
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of well-defined diblock copolymer nano-objects by RAFT non-aqueous emulsion polymerization of N-(2-acryloyloxy)ethyl pyrrolidone in non-polar media†
R. R.
Gibson
a,
A.
Fernyhough
b,
O. M.
Musa
c and
S. P.
Armes
 *a
*a
aDainton Building, Department of Chemistry, University of Sheffield, Brook Hill, Sheffield, South Yorkshire S3 7HF, UK. E-mail: s.p.armes@sheffield.ac.uk
bAshland Inc., Listers Mills, Heaton Rd, Bradford, West Yorkshire BD9 4SH, UK
cAshland Inc., 1005 US 202/206, Bridgewater, New Jersey 08807, USA
First published on 23rd June 2021
Abstract
Polymerization-induced self-assembly (PISA) is widely recognized to be a powerful technique for the preparation of diblock copolymer nano-objects in various solvents. Herein a highly unusual non-aqueous emulsion polymerization formulation is reported. More specifically, the reversible addition–fragmentation chain transfer (RAFT) polymerization of N-(2-acryloyloxy)ethyl pyrrolidone (NAEP) is conducted in n-dodecane using a poly(stearyl methacrylate) (PSMA) precursor to produce sterically-stabilized spherical nanoparticles at 90 °C. This relatively high polymerization temperature was required to ensure sufficient background solubility for the highly polar NAEP monomer, which is immiscible with the non-polar continuous phase. A relatively long PSMA precursor (mean degree of polymerization, DP = 36) was required to ensure colloidal stability, which meant that only kinetically-trapped spheres could be obtained. Dynamic light scattering (DLS) studies indicated that the resulting PSMA36–PNAEPx (x = 60 to 500) spheres were relatively well-defined (DLS polydispersity <0.10) and the z-average diameter increased linearly with PNAEP DP up to 261 nm. Differential scanning calorimetry studies confirmed a relatively low glass transition temperature (Tg) for the core-forming PNAEP block, which hindered accurate sizing of the nanoparticles by TEM. However, introducing ethylene glycol diacrylate (EGDA) as a third block to covalently crosslink the nanoparticle cores enabled a spherical morphology to be identified by transmission electron microscopy studies. This assignment was confirmed by small angle X-ray scattering studies of the linear diblock copolymer nanoparticles. Finally, hydrophobic linear PSMA36–PNAEP70 spheres were evaluated as a putative Pickering emulsifier for n-dodecane–water mixtures. Unexpectedly, addition of an equal volume of water followed by high-shear homogenization always produced oil-in-water (o/w) emulsions, rather than water-in-oil (w/o) emulsions. Moreover, core-crosslinked PSMA36–PNAEP60–PEGDA10 spheres also produced o/w Pickering emulsions, suggesting that such Pickering emulsions must be formed by nanoparticle adsorption at the inner surface of the oil droplets. DLS studies of the continuous phase obtained after either creaming (o/w emulsion) or sedimentation (w/o emulsion) of the droplet phase were consistent with this interpretation. Furthermore, certain experimental conditions (e.g. ≥0.5% w/w copolymer concentration for linear PSMA36–PNAEPx nanoparticles, ≥0.1% w/w for core-crosslinked nanoparticles, or n-dodecane volume fractions ≤0.60) produced w/o/w double emulsions in a single step, as confirmed by fluorescence microscopy studies.
Introduction
Polymerization-induced self-assembly (PISA) enables the highly convenient preparation of a wide range of diblock copolymer nano-objects in various solvents at much higher copolymer concentrations1 than those typically employed for traditional post-polymerization processing techniques.2 Reversible addition–fragmentation chain transfer (RAFT) polymerization3–6 is the most widely used pseudo-living polymerization technique for PISA formulations.2,7,8 This is no doubt owing to its well-known tolerance of monomer functionality and its compatibility with water,9–11 non-polar solvents12,13 and polar solvents.14–16Recently, Cunningham et al.10,17,18 used RAFT polymerization to demonstrate that N-(2-methacryloyloxy)ethyl pyrrolidone (NMEP) is a more readily copolymerizable methacrylic analogue of N-vinyl pyrrolidone (NVP), which is used to prepare a range of water-soluble homopolymers and statistical copolymers on an industrial scale.19–22 For example, the RAFT dispersion polymerization of NMEP in non-polar media was conducted using a poly(stearyl methacrylate) (PSMA) precursor: such PISA formulations provide convenient access to spherical, worm-like and vesicular nano-objects.17 The rate of polymerization of NMEP proved to be much faster than that of benzyl methacrylate (BzMA) for syntheses conducted under precisely the same conditions; this striking difference was attributed to the much greater polarity of the former monomer. PNMEP has also been used as a steric stabilizer block for the RAFT dispersion polymerization of BzMA in ethanol.18,23 Unlike PNVP, PNMEP exhibits a lower critical solution temperature (LCST) in aqueous solution. Cunningham et al. exploited this inverse temperature solubility behavior to devise a low-viscosity route to high molecular weight PNMEP via RAFT aqueous dispersion polymerization.10 Thus, PNMEP chains are weakly hydrophobic when prepared at 70 °C and hence form the dehydrated cores of relatively large sterically-stabilized spheres. On cooling to ambient temperature (i.e. below the LCST of PNMEP), molecular dissolution occurs as the PNMEP chains become hydrophilic. Subsequently, Gibson et al. showed that PNMEP can be used as an electrosteric stabilizer block for aqueous PISA formulations. In this case, a carboxylic acid-functionalized RAFT agent had to be used to introduce anionic charge by adjusting the solution pH so as to ensure end-group ionization.24
In 2009 Shi et al. reported an acrylic analogue of NMEP, N-(2-acryloyloxy)ethyl pyrrolidone (NAEP).25 PNAEP homopolymer exhibits no LCST behavior in aqueous solution at elevated temperature so it is clearly much more hydrophilic than PNMEP.26 Indeed, Deane and co-workers recently used PNAEP as a steric stabilizer for the RAFT aqueous emulsion polymerization of styrene and/or n-butyl acrylate.27 Moreover, the rate of homopolymerization of NAEP is significantly faster than that of NMEP.26 For example, using an ascorbic acid/potassium persulfate redox initiator in combination with a trithiocarbonate-based RAFT agent in aqueous solution led to more than 99% NAEP conversion within 5 min at 30 °C when targeting a mean degree of polymerization (DP) of 80 for the PNAEP block. Moreover, gel permeation chromatography (GPC) analysis indicated a final number-average molecular weight (Mn) of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 g mol−1 and a dispersity (or Mw/Mn, where Mw is the weight-average molecular weight) of 1.15, suggesting a well-controlled RAFT polymerization.
300 g mol−1 and a dispersity (or Mw/Mn, where Mw is the weight-average molecular weight) of 1.15, suggesting a well-controlled RAFT polymerization.
Currently, there are no literature examples of PISA syntheses that use PNAEP as a core-forming block. Indeed, there are far fewer examples of PISA syntheses involving acrylic monomers compared to methacrylic monomers, particularly for non-aqueous formulations. This is somewhat surprising, because low glass transition temperature (Tg) film-forming nanoparticles are potentially useful for paints and coatings applications.28,29 Charleux and co-workers were the first to report an all-acrylic PISA formulation in non-polar media: in this case, poly(2-ethylhexyl acrylate)–poly(methyl acrylate) (PEHA–PMA) diblock copolymer nanoparticles were targeted in iso-dodecane.30–32 Spherical nanoparticles were obtained and GPC analysis indicated relatively good control over the molecular weight distribution (MWD) when using a trithiocarbonate-based macro-CTA (Mw/Mn = 1.21 at 100% conversion). Similarly, Ratcliffe and co-workers prepared all-acrylic diblock copolymer nano-objects by RAFT dispersion polymerization of benzyl acrylate (BzA) using a relatively short poly(lauryl acrylate) precursor in various alkanes.33 More specifically, spheres, worms or vesicles were obtained in n-heptane, n-dodecane or iso-hexadecane at 80 °C. Broader MWDs were observed when such PISA syntheses were conducted at lower copolymer concentrations; this was attributed to chain transfer to solvent, as reported by Veloso and co-workers.34 However, it is well-documented that acrylates can undergo significant branching via chain transfer to polymer, which also broadens the MWD.35–37
Herein, a PSMA precursor is used to conduct the RAFT non-aqueous emulsion polymerization of NAEP in n-dodecane to produce spherical nanoparticles. Genuine non-aqueous emulsion polymerizations, in which the vinyl monomer exhibits minimal solubility in an organic solvent (rather than water) are rather rare:38–41 as far as we are aware, this is the first example of such a PISA formulation. The resulting PSMA36–PNAEPx nanoparticles were sized by dynamic light scattering (DLS), transmission electron microscopy (TEM) and small angle X-ray scattering (SAXS) while GPC, 1H NMR spectroscopy and differential scanning calorimetry (DSC) studies provided additional characterization of the diblock copolymer chains. Finally, PSMA36–PNAEP70 spheres were evaluated for their performance as a putative Pickering emulsifier for n-dodecane–water mixtures.
Experimental
Materials
Chloroform and triethylamine (TEA) were purchased from Alfa Aesar (Haysham, UK). tert-Butyl peroxy-2-ethylhexanoate (T21s) initiator was purchased from AkzoNobel (The Netherlands). n-Dodecane, toluene, ethanol, CDCl3, stearyl methacrylate (SMA), benzyl methacrylate (BzMA) and azobisisobutyronitrile (AIBN) were purchased from Sigma Aldrich (Dorset, UK). N-(2-Acryloyloxy)ethyl pyrrolidone (NAEP; 95% purity) was donated by Ashland (New Jersey, USA) and was further purified in-house by dissolution in chloroform followed by sequential washes with 5% Na2CO3 solution, saturated NaCl solution, and finally deionized water. Repeated washes with water were performed until the aqueous phase exhibited neutral pH. This solution was then dried over anhydrous MgSO4 to remove the water. 4-Cyano-4-(2-phenylethanesulfanylthiocarbonyl)sulfanylpentanoic acid (PETTC) RAFT agent was prepared as previously reported.42 CD2Cl2 was purchased from Goss Scientific Instruments Ltd (Cheshire, UK). Ethylene glycol diacrylate (EGDA) was purchased from Santa Cruz Biotechnology (Dallas, USA).Synthesis of a PSMAx precursor by RAFT solution polymerization in toluene
The PSMA36 precursor was prepared using the following protocol. SMA (36.0 g, 0.11 mol), PETTC RAFT agent (0.60 g, 1.77 mmol; target DP = 60), AIBN (58.1 mg, 0.35 mmol; PETTC/AIBN molar ratio = 5.0) and toluene (36.6 g, 50% w/w solids) were weighed into a 250 mL round-bottom flask and degassed under N2 with continuous magnetic stirring for 30 min. The SMA polymerization was allowed to proceed for 260 min in an oil bath set to 70 °C, resulting in a final monomer conversion of 71% as judged by 1H NMR spectroscopy. Quenching was achieved by exposing the hot reaction solution to air and cooling to 20 °C. The crude polymer was precipitated into excess cold ethanol to remove residual monomer before placing in a vacuum oven at 30 °C for 72 h to afford an orange waxy solid. The mean DP was calculated to be 36 by comparing the integrated aromatic protons of the RAFT end-group at 7.3 ppm to the two oxymethylene protons assigned to the SMA repeat units at 3.8–4.1 ppm. Chloroform GPC analysis indicated an Mn of 10200 g mol−1 and an Mw/Mn of 1.18 expressed relative to a series of ten near-monodisperse PMMA calibration standards using a refractive index detector. A second PSMA precursor with a mean DP of 8 was also prepared using the same synthetic protocol by adjusting the SMA/PETTC molar ratio to 5.0. In this case, chloroform GPC analysis indicated an Mn of 2500 g mol−1 and an Mw/Mn of 1.26.
Synthesis of PSMAx–PNAEPy diblock copolymer nanoparticles by RAFT non-aqueous emulsion polymerization of NAEP in n-dodecane
The synthesis of PSMA36–PNAEP60 nano-objects via RAFT non-aqueous emulsion polymerization of NAEP in n-dodecane was conducted as follows. The PSMA36 precursor (0.15 g, 12.0 μmol), NAEP (0.12 g, 0.72 mmol; target DP = 60) and T21s initiator (0.50 mg, 2.99 μmol; 0.05 g of a 10 mg g−1 T21s stock solution dissolved in n-dodecane; PSMA36/T21s molar ratio = 4.0) were dissolved in n-dodecane (1.10 g). The reaction vial was sealed and degassed under N2 for 20 min before being placed in a pre-heated oil bath set at 90 °C. After 5 h, the NAEP polymerization was quenched by exposing the hot reaction solution to air and cooling to 20 °C. The resulting diblock copolymer chains were characterized by 1H NMR spectroscopy and chloroform GPC while 0.1% w/w dispersions of the nano-objects were prepared by dilution with n-dodecane prior to analysis by DLS and TEM. Chloroform GPC analysis indicated an Mp of 23000 g mol−1 and an Mw/Mn of 1.50 (calculated using a series of ten near-monodisperse PMMA calibration standards and a refractive index detector). Other diblock copolymer compositions were prepared by adjusting the amount of NAEP monomer to target a range of DPs. For these syntheses, the volume of the continuous phase was adjusted to maintain an overall copolymer concentration of 20% w/w solids. 1H NMR analysis indicated that at least 99% NAEP conversion was achieved in all cases. In addition, a series of PSMA8–PNAEPx nanoparticles were also prepared in n-dodecane using the PSMA8 precursor. In this case, the target DP of the insoluble PNAEP block was varied from 20 to 100. Finally, a series of PSMA8–PBzMAy nanoparticles were prepared by RAFT dispersion polymerization of BzMA in n-dodecane at 90 °C, as described by Smith and co-workers.43 In this case, the DP of the PBzMA block was varied from 20 to 100.
The RAFT solution polymerization of NAEP was also conducted at 20% w/w solids in toluene targeting PSMA36–PNAEP60 diblock copolymers at 90 °C within 6 h (>99% NAEP conversion, Mn of 13400 g mol−1 and an Mw/Mn of 2.13).
Synthesis of core-crosslinked PSMA36–PNAEP60–PEGDA10 triblock copolymer nanoparticles
A typical protocol for the synthesis of core-crosslinked PSMA36–PNAEP60–PEGDA10 spherical nanoparticles was conducted as follows. The PSMA36 precursor (0.50 g, 40.0 μmol), NAEP (0.44 g, 2.39 mmol; target DP = 60) and T21s initiator (1.60 mg, 9.99 μmol; 0.16 g of a 10 mg g−1 T21s stock solution dissolved in n-dodecane; PSMA36/T21s molar ratio = 4.0) were dissolved in n-dodecane (4.03 g). The reaction vial was sealed and degassed under N2 for 20 min before being placed in a pre-heated oil bath set at 90 °C for 1 h. EGDA (0.07 g, 0.40 mmol; target DP = 10; previously degassed with N2 gas at 20 °C) was then added using a deoxygenated syringe/needle. The EGDA polymerization was allowed to proceed for 5 h and then quenched by exposing the hot reaction mixture to air while cooling to 20 °C. The resulting core-crosslinked triblock copolymer nanoparticles were diluted with n-dodecane to afford a 0.1% w/w dispersion prior to characterization by DLS and TEM.Estimation of NAEP solubility in n-dodecane as a function of temperature
The solubility of NAEP in n-dodecane was assessed by visual inspection. n-Dodecane was placed in a series of glass vials with continuous stirring at various temperatures ranging from 20 to 90 °C. A known volume of NAEP monomer was added to each vial until a two-phase system was observed.Preparation of o/w (and w/o/w) emulsions using either linear PSMA36–PNAEP70 spheres or core-crosslinked PSMA36–PNAEP60–PEGDA10 spheres
Water (2.0 mL) was homogenized with 2.0 mL of a dispersion containing 0.025–1.0% w/w PSMA36–PNAEP70 (or PSMA36–PNAEP60–PEGDA10) spheres in n-dodecane to afford an emulsion with equi-volume oil and water for 2.0 min at a shear rate of 13500 rpm using an IKA Ultra-Turrax homogenizer at 20 °C. Emulsions were also prepared by varying the relative volume fraction of oil whilst maintaining an overall emulsion volume of 4.0 mL.
Copolymer characterization
000 g mol−1) were used for calibration and data were analyzed using Varian Cirrus GPC software supplied by the instrument manufacturer.
sinθ/λ is the length of the scattering vector and θ is one-half of the scattering angle). A glass capillary of 2.0 mm diameter was used as a sample holder and all measurements were conducted on 1.0% w/w copolymer dispersions in n-dodecane. X-ray scattering data were reduced and normalized using standard routines by the beamline or using the Foxtrot software package supplied with the Xeuss 2.0 instrument and further analyzed (background subtraction and data modelling) using Irena SAS macros for Igor Pro.44
Laser diffraction
Each emulsion was sized by laser diffraction using a Malvern Mastersizer 3000 instrument equipped with a hydro EV wet sample dispersion unit, a red He–Ne laser (λ = 633 nm) and a LED blue light source (λ = 470 nm). The stirring rate was adjusted to 1500 rpm to avoid creaming of the emulsion droplets during analysis. The volume-average (De Brouckere) diameter was determined for each emulsion. After each measurement, the cell was rinsed once with ethanol and twice with deionized water and the laser was aligned centrally to the detector prior to data acquisition.Fluorescence microscopy
Fluorescence microscopy images of the w/o/w double Pickering emulsions were recorded using a Zeiss Axio Scope A1 microscope equipped with an AxioCam 1Cm1 monochrome camera. Nile Red dye was dissolved in n-dodecane prior to high-shear homogenization and the resulting oil droplets were imaged using Zeiss filter set 43 HE (excitation 550/25 nm and emission 605/70 nm). Images were captured and processed using ZEN lite 2012 software.Results and discussion
Optimization to prepare PSMA–PNAEP nanoparticles
Cunningham and co-workers reported that PSMA14 is an effective steric stabilizer block for the RAFT dispersion polymerization of NMEP in n-dodecane, with this PISA formulation providing access to spheres, worms and vesicles.17 Accordingly, PSMA8–PNAEPx diblock copolymer nano-objects were targeted in n-dodecane using a PSMA8 oligomer prepared using PETTC as a RAFT agent. Chloroform GPC curves indicated reasonably high blocking efficiencies and a linear increase in Mn was observed on increasing the target PNAEP DP (Fig. S1a†). However, the relatively broad MWDs (Mw/Mn > 1.40) indicated relatively poor RAFT control and the presence of a high molecular weight shoulder suggested some degree of branching via chain transfer to polymer. Moreover, nanoparticle aggregation was confirmed by DLS analysis after dilution of such copolymer dispersions using n-dodecane, particularly when targeting higher DPs (Fig. S1b†). These results were corroborated by visual inspection of the as-synthesized copolymer dispersions (see Fig. S2†). In contrast, control experiments involving chain extension of the same PSMA8 precursor via RAFT dispersion polymerization of benzyl methacrylate (BzMA) in n-dodecane at 90 °C resulted in the formation of colloidally stable nano-objects (see Fig. S2†). This discrepancy is consistent with the relatively uncontrolled nature of the RAFT polymerization of NAEP (see GPC data) compared to that of BzMA.43 However, our prior PISA experience suggested that selecting a sufficiently long PSMA stabilizer should confer colloidal stability.13,45–47Aliquots were periodically extracted during the RAFT solution homopolymerization of SMA in toluene when targeting a DP of 60 at 70 °C, with monomer conversions being determined by 1H NMR spectroscopy (Fig. S3a†) and molecular weight data being obtained by GPC analysis using chloroform as an eluent (Fig. S3b†). These experiments indicated that the RAFT solution polymerization of SMA is well-controlled, as expected. In a preliminary experiment, a PSMA36 precursor was then chain-extended via RAFT polymerization of NAEP in n-dodecane at 90 °C using T21s initiator when targeting a DP of 60. This precursor was also chain-extended via RAFT polymerization of NAEP in toluene at 90 °C targeting the same DP (Fig. S4†).
NAEP is fully miscible with toluene, so employing this solvent simply leads to a RAFT solution polymerization. In contrast, visual inspection suggests that NAEP is essentially immiscible with n-dodecane at 20 °C (NAEP solubility = 0.25% v/v under such conditions, see Fig. S5†). At 70 °C, the solubility of NAEP in n-dodecane is around 2.9% v/v. However, this seems to be insufficient to enable its efficient non-aqueous polymerization since 1H NMR studies indicated no discernible conversion within 6 h at this temperature. Similar observations were made at 80 °C. However, the solubility of NAEP in n-dodecane is around 4.9% v/v at 90 °C, which is sufficient to enable its efficient non-aqueous emulsion polymerization at this temperature (see Fig. S6†).48 As far as we are aware, such formulations are very rare in the literature. A relatively high blocking efficiency was observed for both the RAFT solution polymerization of NAEP in toluene at 90 °C and the RAFT non-aqueous emulsion polymerization of NAEP in n-dodecane at 90 °C. High temperatures are usually avoided when polymerizing acrylic monomers because this typically leads to poor MWD control but clearly such reaction conditions appear to be unavoidable, at least in the case of the RAFT non-aqueous emulsion polymerization formulation.36,37,49
In view of this constraint, the PSMA36/T21s molar ratio was varied in an attempt to optimize this PISA formulation (Fig. 1). For example, the PSMA36/T21s molar ratio was increased when targeting PSMA36–PNAEP60 diblock copolymers because this should provide greater RAFT control and hence narrower MWDs.50,51 Indeed, GPC analysis indicates that the Mw/Mn is reduced from 1.50 to 1.40 when the PSMA36/T21s molar ratio is raised from 4.0 to 10.0 at 90 °C. However, this also led to a significantly slower rate of polymerization: the final NAEP conversion was reduced from more than 99% within 2 h to only 94% within 4 h. Indeed, when using a PSMA36/T21s molar ratio of 20.0, only 24% NAEP conversion could be achieved within 6 h. Clearly, this approach is rather limited in scope if efficient polymerizations are desired. Moreover, higher molar ratios led to broader particle size distributions. For example, when using a PSMA36/T21s molar ratio of 4.0, DLS studies indicated a z-average diameter of 52 nm and a DLS polydispersity of 0.10, suggesting relatively well-defined spheres. However, the DLS polydispersity increased to 0.23 for approximately the same z-average diameter when using a PSMA36/T21s molar ratio of 10.0. For the current study, the production of well-defined nanoparticles was considered to be more important than achieving control over the MWD. Thus, on the basis of these preliminary studies, it was concluded that the best compromise between MWD control, NAEP conversion and DLS polydispersity when targeting PSMA36–PNAEP60 nanoparticles was achieved when using a PSMA36/T21s molar ratio of 4.0 at 90 °C.
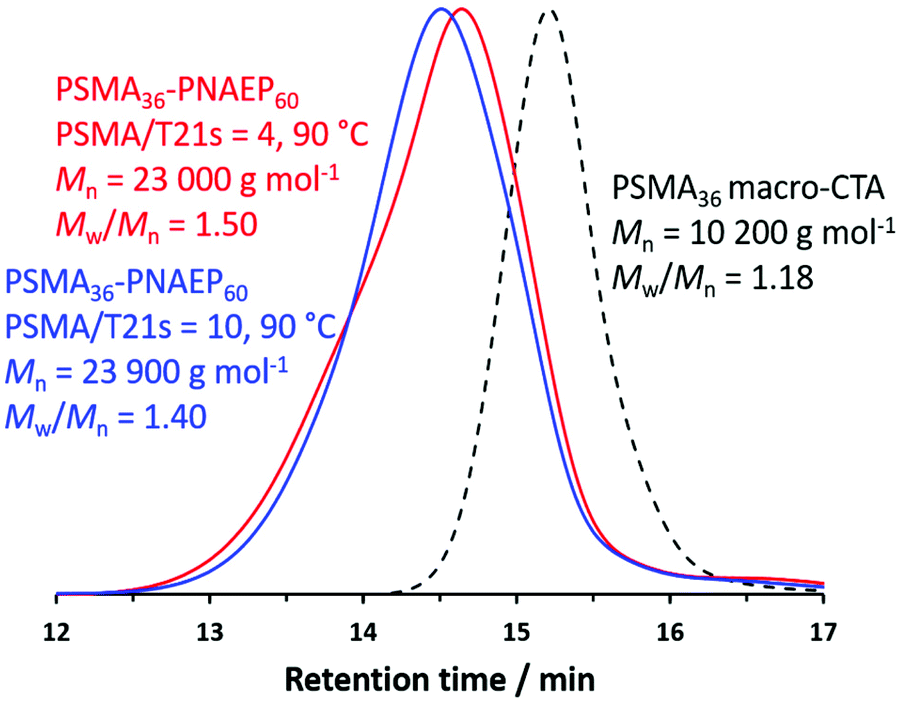 | ||
| Fig. 1 Chloroform GPC curves recorded for a PSMA36 precursor and two corresponding PSMA36–PNAEP60 diblock copolymers prepared by RAFT non-aqueous emulsion polymerization of NAEP in n-dodecane using a PSMA36/T21s molar ratio of 4.0 at 90 °C (red trace) and a PSMA36/T21s molar ratio of 10.0 at 90 °C (blue trace). | ||
The kinetics for the RAFT non-aqueous emulsion polymerization of NAEP in n-dodecane at 90 °C were monitored using 1H NMR spectroscopy when targeting PSMA36–PNAEP60 nanoparticles at 20% w/w solids (Fig. 2a). Chloroform GPC was used to monitor the evolution in Mn and Mw/Mn during this experiment (Fig. 2b). Essentially full NAEP conversion was achieved within 1 h and a linear increase in Mn was observed. The final PSMA36–PNAEP60 diblock copolymer had an Mn of 24100 g mol−1 and an Mw/Mn of 1.48. Relatively little NAEP was consumed within the first 20 min, indicating an initial induction period. However, the subsequent polymerization proceeded rapidly, with 69% conversion being observed after 30 min.
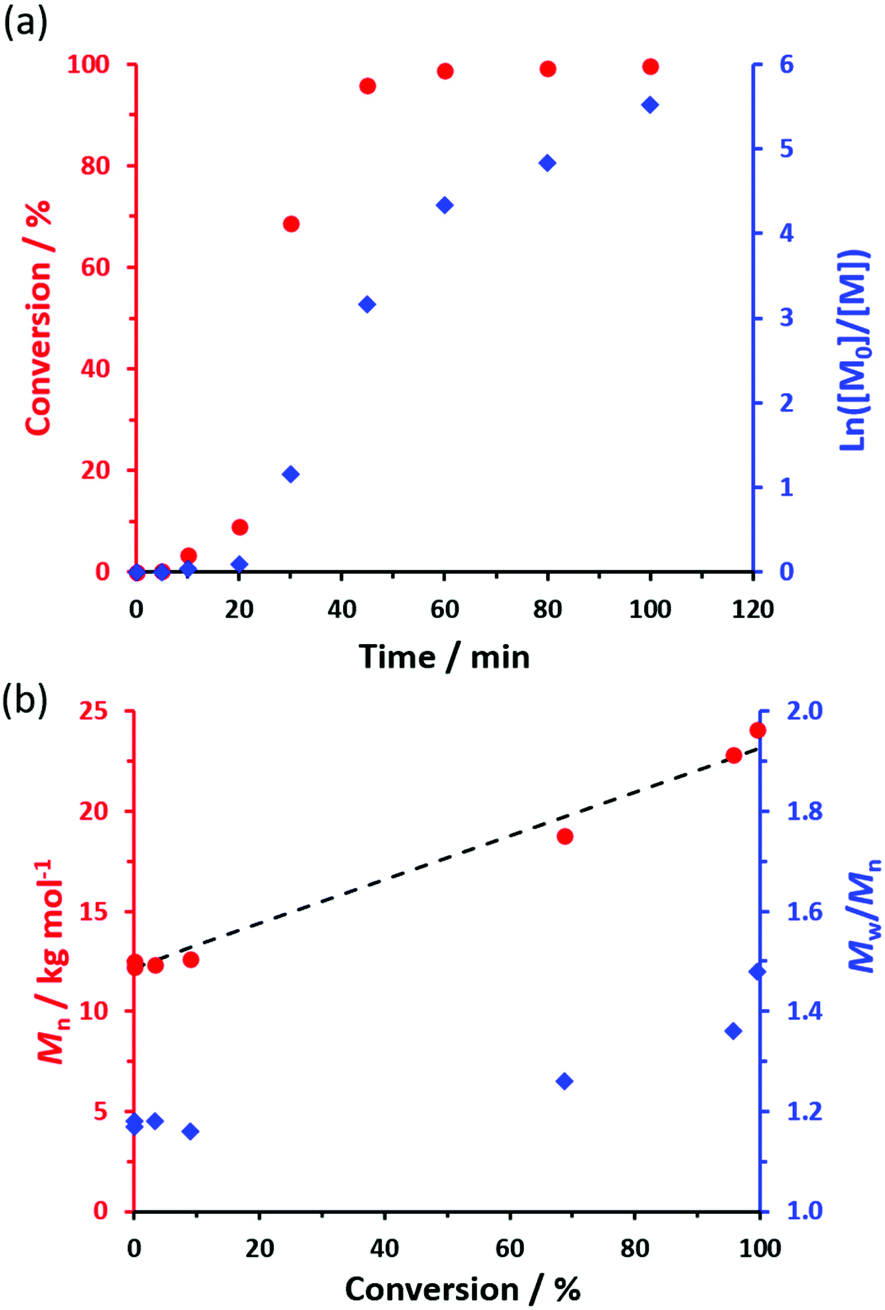 | ||
| Fig. 2 (a) Conversion vs. time curve and corresponding semi-logarithmic plot obtained for the RAFT non-aqueous emulsion polymerization of NAEP using a PSMA36 precursor at 90 °C in n-dodecane and targeting a PNAEP DP of 60 at 20% w/w solids (PSMA36/T21s molar ratio = 4.0). (b) Evolution of Mn and Mw/Mn with NAEP conversion for the same PISA formulation. The dashed line indicates the theoretical Mn data. Given that this GPC protocol uses a series of PMMA calibration standards, the good agreement between the experimental Mn data set and the theoretical line is merely fortuitous in this case. | ||
Characterization of PSMA–PNAEP spherical nanoparticles
A series of PSMA36–PNAEPx (where x = 10–500) nanoparticles were prepared in n-dodecane using the optimized protocol outlined above (Scheme 1). In all cases, high NAEP conversions (≥98%) were achieved as indicated by 1H NMR spectroscopy (Table S1†). As expected, a monotonic relationship was obtained between the z-average diameter of the nanoparticles determined by DLS and the PNAEP DP (Fig. 3a). In particular, systematic variation of the PNAEP DP enables reasonably well-defined nanoparticles (DLS polydispersity ≤0.10) to be prepared over a relatively wide size range (from around 52 to 260 nm) when targeting DPs ranging from 60 to 500. Moreover, a linear increase in Mn (and Mp) was observed when targeting PNAEP DPs up to DP 70 (Fig. 3b) and reasonably good RAFT control was achieved for target PNAEP DPs up to 60 (Mw/Mn < 1.50). Above this DP, inflection points are observed in both the Mp and Mw/Mn data sets. In this context, it is well-known that chain transfer to an acrylic polymer backbone becomes more evident when targeting higher DPs.36,37 Indeed, GPC analysis was not attempted on PSMA36–PNAEPx diblock copolymers when targeting DPs above 110 owing to their incomplete solubility in the GPC eluent (chloroform). Presumably, this problem is related to inter-chain crosslinking via chain transfer to polymer, which leads to nanogel formation.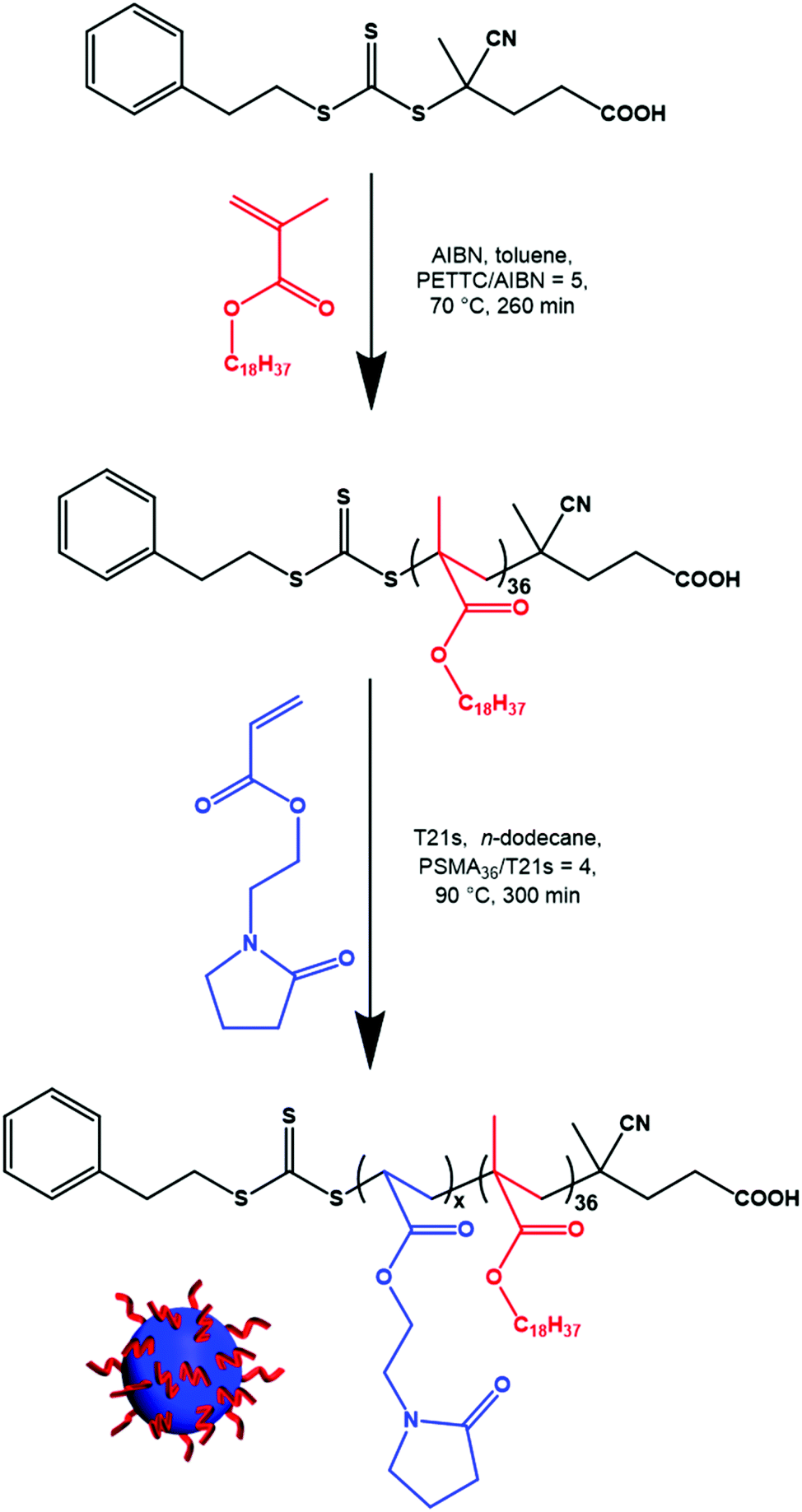 | ||
| Scheme 1 Synthesis of a PSMA36 precursor by RAFT solution polymerization of SMA in toluene at 70 °C and its subsequent chain extension via RAFT non-aqueous emulsion polymerization of NAEP in n-dodecane at 90 °C when targeting 20% w/w solids. | ||
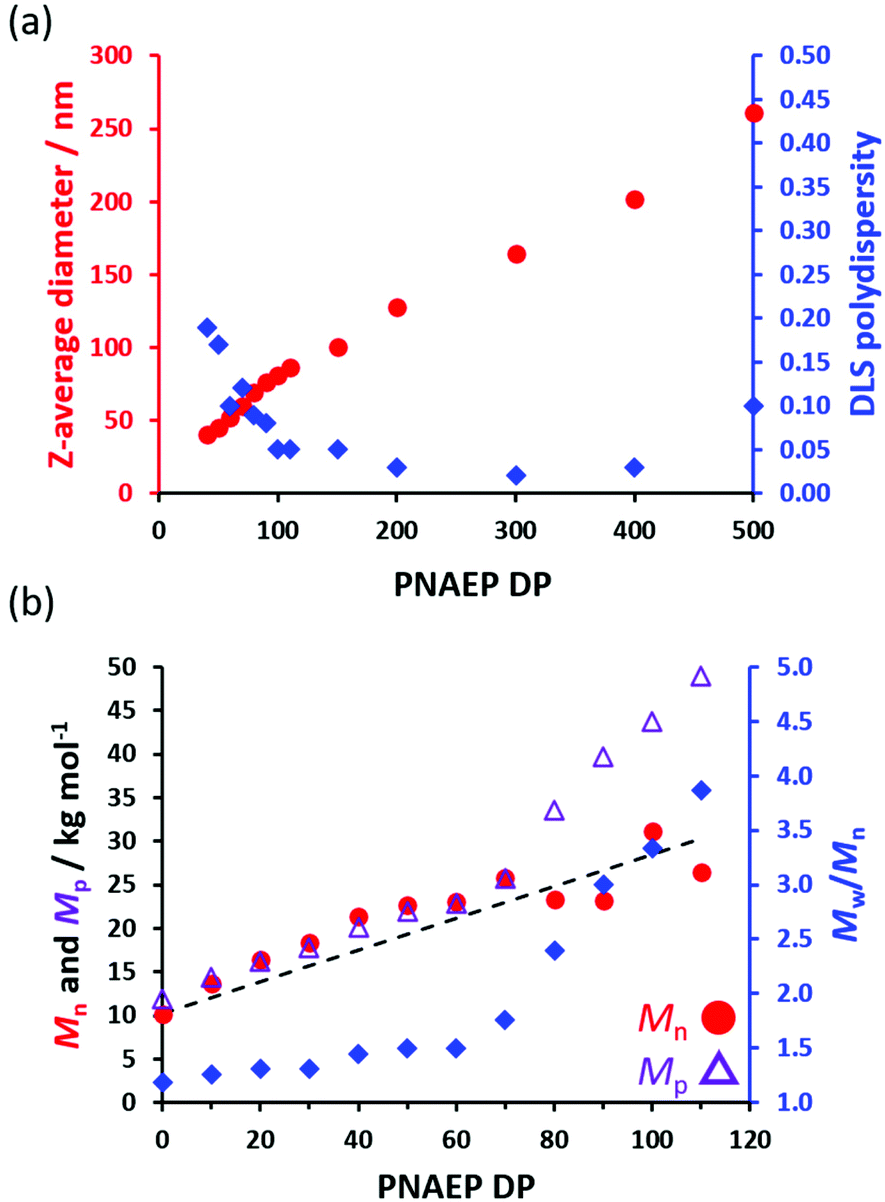 | ||
| Fig. 3 (a) Variation in z-average diameter (and DLS polydispersity) with target PNAEP DP for a series of PSMA36–PNAEPx nanoparticles prepared by RAFT non-aqueous emulsion polymerization of NAEP at 90 °C in n-dodecane using a PSMA36/T21s molar ratio of 4.0 (after dilution from 20% to 0.1% w/w solids using n-dodecane). (b) Evolution in Mn (red filled circles), Mp (purple open triangles) and Mw/Mn with target PNAEP DP for the same series of PSMA36–PNAEPx diblock copolymers and also the corresponding PSMA36 precursor (refractive index detector; near-monodisperse PMMA calibration standards). The dashed line indicates the theoretical Mn data. The experimental Mn data set differs from this theoretical line owing to a small systematic GPC calibration error. | ||
Given their relatively low Tg, TEM studies of acrylic nanoparticles can be problematic.23,52,53 For example, the poly(lauryl acrylate) (PLA)-PBzA nano-objects reported by Ratcliffe and co-workers had to be imaged by cryo-TEM owing to the film-forming nature of the PBzA block (Tg = 6 °C).33 To overcome this problem, an acrylic polymer with a relatively high Tg can be targeted, at least for model studies. Suitable examples here include poly(phenyl acrylate) (PPhA) (Tg = 50 °C),54 and poly(isobornyl acrylate) (PIBOA) (Tg = 94 °C).55 Thus Canning et al. chain-extended a PLA precursor via RAFT dispersion polymerization of PhA in n-heptane.54 In this case, high-quality images of the resulting high Tg nano-objects could be obtained using conventional TEM. Spheres, worms and vesicles could be prepared at 25% w/w solids simply by varying the target degree of polymerization (DP) of the structure-directing PPhA block. However, GPC analysis revealed a high molecular weight shoulder for each copolymer MWD, which suggested that chain transfer to polymer occurred during such PISA syntheses.54
The PSMA36 precursor and four PSMA36–PNAEPx diblock copolymers were characterized using DSC (Fig. 4a). The semi-crystalline PSMA36 homopolymer had a melting transition (Tm) at 30 °C (black trace), similar to that reported by Semsarilar and co-workers.56 For the four diblock copolymers, this Tm slightly shifted to lower temperature as the PNAEP DP was increased. More importantly, the PSMA36–PNAEP60 diblock copolymer exhibited a very weak Tg for the PNAEP block (red trace). This feature becomes much more pronounced on increasing the PNAEP DP from x = 60 to x = 500, albeit with minimal molecular weight dependence (Tg = –7 and −6 °C respectively).26 Such low Tg values mean that TEM analysis is problematic for sizing PNAEP-core nanoparticles owing to their (partial) deformation during TEM grid preparation (Fig. 4b).
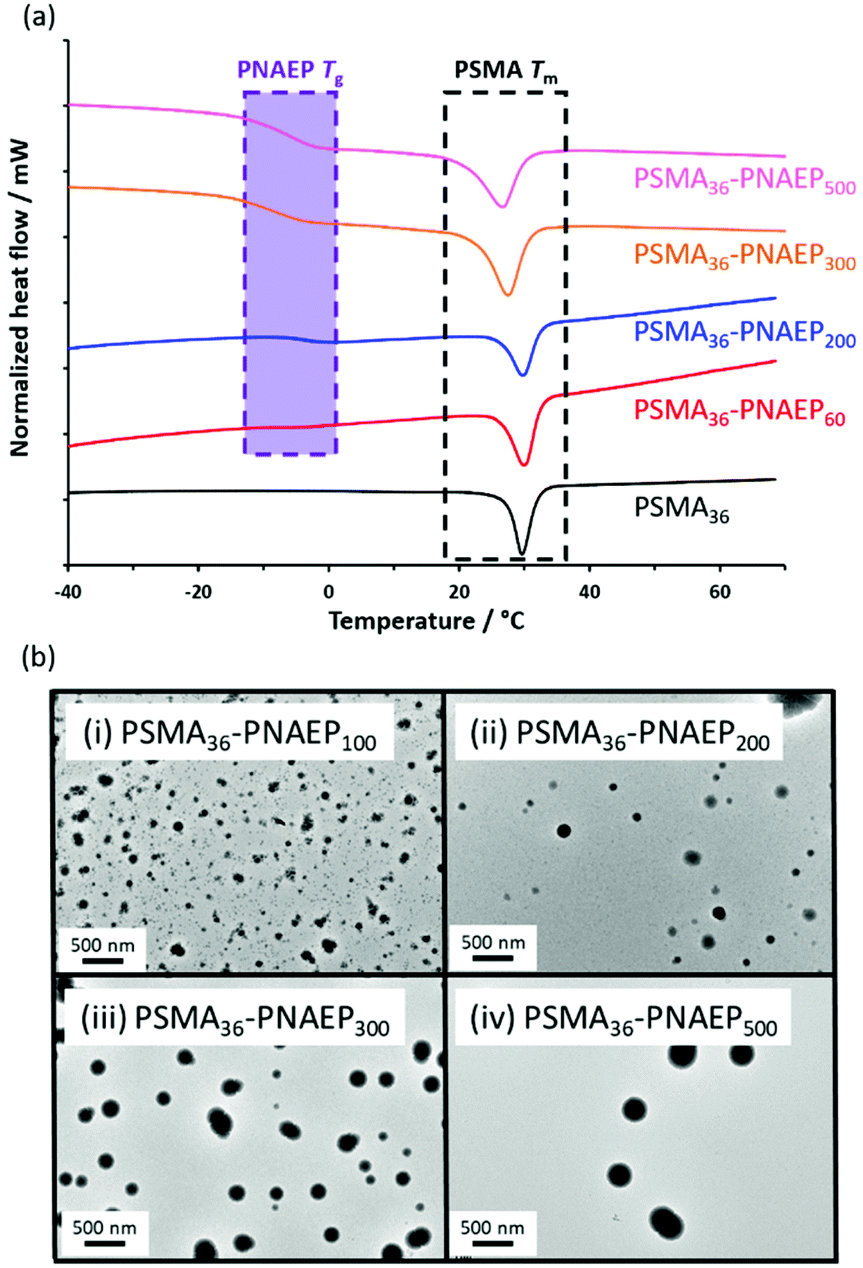 | ||
| Fig. 4 (a) Differential scanning calorimetry (DSC) curves recorded at a heating rate of 10 °C min−1 for the PSMA36 precursor (black trace), PSMA36–PNAEP60 diblock copolymer (red trace), PSMA36–PNAEP200 diblock copolymer (blue trace), PSMA36–PNAEP300 diblock copolymer (orange trace) and PSMA36–PNAEP500 diblock copolymer (pink trace). (b) Representative TEM images recorded for PSMA36–PNAEP100, PSMA36–PNAEP200, PSMA36–PNAEP300 and PSMA36–PNAEP500 diblock copolymer spheres. Such nanoparticles most likely undergo partial deformation (flattening) during TEM grid preparation owing to the relatively low Tg of the core-forming PNAEP block. | ||
Small-angle X-ray scattering (SAXS) patterns were recorded for a series of 1.0% w/w dispersions of PSMA36–PNAEPx nanoparticles in n-dodecane. Satisfactory data fits could be obtained using a well-known spherical micelle model57 (see Fig. 5a) in all five cases. Moreover, the low q gradient (Guinier region) tended to zero, which is consistent with the formation of isotropic spheres. The position of the local minima observed at low q is inversely proportional to the particle radius, so such minima shift to lower q for larger particles.58
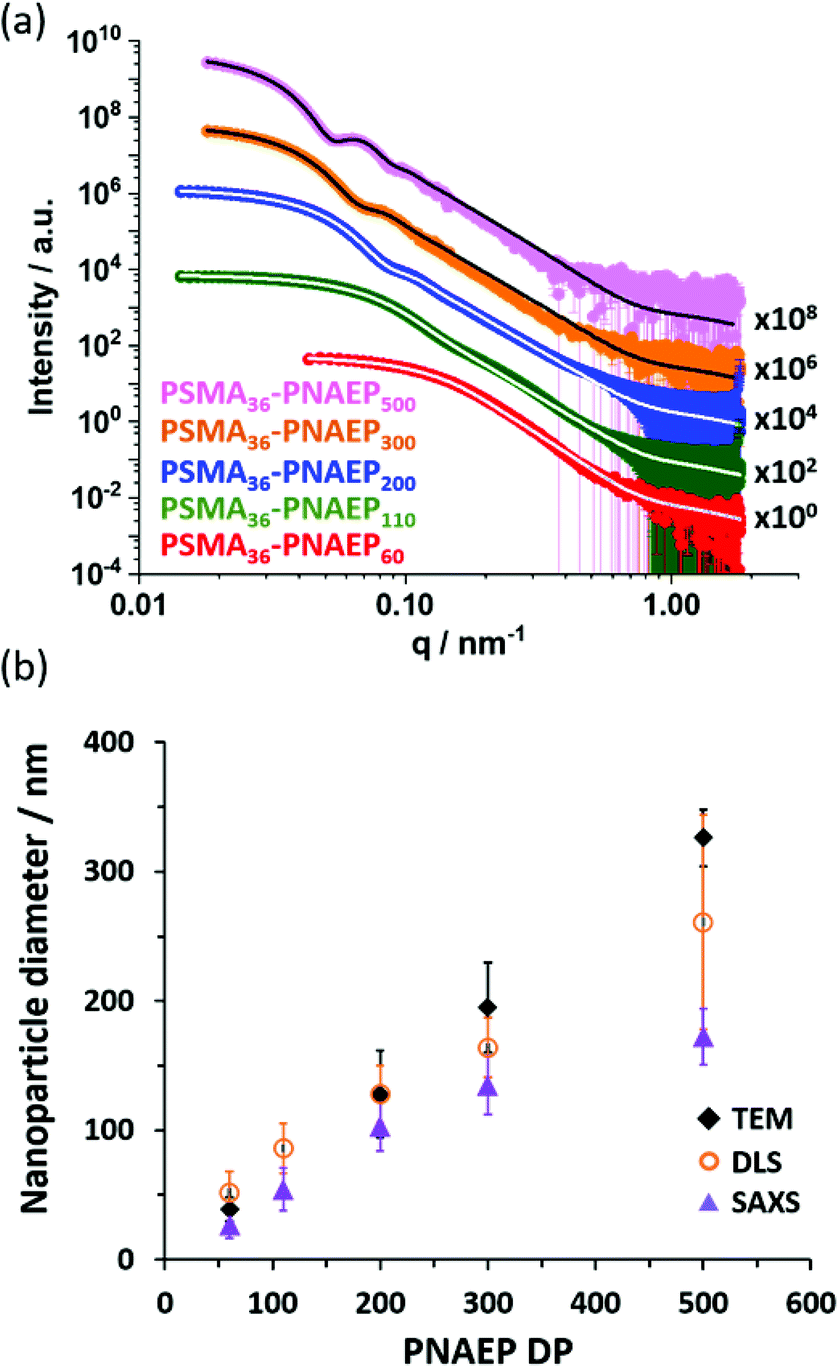 | ||
| Fig. 5 (a) SAXS patterns recorded for 1.0% w/w diblock copolymer dispersions in n-dodecane at 20 °C: PSMA36–PNAEP60 (red), PSMA36–PNAEP110 (green), PSMA36–PNAEP200 (blue), PSMA36–PNAEP300 (orange) and PSMA36–PNAEP500 (pink). Data fits obtained using a well-known spherical micelle model for each of these three patterns are indicated by either white lines (for satisfactory fits) or black lines (for unsatisfactory fits at high q).60 Each SAXS pattern is offset by an arbitrary factor for the sake of clarity. (b) Corresponding sphere diameters determined by DLS (open orange circles), TEM (black diamonds) and SAXS (purple triangles) analysis, respectively. Error bars refer to standard deviations for the nanoparticle diameter and hence indicate the width of each particle size distribution, rather than the experimental error. | ||
Each of these scattering patterns was fitted using a well-known spherical micelle model.57 The Rg for the PSMA36 stabilizer block was fixed using an estimated theoretical value of 1.5 nm. Given the rather low solubility of NAEP monomer in n-dodecane, the mean solvent volume fraction within the PNAEP cores (xsol) was taken to be zero. As expected, SAXS analysis indicated an increase in the volume-average sphere diameter with PNAEP DP. However, satisfactory data fits could not be obtained at high q for PNAEP DPs above 300. For these larger spheres, SAXS patterns were recorded using camera lengths of 1.84 m and 6.25 m and these two data sets were combined prior to modeling, which might contribute to this problem. DLS diameters were significantly greater than those determined by SAXS (Fig. 5b). This is not unexpected, since the former technique reports a z-average diameter whereas the latter technique reports a volume-average diameter. However, the DLS diameter was significantly larger than the SAXS diameter for the PSMA36–PNAEP500 nanoparticles. This suggests the presence of aggregates in this particular case, although further studies would be required to confirm this hypothesis. This is because DLS is particularly sensitive to larger particles because the scattered light intensity scales as r,6 where r is the particle radius.59 The nanoparticle sphere diameters determined by TEM for PNAEP DPs above 200 were even larger than those determined by SAXS. This strongly suggests nanoparticle deformation (flattening) during TEM grid preparation owing to the low Tg of the PNAEP block (Fig. 4). Thus TEM oversizes such deformed nanoparticles.
Evaluation of PSMA–PNAEP nanoparticles as Pickering emulsifiers
Cunningham and co-workers prepared a series of PSMA14–PNMEPx spherical nanoparticles via RAFT dispersion polymerization of NMEP in n-dodecane and examined their performance as putative Pickering emulsifiers.17 For example, addition of an equal volume of water to a 1.0% w/w dispersion of 23 nm diameter PSMA14–PNMEP49 spheres in n-dodecane followed by high-shear homogenization led to the formation of oil-in-water (o/w) emulsions when employing oil volume fractions below 0.50. This was unexpected because such hydrophobic nanoparticles should normally favor the formation of water-in-oil (w/o) emulsions.60 After further investigation, it was concluded that nanoparticle inversion most likely occurred during homogenization, leading to the formation of hydrophilic PNMEP49–PSMA14 nanoparticles that subsequently acted as a Pickering emulsifier.Bearing in mind the above literature precedent, PSMA36–PNAEP70 nanoparticles were prepared in n-dodecane for evaluation as a putative Pickering emulsifier. The copolymer concentration was systematically lowered from 1.0% w/w to 0.025% w/w by dilution with n-dodecane to produce a series of 2.0 mL copolymer dispersions. Then deionized water (2.0 mL) was added to each dispersion in turn to obtain a constant n-dodecane volume fraction of 0.50 and high-shear homogenization was conducted in each case (Fig. 6). The electrical conductivity for an emulsion obtained using 1.0% w/w PSMA36–PNMEP70 diblock copolymer nanoparticles was determined to be 3.2 × 10–4 S m−1, which is close to that of deionized water alone (3.8 × 10–4 S m−1). In contrast, the conductivity of n-dodecane is 1.1 × 10–11 S m−1.61 Thus, these conductivity data indicate the formation of an o/w emulsion, rather than a w/o emulsion. The so-called ‘drop test’ method (which involves taking an aliquot of the emulsion and determining whether it disperses more readily when added to either water or n-dodecane) was used to confirm that o/w emulsions were always produced regardless of the nanoparticle concentration.
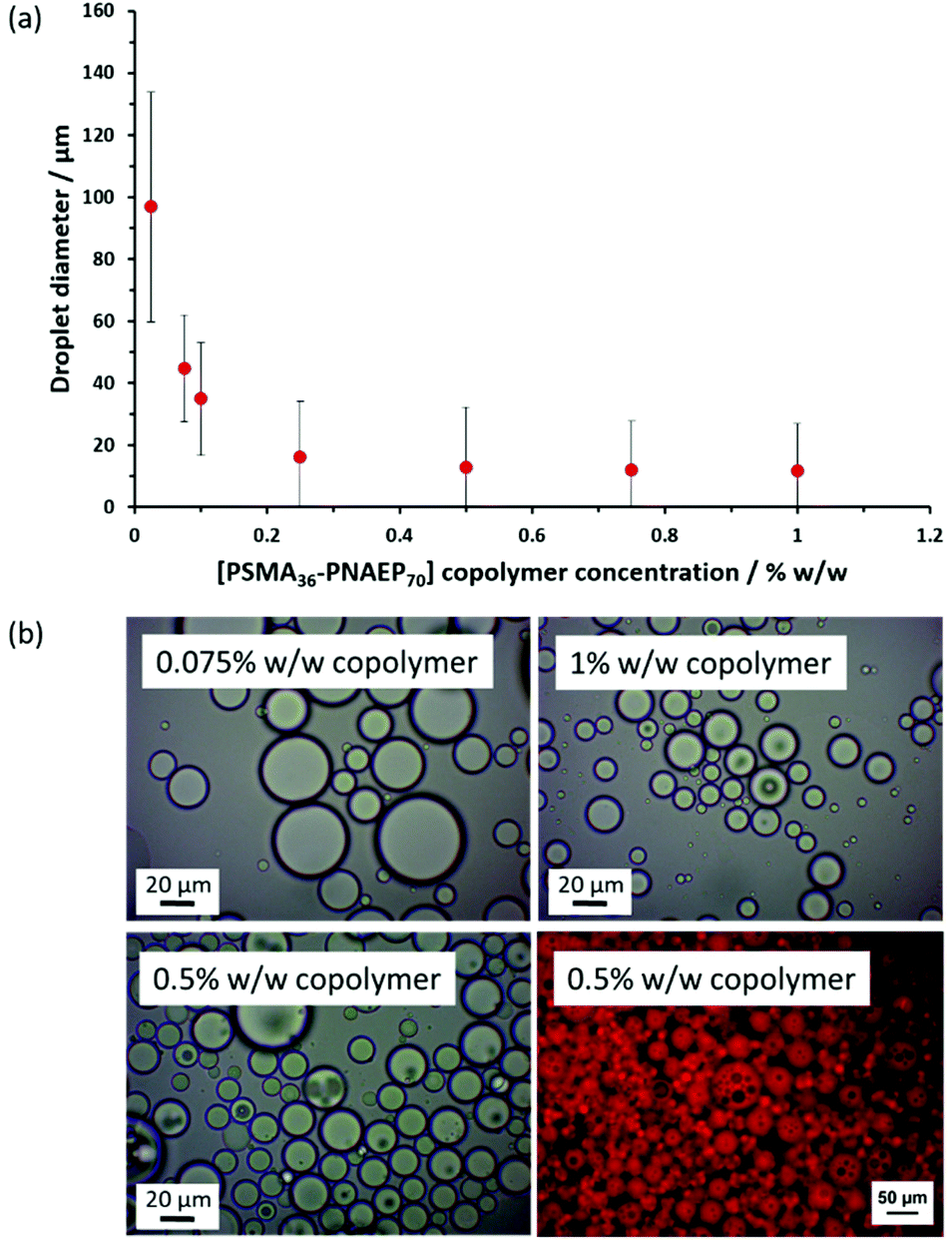 | ||
| Fig. 6 (a) Variation in volume-average droplet diameter (as determined by laser diffraction) for a series of o/w Pickering emulsions obtained by high-shear homogenization when systematically varying the PSMA36–PNAEP70 copolymer concentration at a constant n-dodecane volume fraction of 0.50. The standard deviations indicate the width of the droplet size distribution, rather than the experimental error. (b) Optical micrographs recorded at copolymer concentrations of 0.075%, 0.50% and 1.00% w/w, respectively. When using a copolymer concentration of 0.50% w/w, a water-insoluble dye (Nile Red) was dissolved in the oil phase to aid the identification of w/o/w double emulsions by fluorescence microscopy. | ||
It should be noted that the PETTC RAFT agent that was used to synthesize these diblock copolymer nanoparticles confers a carboxylic acid end-group on the PSMA stabilizer chains (Scheme 1). Depending on the solution pH of the aqueous phase, these hydrophilic end-groups can become ionized, which significantly reduces the particle contact angle. This results in stabilization of oil droplets in water rather than the expected aqueous droplets in oil. Moreover, varying the solution pH also affects the mean droplet diameter. When conducting homogenization using an n-dodecane volume fraction of 0.50 and 1.0% w/w nanoparticles, laser diffraction studies indicated a volume-average droplet diameter of 12 μm at pH 7 and 35 μm at pH 3.
Optical micrographs recorded for Pickering emulsions prepared using a copolymer concentration of ≥0.50% w/w indicate the presence of small droplets within larger droplets, suggesting the formation of double emulsions. Fluorescence microscopy studies were undertaken to corroborate this hypothesis. Accordingly, Nile Red was dissolved in n-dodecane containing 0.50% w/w copolymer prior to homogenization with an equal volume of water (inset of Fig. 6). This water-insoluble dye label is exclusively located within the droplet phase, demonstrating that an o/w emulsion is obtained in this case. However, close inspection revealed that aqueous domains were present within these oil droplets, confirming the formation of a w/o/w double emulsion. This is somewhat surprising: normally such double emulsions are formed by preparing a w/o emulsion first, followed by homogenization in the presence of water to obtain the w/o/w emulsion.62 Moreover, the formation of Pickering double emulsions usually requires two types of particles of differing wettability (i.e. hydrophilic particles to produce an o/w emulsion and hydrophobic particles to stabilize a w/o emulsion).63–66 Nevertheless, there are a few literature reports of Pickering double emulsions being generated during a single emulsification step.67–71 For example, György and co-workers recently obtained w/o/w double emulsions when using hydrophobic PSMA9-poly(2-hydroxypropyl methacrylate)50 (PHPMA) diblock copolymer nanoparticles prepared in mineral oil.71 More specifically, high oil volume fractions (>0.50), high shear rates (>13500 rpm) and relatively high copolymer concentrations (>0.50% w/w) enabled the direct formation of a w/o/w double emulsion in a single step. Similarly, optical microscopy studies of the present system confirm that w/o/w double emulsions are only obtained when using PSMA36–PNAEP70 copolymer concentrations at or above 0.50% w/w.
Laser diffraction was used to size the emulsion droplets (see Fig. 6). At higher copolymer concentrations, the volume-average droplet diameter remained constant at around 12 μm. However, both the mean diameter and the standard deviation increased as the copolymer concentration was lowered to 0.25% w/w. Large, polydisperse droplets with relatively poor stability towards coalescence were obtained at or below 0.10% w/w copolymer concentration. This upturn in droplet size at lower copolymer concentrations is characteristic of Pickering emulsions because there are fewer nanoparticles to stabilize the additional interfacial area created during high-shear homogenization.72–75 This implies that the original linear PSMA36–PNAEP70 nanoparticles survive the high-shear homogenization conditions intact. However, given their highly hydrophobic nature such nanoparticles had been expected to form w/o Pickering emulsions, rather than o/w emulsions. There are two possible explanations for this surprising observation. In principle, in situ nanoparticle inversion might have occurred during homogenization, thus converting the initial hydrophobic PSMA36–PNAEP70 nanoparticles into hydrophilic PNAEP70–PSMA36 nanoparticles. Alternatively, the former nanoparticles may simply adsorb at the inner surface of the oil droplets (Fig. S7†).76
To distinguish between these two scenarios, core-crosslinked PSMA36–PNAEP60–PEGDA10 nanoparticles were used to prepare Pickering emulsions via high-shear homogenization. A bifunctional comonomer, EGDA, was added towards the end of the NAEP polymerization when targeting a PSMA36–PNAEP60 diblock copolymer. As expected, the resulting core-crosslinked PSMA36–PNAEP60–PEGDA10 triblock copolymer nano-objects were somewhat less prone to deformation during TEM grid preparation and hence exhibited a more well-defined spherical morphology (Fig. S8†). Moreover, the z-average diameter indicated by DLS studies of these crosslinked nanoparticles was consistent with that observed for the linear non-crosslinked nanoparticles (57 nm vs. 52 nm, respectively). The DLS diameter for the core-crosslinked PSMA36–PNAEP60–PEGDA10 spheres was also determined in chloroform as well as n-dodecane (see Table S2†). The former solvent is a good solvent for both blocks, so nanoparticle swelling was anticipated under such conditions. Indeed, DLS studies indicated that, unlike the corresponding linear diblock copolymer nanoparticles, such covalently-stabilized nanoparticles swelled appreciably but did not dissolve when diluted with chloroform, which is a good solvent for both blocks (Table S2†). This suggests that the degree of core-crosslinking was sufficient to ensure their structural integrity. Accordingly, the copolymer concentration was systematically lowered from 1.0% w/w to 0.025% w/w when performing high-shear homogenization at a constant oil volume fraction of 0.50. The upturn in droplet diameter observed at lower copolymer concentrations confirmed their Pickering-type character (see Fig. 7). The ‘drop test’ method confirmed the formation of o/w emulsions in all cases. Importantly, such hydrophobic core-crosslinked nanoparticles cannot undergo in situ inversion to form hydrophilic PNAEP-stabilized nanoparticles that could in principle adsorb at the outer surface of the oil droplets. Thus this suggests that both these crosslinked nanoparticles and the linear PSMA36–PNAEP70 nanoparticles must instead adsorb at the inner surface of the oil droplets. Optical microscopy studies indicated the formation of w/o/w double emulsions when using the core-crosslinked PSMA36–PNAEP60–PEGDA10 nanoparticles for the high-shear homogenization of n-dodecane-water mixtures. This was confirmed by fluorescence microscopy studies performed after addition of Nile Red dye to the oil phase (inset of Fig. 7). However, such complex emulsions were formed at somewhat lower copolymer concentrations when using these core-crosslinked nanoparticles; close inspection of the relevant optical micrographs suggest that w/o/w double emulsions can be obtained at copolymer concentrations as low as 0.10% w/w.
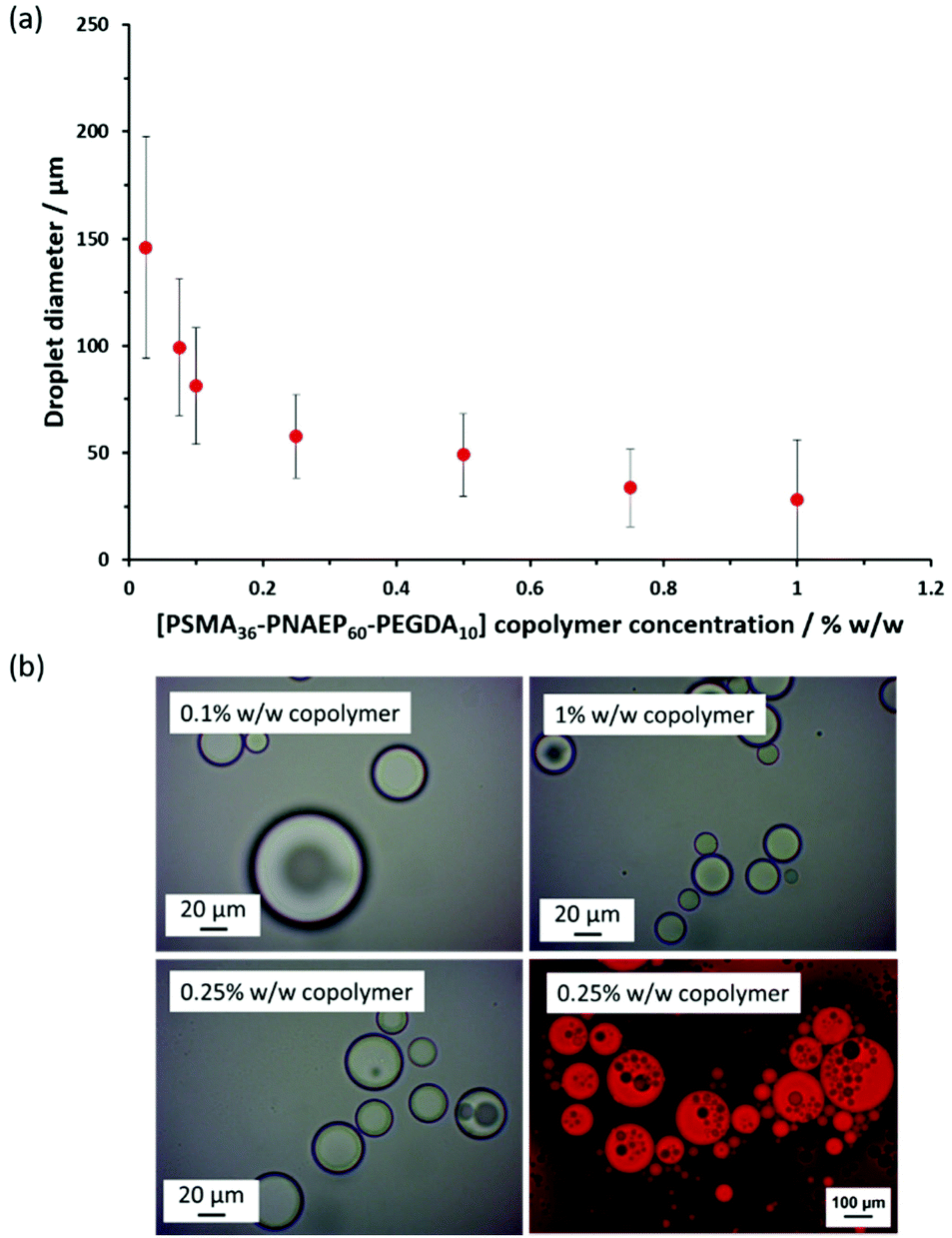 | ||
| Fig. 7 (a) Variation in volume-average droplet diameter (as determined by laser diffraction) for a series of o/w Pickering emulsions obtained by high-shear homogenization when systematically varying the PSMA36–PNAEP60–PEGDA10 copolymer concentration at a constant n-dodecane volume fraction of 0.50. The standard deviations indicate the width of the droplet size distribution, rather than the experimental error. (b) Optical micrographs recorded at copolymer concentrations of 0.10%, 0.25% and 1.00% w/w. When using a copolymer concentration of 0.25% w/w, a water-insoluble dye (Nile Red) was dissolved in the oil phase to aid the identification of w/o/w double emulsions using fluorescence microscopy. | ||
Finally, the relative volume fraction of n-dodecane was varied while fixing the linear PSMA36–PNAEP70 copolymer concentration at 1.0% w/w with respect to the oil phase (Fig. 8). In this series of experiments, w/o/w Pickering double emulsions were obtained for n-dodecane volume fractions up to 0.60, with mean droplet diameters increasing from 5 μm to 19 μm when adjusting the oil volume fraction from 0.10 to 0.60. However, a w/o emulsion was produced when attempting homogenization at an n-dodecane volume fraction of either 0.65 or 0.70. Nevertheless, it is clear that relatively concentrated w/o/w Pickering double emulsions can be obtained using this simple protocol.
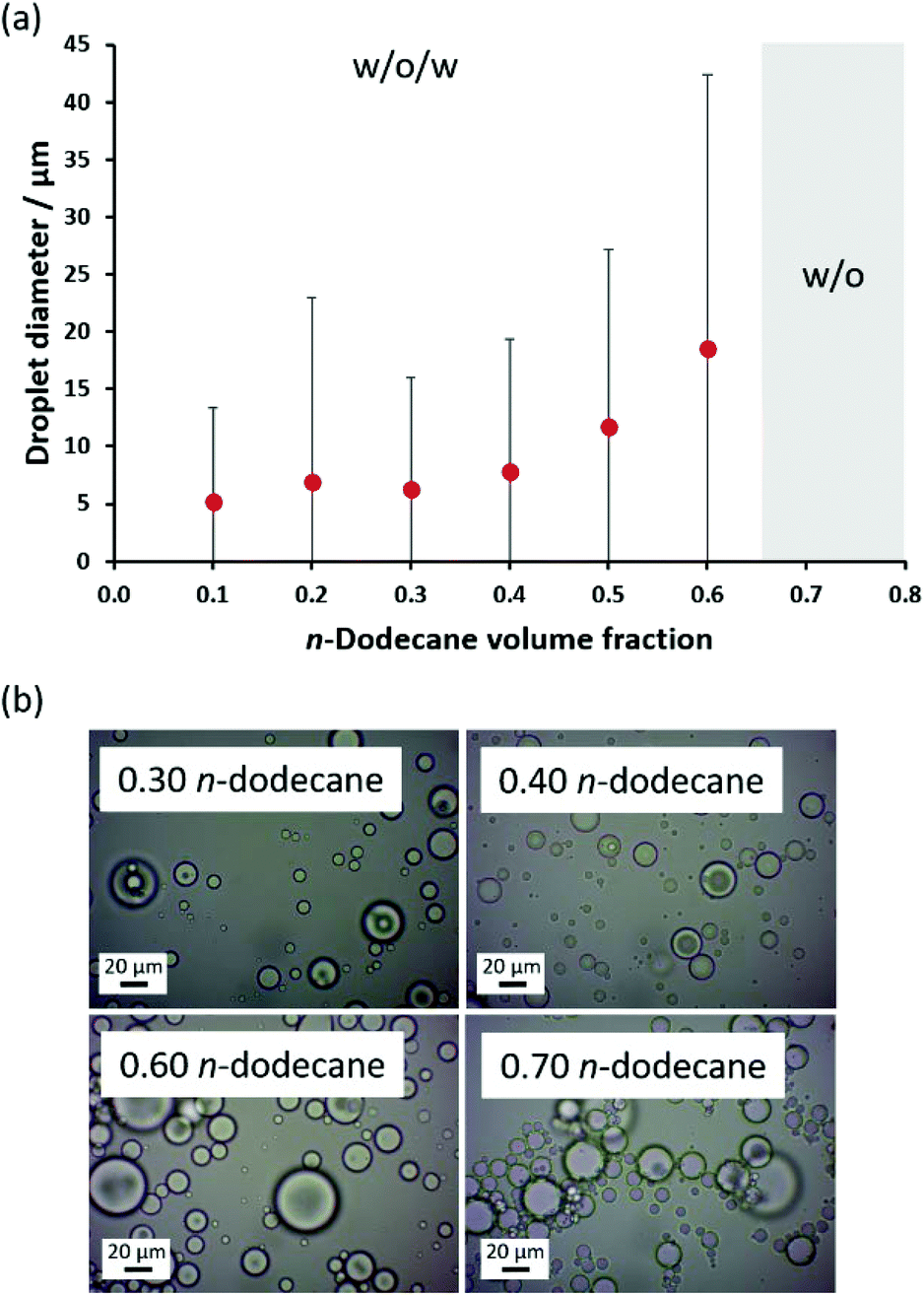 | ||
| Fig. 8 (a) Variation in volume-average droplet diameter (as determined by laser diffraction) for a series of w/o/w Pickering emulsions obtained by high-shear homogenization when systematically varying the volume fraction of n-dodecane at a constant copolymer concentration of 1.0% w/w when using the linear PSMA36–PNAEP70 nanoparticles. The standard deviations indicate the droplet polydispersity, rather than the error in the measurements. (b) Optical micrographs recorded for Pickering emulsions prepared at n-dodecane volume fractions of 0.30, 0.40, 0.60 and 0.70, respectively. | ||
Emulsions prepared using 1.0% w/w linear PSMA36–PNAEP70 nanoparticles at oil volume fractions of either 0.50 or 0.65 were allowed to stand for 72 h to enable the droplets to either cream or sediment, respectively. These two emulsion formulations were chosen because an oil volume fraction of 0.50 gives a w/o/w emulsion and a volume fraction of 0.65 produces a w/o emulsion (see Fig. 8a). DLS analysis was performed on each continuous phase after careful removal of the droplet phase. For the oil-continuous phase derived from the w/o emulsion, DLS studies confirmed strong light scattering (derived count rate = 10000 kcps) and the presence of nanoparticles (z-average diameter = 53 nm; DLS polydispersity = 0.14) that were comparable in size compared to the original nanoparticles (z-average diameter = 60 nm and DLS polydispersity = 0.12). Conversely, DLS studies of the aqueous continuous phase isolated from the w/o/w emulsion confirmed that essentially no nanoparticles were present (only a much lower derived count rate of only 600 kcps was observed in this case). Thus, these DLS experiments confirm that the w/o/w Pickering emulsion is indeed stabilized by hydrophobic PSMA36–PNAEP70 nanoparticles adsorbing at the oil/water interface from within the droplets, rather than undergoing in situ inversion to form hydrophilic PNAEP70–PSMA36 nanoparticles.
Conclusions
A PSMA36 precursor was employed for the RAFT non-aqueous emulsion polymerization of NAEP in n-dodecane to produce a series of sterically-stabilized PSMA36–PNAEPx diblock copolymer spheres with high NAEP conversions being achieved in all cases (≥98% within 5 h at 90 °C). Systematic increases in both z-average diameter and Mp were observed for this unusual PISA formulation when targeting higher PNAEP DPs, but only relatively poor RAFT control could be achieved. This is because this unusual PISA formulation requires a relatively high reaction temperature to ensure sufficient NAEP solubility in n-dodecane, which inevitably leads to chain transfer to the acrylic polymer backbone. TEM studies of the linear diblock copolymer nanoparticles were somewhat problematic owing to film formation during grid preparation. Thus, EGDA was employed as a bifunctional crosslinker and added towards the end of the NAEP polymerization to produce covalently-stabilized nanoparticles. This enabled a well-defined spherical morphology to be confirmed by TEM while also producing core-crosslinked nanogels that swelled when dispersed in chloroform, which is a good solvent for both blocks. Furthermore, this spherical morphology was confirmed for the linear diblock copolymer nanoparticles by SAXS since each scattering pattern could be satisfactorily fitted using a well-known spherical micelle model.PSMA36–PNAEP70 spheres prepared in n-dodecane were evaluated as a putative Pickering emulsifier. In principle, employing such hydrophobic nanoparticles should favor the formation of w/o emulsions. Unexpectedly, addition of an equal volume of water followed by high-shear homogenization produced o/w emulsions instead. Laser diffraction and optical microscopy studies indicated that larger droplets were formed on lowering the copolymer concentration. This indicates that the nanoparticles remain intact after homogenization, thus producing genuine Pickering emulsions. Thus, either the hydrophobic PSMA36–PNAEP70 spheres are adsorbed at the inner surface of the oil droplets or nanoparticle inversion occurred during high-shear homogenization to form hydrophilic PNAEP70–PSMA36 spheres that then adsorb at the outer surface of the oil droplets. Accordingly, core-crosslinked PSMA36–PNAEP60–PEGDA10 spheres were prepared in n-dodecane to discriminate between these two possibilities. In this case, high-shear homogenization at various copolymer concentrations always produced o/w Pickering emulsions. As these covalently-stabilized hydrophobic nanoparticles cannot undergo inversion to form hydrophilic nanoparticles, this suggests that such Pickering emulsions must be formed by nanoparticle adsorption at the inner surface of the oil droplets. This interpretation is supported by DLS studies of the continuous phase, which reveals the absence of any nanoparticles for the o/w emulsion but the presence of excess nanoparticles for the w/o emulsion. For both types of Pickering emulsifiers, fluorescence microscopy studies confirmed the formation of w/o/w double emulsions under certain conditions, rather than o/w emulsions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Mr Saul J. Hunter is thanked for imaging the w/o/w emulsions by fluorescence microscopy. EPSRC is thanked for funding a CDT PhD studentship for the first author (EP/L016281). Ashland Specialty Ingredients (Bridgewater, New Jersey, USA) is thanked for partial financial support of this PhD project, for supplying the OAA monomer and for permission to publish this work. S. P. A. also thanks the EPSRC for an Established Career Particle Technology Fellowship (EP/R003009).References
- M. Lansalot and J. Rieger, Macromol. Rapid Commun., 2019, 40, 1800885 CrossRef PubMed.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- J. Chiefari, Y. K. B. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., 2020, 59, 8368–8392 CrossRef PubMed.
- X. Wang and Z. An, Macromol. Rapid Commun., 2019, 40, 1800325 CrossRef PubMed.
- V. J. Cunningham, M. J. Derry, L. A. Fielding, O. M. Musa and S. P. Armes, Macromolecules, 2016, 49, 4520–4533 CrossRef CAS PubMed.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- M. J. Derry, L. A. Fielding, N. J. Warren, C. J. Mable, A. J. Smith, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2016, 7, 5078–5090 RSC.
- W. Zhao, G. Gody, S. Dong, P. B. Zetterlund and S. Perrier, Polym. Chem., 2014, 5, 6990–7003 RSC.
- D. Zehm, L. P. D. Ratcliffe and S. P. Armes, Macromolecules, 2013, 46, 128–139 CrossRef CAS.
- E. R. Jones, O. O. Mykhaylyk, M. Semsarilar, M. Boerakker, P. Wyman and S. P. Armes, Macromolecules, 2016, 49, 172–181 CrossRef CAS PubMed.
- V. J. Cunningham, S. P. Armes and O. M. Musa, Polym. Chem., 2016, 7, 1882–1891 RSC.
- V. J. Cunningham, Y. Ning, S. P. Armes and O. M. Musa, Polymer, 2016, 106, 189–199 CrossRef CAS.
- M. Teodorescu and M. Bercea, Polym.-Plast. Technol. Eng., 2015, 54, 923–943 CrossRef CAS.
- J. S. Thomaides, K. A. Rodrigues, S. A. Vona, G. T. Martino and A. J. Adamo, US9321873B2, 2010, 1–27.
- L. Zaidel, G. Pan, S. K. Chopra, P. Mandadi and M. Prencipe, CA2576731C, 2005, 1–44.
- J. D. Moon, EP0069775B1, 1982, 1–10.
- R. R. Gibson, E. J. Cornel, O. M. Musa, A. Fernyhough and S. P. Armes, Polym. Chem., 2020, 11, 1785–1796 RSC.
- R. R. Gibson, S. P. Armes, O. M. Musa and A. Fernyhough, Polym. Chem., 2019, 10, 1312–1323 RSC.
- Y. Shi, G. Liu, H. Gao, L. Lu and Y. Cai, Macromolecules, 2009, 42, 3917–3926 CrossRef CAS.
- O. J. Deane, J. R. Lovett, O. M. Musa, A. Fernyhough and S. P. Armes, Macromolecules, 2018, 51, 7756–7766 CrossRef CAS PubMed.
- O. J. Deane, O. M. Musa, A. Fernyhough and S. P. Armes, Macromolecules, 2020, 53, 1422–1434 CrossRef CAS.
- M. Chenal, L. Bouteiller and J. Rieger, Polym. Chem., 2013, 4, 752–762 RSC.
- X. Zhang, S. Boissé, C. Bui, P. A. Albouy, A. Brûlet, M. H. Li, J. Rieger and B. Charleux, Soft Matter, 2012, 8, 1130–1141 RSC.
- L. Houillot, C. Bui, M. Save, B. Charleux, C. Farcet, C. Moire, J.-A. Raust and I. Rodriguez, Macromolecules, 2007, 40, 6500–6509 CrossRef CAS.
- J. A. Raust, L. Houillot, M. Save, B. Charleux, C. Moire, C. Farcet and H. Pasch, Macromolecules, 2010, 43, 8755–8765 CrossRef CAS.
- L. Houillot, C. Bui, C. Farcet, C. Moire, J. A. Raust, H. Pasch, M. Save and B. Charleux, ACS Appl. Mater. Interfaces, 2010, 2, 434–442 CrossRef CAS PubMed.
- L. P. D. Ratcliffe, B. E. McKenzie, G. M. D. Le Bouëdec, C. N. Williams, S. L. Brown and S. P. Armes, Macromolecules, 2015, 48, 8594–8607 CrossRef CAS.
- A. Veloso, W. García, A. Agirre, N. Ballard, F. Ruipérez, J. C. De La Cal and J. M. Asua, Polym. Chem., 2015, 6, 5437–5450 RSC.
- N. M. Ahmad, F. Heatley, D. Britton and P. A. Lovell, Macromol. Symp., 1999, 143, 231–241 CrossRef CAS.
- F. Heatley, P. A. Lovell and T. Yamashita, Macromolecules, 2001, 34, 7636–7641 CrossRef CAS.
- N. M. Ahmad, B. Charleux, C. Farcet, C. J. Ferguson, S. G. Gaynor, B. S. Hawkett, F. Heatley, B. Klumperman, D. Konkolewicz, P. A. Lovell, K. Matyjaszewski and R. Venkatesh, Macromol. Rapid Commun., 2009, 30, 2002–2021 CrossRef CAS.
- K. Landfester, M. Willert and M. Antonietti, Macromolecules, 2000, 33, 2370–2376 CrossRef CAS.
- X. You, V. L. Dimonie and A. Klein, J. Appl. Polym. Sci., 2001, 80, 1951–1962 CrossRef CAS.
- K. Hariri, S. Al Akhrass, C. Delaite, P. Moireau and G. Riess, Polym. Int., 2007, 56, 1200–1205 CrossRef CAS.
- O. M. Daraba, A. N. Cadinoiu, D. M. Rata, L. I. Atanase and G. Vochita, Polymers, 2020, 12, 1018–1034 CrossRef CAS PubMed.
- E. R. Jones, M. Semsarilar, A. Blanazs and S. P. Armes, Macromolecules, 2012, 45, 5091–5098 CrossRef CAS.
- G. N. Smith, S. L. Canning, M. J. Derry, O. O. Mykhaylyk, S. E. Norman and S. P. Armes, Polym. Chem., 2020, 11, 2605 RSC.
- J. Ilavsky and P. R. Jemian, J. Appl. Crystallogr., 2009, 42, 347–353 CrossRef CAS.
- Y. Pei, O. R. Sugita, L. Thurairajah and A. B. Lowe, RSC Adv., 2015, 5, 17636–17646 RSC.
- Y. Pei, L. Thurairajah, O. R. Sugita and A. B. Lowe, Macromolecules, 2015, 48, 236–244 CrossRef CAS.
- B. R. Parker, M. J. Derry, Y. Ning and S. P. Armes, Langmuir, 2020, 36, 3730–3736 CrossRef CAS PubMed.
- One reviewer of this manuscript suggested that our results might be related to the initiator half-life rather than to the temperature-dependent solubility of the NAEP monomer. To test this hypothesis, we attempted the same PISA syntheses targeting PSMA36–PNAEP60 using an azo initiator (AIBN) instead of T21s. However, no polymerization was observed at either 70 °C or 80 °C, whereas 99% conversion was achieved within 6 h at 90 °C (see entry 7 in Table S1†). Thus the polymerization of NAEP in n-dodecane depends strongly on the temperature-dependent monomer solubility but not on the type of initiator.
- D. Britton, F. Heatley and P. A. Lovell, Macromolecules, 1998, 31, 2828–2837 CrossRef CAS.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 CrossRef PubMed.
- T. R. Guimarães, M. Khan, R. P. Kuchel, I. C. Morrow, H. Minami, G. Moad, S. Perrier and P. B. Zetterlund, Macromolecules, 2019, 52, 2965–2974 CrossRef.
- S. J. Byard, M. Williams, B. E. McKenzie, A. Blanazs and S. P. Armes, Macromolecules, 2017, 50, 1482–1493 CrossRef CAS PubMed.
- S. J. Byard, C. T. O'Brien, M. J. Derry, M. Williams, O. O. Mykhaylyk, A. Blanazs and S. P. Armes, Chem. Sci., 2020, 11, 396–402 RSC.
- S. L. Canning, V. J. Cunningham, L. P. D. Ratcliffe and S. P. Armes, Polym. Chem., 2017, 8, 4811–4821 RSC.
- J. Tan, C. Huang, D. Liu, X. Zhang, Y. Bai and L. Zhang, ACS Macro Lett., 2016, 5, 894–899 CrossRef CAS.
- M. Semsarilar, N. J. W. Penfold, E. R. Jones and S. P. Armes, Polym. Chem., 2015, 6, 1751–1757 RSC.
- J. S. Pedersen, J. Appl. Crystallogr., 2000, 33, 637–640 CrossRef CAS.
- M. Li, Y. Liu, H. Nie, R. Bansil and M. Steinhart, Macromolecules, 2007, 40, 9491–9502 CrossRef CAS.
- J. Stetefeld, S. A. McKenna and T. R. Patel, Biophys. Rev., 2016, 8, 409–427 CrossRef CAS PubMed.
- K. L. Thompson, L. A. Fielding, O. O. Mykhaylyk, J. A. Lane, M. J. Derry and S. P. Armes, Chem. Sci., 2015, 6, 4207–4214 RSC.
- S. E. Taylor and H. Zeng, Colloids Interfaces, 2020, 4, 44 CrossRef CAS.
- N. Garti, Colloids Surf., A, 1997, 123–124, 233–246 CrossRef CAS.
- S. Zou, C. Wang, Q. Gao and Z. Tong, J. Dispersion Sci. Technol., 2013, 34, 173–181 CrossRef CAS.
- M. Williams, N. J. Warren, L. A. Fielding, S. P. Armes, P. Verstraete and J. Smets, ACS Appl. Mater. Interfaces, 2014, 6, 20919–20927 CrossRef CAS PubMed.
- M. Williams, S. P. Armes, P. Verstraete and J. Smets, Langmuir, 2014, 30, 2703–2711 CrossRef CAS PubMed.
- K. L. Thompson, C. J. Mable, J. A. Lane, M. J. Derry, L. A. Fielding and S. P. Armes, Langmuir, 2015, 31, 4137–4144 CrossRef CAS PubMed.
- Y. Nonomura, N. Kobayashi and N. Nakagawa, Langmuir, 2011, 27, 4557–4562 CrossRef CAS PubMed.
- L. Hong, G. Sun, J. Cai and T. Ngai, Langmuir, 2012, 28, 2332–2336 CrossRef CAS PubMed.
- F. Tu and D. Lee, Chem. Commun., 2014, 50, 15549–15552 RSC.
- P. S. Clegg, J. W. Tavacoli and P. J. Wilde, Soft Matter, 2016, 12, 998–1008 RSC.
- C. György, S. J. Hunter, C. Girou, M. J. Derry and S. P. Armes, Polym. Chem., 2020, 11, 4579–4590 RSC.
- B. P. Binks and C. P. Whitby, Langmuir, 2004, 20, 1130–1137 CrossRef CAS PubMed.
- A. J. Morse, S. P. Armes, K. L. Thompson, D. Dupin, L. A. Fielding, P. Mills and R. Swart, Langmuir, 2013, 29, 5446–5475 Search PubMed.
- C. J. Mable, N. J. Warren, K. L. Thompson, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2015, 6, 6179–6188 RSC.
- K. L. Thompson, C. J. Mable, A. Cockram, N. J. Warren, V. J. Cunningham, E. R. Jones, R. Verber and S. P. Armes, Soft Matter, 2014, 10, 8615–8626 RSC.
- S. A. F. Bon, S. D. Mookhoek, P. J. Colver, H. R. Fischer and S. van der Zwaag, Eur. Polym. J., 2007, 43, 4839–4842 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00572c |
| This journal is © The Royal Society of Chemistry 2021 |