
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterobimetallic Au(I)/Y(III) single chain nanoparticles as recyclable homogenous catalysts†
Josina L.
Bohlen‡
a,
Bragavie
Kulendran‡
a,
Hannah
Rothfuss
b,
Christopher
Barner-Kowollik
 *bc and
Peter W.
Roesky
*a
*bc and
Peter W.
Roesky
*a
aInstitute for Inorganic Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstrasse 15, 76131 Karlsruhe, Germany. E-mail: roesky@kit.edu
bInstitute of Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: christopher.barner-kowollik@kit.edu
cCentre for Materials Science, School of Chemistry and Physics, Queensland University of Technology (QUT), 2 George Street, Brisbane, QLD 4000, Australia. E-mail: christopher.barnerkowollik@qut.edu.au
First published on 28th June 2021
Abstract
Heterobimetallic single chain nanoparticles were synthesized and applied as recyclable homogenous catalysts. A terpolymer containing two orthogonal ligand moieties, phosphines and carboxylates, was obtained via nitroxide-mediated polymerization. Single chain nanoparticle (SCNP) formation is induced by selective metal complexation of Y(III) by the carboxylate functions, while Au(I) is selectively coordinated to phosphine moieties. In contrast to previous work, the two functionalities, SCNP folding and formation of a catalytic center, were distributed over two metals, which critically increases the flexibility of the system. The formation of Au(I)/Y(III)-SCNPs is evidenced by size exclusion chromatography, dynamic light scattering, nuclear magnetic resonance (1H, 31P{1H}) and infrared spectroscopy. Importantly, the activity of the Au(I)/Y(III)-SCNPs as homogenous, yet recyclable catalyst, bridging the gap between homogenous and heterogeneous catalysis, was demonstrated using the hydroamination of aminoalkynes as an example.
Introduction
In recent years, single chain nanoparticles (SCNPs), i.e. intramolecularly cross-linked single polymer chains,1–3 emerged as a highly prominent research topic due to their manifold emerging applications.1–17 In the reported systems, the intramolecular linkage was mostly realized by hydrogen- or covalent bonding, taking inspiration from biomolecules.17–19 More recently, however, metal–ligand coordination was introduced to form intramolecular crosslinks.1,7,20–28 The special feature of this binding strategy enables the simultaneous embedding of functional centers, e.g. for catalysis.6,7,28 One of the major challenges in catalysis is to bridge the gap between homogenous (high catalytic activities, but difficult catalyst recovery) and heterogeneous catalysis (easy recovery, but lower catalytic activities). We recently reported Pt(II)-SCNPs that enabled homogenous catalysis but were easily separated from the reaction mixture by change of solvent polarity, thus bridging the gap between homogenous and heterogeneous catalysis.7 To make the SCNPs even more versatile, the introduction of different metal species – though synthetically challenging – is desirable. This allows for the design of metal containing SCNPs with specific morphologies and functions, mimicking metalloenzymes.17,27 Initial progress was made by Terashima and coworkers10 in water and Lemcoff and colleagues in organic solvents,25 who crosslinked diene functionalized polymer chains with catalytically active Rh(I) and Ir(I) centers. However, as only one ligand type was present, no targeted metal placement was possible. With the synthesis of heterobimetallic Eu(III)/Pt(II)-SCNPs, our group pioneered the selective and controlled introduction of two metal species exhibiting different functionalities (phosphine and phosphine oxide) by selective coordination to orthogonal ligand moieties.28 Thereby, the catalytically active Pt(II) precursor induced the chain collapse, whereas the orthogonally introduced Eu(III) centers are luminescent and allow for the visualization and tracking of the SCNPs during catalysis. However, by using the catalytic active metal for chain collapse, the variation of metals and the catalytic scope are very limited. For a higher flexibility in terms of catalytic applications, we are therefore interested in bimetallic SCNPs with one metal forming the structure, while the other one is involved in the catalytic conversion. Moreover, we were challenged to use a rare earth element to induce the chain collapse to broaden the method of metal induced SCNP formation significantly.29 Rare earth ions significantly differ in their synthetic chemistry from main group and transition metals by their higher coordination numbers, which are a result of their larger ion radii. While most transition metal complexes have coordination numbers of 4–6, coordination numbers of 7–9 or even higher are frequently seen for rare earth metals. This property may facilitate the chain collapse of metal functionalized SCNPs in the presence of several multidentate ligands. Moreover, rare earth elements feature unique optical and magnetic properties, which can be potentially explored once the polymer SCNP synthesis is established. While the rare earth ions selectively coordinate to a hard donor, we choose Au(I) for the selective binding to a soft donor. As result of this strategy, orthogonal coordination is possible. Due to the linear coordination of Au(I), only one donor is needed for anchoring the metal. Thus, no further compaction of the polymer is required. Therefore, we avoid any strain in the system by coordination of the second metal and clearly assign the folding to the rare earth ion.As result, we herein present the formation of heterobimetallic Au(I)/Y(III)-SCNPs and their application as catalysts. We demonstrate that selective embedding of any two metal species and thus fine-tuning of the resulting SCNP properties is possible.
A linear polymer precursor served as a basis for the targeted heterobimetallic SCNPs. In a terpolymer containing both carboxylate and phosphine units, the hard carboxylates enable the selective complexation of Y(III) centers, whereas the soft phosphine units coordinate to the catalytically active Au(I) ions. The Y(III) atoms thus act as structure forming units inducing the SCNP formation, while the two-fold coordinated Au(I) atoms are the catalytically active centers.
Results and discussion
Polymer synthesis
To synthesize a suitable terpolymer following the “repeat unit approach”,2,30 the monomers 4-vinylbenzoic acid, diphenyl(4-vinylphenyl)-phosphine and styrene, which functions as a spacer, were copolymerized via nitroxide-mediated polymerization (NMP) yielding the polymer P1 (Scheme 1), which can easily be separated by filtration. The monomers are distributed statistically along the polymer chain. The exact monomer composition was determined by 1H and 31P{1H} NMR spectroscopy (in THF–d8), comparing the resonance integrals of the respective monomer species (ESI, Fig. S15†). The terpolymer contains approximately 3% of phosphine units (0.27 mmol per 1.00 g of polymer) and 6% of benzoic acid units (0.57 mmol per 1.00 g of polymer). In the 1H NMR spectrum a broad singulet at δ = 11.2 ppm is seen, which is assigned to the acidic protons of the benzoic acid units. The ratio of the functional groups was adjusted by the stoichiometric ratio of the precursors.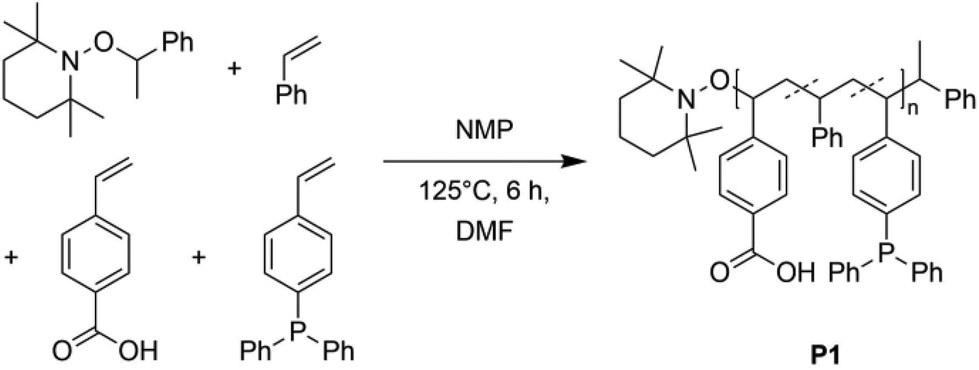 | ||
| Scheme 1 Synthesis of the terpolymer P1 by nitroxide-mediated radical polymerization of styrene, 4-vinylbenzoic acid and diphenyl(4-vinylphenyl)phosphine. The alkoxyamine was used as initiating material. | ||
In the 31P{1H} NMR spectrum a resonance at δ = −6.24 ppm, attributed to the phosphine units, is observed. The chemical shift is in accordance with literature values for similar triarylphosphine functionalized copolymers,7,31 confirming the successful implementation of the functional units into the terpolymer P1. Size exclusion chromatography (SEC) analysis [THF, RI] of P1 indicates a number average molecular weight Mn of close to 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 g mol−1 and a dispersity of Đ = 1.1. Combining the NMR and SEC results, 22 benzoic acid units and 11 phosphine units are estimated per polymer chain (see ESI, p. S4 and Fig. S14†).
500 g mol−1 and a dispersity of Đ = 1.1. Combining the NMR and SEC results, 22 benzoic acid units and 11 phosphine units are estimated per polymer chain (see ESI, p. S4 and Fig. S14†).
To evidence the suitability of our system for catalytic applications first, P1 was functionalized with a gold precursor, without forming SCNPs. Using [AuCl(tht)] (tht = tetrahydothiophene) as suitable Au(I) reagent, the phosphine moieties of terpolymer P1 were functionalized by Au(I) complexation, yielding the metallopolymer P1–Au(I). The successful and quantitative coordination of AuCl was proved by 31P{1H} NMR spectroscopy (in THF-d8). In the respective spectrum one resonance at δ = 31.6 ppm is observed and is attributed to the phosphine-gold(I) moieties.32 Compared to the resonance of the phosphine units in P1 (δ = −6.24 ppm), this is a significant downfield shift. 1H NMR spectroscopy further confirms the selective coordination of AuCl to the phosphor atoms (Fig. 1). The 1H NMR spectrum still contains a broad singulet at δ = 11.0 ppm, attributed to the acidic protons of the benzoic acid units. Furthermore, a downfield shift is only observed for the resonance of the aromatic phosphine unit protons. In addition, the metallopolymer P1-Au(I) was analysed by IR spectroscopy (ESI, Fig. S28†). Among others, a characteristic band attributed to the P–C vibrational stretching is observed. Compared to the free polymer, this band is shifted from ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 1090 cm−1 for free phosphine to 1102 cm−1 for the Au(I) complexed phosphine moieties.33 In the IR spectra of P1 as well as P1-Au(I) bands attributed to the C
= 1090 cm−1 for free phosphine to 1102 cm−1 for the Au(I) complexed phosphine moieties.33 In the IR spectra of P1 as well as P1-Au(I) bands attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrational stretching are observed at = 1660–1750 cm−1, confirming the presence of free carboxylic acid groups.34 SEC measurements [THF, RI] indicated a molecular weight of close to Mn = 39000 g mol−1 and a dispersity of Đ = 1.2. As the incorporation of AuCl leads to slightly heavier and larger particles, the SEC elugram of P1-Au(I) is shifted towards shorter retention times compared to P1 (Fig. 2).
O vibrational stretching are observed at = 1660–1750 cm−1, confirming the presence of free carboxylic acid groups.34 SEC measurements [THF, RI] indicated a molecular weight of close to Mn = 39000 g mol−1 and a dispersity of Đ = 1.2. As the incorporation of AuCl leads to slightly heavier and larger particles, the SEC elugram of P1-Au(I) is shifted towards shorter retention times compared to P1 (Fig. 2).
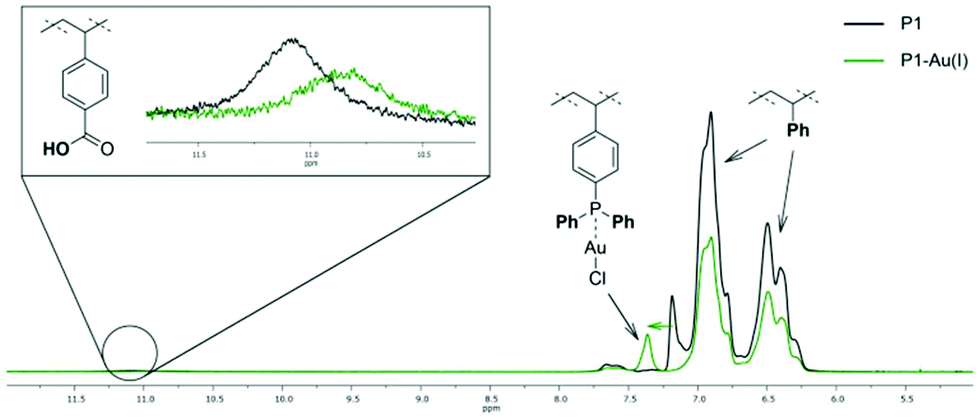 | ||
| Fig. 1 Superimposed 1H NMR spectra of P1 (black) and P1-Au(I) (green) (not normalized). Due to the coordination of AuCl, the resonances of the aromatic protons are shifted towards higher values of δ (lower field). The broad singulet attributed to the acidic protons of the benzoic acid units is visible for both, P1 and P1-Au(I), respectively, proving that AuCl selectively coordinates to the phosphine units. | ||
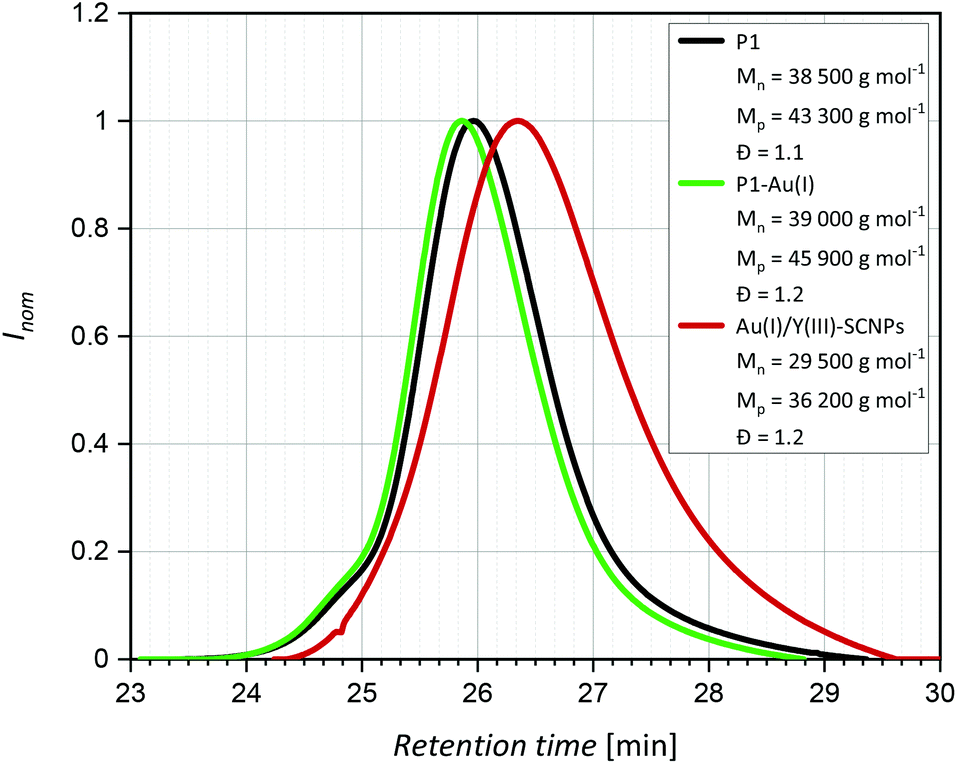 | ||
| Fig. 2 SEC-traces of P1, P1-Au(I) and Au(I)/Y(III)-SCNPs. Showing the decrease of the hydrodynamic radius, the trace of the Au(I)/Y(III) SCNPs is significantly shifted towards longer retention times compared to P1 and P1-Au(I). | ||
In a similar fashion to P1-Au(I), we prepared the metallopolymer P2-Au(I), which does not contain any benzoic acid units, establishing a benchmark for the catalytic activity of the target SCNPs. In a first step, the copolymer P2 was synthesized via NMP of styrene and diphenyl(4 vinylphenyl)phosphine (monomer ratio of 23:1), the phosphine units are thereby statistically distributed along the polymer chain. SEC analysis [THF, RI] indicates a molecular weight of approximately Mn = 34000 g mol−1 and a dispersity of Đ = 1.2 The successful incorporation of the phosphine units was proved by 31P{1H} NMR spectroscopy (ESI, Fig. S19†). By 1H NMR spectroscopy an amount of 5% phosphine units was determined (0.51 mmol per 1.00 g of polymer). P2 was converted to the desired metallopolymer P2-Au(I) using the Au(I) precursor [AuCl(tht)]. The successful coordination of AuCl to the phosphine units was verified with SEC [THF, RI], 31P{1H} NMR and IR spectroscopy. The metallopolymer showed a molecular weight of close to Mn = 35100 g mol−1 and a dispersity of Đ = 1.2.
Synthesis of Au(I)/Y(III)-SNCPs
The free benzoic acid units of P1-Au(I) enable the incorporation of a second metal species. Coordination of several benzoate units to one metal atom allows for an intramolecular crosslink of the polymer chain. The intramolecular collapse of P1-Au(I) was induced by addition of [Y(NO3)3]·6 H2O as precursor for the diamagnetic rare earth ion Y(III) (Scheme 2). To confirm the folding of the polymer chain into well-defined SCNPs, SEC analysis [THF, RI] was carried out. Compared to the SEC traces of the polymer P1 and the metallopolymer P1-Au(I), the trace of the Au(I)/Y(III)-SCNPs (Mn = 29500 g mol−1, Đ = 1.2) is significantly shifted towards longer retention times (Fig. 2). This indicates a strongly decreased hydrodynamic radius and thus the exclusive formation of the SCNPs without intermolecular crosslinks. Although we cannot determine the exact composition of the Y(III) complex, we suggest that its structure is similar to Y(III)-benzoate complexes, which have been synthesized and characterized by Deacon and colleagues.35 It was found that the majority of these complexes exist as dimeric structures, in which the Y(III) ions are coordinated by at least two bidentate bridging benzoate ligands. Moreover, in some Y(III)-benzoate complexes, multiple coordination modes occurred simultaneously. Therefore, the benzoic acid moieties in the Au(I)/Y(III)-SCNPs are thought to coordinate to the Y(III) ions in a similar manner, which accounts for the intramolecular chain collapse of the polymer chains.
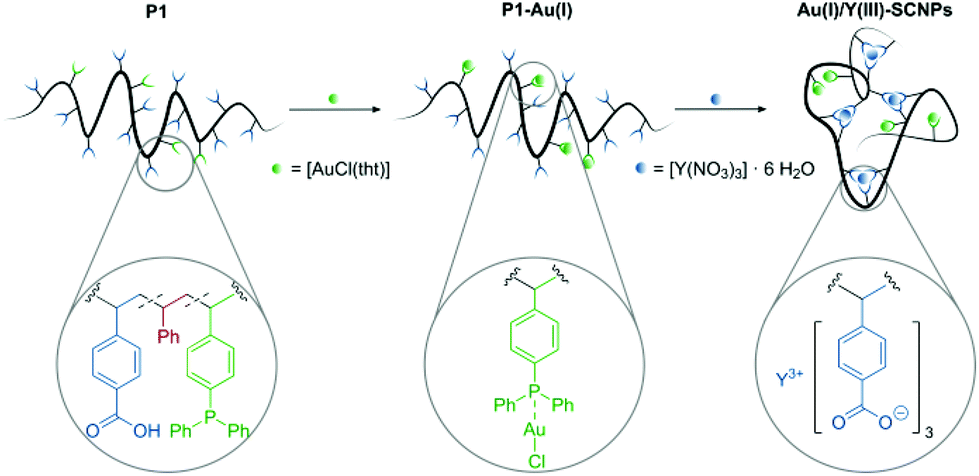 | ||
| Scheme 2 Au(I) functionalization of P1 using [AuCl(tht)] and subsequent folding of the obtained metallopolymer P1-Au(I) with [Y(NO3)3]·6 H2O. The metal species coordinate orthogonally to the respective ligands in the polymer, yielding well-defined catalytically active Au(I)/Y(III)-SCNPs. | ||
The SCNPs were further investigated by 1H and 31P{1H} NMR spectroscopy (in THF-d8) and IR spectroscopy. The 1H NMR spectrum does not show a broad singulet at δ = 11.0 ppm, attributed to the acidic protons of the benzoic acid units, proving a quantitative complexation with Y(III) ions.
The resonances at δ = 6.80 and 6.40 ppm, attributed to the phosphine moieties, are still present. In the 31P{1H} NMR spectrum of the SCNPs, the resonance of the phosphine AuCl units remains almost unchanged (δ = 31.6 ppm in P1-Au(I)vs. 32.4 ppm in the SCNPs), further confirming the intact AuCl coordination. In the IR spectra (ESI, Fig. S27 and S28†), the bands of the CO vibrational at = 1660 and 1750 cm−1 attributed to the free carboxylic acid units, are not detected anymore. Instead, two new bands at a = 1514 and s = 1417 cm−1 are observed, consistent with literature values of yttrium(III) benzoate complexes, which demonstrates the carboxylate complexation of the Y(III) ions.36,37 The unchanged νP–C band at = 1103 cm−1 further proves the [ArPPh2P-AuCl] moieties to be intact.
Catalytic application
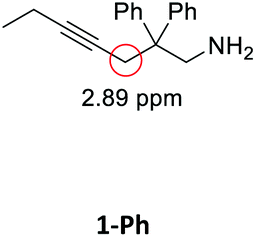
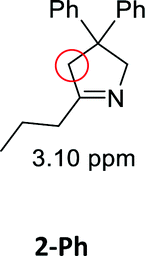
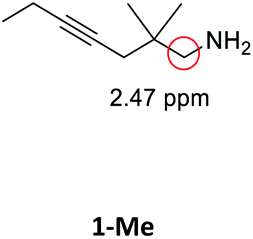
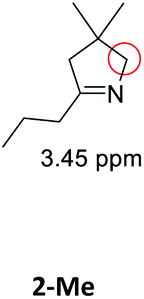
Having synthesized the SCNPs, we investigated them as recyclable homogenous catalysts in the intramolecular hydroamination of aminoalkynes. Although there are many established synthetic procedures towards nitrogen-containing heterocycles, hydroamination reactions are considered as atom economical and green because neither waste nor any side products are formed and have therefore been intensely studied.38 This makes this transformation especially valuable as benchmark reaction for recyclable catalysts. For this benchmark reaction we required a simple homogenous catalyst with a coordination sphere, which is very similar to our polymer supported catalysts for comparison. As Au(I) complexes are known to transform alkynes efficiently, we selected the hydroamination of aminoalkynes as benchmark reaction and [AuCl(PPh3)] as benchmark catalyst.The molecular catalyst [AuCl(PPh3)] and the polymeric catalyst P2-Au(I) were used to determine suitable reaction conditions and function as benchmark system for the catalytic activity of the Au(I)/Y(III)-SCNPs (ESI, Fig. S5–S7†). Different cocatalysts were tested and blind tests conducted. NaBArF was chosen as cocatalyst for all catalyses, not showing any catalytic activity in the performed tests (ESI, pp S12–14†). We investigated the hydroamination of two selected substrates, which were synthesized by variations of literature procedures (1-Ph;391-Me40) and dried and degassed prior to usage. The catalyses were carried out under inert conditions in CDCl3. 2 mol% Au(I) catalyst (ccat = 2.6 μM), 2 mol% of NaBArF as cocatalyst, and 10 mol% of ferrocene (internal standard) were dissolved in dry CDCl3 at 20 °C. After 30 min (to allow for the activation of the catalyst), the respective hydroamination substrate 1-Ph or 1-Me was added. This mixture was thawed right before the insertion into the NMR machine. To monitor substrate conversion, 1H NMR spectra were recorded at preselected times. Under these mild reaction conditions, all substrates were quantitively converted into the respective heterocyclic products. Conversion rates for each substrate and catalyst are summarized in Table 1.
| Substrate | Product | Catalyst | Time [h] | Conv.b [%] |
|---|---|---|---|---|
| a Conditions: 1.00 eq. substrate, 2 mol% Au(I)-catalyst, 2 mol% NaBArF, 10 mol% ferrocene, CDCl3, 20 °C. b Calculated by 1H NMR spectroscopy with ferrocene as internal standard. | ||||
|
|
[AuCl(PPh3)] | 4 | 98 |
| 24 | 100 | |||
| P2-Au(I) | 4 | 98 | ||
| 24 | 100 | |||
| Au(I)/Y(III)-SCNPs | 4 | 96 | ||
| 24 | 100 | |||
|
|
[AuCl(PPh3)] | 4 | 58 |
| 24 | 87 | |||
| P2-Au(I) | 4 | 83 | ||
| 24 | 98 | |||
| Au(I)/Y(III)-SCNPs | 4 | 41 | ||
| 24 | 78 | |||
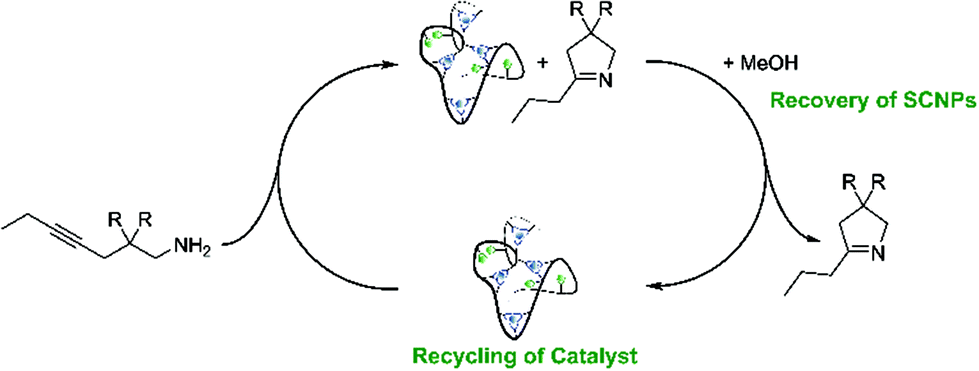
As can be seen from the differing conversion rates, the Thorpe Ingold effect has a strong influence on the reactivity of the substrates:41 Whereas for 1-Ph (high steric demand in the backbone) quantitative conversion is reached with all catalysts under mild conditions and after comparably short reaction times, the conversion of 1-Me proceeds significantly slower. Compared to the benchmark systems [AuCl(PPh3)] and P2-Au(I), a slightly decreased catalytic activity was observed for the Au(I)/Y(III)-SCNPs. However, the SCNPs proved to be active homogenous catalysts for the intramolecular hydroamination, yielding conversions of up to 100% in only 10 h (ESI, Fig. S8 and S9†). The chosen reaction conditions enable the recovery of the Au(I)/Y(III)-SCNPs after catalysis by applying dialysis in methanol (Scheme 3). We investigated the structure of the post-catalytic SCNPs by SEC, IR spectroscopy and NMR spectroscopy (Fig. 3). The SEC traces of the nanoparticles before and after catalysis match closely, indicating that the folded structure is maintained. In the 31P{1H} NMR spectrum one resonance at δ = 32.3 ppm attributed to the phosphine AuCl units is observed. No resonance for free phosphine units was observed, pointing to the ongoing quantitative coordination of AuCl.
 | ||
| Scheme 3 The Au(I)/Y(III) SCNPs were successfully recovered and recycled as catalysts, closing the gap between homogeneous and heterogeneous catalysis. | ||
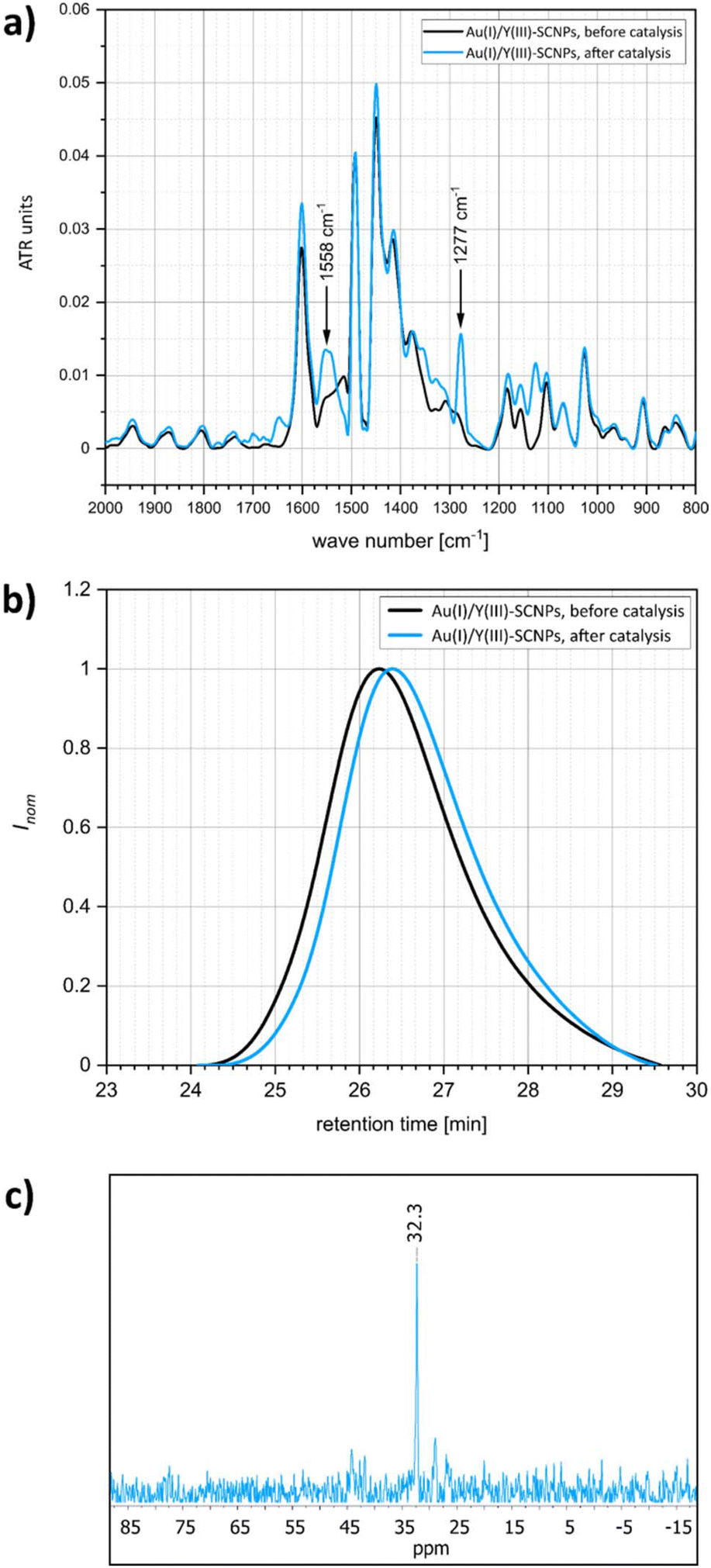 | ||
| Fig. 3 Analytics of the Au(I)/Y(III)-SCNPs before catalysis (black) and after catalysis (blue). (a) Superimposed IR spectra. (b) SEC traces. (c) 31P{1H} spectrum of the SCNPs after catalysis. | ||
The almost identical IR spectra are further evidence for intact folding units. Two additional bands are observed at = 1558 cm−1 and at = 1277 cm−1, which are possibly caused by complexed substrate molecules (Fig. 3a).
As we demonstrate that the Au(I)/Y(III)-SCNPs are intact after recovery, we subjected them to a second catalytic cycle, in order to investigate their reusability (ESI, Fig. S10 and S11†). For the catalysis, the same procedure as described above was followed. Comparison of the conversions reached with new and with recovered Au(I)/Y(III)-SCNPs, i.e. in the first and second catalysis cycle, shows a slight decrease in the catalytic activity (Fig. 4). The highest conversion observed with the recovered Au(I)/Y(III)-SCNPs after 24 h was 95%. A possible explanation for this observation is the decreased solubility, which leads to an effective catalyst concentration lower than 2 mol%. Furthermore, from the additional bands observed in the IR spectra, we suspect complexed substrate molecules in the recovered Au(I)/Y(III)-SCNPs. In this case, some of the functional units of the Au(I)/Y(III)-SCNPs would remain inaccessible.
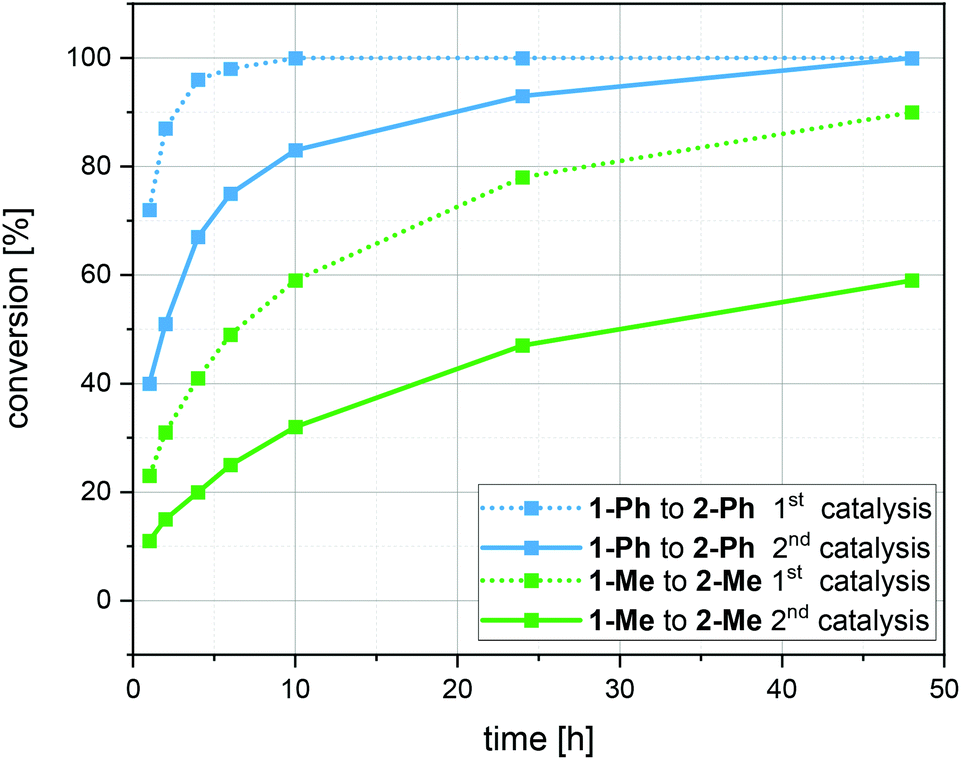 | ||
| Fig. 4 Comparison of the conversions reached with the Au(I)/Y(III) SCNPs in the 1st and 2nd catalysis cycles (first catalysis dashed lines; second catalysis solid lines) for each substrate (1 Ph (blue), 1 Me (green)). | ||
Kinetic measurements were performed for substrate 1-Me, which features the slowest conversion rates. A first order of the reaction in the starting material (excluding a short induction period) was determined (ESI, Fig. S14†).42
Conclusions
In summary, we introduce the synthesis and in-depth characterization of well-defined heterobimetallic Au(I)/Y(III)-SCNPs. In contrast to previous system, the catalytic active metal is not involved in the SCNP formation, which potentially allows more flexibility in exchanging this metal. The formation of the nanoparticles was confirmed by SEC, NMR spectroscopy (1H, 31P{1H}) and IR spectroscopy and proved to be selective in metal coordination. Featuring catalytic centres, the Au(I)/Y(III)-SCNPs were found to be an active homogeneous catalyst for the hydroamination of aminoalkynes. Additionally, recovery of the nanoparticles and application in a second catalysis cycle was possible. Thus, the SCNPs represent a catalyst system that is combining the advantages of homogenous and heterogeneous catalysis. Furthermore, we show that any two metal species with different functionalities (that are e.g. catalytically active or luminescent) can be incorporated orthogonally, yielding highly variable and versatile SCNPs and thus manifold potential applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr. N. Knöfel is acknowledged for helpful discussions. C.B.-K. acknowledges funding by the Australian Research Council (ARC) in the form of a Laureate Fellowship as well as the Queensland University of Technology (QUT).References
- A. Sanchez-Sanchez, A. Arbe, J. Colmenero and J. A. Pomposo, ACS Macro Lett., 2014, 3, 439–443 CrossRef CAS.
- O. Altintas, T. S. Fischer and C. Barner-Kowollik, in Single–Chain Polymer Nanoparticles, 2017, pp. 1–45, DOI:10.1002/9783527806386.ch1.
- E. Blasco, B. T. Tuten, H. Frisch, A. Lederer and C. Barner-Kowollik, Polym. Chem., 2017, 8, 5845–5851 RSC.
- J. He, L. Tremblay, S. Lacelle and Y. Zhao, Soft Matter, 2011, 7, 2380–2386 RSC.
- I. Perez-Baena, I. Loinaz, D. Padro, I. García, H. J. Grande and I. Odriozola, J. Mater. Chem., 2010, 20, 6916–6922 RSC.
- H. Rothfuss, N. D. Knöfel, P. W. Roesky and C. Barner-Kowollik, J. Am. Chem. Soc., 2018, 140, 5875–5881 CrossRef CAS PubMed.
- N. D. Knöfel, H. Rothfuss, J. Willenbacher, C. Barner-Kowollik and P. W. Roesky, Angew. Chem., Int. Ed., 2017, 56, 4950–4954 CrossRef.
- J. Rubio-Cervilla, E. González and J. A. Pomposo, Nanomaterials, 2017, 7, 341 CrossRef.
- M. Artar, T. Terashima, M. Sawamoto, E. W. Meijer and A. R. A. Palmans, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 12–20 CrossRef CAS.
- T. Terashima, T. Mes, T. F. A. De Greef, M. A. J. Gillissen, P. Besenius, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2011, 133, 4742–4745 CrossRef CAS PubMed.
- Y. Bai, X. Feng, H. Xing, Y. Xu, B. K. Kim, N. Baig, T. Zhou, A. A. Gewirth, Y. Lu, E. Oldfield and S. C. Zimmerman, J. Am. Chem. Soc., 2016, 138, 11077–11080 CrossRef CAS.
- M. A. J. Gillissen, I. K. Voets, E. W. Meijer and A. R. A. Palmans, Polym. Chem., 2012, 3, 3166–3174 RSC.
- A. Sanchez-Sanchez, S. Akbari, A. Etxeberria, A. Arbe, U. Gasser, A. J. Moreno, J. Colmenero and J. A. Pomposo, ACS Macro Lett., 2013, 2, 491–495 CrossRef CAS.
- R. J. Passarella, D. E. Spratt, A. E. van der Ende, J. G. Phillips, H. Wu, V. Sathiyakumar, L. Zhou, D. E. Hallahan, E. Harth and R. Diaz, Cancer Res., 2010, 70, 4550 CrossRef CAS PubMed.
- G. Hariri, A. D. Edwards, T. B. Merrill, J. M. Greenbaum, A. E. van der Ende and E. Harth, Mol. Pharm., 2014, 11, 265–275 CrossRef CAS PubMed.
- A. P. P. Kröger, N. M. Hamelmann, A. Juan, S. Lindhoud and J. M. J. Paulusse, ACS Appl. Mater. Interfaces, 2018, 10, 30946–30951 CrossRef PubMed.
- A. Latorre-Sánchez and J. A. Pomposo, Polym. Int., 2016, 65, 855–860 CrossRef.
- B. T. Tuten, D. Chao, C. K. Lyon and E. B. Berda, Polym. Chem., 2012, 3, 3068–3071 RSC.
- N. Hosono, M. A. J. Gillissen, Y. Li, S. S. Sheiko, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2013, 135, 501–510 CrossRef CAS.
- S. Mavila, C. E. Diesendruck, S. Linde, L. Amir, R. Shikler and N. G. Lemcoff, Angew. Chem., Int. Ed., 2013, 52, 5767–5770 CrossRef CAS PubMed.
- S. Basasoro, M. Gonzalez-Burgos, A. J. Moreno, F. L. Verso, A. Arbe, J. Colmenero and J. A. Pomposo, Macromol. Rapid Commun., 2016, 37, 1060–1065 CrossRef CAS PubMed.
- R. Lambert, A.-L. Wirotius, S. Garmendia, P. Berto, J. Vignolle and D. Taton, Polym. Chem., 2018, 9, 3199–3204 RSC.
- J. Willenbacher, O. Altintas, V. Trouillet, N. Knöfel, M. J. Monteiro, P. W. Roesky and C. Barner-Kowollik, Polym. Chem., 2015, 6, 4358–4365 RSC.
- Z. Zhu, N. Xu, Q. Yu, L. Guo, H. Cao, X. Lu and Y. Cai, Macromol. Rapid Commun., 2015, 36, 1521–1527 CrossRef CAS PubMed.
- S. Mavila, I. Rozenberg and N. G. Lemcoff, Chem. Sci., 2014, 5, 4196–4203 RSC.
- K. Freytag, S. Säfken, K. Wolter, J. C. Namyslo and E. G. Hübner, Polym. Chem., 2017, 8, 7546–7558 RSC.
- W. Wang, J. Wang, S. Li, C. Li, R. Tan and D. Yin, Green Chem., 2020, 22, 4645–4655 RSC.
- N. D. Knöfel, H. Rothfuss, P. Tzvetkova, B. Kulendran, C. Barner-Kowollik and P. W. Roesky, Chem. Sci., 2020, 11, 10331–10336 RSC.
- L. N. Neumann, D. A. Urban, P. Lemal, S. Ramani, A. Petri-Fink, S. Balog, C. Weder and S. Schrettl, Polym. Chem., 2020, 11, 586–592 RSC.
- O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2016, 37, 29–46 CrossRef CAS.
- J. R. Van Wazer, C. F. Callis, J. N. Shoolery and R. C. Jones, J. Am. Chem. Soc., 1956, 78, 5715–5726 CrossRef CAS.
- C. Nieto-Oberhuber, S. López and A. M. Echavarren, J. Am. Chem. Soc., 2005, 127, 6178–6179 CrossRef CAS PubMed.
- R. Faggiani, H. E. Howard-Lock, C. J. L. Lock and M. A. Turner, Can. J. Chem., 1987, 65, 1568–1575 CrossRef CAS.
- H. Günzler and H.-U. Gremlich, in IR–Spektroskopie, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2003, vol. 4, pp. 157–264 Search PubMed.
- G. B. Deacon, S. Hein, P. C. Junk, T. Jüstel, W. Lee and D. R. Turner, CrystEngComm, 2007, 9, 1110–1123 RSC.
- J. R. Locatelli, E. C. Rodrigues, A. B. Siqueira, E. Y. Ionashiro, G. Bannach and M. Ionashiro, J. Therm. Anal. Calorim., 2007, 90, 737–746 CrossRef CAS.
- M. Iwan, A. Kula, Z. Rzączyńska, S. Pikus, D. Flisiuk and M. Gomoła, Chem. Pap., 2007, 61, 376–382 CAS.
- R. A. Widenhoefer and X. Han, Eur. J. Org. Chem., 2006, 4555–4563 CrossRef CAS.
- J. Luo, Q. Cao, X. Cao and X. Zhao, Nat. Commun., 2018, 9, 527 CrossRef.
- M. Oishi, Y. Nakanishi and H. Suzuki, Inorg. Chem., 2017, 56, 9802–9813 CrossRef CAS PubMed.
- R. M. Beesley, C. K. Ingold and J. F. Thorpe, J. Chem. Soc., Trans., 1915, 107, 1080–1106 RSC.
- J. I. Steinfeld, J. S. Francisco and C. K. a. D. L. Hase, Chemical Kinetics and Dynamics, Pearson, Pearson, 1998, vol. 2, pp. 7–8., 1998 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00552a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |