
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homo- and co-polymerisation of di(propylene glycol) methyl ether methacrylate – a new monomer†
Anna P.
Constantinou
a,
Georgios
Patias
 b,
Birsen
Somuncuoğlu
a,
Toby
Brock
a,
Daniel W.
Lester
b,
David M.
Haddleton
b and
Theoni K.
Georgiou
*a
b,
Birsen
Somuncuoğlu
a,
Toby
Brock
a,
Daniel W.
Lester
b,
David M.
Haddleton
b and
Theoni K.
Georgiou
*a
aDepartment of Materials, Imperial College London, Exhibition Road, SW7 2AZ, UK. E-mail: t.georgiou@imperial.ac.uk
bDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK
First published on 12th May 2021
Abstract
In this study, a new methacrylate monomer with two propylene glycol groups on the side chain, di(propylene glycol) methyl ether methacrylate (diPGMA), was synthesised via an esterification reaction. This new monomer was homo- and co-polymerised for the first time via group transfer polymerisation (GTP). Nine ABA triblock copolymers were synthesised via a “one-pot” GTP, with A and B blocks being based on the hydrophilic and the thermoresponsive oligo(ethylene glycol) methyl ether methacrylate, average Mn 300 g mol−1 (OEGMA300), and the hydrophobic diPGMA, respectively. The molar mass (MM) and OEGMA300/diPGMA content was systematically varied and the effect of this on the self-assembly and thermoresponsive properties was investigated. All copolymers were shown to self-assemble into aggregates and the size of these aggregates increased with both the MM of the polymer and the polymer content in diPGMA. A thermoresponse was observed in aqueous media, with the cloud point (CP) decreasing as the hydrophobic content increases, and the MM decreases. In concentrated aqueous solutions, the polymer with the highest MM and highest diPGMA content formed gels, whose storage modulus increases as a function of the concentration. This study reports a promising alternative hydrophobic monomer to be used in the fabrication of thermogelling materials.
Introduction
Thermo-responsive polymers (temperature-responsive polymers) are polymers that respond to temperature.1–11 When aqueous solvents are present, the response is seen as a change in the hydrophilicity of the structure. When the hydrophilicity, and thus the water solubility, increases or decreases when the solution is heated, the polymers are characterised by either an upper critical or lower critical solution temperature, UCST or LCST, respectively. LSCT polymers have attracted much scientific interest for potential in biological applications, as they are soluble at low temperatures, at which they can be easily mixed with drugs and cells, without compromising their structural integrity. Under the appropriate external conditions, solvent, pH and temperature, depending on the molecular structure, the incompatibility with the solvent can be manifest by the formation of a 3D physical network. These networks are known as thermoresponsive gels (TRGs) and find numerous applications in biomedical engineering as injectable gels for tissue engineering and drug delivery, and 3D printing.1–6,12,13One of the most extensively studied families of thermoresponsive polymers are poloxamers, i.e. ABA triblock copolymers with A and B blocks being based on ethylene glycol (EG) and propylene glycol (PG), respectively.14,15 Whilst poly(ethylene glycol) (PEG) is highly hydrophilic and thermoresponsive at high temperatures depending on its molar mass (MM),16,17 poly(propylene glycol) (PPG) is thermoresponsive at lower temperatures.18,19 More specifically, PPG is hydrophilic below room temperature, depending on the concentration, whilst it is hydrophobic at higher temperatures, thus promoting self-assembly.18,19 Numerous registered tradenames are available for both the solid polymers and solutions, including Pluronic® (BASF), Lutrol® (BASF), Kolliphor® (BASF), Synperonic® (Croda) and Antarox® (Rhodia).14,15,20 Various poloxamers are commercially available with differences in the EG/PG ratio and the total MM.
Poloxamers have been previously reported in studies concerning injectable gels, including poloxamer 407 (P407, Pluronic® F127),21–23 poloxamer 188 (P188, Pluronic® F68),24,25 poloxamer 124 (P124, Lutrol® L44),22 and poloxamer 105 (P105, Pluronic® L35).26 Amongst these, an extensively used material in thermogelling systems is Pluronic® F127. This polymer has a total MM of approximately 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 g mol−1, and a composition of EG-PG = 70–30 w/w%.14 Its aqueous solutions form gels at a concentration of at least 15 w/w%, with gels being formed at room temperature at only 20 w/w%. Owing to its commercial availability and interesting thermogelling properties, many studies have been focused on applying it either as in vivo injectable gel21–23 and as 3D printable material.27,28
600 g mol−1, and a composition of EG-PG = 70–30 w/w%.14 Its aqueous solutions form gels at a concentration of at least 15 w/w%, with gels being formed at room temperature at only 20 w/w%. Owing to its commercial availability and interesting thermogelling properties, many studies have been focused on applying it either as in vivo injectable gel21–23 and as 3D printable material.27,28
As PEG is highly hydrophilic and largely biocompatible, it has often been incorporated into biological systems to enhance biocompatibility.29,30 (Meth)acrylate derivatives of PEG are commercially available and have gained significant scientific interest in the last two decades.31–42 When polymerised, the backbone of the polymer consists of carbon–carbon bonds, with the ether bonds (originating from the EG units) being on the side chain, and their thermoresponsive ability is affected by the number of EG repeating units.34,40,41,43 Poloxamers contain both EG and PG repeating units in a linear polymer consisting of ether bonds along the backbone. Using (meth)acrylate PEG and PPG derivatives is advantageous as a wider variety of monomers can be used, including functional groups at which peptides or fluorescent groups might be attached post-polymerisation. In addition, these functional monomers could be polymerised at different points within the polymer chain with appropriate polymerisation methodology, unlike in poloxamers, in which the functionalisation can only take place at the ends of the polymer chain.
Inspired by poloxamers, this study focused on synthesising methacrylate ABA block copolymers with PEG and PPG side chains. For this, oligo(ethylene glycol) methyl ether methacrylate with average Mn 300 g mol−1 (OEGMA300, OEG, giving average number of EG units = 4.5) was used to form the outer hydrophilic A blocks. For the central block, an in-house synthesised methacrylate monomer with two PG groups on the side chain, namely di(propylene glycol) methyl ether methacrylate (diPGMA, diPG), was used, Fig. 1. This monomer exists as a mixture of four isomers: (i) 2-(2-methoxy-2-methylethoxy)-1-propyl methacrylate [2-(2-M-2-ME)-1-P]MA, (ii) 1-(2-methoxy-1-methylethoxy)-2-propyl methacrylate [1-(2-M-1-ME)-2-P]MA, (iii) 2-(2-methoxy-1-methylethoxy)-1-propyl methacrylate [2-(2-M-1-ME)-1-P]MA, and (iv) 1-(2-methoxy-2-methylethoxy)-2-propyl methacrylate [1-(2-M-2-ME)-2-P]MA, Fig. 1. This newly synthesised hydrophobic monomer was both homo- and co-polymerised via Group Transfer Polymerisation (GTP). Nine copolymers were synthesised in total which differ in the MM (5100, 10100, and 13100 g mol−1) and OEGMA300-diPGMA composition (80–20, 70–30, and 60–40 w/w%). The monomer and polymer synthesis, as well as the investigation of the aqueous solution properties of the polymers are presented and discussed below.
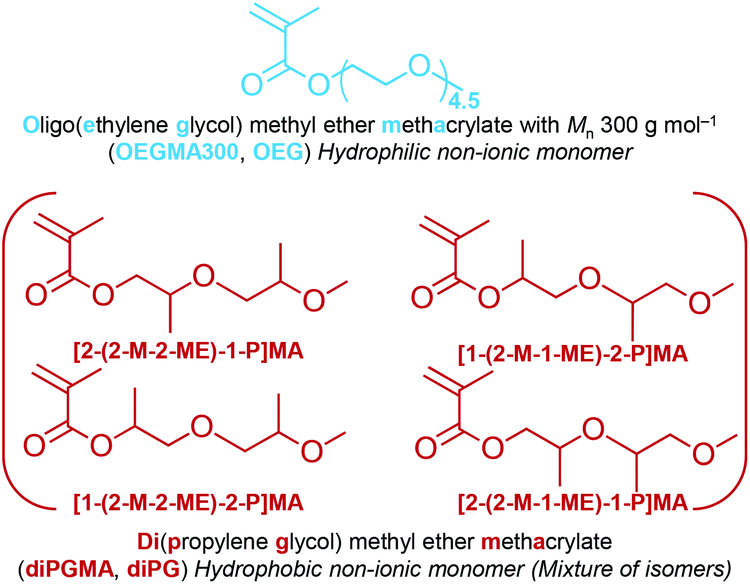 | ||
| Fig. 1 Chemical structures, names, and abbreviations of the monomers. OEGMA300 (OEG) and diPGMA (diPG) are the abbreviations of oligo(ethylene glycol) methyl ether methacrylate, average Mn 300 g mol−1, and di(propylene glycol) methyl ether methacrylate (mixture of isomers), respectively. | ||
Results and discussion
Monomer characterisation
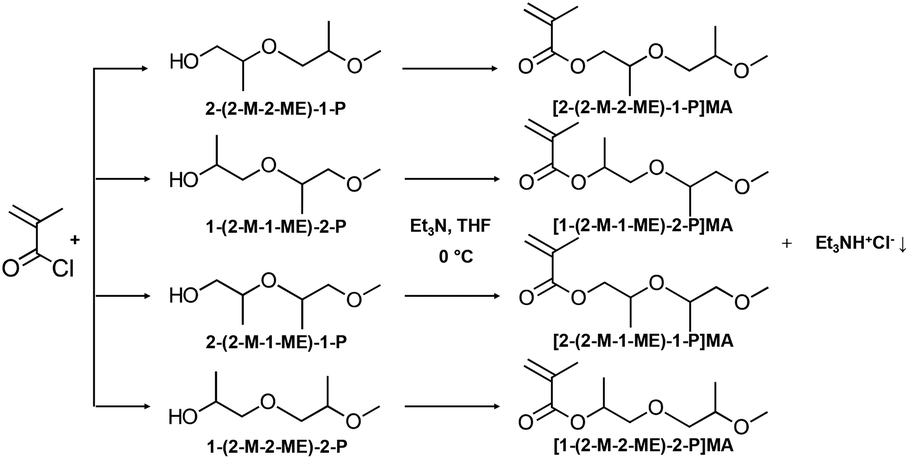 | ||
| Fig. 2 Chemical reaction of the synthesis of diPGMA monomer (mixture of isomers, Fig. 1), which is an esterification reaction between methacryloyl chloride and di(propylene glycol) monomethyl ether (diPGOH, mixture of isomers – from top to bottom: (i) 2-(2-methoxy-2-methylethoxy)-1-propanol [2-(2-M-2-ME)-1-P], (ii) 1-(2-methoxy-1-methylethoxy)-2-propanol [1-(2-M-1-ME)-2-P], (iii) 2-(2-methoxy-1-methylethoxy)-1-propanol [2-(2-M-1-ME)-1-P], and (iv) 1-(2-methoxy-2-methylethoxy)-2-propanol [1-(2-M-2-ME)-2-P]. The esterification was catalysed by triethylamine, in THF as solvent and the reaction was carried out in an ice-bath. | ||
The FT-IR analysis supports the successful esterification of the alcohol, diPGOH, to the corresponding methacrylate ester, diPGMA, Fig. 3, Table S1,† with the broad peaks of the νO–H stretching and bending at 3433.1 cm−1 and 1373.8 cm−1, respectively, and νC–O stretching at 1202.5 cm−1, disappearing when the esterification takes place. In addition to the disappearance of the peaks corresponding to the alcohol precursor, additional peaks corresponding to the ester group are present in the spectrum of diPGMA. Specifically, a strong peak corresponding to the νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching appears at 1715.2 cm−1, whereas the νC–O stretching of the ester appears at 1293.7 cm−1 and 1165.4 cm−1, and the CC stretching appears at 1637.1 cm−1.
O stretching appears at 1715.2 cm−1, whereas the νC–O stretching of the ester appears at 1293.7 cm−1 and 1165.4 cm−1, and the CC stretching appears at 1637.1 cm−1.
 | ||
| Fig. 3 FT-IR spectra of (a) diPGOH precursor, in black and (b) diPGMA, in dark red, with assignment of the corresponding functional groups. | ||
The 1H NMR analysis, Fig. 4, confirms the successful esterification, as the peaks corresponding to the methacrylate vinyl bond appear between 5.5 to 6.0 ppm; the corresponding protons are denoted by a and b respectively. In addition, the peak which corresponds to the methyl group of the methacrylate group appears at 1.8 ppm. Concerning the protons derived from the alcohol precursor, their peaks appear at similar chemical shifts with the ones of the alcohol, as expected (Fig. 4).
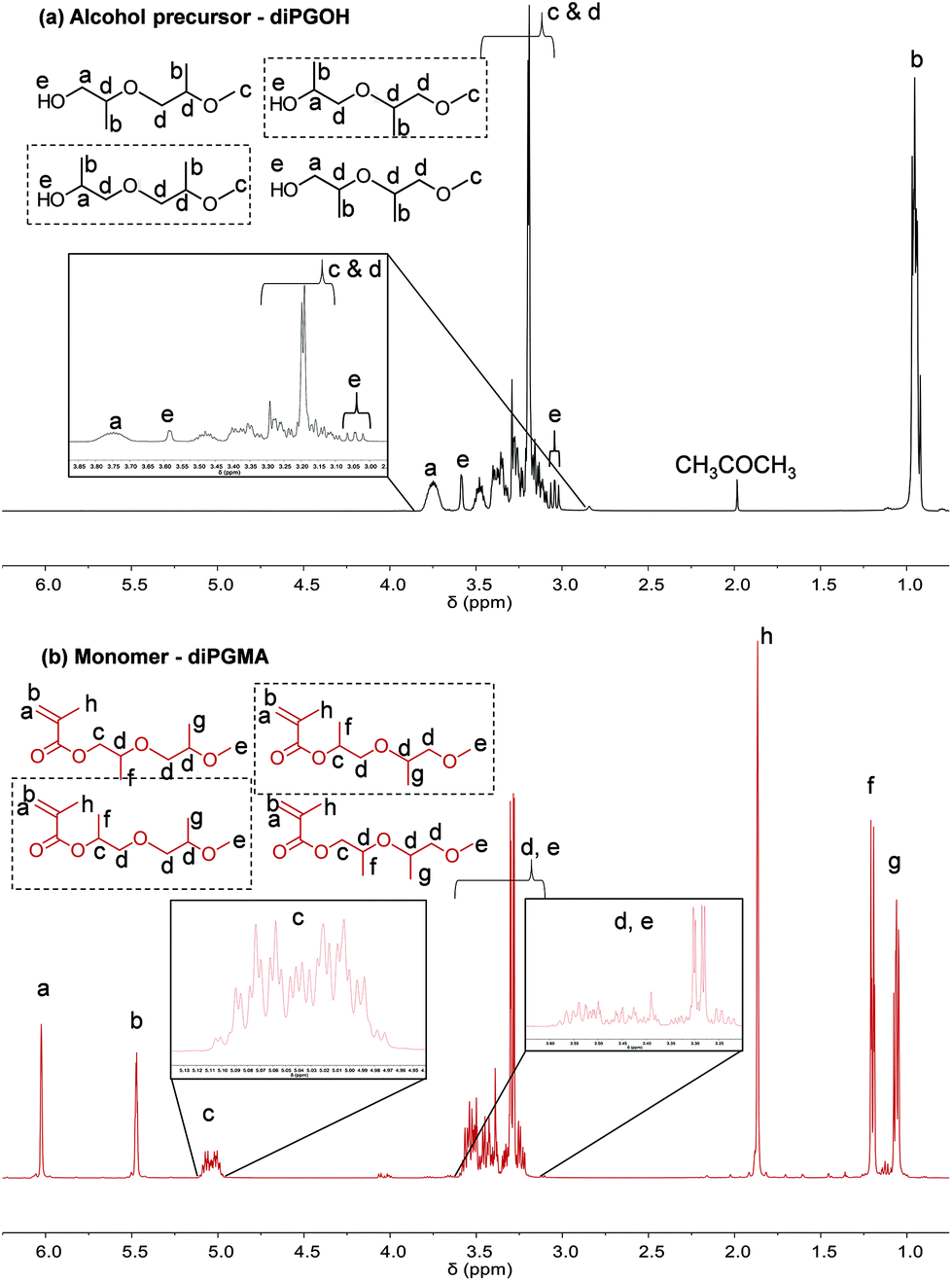 | ||
| Fig. 4
1H NMR spectra of (a) diPGOH: 1H NMR (CDCl3) δ 0.92–0.97 [m, 6.00H, CH3CH–], δ 3.02 (s, 0.06H, –OH), δ 3.04 (s, 0.12H, –OH), δ 3.07 (s, 0.07H, –OH), δ 3.09–3.52 [m, 8.29H, CH3O–, –OCH(CH3)CH2O–, –CH2CH(CH3)OH and HOCH2CH(CH3)–], δ 3.59 (s, 0.27H, –OH) and δ 3.72–3.81 [m, 0.95H, HOCH(CH3)– and HOCH2–], and (b) diPGMA: 1H NMR (CDCl3) δ 1.02–1.05 [m, 3.04H, –OCH2CH(CH3)O–], δ 1.16–1.18 [dd, 2.99H, J = 6.5, 2.2Hz, –(CO)OCH(CH3)CH2O– and –(CO)OCH2CH(CH3)O–], δ 1.84 [s, 2.98H, CH3C(CH2)–], δ 3.19–3.54 [m, 8.11H, CH3O–, –OCH(CH3)CH2O–, –CH2CH(CH3)O(CO)– and –(CO)OCH2CH(CH3)–], δ 4.96–5.06 [m, 0.95H, –(CO)OCH(CH3)– and –(CO)OCH2–], δ 5.44 [s, 1.00H, CH2C(CH3)–] and δ 6.00 [s, 1.00H, CH2C(CH3)–]. The two most abundant isomers are indicated by a square – this will be discussed in the relevant section in GC analysis. | ||
Similar observations are made by 13C NMR analysis (Fig. 5). Three additional peaks are observed in the spectrum of the monomer, at δ ≥100 ppm, which correspond to the carbons of the methacrylate part (double bond and ester/carbonyl bond).
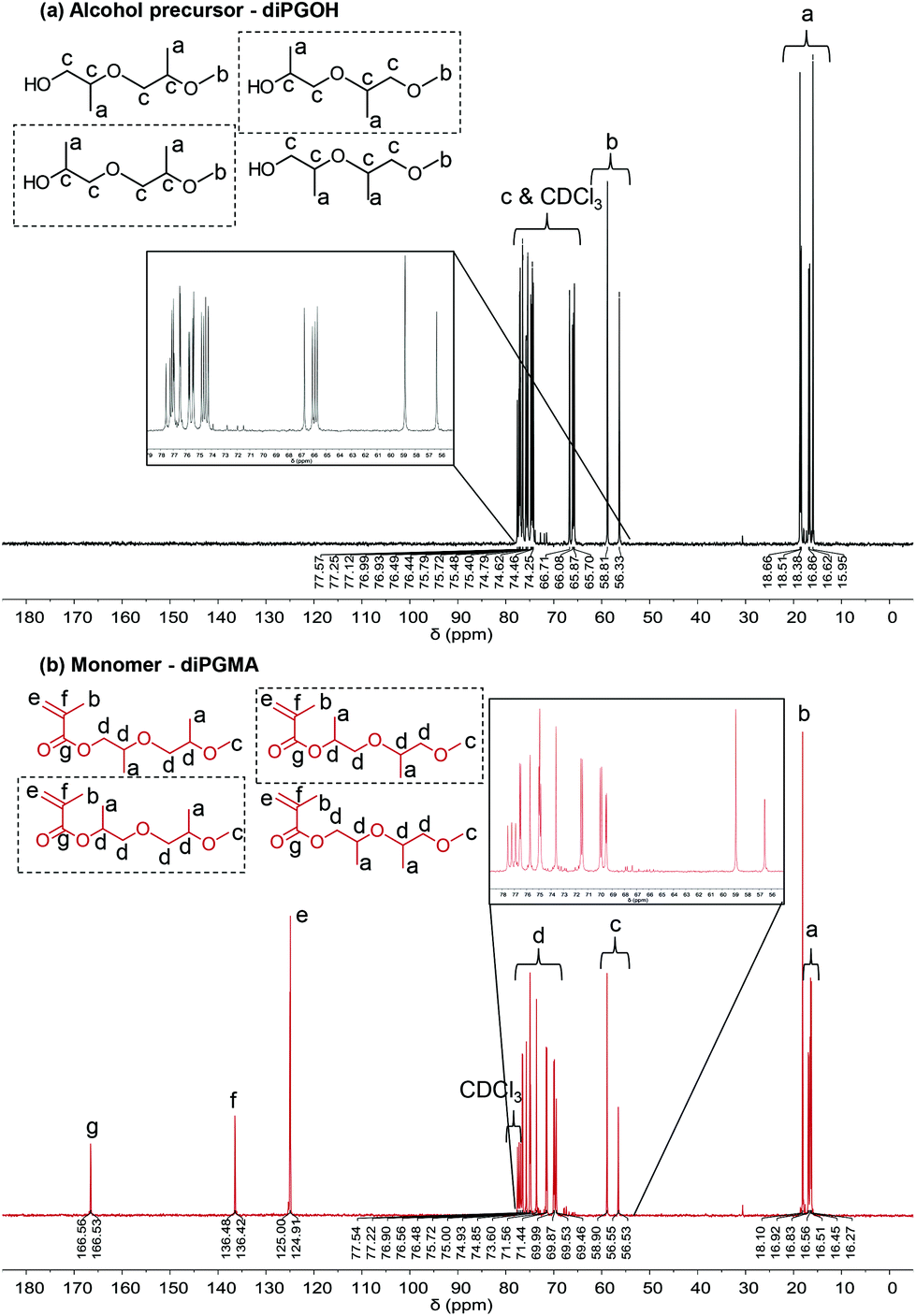 | ||
| Fig. 5 13C NMR spectra of (a) diPGOH, in black, and (b) diPGMA, after purification, in dark red. The corresponding chemical structures are also presented, with the two most abundant isomers indicated by a square – this will be discussed in the relevant section in GC analysis. | ||
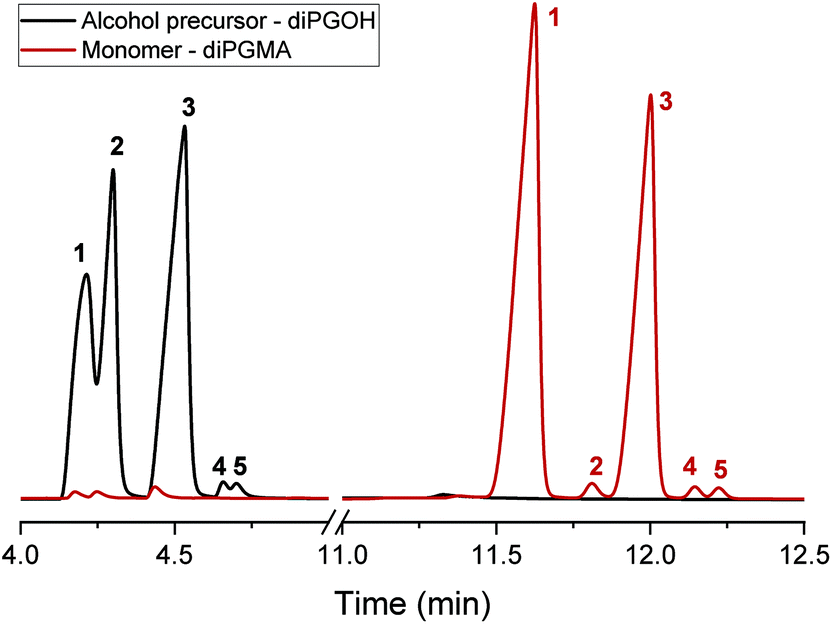 | ||
| Fig. 6 GC-FID chromatograms of the alcohol, diPGOH (mixture of isomers), and the monomer, diPGMA (mixture of isomers), in black and dark red, respectively. | ||
The alcohol elutes first due to its higher volatility. Five peaks are identified in each chromatogram, indicating the presence of five isomers. In the case of diPGOH, the number of peaks and their relative percentages agree with what has been previously reported in official studies on its airborne toxicity.47 In more detail, the first and second peaks correspond to configurational isomers of 1-(2-M-1-ME)-2-P (second isomer in Fig. 2, 28.22 w/w% and 24.36 w/w%), while the third peak corresponds to 1-(2-M-2-ME)-2-P (fourth isomer, 46.72 w/w%). The fourth and fifth peaks correspond to 2-(2-M-2-ME)-1-P (first isomer, 0.40 w/w%) and 2-(2-M-1-ME)-1-P (third isomer, 0.31 w/w%), respectively.47 Regarding diPGMA, there is lower resolution due to the longer elution times with the first peak corresponding to both configurational isomers of [1-(2-M-1-ME)-2-P]MA (second isomer in Fig. 2, 56.92 w/w%), while the third peak corresponds to [1-(2-M-2-ME)-2-P]MA (fourth isomer, 40.56 w/w%). Similarly to the analysis of the alcohol, the fourth and fifth peaks correspond to [2-(2-M-2-ME)-1-P]MA (first isomer, 0.76 w/w%) and [2-(2-M-1-ME)-1-P]MA (third isomer, 0.70 w/w%), respectively, while the second peak might be attributed to contamination or another isomer. To conclude, [1-(2-M-1-ME)-2-P]MA is the most abundant isomer, whereas [1-(2-M-2-ME)-2-P]MA is the second most abundant isomer. For simplicity in the rest of the text, the monomer will be referred as di(propylene glycol) methyl ether methacrylate, and in relevant figures, only the most abundant isomer will be presented.
In order to investigate more complex architectures containing diPGMA, ABA triblock bipolymers were synthesised, where A is OEGMA300 and B is diPGMA. Nine copolymers which differ in MM and composition were successfully synthesised via a one-pot GTP. The synthetic route followed is shown in Fig. 7(a), along with the schematic of the polymer structures (Fig. 7(b)), in which the OEGMA300 and diPGMA units are shown in light blue and red, respectively. Regarding the structures targeted, the MM increases from 5100 g mol−1 (Polymers 2 to 4) to 10100 g mol−1 (Polymers 5 to 7), to 13100 g mol−1 (Polymers 8 to 10). In each set of constant MM, the OEGMA300-diPGMA composition has been varied by increasing the diPGMA content as follows: 80–20 w/w% (P2, P5, and P8), (ii) 70–30 w/w% (P3, P6, and P9), and (iii) 60–40 w/w% (P4, P7, and P10).
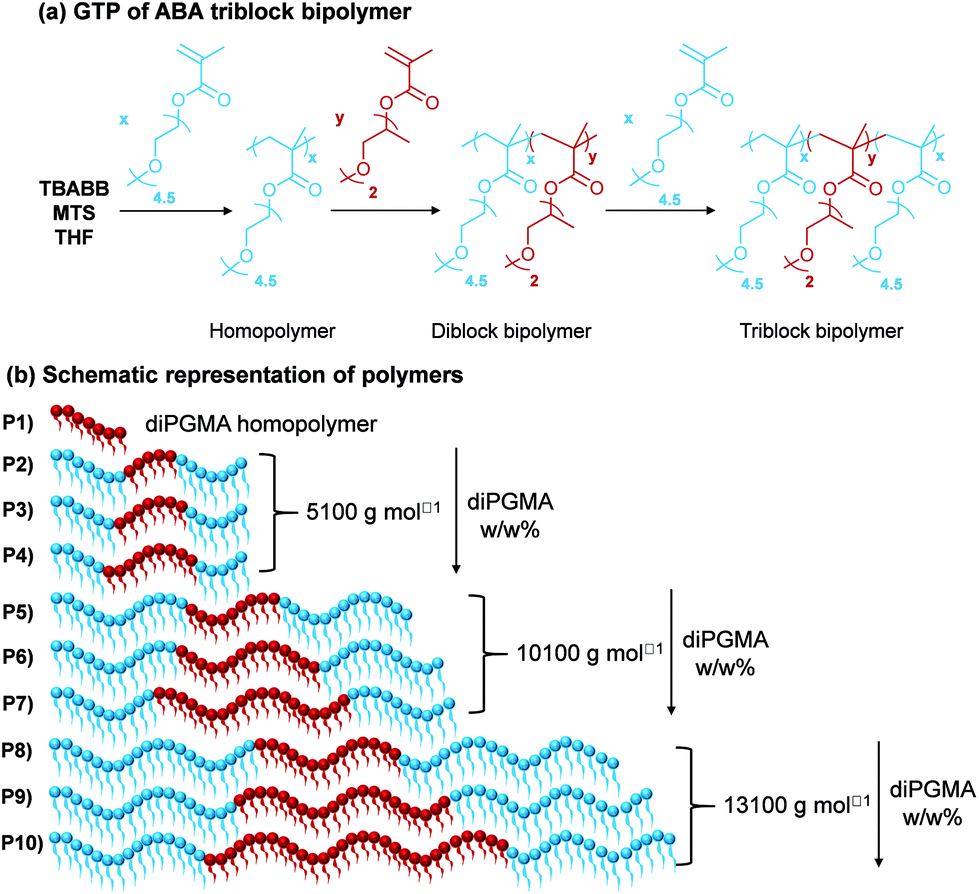 | ||
| Fig. 7 (a) Reaction scheme for the GTP to the ABA triblock polymer based on OEGMA300 (A), and diPGMA (B), with tetrabutyl ammoninum bibenzoic acid (TBABB), methyl trimethylsilyl dimethylketene acetal (MTS), and THF used as the catalyst, initiator and solvent, respectively. The most abundant isomer of diPGMA is used in the scheme. (b) Schematic representation of the diPGMA homopolymer (P1) and the ABA triblock bipolymers of various MMs and compositions: P2–P4 with MM 5100 g mol−1, P5–P7 with MM 10100 g mol−1, and P8–P10 13100 g mol−1. Light blue and dark red spheres represent the hydrophilic OEGMA300 (A), and the hydrophobic diPGMA (B), respectively. | ||
The polymerisations were followed by GPC, and the experimental MM and dispersity (Đ) of the final polymers and their precursors (in the case of the triblock copolymers) have been determined, Table 1. The GPC traces of Polymer 2, OEGMA3007-b-diPGMA5-b-OEGMA3007, are shown in Fig. 8 with dark blue dashed line, along with the GPC traces of its precursors; the GPC chromatograms of the homopolymer and diblock copolymer are shown in light blue solid and red dotted lines, respectively. The trends observed, i.e. peak moving to higher MM values, confirm the successful sequential GTP with diPGMA as a central block, and OEGMA300 as outer blocks. The GPC traces of other triblock copolymers can be found in Fig. S3.†
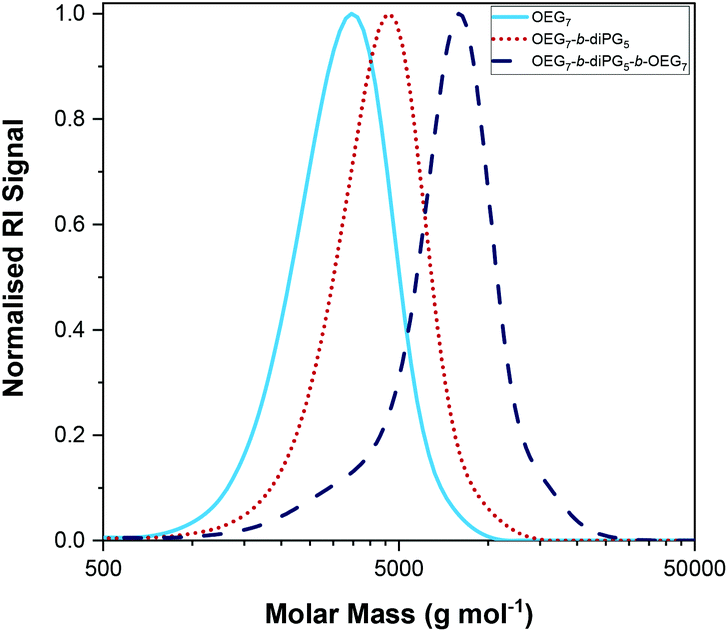 | ||
| Fig. 8 The GPC traces of the triblock bipolymer OEGMA3007-b-diPGMA5-b-OEGMA3007 (P2) are shown in dark blue dotted line. The GPC chromatograms of its precursors are also shown in light blue solid line for OEGMA3007, and in red dotted line for OEGMA3007-b-diPGMA5. | ||
| No. | Theoretical polymer structurea | MMtheor.b (g mol−1) |
M
nc (g mol−1) |
Đ | w/w% OEGMA300-diPGMA | |
|---|---|---|---|---|---|---|
| Theoretical | 1H NMR | |||||
a OEGMA300 and diPGMA are the abbreviations for oligo(ethylene glycol) methyl ether methacrylate with average Mn 300 g mol−1, and di(propylene glycol) methyl ether methacrylate, respectively.
b The theoretical molar mass is calculated using the following equation: 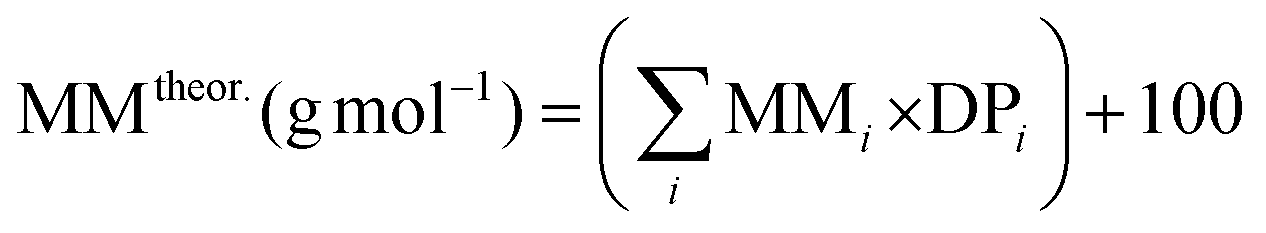 .
c The number-average molar mass (Mn) and the dispersity index (Đ) of the polymers and their linear precursors (if any) were determined by gel permeation chromatography (GPC). The calibration was based on well-defined linear poly(methyl methacrylate) (PMMA) standard samples with MM equal to 2, 4, 8, 20, 50, and 100 kg mol−1. .
c The number-average molar mass (Mn) and the dispersity index (Đ) of the polymers and their linear precursors (if any) were determined by gel permeation chromatography (GPC). The calibration was based on well-defined linear poly(methyl methacrylate) (PMMA) standard samples with MM equal to 2, 4, 8, 20, 50, and 100 kg mol−1.
|
||||||
| 1 | diPGMA7 | 1600 | 2200 | 1.19 | 00–100 | 00–100 |
| 2 | OEGMA3007 | 2100 | 2900 | 1.17 | 100–00 | 100–00 |
| OEGMA3007-b-diPGMA5 | 3100 | 3900 | 1.15 | 67–33 | 74–26 | |
| OEGMA3007-b-diPGMA5-b-OEGMA3007 | 5100 | 6600 | 1.21 | 80–20 | 87–13 | |
| 3 | OEGMA3006 | 1850 | 2600 | 1.16 | 100–00 | 100–00 |
| OEGMA3006-b-diPGMA7 | 3350 | 4400 | 1.14 | 54–46 | 60–40 | |
| OEGMA3006-b-diPGMA7-b-OEGMA3006 | 5100 | 7100 | 1.21 | 70–30 | 74–26 | |
| 4 | OEGMA3005 | 1600 | 2500 | 1.17 | 100–00 | 100–00 |
| OEGMA3005-b-diPGMA9 | 3600 | 5000 | 1.17 | 43–57 | 44–56 | |
| OEGMA3005-b-diPGMA9-b-OEGMA3005 | 5100 | 7100 | 1.21 | 60–40 | 57–43 | |
| 5 | OEGMA30013 | 4100 | 4900 | 1.13 | 100–00 | 100–00 |
| OEGMA30013-b-diPGMA9 | 6100 | 6700 | 1.15 | 67–33 | 69–31 | |
| OEGMA30013-b-diPGMA9-b-OEGMA30013 | 10100 |
12000 |
1.28 | 80–20 | 81–19 | |
| 6 | OEGMA30012 | 3600 | 4700 | 1.13 | 100–00 | 100–00 |
| OEGMA30012-b-diPGMA14 | 6600 | 8000 | 1.17 | 54–46 | 57–43 | |
| OEGMA30012-b-diPGMA14-b-OEGMA30012 | 10100 |
12900 |
1.29 | 70–30 | 72–28 | |
| 7 | OEGMA30010 | 3100 | 3000 | 1.14 | 100–00 | 100–00 |
| OEGMA30010-b-diPGMA19 | 7100 | 6500 | 1.14 | 43–57 | 40–60 | |
| OEGMA30010-b-diPGMA19-b-OEGMA30010 | 10100 |
10000 |
1.19 | 60–40 | 59–41 | |
| 8 | OEGMA30017 | 5300 | 6200 | 1.19 | 100–00 | 100–00 |
| OEGMA30017-b-diPGMA12 | 7900 | 9100 | 1.18 | 67–33 | 68–32 | |
| OEGMA30017-b-diPGMA12-b-OEGMA30017 | 13100 |
15200 |
1.16 | 80–20 | 79–21 | |
| 9 | OEGMA30015 | 4650 | 6000 | 1.18 | 100–00 | 100–00 |
| OEGMA30015-b-diPGMA18 | 8550 | 10600 |
1.20 | 54–46 | 56–44 | |
| OEGMA30015-b-diPGMA18-b-OEGMA30015 | 13100 |
17000 |
1.16 | 70–30 | 71–29 | |
| 10 | OEGMA30013 | 4000 | 5100 | 1.17 | 100–00 | 100–00 |
| OEGMA30013-b-diPGMA24 | 9200 | 11200 |
1.23 | 43–57 | 43–57 | |
| OEGMA30013-b-diPGMA24-b-OEGMA30013 | 13100 |
15400 |
1.21 | 60–40 | 60–40 | |
The measured Mn values are slightly higher than the theoretical ones, similarly to previous GTP studies, due to the slight deactivation caused by humidity and acidic impurities.43,54–59 In addition, the Đ values are ≤1.30, and they are satisfactorily low for the purpose of this study, while the experimental compositions agree with the targeted values. Generally, the data from GPC and 1H NMR analysis support successful GTP formation of triblock copolymers with diPGMA as a central block. The 1H NMR spectra of Polymer 2 and its precursors are shown in Fig. S4,† with a complete assignment of the peaks.
| No. | MMtheor. (g mol−1) | diPGtheor. (w/w%) | T g ± 1 (°C) |
|---|---|---|---|
| 1 | 1600 | 100 | −24 |
| 8 | 13100 |
20 | −55 |
| 9 | 30 | −51 | |
| 10 | 40 | −52 |
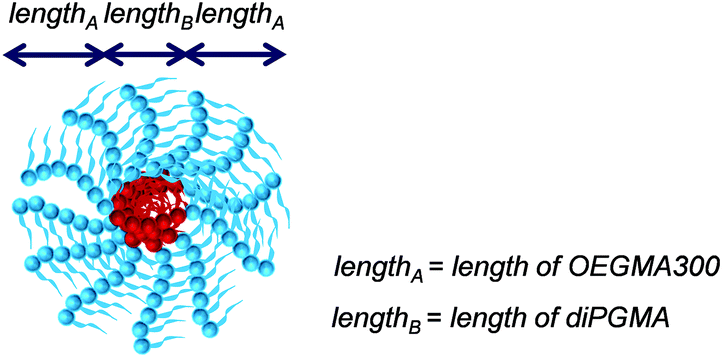 | ||
| Fig. 9 Schematic illustration of the theoretical self-assembled structure, namely core–shell spherical micelle, adopted by OEGMA3007-b-diPGMA5-b-OEGMA3007 (P2). The OEGMA300 units form the corona of the micelle and they are shown in light blue spheres, while the diPGMA units, which form the core of the micelle, are indicated by red spheres. All ABA triblock bipolymers (P2–P10) are assumed to self-assemble in the same way as shown. | ||
| No. | MMtheor. (g mol−1) | diPGtheor. (w/w%) | Hydrodynamic diameter (dh, nm) | |||
|---|---|---|---|---|---|---|
| Theor.a | Experimental ± 1b | |||||
| By Int. | By No | PDI | ||||
| a Calculations of the theoretical diameters are based on the experimental degrees of polymerisation (DPs); these were calculated by using the number-average molar mass (Mn) after precipitation, as determined by GPC, and the experimental OEGMA300/diPGMA content, as determined by 1H NMR spectroscopy. The theoretical hydrodynamic diameter of the ABA diblock bipolymers was calculated by assuming formation of traditional core–shell micelles and the following equation was used: dh (nm) = [DPOEGMA300 + DPdiPGMA] × 0.254 nm. b Experimental hydrodynamic diameters reported are the mean diameters which correspond to the maximum of the peak by (maximum) intensity or by number in the corresponding DLS histograms. c Polymer 1, diPGMA7, was insoluble in water. | ||||||
| 1 | 1600 | 100 | —c | NS | ||
| 2 | 5100 | 20 | 5.9 | 6 | 3 | 0.335 |
| 3 | 30 | 6.6 | 10 | 6 | 0.149 | |
| 4 | 40 | 7.2 | 14 | 8 | 0.098 | |
| 5 | 10100 |
20 | 10.9 | 14 | 7 | 0.113 |
| 6 | 30 | 12.1 | 16 | 12 | 0.047 | |
| 7 | 40 | 9.8 | 16 | 10 | 0.070 | |
| 8 | 13100 |
20 | 13.9 | 12 | 7 | 0.350 |
| 9 | 30 | 16.0 | 18 | 12 | 0.065 | |
| 10 | 40 | 15.0 | 21 | 14 | 0.071 | |
The self-assembly of the triblock copolymers were also confirmed with fluorescence spectroscopy measurements using pyrene as the probe, form which the critical micelle concentrations (CMCs) were determined. These ranged from 8.5 × 10–5 to 1.2 × 10–3 mmol L−1 (see ESI† for details) and some trends were observed. Specifically, the CMC decreased (i) as the MM increased when the experimental composition was the similar and (ii) as the wt% diPGMA was increased, as expected.
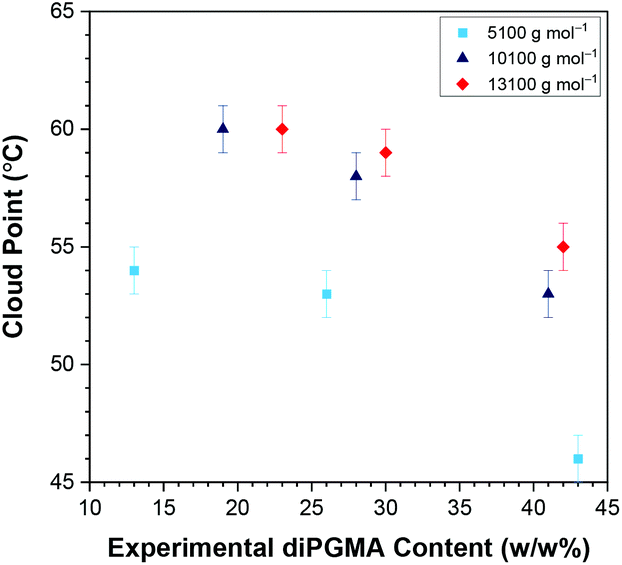 | ||
| Fig. 10 Effect of content in diPGMA on the cloud points, by UV–Vis, of solutions of the ABA triblock copolymers (1 w/w% in deionised water); where A and B are OEGMA300 and diPGMA, respectively. The effect of the molar mass is also shown in light blue squares (5100 g mol−1), dark blue triangles (10100 g mol−1), and red rhombi (13100 g mol−1). | ||
| No. | MMtheor. (g mol−1) | diPGtheor. (w/w%) | Cloud point (CP, °C) | ||
|---|---|---|---|---|---|
| Visual test (±2 °C)a | UV–Vis (±1 °C)b | DLS (±1 °C)c | |||
| a The CP is determined visually as the temperature at which the solution turns cloudy. b The CP is determined via UV–Vis as the temperature at 50% transmittance. c The CP is determined via DLS as the temperature of 50% aggregation. | |||||
| 2 | 5100 | 20 | 54 | 54 | 56 |
| 3 | 30 | 52 | 53 | 53 | |
| 4 | 40 | 45 | 46 | 47 | |
| 5 | 10100 |
20 | 60 | 60 | 63 |
| 6 | 30 | 60 | 58 | 59 | |
| 7 | 40 | 53 | 53 | 55 | |
| 8 | 13100 |
20 | 60 | 60 | 62 |
| 9 | 30 | 59 | 59 | 61 | |
| 10 | 40 | 55 | 55 | 55 | |
Generally, the CPs resulted by the three techniques are equal within experimental error, and the same trends are observed. When copolymers of the same MM are considered, it is observed that the CP decreases as the content in the hydrophobic diPGMA increases, as expected.58 This trend has been previously observed and reported,58,60–62 and it is attributed to the enhancement of the “hydrophobic effect” when the hydrophobicity of the copolymers increases. Regarding the effect of MM, while the copolymers with comparable content in diPGMA and 10100 and 13100 g mol−1 do not show much difference in CP, as the MM difference is not significant enough, but the CPs of the copolymers with 5100 g mol−1 are much lower. This is in contrast with some previous studies, which observed the CP decreasing when increasing the polymer MM.43,56,63–65 These studies investigated: (i) thermoresponsive homopolymers based on amine56,64 and PEG-containing methacrylate units,43 oxazoline63 and N-vinylpiperidone,65 (ii) statistical bipolymers consisting of two different thermoresponsive oxazoline units,63 and (iii) triblock terpolymers consisting of a thermo- and pH-responsive block, one block which is hydrophilic and thermoresponsive at higher temperatures, and a hydrophobic block.56 Thus, it is more accurate to compare the synthesised polymers with their Poloxamers counterparts, whose thermoresponse is attributed to both PPG and PEG blocks. As expected, the current trend of this study, i.e. decrease in CP with decrease in MM, is observed in Pluronics®, when the MM difference is profound.14 For example, when Pluronics® of 30 w/w% EG are compared, the CP increases from 42 °C (1850 g mol−1, Pluronic® L43), to 86 °C (4950 g mol−1, Pluronic® P103), to 90 °C (5750 g mol−1, Pluronic® P123). This is also the case when Pluronics® with 40 w/w% EG are compared; the CP increases from 58 °C (2900 g mol−1, Pluronic® L64), to 74 °C (4200 g mol−1, Pluronic® P84), to 81 °C (5900 g mol−1, Pluronic® P104).14 Interestingly, systems based on diblock copolymers of (i) methyl tri(ethylene glycol) vinyl ether and isobutyl vinyl ether,66 and (ii) OEGMA300 and n-hexyl methacrylate,67 in which only one block was thermoresponsive, also reported increase in the CP by increase in the MM. Patrickios et al. attributed this trend to the increased micelle size with increased MM, due to steric stabilisation of these colloidal systems.66
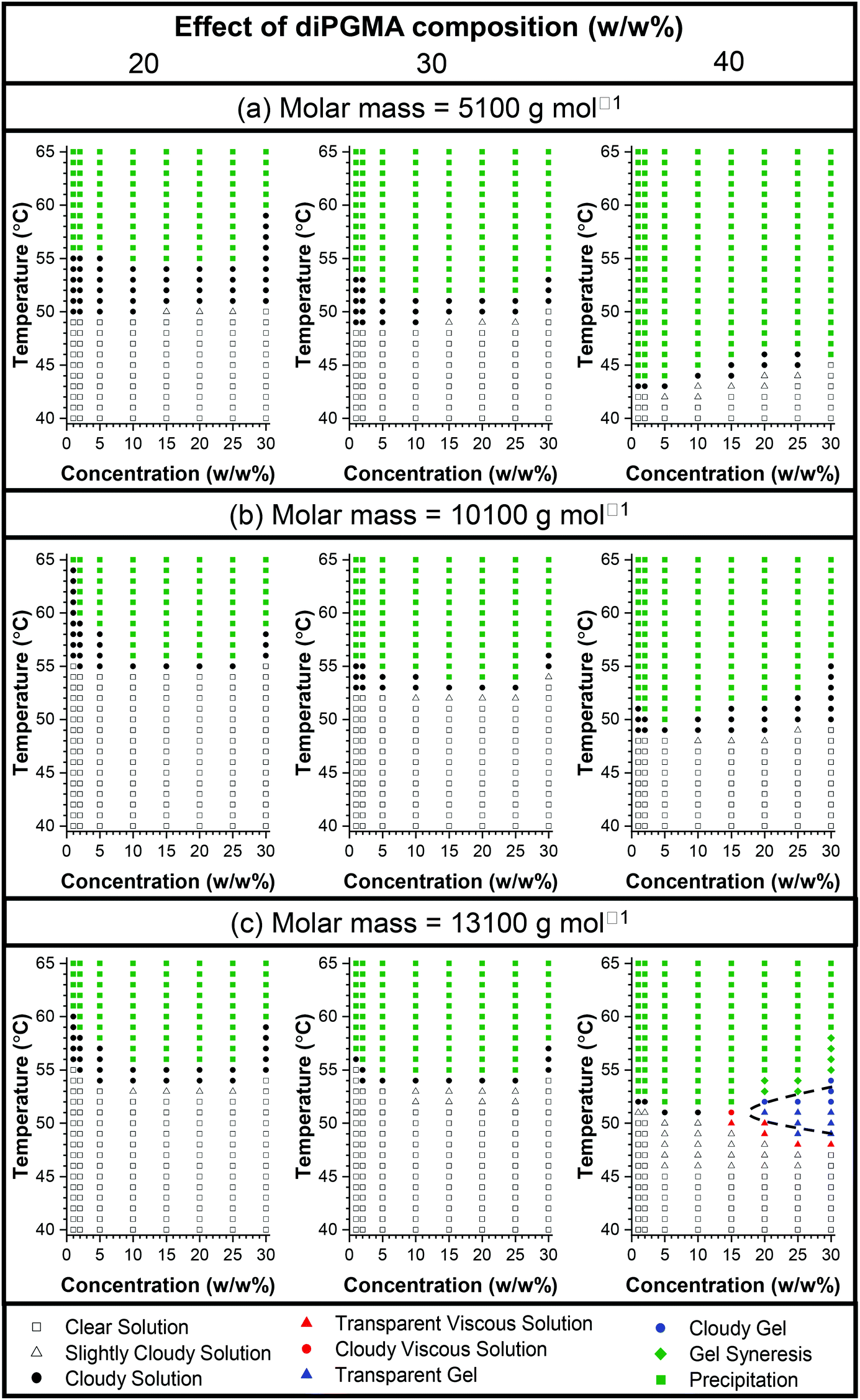 | ||
| Fig. 11 Phase diagrams of the ABA triblock copolymers (P2–P10) in phosphate buffered saline (PBS). The effect of diPGMA composition is shown from left to right, whereas the effect of the MM is shown from the top to bottom: (a) MM 5100 g mol−1 (top), (b) MM 10100 g mol−1 (middle), and (c) MM 13100 g mol−1 (bottom). Four phases are observed: (i) runny solution coloured in white (square: clear, triangle: slightly cloudy) and black circles for cloudy, (ii) viscous solution coloured in red (triangle and circle for transparent and cloudy, respectively), (iii) the gel phase is shown in blue (triangle and circle for transparent and cloudy, respectively), the two-phase system is coloured in light green (rhombus and square for gel syneresis and precipitation, respectively). | ||
The diluted solutions at 1 w/w% in PBS present a CP, which (a) decreases as the diPGMA content increases (for polymers with similar MM), and (b) increases when the increase in MM is profound (for polymer with the similar composition). As expected, the CPs are lower in PBS than in DI water, which is caused by the presence of salt in solution.59,68
Similarly, in more concentrated solutions, both a composition effect and a MM effect are observed. For the families of low and intermediate MM, all the polymers precipitate out of solution as the temperature increases, however, it is clearly observed that the temperature of phase separation decreases as the content in the hydrophobic diPGMA increases. This shows a clear effect of composition, as the increase in the hydrophilic OEGMA300 content shifts the thermoresponsiveness to higher temperatures and diminishes gelation if used at high content, in agreement with previous studies.55,58 Interestingly, in the case of the polymers with high MM, gelation is observed at around 50 °C, shown in blue symbols, when the content of diPGMA is increased to 40 w/w%. This is in consistent with previous studies, which showed that increasing the MM promotes gelation.56 Nevertheless, even though in most of the cases gelation was not observed, the results are promising, with diPGMA serving as an alternative hydrophobic monomer, to be used on promoting self-assembly and/or designing novel thermogelling systems.
 | ||
| Fig. 12 Rheological properties as a function of temperature for the solution of Polymer 10, OEGMA30013-b-diPGMA24-b-OEGMA30013, which forms a gel rheologically at 15 w/w% in phosphate buffered saline (PBS). The changes in shear storage modulus (elastic modulus, G′) are represented by blue triangles, while the changes in shear loss modulus (viscous modulus, G′′) are shown in red circles. The OEGMA300 and diPGMA units on the polymer structures are coloured in light blue and dark red, respectively. | ||
The solutions of Polymer 10 at 20 w/w%, 25 w/w% and 30 w/w% in PBS were also studied by rheology. The changes in shear storage and shear loss moduli as a function of temperature are shown in Fig. S13.† All solutions form a gel as the temperature increases (G′ > G′′), with the maximum storage modulus increasing from 21 Pa to 110 Pa as the concentration of polymer in solution increases from 15 w/w% to 30 w/w%; this trend is depicted in Fig. 13. It is noted that while the moduli drop rapidly as the gel destabilises for the 15 w/w%, this drop diminishes as the polymer concentration increases. This matches the visual observations, during which the viscous solution formed by the 15 w/w% solution directly precipitated, while the more concentrated solutions formed a stable gel, which destabilised by presenting gel syneresis, i.e. slight exclusion of the solvent, followed by complete phase separation.
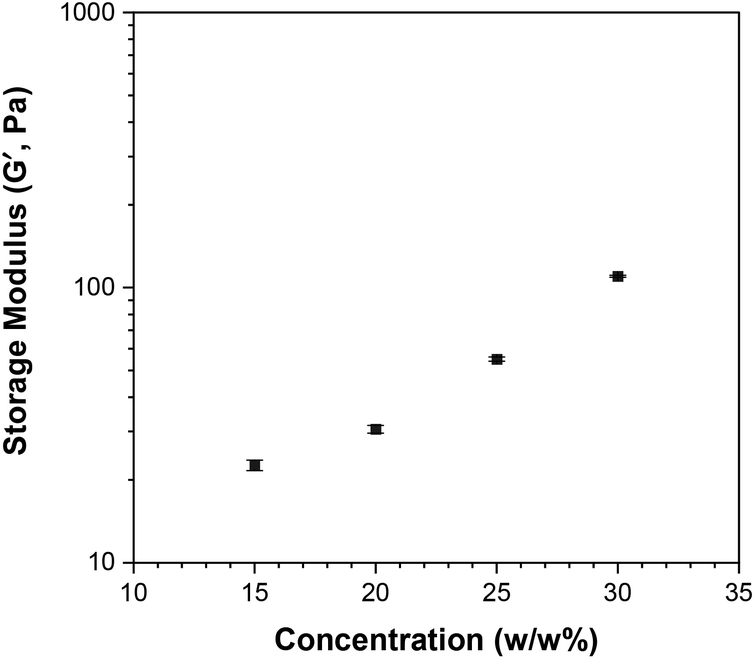 | ||
| Fig. 13 Effect of polymer concentration on the maximum storage modulus (Pa) of the thermogelling solutions of Polymer 10 in phosphate buffered saline (PBS). | ||
Conclusions
In summary, a hydrophobic monomer based on propylene glycol side chain, diPGMA, was successfully synthesised and homopolymerised for the first time. In addition, it was copolymerised to produce nine ABA triblock copolymers with B block being based on diPGMA and A block being based on OEGMA300. The block copolymers MM and composition were systematically varied. Specifically, MM ranged from 5100 g mol−1, to 10100 g mol−1, to 13100 g mol−1, while the content in diPGMA changed from 20 w/w%, to 30 w/w%, to 40 w/w%. While poly(diPGMA) is hydrophobic and thus water-insoluble, all the copolymers are soluble in aqueous solvents, due to the high content in the hydrophilic OEGMA300. When assembled in aqueous media, the copolymers self-assemble into micelles. The size of the micelles increases with increased MM and content in diPGMA. The copolymers were also thermoresponsive and it was observed that the CP decreases by decreasing the overall MM of the polymer and increasing its hydrophobic content. Interestingly, gelation is controlled by the hydrophobicity and the MM of the structure. Specifically, gelation was observed for Polymer 10, which is the one with the highest MM (13100 g mol−1), and highest hydrophobic diPGMA content (40 w/w%). In conclusion, diPGMA is a promising monomer that could be used to promote self-assembly and gelation in thermogelling systems.
Author contributions
APC synthesised the monomer and the polymers, performed the characterisation of the monomer via FT-IR and NMR, and of the polymers in organic solvents, contributed to the characterisation of the polymers in aqueous media, and wrote the first draft of the manuscript. GP performed the CMC experiments. BS assisted with the monomer synthesis, monomer purification, and polymerisation. TB assisted with monomer purification, and the characterisation of the polymers in aqueous solvent during his MSc project. GP and DMH performed the characterisation of the monomer via GC. DWL performed the DSC experiments. TKG secured the funding and was the project's supervisor, manager, and coordinator.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. P. C. and B. S. acknowledge the Engineering and Physical Sciences Research Council (EPSRC) and the Department of Materials at Imperial College London (ICL) for the PhD scholarships. EPSRC is also acknowledged for Dr Constantinou's Doctoral Prize Fellowship (EP/M506345/1), as well as the EPSRC Impact Acceleration Grant EP/R511547/1.References
- A. P. Constantinou and T. K. Georgiou, Eur. Polym. J., 2016, 78, 366–375 CrossRef CAS.
- A. P. Constantinou and T. K. Georgiou, Thermoresponsive Multiblock Copolymers: Chemistry, Properties and Applications. Temperature-Responsive Polymers: Chemistry, Properties, and Applications, John Wiley & Son Ltd, United Kingdom, 2018 Search PubMed.
- L. Klouda and A. G. Mikos, Eur. J. Pharm. Biopharm., 2008, 68, 34–45 CrossRef CAS PubMed.
- L. Klouda, Eur. J. Pharm. Biopharm., 2015, 97, 338–349 CrossRef CAS PubMed.
- Y. Kim and Y. T. Matsunaga, J. Mater. Chem. B, 2017, 5, 4307–4321 RSC.
- F. Doberenz, K. Zeng, C. Willems, K. Zhang and T. Groth, J. Mater. Chem. B, 2020, 8, 607–628 RSC.
- M. T. Cook, P. Haddow, S. B. Kirton and W. J. McAuley, Adv. Funct. Mater., 2021, 31, 2008123 CrossRef CAS.
- C. Chassenieux and C. Tsitsilianis, Soft Matter, 2016, 12, 1344–1359 RSC.
- J. C. Foster, I. Akar, M. C. Grocott, A. K. Pearce, R. T. Mathers and R. K. O’Reilly, ACS Macro Lett., 2020, 1700–1707 CrossRef CAS PubMed.
- R. Hoogenboom and H. Schlaad, Polym. Chem., 2017, 8, 24–40 RSC.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS PubMed.
- M. A. Ward and T. K. Georgiou, Polymers, 2011, 3, 1215–1242 CrossRef CAS.
- S. J. Buwalda, K. W. M. Boere, P. J. Dijkstra, J. Feijen, T. Vermonden and W. E. Hennink, J. Controlled Release, 2014, 190, 254–273 CrossRef CAS PubMed.
- M. Almeida, M. Magalhães, F. Veiga and A. Figueiras, J. Polym. Res., 2017, 25, 31 CrossRef.
- G. Dumortier, J. L. Grossiord, F. Agnely and J. C. Chaumeil, Pharm. Res., 2006, 23, 2709–2728 CrossRef CAS PubMed.
- S. Hocine and M. Li, Soft Matter, 2013, 9, 5839–5861 RSC.
- S. Saeki, N. Kuwahara, M. Nakata and M. Kaneko, Polymer, 1976, 17, 685–689 CrossRef CAS.
- A. A. Barba, M. d'Amore, M. Grassi, S. Chirico, G. Lamberti and G. Titomanlio, J. Appl. Polym. Sci., 2009, 114, 688–695 CrossRef CAS.
- C. Branca, K. Khouzami, U. Wanderlingh and G. D'Angelo, J. Colloid Interface Sci., 2018, 517, 221–229 CrossRef CAS PubMed.
- S. E. Piechota, Colgate-Palmolive Company and N. J. Piscataway, US5256396A, 1993.
- J. Xuan, P. Balakrishnan, D. H. Oh, W. H. Yeo, S. M. Park, C. S. Yong and H. Choi, Int. J. Pharm., 2010, 395, 317–323 CrossRef CAS PubMed.
- S. Baldassari, A. Solari, G. Zuccari, G. Drava, S. Pastorino, C. Fucile, V. Marini, A. Daga, A. Pattarozzi, A. Ratto, A. Ferrari, F. Mattioli, F. Barbieri, G. Caviglioli and T. Florio, Sci. Rep., 2018, 8, 3929 CrossRef.
- S. Bobbala, B. Gibson, A. B. Gamble, A. McDowell and S. Hook, Immunol. Cell Biol., 2018, 96, 656–665 CrossRef CAS.
- Y. Mao, X. Li, G. Chen and S. Wang, J. Pharm. Sci., 2016, 105, 194–204 CrossRef CAS PubMed.
- Z. Yu, F. Guo, Y. Guo, Z. Zhang, F. Wu and X. Luo, PLoS One, 2017, 12, e0173949 CrossRef PubMed.
- X. Li, R. Fan, Y. Wang, M. Wu, A. Tong, J. Shi, M. Xiang, L. Zhou and G. Guo, RSC Adv., 2015, 5, 101494–101506 RSC.
- M. Müller, J. Becher, M. Schnabelrauch and M. Zenobi-Wong, J. Visualized Exp., 2013, 10(77), e50632 Search PubMed.
- T. Lorson, S. Jaksch, M. M. Lübtow, T. Jüngst, J. Groll, T. Lühmann and R. Luxenhofer, Biomacromolecules, 2017, 18, 2161–2171 CrossRef CAS PubMed.
- H. Y. Cho, A. Srinivasan, J. Hong, E. Hsu, S. Liu, A. Shrivats, D. Kwak, A. K. Bohaty, H. Paik, J. O. Hollinger and K. Matyjaszewski, Biomacromolecules, 2011, 12, 3478–3486 CrossRef CAS PubMed.
- T. Kim, H. J. Seo, J. S. Choi, H. Jang, J. Baek, K. Kim and J. Park, Biomacromolecules, 2004, 5, 2487–2492 CrossRef CAS PubMed.
- J. Lutz, K. Weichenhan, Ö. Akdemir and A. Hoth, Macromolecules, 2007, 40, 2503–2508 CrossRef CAS.
- J. Lutz, A. Hoth and K. Schade, Des. Monomers Polym., 2009, 12, 343–353 CrossRef CAS.
- J. Lutz, J. Andrieu, S. Üzgün, C. Rudolph and S. Agarwal, Macromolecules, 2007, 40, 8540–8543 CrossRef CAS.
- J. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- J. Lutz and A. Hoth, Macromolecules, 2006, 39, 893–896 CrossRef CAS.
- J. Lutz, Adv. Mater., 2011, 23, 2237–2243 CrossRef CAS.
- N. Badi and J. Lutz, J. Controlled Release, 2009, 140, 224–229 CrossRef CAS PubMed.
- N. Fechler, N. Badi, K. Schade, S. Pfeifer and J. Lutz, Macromolecules, 2009, 42, 33–36 CrossRef CAS.
- W. Steinhauer, R. Hoogenboom, H. Keul and M. Moeller, Macromolecules, 2013, 46, 1447–1460 CrossRef CAS.
- C. R. Becer, S. Hahn, M. W. M. Fijten, H. M. L. Thijs, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7138–7147 CrossRef CAS.
- G. Vancoillie, D. Frank and R. Hoogenboom, Prog. Polym. Sci., 2014, 39, 1074–1095 CrossRef CAS.
- D. Fournier, R. Hoogenboom, H. M. L. Thijs, R. M. Paulus and U. S. Schubert, Macromolecules, 2007, 40, 915–920 CrossRef CAS.
- Q. Li, A. P. Constantinou and T. K. Georgiou, J. Polym. Sci., 2021, 59, 230–239 CrossRef CAS.
- M. Elladiou and C. S. Patrickios, Polym. Chem., 2012, 3, 3228–3231 RSC.
- M. Elladiou and C. S. Patrickios, Macromolecules, 2015, 48, 7503–7512 CrossRef CAS.
- A. P. Constantinou, M. Elladiou and C. S. Patrickios, Macromolecules, 2016, 49, 3869–3880 CrossRef CAS.
- United States Department of Labour, http://www.osha.gov/dts/sltc/methods/organic/org101/org101.html#ref51, (accessed 2019).
- P. Maksym-Bębenek, T. Biela and D. Neugebauer, RSC Adv., 2015, 5, 3627–3635 RSC.
- E. Suljovrujic and M. Micic, Nucl. Instrum. Methods Phys. Res., Sect. B, 2015, 342, 206–214 CrossRef CAS.
- Z. R. Miladinovic, M. Micic and E. Suljovrujic, J. Polym. Res., 2016, 23, 77 CrossRef.
- Z. R. Miladinovic, M. Micic, A. Mrakovic and E. Suljovrujic, J. Polym. Res., 2017, 25, 1 Search PubMed.
- C. Cha, E. Kim, I. W. Kim and H. Kong, Biomaterials, 2011, 32, 2695–2703 CrossRef CAS PubMed.
- X. J. Loh, S. J. Ong, Y. T. Tung and H. T. Choo, Mater. Sci. Eng., C, 2013, 33, 4545–4550 CrossRef CAS PubMed.
- M. A. Ward and T. K. Georgiou, Polym. Chem., 2013, 4, 1893–1902 RSC.
- M. A. Ward and T. K. Georgiou, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 775–783 CrossRef CAS.
- M. A. Ward and T. K. Georgiou, Soft Matter, 2012, 8, 2737–2745 RSC.
- A. P. Constantinou, N. Sam-Soon, D. R. Carroll and T. K. Georgiou, Macromolecules, 2018, 51, 7019–7031 CrossRef CAS.
- A. P. Constantinou and T. K. Georgiou, Polym. Chem., 2016, 7, 2045–2056 RSC.
- A. P. Constantinou, T. Lan, D. R. Carroll and T. K. Georgiou, Eur. Polym. J., 2020, 130, 109655 CrossRef CAS.
- A. P. Constantinou, B. Zhan and T. K. Georgiou, Macromolecules, 2021, 54(4), 1943–1960 CrossRef CAS.
- J. A. Jones, N. Novo, K. Flagler, C. D. Pagnucco, S. Carew, C. Cheong, X. Z. Kong, N. A. D. Burke and H. D. H. Stöver, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6095–6104 CrossRef CAS.
- C. Diehl and H. Schlaad, Macromol. Biosci., 2009, 9, 157–161 CrossRef CAS PubMed.
- R. Hoogenboom, H. M. L. Thijs, M. J. H. C. Jochems, B. M. van Lankvelt, M. W. M. Fijten and U. S. Schubert, Chem. Commun., 2008, 5758–5760 RSC.
- V. Bütün, S. P. Armes and N. C. Billingham, Polymer, 2001, 42, 5993–6008 CrossRef.
- N. S. Ieong, M. Hasan, D. J. Phillips, Y. Saaka, R. K. O'Reilly and M. I. Gibson, Polym. Chem., 2012, 3, 794–799 RSC.
- C. S. Patrickios, C. Forder, S. P. Armes and N. C. Billingham, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 1529–1541 CrossRef CAS.
- N. H. Raduan, T. S. Horozov and T. K. Georgiou, Soft Matter, 2010, 6, 2321–2329 RSC.
- J. de Souza Carlos Perbone, A. F. Naves and F. Florenzano, Colloid Polym. Sci., 2012, 290, 1285–1291 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and supplementary data. See DOI: 10.1039/d1py00444a |
| This journal is © The Royal Society of Chemistry 2021 |