
Synthesis and characterization of β,β′-dimethylated dithieno[3,2-b:2′,3′-d]pyrroles and their corresponding regioregular conducting electropolymers†
Sebastian
Förtsch‡
,
Elena
Mena-Osteritz
and
Peter
Bäuerle
 *
*
Institute of Organic Chemistry II and Advanced MaterialsUniversity of Ulm, Albert-Einstein-Allee 11, 89081 Ulm, Germany. E-mail: elena.mena-osteritz@uni-ulm.de; peter.baeuerle@uni-ulm.de
First published on 12th May 2021
Abstract
β,β‘-Dimethyl-substituted dithieno[3,2-b:2′,3′-d]pyrroles (Me-DTP) 2–7 represent novel electron-rich building blocks, which were converted to corresponding conducting polymers p(Me-DTP) and to func-tional dyes for organic electronic applications. Supported by quantum chemical calculations, structural and optoelectronic properties of the three types of π-conjugated materials were investigated and led to valuable structure–property relationships.
1. Introduction
S,N-Heterotriacene dithieno[3,2-b:2′,3′-d]pyrrole (DTP) comprises two thiophenes fused to one central pyrrole ring and represents a coplanar, electron-rich π-conjugated molecule, which has been used in donor–acceptor polymers, oligomers, or S,N-heteroacenes for application in organic electronics, in particular organic solar cells and perovskite solar cells.1,2 DTPs have also been polymerized to conducting DTP-polymers (pDTP) with extended conjugation and relatively rigid structure. Polymerization methods include transition metal-catalyzed condensation polymerizations such as Yamamoto, Suzuki, or Stille-type coupling, but most of the DTPs afforded only low molecular weight materials.3 Due to their reactive α-positions, DTPs can be polymerized by oxidation either electrochemically or by chemical oxidants such as iron trichloride or ruthenium trichloride.1,4,5 The resulting pDTPs are typically obtained as insoluble powders or as thin films on the working electrode and exhibit reversible doping, good electrical conductivity, and electrochromisms.6 Soluble fractions of rather low molecular weight were isolated in particular for pDTPs with long solubilizing alkyl side chains and allowed for solution studies.7 Likewise, the DTP structure allows for N-functionalization without affecting coplanarity and pDTPs bearing acyls,8 amines,9 ethers,10 or chiral side groups11 were prepared. Applications such as cathode active material in rechargeable batteries10,12 or as sensors come into play.9 Whereas in condensation polymerizations selectively α-coupled pDTPs are obtained, couplings defects via α–β or β–β couplings arise with an oxidative polymerization mechanism as a consequence of significant spin-density at the β-position of the intermediate DTP radical cations.7 Such interruptions of the conjugated pathway significantly influence the optoelectronic properties of afforded pDTPs. In this respect, we found 22–32% coupling defects in soluble alkylated pDTPs, which were analysed by NMR spectroscopy.13Herein, we disclose synthesis, structural characterization, and oxidative polymerization of β,β′-dimethyl-DTP 2 and its N-functionalized analogues (R = propyl, hexyl, phenyl, 9-anthryl, benzoyl) 3–7 as novel monomers, which by oxidative polymerization lead to linear, strictly regioregular pDTPs with-out structural defects due to the blocked β-positions (Fig. 1). Optical and redox properties of the prepared monomers were investigated by UV-vis absorption spectroscopy and cyclic voltammetry. The obtained structure–property relationships were supported by quantum chemical calculations. The electrochemically obtained pDTP-films were characterized by cyclic voltammetry and spectroelectrochemistry (Scheme 1).
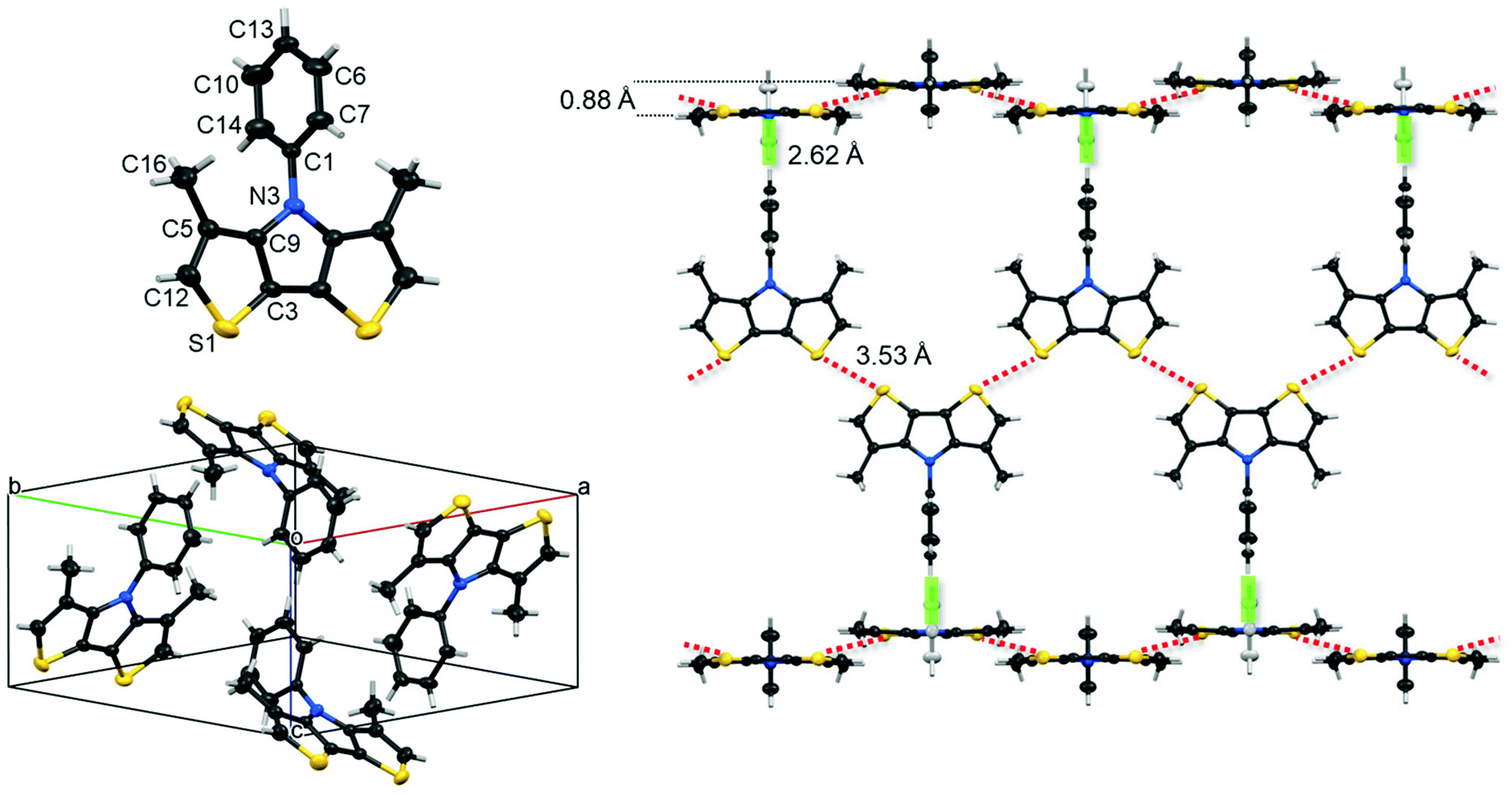 | ||
| Fig. 1 Crystal structure analysis of Me-DTP 5: individual molecule and atom label of the asymmetric unit (top left) and herringbone-type packing structure of the four molecules in the unit cell (bottom left). Packing motif perpendicular to the (9 0 4) plane (non-interacting molecules have been avoided for clarity) (right): Intermolecular close S–S atom contacts (red dotted lines) forming a dimeric ((double molecule)) band and H–π interactions (green rods) between the perpendicular ((dimeric)) bands (right). Thermal ellipsoids are set at a 50% probability level. | ||
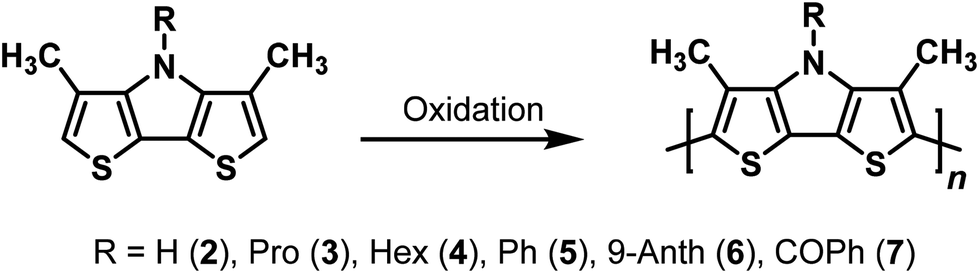 | ||
| Scheme 1 Molecular structure of Me-DTPs 2–7 and corresponding polymers P2–P7. | ||
2. Results and discussion
2.1 Synthesis of Me-DTPs 2–7
The synthesis of N-substituted DTPs has been facilitated and vastly extended by the elaboration of the efficient Pd-catalyzed amination of 3,3′-dibromo-2,2′-bithiophene with respective amines, mostly alkyl and aryl amines, or Cu-catalyzed acylation and benzoylation.1,14 As an alternative, we developed the versatile N-substitution of 4H-DTP by alkylation and arylation reactions.13,15 Regardless, the related class of β,β′-disubstituted DTPs is practically unknown and just very recently the first example was published by Heeney et al., who implemented 3,3′-didodecyl-4H-DTP as a core unit into a conjugated oligomer, which was then used as a UV-vis spectroscopic pH-sensor.16Our synthesis of Me-DTPs involved the preparation of dibromobithiophene precursor 1 from 3-methylthiophene in three steps according to a literature procedure.17 This precursor was subsequently used to prepare Me-DTPs 2–7 bearing hydrogen, propyl, hexyl, phenyl, 9-anthracenyl, or benzoyl residue at the nitrogen, respectively (Scheme 2). The alkyl and aryl-substituted Me-DTPs 3–6 were synthesized in Pd-catalysed Buchwald–Hartwig aminations of dibromobithiophene 1 with the respective amines and the catalytic system Pd(dba)2/dppf in 79–85% and 74–95% yield, respectively. Hence, the β-methyl groups in 1 had no detrimental influence on the two-fold amination and ring-closure reaction in comparison to the DTP-analogues.5 For the reaction of the sterically demanding 9-anthrylamine to DTP-derivative 6 (74% yield) the catalyst Pd2dba3 and monodentate ligand RuPhos was used (Scheme 2). Benzoylated Me-DTP 7 was obtained from an Ullmann-type coupling of 1 with benzamide using the catalytic system CuI and DMEDA. After 24 hours reaction time, the desired benzoyl-substituted Me-DTP 7 was isolated in a yield of 36% besides 22% of the unsubstituted analogue 2 originating from the in situ hydrolysis of the benzoyl residue in Me-DTP 7. By prolongation of the reaction time to overall six days, the yield of parent Me-DTP 2 could be increased to 63%. The analogous reaction was utilized for the preparation of H-DTP from corresponding benzoyl-DTP and a maximum yield of 59% was achieved after two days.18 From these findings we conclude that the two methyl groups in the β-positions exhibited a pronounced sterical influence towards the substituent at the nitrogen, thus shielding the carbonyl group and decele-rating the benzoyl cleavage by nucleophilic attack from water or hydroxide anions.
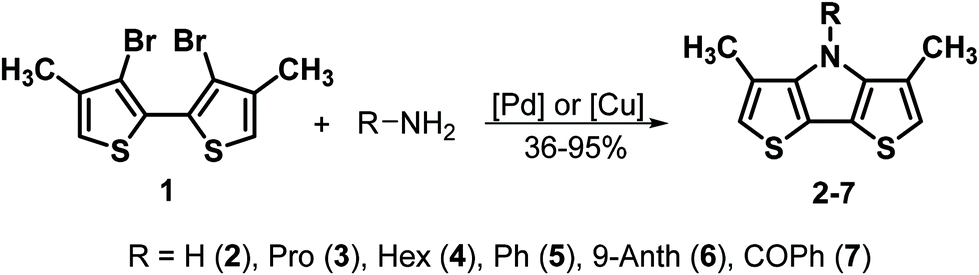 | ||
| Scheme 2 Synthesis of Me-DTPs 2–7. | ||
Structure and purity of Me-DTPs 2–7 were confirmed by melting points (Table 3), NMR spectroscopy, mass spectrometry, and elemental analysis. In the case of phenylated Me-DTP 5 a single crystal X-ray structure analysis was performed (vide infra).
2.2 Structural properties of Me-DTP 5
Single crystals of phenyl-substituted Me-DTP 5 were obtained by cooling a solution of 5 in n-hexane to −20 °C to afford colourless plate-shaped crystals suitable for X-ray structure analysis. Me-DTP 5 crystallized in the orthorhombic space group Pnma with four molecules per unit cell (a = 14.1117(3), b = 14.1998(3), c = 6.88772(14) Å, α, β, γ = 90°, V = 1380.18(5) Å3) (Table S1, ESI†). The geometry of Me-DTP 5 is shown in Fig. 1 and selected bond lengths, bond angles, and torsion angles are summarized in Tables S2–S4 (ESI†). Bond distances and angles of the central DTP units correspond to typical fused π-conjugated systems with reduced bond length alternation. The striking difference to the structure analysis of corresponding phenyl-substituted DTP-Ph19 is represented by the substantially increased dihedral angle (C1N3C9C5) between the DTP and the N-phenyl unit of 37° for DTP-Ph to 88° for Me-DTP 5.The almost orthogonal (88°) torsion angle of the phenyl residue with respect to the Me-DTP plane is entirely due to repulsive interactions with the methyl groups which are sufficeent to enforce a cruci-form-type geometry. Intramolecular short contacts (below the sum of the van der Waals radii) between atoms C1 and C16 support this conclusion (Table S5, ESI†). Like the symmetry group and unit cell angles indicate, molecules of 5 arrange in fully perpendicular herringbone geometry. Nevertheless, persistent intermolecular short contacts of 3.53 Å between sulphur atoms far below the sum of van der Waals radii (red dotted lines in Fig. 1, right) allows Me-DTP 5 to organize in almost flat dimeric bands. Further intermolecular edge-to-face H–π interactions of the phenyl residues with adjacent Me-DTP units of the perpendicular ongoing bands at distances of 2.62 Å have been identified (green rods in Fig. 1, right). In spite of the fully perpendicular herringbone geometry of Me-DTP 5, weak π–π stacking (perpendicular to the planes (9 0 4) and (9 0 −4)) between the dimeric bands (Me-DTP units) at distances of 3.507 Å can be extracted from the X-ray structure analysis (Fig. S1, ESI†).
2.3 Quantum chemical calculations on Me-DTPs 2–7
The methyl groups in the β-positions of Me-DTPs 2–7 are likely to exhibit sterical repulsion on the residues attached at the nitrogen. Therefore, we carried out quantum chemical DFT calculations using the M06-2X functional20 and the obtained frontier molecular orbital surfaces are depicted in Table 1. Similarly to the investigated DTP analogues without methyl residues, all derivatives possess a node at the nitrogen in the highest occupied molecular orbital (HOMO).5,21 Thus, only the DTP unit contributes to the electronic distribution in the HOMO with minor participation of the methyl groups. The attached residues at the central nitrogen may only indirectly influence the HOMO energy level via inductive effects. In contrast to that, the lowest unoccupied molecular orbitals (LUMO) of unsubstituted and alkyl-substituted Me-DTPs 2–4 are mainly localized at the N-substituent and thus differ strongly from the LUMOs of their DTP-analogues. With a dihedral angle of 39° the benzoyl residue of Me-DTP 7 is bent out of the DTP plane and still exhibits small contributions to the Me-DTP backbone.
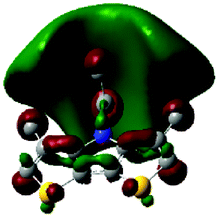
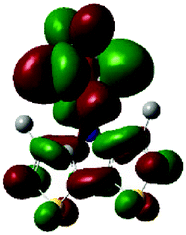
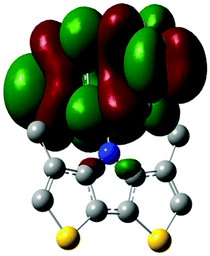
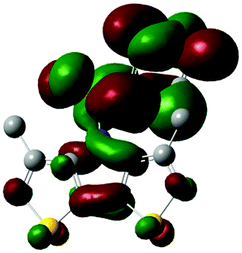

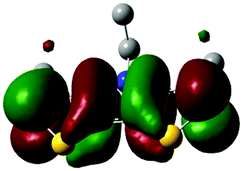
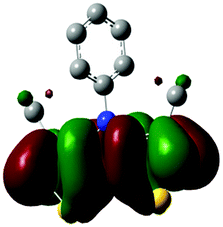
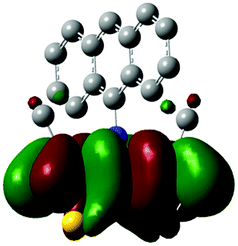
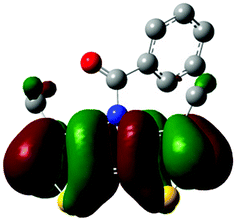
Due to the significantly larger sterical interactions of the methyl groups with the phenyl residue in Me-DTP 5 a dihedral angle of 67° was obtained in the optimized calculated geometry, which is lower than this obtained in the single crystal X-ray structure analysis (88°), in which also packing effects play a role (vide ultra). Compared to the DTP analogue5 this altered structure leads to a more pronounced dominance of the phenyl substituent in the LUMO. This trend is even stronger in Me-DTP 6, as almost only the 9-anthracenyl residue contributes to the LUMO because it exhibits a large sterical demand and therefore enforces an orthogonal geometry, in which the π-orbitals of the Me-DTP unit and the 9-anthryl substituent cannot overlap anymore.
2.4 Optical properties of Me-DTPs 2–7
The optical properties of the synthesized Me-DTPs 2–7 were determined by UV-vis absorption spectroscopy and were recorded in THF solution. The UV-vis spectra of Me-DTPs 2–5 consist of one strong absorption band with a vibrational fine-structure and exhibit very similar maxima (292–296 nm), extinction coefficients, and onset absorptions (Fig. 2, left, Table 2). Hence, alkyl and aryl substituents exert only a marginal influence and therefore almost the same optical energy gaps (Eg = 3.91–3.94 eV) were obtained compared to those of the DTP analogues without β,β′-substitution. In contrast to that, the absorption of benzoyl-substituted Me-DTP 6 exhibited a low energy shoulder extending to nearly 400 nm which we attribute to a charge transfer contribution due to the electron-withdrawing benzoyl substituent (Fig. 2, right).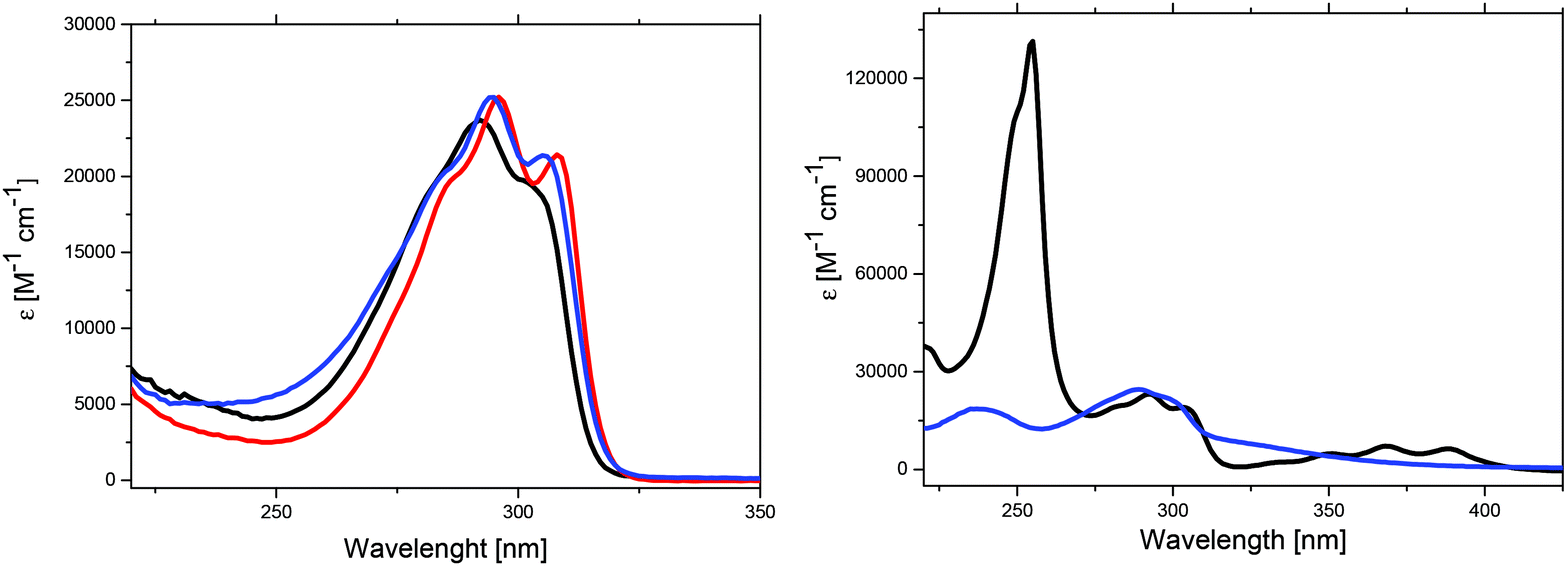 | ||
| Fig. 2 UV-vis absorption spectra of Me-DTPs 2 (black curve), 4 (red), and 5 (blue), left, and of 6 (black) and 7 (blue), right, measured in THF solution. | ||
| Me-DTP | Mp [°C] | λ max [nm] | ε [M−1 cm−1] | λ onset [nm] | E optg [eV] |
|---|---|---|---|---|---|
| 2 | 160–163 | 292 | 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 700 |
315 | 3.94 |
| 3 | 122–124 | 296 | 20310 |
317 | 3.91 |
| 4 | 103–104 | 296 | 25200 |
317 | 3.91 |
| 5 | 162–165 | 295 | 25200 |
317 | 3.91 |
| 6 | 301–305 | ![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) ![[5 with combining low line]](https://www.rsc.org/images/entities/char_0035_0332.gif) , 292, 368 , 292, 368 |
131400 |
406 | 3.05 |
| 7 | 123–124 | 237, ![[8 with combining low line]](https://www.rsc.org/images/entities/char_0038_0332.gif) ![[9 with combining low line]](https://www.rsc.org/images/entities/char_0039_0332.gif) |
24600 |
373 | 3.32 |
In the UV-vis spectrum of 9-anthryl-substituted Me-DTP 7 both molecular moieties contribute and three main bands can be identified. In the range of 270–320 nm, the vibrationally resolved band with a maximum at 292 nm is similar to those of Me-DTPs 2–5 and can be attributed to the Me-DTP unit, whereas the very intense band at 255 nm and the fine-structured band in the range of 320–400 nm are characteristic of the 9-anthryl substituent (Fig. 2, right) and well coincide with those of pure anthracene.22 For the latter two derivatives, consequently the optical energy gaps are substantially reduced and originate from lower LUMO energy levels (vide infra).
2.5 Electrochemical properties and electropolymerization of Me-DTPs
The redox properties of Me-DTPs 2 and 4–7 were investigated by means of cyclic voltammetry in the electrolyte acetonitrile/tetrabutylammonium hexafluorophosphate (TBAPF6, 0.1 M). Potentials are referenced against the ferrocene/ferricenium (Fc/Fc+) redox couple (Table 3). The cyclic voltammograms of the Me-DTPs revealed irreversible oxidation waves with peak potentials in the range of 0.43–0.64 V depending on the electron-withdrawing character of the substituent at the Me-DTP nitrogen. These oxidation potentials are slightly decreased compared to corresponding DTPs indicating inductive stabilization of the formed radical cations by the β-methyl substituents.5 The HOMO energy levels were determined from the onset of the oxidation wave and accordingly gradually decreased from Me-DTPs 2 to 7 (−5.41 eV to −5.65 eV). Due to the absence of reduction waves in the cyclic voltammograms (CV), LUMO energy levels were calculated taking Eoptg into account. An energy level diagram of the frontier orbitals is shown in Fig. 4, left.| Me-DTP |
E
pa
[V] |
E onset [V] | E HOMO [eV] | E LUMO [eV] | p(Me-DTP) |
E
pa
[V] |
E
pc
[V] |
E onset [V] | E HOMO [eV] | Film lossd [%] |
|---|---|---|---|---|---|---|---|---|---|---|
| Potentials are referenced vs. Fc/Fc+.a Epa: anodic peak potential (scan rate 100 mV s−1).b Epc: cathodic peak potential (scan rate 100 mV s−1).c Redox potential of Fc/Fc+ is −5.1 eV on the Fermi scale.23d Determined as the difference of exchanged charges during scan 2 and scan 30.e Difficult to determine due to very broad wave. | ||||||||||
| 2 | 0.43 | 0.31 | −5.41 | −1.47 | P2 | 0.26 | −0.08 | −0.56e | −4.54 | 4 |
| 4 | 0.43 | 0.33 | −5.43 | −1.52 | P4 | 0.24 | 0.07 | −0.03 | −5.07 | 5 |
| 5 | 0.46 | 0.35 | −5.45 | −1.54 | P5 | 0.17 | −0.05 | −0.19 | −4.91 | 6 |
| 6 | 0.52 | 0.44 | −5.54 | −2.49 | ||||||
| 7 | 0.64 | 0.55 | −5.65 | −2.33 | P7 | 0.56 | 0.38 | 0.11 | −5.21 | 6 |
Due to the reactive α-positions of the Me-DTP-units, the formed radical cations undergo follow-up reactions and couple to larger entities according to the well accepted mechanism for the oxidative polymerization of five-membered heterocycles and related monomers (Scheme S1, ESI†).24 By potentio-dynamic cycling of Me-DTP 2 and 4–7 monomers in acetonitrile/TBAPF6 corresponding conducting polymers p(Me-DTP) P2, P4, P5, and P7 were deposited on the platinum working electrode. The regioregular structure of the polymers is shown in Scheme 3. In contrast to the other Me-DTP monomers, 9-anthryl Me-DTP 6 did not show the typical evolution of the cyclic voltammograms during multiple scanning and only very thin unstable polymer films were obtained (Fig. S2, ESI†). Thus, the polymerization process was most likely hampered by the significant sterical demand of the 9-anthryl substituent and only lower molecular weight material was formed.
 | ||
| Scheme 3 Electropolymerization of Me-DTPs 2, 4, 5, and 7 to the corresponding p(Me-DTP)s P2, P4, P5, and P7. | ||
In Fig. 3, the potentiodynamic electropolymerization of phenyl-substituted Me-DTP 5 (a) and the subsequent characterization of the formed p(Me-DTP) film P5 at different scan rates (b) is shown as a representative example for the Me-DTP series. From the first scan in the polymerization, the oxidation of 5 was observed as an irreversible anodic signal at Epa = 0.46 V. With increasing number of cycles during the polymerization, a new redox wave at lower potentials appeared and the currents were continuously increasing, because the formed radical cations initialize the polymerization of 5 and a dark blue film of p(Me-DTP) P5 was formed on the surface of the working electrode. In subsequent scans the oxidation and reduction of the polymer at lower potentials was observed. With increasing number of scans, the currents of these signals increased indicating the growth of the film. The polymer film was then subjected to several scans in a monomer-free electrolyte solution until a stable current response was obtained. At this ‘conditioning’ phase residual oligomeric parts in the film were further polymerized. Subsequently, the polymer film was characterized via cyclic voltammetry applying different scan rates between 10 mV s−1 and 100 mV s−1 (Fig. 3b) whereby an anodic peak potential of Epa = 0.17 V and a cathodic peak potential of Epc = −0.05 V were determined. The linear dependency of the corres-ponding anodic (Ipa) and the cathodic peak currents (Ipc) from the applied scan rate indicated good adherence of the films on the working electrode (Fig. 3d). Finally, 30 cycles were measured with a scan rate of 100 mV s−1 in order to investigate film stability. The exchanged charge just slightly decreased indicating minor losses of 6% of electroactive material (Fig. 3c, Table 4).
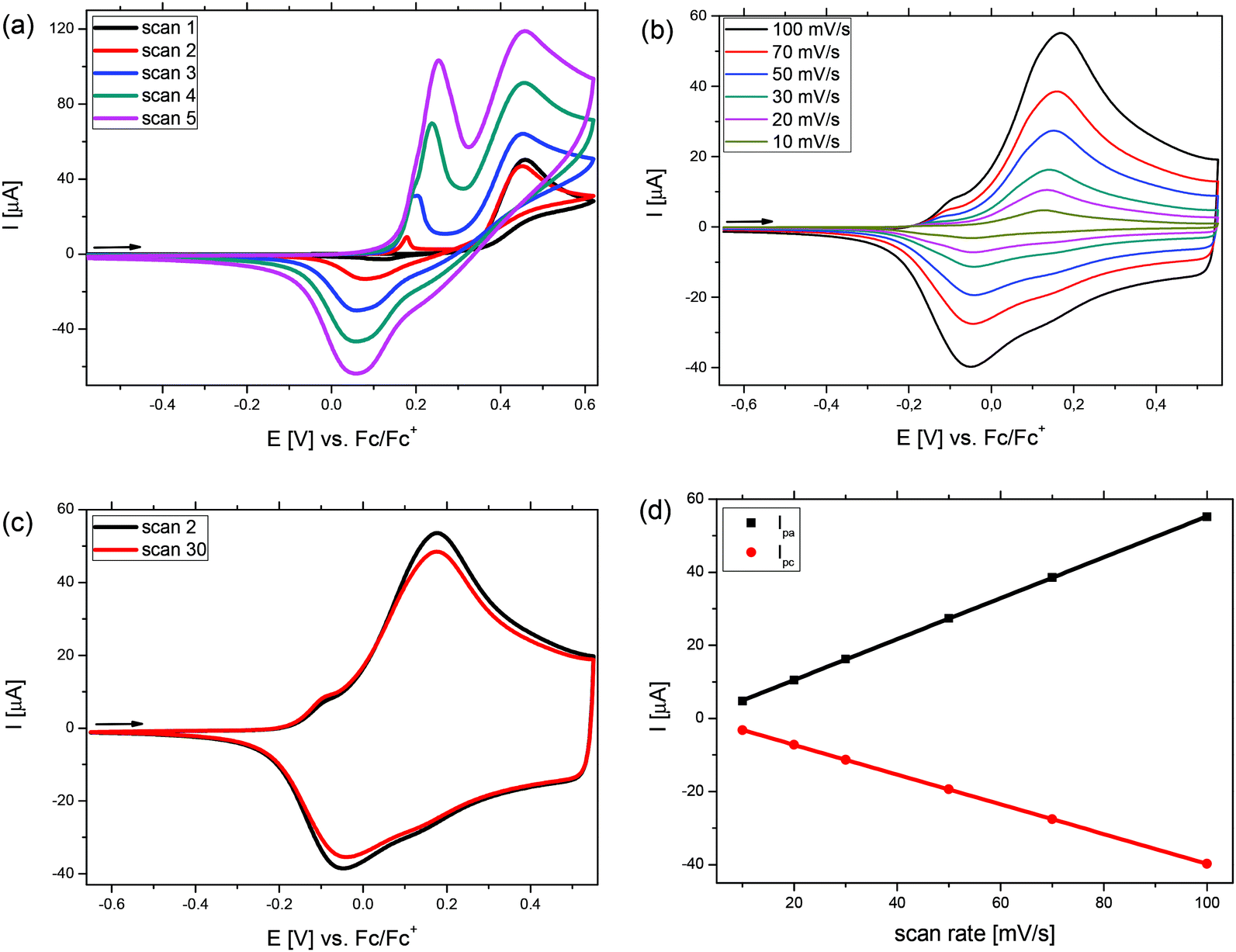 | ||
| Fig. 3 Electropolymerization of phenyl Me-DTP monomer 5 (a) and the characterization of the obtained film P5 using different scan rates (b). Stability test by carrying out 30 cycles at a rate of 100 mV s−1 (c). Dependency of the anodic peak currents Ipa and the cathodic peak currents Ipc from the applied scan rate (d). | ||
The onset oxidation (Eonset) of p(Me-DTP) P5 occurred at −0.19 V (vs. Fc/Fc+) and characterizes the transition from the semiconducting, neutral state to the charged conducting state of the polymer. From this value a HOMO energy level of −4.91 eV was calculated. This is substantially lower than the corresponding HOMO energy level of monomer 5 due to the largely extended π-conjugation in the polymer (Fig. 3, right). The respective electropolymerization data for p(Me-DTP)s P2, P4, and P7 are shown in Fig. S3–S5 (ESI†) and are collected in Table 3. The corresponding HOMO–LUMO energy level diagram for p(Me-DTP)s P2, P4, P5, and P7 is shown in Fig. 4, right. As well the chemically prepared p(Me-DTP)s P4′ is included in this diagram for comparison.
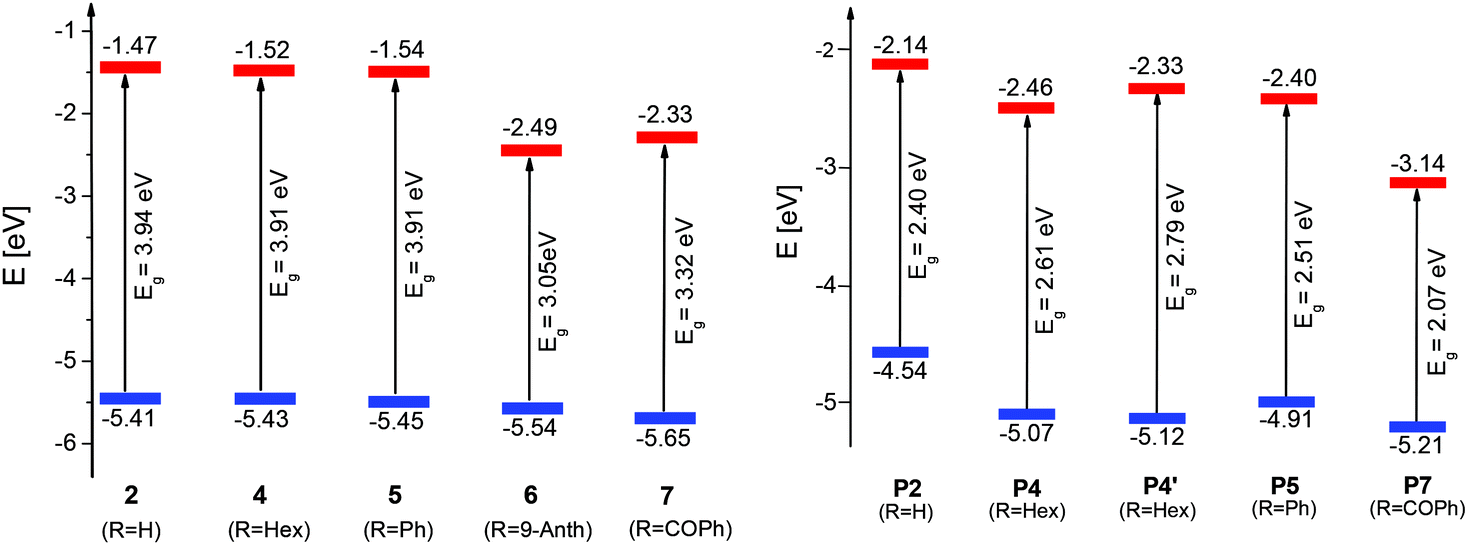 | ||
| Fig. 4 Experimentally determined HOMO (blue) and LUMO (red) energy levels of Me-DTPs 2, 4–7. The energy levels of propyl-substituted Me-DTP 3 are almost identical to the values of hexyl-substituted Me-DTP 4 (left). Energy diagram for the corresponding polymers p(Me-DTP) P2, P4, P5, P7, and P4′. The LUMO energy levels were calculated from the HOMO energy levels and the energy gaps Eg (right). | ||
Except for p(Me-DTP) P2 the onset potentials of P4, P5, and P7 are positively shifted and consequently the HOMO energy levels are lowered by ca. 0.5 eV compared to the analogous polymers pDTPs,5 indicating a decreased mean conjugation length. This finding rationalizes that despite the linear and regioregular structure of the p(Me-DTP)s without α–β or β–β coupling defects, the sterical hindrance of β-methyl groups of adjacent monomer units causes substantial distorsions in the conjugated chain (Scheme 3). In this respect, DFT-calculations on a Me-DTP-trimer revealed dihedral angles of 60° between the repeating units (Fig. S6, ESI†), whereby in a corresponding DTP-trimer (without β-methyl groups) only an angle of 18° relative to each other was found.13
The onset potential and the corresponding HOMO energy level are important parameters of conducting polymers for the stability of the neutral semiconducting and the charged conducting state, respectively, and hence determine their applicability. Thus, the novel p(Me-DTP)s with an onset potential of around −0.2 V indicate a balanced stability in both oxidation states under ambient conditions like pDTPs and polythiophene (PT). On the other hand, the neutral form of poly(3,4-ethylenedioxythiophene) (PEDOT) is immediately oxidized from oxygen or air at around −1 V to the highly stable conducting form, which allows for a wide range of applications such as a transparent electrode material in the field of organic electronics (Table 4).25,27
2.6 Spectroelectrochemical characterization of p(Me-DTP)s
In addition to the electrochemical characterization, the polymer films were investigated via spectroelectrochemistry. A previously described setup28 with a platinum disk working electrode was applied to ensure that the same materials were obtained for both, the electrochemical and the spectroelectrochemical characterization, respectively, and absorption spectra were recorded in reflectance mode by applying potentials from −0.5 V up to 1.4 V vs. Ag/AgCl in acetonitrile/tetrabutylammonium hexafluorophosphate (0.1 M). The sequence of UV-Vis-NIR spectra for p(Me-DTP) P2 are shown in Fig. 5 as a representative example for the whole series. Corresponding spectra for the other polymers are depicted in Fig. S7–S9 (ESI†). By setting the potential to −0.5 V in the beginning of the experiment, the neutral polymer film was obtained without any oxidized residues. A strong absorption band with a maximum at 409 nm and an absorption onset of 514 nm was detected, which correlates to an optical gap of 2.4 eV and explains the dark blue colour of the neutral film. Increasing the applied potential step by step from −0.5 V to 0.9 V led to the partial oxidation of polymer P2 concomitant with the formation of polaronic states. This was indicated by the emergence of two new bands at 628 nm and 1320 nm at the expense of the absorption band of the neutral film. Further oxidation to 1.2 V resulted in full formation of bipolarons, which possessed a new very broad absorption band with a maximum at 1200 nm covering the spectral range from 500 nm to 1600 nm (Table 5).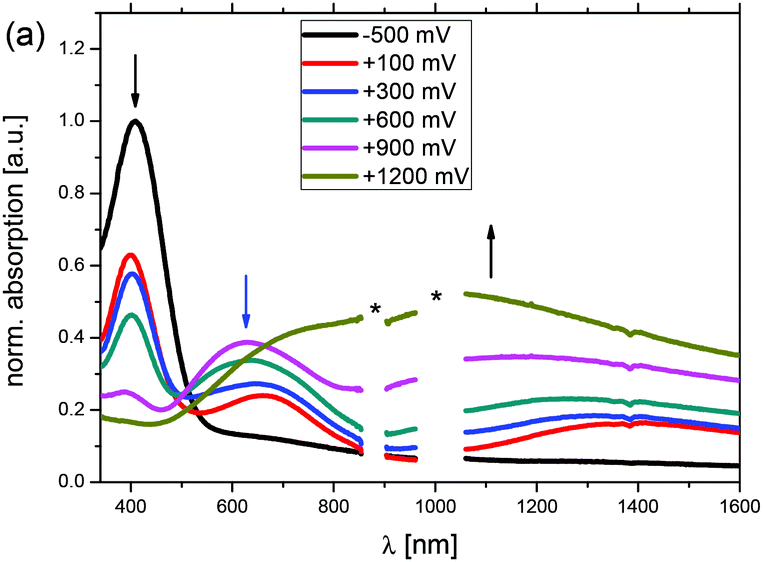 | ||
| Fig. 5 UV-Vis-NIR spectra obtained from spectroelectrochemical measurements of films of p(Me-DTP) P2. Applied voltages are given vs. Ag/AgCl. Black arrows show the changes starting at low potentials. The blue arrow shows the further changes at high potentials. Artefacts due to switching of the detectors in the spectrometer are marked with * in the spectra. | ||
| p(Me-DTP) | λ max [nm] | λ onset [nm] | E g [eV] | E LUMO [eV] | λ max, p [nm] | λ max, bp [nm] |
|---|---|---|---|---|---|---|
| a Maximum was outside of the measured range. p = polaron; bp = bipolaron. | ||||||
| P2 | 409 | 516 | 2.40 | −2.77 | 628, 1320 (br) | 1200 (br) |
| P4 | 400 | 475 | 2.61 | −2.62 | 704, 1330 (br) | |
| P5 | 414 | 494 | 2.51 | −2.66 | 648 | |
| P7 | 459 | 600 | 2.07 | −3.31 | 664 | |
The spectroelectrochemical behaviour of the other p(Me-DTP)s in the series is roughly the same except that the absorption of the neutral P7-film is red-shifted. Like in the corresponding Me-DTP 7, this effect is caused by the electron-withdrawing effect of the benzoyl groups and leads to a decreased energy gap and a stabilized LUMO energy. In comparison to the p(DTP) series,5 absorption maxima of the neutral p(Me-DTP) films are substantially blue-shifted concomitant with larger optical gaps. This is in accordance with the findings from electrochemistry and decreased mean conjugation length. However, in the oxidized states obviously planarization of the monomeric units relative to each other takes place and smears over the differences between the two series.
2.7 Chemical oxidative polymerization of Me-DTP 4
The insolubility of the electrochemically prepared p(Me-DTP) films impeded analysis of their molecular weights and degree of polymerization via gel permeation chromatography (GPC). Hence, Me-DTP 4 bearing a solubilizing hexyl side chain at the DTP-nitrogen was oxidatively polymerized applying four equivalents of iron trichloride as oxidant (Scheme 4). The resulting raw polymer was dedoped by treatment with aqueous hydrazine, washed with n-hexane to remove short polymers, and extracted with THF to afford P4′ in a yield of 21%.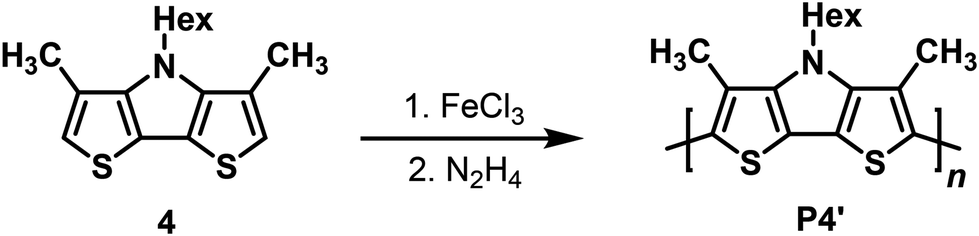 | ||
| Scheme 4 Chemical oxidative polymerization of Me-DTP 4 to soluble (Me-DTP) P4′ (21% yield). | ||
The THF-soluble fraction of p(Me-DTP) P4′ was subjected to GPC and a number average molar mass Mn of 2600 g mol−1 (Mw = 4300 g mol−1) was determined corresponding to a moderate average chain length of 9 repeat units and a polydispersity of 1.7. Thus, rather oligomeric material was obtained which, however, allowed for structural analysis by NMR-spectroscopy. In the 1H-NMR spectrum, the signal for the terminal α–protons was observed at δ = 6.80 ppm as a singlet (Fig. S10, ESI†). Comparison of the integral to the other signals revealed a mean chain length of 8.7 repeat units and hence was in good agreement with GPC data. By analogous oxidation with FeCl3 the hexyl derivative of corresponding (non-methylated) DTP yielded a polymer with higher Mn (8100 g mol−1) and there-fore increased mean chain length.13 However, NMR analysis revealed that the soluble p(DTP)s contained 22–32% α–β or β–β coupling defects. Most likely, such defects increase the solubility of formed oligomeric DTP species and thus enable a larger final degree of polymerization. On the other hand, oligomeric Me-DTP species quickly became insoluble and precipitated due to the absence of any defects. Therefore, regioregular but shorter p(Me-DTP) chains were afforded.
Subsequently, the optical and redox properties of p(Me-DTP) P4′ were determined by cyclic voltammetry and UV-Vis spectroscopy in solution. The absorption spectrum showed a broad band in the range of 250–500 nm with a maximum of 383 nm and an onset absorption at 444 nm resulting in an optical gap of 2.79 eV (Fig. S11, ESI†). The onset oxidation occurred at 0.02 V vs. Fc/Fc+ and corresponded to a HOMO energy level of −5.12 eV whereas the LUMO energy was calculated to −2.33 eV. A rough comparison of the data of soluble polymer P4′ with this of p(Me-DTP) P4 (film) reveals that in polymer P4′ the redox potentials are more positive and the absorptions blue-shifted due to reduced mean conjugation length (Table S6, ESI†).
2.8 Synthesis of acceptor-capped Me-DTPs as dyes for organic electronic application
As already mentioned, DTP-units have frequently been used as building block in conjugated materials for application in organic electronics,1,2 whereby related β,β′-disubstituted DTPs appeared only once in literature and were implemented into a conjugated oligomer for a colorimetric sensor.16 In order to further develop our β,β′-dimethyl-DTPs into convenient molecules for organic electronic applications, light absorption in the visible range of the solar spectrum was targeted. Therefore, we implemented Me-DTPs 3 and 5 as donor (D) into conjugated oligomers with end-capped acceptor (A) groups to yield an A–D–A-type structure. Tailored oligomers of this type have been successfully implemented as photoactive component into organic solar cells reaching power conversion efficiencies (PCE) of over 8%.29 DTP units were also implemented as central core into conjugated hole-transport materials for perovskite solar cells and reached efficiencies over 21%.30Hence, for the envisioned Pd-catalyzed C–C coupling of the basic Me-DTP building blocks with an acceptor-substituted thiophene, firstly, stannylation of Me-DTP 5 was investigated. Neither lithiation of Me-DTP 5 with n-BuLi or t-BuLi and subsequent quenching with trimethylstannyl chloride nor the Pd-catalyzed reaction of 2,6-diiodinated Me-DTP 11 with bis(tributyltin) gave satisfactory results. Instead, always inseparable product mixtures containing starting material and the corresponding monostannyl and distannyl derivatives were isolated. Therefore, we directly used diiodide 11, which was prepared by iodination of Me-DTP 5 with N-iodosuccinimide (NIS) in quantitative yield, in a Stille-type reaction with stannylated dicyanovinylene (DCV)-substituted thiophene 12.15,31 Me-DTP-based A–D–A oligomer 14 was finally separated from several side products, which mostly originated from homocoupling of precursor 11, by high-vacuum gradient sublimation. The product was obtained in pure form but in a low yield of only 5% due to large amounts of decomposed sample remaining in the source.
Hence, the synthetic procedure for propyl-substituted oligomer 13 was slightly adapted. Twofold iodination of Me-DTP 3 with NIS gave diiodide 10 in 90% yield, which was now coupled with stannyl 12 and the less active palladium catalyst Pd(PPh3)2Cl2 instead of Pd(PPh3)4 in order to avoid larger fractions of homocoupling products. A–D–A oligomer 13 could be purified via column chromatography and was obtained in 63% yield. Subsequent gradient sublimation worked more efficiently than in the case of the raw phenyl-substituted analogue 14 and a sublimation yield of 73% for very pure oligomer 13 was achieved. Structures and purity were confirmed by NMR spectroscopy and high-resolution MALDI mass spectrometry (Scheme 5).
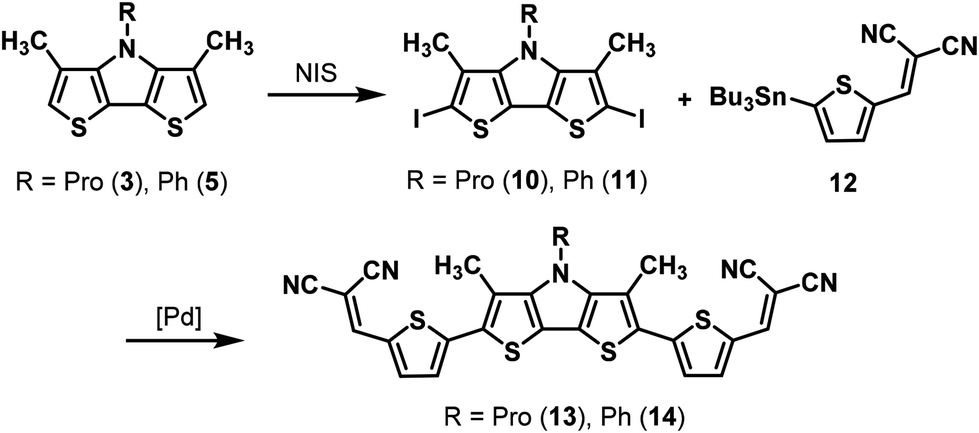 | ||
| Scheme 5 Synthesis of Me-DTP-based A–D–A oligomers 13 and 14via Stille-type cross-coupling. | ||
The thermal properties of the synthesized oligomers 13 and 14 were determined by differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA). Corresponding melting points and decomposition temperatures, respectively, are listed in Table 6. We assume that due to the almost perpendicular distortion of the phenyl ring in oligomer 14 the π–π stacking of the conjugated backbone in the solid state is reduced and therefore the melting point is lower compared to propyl-substituted oligomer 13. Both derivatives are stable up to around 350 °C.
| Oligomer | Mp [°C] |
T
d
[°C] |
λ max, abs [nm] | ε [M−1 cm−1] | λ onset, abs [nm] | E optg [eV] | λ max, em [nm] |
|---|---|---|---|---|---|---|---|
| a Decomposition temperature Td determined from the onset of the mass loss in the TGA curves. b E optg was determined from λonset, abs. | |||||||
| 13 | 323–325 | 351 | 573 | 64100 |
649 | 1.91 | 676 |
| 14 | 305–310 | 356 | 573 | 73400 |
646 | 1.92 | 670 |
Absorption and fluorescence spectra were measured in dichloromethane solution and are shown in Fig. 6 (left), while results are summarized in Table 6. The most intense absorption band in the visible range with a maximum at 573 nm for both derivatives correspond to a π–π* transition with charge-transfer contribution and explains their deep blue colour. Compared to the Me-DTP core molecules 3 and 5, the elongation of the conjugated π-system in the A–D–A structures 13 and 14 leads to a strong intensification of the absorption and to bathochromic shifts of nearly 280 nm. At the same time, the optical gaps Eoptg, which are determined from the onset absorption, are substantially reduced from 3.91 eV to 1.91 eV. Similar to the core molecules, alkyl and aryl substituents therefore exert only a negligible direct influence on the optical properties of the A–D–A-type oligomers.
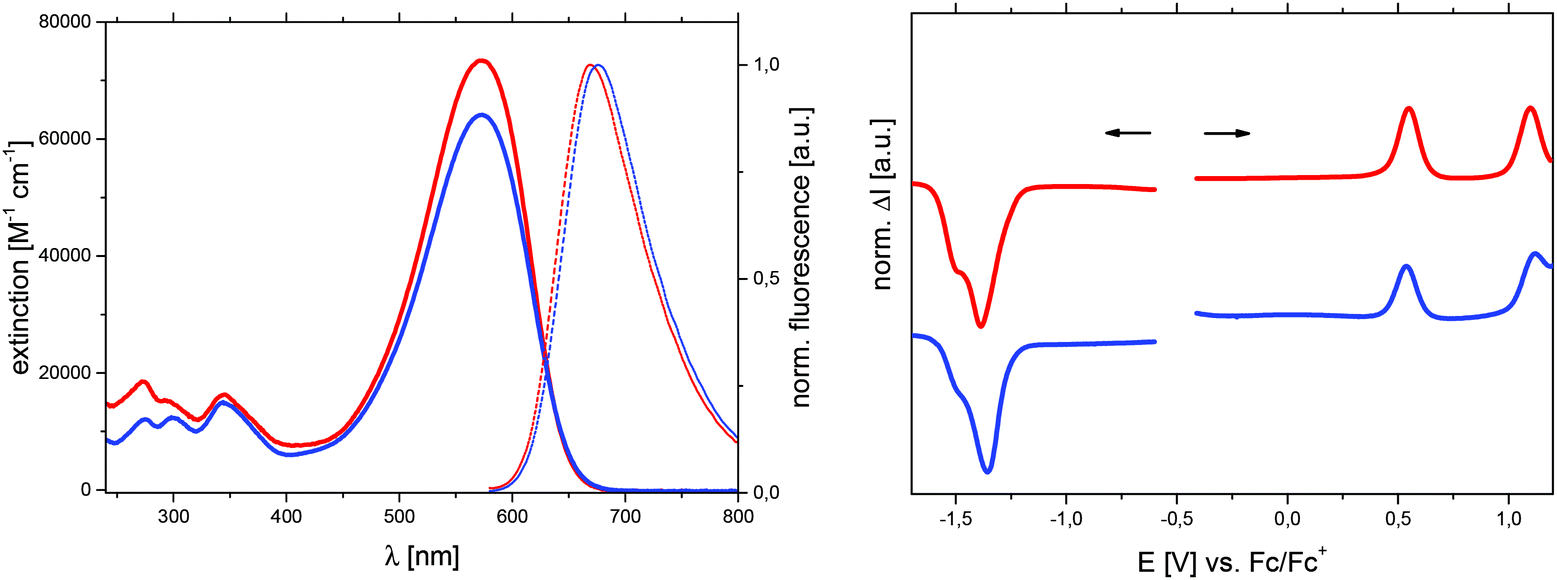 | ||
| Fig. 6 UV-Vis absorption (solid lines) and normalized fluorescence spectra (dotted lines) of Me-DTP-based oligomers 13 (red curves) and 14 (blue) recorded in dichloromethane solution. For the fluorescence spectra the compounds were excited at 560 nm at a concentrations of 10−7 M (left). Square wave voltammograms of oligomers 13 and 14 recorded in dichloromethane/TBAPF6 (0.1 M) at a concentration of 0.5 μM, potentials vs. Fc/Fc+ (right). | ||
Because of their poor solubility, the redox properties of Me-DTP oligomers 13 and 14 were determined by square wave voltammetry at a concentration of 0.5 μM (Fig. 6, right). The redox potentials are listed in Table 7 and the energy levels of the frontier molecular orbitals were determined from the onset of the first oxidation and reduction wave. The energy gap Eecg was calculated from the difference of the HOMO and LUMO energies. As discussed for the optical properties, the substituent at the nitrogen does not significantly influence the electrochemical properties as well. Thus, independent from the propyl or phenyl group, both oligomers are oxidized at 0.54–0.55 V and 1.10–1.12 V to radical cations and dications, respectively, which are delocalized on the core donor unit. The terminal DCV groups are reduced at potentials of −1.36 to −1.38 V. Thereof, HOMO and LUMO energy levels of ca. −5.55 eV and −3.85 eV were calculated, respectively, leading to a HOMO–LUMO energy gap of 1.70 eV which is in good accordance with comparable DTP-based A–D–A-type oligomers.32 First vacuum-processed bulk-heterojunction solar cells with Me-DTP-based oligomer 13 as photoactive donor in conjunction with fullerene C60 as acceptor yielded promising photovoltaic properties.33
| Oligomer |
E
ox1a [V] |
E
ox2b [V] |
E
redc [V] |
HOMO d [eV] | LUMO d [eV] |
E
ecge [eV] |
|---|---|---|---|---|---|---|
| Potentials are referenced vs. Fc/Fc+.a Eox1: first oxidation potential.b Eox2: second oxidation potential.c Ered: reduction potential.d Redox potential of Fc/Fc+ is −5.1 eV on the Fermi scale.23e Difference of electrochemically determined HOMO and LUMO energy levels. | ||||||
| 13 | 0.54 | 1.12 | −1.36 | −5.54 | −3.84 | 1.70 |
| 14 | 0.55 | 1.10 | −1.38 | −5.55 | −3.86 | 1.69 |
3. Conclusions
We synthesized a series of novel β,β′-dimethyl-substituted dithieno[3,2-b:2′,3′-d]pyrroles (Me-DTP) 2–7 with various substituents (H, alkyl, aryl, and benzoyl) at the DTP nitrogen in good yields. Compared to analogous DTPs,5 the anchoring of methyl groups at the β,β′-positions of the DTP unit significantly changed structural and optoelectronic properties of the molecules. Various analyses corroborated by quantum chemical calculations showed that the dihedral angle between the DTP core and the substituent substantially increases due to the steric interaction with the adjacent methyl groups. Therefore, UV-vis absorptions of Me-DTPs are slightly blue-shifted, whereas oxidation is facilitated due to the inductive effect of the methyl groups.By oxidative polymerization of Me-DTP monomers, the corresponding regioregular conducting polymers p(Me-DTP) were obtained as adhering films on the working electrode or as soluble fractions. Compared to the series of p(DTP)s, onset potentials were shifted to more positive values concomitant with a stabilization and decrease of the HOMO energies. Two effects were responsible for this finding: firstly, the steric demand of the methyl groups leads to distortions of adjacent repeating units which diminishes the mean conjugation length. Secondly, the mean degree of polymerization is lower for the p(Me-DTP) series, presumably due to lower solubilities of intermediate defect-free oligomers. Notably, the steric impact of the methyl groups is however cancelled in the oxidized conducting state of the polymers. Here, the absorptions of the respective p(Me-DTP) polarons and bipolarons were found at lower energies compared to analogous p(DTP)s.
In a third series, Me-DTP building blocks were integrated into A–D–A-type co-oligomers representing functional dyes for organic electronic applications. Their absorption is covering a wide range of the visible spectrum showing an energy gap of 1.70 eV, which is in particular attractive for application in organic solar cells. In summary, on the basis of novel dithieno[3,2-b:2′,3′-d]pyrroles (Me-DTP) 2–7, three types of π-conjugated materials were prepared and investigated leading to valuable structure–property relationships.
4. Experimental section
4.1 Instruments and measurements
Thin layer chromatography was carried out on aluminium plates, precoated with silica gel, Merck Si60 F254. Preparative column chromatography was performed on glass columns packed with silica gel (particle size 40–63 μm) from Macherey-Nagel. Melting points were determined using a Büchi Melting Point B-545 or a Mettler Toledo DSC 823e under argon flow (heating rate 10 °C min−1). UV-Vis absorption spectra were recorded in THF solutions on a PerkinElmer Lambda 19 spectrometer. NMR spectra were recorded on an Avance 400 spectrometer (1H NMR: 400 MHz, 13C NMR: 101 MHz). Chemical shifts (δ) are reported in ppm using residual solvent protons (1H NMR: δH = 7.26 for CDCl3; δH = 5.32 for CD2Cl2; δH = 3.58 for THF-d8; 13C NMR: δC = 77.16 for CDCl3; δC = 53.84 for CD2Cl2) as internal standard. The splitting patterns are designated as follows: s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Coupling constants J relate to proton-proton couplings. Protons at the α-positions of DTP were assigned as Th-Hα. Elemental analyses were performed on an Elementar Vario EL. GC/EI-MS (70 eV) measurements were performed on a Shimadzu GCMS-QP2010 SE. Chemical ionisation (CI) mass spectra were measured on a Finnigan MAT SSQ-7000.X-ray diffraction data of a colourless plate-shaped single crystal of Me-DTP 5 were collected in a stream of nitrogen at 150 K on an Agilent SuperNova, Cu at zero, Atlas CCD using graphite-monochromated Cu Kα radiation. Data collection, data reduction, and cell refinement were performed using the CrysAlisPro software.34 An absorption correction based on the semi-empirical “multi-scan” approach was performed using the SCALE3 ABSPACK scaling algorithm.34 The structure was solved by charge flipping using Superflip.35–37 For the final model all non-hydrogen atoms were refined anisotropically using SHELXL.38
Cyclic voltammetry experiments were performed using a computer-controlled Autolab PGSTAT 30 potentiostat in a three-electrode single-compartment cell with a platinum working electrode, a platinum wire counter electrode, and a Ag/AgCl reference electrode. All potentials were internally referenced to the ferrocene/ferricenium (Fc/Fc+) couple. Deoxygenated acetonitrile dried over molecular sieves or THF dried via a MB SPS-800 solvent purifying system (MBraun) was used as the solvent. Solutions of tetrabutylammonium hexafluorophosphate (Sigma Aldrich) as the supporting electrolyte with a concentration of 0.1 M were applied and were blanketed with argon during the measurements. For electropolymerizations the respective monomer was used in a concentration of 0.01 M in acetonitrile. The obtained film was washed with dry acetonitrile and subjected to several scans in a monomer-free electrolyte solution until a stable current response was recorded (“conditioning”). Subsequently, the respective film was characterized by cyclic voltammetry using different scan rates between 100 mV s−1 and 10 mV s−1. At every scan rate three scans were conducted. Additionally, 30 scans were measured with a scan rate of 100 mV s−1.
Spectroelectrochemical measurements of the polymer films were carried out in a 0.1 M solution of tetrabutylammonium hexafluorophosphate in dry acetonitrile. The applied setup has been described in detail in literature.28 A Princeton Applied Research PAR 363 potentiostat was used together with a platinum working electrode, an Ag/AgCl reference electrode, and a platinum sheet as the counter electrode. Polymer films were prepared electrochemically as stated above, including the “conditioning” phase. During the recording of the UV-Vis-NIR spectra the applied potential was kept constant. In order to obtain the UV-Vis-NIR spectrum of the neutral polymer film without any oxidized parts, a potential of −500 mV vs. Ag/AgCl was applied at the beginning of the spectroelectrochemical measurements. Instrumental artefacts were removed and marked in the spectra. Moreover, the absorption in the UV-Vis region below 860 nm was adjusted to the absorption at higher wavelengths to obtain continuous spectra.
GPC-UV analysis with THF as eluent was performed at 1 mL min−1 on a Merck Hitachi LaChrom GPC system equipped with an L-7100 HPLC pump, L-7420 UV-Vis detector, and three columns (PSS SDV 103 Å, 104 Å, and 105 Å). The columns were kept in a column heater at 35 °C and were calibrated with polystyrene standards (Polymer Standards Service).
Quantum chemical calculations were carried out with the Gaussian 09 program.39 Alkyl residues were shortened to ethyl in order to save computational time. Geometry optimizations and energy calculations were performed via DFT methods using the M06-2X20 correlation/exchange functional and the 6-311+G(d) or the 6-311G basis set. Molecular orbital surfaces were generated utilizing the Gauss View 5.0 program with an isovalue of 0.02.
4.2 Materials
Toluene (VWR) and THF (Sigma Aldrich) were dried and purified by a MB SPS-800 (MBraun). Chloroform, methanol, ethanol, diethyl ether, petroleum ether, toluene, dichloromethane, and n-hexane were purchased from VWR and distilled prior to use. Diisopropylamine and trimethylamine were purchased from VWR and dried over calcium hydride. Propylamine, hexylamine, and aniline were purchased from Merck and dried over calcium hydride. CuCl2 was purchased from Merck and dried in vacuo at 150 °C prior to use. Sodium tert-butoxide, copper(I) iodide, acetic acid, 3-methylthiophene, ammonium acetate, and anhydrous iron trichloride were purchased from Merck. N,N′-Dimethylethylenediamine (DMEDA) was purchased from Alfa Aesar. Potassium carbonate, Pd(dba)2, Pd2(dba)3, N-bromosuccinimide, N-iodosuccinimide, trimethyltin chloride, 2-(tributylstannyl)thiophene, malono-dinitrile, hydrochloric acid (35%), and 2-dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl (RuPhos) were purchased from Sigma Aldrich. Benzamide, zinc, and n-butyl lithium were purchased from Acros Organics. 1,1′-Bis(diphenylphosphino)ferrocene (dppf) and bis(triphenylphosphine)palladium dichloride was purchased from Fluorchem. 9-Aminoanthracene,40 3,3′-dibromo-4,4′-dimethyl-2,2′-bithiophene 1,17 and 2-{[5-(tributylstannyl)thiophen-2-yl]methylene}malonodinitrile 1215 were synthesized according to published procedures. All synthetic steps were carried out under argon atmosphere.4.3 Synthesis
:1) to yield Me-DTP 2 as a colourless solid (391 mg, 1.89 mmol, 63%). mp 160–163 °C; 1H NMR (CD2Cl2, 400 MHz): δ = 8.30 (s, 1 H, NH), 6.77 (q, J = 1.1 Hz, 2 H, Th-Hα), 2.38 (d, J = 1.2 Hz, 6 H, CH3) ppm; 13C NMR (CD2Cl2, 101 MHz): δ = 143.9, 122.7, 118.9, 116.5, 13.4 ppm; CI-MS: m/z (%) = 207 (100) [M]+; elemental analysis: calc. (%) for C10H9NS2: C 57.94, H 4.38, N 6.76, S 30.93; found: C 57.70, H 4.58, N 6.67, S 30.95; UV-Vis (THF) λmax (ε): 301 (sh, 19800), 292 (23700), 281 (sh, 18700) nm.
200), 296 (20300), 286 (sh, 16100) nm.
400), 296 (25200), 286 (sh, 19700) nm.
400), 295 (25200), 285 (sh, 20300) nm.
:DCM = 6:1) and recrystallized from n-hexane and toluene to yield Me-DTP 6 as a yellow solid (284 mg, 0.74 mmol, 74%). mp 301–305 °C (DSC); 1H NMR (CD2Cl2, 400 MHz): δ = 8.68 (s, 1 H, H10′), 8.14 (d, J = 8.4 Hz, 2 H, H1′, H8′), 7.55–7.50 (m, 2 H, H4′, H5′), 7.45–7.38 (m, 4 H, H2′, H3′, H6′, H7′), 6.67 (q, J = 1.1 Hz, 2 H, Th-Hα), 1.20 (d, J = 1.2 Hz, 6 H, CH3) ppm; 13C NMR (CD2Cl2, 101 MHz): δ = 145.9, 131.5, 131.3, 130.2, 128.9, 128.6, 127.8, 126.2, 123.7, 123.4, 119.5, 116.4, 12.8 ppm; GC/EI-MS (70 eV): tR = 27.9 min; m/z (%) = 383 (100) [M]+; CI-MS: m/z (%) = 384 (100) [M + H]+; elemental analysis: calc. (%) for C24H17NS2: C 75.16, H 4.47, N 3.65, S 16.72; found: C 75.29, H 4.55, N 3.79, S 16.61; UV-Vis (THF) λmax (ε): 388 (6 400), 368 (7 200), 351 (4 900), 334 (sh, 2 300), 303 (19100), 292 (23100), 282 (sh, 19400), 255 (131300), 250 (sh, 109700) nm.
:DCM = 3:1) to yield unsubstituted Me-DTP 2 as a colourless solid (135 mg, 0.65 mmol, 22%) and benzoyl-substituted Me-DTP 7 as a yellow solid (337 mg, 1.08 mmol, 36%). The analytical data of unsubstituted Me-DTP 2 was in accordance with the data above. Analytical data of benzoyl-substituted Me-DTP 7: mp 123–124 °C; 1H NMR (CD2Cl2, 400 MHz): δ = 7.84–7.80 (m, 2 H, o-Ph-H), 7.70–7.64 (m, 1 H, p-Ph-H), 7.53–7.48 (m, 2 H, m-Ph-H), 6.79 (q, J = 1.1 Hz, 2 H, Th-Hα), 1.84 (d, J = 1.1 Hz, 6 H, CH3) ppm; 13C NMR (CD2Cl2, 101 MHz): δ = 167.6, 143.1, 135.6, 134.3, 131.2, 129.3, 125.7, 121.6, 120.9, 15.9 ppm; CI-MS: m/z (%) = 311 (52) [M]+; elemental analysis: calc. (%) for C17H13NOS2: C 65.57, H 4.21, N 4.50, S 20.59; found: C 65.41, H 4.05, N 4.65, S 20.59; UV-Vis (THF) λmax (ε): 324 (sh, 7 200), 297 (sh, 21000), 289 (23000), 241 (sh, 16700) nm.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) cm−1.
100), 344 (15000), 298 (12400), 275 (12100) nm.
400), 346 (16300), 295 (sh, 15100), 273 (18500) nm.
C) cm−1.
100), 344 (15000), 298 (12400), 275 (12100) nm.
400), 346 (16300), 295 (sh, 15100), 273 (18500) nm.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge B. Müller (Institute of Inorganic Chemistry II, Ulm University) for the single crystal X-ray structure analysis.Notes and references
- S. C. Rasmussen and S. J. Evanson, Prog. Polym. Sci., 2013, 38, 1773–1804 CrossRef CAS.
- J. Cao, F. Du, L. Yang and W. Tang, J. Mater. Chem. A, 2020, 8, 22572–22592 RSC.
- W. Zhang, J. Li, B. Zhang and J. Qin, Macromol. Rapid Commun., 2008, 29, 1603–1608 CrossRef CAS.
- A. Berlin, G. Pagani, G. Zotti and G. Schiavon, Macromol. Chem., 1992, 193, 399–409 CrossRef CAS.
- S. Förtsch and P. Bäuerle, Polym. Chem., 2017, 8, 3586–3595 RSC.
- X. Zhang, T. T. Steckler, R. R. Dasari, S. Ohira, W. J. Potscavage Jr. and S. P. Tiwari, J. Mater. Chem., 2010, 20, 123–124 RSC.
- K. Ogawa and S. C. Rasmussen, Macromolecules, 2006, 39, 1771–1778 CrossRef CAS.
- S. J. Evenson and S. C. Rasmussen, Org. Lett., 2010, 12, 4054–4057 CrossRef CAS PubMed.
- Y. A. Udum, H. B. Yildiz, H. Azak, E. Sahin, O. Talaz, A. Cirpan and L. Toppare, J. Appl. Polym. Sci., 2014, 131, 40701 CrossRef.
- J. F. Mike, L. Shao, J.-W. Jeon and J. F. Lutkenhaus, Macromolecules, 2014, 47, 79–88 CrossRef CAS.
- G. Koeckelberghs, L. De Cremer, A. Persoons and T. Verbiest, Macromolecules, 2007, 40, 4173–4181 CrossRef CAS.
- P.-O. Schwartz, S. Förtsch, E. Mena-Osteritz, D. Weirather-Köstner, M. Wachtler and P. Bäuerle, RSC Adv., 2018, 8, 14193–14200 RSC.
- S. Förtsch, PhD thesis, University of Ulm, Germany, 2018.
- S. C. Rasmussen, E. J. Uzelac and E. W. Cluiver, Adv. Heterocycl. Chem., 2020, 130, 75–144 CrossRef CAS.
- A. Vogt, F. Schwer, S. Förtsch, C. Lorenz, E. Mena-Osteritz, A. Aubele, T. Kraus and P. Bäuerle, Eur. J. Org. Chem. Search PubMed , submitted.
- J. P. Green, H. Dai, F. Aniés and M. Heeney, Macromolecules, 2020, 53, 6849–6855 CrossRef.
- K. Kawabata, M. Takeguchi and H. Goto, Macromolecules, 2013, 46, 2078–2091 CrossRef CAS.
- S. Förtsch and P. Bäuerle, J. Phys. Org. Chem., 2017, 30, e3743 CrossRef.
- S. J. Evenson, T. M. Pappenfus, M. C. R. Delgado, K. R. Radke-Wohlers, J. T. L. Navarrete and S. C. Ramussen, Phys. Chem. Chem. Phys., 2012, 14, 6101–6111 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- K. Ogawa and S. C. Rasmussen, J. Org. Chem., 2003, 68, 2921–2928 CrossRef CAS PubMed.
- M. Taniguchi and J. S. Lindsey, Photochem. Photobiol., 2018, 94, 290–327 CrossRef CAS PubMed.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- P. Audebert and F. Miomandre, Electrochemistry of Conducting Polymers, in Handbook of Conducting Polymers, ed. T. A. Skotheim and J. R. Reynolds, CRC Press, Boca Raton, USA, 3rd edn, 2007, pp. 1–18 Search PubMed.
- A. V. Volkov, K. Wijeratne, E. Mitraka, U. Ail, D. Zhao, K. Tybrandt, J. W. Andreasen, M. Berggren, X. Crispin and I. V. Zozulenko, Adv. Funct. Mater., 2017, 27, 1700329 CrossRef.
- R. B. Araujo, A. Banerjee, P. Panigrahi, L. Yang, M. Sjödin, M. Strømme, C. M. Araujo and R. Ahuja, Phys. Chem. Chem. Phys., 2017, 19, 3307–3314 RSC.
- S. Kirchmeyer, A. Elschner, W. Lovenich and K. Reuter, PEDOT: Principles and Applications of an Intrinsically Conductive Polymer, CRC Press, Taylor & Francis Inc, 2010 Search PubMed.
- J. Salbeck, J. Electroanal. Chem., 1992, 340, 169–195 CrossRef CAS.
- I. Ata, S. B. Dkhil, M. Pfannmöller, S. Bals, D. Duché, J.-J. Simon, C. Videlot-Ackermann, O. Margeat, J. Ackermann and P. Bäuerle, Org. Chem. Front., 2017, 4, 1561–1573 RSC.
- J. Cao, F. Du, L. Yang and W. Tang, J. Mater. Chem. A, 2020, 8, 22572–22592 RSC.
- M. Yuchao, W. Jianchang, G. Wei, Y. Ni, W. Lilei and M. Changqi, Patent CN, 105646559, Jun 08, 2016 Search PubMed.
- G. L. Schulz, P. Kar, M. Weidelener, A. Vogt, M. Urdanpilleta, M. Linden, E. Mena-Osteritz, A. Mishra and P. Bäuerle, J. Mater. Chem. A, 2016, 4, 10514–10523 RSC.
- Unpublished results.
- CryaAlisPro, Agilent Technologies XRD Products.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- L. Palatinus and A. van der Lee, J. Appl. Crystallogr., 2008, 41, 975–984 CrossRef CAS.
- L. Palatinus, S. J. Prathapa and S. van Smaalen, J. Appl. Crystallogr., 2012, 45, 575–580 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- H. Adams, R. A. Bawa, K. G. McMillan and S. Jones, Tetrahedron: Asymmetry, 2007, 18, 1003–1012 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2070707. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1py00479d |
| ‡ New address: IFF, August-Wolff-Straße 13, 29699 Walsrode, Germany. |
| This journal is © The Royal Society of Chemistry 2021 |