannot initiate polymerisation, so the conversion is smaller. Besides the solvent boiling, initiator decomposition can also lead to the formation of bubbles.4,50 To avoid this phenomenon, the reaction must be conducted under pressure,1,51 avoiding oxygen in the system or using a special blowing agent.50
Many acrylates have a different function group (e.g., N-methylolacrylamide). After the initiation step, some of this group has the ability to self-condensation or even react with groups from other polymers.41 Crosslinking can lead to achieving a product with properties unusual for “mono systems”, e.g., increased tensile strength, impact resistance, abrasion resistance, peel strength, solvent resistance and gloss.52 Acrylates also have a lower gel point compared to other systems such as thiol-enes.15
Analysing the conversion process of acrylic systems shows that the surface layer is almost completely reacted but then drops at some point, which may be due to that the propagation reaction becomes diffusion controlled during the auto deceleration regime. Moreover, crosslinking could have a negative impact on conversion because as the degree of crosslinking increases, the mobility of the radicals decreases.15 The creation of bubbles could also play a crucial role during the process. Several approaches allow eliminating this effect, such as high-pressure polymerisation,1 solvent-free synthesis,41 or carrying out the reaction in an aqueous medium with the use of water-soluble initiators.4,53
Thiol–ene systems in radical frontal photopolymerisation
Thiols are reacting with ene compounds giving polymers. This process is based on a free radical step growth mechanism (Fig. 6). In typical free-radical systems, radicals formed after cleavage react with oxygen, which is dissolved in the monomer formulation, or with oxygen that is diffused into the system during polymerisation. This process is very efficient, the affinity of radicals for oxygen is much greater than for propagation, what strongly inhibits the conversation of the monomer. Due to the unique propagation mechanism, thiol–ene systems are much more resistant to the influence of oxygen. Reacting radicals in contact with oxygen create peroxyl radicals that induce hydrogen from the thiol group, a thiol radical is formed, and the polymerisation is continued (Fig. scavenging).54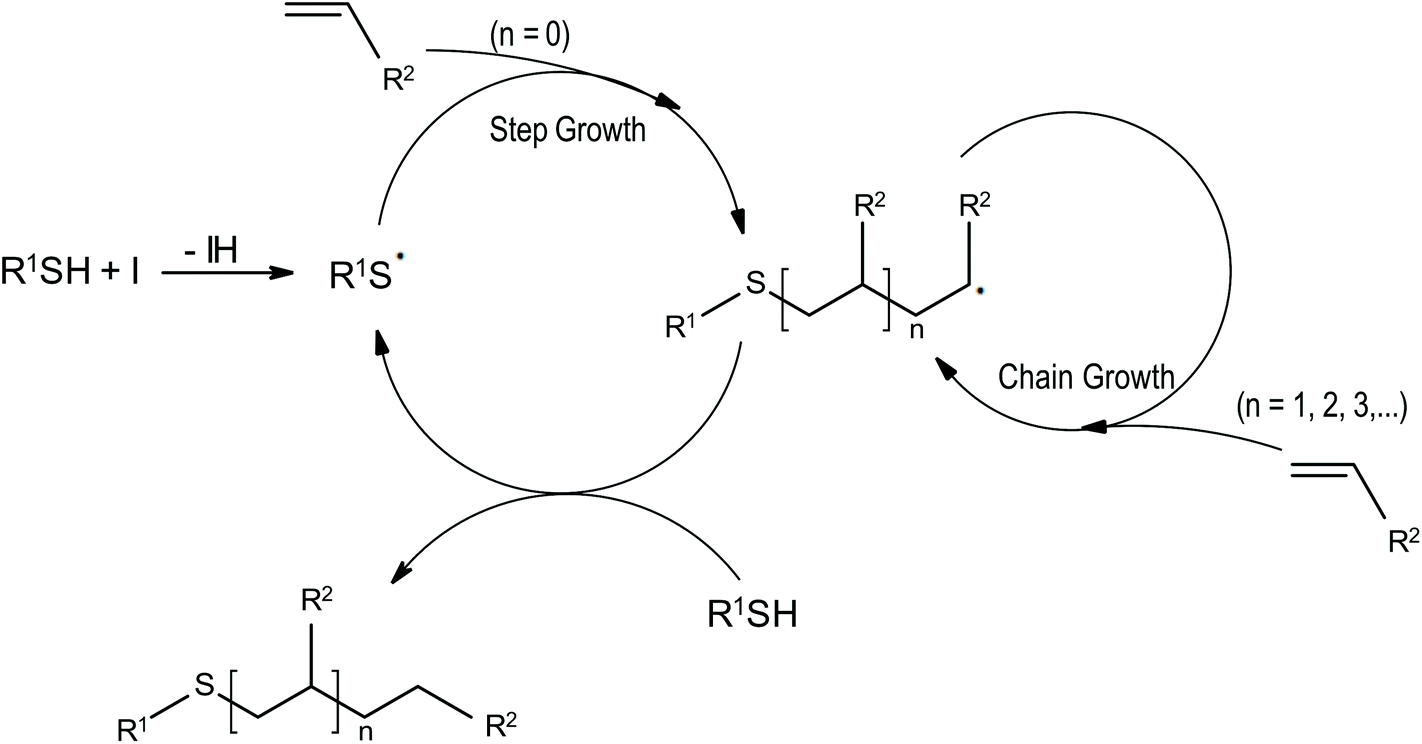 | ||
| Fig. 6 Mechanism of thiol–ene polymerisation. | ||
Thiol–ene system was used for the first time in frontal polymerisation by the Pojman team.51 Two thiol–ene systems were examined: dithiol with triallyl ether and trithiol with diacrylate. Based on the research, it was found that the properties of the propagating front depend not only on the initiator and monomer concentrations but also on the ratio of the monomer components.51 Besides Pojman, there are also others reports of thiol–ene FPP systems.15–17,55–59
The advantage of thiol–ene systems lies in rapid polymerisation with minimal oxygen inhibition. In addition, the depth of cure is larger than for acrylic monomers.55 Moreover, the shrinkage and shrinking stress are smaller.16 Polymerisation with thiol–ene monomers can proceed without photoinitiator molecules. Thiols polymerise vinyl systems to obtain products with unique properties – longer lifetime caused by higher sunlight resistance (because photosensitive molecules are absent) and no yellowing phenomenon of the polymer caused by the degradation of initiator molecules.57
Thiol monomers can be used in acrylate systems. The addition of thiol comonomer allows the polymerisation's control to start delay time and lower the front's temperature. The thiol lower the time needed to initiate the process. Due to thiols, polymerisation of highly filled acrylate systems that are otherwise very sensitive to such high concentration of oxygen is possible. FPP of highly filled systems is needed to obtain a polymer with enhanced mechanical properties. The main disadvantage of thiol-acrylate systems is a decrease in the front initiation rate with an increase in the concentration of thiol monomers, up to a specific critical value in which initiation does not occur at all. The reason is that the less exothermic thiol-acrylate copolymerisation process is much slower than the homopolymerisation of acrylate. In addition, besides of a faster initiation step, with too much thiol addition, the propagation may be slower. Therefore, these systems have not found application in highly bubbling systems.56
Application of radical frontal photopolymerisation
Frontal photopolymerisation has found wide application in many areas. It is used in lithography,58,59 rapid prototyping,60,61 dentistry,62 biomedicine,63 coatings64 and many others.24,65–67 Pojman company using frontal polymerisation to obtain different materials such as clay for modelling application or “woodFiller” and putty as repair materials.68From an interesting point of view, tissue engineering seems like the perfect place to use polymers created by FPP, especially acrylate systems. One idea of tissue engineering is to seek a material that could replace or restore damaged or diseased tissues. This material needs to be biocompatible, biodegradable, and meet bifunctional requirements. At the same time, it must have good mechanical properties.69 FPP is the perfect solution for these needs. During the process, thick, multilayer hydrogels can be created. Many acryls which have confirmed nontoxic properties are very popular in use as biomaterials. They minimalize protein adsorption, having high mechanical and biological resistance, and low hysteresis.64 FPP can quickly prepare uniform materials and vary hydrogels’ morphology in a controlled manner.61
Other interesting application is forming materials with wrinkled patterns on the surface. Two-stage polymerisation is necessary to make such a polymer. In the first step, an elastomer is synthesized, for example, using a stoichiometric imbalance of tetra-thiol and diacrylate monomers with an excess of acrylate. Next, the material is once again exposure to UV. The polymerization front advance, cross-link density increases, and the compressive stresses occurring in the process lead to forming a wrinkling pattern.70 One-pot approach is also presented in literature. Crosby and Chandra formed UV-curable composites with an uncured liquid layer, and then, induced by oxygen inhibition, photo frontal polymerisation was conducted. In this case, swelling of the uncured liquid layer occurred and a wrinkling pattern was formed.71 This phenomenon can be used in many ways, but the most important is the use of special photomasks to selectively create wrinkled and unwrinkled patterns on the surface, which allows to create inscriptions and other designed shapes.70
FPP can be used for obtaining three-dimensional (3D) origami structures.18,27,72,73 The mechanism of creating 3D structures from photosensitive polymers is based on photopolymerisation-induced volume shrinking (Fig. 7). Light irradiation of the initiator monomer system leads to a photopolymerisation process. A solid front is created at the surface. Covalent bonds are formed between monomers and cross-linkers with the continuous illumination front propagate which causes volume decrease. Essential is that in the FPP process, the phenomenon of volume shrinking is nonuniform. It proceeds as the front is moving into the depth of the system. Volume shrinkage can be controlled and used to create bending structures of the desired shape.27
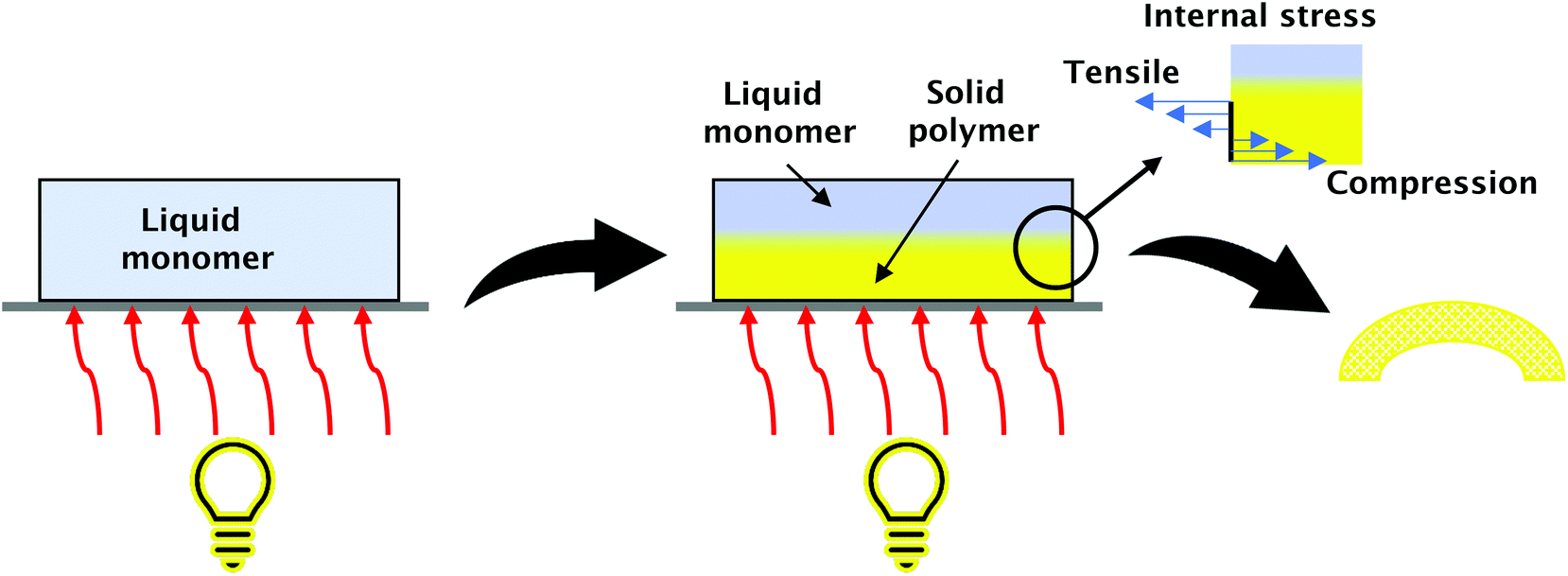 | ||
| Fig. 7 Obtaining 3D structures by FPP. Forces working on cured layers lead to material bending, where the degree of bend can be controlled by the process's conditions. | ||
Light gradient and the depth of its penetration can be controlled by the addition of photo absorbers.18 To explain the forming of 3D origami, we need to consider the reacting system as layers laying one on another. The surface layer affected by light will be cured immediately, but the layers that are adjective remain in a liquid state, together with the front propagating thickness of the polymer increases inwards. During further polymerisation, the layers start shrinking from external to internal, besides the first layer which is considered stress-free because the interactions between the polymer first layer and the substrate are not fully established initially. As a result, the obtained system is nonuniform; compressive stress works on layers near the surface and tensile stress on the newly created. The opposing forces lead to the bending of the resulting material. The degree and form of the bending depend on the process's conditions, so appropriate control allows for obtaining various structures.27 These phenomena are used, for example, in soft machines,74 artificial DNA structures,75 mechanical materials,76 sensors,77 and stretchable electronics.78
Ionic frontal photopolymerisation
Another commonly used type of photopolymerisation is ionic polymerisation, especially cationic ring-opening polymerisation of epoxides, oxetanes, and glycidyls.79 This polymerisation type has found applications in areas such as protective coatings, graphic arts, microelectronics, dental applications, security inks, and 3D printing.80 The main advantage of ionic photopolymerisation is its ability to continue even after the light is turned off. This phenomenon is called dark curing.81 Therefore, under certain conditions, ionic polymerisation may be considered as living polymerisation.82 Another crucial property of such kind of polymerisation is exothermic chain elongation. The heat generated during the ring-opening process is able to sustain polymerisation. It can manifest as self-propagation of the front.83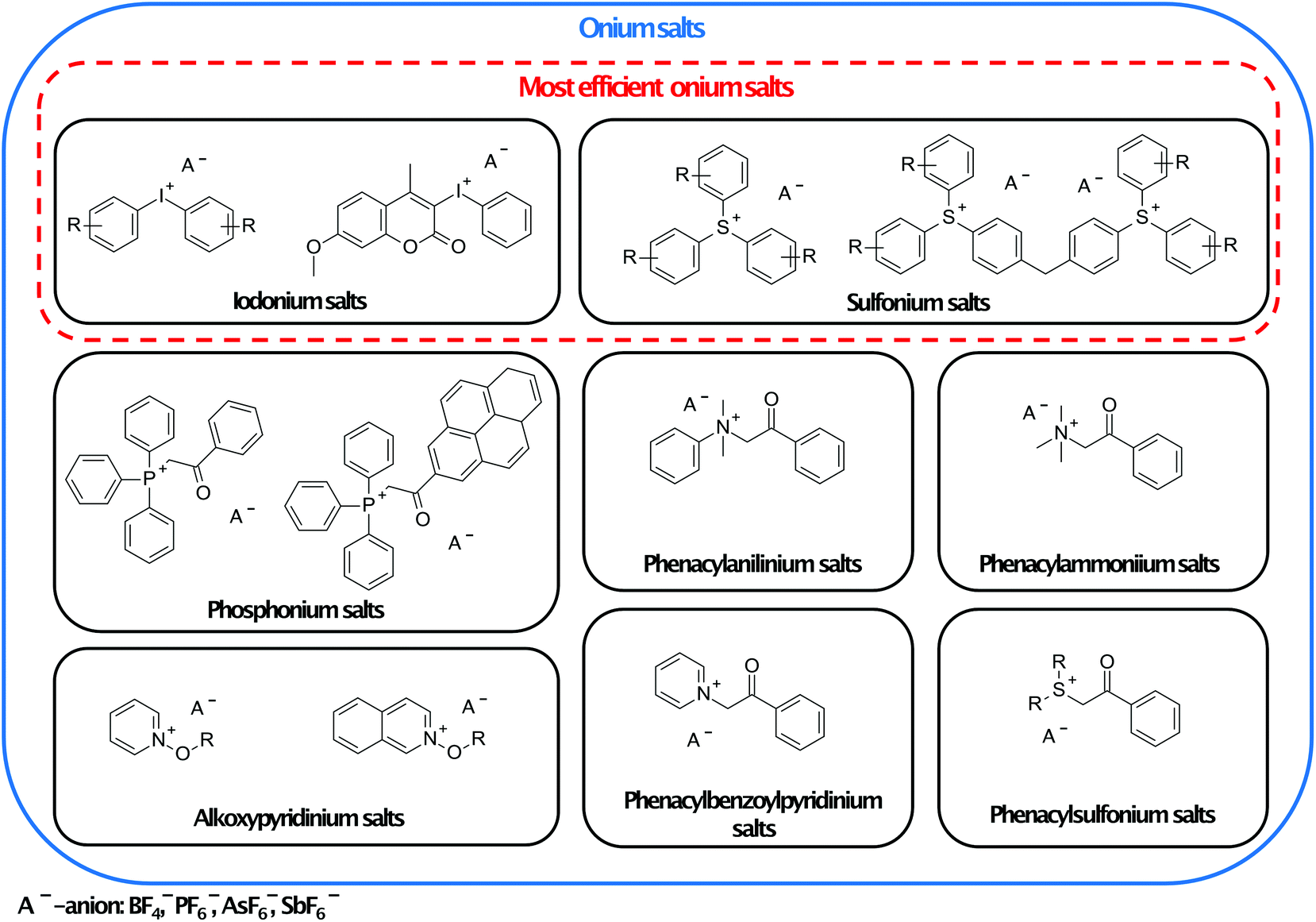 | ||
| Fig. 8 Families of photoinitiators and examples of their representatives. The most efficient ring-opening cationic polymerisation photoinitiators are marked in the red frame.88–90 | ||
The mechanism of action of onium salts during ring-opening cationic polymerisation is shown in Fig. 9. In the first step, photolysis of the cationic photoinitiator (e.g., onium salt) occurs, followed by the generation of Brønsted super acid through the reaction of the fragmentation product of the photoinitiator molecule with photogenic components of the composition. Generated super acid takes part in monomer molecules’ protonation (oxirane or oxetane molecules) to form a secondary oxonium ion which is the active form of monomer (cation). In the following steps, this secondary oxonium species is attacked by a monomer molecule that creates a tertiary oxonium ion active polymerisation centre. This centre is attacked by monomer molecules that elongate the polymer chain. Elongation occurs until all monomer molecules react, or the termination reaction occurs as other nucleophile species react with the active centre. The strength of photogenerated acid is crucial in terms of induction time and polymerisation rate.91 Only non-nucleophilic super acid as hexafluoroantimonate or hexafluoroarsenate is strong enough. The protonation of oxirane or oxetane moieties is nearly instantaneous in their presence, but the following steps’ rate strongly depends on the monomer structure.
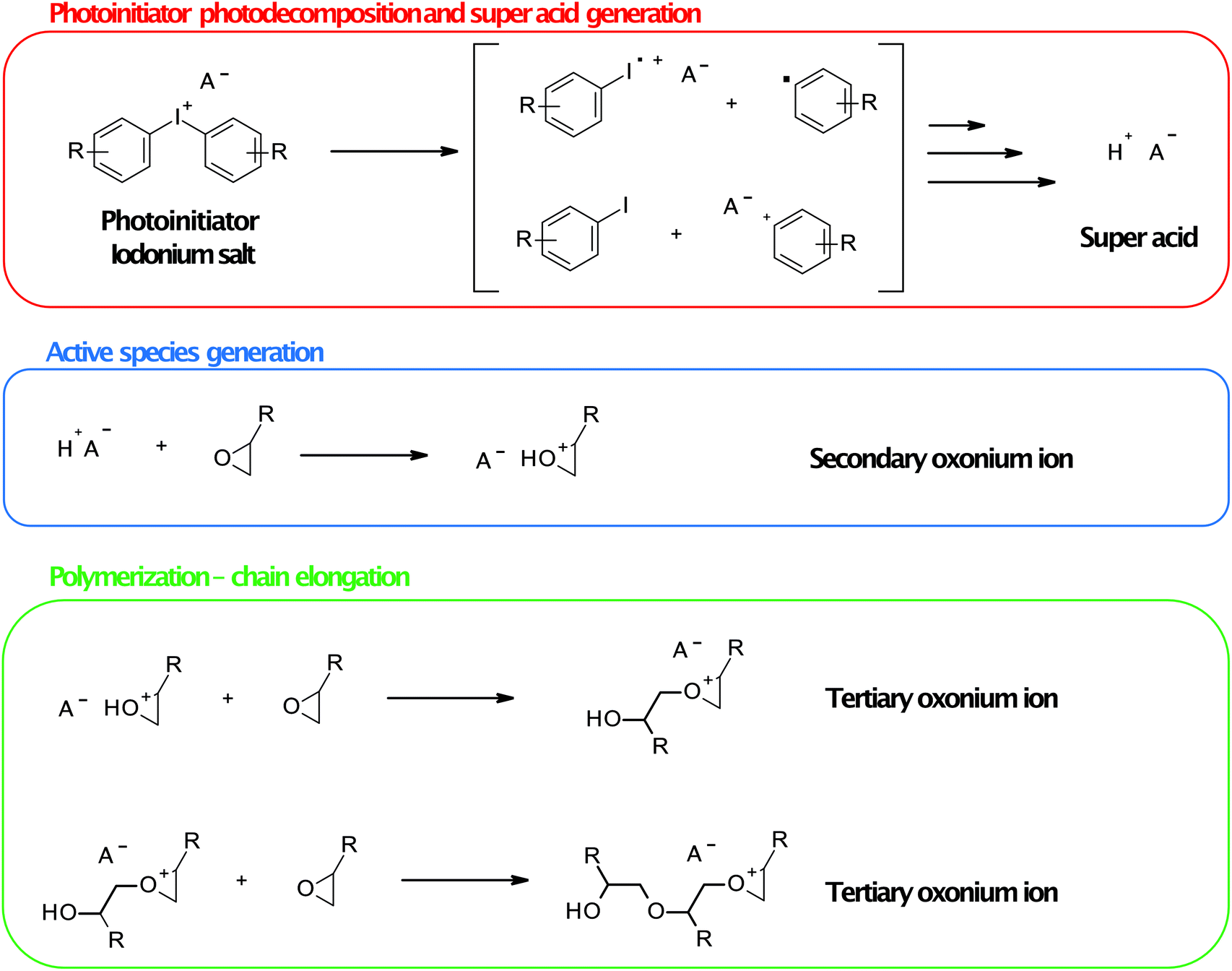 | ||
| Fig. 9 The mechanism of action of onium salts during ring-opening cationic polymerisation. Iodonium salt and oxirane monomers were shown for simplicity. | ||
In ring-opening cationic polymerisation, monomers are equipped with oxirane and oxetane moieties. These groups exhibit high ring tension, 18–24 kcal mol−1 for oxirane and 19–20 kcal mol−1 for oxetanes, respectively.92,93 This energy is released during the polymerisation process (with ring-opening phenomenon), leading to warm-up the polymerised samples. It can be used to sustain the polymerisation process in a frontal manner.
An overview of the process has been presented in Fig. 10. It has been confirmed that pre-irradiation is necessary to form active species of monomers. The masks’ experiments show that the frontal polymerisation occurs only at the boundary of the pre-irradiated surface and the rest of the monomer remains unpolymerized (Fig. 10B). This is evidence that the new active species are not formed during the thermal reaction.95
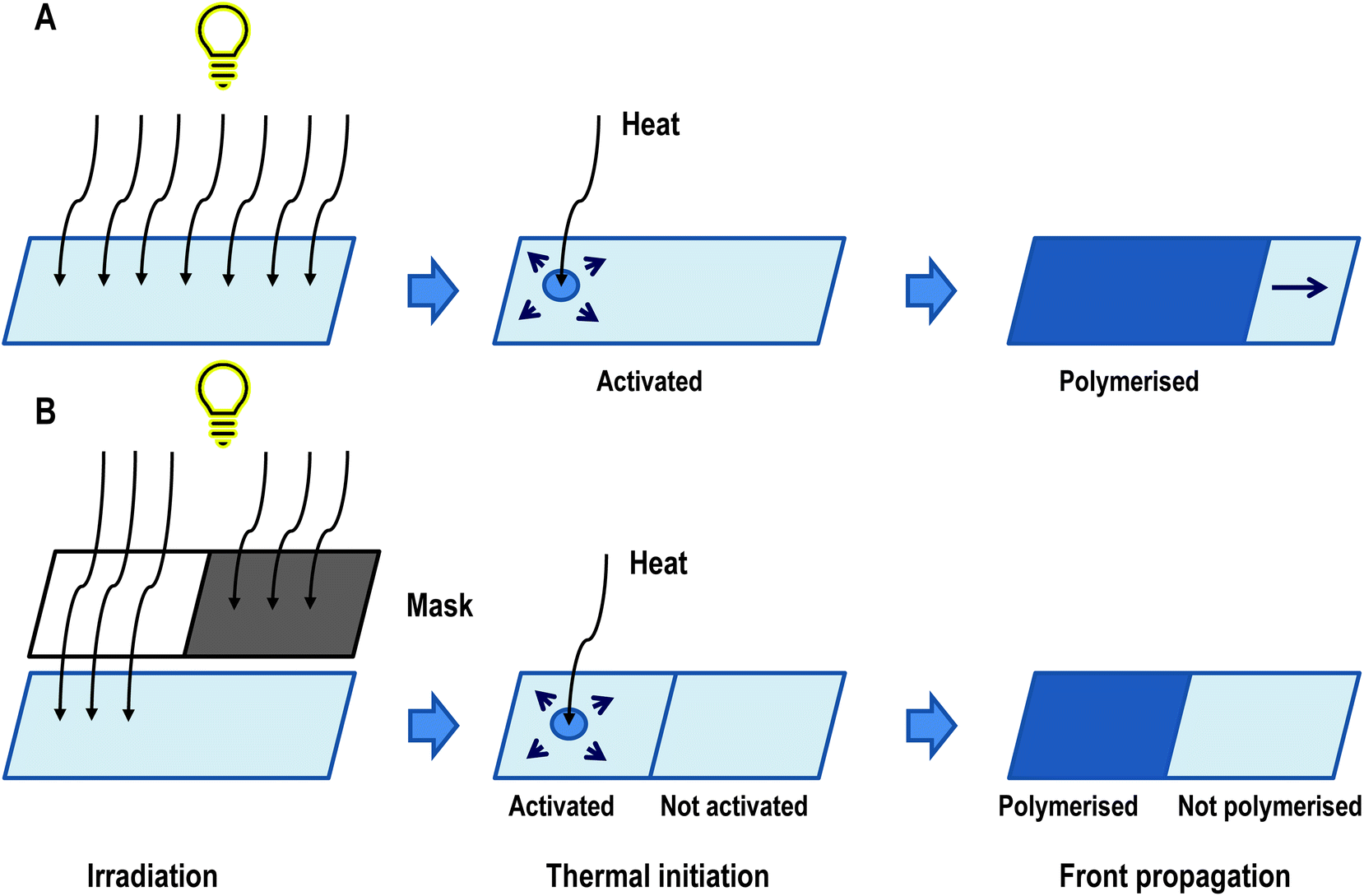 | ||
| Fig. 10 Conception of photoactivated cationic ring-opening frontal polymerisation divided into three stages of process. (A) Experiment without mask, where all sample was activated and polymerised. (B) Experiment with a mask, where only the irradiated sample was polymerised. This is evidence that the sample needs to be photoactivated.83 | ||
Irradiation is necessary by itself, but an appropriate dose of radiation is also required. The process was observed after irradiation with doses of 2936 and 1468 mJ cm−2, but not with a dose of 734 mJ cm−2.83 The explanation for this observation most likely lies in the number of active species generated during irradiation. The lowest flux generates an insufficient number of them, and the exothermic heat generated during polymerisation is not high enough to sustain the self-propelling polymerisation process.
Another factor that must be met is the type of acid generated by the photodecomposition of the initiator. It was observed that the photoactivated cationic frontal polymerisation is independent of the structure of the cation.96 However, the process is strongly dependent on the type and strength of the photogenerated acid. In Fig. 11, the different acids and super acids were presented in terms of their protonation ability shown in and the logarithmic Hammett acidity scale.91 Experiments have shown that super acids generated from anions like PF6− or BF4− are too weak in terms of protonating ability and reactivity of ion pairs formed in the first steps of the polymerisation process, to support a self-sustaining frontal polymerisation.96 The type of generated super acid must be appropriate, but the initiator's concentration must be high enough to generate enough number of active species, leading to a self-sustaining polymerisation process.96,97
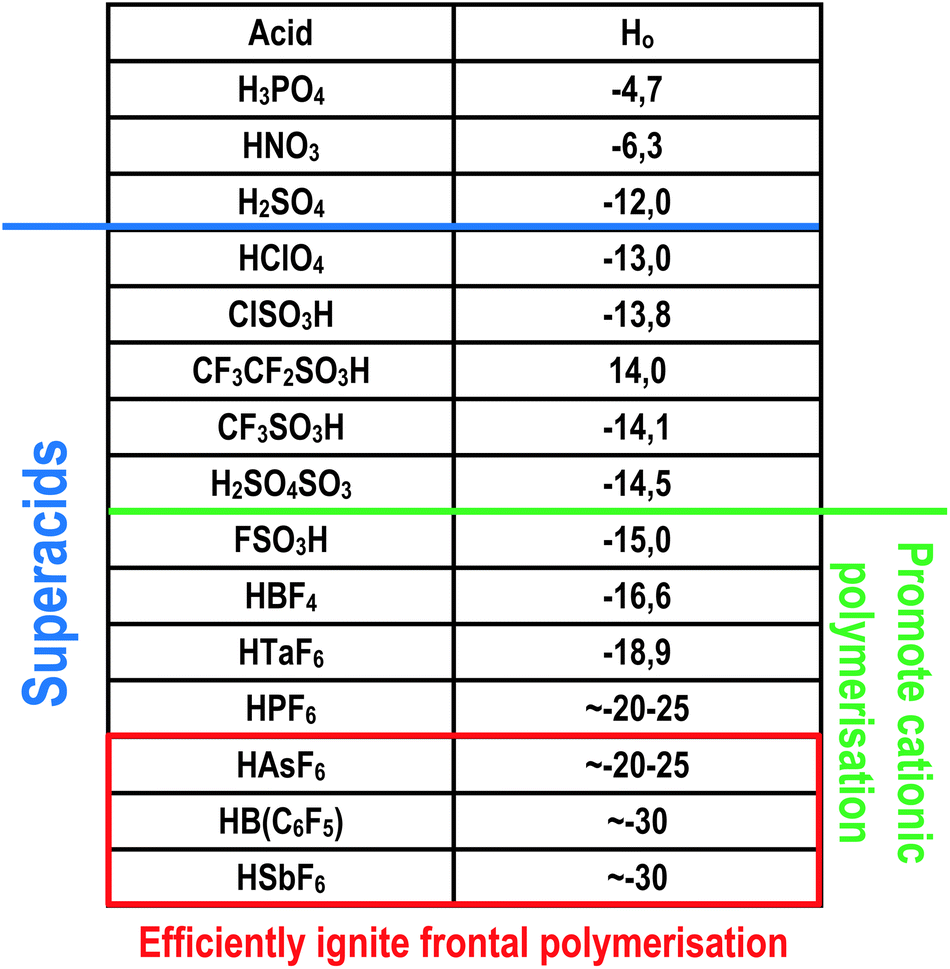 | ||
| Fig. 11 Acid strength comparison. H0 is the acid Hammett constant, the lower value the stronger acid.91 | ||
The induction time of polymerisation is a factor that is crucial for photoactivated cationic frontal polymerisation. Longer induction time allows longer irradiation, ensuring that all initiator molecules will be photodissociation.83 This parameter depends not only on the strength of the acid generated after photodissociation of the onium salt but also on the monomer structure. Not all monomers with oxirane and oxetane moieties exhibit a sufficiently long induction time for photoactivation to be possible.83 A large group of monomers were investigated, and their structures were depicted in Fig. 12.
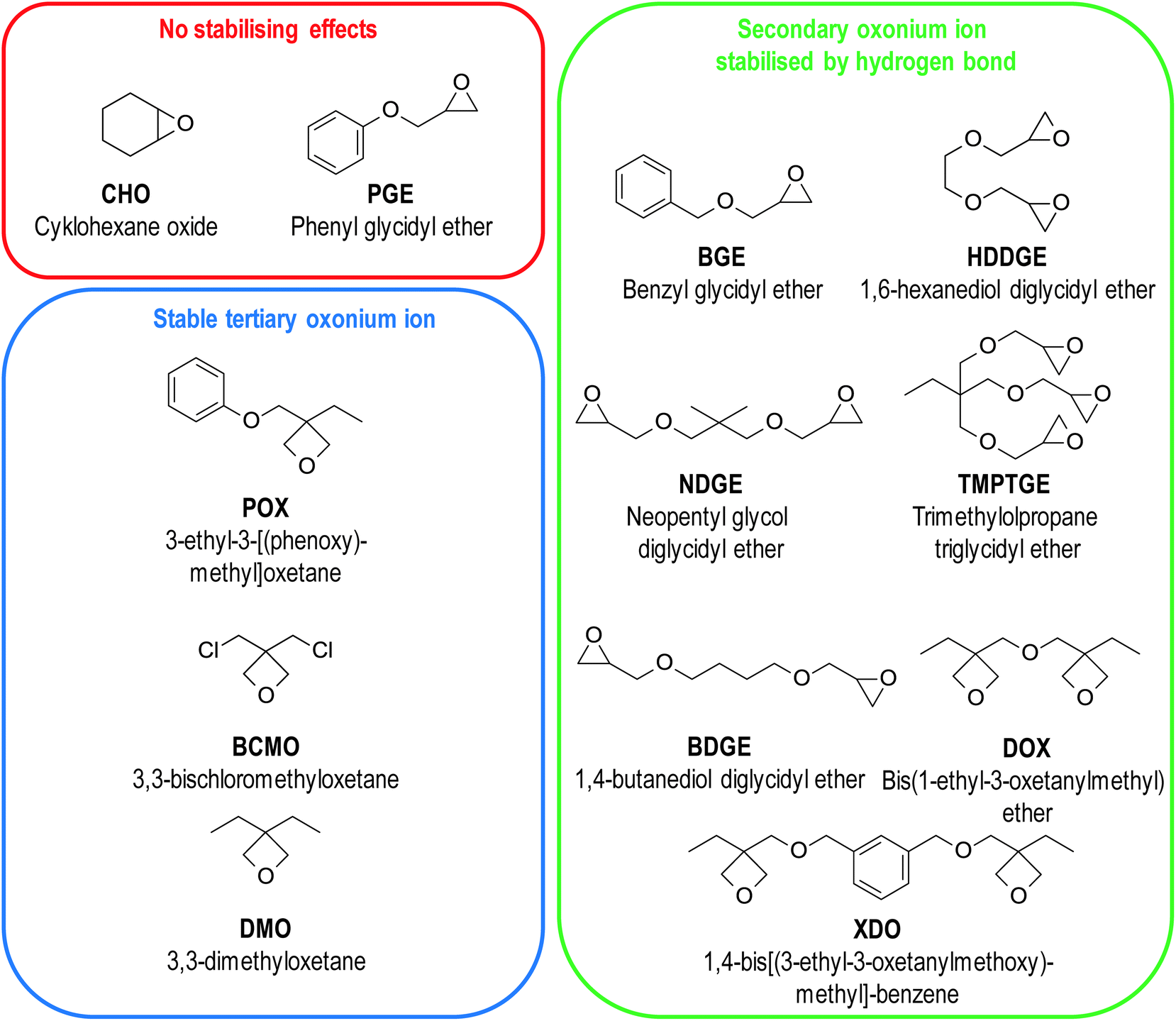 | ||
| Fig. 12 Structures of monomers investigated by Crivello group. They were divided due to the type of stabilising effect they possess or do not possess. | ||
Many factors can influence the induction time of monomer polymerisation. They are all related to the stability of oxonium ions produced during the first polymerisation steps.83 Stabilisation mechanisms are different for different monomer groups, and these stabilisation effects determine the energy activation of the next steps in the polymerisation process. Therefore, cationic polymerisation is strongly dependent on the reaction temperature, contrary to radical polymerisation.98
For alkyl oxiranes and cycloaliphatic ones (like cyclohexane oxide, CHO) with high ring strain, there are no stabilisation effects of oxonium ions, and they undergo rapid polymerisation with no observable induction time because of low energy activation of polymerisation. This type of monomer is not suitable for photoactivated frontal polymerisation. For the rest types of monomers, secondary and tertiary oxonium ions can be stabilised, so their ring-opening reaction is slow.83 The polymerisation process's energy activation is too high to be reached at room temperature, and an external heat source is needed. This ensures an appropriate induction time in most, but not in all cases. This was proved by raising the temperature during irradiation, which led to shorter induction time and high temperature, which led to the induction time disappearing.99
For monomers like BGE or DOX bearing ether moiety near the oxirane or oxetane groups, the secondary oxonium ion can be stabilised through a hydrogen bond structure depicted in Fig. 13. This hydrogen bond formation is possible because both oxygen atoms exhibit similar low basicity. Additional energy is essential to overcome the activation barrier of tertiary oxonium ion formation to start the rapid polymerisation process. Monomers that bear more than two glycidyl ether groups like HDDGE are even more stabilised because of more oxygen atoms with similar basicity.100,101
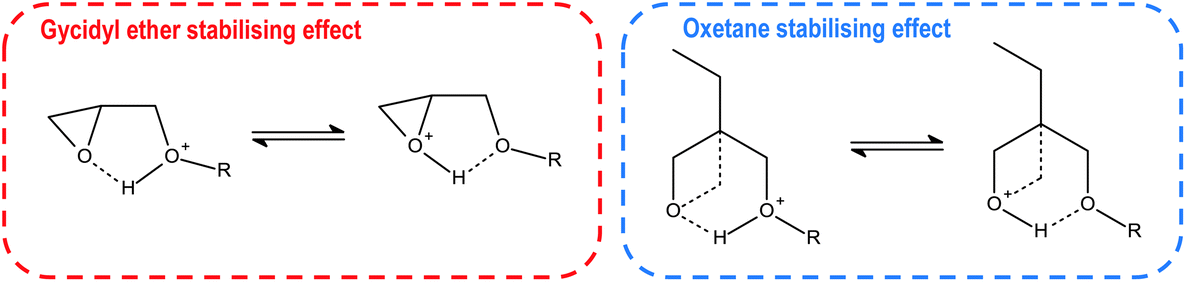 | ||
| Fig. 13 Resonance structures of stabilising hydrogen bond between oxirane/oxetane moiety and ether oxygen in the described monomers.93 | ||
On the other hand, especially for oxiranes with an ether moiety near to the aryl ring (like glycidyl ethers, PGE), this hydrogen bond formation is prevented by the resonance interaction between ether oxygen and aryl ring reducing the basicity of this oxygen.83 For this type of monomer, there is no induction time. The polymerisation process is sluggish but not latent.
The special group of monomers are 3,3-disubstituted oxetanes like DMO. Although Kato and Sasaki suggest that in this case, the secondary oxonium ion is most stable like previous cases,99 Bulut et al. proved experimentally that another effect occurs for this type of monomers.102 They do not have ether oxygen, so hydrogen bond stabilisation is impossible, and the secondary oxonium ion is unstable. However, the tertiary oxonium ions of this type of monomers exhibit above-average stability. They have a relatively high activation barrier of chain elongation. Therefore, the surface remains latent for hours after irradiation, but polymerisation becomes rapid when the composition's temperature is raised by 10–30 °C. This rapid polymerisation is caused by the negative enthalpy of monomer protonation and monomer nucleophilic attack on secondary oxonium ions, which are fast processes generating a significant amount of heat.103 Described properties, especially in the case of POX, DOX, and XDO, are greatly inconvenient in average industrial application but perfect in terms of photoactivated cationic frontal polymerisation.104 The induction time of these monomers can be really long because of two stabilisation effect occurs in their cases (Fig. 14).105,106
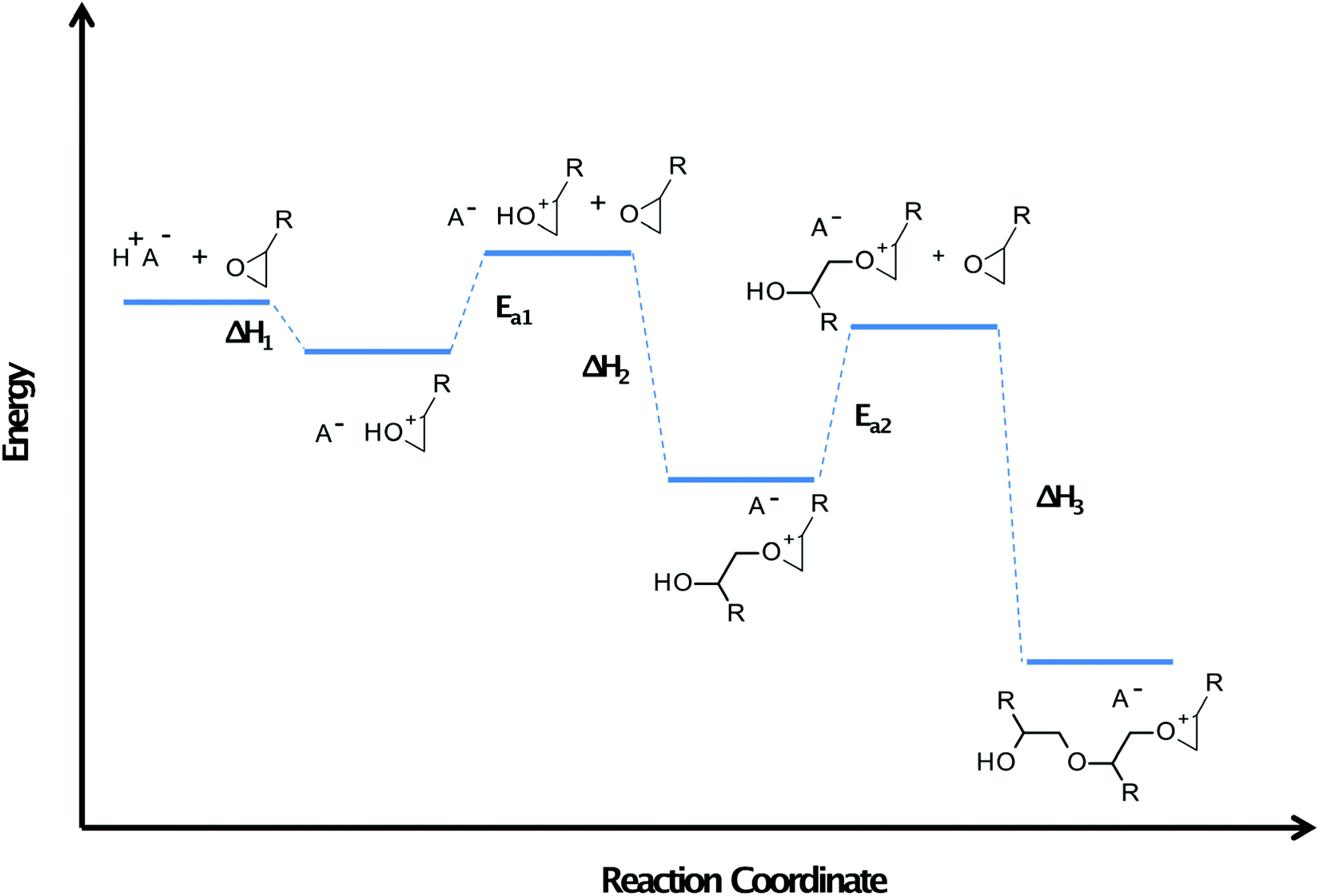 | ||
| Fig. 14 Schematic energy diagram for the photoactivated cationic ring-opening frontal polymerisation. Ea – activation energy. Monomers with higher values of this physical quantity exhibit a longer induction time appropriate for photoactivated cationic ring-opening frontal polymerisation.83 | ||
Photoactivated cationic frontal polymerisation is an interesting but not energy efficient process. Prior activation leads to savings in thermal energy. However, irradiation of all polymerised surfaces leads to a waste of irradiation energy. Ideal frontal polymerisation defined by Pojman should be spontaneous after a local application of energy.1 Parallel to the investigation described above, a new cationic frontal photopolymerisation technique was invented.107 Dual-cure systems of photo-and thermal cationic initiators were employed to obtain a stable front of polymerisation. Furthermore, the cationic radical hybrid system based on radical-induced cationic polymerisation was strongly investigated.
Recently, much research has been done utilising pyrylium salts as cationic photoinitiators.112 This type of compounds exhibits relevant absorption in the visible range, making them useful in LED irradiation applications. However, they can also be used as thermal cationic photoinitiators due to their low thermal stability.113,114 In the described system, pyrylium salts act as both photo and thermal initiators. Lecompere et a.l investigated 2,4,6-triphenylpyrylium tetrafluoroborate (TPP+) as efficient cationic frontal polymerisation photo/thermal initiator111 (Fig. 15).
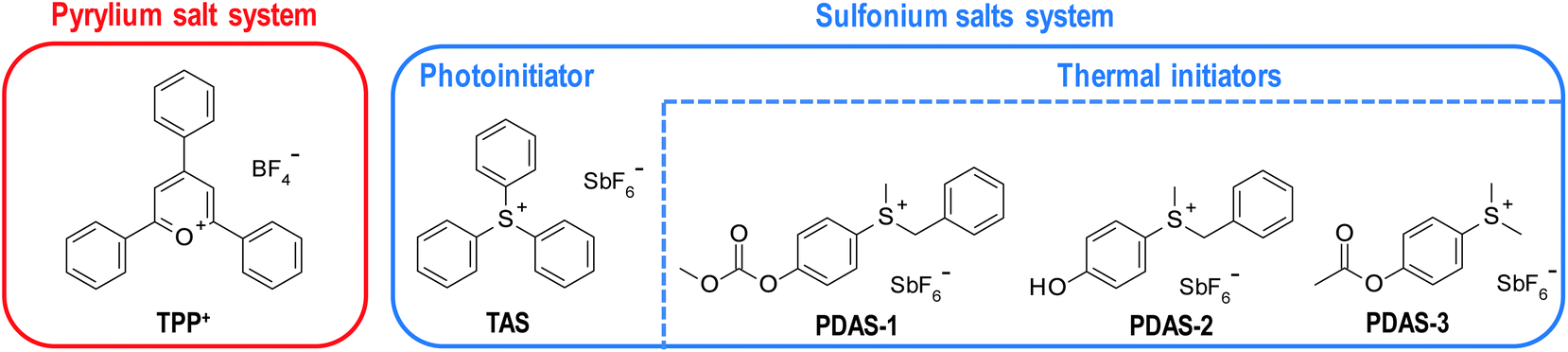 | ||
| Fig. 15 Structures of photo and thermal initiators used in both described dual-cure frontal polymerisation systems. | ||
TPP+ was used to cure CADE and OXT-101 monomers. However, to obtain a stable polymerising front under 395nm LED irradiation, two co-initiators were used. They were used to raise the released heat of the front.115 Similar effects were obtained for 1 wt% of hydrogen peroxide and 3 wt% isobutylvinylether. The latter was efficient, especially when used with an oxetane monomer.
Another interesting phenomenon observed in the TPP+ initiating system was the decrease of the energy required to support the front in depth and raise its velocity. It is considered a great advantage when the formulation contains fillers.111 However, the pyrylium salt is chemically unstable, leading to relatively poor pot-life.109 Therefore, a need arose for a better solution.
Gachet et al. proposed a photo/thermal initiating system based on sulfonium salts.109 Triarylsulfonium salts were described above as one of the more efficient types of onium salts (Fig 15), but they do not exhibit any particular thermal activity, making them thermally stable. On the other hand, the exchange of one or two aryl rings with benzyl or alkyl substituents lowers this stability, which makes these salts efficient thermal initiators of epoxy monomers.116,117 However, these modifications blue-shift sulfonium salts absorption spectra and make them photochemically inactive.118,119
Therefore, a combination of triarylsulfonium hexafuoroantimonate (TAS) with aliphatic modified sulfonium salts (depicted in Fig. 15) provides an efficient initiating system of epoxy resin cationic frontal photopolymerisation where the photoactive one initiates the polymerisation process on the surface of the sample and generates the heat necessary to decomposed thermally active one and curing the sample throughout its depth.109
In the system proposed by Gachet et al., 1 wt% of TAS was sufficient to generate a front of 250 °C. At this concentration of TAS, three PDAS were tested with CADE monomer. As a result, only PDAS-1 and PDAS-2 exhibit a stable front and allow the desired depth curing. The best of both initiators was PDAS-1, which generates a stable front even at a concentration of 0.3 wt%.
The described system was found to be very efficient, especially in photopolymerise thick and opaque carbon fibre reinforced polymers. However, it has some drawbacks. One of them is severely decreasing the temperature jump in resins with a high concentration of fillers.109 Another is utilising a thermally stable photoinitiator in full sample volume, despite being used only on the sample surface to initiate a front.
It was proposed to utilise the RICP process in a frontal polymerisation manner.107 In this approach, iodonium salt is combined with a thermal radical initiator. In the first step of this process, the iodonium salt dissociates after irradiation generating super acid, protonating the monomer molecules leading to ring-opening reaction and starts polymerisation. This process can release heat which warms up the sample causing the decomposition of the radical thermal initiator. Radicals generated in this process easily undergo oxidation to carbocation by iodonium salts, what causes the decomposition of iodonium salts and the release of another molecule of super acid. Especially carbon-centred radicals undergo this process efficiently,127 and in some cases, carbocations generated in this way can promote ring-opening cationic polymerisation. This mechanism allows the front to form and sustain until all samples are cured and called Radical Induced Cationic Frontal Polymerisation (RICFP, Fig. 16).
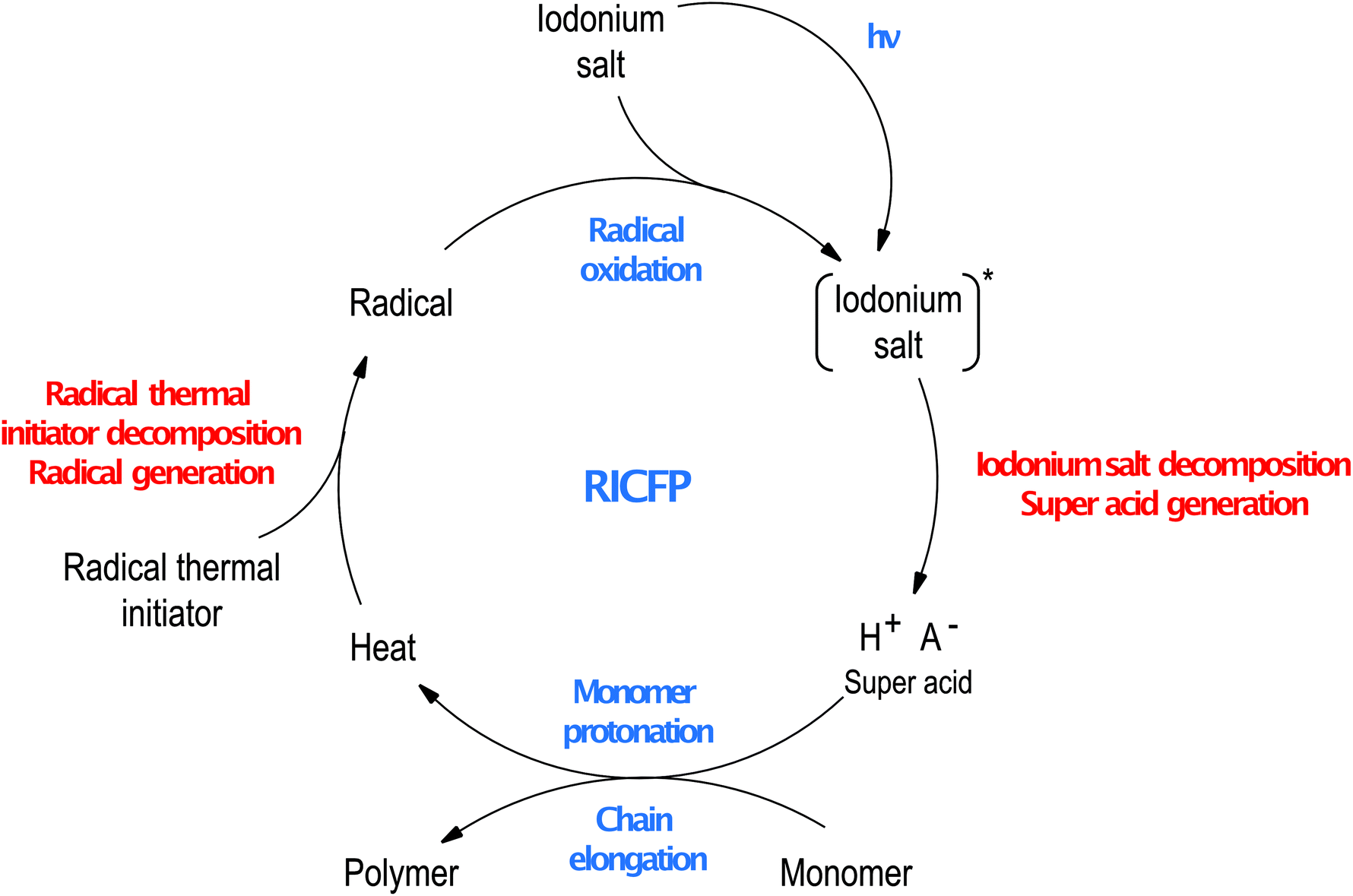 | ||
| Fig. 16 Scheme of radical-induced cationic frontal polymerisation process. | ||
The photo-induced RICFP process was utilised to cure epoxide monomers by Mariani et al. in 2004 for the first time.107 In this work, a combination of dibenzyl peroxide (BPO, thermal initiator) and iodonium salt (photoinitiator) were used to cure samples containing CADE monomer. They obtained a large number of initiating cationic species in the whole sample in the non-photochemical process, after point exposure of the sample to the irradiation. Simultaneously, they proved that both thermal and photoinitiator are simultaneously necessary to create a self-sustaining front. Formed carbocations were able to induce cationic polymerisation of the thicker layers of the sample.
In 2015, Bomze et al. extended this method to cure bisphenol A diglycidyl ether (BADGE) with iodonium salt and 1,1,2,2-tetraphenylethanediol (TPED),108 which is known as C–C labile compound.128 They tested different peroxides and azobis(isobutyronitrile) (AIBN) as thermal initiators, but only the carbon-centred radicals generated by compounds like TPED were efficiently oxidised to carbocations that sustain frontal polymerisation of BADGE. This system was further investigated and proved to be efficient in preparing composite materials to successfully cure BADGE with even 20% of mica powder.129 RICFP approach overcomes the common disadvantages of thermal and photoinitiation of thick samples containing heterogeneous fillers. Local application of irradiance is sufficient to ignite the frontal polymerisation process that eliminates the problem with light penetration of the sample. In contrast, the heat generated during polymerisation sustains a stable front that eliminates the problem with heating of all volumes of sample.
However, a filler can change the conditions of RICFP that was investigated with silica filler by Klikovits et al. in the same system as proposed by Bomze et al.130 SiO2 nanoparticles are thermally low conductive that can influence the sustainability of the propagating front. It was proved that a stable front was formed at 1–3% phr (parts per hundred resin) of silica filler. At higher values, such as 4% and 5%, it was only possible at intense irradiation because of strong light scattering caused by filler particles. The samples with a higher concentration of fillers need to be irradiated with higher intensity. Ignition of frontal polymerisation strongly depends on the irradiation system. This investigation also proved that the front is formed when the molecules of thermal initiators start to decompose.
Nevertheless, in recent years, the composites with fillers such as zinc oxide and carbon fibres,131 multiwalled carbon nanotubes,107 glass fibres,132 glass microspheres and graphite powder133 were successfully prepared using the RCIFP method. This self-sustaining frontal polymerisation system was also successfully used in prepreg technology134 and interpenetrating phase composites.135 The convenience and versatility of this method will undoubtedly result in more applications in the future.
The system proposed by Bomze et al. was further developed by Knaack et al.131 They used tetrakis(perfluoro-tert-butyloxy)aluminate anions instead of the hexafluoroantimonate anion used in the original system. Iodonium salt with this anion is more reactive and can generate a front of polymerisation with higher stability. Due to the BADGE monomer's high viscosity, the use of oxetane monomer (3-ethyloxetan-3-yl)methanol (EOM) was also investigated. It strongly raised the reactivity of the formulation and facilitated the formation of a stable front. There were 1,3-bis(2,3-epoxypropoxy)-2,2-dimethylpropane (NPDGE), 3,4-epoxycyclohexanecarboxylate (CADE), 1,6-hexanediol diglycidylether (HDDGE) also tested as diluents but found to be less reactive. They increase the front velocity only slightly.133
Photo-induced RICFP was also compared with “classically” thermally induced RICFP.136 This investigation revealed that the phototriggered propagation is twice as fast as the thermally induced one with a comparable maximal reaction temperature and heat conductivity. Additionally, materials obtained with thermally-induced RICFP exhibit high microstructural heterogeneity, which confirmed the photo-induced process's predominance.
RICFP was also investigated in photoactivated cationic frontal polymerisation with oxetane monomer. Bulut et al. used 2,2-dimethoxy-2-phenyl-acetophenone (DMPA) – radical photoinitiator – instead of thermal one.102 Iodonium salts easily oxidise carbon-centred radicals produced during photodecomposition of DMPA into carbocations which can attack oxetane oxygen leading to tertiary oxonium ions. This reaction is strongly exothermic and produces heat which can help the active a self-sustaining front. Additionally, this type of oxetane oxonium ion is much more reactive than the one for neat oxetane, leading to a shorter induction time and a more stable front. Another advantage of this system is the absorption spectrum of the radical photoinitiator. DMPA absorb efficiently above 300 nm, where iodonium salts remain photoinactive.84 Using DMPA sensitises the sample to a longer wavelength range. In this system, molecules of iodonium salt are decomposed in a non-photochemical reaction.
In 2007, Crivello utilised this system in interpenetrating polymer network preparation.137 In this approach mixture of two monomers, cationic and radical, was irradiated in the presence of cationic and radical photoinitiators. Radicals generated during the decomposition of both types of photoinitiators made a polyacrylate scaffold. Simultaneously, the ring-opening polymerisation of oxirane or oxetanes was suppressed by the surface of an efficient heat sink on which the sample was placed. After local application of heat, the cationic polymerisation of the unpolymerized phase was ignited, and an interpenetrating polymer network was made in a frontal manner. It was essential to avoid the densely crosslinked polyacrylate film, which prevents a stable front.
Other photoinitiating systems
Besides the systems described above based on radical and cationic photoinitiators, there are many other photo triggering frontal polymerisations systems. The most useful and promising ones are shortly reviewed below.Use of an appropriate ruthenium catalyst, for example, second-generation Grubb's catalyst, enables photo-igniting the start of the reaction by photodissociation of the phosphine ligand (Fig. 17).144 This leads to phototriggered FROMP, where the process starts after irradiation of a sample part and then a stable thermal front initiates the polymerisation process in whole sample volume thermally.145 This system can be extended to carbon nanoparticles, including carbon black, multiwall carbon nanotubes, and vapour-grown carbon nanofibers leading to composite materials.146 This additive enables using a broader irradiation spectrum and photothermally initiation of the reaction.
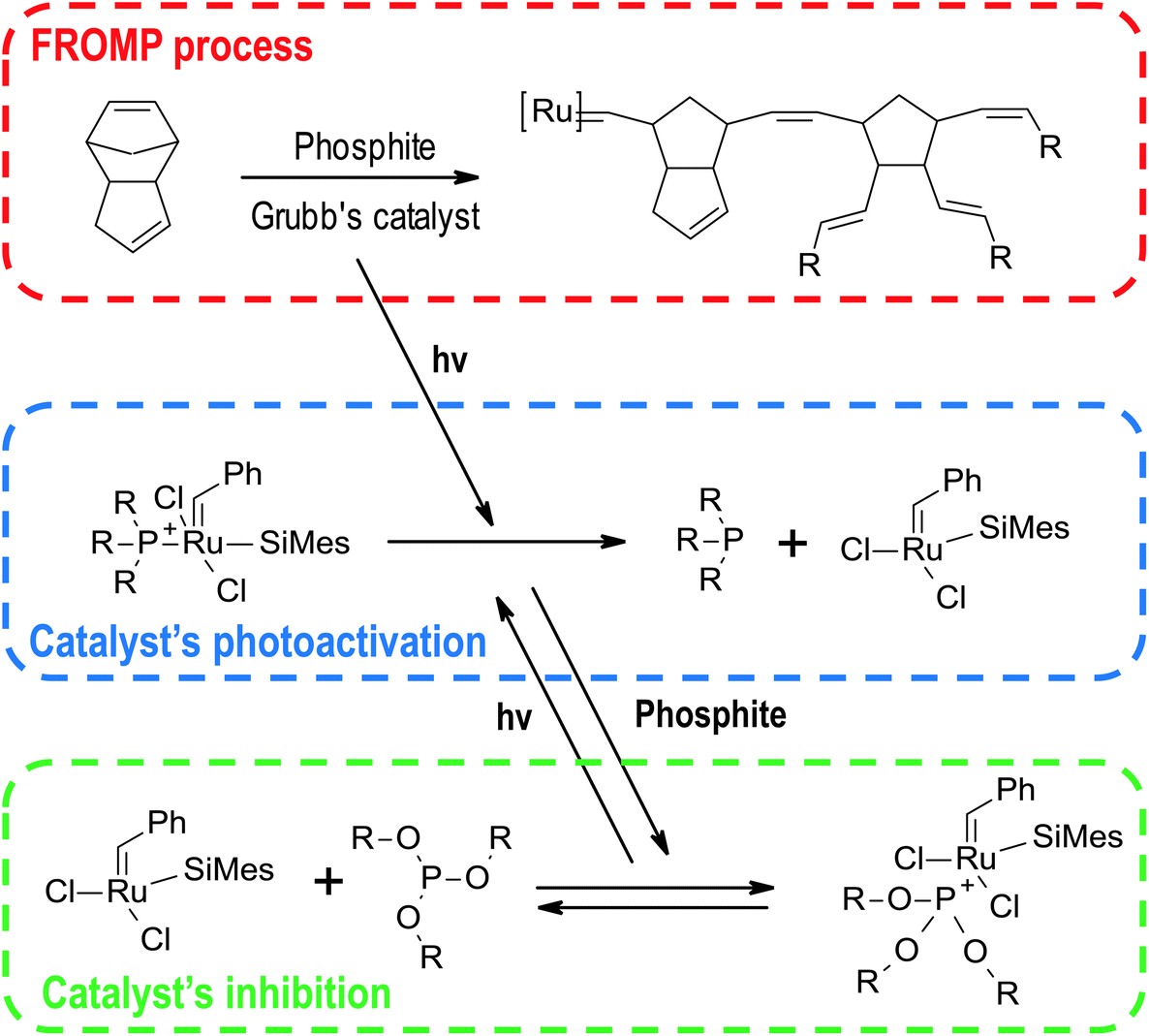 | ||
| Fig. 17 Scheme of photoinitiation of FROMP process in Grubb's catalyst/phoshite system. | ||
Conclusion
Frontal photopolymerisation and photo-triggered thermal frontal polymerisation every year find growing interest. This is because of their great advantages in comparison with standard polymerising systems. Frontal photopolymerisation with continuous radiation flux makes it possible to obtain objects with different three-dimensional shapes based on acrylate monomer shrinkage. These shapes are difficult to obtain in that scale in other methods. On the other hand, photo-triggered thermal frontal polymerisation is greatly energy-saving. After local light application, it allows curing thick and opaque samples where both standard thermal and photopolymerisation are problematic. It makes this approach environmentally friendly. However, both techniques meet some limitations. Samples of any volume cannot be cured with continuous radiation flux because of the penetration limit, also, the high temperature of the front limits its application, for example, prevents the use of this method in dentistry. Nevertheless, using frontal photopolymerisation and phototriggered frontal polymerisation, it is possible to obtain a wide range of materials, including composites.This review attempts to summarise previous achievements mainly in the fields of radical and cationic frontal photopolymerisation, but some other kinds of polymerisation were mentioned. Presented systems are early developed and need to be optimised. However, even at this stage of development, they are very promising because of their convenience and versatility. New solutions in this topic appear every year, and each year we observe a growing number of new papers and reports of progress. This creates a bright future for frontal photopolymerization.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the National Centre for Research and Development (Warsaw, Poland) – INNOCHEM project contract number POIR.01.02.00-00-0038/16-00; project title: “A new generation of photoinitiators for photopolymerization processes dedicated to the coating-forming industry and adhesives”.References
- J. A. Pojman, V. M. Ilyashenko and A. M. Khan, Free-radical frontal polymerization: Self-propagating thermal reaction waves, J. Chem. Soc., Faraday Trans., 1996, 92, 2825–2837, 10.1039/ft9969202825.
- N. Chechilo, R. Khivilivitskii and N. Enikolopyan, On the Phenomenon of Polymerization Reaction Spreading, Dokl. Akad. Nauk BSSR, 1972, 204, 1180–1181 CAS.
- J. A. Pojman, Frontal Polymerization, Elsevier B.V., 2012, DOI:10.1016/B978-0-444-53349-4.00124-2.
- P. M. Potzmann, F. J. López-Villanueva and R. Liska, UV-Initiated, Bubble-Free Frontal Polymerization in Aqueous Conditions, Macromolecules, 2015, 48, 8738–8745, DOI:10.1021/acs.macromol.5b02348.
- T. Hu, Y. Fang, H. Yu, L. Chen and S. Chen, Synthesis of poly(N-methylolacrylamide)/polymethylacrylamide hybrids via frontal free-radical polymerization, Colloid Polym. Sci., 2007, 285, 891–898, DOI:10.1007/s00396-006-1635-z.
- K. Kostrzewska, J. Ortyl, R. Dobosz and J. Kabatc, Squarylium dye and onium salts as highly sensitive photoradical generators for blue light, Polym. Chem., 2017, 8, 3464–3474, 10.1039/c7py00621g.
- D. Nowak, J. Ortyl, I. Kamińska-Borek, K. Kukuła, M. Topa and R. Popielarz, Photopolymerization of hybrid monomers, Part II: Determination of relative quantum efficiency of selected photoinitiators in cationic and free-radical polymerization of hybrid monomers, Polym. Test., 2018, 67, 144–150, DOI:10.1016/j.polymertesting.2018.02.025.
- J. Ortyl and R. Popielarz, The performance of 7-hydroxycoumarin-3-carbonitrile and 7-hydroxycoumarin-3-carboxylic acid as fluorescent probes for monitoring of cationic photopolymerization processes by FPT, J. Appl. Polym. Sci., 2013, 128, 1974–1978, DOI:10.1002/app.38378.
- J. Ortyl, P. Milart and R. Popielarz, Applicability of aminophthalimide probes for monitoring and acceleration of cationic photopolymerization of epoxides, Polym. Test., 2013, 32, 708–715, DOI:10.1016/j.polymertesting.2013.03.009.
- J. Ortyl, J. Wilamowski, P. Milart, M. Galek and R. Popielarz, Relative sensitization efficiency of fluorescent probes/sensitizers for monitoring and acceleration of cationic photopolymerization of monomers, Polym. Test., 2015, 48, 151–159, DOI:10.1016/j.polymertesting.2015.10.006.
- M. G. Hennessy, A. Vitale, O. K. Matar and J. T. Cabral, Controlling frontal photopolymerization with optical attenuation and mass diffusion, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2015, 91, 14–18, DOI:10.1103/PhysRevE.91.062402.
- C. Ebner, J. Mitterer, P. Eigruber, S. Stieger, G. Riess and W. Kern, Ultra-High Through-Cure of (Meth)Acrylate Copolymers via Photofrontal Polymerization, Polymers, 2020, 12, 1291, DOI:10.3390/polym12061291.
- K. Asakura, E. Nihei, H. Harasawa, A. Ikumo and S. Osanai, Spontaneous frontal polymerization: Propagating front spontaneously generated by locally autoaccelerated free-radical polymerization, Am. Chem. Soc., 2003, 869, 135–146, DOI:10.1021/bk-2004-0869.ch011.
- T. J. Tulig and M. Tirrell, Toward a Molecular Theory of the Trommsdorff Effect, Macromolecules, 1981, 14, 1501–1511, DOI:10.1021/ma50006a070.
- A. Vitale, M. G. Hennessy, O. K. Matar and J. T. Cabral, A Unified Approach for Patterning via Frontal Photopolymerization, Adv. Mater., 2015, 27, 6118–6124, DOI:10.1002/adma.201502607.
- A. Vitale, M. G. Hennessy, O. K. Matar and J. T. Cabral, Interfacial profile and propagation of frontal photopolymerization waves, Macromolecules, 2015, 48, 198–205, DOI:10.1021/ma5021215.
- M. G. Hennessy, A. Vitale, O. K. Matar and J. T. Cabral, Monomer diffusion into static and evolving polymer networks during frontal photopolymerisation, Soft Matter, 2017, 13, 9199–9210, 10.1039/c7sm01279a.
- J. Wang, N. Dai, C. Jiang, X. Mu, B. Zhang, Q. Ge and D. Wang, Programmable shape-shifting 3D structures via frontal photopolymerization, Mater. Des., 2021, 198, 109381, DOI:10.1016/j.matdes.2020.109381.
- J. H. Lee, R. K. Prud'homme and I. A. Aksay, Cure depth in photopolymerization: Experiments and theory, J. Mater. Res., 2001, 16, 3536–3544, DOI:10.1557/JMR.2001.0485.
- A. Khan and J. A. Pojman, The Use of Frontal Polymerization in Polymer Synthesis, in Trends Polym. Sci., Elsevier Trends Journals, Cambridge, 1996, pp. 253 Search PubMed.
- M. Belk, K. G. Kostarev, V. Volpert and T. M. Yudina, Frontal photopolymerization with convection, J. Phys. Chem. B, 2003, 107, 10292–10298, DOI:10.1021/jp0276855.
- M. He, X. Huang, Z. Zeng and J. Yang, Photo-triggered redox frontal polymerization: A new tool for synthesizing thermally sensitive materials, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4515–4521 CAS.
- F. Li, F. Li, Y. Song, M. Yao, J. Nie, J. Nie, Y. He and Y. He, Design and properties of novel photothermal initiators for photoinduced thermal frontal polymerization, Polym. Chem., 2020, 11, 3980–3986, 10.1039/d0py00305k.
- D. Nuvoli, V. Alzari, J. A. Pojman, V. Sanna, A. Ruiu, D. Sanna, G. Malucelli and A. Mariani, Synthesis and characterization of functionally gradient materials obtained by frontal polymerization, ACS Appl. Mater. Interfaces, 2015, 7, 3600–3606, DOI:10.1021/am507725k.
- F. Zhu, Y. Song, S. Liu, J. Nie and Y. He, Preparation of polymerizable thermal initiator and its application in photo-induced thermal frontal polymerization, Eur. Polym. J., 2019, 118, 107–112, DOI:10.1016/j.eurpolymj.2019.05.044.
- A. Gugg, C. Gorsche, N. Moszner and R. Liska, Frontal polymerization: Polymerization induced destabilization of peracrylates, Macromol. Rapid Commun., 2011, 32, 1096–1100, DOI:10.1002/marc.201100188.
- Z. Zhao, J. Wu, X. Mu, H. Chen, H. J. Qi and D. Fang, Origami by frontal photopolymerization, Sci. Adv., 2017, 3, 1–8, DOI:10.1126/sciadv.1602326.
- G. G. Aleksanyan, K. A. Arutyunyan, V. L. Bodneva, S. P. Davtyan, E. V. Prut, B. A. Rozenberg, K. G. Shkadinskii and N. S. Yenikolopyan, Some rules governing the extension of the front of radical polymerization of vinyl monomers, Polym. Sci. USSR, 1975, 17, 1052–1059, DOI:10.1016/0032-3950(75)90283-X.
- M. V. Turturro, D. M. V. Rendón, F. Teymour and G. Papavasiliou, Kinetic Investigation of Poly(ethylene glycol) Hydrogel Formation via Perfusion-Based Frontal Photopolymerization: Influence of Free-Radical Polymerization Conditions on Frontal Velocity and Swelling Gradients, Macromol. React. Eng., 2013, 7, 107–115, DOI:10.1002/mren.201200063.
- J. Wang, X. Mu, D. Li, C. Yu, X. Cheng and N. Dai, Modeling and Application of Planar–to–3D Structures via Optically Programmed Frontal Photopolymerization, Adv. Eng. Mater., 2019, 21, 1801279, DOI:10.1002/adem.201801279.
- S. A. Chesnokov, V. M. Treushnikov, Y. V. Chechet, V. K. Cherkasov and O. N. Mamysheva, General conditions and experimental design of sustained frontal photopolymerization in photopolymerizable liquid compositions, Polym. Sci., Ser. A, 2008, 50, 291–298, DOI:10.1134/s0965545x08030073.
- S. P. Davtyan and A. O. Tonoyan, The Frontal Polymerization Method in High Technology Applications, Rev. J. Chem., 2019, 9, 71–94, DOI:10.1134/s2079978018040039.
- A. R. Purnama, M. G. Hennessy and A. Vitale, Evolving Conversion Profile, 2017 Search PubMed.
- Y. Tao, J. Yang, Z. Zeng, Y. Cui and Y. Chen, Study on spatial–temporal kinetics of photo-initiated frontal polymerization in stacked reaction cells, Polym. Int., 2006, 55, 418–425, DOI:10.1002/pi.1988.
- C. Sheppard, Azo compounds, in Encycl. Polym. Sci. Eng., Wiley-Interscience, New York, 1985, pp. 143–157 Search PubMed.
- H. Yu, Y. Fang, L. Chen and S. Chen, Investigation of redox initiators for free radical frontal polymerization, Polym. Int., 2009, 58, 851–857, DOI:10.1002/pi.2590.
- A. Mariani, D. Nuvoli, V. Alzari and M. Pini, Phosphonium-based ionic liquids as a new class of radical initiators and their use in gas-free frontal polymerization, Macromolecules, 2008, 41, 5191–5196, DOI:10.1021/ma800610g.
- J. A. Yoon, J. Kamada, K. Koynov, J. Mohin, R. Nicolaÿ, Y. Zhang, A. C. Balazs, T. Kowalewski and K. Matyjaszewski, Self-healing polymer films based on thiol-disulfide exchange reactions and self-healing kinetics measured using atomic force microscopy, Macromolecules, 2012, 45, 142–149, DOI:10.1021/ma2015134.
- G. Odian, Radical chain polymerization, in Princ. Polym., Wiley-Interscience, Hoboken, 4th edn, 2004, pp. 198–349 Search PubMed.
- R. P. Washington and O. Steinbock, Frontal polymerization synthesis of isopropylacrylamide hydrogels, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2002, 43, 726–727 CAS.
- L. Chen, T. Hu, H. Yu, S. Chen and J. A. Pojman, First Solvent-Free Synthesis of Poly(N-methylolacrylamide) via Frontal Free-Radical Polymerization, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4322–4330, DOI:10.1002/pola.22176.
- S. Chen, T. Hu, Y. Tian, L. Chen and J. A. Pojman, Facile Synthesis of Poly(hydroxyethyl Acrylate) by Frontal Free-Radical Polymerization, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 873–881, DOI:10.1002/pola.
- G. Lu, Q. Yan and C. Ge, Preparation of porous polyacrylamide hydrogels by frontal polymerization, Polym. Int., 2007, 56, 1016–1020 CrossRef CAS.
- Q. Feng, Q. Yan and C. Ge, Synthesis of Macroporous Polyacrylamide and Poly(N-Isopropylacrylamide) Monoliths Via Frontal Polymerization and Investigation of Pore Structure Variation of Monoliths, Chin. J. Polym. Sci., 2009, 27, 743–753 CrossRef.
- Y. Cui, J. Yang, Z. Zeng, Z. Zeng and Y. Chen, Unique morphology and properties study of polyacrylate obtained via frontal photopolymerization, Polymer, 2007, 48, 5994–6001, DOI:10.1016/j.polymer.2007.08.013.
- M. V. Turturro and G. Papavasiliou, Generation of mechanical and biofunctional gradients in PEG diacrylate hydrogels by perfusion-based frontal photopolymerization, J. Biomater. Sci., Polym. Ed., 2012, 23, 917–939, DOI:10.1163/092050611X566450.
- J. Su, Y. Yang, Z. Chen, J. Zhou, X. Liu, Y. Fang and Y. Cui, Preparation and performance of thermosensitive poly(N-isopropylacrylamide) hydrogels by frontal photopolymerization, Polym. Int., 2019, 68, 1673–1680, DOI:10.1002/pi.5868.
- W. F. Schroeder, S. L. Asmussen, W. D. Cook and C. I. Vallo, Efficiency of 4,4′-bis(N,N-diethylamino) benzophenone for the polymerization of dimethacrylate resins in thick sections, Polym. Int., 2011, 60, 1362–1369, DOI:10.1002/pi.3089.
- C. Nason, T. Roper, C. Hoyle and J. A. Pojman, UV-induced frontal polymerization of multifunctional (meth)acrylates, Macromolecules, 2005, 38, 5506–5512, DOI:10.1021/ma048569y.
- W. J. Ainsworth, J. A. Pojman, Y. A. Chekanov and J. Masere, Bubble behavior in frontal polymerization: Results from KC-135 parabolic flights, ACS Symp. Ser., 2001, 793, 112–125, DOI:10.1021/bk-2001-0793.ch008.
- J. A. Pojman, B. Varisli, A. Perryman, C. Edwards and C. Hoyle, Frontal polymerization with thiol-ene systems, Macromolecules, 2004, 37, 691–693, DOI:10.1021/ma035751a.
- S. Krishnan, A. Klein, M. S. El-Aasser and E. D. Sudol, Influence of chain transfer agent on the cross-linking of poly(n-butyl methacrylate-co-N-methylol acrylamide) latex particles and films, Macromolecules, 2003, 36, 3511–3518, DOI:10.1021/ma021121h.
- W. Tomal and J. Ortyl, Water-soluble photoinitiators in biomedical applications, Polymers, 2020, 12, 1–30, DOI:10.3390/POLYM12051073.
- N. B. Cramer and C. N. Bowman, Thiol–ene and Thiol–yne Chemistry in Ideal Network Synthesis, in Radiat. Curing Polym. Sci. Technol, Elsevier, New York, 1993, pp. 219–226, 10.1039/9781849736961-00001.
- J. A. Warren, J. T. Cabral and J. F. Douglas, Solution of a field theory model of frontal photopolymerization, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 72, 021801 CrossRef PubMed.
- N. Charles, J. A. Pojman and C. Hoyle, The Effect of a Trithiol and Inorganic Fillers on the Photo-Induced Thermal Frontal Polymerization of a Triacrylate, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 8091–8096, DOI:10.1002/pola.
- N. B. Cramer, J. P. Scott and C. N. Bowman, Photopolymerizations of thiol-ene polymers without photoinitiators, Macromolecules, 2002, 35, 5361–5365, DOI:10.1021/ma0200672.
- J. T. Cabral, S. D. Hudson, C. Harrison and J. F. Douglas, Frontal photopolymerization for microfluidic applications, Langmuir, 2004, 20, 10020–10029, DOI:10.1021/la049501e.
- J. McKechnie and D. Sinton, Dynamic microfluidic photomasking, J. Microelectromech. Syst., 2007, 16, 1145–1151, DOI:10.1109/JMEMS.2007.900878.
- T. Wu, Y. Mei, J. T. Cabral, C. Xu and K. L. Beers, A new synthetic method for controlled polymerization using a microfluidic system, J. Am. Chem. Soc., 2004, 126, 9880–9881, DOI:10.1021/ja048432n.
- R. P. Washington and O. Steinbock, Frontal free-radical polymerization: Applications to materials synthesis, Polym. News, 2003, 28, 303–310 CAS.
- G. B. dos Santos, R. V. M. Alto, H. R. S. Filho, E. M. da Silva and C. E. Fellows, Light transmission on dental resin composites, Dent. Mater., 2008, 24, 571–576, DOI:10.1016/j.dental.2007.06.015.
- J. Elisseeff, K. Anseth, D. Sims, W. McIntosh, M. Randolph, M. Yaremchuk and R. Langer, Transdermal photopolymerization of poly(ethylene oxide)-based injectable hydrogels for tissue-engineered cartilage, Plast. Reconstr. Surg., 1999, 104, 1014–1022, DOI:10.1097/00006534-199909020-00018.
- X. Qin, F. Zhao, Y. Liu and S. Feng, Frontal photopolymerization synthesis of multilayer hydrogels with high mechanical strength, Eur. Polym. J., 2011, 47, 1903–1911, DOI:10.1016/j.eurpolymj.2011.07.001.
- C. Decker, Kinetic study and new applications of UV radiation curing, Macromol. Rapid Commun., 2002, 23, 1067–1093, DOI:10.1002/marc.200290014.
- V. M. Treushnikov and S. A. Chesnokov, Single-stage processes of polymer products photochemical synthesis with optical accuracy, J. Photochem. Photobiol., A, 2008, 196, 201–209, DOI:10.1016/j.jphotochem.2007.07.030.
- P. Carion, A. Ibrahim, X. Allonas, C. Croutxé-Barghorn and G. L'Hostis, Frontal free-radical photopolymerization of thick samples: Applications to LED-induced fiber-reinforced polymers, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 898–906, DOI:10.1002/pola.29341.
- J. A. Pojman, Pojman Polymer Products, LLC, n.d. Search PubMed.
- M. V. Turturro, M. C. Christenson, J. C. Larson, D. A. Young, E. M. Brey and G. Papavasiliou, MMP-Sensitive PEG Diacrylate Hydrogels with Spatial Variations in Matrix Properties Stimulate Directional Vascular Sprout Formation, PLoS One, 2013, 8, e58897, DOI:10.1371/journal.pone.0058897.
- C. M. González-Henríquez, M. A. Sarabia-Vallejos and J. Rodríguez-Hernández, Wrinkles Obtained by Frontal Polymerization/Vitrification, in Wrinkled Polym. Surfaces Strateg. Methods Appl, Springer, Cham, 2019, pp. 63–84, DOI:10.1007/978-3-030-05123-5_3.
- D. Chandra and A. J. Crosby, Self-wrinkling of UV-cured polymer films, Adv. Mater., 2011, 23, 3441–3445, DOI:10.1002/adma.201101366.
- W. Cho, D. Y. Hahm, J. H. Yim, J. H. Lee, Y. J. Lee, D. Kim, Y. S. Kim and J. J. Wie, Programmable Building Blocks via Internal Stress Engineering for 3D Collective Assembly, Adv. Mater. Technol., 2020, 5, 2000758, DOI:10.1002/admt.202000758.
- Z. Zhao, J. Wu, X. Mu, H. Chen, H. J. Qi and D. Fang, Desolvation Induced Origami of Photocurable Polymers by Digit Light Processing, Macromol. Rapid Commun., 2017, 38, 1–6, DOI:10.1002/marc.201600625.
- S. Felton, M. Tolley, E. Demaine, D. Rus and R. Wood, A method for building self-folding machines, Science, 2014, 644–646 CrossRef CAS PubMed.
- B. Saccà and C. M. Niemeyer, DNA origami: The art of folding DNA, Angew. Chem., Int. Ed., 2012, 51, 58–66, DOI:10.1002/anie.201105846.
- L. H. Dudte, E. Vouga, T. Tachi and L. Mahadevan, Programming curvature using origami tessellations, Nat. Mater., 2016, 15, 583–588, DOI:10.1038/nmat4540.
- Z. Chen, G. Huang, I. Trase, X. Han and Y. Mei, Mechanical Self-Assembly of a Strain-Engineered Flexible Layer: Wrinkling, Rolling, and Twisting, Phys. Rev. Appl., 2016, 5, 1–33, DOI:10.1103/PhysRevApplied.5.017001.
- J. Rogers, Y. Huang, O. G. Schmidt and D. H. Gracias, Origami MEMS and NEMS, MRS Bull., 2016, 41, 123–129, DOI:10.1557/mrs.2016.2.
- J. V. Crivello, Photopolymerization, in Polym. Sci. A Compr. Ref. 10 Vol. Set, Elsevier, 2012, pp. 919–955, DOI:10.1016/B978-0-444-53349-4.00123-0.
- M. Sangermano, I. Roppolo and A. Chiappone, New horizons in cationic photopolymerization, Polymers, 2018, 10, 136, DOI:10.3390/polym10020136.
- S. Kaalberg and J. L. P. Jessop, Enhancing cationic ring-opening photopolymerization of cycloaliphatic epoxides via dark cure and oxetanes, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1436–1445, DOI:10.1002/pola.29024.
- M. Sawamoto, Modern cationic vinyl polymerization, Prog. Polym. Sci., 1991, 16, 111–172, DOI:10.1016/0079-6700(91)90008-9.
- B. Falk, M. R. Zonca and J. V. Crivello, Photoactivated cationic frontal polymerization, in Macromol. Symp, Wiley-VCH Verlag, 2005, pp. 97–108, DOI:10.1002/masy.200550810.
- A. W. Green, in Industrial Photoinitiators: A Technical Guide, CRC Press, 1st edn, 2010 Search PubMed.
- X. Allonas, Photopolymerization, Cationic, in Encycl. Polym. Sci. Technol., Wiley, 2019, pp. 1–30, DOI:10.1002/0471440264.pst491.pub2.
- J. V. Crivello and J. H. W. Lam, Diaryliodonium Salts., A New Class of Photoinitiators for Cationic Polymerization, Macromolecules, 1977, 10, 1307–1315, DOI:10.1021/ma60060a028.
- J. V. Crivello and J. H. W. Lam, Photoinitiated cationic polymerization with triarylsulfonium salts, J. Polym. Sci., Polym. Chem. Ed., 1979, 17, 977–999, DOI:10.1002/pol.1979.170170405.
- J. Ortyl, Cationic Photoinitiators, in RSC Polym. Chem. Ser, Royal Society of Chemistry, 2018, ch. 3, pp. 74–130, 10.1039/9781788013307-00074.
- J. Ortyl and R. Popielarz, New photoinitiators for cationic polymerization, Polimery, 2012, 57(7–8), 510–517 CrossRef CAS.
- H. Mokbel, J. Toufaily, T. Hamieh, F. Dumur, D. Campolo, D. Gigmes, J. Pierre Fouassier, J. Ortyl and J. Lalevée, Specific cationic photoinitiators for near UV and visible LEDs: Iodonium versus ferrocenium structures, J. Appl. Polym. Sci., 2015, 132, 42759, DOI:10.1002/app.42759.
- J. V. Crivello, Deslgn of photoacid generatlng systems, J. Photopolym. Sci. Technol., 2009, 22, 575–582, DOI:10.2494/photopolymer.22.575.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, Wiley, 4th edn, 2003 Search PubMed.
- J. V. Crivello, B. Falk and M. R. Zonca, Photoinduced cationic ring-opening frontal polymerizations of oxetanes and oxiranes, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1630–1646, DOI:10.1002/pola.20012.
- S. Penczek, P. Kubisa and K. Matyjaszewski, Cationic Ring-Opening Polymerization of Heterocyclic Monomers, Springer Berlin Heidelberg, Berlin, Heidelberg, 1980, DOI:10.1007/BFb0023043.
- J. V. Crivello and U. Bulut, Photoactivated cationic ring-opening frontal polymerizations of oxetanes, Des. Monomers Polym., 2005, 8, 517–531, DOI:10.1163/156855505774597803.
- J. V. Crivello, Investigation of the photoactivated frontal polymerization of oxetanes using optical pyrometry, Polymer, 2005, 46, 12109–12117, DOI:10.1016/j.polymer.2005.10.087.
- G. A. Olah, G. K. S. Prakash, J. Sommer and A. Molnar, Superacid Chemistry, Wiley, 2nd edn, 2009 Search PubMed.
- G. Odian, in Principles of Polymerization, Wiley-Interscience, 3rd edn, 1992 Search PubMed.
- H. Kato and H. Sasaki, Cationic Photopolymerization of 2-Phenyloxetanes, 2003, pp. 285–295, DOI:10.1021/bk-2003-0847.ch025.
- J. V. Crivello, Design and synthesis of multifunctional glycidyl ethers that undergo frontal polymerization, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6435–6448, DOI:10.1002/pola.21761.
- J. V. Crivello, Cationic photopolymerization of alkyl glycidyl ethers, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3036–3052, DOI:10.1002/pola.21419.
- U. Bulut and J. V. Crivello, Reactivity of oxetane monomers in photoinitiated cationic polymerization, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3205–3220, DOI:10.1002/pola.20723.
- E. M. Arnett and G. Scorrano, Protonation and Solvation in Strong Aqueous Acids, Adv. Phys. Org. Chem., 1976, 13, 83–153, DOI:10.1016/S0065-3160(08)60213-0.
- H. Sasaki, J. M. Rudzinński and T. Kakuchi, Photoinitiated cationic polymerization of oxetane formulated with oxirane, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 1807–1816, DOI:10.1002/pola.1995.080331107.
- D. B. Yang, Kinetic studies of photopolymerization using real time FT–IR spectroscopy, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 199–208, DOI:10.1002/pola.1993.080310124.
- C. Decker, Recent Developments in Radiation Curing Chemistry, in Process. Photoreact. Polym, Springer US, 1995, pp. 34–55, DOI:10.1007/978-1-4615-1767-2_2.
- A. Mariani, S. Bidali, S. Fiori, M. Sangermano, G. Malucelli, R. Bongiovanni and A. Priola, UV-ignited frontal polymerization of an epoxy resin, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2066–2072, DOI:10.1002/pola.20051.
- D. Bomze, P. Knaack and R. Liska, Successful radical induced cationic frontal polymerization of epoxy-based monomers by C-C labile compounds, Polym. Chem., 2015, 6, 8161–8167, 10.1039/c5py01451d.
- B. Gachet, M. L. Lecompère, C. Croutxé-Barghorn, D. Burr, G. L’Hostis and X. Allonas, Highly reactive photothermal initiating system based on sulfonium salts for the photoinduced thermal frontal cationic polymerization of epoxides: a way to create carbon-fiber reinforced polymers, RSC Adv., 2020, 10, 41915–41920, 10.1039/d0ra07561b.
- A. H. Bonardi, F. Dumur, T. M. Grant, G. Noirbent, D. Gigmes, B. H. Lessard, J. P. Fouassier and J. Lalevée, High Performance Near-Infrared (NIR) Photoinitiating Systems Operating under Low Light Intensity and in the Presence of Oxygen, Macromolecules, 2018, 51, 1314–1324, DOI:10.1021/acs.macromol.8b00051.
- M. Lecompere, X. Allonas, D. Marechal and A. Criqui, Dual-cure Photo-thermal Initiating System for Cationic Polymerization of Epoxy under LED Visible Light, J. Photopolym. Sci. Technol., 2017, 30, 399–404, DOI:10.2494/photopolymer.30.399.
- E. Hola and J. Ortyl, Pyrylium salt as a visible-light-induced photoredox catalyst for polymer and organic synthesis - perspectives on catalyst design and performance, Eur. Polym. J., 2021, 150, 110365, DOI:10.1016/j.eurpolymj.2021.110365.
- M. Lecompère, X. Allonas, D. Maréchal and A. Criqui, Mechanistic approach to a photochemical/thermal dual-cure initiating system based on pyrylium salt-hydroperoxide for epoxide cationic polymerization, Polym. Chem., 2017, 8, 388–395, 10.1039/c6py01741j.
- D. Maréchal, X. Allonas, M. Lecompère and A. Criqui, Novel Dual-Cure Initiating System for Cationic Polymerization of Epoxides, Macromol. Chem. Phys., 2016, 217, 1169–1173, DOI:10.1002/macp.201500523.
- M. Lecompère, X. Allonas, D. Maréchal and A. Criqui, Versatility of Pyrylium Salt/Vinyl Ether Initiating System for Epoxide Dual-Cure Polymerization: Kick-Starting Effect of the Coinitiator, Macromol. Rapid Commun., 2017, 38, 1600660, DOI:10.1002/marc.201600660.
- S. P. Pappas and L. W. Hill, Kinetic parameter considerations for maximizing stability and minimizing cure temperature of thermosetting coatings, J. Coat. Technol., 1981, 53, 43–51 CAS.
- T. Endo and H. Arita, Polymerization of spiroorthocarbonates by sulfonium salts, as latent thermal catalysts, Die Makromol. Chemie, Rapid Commun., 1985, 6, 137–139, DOI:10.1002/marc.1985.030060304.
- F. Hamazu, S. Akashi, T. Koizumi, T. Takata and T. Endo, Photopolymerization of epoxides with benzyl sulfonium salts as latent photoinitiators, J. Photopolym. Sci. Technol., 1992, 5, 247–254, DOI:10.2494/photopolymer.5.247.
- F. Hamazu, S. Akashi, T. Koizumi, T. Takata and T. Endo, Photopolymerization of Epoxide by benzyl(p-hydroxyphenyl)-methylsulfonium salts as novel photolatent initiators, Makromol. Chem., Rapid Commun., 1992, 13, 203–206, DOI:10.1002/marc.1992.030130402.
- F. A. M. Abdul-Rasoul, A. Ledwith and Y. Yagci, Photochemical and thermal cationic polymerizations promoted by free radical initiators, Polymer, 1978, 19, 1219–1222, DOI:10.1016/0032-3861(78)90074-5.
- A. Ledwith, Possibilities for promoting cationic polymerization by common sources of free radicals, Polymer, 1978, 19, 1217–1219, DOI:10.1016/0032-3861(78)90073-3.
- A. Ledwith, Formation and reactivitie soffreeion sincationic polymerisation, Pure Appl. Chem., 1979, 51, 159–171, DOI:10.1351/pac197951010159.
- J. V. Crivello and J. L. Lee, Redox-initiated cationic polymerization: the diaryliodonium salt/benzoin redox couple, J. Polym. Sci., Part A-1: Polym. Chem., 1983, 21, 1097–1110, DOI:10.1002/pol.1983.170210417.
- J. V. Crivello, Unconventional methods of initiating cationic polymerization using onium salts, in Macromol. Symp, 2002, pp. 65–70, DOI:10.1002/1521-3900(200207)183:1<65::AID-MASY65>3.0.CO;2-C.
- B. Falk, M. R. Zonca and J. V. Crivello, Modification of photoinitiated cationic epoxide polymerizations by sulfides, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2504–2519, DOI:10.1002/pola.20726.
- Y. Yaǧci and I. Reetz, Externally stimulated initiator systems for cationic polymerization, Prog. Polym. Sci., 1998, 23, 1485–1538, DOI:10.1016/S0079-6700(98)00010-0.
- H. J. Timpe and A. G. Rajendran, Light-induced polymer and polymerization reactions-42. A kinetic study of cationic photopolymerization with (benzoin derivative)/(onium salt) systems, Eur. Polym. J., 1991, 27, 77–83, DOI:10.1016/0014-3057(91)90129-C.
- D. Braun, T. Skrzek, S. Steinhauer-Beißer, H. Tretner and H. J. Lindner, Initiation of polymerization with substituted ethanes, 13. Free radical polymerization of methyl methacrylate and styrene with substituted succinonitriles, Macromol. Chem. Phys., 1995, 196, 573–591, DOI:10.1002/macp.1995.021960213.
- D. Bomze, P. Knaack, T. Koch, H. Jin and R. Liska, Radical induced cationic frontal polymerization as a versatile tool for epoxy curing and composite production, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3751–3759, DOI:10.1002/pola.28274.
- N. Klikovits, R. Liska, A. D'Anna and M. Sangermano, Successful UV-Induced RICFP of Epoxy-Composites, Macromol. Chem. Phys., 2017, 218, 1700313, DOI:10.1002/macp.201700313.
- P. Knaack, N. Klikovits, A. D. Tran, D. Bomze and R. Liska, Radical induced cationic frontal polymerization in thin layers, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1155–1159, DOI:10.1002/pola.29375.
- M. Sangermano, A. D'Anna, C. Marro, N. Klikovits and R. Liska, UV-activated frontal polymerization of glass fibre reinforced epoxy composites, Composites, Part B, 2018, 143, 168–171, DOI:10.1016/j.compositesb.2018.02.014.
- A. D. Tran, T. Koch, P. Knaack and R. Liska, Radical induced cationic frontal polymerization for preparation of epoxy composites, Composites, Part A, 2020, 132, 105855, DOI:10.1016/j.compositesa.2020.105855.
- A. D. Tran, T. Koch, R. Liska and P. Knaack, Radical-induced cationic frontal polymerisation for prepreg technology, Monatshefte Fur Chemie, 2021, 152, 151–165, DOI:10.1007/s00706-020-02726-y.
- D. Maugeri, M. Sangermano and Y. Leterrier, Radical photoinduced cationic frontal polymerization in porous media, Polym. Int., 2021, 70, 269–276, DOI:10.1002/pi.6156.
- H. Švajdlenková, A. Kleinová, O. Šauša, J. Rusnák, T. A. Dung, T. Koch and P. Knaack, Microstructural study of epoxy-based thermosets prepared by “classical” and cationic frontal polymerization, RSC Adv., 2020, 10, 41098–41109, 10.1039/d0ra08298h.
- J. V. Crivello, Hybrid free radical/cationic frontal photopolymerizations, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4331–4340, DOI:10.1002/pola.22177.
- M. He, X. Huang, Z. Zeng and J. Yang, Phototriggered base proliferation: A highly efficient domino reaction for creating functionally photo-screened materials, Macromolecules, 2013, 46, 6402–6407, DOI:10.1021/ma400983t.
- M. He, X. Huang, Y. Huang, Z. Zeng and J. Yang, Photoinduced redox initiation for fast polymerization of acrylaytes based on latent superbase and peroxides, Polymer, 2012, 53, 3172–3177, DOI:10.1016/j.polymer.2012.05.031.
- A. Mariani, S. Fiori, Y. Chekanov and J. A. Pojman, Frontal ring-opening metathesis polymerization of dicyclopentadiene [5], Macromolecules, 2001, 34, 6539–6541, DOI:10.1021/ma0106999.
- A. Ruiu, D. Sanna, V. Alzari, D. Nuvoli and A. Mariani, Advances in the frontal ring opening metathesis polymerization of dicyclopentadiene, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2776–2780, DOI:10.1002/pola.27301.
- I. D. Robertson, L. M. Dean, G. E. Rudebusch, N. R. Sottos, S. R. White and J. S. Moore, Alkyl Phosphite Inhibitors for Frontal Ring-Opening Metathesis Polymerization Greatly Increase Pot Life, ACS Macro Lett., 2017, 6, 609–612, DOI:10.1021/acsmacrolett.7b00270.
- I. D. Robertson, M. Yourdkhani, P. J. Centellas, J. E. Aw, D. G. Ivanoff, E. Goli, E. M. Lloyd, L. M. Dean, N. R. Sottos, P. H. Geubelle, J. S. Moore and S. R. White, Rapid energy-efficient manufacturing of polymers and composites via frontal polymerization, Nature, 2018, 557, 223–227, DOI:10.1038/s41586-018-0054-x.
- H. Kunkely and A. Vogler, Photoreactivity of bis(tricyclohexylphosphine)benzylidene ruthenium dichloride (Grubbs's catalyst), Inorg. Chim. Acta, 2001, 325, 179–181, DOI:10.1016/S0020-1693(01)00647-8.
- K. J. Stawiasz, J. E. Paul, K. J. Schwarz, N. R. Sottos and J. S. Moore, Photoexcitation of Grubbs’ Second-Generation Catalyst Initiates Frontal Ring-Opening Metathesis Polymerization, ACS Macro Lett., 2020, 9, 1563–1568, DOI:10.1021/acsmacrolett.0c00486.
- L. M. Dean, A. Ravindra, A. X. Guo, M. Yourdkhani and N. R. Sottos, Photothermal Initiation of Frontal Polymerization Using Carbon Nanoparticles, ACS Appl. Polym. Mater., 2020, 2, 4690–4696, DOI:10.1021/acsapm.0c00726.
| This journal is © The Royal Society of Chemistry 2021 |