
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemically-initiated polymerization of reactive monomers via 4-fluorobenzenediazonium salts†
Edgar
Molle
 ab,
Stefan
Frech
ab,
Tilman
Grüger
a and
Patrick
Theato
*ab
ab,
Stefan
Frech
ab,
Tilman
Grüger
a and
Patrick
Theato
*ab
aInstitute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76131 Karlsruhe, Germany. E-mail: patrick.theato@kit.edu
bInstitute for Biological Interfaces III (IBG-3), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 23rd September 2021
Abstract
The straightforward electrochemically-initiated radical polymerization of three reactive monomers, i.e. 2,6-difluorophenyl acrylate (DFPA), pentafluorophenyl acrylate (PFPA), and glycidyl methacrylate (GMA), using a simple commercially available electrochemical setup for the generation of initiating radical species from 4-fluorobenzenediazonium tetrafluoroborate upon electrochemical cathodic reduction is reported. The use of a fluorine-labelled initiator allowed for the determination of number-average molar masses Mn of fluorine-containing polymers by 19F NMR spectroscopy in addition to size-exclusion chromatography. The obtained polymers were thereafter successfully subjected to post-polymerization modifications in order to emphasize the applicability of this polymerization method in regard to highly reactive monomers.
Introduction
Polymeric materials are predominant in today's life and living without them seems almost unimaginable. Due to their prominent role in the 21st century, polymers have been excessively explored and different polymerization techniques have been established to meet this demand. Among the great variety of polymerization methods, radical polymerization, especially free radical polymerization (FRP), stands out because of its ease of handling1 and its robustness. From an industrial viewpoint, polymers obtained by radical polymerization roughly made up 40 to 45 percent of all industrially produced polymers in 2012.2 Thus, radical polymerization represents a main pillar of industrial production of polymers. Nonetheless, radical polymerizations are not only of interest for industrial production, but are also of scientific interest in academic research. For instance, different initiation methods have been reported, such as ultrasound-initiated polymerizations3,4 or photo-initiated polymerizations,5,6 the latter being extensively studied and finding application in dental medicine for instance.7,8 Additionally, reversible-deactivation radical polymerization (RDRP)9 techniques have been established as powerful polymerization toolbox, allowing for the preparation of well-defined macromolecules from a broad range of vinyl monomers featuring high end group fidelity, low dispersities, predictable molar masses, and different end groups attached to the polymers for further functionalization, depending on the polymerization method employed.10 Reversible addition–fragmentation chain transfer (RAFT)11–13 polymerization, atom-transfer polymerization (ATRP),14,15 and nitroxide-mediated polymerization (NMP)16 are the most prominent examples among the RDRP methods. Unlike ATRP and NMP, the RAFT process relies on degenerate chain transfer, resulting in enhanced polymerization rates compared to its contenders, which are based on equilibria between active and dormant species. RDRP methods have been excessively explored, electrochemical approaches were utilized among others in RAFT polymerization17–20 and ATRP (e.g. eATRP).21–25 Moreover, the latter has been realized using the same simple and commercially available undivided cell setup from IKA as employed in this report, enabling the plug-and-play aqueous eATRP.26Synthetic (organic) electrochemistry27–32 is a growing field attracting increasing interest as a powerful and impactful methodological tool for the clean and sustainable preparation of a great diversity of chemical compounds.28 Ideally, electrons themselves serve as reagents, thereby overcoming the necessity to add chemical reagents and/or catalysts to the reaction, which might be difficult to remove afterwards. Moreover, the necessary potentiostat can be operated in a climate-neutral fashion, making synthetic (organic) electrochemistry an important tool for the environmentally friendly synthesis of compounds and materials, especially in times of raw material shortage. The direct use of electrons has already aroused scientific interest in early times and led to a variety of reactions, naming the Kolbe electrolysis as one of the first reactions involving synthetic electrochemistry29 reported in 1834 by Faraday33 and further explored by Kolbe in 1849.34
Besides common polymerization initiations, electrochemistry has been employed due the ability to start polymerizations at the push of a button. On the one hand, electropolymerizations of different kinds of monomers (e.g. thiophene35 and pyrrole,36 but also styrene37 and acrylonitrile38) have been reported by direct oxidation or reduction of the monomers. On the other hand, electrochemically-mediated polymerizations use mediating moieties, which form inter alia species being able to initiate a polymerization upon application of a potential, as in the case of the initiation of a RAFT polymerization by reduction of an aromatic diazonium salt.17
Also, functional polymers, especially based on poly(acrylates), are of scientific and industrial interest. For instance, active ester polymers, derived from the respective acrylate monomer structures among others, allow for an efficient and straightforward functionalization via post-polymerization modification (PPM).39 Only recently, the direct transesterification of poly(methyl acrylate) towards functional polyacrylates was shown.40 Furthermore, the full and partial amidation of poly(methyl acrylate) was reported as basis for functional polyacrylamide (co)polymers.41
Herein, we report on the free radical polymerization (FRP) of 2,6-difluorophenyl acrylate (DFPA), pentafluorophenyl acrylate (PFPA), and glycidyl methacrylate (GMA) by electrochemical reduction of 4-fluorobenzenediazonium tetrafluoroborate as fluorine-labelled initiator using a simple, fully commercially available electrochemical setup, and the subsequent PPM of the obtained functional polymers with a reactive amine featuring another fluorine-label (Scheme 1).
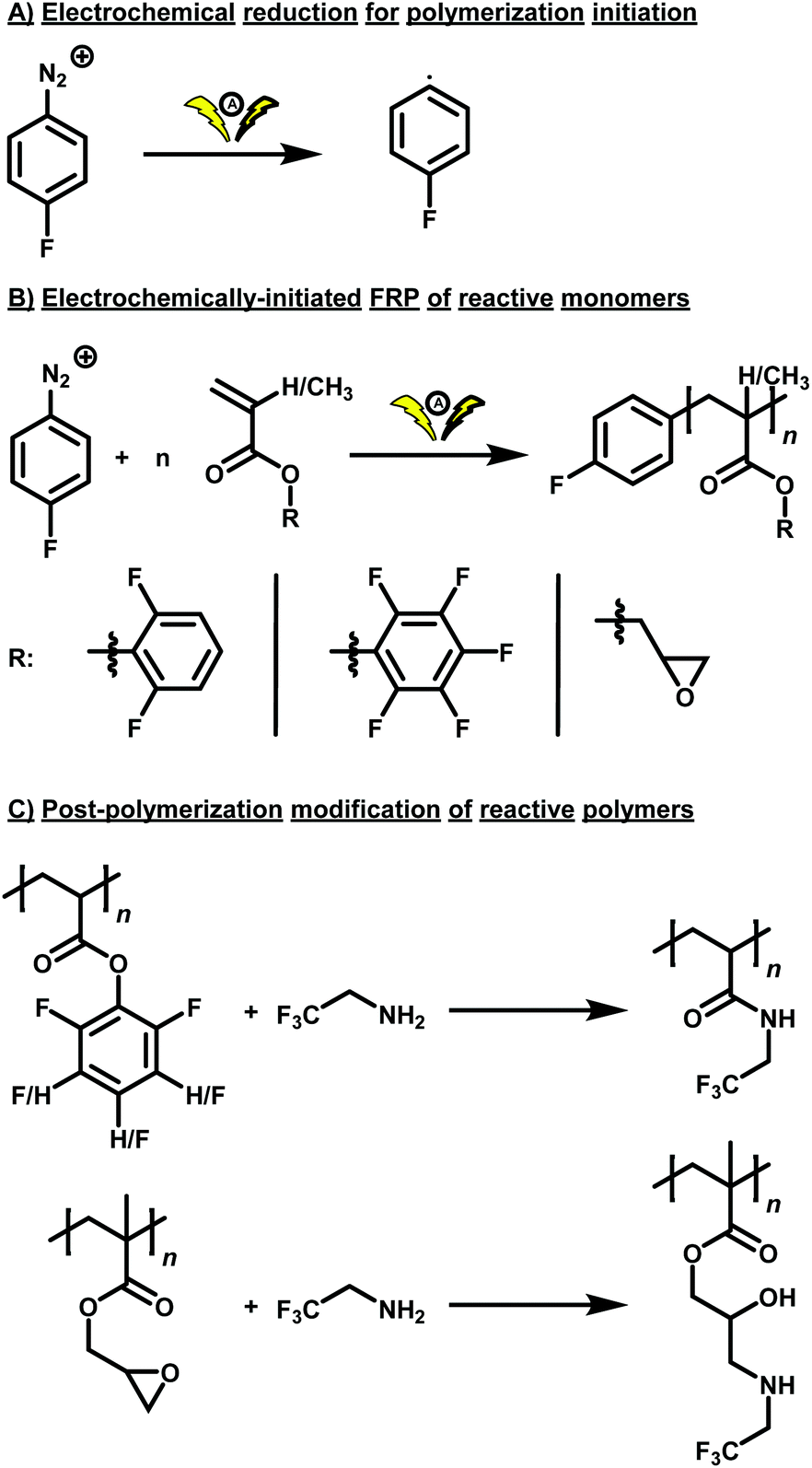 | ||
| Scheme 1 Schematic of the electrochemical reduction of a fluorine-labelled aromatic diazonium salt ((A)) for the FRP of DFPA, PFPA, and GMA as reactive monomers ((B)). The obtained polymers were subjected to PPM reactions using a fluorine-labelled amine for the preparation of polyamides and poly(β–amino alcohols) ((C)). | ||
Experimental
Materials
Glycidyl methacrylate (Sigma Aldrich, 97%) was passed through basic aluminum oxide prior to use. Acetone (AnalaR NORMAPUR), acryloyl chloride (Alfa Aesar, 96%), cyclohexane (AnalaR NORMAPUR), diethyl ether (Fisher, ≥99%), ethyl acetate (AnalaR NORMAPUR), 2,6-difluorophenol (Sigma Aldrich, 98%), N,N-dimethylformamide (Acros Organics, 99.8%, anhydrous), 4-fluoroaniline (Sigma Aldrich, 99%), magnesium sulfate (Roth, ≥99%), methanol (AnalaR NORMAPUR), pentafluorophenol (abcr, 99%), petroleum ether (AnalaR NORMAPUR), sodium nitrite (Honeywell, ≥99%), tetrabutylammonium tetrafluoroborate (Sigma Aldrich, 99%), tetrafluoroboric acid (Sigma Aldrich, 48 wt%), tetrahydrofuran (Acros Organics, 99.5%, anhydrous), tetrahydrofuran (AnalaR NORMAPUR), triethylamine (Acros Organics, 99%), and 2,2,2-trifluoroethylamine (TCI, >97%) were used as received.Characterization
1H NMR, 13C NMR, and 19F NMR spectra were recorded on a Bruker Ascend III 400 MHz spectrometer at a frequency of ν = 400 MHz, ν = 101 MHz, and ν = 377 MHz, respectively. All samples were dissolved in deuterated solvents, chemical shifts are reported relative to the residual solvent signals.SEC measurements with THF as eluent were performed on an Agilent 1200 Series System, comprising an autosampler, a differential Refractive Index (RI) detector, three PLgel 5 μm Mixed C columns (300 × 7.5 mm) and a PLgel 3 μm Mixed E column (300 × 7.5 mm). The measurements were performed at a temperature of T = 35 °C and a flow rate of 1.0 mL min−1. Samples were measured at a concentration of 2 mg mL−1 and filtered prior to measurement. All number-average molar mass Mn and dispersity Đ values were extrapolated from a range of linear poly(methyl methacrylate) standards between 800 and 2.2 × 106 g mol−1 and linear polystyrene standards between 370 and 2.52 × 106 g mol−1.
SEC measurements with DMAc as eluent were performed on an Agilent 1200 Series System, comprising an autosampler, a differential Refractive Index (RI) detector and two PLgel 5 μm Mixed C columns (300 × 7.5 mm). The measurements were performed at a temperature of T = 50 °C and a flow rate of 0.5 mL min−1. Samples were measured at a concentration of 2 mg mL−1 and filtered prior to measurement. All number-average molar mass Mn and dispersity Đ values were extrapolated from a range of linear poly(methyl methacrylate) standards between 800 and 2.2 × 106 g mol−1 and linear polystyrene standards between 370 and 2.52 × 106 g mol−1.
ATR-FT-IR spectra were recorded on a Bruker Vertex 80 from 500 to 4000 cm−1 at 25 °C.
GC-MS measurements were performed on a Varian 431 GC instrument with a capillary column FactorFour™ VF-5 ms (30 m × 0.25 mm × 0.25 μm) and a Varian 210 ion trap mass detector.
The commercially available IKA ElectraSyn 2.0 potentiostat as well as the respective IKA vial (5 mL) equipped with a septum on the outlet and an argon-filled balloon on top were used for the electrochemically-initiated polymerization of DFPA, PFPA, and GMA. Commercially available zinc and graphite electrodes from IKA were employed as anode and cathode material, respectively. After completion of the reaction, the electrodes were detached from the vial cap, rinsed with THF and acetone, and wiped with an acetone-soaked wipe. Thereafter, the surfaces of the electrodes were carefully scratched off with an APOLLO Ever-Sharp Blade and eventually wiped with an acetone-soaked wipe again. For the case of DFPA, the electrode was subsequently placed in a vial filled with acetone and placed in an ultrasonic bath. The acetone was changed three times and the electrode was again wiped with an acetone-soaked wipe.
CV measurements were performed on a Metrohm Autolab PGSTAT128N potentiostat featuring a glassy carbon working electrode, a Pt-wire counter electrode and an Ag/AgCl reference electrode with a scan rate of 50 mV s−1. Therefore, the analytes (10 mM) and tetrabutylammonium tetrafluoroborate (0.1 M) were dissolved in anhydrous acetonitrile (10 mL) and the solution was deoxygenated by nitrogen purging for 15 minutes.
Synthesis of 4-fluorobenzenediazonium tetrafluoroborate
4-Fluoroaniline (0.556 g, 5.00 mmol, 1.00 eq.) was dissolved in distilled water (2.5 mL) and 48 wt% tetrafluoroboric acid (HBF4) in water (1.7 mL). The solution was cooled to 0 °C and sodium nitrite (0.345 g, 5.00 mmol, 1.00 eq.) in distilled water (1.0 mL) was added dropwise within 30 minutes. The reaction was stirred for 1.5 hours at 0 °C and the mixture was filtered. The obtained solid was thereafter dissolved in a minimum amount of acetone. Diethyl ether was added until precipitation of the diazonium salt. The precipitate was washed thoroughly with diethyl ether and dried under vacuum yielding a colourless solid (0.33 g, 31%).1H NMR (acetone-d6): δ/ppm = 8.98–9.04 (m, 2H), 7.87–7.94 (m, 2H).
19F NMR (acetone-d6): δ/ppm = −86.66 (tq, J = 8.3, 4.2 Hz, 1F), −151.09 (d, J = 20.3 Hz, 4F).
ATR-FT-IR: ṽ/cm−1 = 3116, 2360, 2295, 1579, 1484, 1432, 1330, 1305, 1260, 1250, 1167, 1118, 1013, 850, 836, 807, 769, 685, 668.
Synthesis of 2,6-difluorophenyl acrylate (DFPA)
2,6-Difluorophenol (2.00 g, 15.37 mmol, 1.00 eq.) was dissolved in anhydrous THF (30 mL). The solution was cooled to 0 °C and triethylamine (2.6 mL, 1.87 g, 18.45 mmol, 1.20 eq.) was added. A solution of acryloyl chloride (1.5 mL, 1.67 g, 18.45 mmol, 1.20 eq.) in anhydrous THF (10 mL) was added slowly and the mixture was stirred for one hour at 0 °C and for 15.5 hours at ambient temperature. The solvent was removed under reduced pressure and diethyl ether (50 mL) was added. The organic phase was washed with water (3 × 50 mL) and with brine (1 × 50 mL). The organic phase was dried over magnesium sulfate and the solvent was removed under reduced pressure. The crude product was purified by column chromatography using a 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of cyclohexane/ethyl acetate as eluent yielding a yellowish liquid (2.19 g, 77%).
:1 mixture of cyclohexane/ethyl acetate as eluent yielding a yellowish liquid (2.19 g, 77%).
1H NMR (acetone-d6): δ/ppm = 7.34–7.43 (m, 1H), 7.15–7.23 (m, 2H), 6.68 (dd, J = 17.3, 1.2 Hz, 1H), 6.48 (dd, J = 17.3, 10.4 Hz, 1H), 6.24 (dd, J = 10.4, 1.2 Hz, 1H).
19F NMR (acetone-d6): δ/ppm = −128.34 to −128.47 (m, 2F).
ATR-FT-IR: ṽ/cm−1 = 1755, 1611, 1479, 1405, 1294, 1246, 1202, 1127, 1069, 1012, 981, 888, 775, 735, 697.
Synthesis of pentafluorophenyl acrylate (PFPA)
Pentafluorophenol (6.00 g, 32.60 mmol, 1.00 eq.) was dissolved in anhydrous THF (40 mL). The solution was cooled to 0 °C and triethylamine (5.5 mL, 3.96 g, 39.12 mmol, 1.20 eq.) was added. A solution of acryloyl chloride (3.2 mL, 3.54 g, 39.12 mmol, 1.20 eq.) in anhydrous THF (10 mL) was added slowly and the mixture was stirred for one hour at 0 °C and 18 hours at ambient temperature. The solvent was removed under reduced pressure and diethyl ether (75 mL) was added. The organic phase was washed with water (3 × 75 mL) and with brine (1 × 75 mL). The organic phase was dried over magnesium sulfate and the solvent was removed under reduced pressure. The crude product was purified by column chromatography using petroleum ether as eluent yielding a colourless liquid (5.20 g, 67%).1H NMR (CDCl3): δ/ppm = 6.72 (dd, J = 17.3, 0.9 Hz, 1H), 6.37 (dd, J = 17.3, 10.5 Hz, 1H), 6.18 (dd, J = 10.5, 1.0 Hz, 1H).
19F NMR (CDCl3): δ/ppm = −152.21 to −152.65 (m, 2F), −157.93 (t, J = 21.8 Hz, 1F), −162.24 to −162.162.42 (m, 2F).
ATR-FT-IR: ṽ/cm−1 = 1771, 1634, 1516, 1472, 1406, 1292, 1218, 1112, 1070, 1030, 994, 870, 796.
Electrochemically-initiated polymerizations
A dry IKA ElectraSyn vial (5 mL) was charged with monomer (DFPA/PFPA/GMA) (equivalents respective to 4–fluorobenzenediazonium tetrafluoroborate as initiator, see Table 1), tetrabutylammonium tetrafluoroborate (0.138 g, 0.40 mmol, 0.2 M), 4-fluorobenzenediazonium tetrafluoroborate (0.025 g, 0.12 mmol, 1.00 eq.) and anhydrous DMF (2 mL). The vial was closed with the respective IKA ElectraSyn cap bearing a sacrificial zinc anode and a graphite cathode and the solution was deoxygenated by argon purging for 15 minutes. Constant currents (–4 mA for DFPA and GMA, −1 mA for PFPA) were applied until 1.1 F mol−1 (DFPA and PFPA) or 2.0 F mol−1 (GMA) passed through the system. The electrodes were rinsed with THF and acetone (for electrode treatment, see ESI†). The mixture was precipitated in cold methanol, the solids obtained by centrifugation were dissolved in acetone and filtered (to remove graphite from the mixture) prior to a second precipitation in cold methanol. The solids were obtained by centrifugation and dried under vacuum.| Entry | Monomer | Eq. of monomers | M n /g mol−1 | M n /g mol−1 | M n /g mol−1 | Đ | M n /g mol−1 | Đ |
|---|---|---|---|---|---|---|---|---|
| a Determined by 1H NMR spectroscopy. b Determined by 19F NMR spectroscopy. c Determined by SEC using THF as eluent. d Determined by SEC using DMAc as eluent. e PMMA calibration. f PS calibration. g Monomer concentration too high, viscous mixture and formation of hardly soluble material between electrodes. h −2 mA instead of −1 mA. | ||||||||
| 1 | DFPA | 10.00 | — | 35250 |
16800e |
1.83e | 14600e |
2.49e |
| 15300f |
1.75f | 17300f |
2.23f | |||||
| 2 | DFPA | 5.00 | — | 21330 |
11300e |
1.58e | 8900e | 2.13e |
| 10500f |
1.50f | 11000f |
1.89f | |||||
| 3g | PFPA | 40.00 | 33240 |
40180 |
41800e |
2.30e | 30300e |
2.48e |
| 37000f |
2.28f | 34300f |
2.29f | |||||
| 4 | PFPA | 10.00 | 22630 |
35030 |
19200e |
1.83e | 13900e |
2.03e |
| 17400f |
1.76f | 16500f |
1.83f | |||||
| 5 | PFPA | 5.00 | 14170 |
24480 |
13600e |
1.61e | 9900e | 1.82e |
| 12500f |
1.53f | 12000f |
1.65f | |||||
| 6h | PFPA | 5.00 | 14290 |
22500 |
13200e |
1.59e | 9500e | 1.72e |
| 12100f |
1.52f | 11500f |
1.57f | |||||
| 7 | GMA | 20.00 | 10350 |
— | 8200e | 1.85e | 6200e | 2.40e |
| 7900f | 1.71f | 8000f | 2.06f | |||||
| 8 | GMA | 40.00 | 14500 |
— | 15700e |
2.40e | 12600e |
3.10e |
| 14700f |
2.24f | 15400f |
2.68f | |||||
| 9 | GMA | 10.00 | 6970 | — | 6800e | 1.60e | 5200e | 2.00e |
| 6500f | 1.50f | 6700f | 1.76f | |||||
PDFPA:
1H NMR (acetone–d6): δ/ppm = 7.15–7.38 (1H), 6.87–7.13 (2H), 3.22–3.42 (1H), 2.16–2.65 (2H).
19F NMR (acetone–d6): δ/ppm = −117.66 to −117.86, −125.08 to –128.10.
ATR-FT-IR: ṽ/cm−1 = 1770, 1609, 1499, 1479, 1450, 1293, 1246, 1200, 1118, 1012, 772, 722, 695.
PPFPA:
1H NMR (acetone–d6): δ/ppm = 7.28–7.39 (2H), 7.02–7.12 (2H), 3.11–3.41 (1H), 2.10–2.68 (2H).
19F NMR (acetone–d6): δ/ppm = −117.42 to −117.60, −153.68 to –154.71, −159.40 to −159.88, −164.30 to −165.04.
ATR-FT-IR: ṽ/cm−1 = 1782, 1515, 1472, 1451, 1227, 1078, 989, 856, 624.
PGMA:
1H NMR (DCM–d2): δ/ppm = 6.90–7.36 (4H), 4.22–4.40 (1H), 3.69–3.85 (1H), 3.17–3.26 (1H), 2.75–2.87 (1H), 2.58–2.67 (1H), 1.63–2.10 (2H), 0.62–1.26 (3H).
19F NMR (DCM–d2): δ/ppm = −116.85 to −117.05.
ATR-FT-IR: ṽ/cm−1 = 2999, 1724, 1483, 1448, 1387, 1340, 1254, 1147, 992, 906, 844, 759.
PPM of PDFPA
PDPFA (0.040 g, 0.217 mmol of repeating units, 1.00 eq.) was dissolved in anhydrous DMF (0.4 mL) and 2,2,2-trifluoroethylamine (0.43 mL, 0.538 g, 5.431 mmol, 25.00 eq.) was added. The solution was stirred for 72 hours at 100 °C and the solvent was removed under reduced pressure. The residue was dissolved in acetone and precipitated in petroleum ether. The solids were obtained by centrifugation, dissolved in acetone again and filtered prior to a second precipitation in petroleum ether. The solids were obtained by centrifugation and dried under vacuum.1H NMR (acetone-d6): δ/ppm = 6.80–7.70, 4.29–4.60, 3.83–4.17, 2.50–3.35, 1.25–1.90.
19F NMR (acetone-d6): δ/ppm = −68.56 to −70.45, −71.20 to –73.38, −117.90 to −118.98.
ATR-FT-IR: ṽ/cm−1 = 3334, 2939, 1689, 1626, 1398, 1341, 1261, 1153, 1116, 834, 668.
PPM of PPFPA
PPFPA (0.040 g, 0.168 mmol of repeating units, 1.00 eq.) was dissolved in anhydrous DMF (1 mL) and 2,2,2-trifluoroethylamine (0.33 mL, 0.416 g, 4.200 mmol, 25.00 eq.) was added. The solution was stirred for 72 hours at ambient temperature and the solvent was removed under reduced pressure. The residue was dissolved in acetone and precipitated in petroleum ether. The solids were obtained by centrifugation, dissolved again in acetone and precipitated a second time in petroleum ether. The solids were obtained by centrifugation and dried under vacuum.1H NMR (acetone-d6): δ/ppm = 7.53–8.06 (1H), 7.12–7.24 (2H), 6.92–7.03 (2H), 3.67–4.26 (2H), 2.15–2.66 (1H), 1.37–1.98 (2H).
19F NMR (acetone-d6): δ/ppm = −69.70 to −70.10, −72.30 to –72.98, −118.63 to −119.00.
ATR-FT-IR: ṽ/cm−1 = 3306, 1662, 1525, 1396, 1268, 1149, 981, 833, 669.
PPM of PGMA
PGMA (0.040 g, 0.281 mmol of repeating units, 1.00 eq.) was dissolved in anhydrous DMF (0.6 mL) and 2,2,2-trifluoroethylamine (0.55 mL, 0.697 g, 7.035 mmol, 25.00 eq.) was added. The solution was stirred for 72 hours at 90 °C and the mixture was precipitated in cold diethyl ether. The solids obtained by centrifugation were dissolved in acetone and precipitated a second time in cold diethyl ether. The solids were obtained by centrifugation and dried under vacuum.1H NMR (acetone-d6): δ/ppm = 6.97–8.08 (4H), 4.28–4.60 (1H), 3.89–4.17 (2H), 3.20–3.48 (2H), 2.74–2.99 (2H), 1.76–2.08 (2H), 0.72–1.45 (3H).
19F NMR (acetone-d6): δ/ppm = −71.10 to −71.76, −71.85 to –72.75, −117.80 to −118.01.
ATR-FT-IR: ṽ/cm−1 = 3365, 2944, 1724, 1455, 1393, 1266, 1139, 837, 748, 668.
Results and discussion
In the present study, functional polymers have been synthesised from DFPA, PFPA, and GMA, respectively, by electrochemical reduction of a fluorine-labelled aromatic diazonium salt. Radical species are generated from the latter when a negative potential is applied under elimination of elemental nitrogen from the diazonium moiety (see ESI† for CV data of the initiator and DFPA and PFPA as monomers). This initiation system was chosen for two reasons: (i) a similar system was employed in an electrochemically-mediated RAFT polymerization,17 where a bromine atom instead of a fluorine atom in para position was used as a commercially available diazonium salt, and (ii) it allows for mild electrochemical reaction conditions. The latter is important for the polymerization of reactive monomers since functional groups could potentially interfere with the redox behaviour of the initiator and undergo side reactions during the reaction. Additionally, the fluorine-label in the initiator as well as in the two reactive monomers DFPA and PFPA permits the straightforward analysis of the resulting polymers by 19F NMR spectroscopy, which also allows for determination of the number-average molar mass of the as-prepared macromolecules.The initiator, i.e. 4-fluorobenzenediazonium tetrafluoroborate, was synthesised according to standard diazotation procedures from literature,42,43 involving the use of sodium nitrite (NaNO2) and aqueous tetrafluoroboric acid (HBF4). 4-Fluoroaniline was used as starting material and not more than 0.5 g of product were synthesised in a single batch for safety reasons, since diazonium salts exhibit inherent potential explosion hazards.44 After the diazotation, the diazonium salt was filtered off, precipitated in cold diethyl ether, and thoroughly washed with cold diethyl ether, yielding in a colourless solid. The obtained diazonium salt was stored in the dark at a temperature of T = −20 °C in a vial by covering it with aluminium foil. The purity was determined by 1H, 13C NMR, 19F NMR, and IR spectroscopy (Fig. S2–S5†).
For the polymerization, a beginner-friendly, fully commercially available, and affordable electrochemical setup (IKA ElectraSyn 2.0) was used with standard electrodes (zinc anode, graphite cathode) as source of electrons. Galvanostatic (i.e. constant current) conditions allowing for the use of a two-electrode cell setup were applied, demonstrating the ease of polymerization and its potential scalability. Polymerizations were conducted in DMF solutions after degassing. Typically, polymerizations were performed with different but constant currents for the respective monomers (–4 mA for DFPA and GMA, −1 mA for PFPA) until 1.1 F mol−1 (DFPA and PFPA) or 2.0 F mol−1 (GMA) were reached.
After successful polymerization resulting in reactive polymers with a fluorine-label arising from the initiating moiety, PPM using 2,2,2-trifluoroethylamine as fluorine-labelled nucleophile was employed. The resulting modified polymers were again analysed by SEC and NMR as well as IR spectroscopy, proving the successful functionalization. In the case of 2,6-difluorophenyl acrylate and PFPA, the reactive polymers are based on active ester side groups, which can be transformed into the respective amide.39,45–47 Poly(glycidyl methacrylate) (PGMA) on the other hand bears an epoxide functional group, which can be ring-opened by a nucleophile.48 Details are discussed in the following.
DFPA as reactive monomer
Two different fluorine-labelled, reactive monomers, i.e. DFPA and PFPA, were polymerized by electrochemical reduction of 4-fluorobenzenediazonium tetrafluoroborate, allowing for a straightforward evaluation of the number-average molar mass of the resulting polymers by fluorine NMR spectroscopy. DFPA was synthesised by reaction of 2,6-difluorophenol with acryloyl chloride using triethylamine as auxiliary base. After purification by column chromatography, the monomer DFPA was employed in an electrochemically-initiated polymerization in N,N-dimethylformamide (DMF) with tetrabutylammonium tetrafluoroborate (Bu4NBF4) (0.2 M) in an undivided cell setup bearing a sacrificial zinc anode and a simple graphite cathode. A constant current of −4 mA was applied for 1.1 F mol−1 (with respect to the initiator), leading to visible evolution of gaseous nitrogen in the reaction setup. The polymer was obtained by two subsequent precipitation steps in cold methanol. The resulting polymer was analysed by NMR and IR spectroscopy as well as SEC. The ratio of monomer to initiator (amount of initiator was kept constant at 0.12 mmol in all experiments) was varied to obtain polymers featuring different chain lengths and thus molar masses. The latter was determined both by SEC and 19F NMR spectroscopy. The polymerization yielded polymers with dispersities around 2, typical for FRP. The number-average molar masses determined by SEC and 19F NMR spectroscopy differed from each other, with the former however not based on a suitable calibration for the determination of absolute molar mass values. In contrast, 19F NMR spectroscopy allows for the straightforward calculation of the number of repeating units with respect to the initiator, enabling the number-average molar mass determination. However, this method is limited to lower molar masses, because the signal arising from the initiator is relatively low in intensity in comparison to the signals arising from the fluorine-containing side groups. Nonetheless, we report both values and the values obtained by 1H NMR spectroscopy for PPFPA and PGMA in Table 1. In the case of PDFPA, the aromatic signals arising from the 2,6-difluorophenyl moiety are overlapping with the initiator signals, and thus no values could be provided. Subsequently, PPM was performed on one of the obtained polymers (Table 1, entry 1) with 2,2,2-trifluoroethylamine. A large excess of 2,2,2-trifluoroethylamine (25.00 eq.) was used due to its volatile nature. The reaction was performed for three days at elevated temperature (T = 100 °C). The resulting polymer was precipitated twice in petroleum ether and analysed. The SEC traces (DMAc as eluent, PMMA calibration) after PPM were slightly shifted from Mn = 14600 g mol−1 and Đ = 2.49 before functionalization to Mn = 13100 g mol−1 and Đ = 2.44 (Fig. 1(A)). 1H NMR spectroscopy revealed the removal of the 2,6–difluorophenol motif after the reaction, which was also confirmed by 19F NMR spectroscopy (Fig. 1(B)). The signals at δ = −126.55 to −127.40 ppm disappeared completely, two new signals at δ = −71.20 to −73.38 ppm and δ = −68.56 to −70.45 ppm arose from the newly formed amide and presumably the imide, respectively. The transformation was also followed by IR spectroscopy, showing the disappearance of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration at ṽ = 1770 cm−1 of the ester, while new CO vibrations of the amide and imide appeared (Fig. 1(C)). The spectrum differs from the one obtained after functionalization of PPFPA, which yielded only the respective amide without formation of significant amounts of imide (Fig. 2(C)). This is in accordance to our assumption that, under the conditions required to modify poly(2,6-difluorophenyl acrylate), the respective imide is formed.
O vibration at ṽ = 1770 cm−1 of the ester, while new CO vibrations of the amide and imide appeared (Fig. 1(C)). The spectrum differs from the one obtained after functionalization of PPFPA, which yielded only the respective amide without formation of significant amounts of imide (Fig. 2(C)). This is in accordance to our assumption that, under the conditions required to modify poly(2,6-difluorophenyl acrylate), the respective imide is formed.
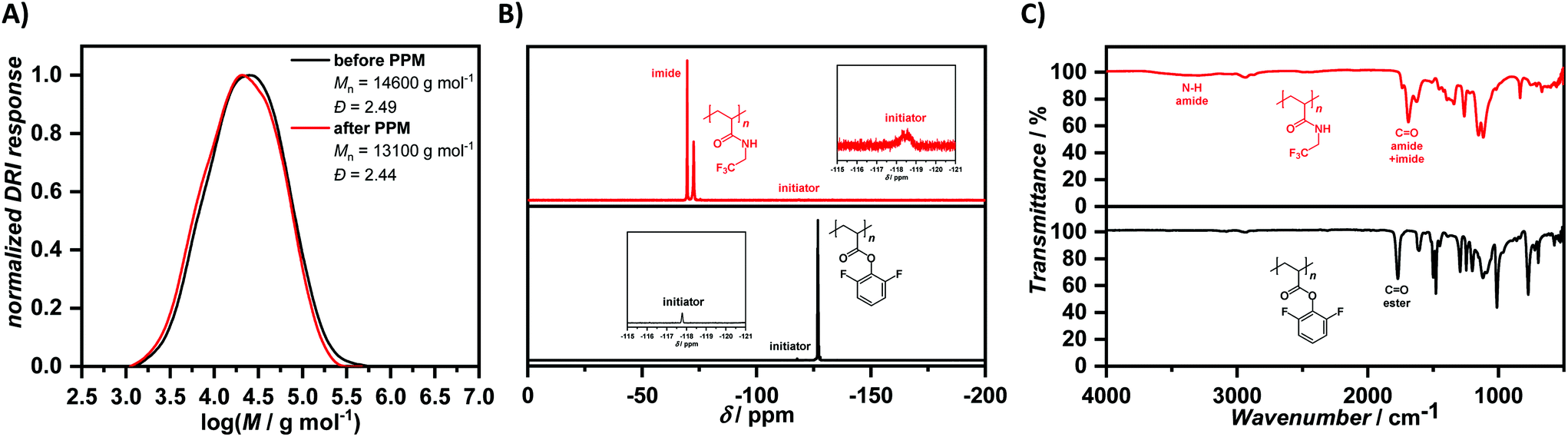 | ||
| Fig. 1 Comparison of SEC, 19F NMR, and ATR-FT-IR data. (A) The SEC trace is slightly shifted after PPM while retaining its original shape. (B) The signals arising from PDFPA in fluorine NMR spectroscopy completely disappeared accompanied by two new signals of the formed amide and imide moieties, respectively. (C) Comparison of the IR spectra shows the disappearance of the CO vibration of the ester and arising CO vibrations. | ||
 | ||
| Fig. 2 Comparison of SEC, 19F NMR, and ATR-FT-IR data. (A) The SEC trace is shifted towards higher molar masses after PPM while retaining its original shape. (B) The signals arising from PPFPA in fluorine NMR spectroscopy completely disappeared accompanied by the appearance of the signal arising from the formation of the respective amide. (C) Comparison of the IR spectra shows the disappearance of the CO vibration of the ester and the appearance of the CO vibration from the amide. | ||
PFPA as reactive monomer
Being the second fluorine-labelled monomer in this study, PFPA features an active ester and has been extensively employed for PPM.39,45–47 It was obtained in a similar fashion to 2,6-difluorophenyl acrylate from acryloyl chloride as mentioned above. First polymerization attempts applying a constant current of −4 mA until 4 F mol−1 passed through the system turned out to deliver polymeric material, however the 19F NMR spectrum exhibited signals arising from potential decomposition reactions (Fig. S43†). Instead of applying a current of −4 mA, we tried currents of −2 mA and −1 mA for 4 F mol−1, respectively. Only at −1 mA, the signals arising from potential decomposition reactions were absent, showing the fragile and reactive nature of the employed PFPA monomer requiring substantially lower currents for a polymerization. Similar to the case of DFPA, the amount of initiator was kept constant at 0.12 mmol in a 0.2 M solution of Bu4NBF4 in anhydrous DMF (2 mL). The reaction mixtures after polymerization were precipitated into cold methanol to remove the electrolyte. The obtained solids after centrifugation were dissolved in acetone and reprecipitated a second time into cold methanol. The prepared polymers were, analogous to PDFPA, analysed by NMR and IR spectroscopy as well as SEC. The number-average molar mass values obtained by 19F NMR spectroscopy and SEC are listed in Table 1 and differed from each other. For comparison, the number-average molar mass values were also calculated by 1H NMR spectroscopy. The intensity of the signal arising from the initiator in 19F NMR spectroscopy is comparatively low when looking at the PPFPA signals. The calculation is based on the integration and the choice of integration limits influences the results dramatically. Nonetheless, a clear trend emerged from both NMR spectroscopy and size-exclusion chromatography, as the degree of polymerization is increasing with increasing monomer to initiator ratio. A ratio of monomer to initiator of 20 and above resulted in the formation of a hardly soluble polymer between the electrodes, with the concentration of PFPA in the solution presumably being too high in these cases to guarantee fully soluble polymers throughout the course of the polymerization. The obtained polymer after polymerization of 10 equivalents of monomer with respect to initiator (Table 1, entry 4) was used as active ester moiety in PPM with 2,2,2-trifluoroethylamine, analogous to DFPA as reactive monomer. Unlike DFPA, PFPA features a higher reactivity towards amines due to the stronger electron-withdrawing nature of the pentafluorophenyl unit. Milder reaction conditions could thus be applied for the PPM, resulting in the formation of the respective polyacrylamide in a quantitative fashion according to 19F NMR spectroscopy. The signals arising from the pentafluorophenyl moiety at δ = −153.80 to −154.64 ppm, δ = −159.42 to −159.82 ppm, and δ = −164.35 to −164.95 ppm were replaced by signals arising from the newly formed fluorine-labelled amine at δ = −72.30 to −72.98 ppm (Fig. 2(B)). In contrast to the PPM of DFPA, only insignificant amounts of imide were formed under these conditions. To prove the shifts of the signals in 19F NMR spectroscopy, the respective amide monomer was prepared and polymerized by FRP. The 19F NMR spectroscopical analysis only revealed one signal (Fig. S79†), whose shift matched the one obtained after PPM of PPFPA and the assigned amide signal for the PPM of PDFPA. In the case of direct polymerization of the amide monomer, no imide formation can take place in the polymerization, whereas the imide formation during PPM reactions is determined by the respective conditions. At first, we applied common conditions for the PPM of active ester polymers, using triethylamine as a base. These conditions favoured the imide formation in comparison to reactions without additional base, with the respective results complied in the ESI.† The SEC traces (DMAc as eluent, PMMA calibration) after PPM were shifted towards higher molar masses from Mn = 13900 g mol−1 and Đ = 2.03 before functionalization to Mn = 16100 g mol−1 and Đ = 2.18 after functionalization (Fig. 2(A)). Furthermore, IR spectroscopy showed the disappearance of the CO vibration at ṽ = 1782 cm−1 of the active ester, while a new CO vibration at ṽ = 1662 cm−1 of the amide arose (Fig. 2(C)). The spectrum exhibited less vibrational signals in comparison to the one of modified PDFPA, due to the almost exclusive formation of the amide in contrast to a mixture of amide and imide obtained in the PPM of PDFPA.
GMA as reactive monomer
After successful polymerization of the fluorine-labelled monomers DFPA and PFPA, we tried to expand the scope of this polymerization method to other functional monomers. GMA is a commercially available monomer that has been extensively studied for PPM.48 Polymerization attempts have been conducted with a constant current of −4 mA for 2 F mol−1. The first reaction directly yielded PGMA, so we next varied the monomer to initiator ratio again to prepare polymeric materials with a varying degree of polymerization. The polymers featured different molar mass values and dispersities, however, the calculation of the number-average molar mass value by 1H NMR spectroscopy in this case (a calculation by 19F NMR spectroscopy was not possible, since no fluorine atoms are present in the monomer) is close to the value obtained by SEC using THF as eluent. The proton NMR spectrum showed all the respective signals assigned to PGMA, as reported in literature.49 The epoxide functionality seemed to be stable towards the polymerization conditions and it did not require a fine-tuning of the polymerization as in the case of PFPA. As in the case of PDFPA and PPFPA, PGMA was functionalized using 2,2,2-trifluoroethylamine, in this case resulting in a ring-opening of the epoxide. First attempts using triethylamine as base with only a slight excess (4.00 eq.) of the fluorine-labelled amine resulted in insoluble material, possibly due to cross-linking reactions,50 which can be avoided by use of a larger excess of the respective amine.51 The functionalization was thus conducted with a large excess of the amine (25 eq.) in DMF at elevated temperature (T = 90 °C) without the addition of a base for three days and the mixture was precipitated into cold diethyl ether. The resulting polymers were isolated by centrifugation, dissolved and reprecipitated into cold diethyl ether again. Proton NMR spectroscopy proved the successful modification of PGMA with the fluorine-labelled amine. The 1H NMR spectra of PGMA before and after PPM differed significantly from each other, with new signals at δ = 4.25–4.55 ppm, δ = 3.86–4.12 ppm, δ = 3.17–3.47 ppm, and δ = 2.70–2.98 ppm after functionalization and signals arising from the initial PGMA structural motif completely disappearing. Additionally, the 19F NMR spectrum (Fig. 3(B)) exhibited signals from incorporated 2,2,2-trifluoroethylamine moieties at δ = −72.35 ppm accompanied by a smaller signal at δ = −71.59 ppm, which could arise from either the ring-opening of one epoxide moiety by an already formed amide functionality next to it or the formation of the other regioisomer. Additionally, SEC comparison exhibited a clear shift towards higher molar masses after PPM (from Mn = 6200 g mol−1, Đ = 2.40 to Mn = 20300 g mol−1, Đ = 1.99; judged by SEC using DMAc as eluent and PMMA calibration) (Fig. 3(A)), proving a successful modification of PGMA obtained by electrochemically-initiated polymerization. Furthermore, the comparison of the IR spectra clearly showed the disappearance of the epoxide vibration of PGMA52 at ṽ = 905 cm−1 and the appearance of vibrations at ṽ = 3200–3600 cm−1, assigned to the formation of the β–amino alcohol structural motif (Fig. 3(C)).
 | ||
| Fig. 3 Comparison of SEC, 19F NMR, and ATR-FT-IR data. (A) The SEC trace is shifted towards higher molar masses after PPM. (B) After PPM, the signals of the incorporated CF3 group are arising, proving the formation of β–amino alcohols. (C) Comparison of the IR spectra shows the disappearance of the epoxide vibration and the appearance of N–H and O–H vibrations. | ||
Conclusions
In conclusion, three reactive monomers (DFPA, PFPA, and GMA) were used in an electrochemically-initiated radical polymerization approach based on the reduction of a fluorine-labelled aromatic diazonium salt. In the cases of DFPA and PFPA as monomers, this fluorine-label allowed for the straightforward calculation of the number-average molar mass values by 19F NMR spectroscopy. Different molar masses could be reached by variation of the monomer to initiator ratio based on a fully commercially available electrochemical setup, enhancing reproducibility of electrochemical synthesis procedures. The obtained polymers were subsequently used in PPM reactions with a fluorine-labelled amine, resulting in the formation of the respective polyacrylamide in the case of PDFPA and PPFPA (for the former, considerable amounts of the respective imide were formed) and β-amino alcohol for PGMA. The reactions were followed by NMR and IR spectroscopy as well as SEC, proving the successful functionalization of the obtained polymers and thus their untouched reactivity after the electrochemically-initiated polymerization of the respective reactive monomers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support via the Helmholtz Association.Notes and references
- D. Colombani, Prog. Polym. Sci., 1997, 22, 1649–1720 CrossRef CAS.
- P. Nesvadba, Radical Polymerization in Industry, Wiley, New Jersey, 2012 Search PubMed.
- T. G. McKenzie, F. Karimi, M. Ashokkumar and G. G. Qiao, Chem. – Eur. J., 2019, 25, 5372–5388 CrossRef CAS PubMed.
- O. Lindström and O. Lamm, J. Phys. Chem., 1951, 55, 1139–1146 CrossRef.
- A. Eibel, D. E. Fast and G. Gescheidt, Polym. Chem., 2018, 9, 5107–5115 RSC.
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245–6260 CrossRef CAS.
- S. H. Dickens, J. W. Stansbury, K. M. Choi and C. J. E. Floyd, Macromolecules, 2003, 36, 6043–6053 CrossRef CAS.
- A. Kowalska, J. Sokolowski and K. Bociong, Polymers, 2021, 13, 470 CrossRef CAS PubMed.
- A. D. Jenkins, R. G. Jones and G. Moad, Pure Appl. Chem., 2010, 82, 483–491 CAS.
- D. A. Shipp, Polym. Rev., 2011, 51, 99–103 CrossRef CAS.
- J. Chiefari, Y. K. B. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo, S. H. Thang and C. South, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- P. Corpart, D. Charmot, T. Biadatti, S. Zard and D. Michelet, WO1998058974A1, 1998.
- T. P. Le, G. Moad, E. Rizzardo and S. H. Thang, WO1998001478A1, 1998.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- D. H. Solomon, E. Rizzardo and P. Cacioli, US4581429A, 1986.
- Y. Wang, M. Fantin, S. Park, E. Gottlieb, L. Fu and K. Matyjaszewski, Macromolecules, 2017, 50, 7872–7879 CrossRef CAS PubMed.
- F. Lorandi, M. Fantin, S. Shanmugam, Y. Wang, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2019, 52, 1479–1488 CrossRef CAS.
- W. Sang, M. Xu and Q. Yan, ACS Macro Lett., 2017, 6, 1337–1341 CrossRef CAS.
- C. Bray, G. Li, A. Postma, L. T. Strover, J. Wang and G. Moad, Aust. J. Chem., 2021, 74, 56–64 CrossRef CAS.
- P. Chmielarz, M. Fantin, S. Park, A. A. Isse, A. Gennaro, A. J. D. Magenau, A. Sobkowiak and K. Matyjaszewski, Prog. Polym. Sci., 2017, 69, 47–78 CrossRef CAS.
- A. J. D. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81–85 CrossRef CAS PubMed.
- F. Lorandi, M. Fantin, A. A. Isse and A. Gennaro, Polym. Chem., 2016, 7, 5357–5365 RSC.
- F. De Bon, M. Fantin, A. A. Isse and A. Gennaro, Polym. Chem., 2018, 9, 646–655 RSC.
- F. Lorandi, M. Fantin, A. A. Isse and A. Gennaro, Curr. Opin. Electrochem., 2018, 8, 1–7 CrossRef CAS.
- B. Zhao, M. Mohammed, B. A. Jones and P. Wilson, Chem. Commun., 2021, 57, 3897–3900 RSC.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed.
- M. Ghosh, V. S. Shinde and M. Rueping, Beilstein J. Org. Chem., 2019, 15, 2710–2746 CrossRef CAS PubMed.
- C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Green Chem., 2020, 22, 3358–3375 RSC.
- C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed.
- M. Faraday, Ann. Phys., 1834, 109, 433–451 CrossRef.
- H. Kolbe, Justus Liebigs Ann. Chem., 1849, 69, 257–294 CrossRef.
- K. Tanaka, T. Shichiri, S. Wang and T. Yamabe, Synth. Met., 1988, 24, 203–215 CrossRef CAS.
- S. Sadki, P. Schottland, N. Brodie and G. Sabouraud, Chem. Soc. Rev., 2000, 29, 283–293 RSC.
- J. M. Ortega, S. Menolasina, O. P. de Márquez and J. Márquez, Polymer, 1986, 27, 1304–1306 CrossRef CAS.
- M. Mertens, C. Calberg, L. Martinot and R. Jérôme, Macromolecules, 1996, 29, 4910–4918 CrossRef CAS.
- A. Das and P. Theato, Chem. Rev., 2016, 116, 1434–1495 CrossRef CAS PubMed.
- J. G. Kim, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2554–2560 CrossRef CAS.
- J. F. R. Van Guyse, J. Verjans, S. Vandewalle, K. De Bruycker, F. E. Du Prez and R. Hoogenboom, Macromolecules, 2019, 52, 5102–5109 CrossRef CAS.
- D. Prasad Hari, T. Hering and B. König, Angew. Chem., Int. Ed., 2014, 53, 725–728 CrossRef CAS PubMed.
- S. Shaaban, A. Jolit, D. Petkova and N. Maulide, Chem. Commun., 2015, 51, 13902–13905 RSC.
- S. P. Green, K. M. Wheelhouse, A. D. Payne, J. P. Hallett, P. W. Miller and J. A. Bull, Org. Process Res. Dev., 2020, 24, 67–84 CrossRef CAS PubMed.
- Y. Pinyakit, T. Palaga, S. Kiatkamjornwong and V. P. Hoven, J. Mater. Chem. B, 2020, 8, 454–464 RSC.
- L. He, J. Shang and P. Theato, Eur. Polym. J., 2015, 69, 523–531 CrossRef CAS.
- W. Xue, H. Mutlu and P. Theato, Eur. Polym. J., 2020, 130, 109660 CrossRef CAS.
- E. M. Muzammil, A. Khan and M. C. Stuparu, RSC Adv., 2017, 7, 55874–55884 RSC.
- D. Pranantyo, L. Q. Xu, K. G. Neoh, E. T. Kang, W. Yang and S. L. M. Teo, J. Mater. Chem. B, 2014, 2, 398–408 RSC.
- M. Ma, F. Li, F. J. Chen, S. X. Cheng and R. X. Zhuo, Macromol. Biosci., 2010, 10, 183–191 CrossRef CAS PubMed.
- F. J. Xu, Y. Zhu, M. Y. Chai and F. S. Liu, Acta Biomater., 2011, 7, 3131–3140 CrossRef CAS PubMed.
- L. M. Meng, Y. C. Yuan, M. Z. Rong and M. Q. Zhang, J. Mater. Chem., 2010, 20, 6030–6038 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00536g |
| This journal is © The Royal Society of Chemistry 2021 |