
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProgress, challenges and future directions of heterocycles as building blocks in iterative methodologies towards sequence-defined oligomers and polymers
Stephen A.
Hill
 *,
Robert
Steinfort
and
Laura
Hartmann
*
*,
Robert
Steinfort
and
Laura
Hartmann
*
Institute of Organic and Macromolecular Chemistry, Heinrich Heine University Düsseldorf, Universitätsstraße 1, 40225 Düsseldorf, Germany. E-mail: stephen.hill@hhu.de; laura.hartmann@hhu.de
First published on 14th July 2021
Abstract
The synthesis of sequence-defined oligomers and polymers is an important contemporary research topic due to the potential for creating materials with function tailored directly into the primary structure. Sequence-defined materials have so far been applied to biomimicry, data storage and sensing. Heterocyclic building blocks such as thiolactones and epoxides are key building blocks for producing sequence-defined macromolecules using iterative methodologies. In this review we critically evaluate: the current literature on different heterocyclic strategies using solid- or solution-phase methods for the iterative synthesis of sequence-defined polymers; the challenges surrounding these systems; and the potential future direction of heterocycles, so far not used in this field.
1. Introduction
Sequence definition is a topic of contemporary interest given the critical relationship between a macromolecule's primary sequence and its shape and function. The importance is clear in Nature with the successful function of polynucleotides e.g. DNA and RNA, proteins and polysaccharides allied to the defined order of the individual building blocks (BBs) in a macromolecule's chain.1 For the purposes of this review, sequence definition applied to macromolecules is defined as a “uniform macromolecule with an exact chain-length and a perfectly defined sequence of monomers”.2 Aside from sequence definition studies directed towards protein folding,3 focus within the polymer sciences over the past two decades has turned to understanding sequence definition and its effect on synthetic macromolecular properties. The attention has led to the burgeoning field of sequence-defined macromolecules (SDMs) with chemists searching for protocols, motifs and architectures that diverge from the well-studied biomacromolecule classes.4Various synthetic strategies have been applied to obtain SDMs,5 however, in this review, we will focus exclusively on methods based on iterative processes. When considering SDM synthesis, methods employing the iterative coupling of BBs on to a growing macromolecular backbone have clear advantages. Ever since R. B. Merrifield reported the first synthesis of a tetrapeptide on a solid-phase (SP) support,6 the inherent benefits of SP and iterative coupling strategies have been realized for polysaccharide,7,8 polypeptide,9 and polynucleotide construction.10 The benefits of polymer chain immobilization include: (1) ease of purification; (2) coupling iteration; and (3) high yield acquisition through excess reagent usage. Despite time and efficiency advantages SP protocols have limitations too: (1) scale-up challenge; (2) optimising new processes i.e., solvents, linker, concentrations etc.; and (3) potential requirement for protecting groups (PGs). Solution-phase strategies circumvent scale limitations but instead introduce purification requirements, which have, in part, been addressed by soluble fluorous, PEG or polystyrene supports.11,12
Many successful SDM syntheses have been based upon the iterative assembly of acyclic BBs. Using tailored, acyclic BBs holds many advantages as the chemist has control over backbone length and composition e.g., tuning hydrophilic or hydrophobic properties; spacing between side-chains, etc. Custom synthesis allows libraries of acyclic BBs to be created: with demarcated lengths, elemental composition, and side- and end-groups, including BBs with or without PGs.13 However, given the contemporary drive for ever increasing efficiencies, non-commercial, tailored acyclics may be disfavoured due to “atom economy” considerations or the time investment often required for the multi-step syntheses.14 Solutions to overcoming inefficiencies, particularly at large scale, are being addressed i.e. we recently published an automation-compatible protocol to recover unreacted BBs.15
Alongside acyclic BB development, cyclic BBs have been developed too. Although not entirely exclusive, (hetero)cyclic-based strategies have been developed as they can offer the following benefits: (1) yielding products with bi- or multi-functionality; (2) proceeding with high regio- and/or stereoselectivity; and (3) being ring-opened with a range of nucleophiles, typically avoiding the need for activation or catalytic agents. Nevertheless, ring openings bring challenges too, namely (un)foreseen cascade/side reactions and product mixtures.
In this review, we shall highlight and discuss strategies and techniques which iteratively construct monodisperse SDMs by employing heterocyclic BBs. The scope of this review will encompass SP and in-solution approaches which specifically employ heterocyclic BBs, that are restricted to cyclic compounds with 3–6 endocyclic atoms, with at least one endocyclic atom being a heteroatom, and the reactivity is directly associated with the cyclic motif's inherent chemistry, and not that of residual groups. Although much literature exists regarding carbocyclic (aliphatic and aromatic) ring synthesis and transformation,16–18 there exists a rich set of diverse heterocycles which offer distinct advantages including, but not limited to: (1) their synthesis/sourcing from renewable resources; and [2] their reactivity generated from endo- and exocyclic polarity differences. Heterocycles which have been reported for SDM synthesis shall be addressed, but also potential future direction and opportunities e.g. of building blocks so far not used within the SDM field shall be presented.
For the purposes of this review, we shall introduce a distinction to differentiate cyclic BBs under the categories of “active” and “latent”. These titles designate the outcome of a given BB's coupling to a macromolecule (Fig. 1). If a BB is “active”, the heterocycle is chemically transformed upon SDM incorporation. “Latent” BBs have an intact, unreacted heterocycle after SDM incorporation, retaining their specific reactivity until a later juncture.
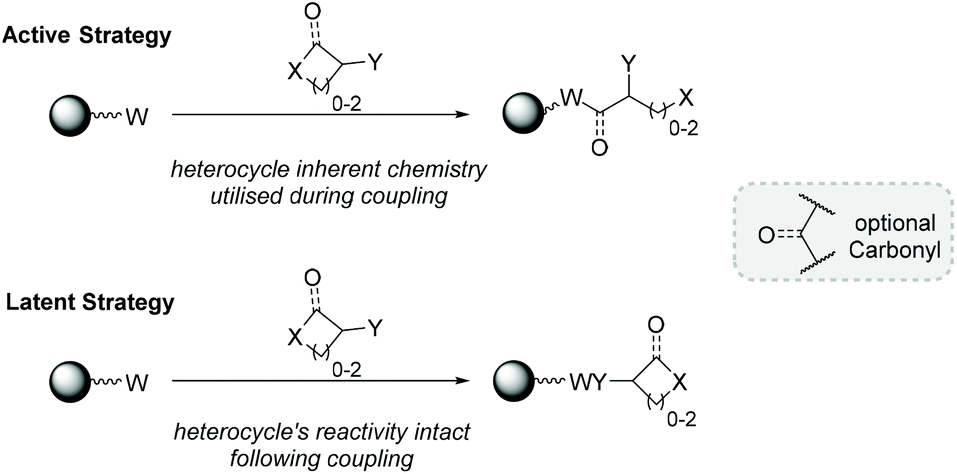 | ||
| Fig. 1 Illustration of active and latent strategies of generic heterocycle addition to a growing sequence-defined macromolecule. | ||
Throughout this review automation shall be highlighted given its importance in offering productivity and safety advantages. Given the topic's importance, automated SP syntheses for biomacromolecules,7,19 and SDMs have already been reviewed.12 It should be noted, larger macrocyclic rings are also known to have been utilized to generate polymeric sequence-control and sequence-definition. Macrocyclic ring opening to generate polymers bring the advantage of rapidly accessing higher molecular weight polymers. For example, Gutekunst and Hawker in 2015 reported the generation of an unstrained, cyclic macromonomer with a defined BB order encoded into the macromonomer ring.20 The macrocycle also contained an enyne trigger which facilitated ring-opening metathesis polymerization after exposure to Grubbs catalyst, producing high molecular weight, sequence-defined polymers. Similar macrocycle strategies to achieve sequence-definition or control are known, but are beyond the scope of this review.21–24
2. Heterocycles applied for SDM synthesis
2.1. N-Heteroaromatics
N-Heteroaromatics bearing halogen leaving groups e.g., chloride are known to undergo nucleophilic aromatic substitutions (SNAr) with nucleophiles like alcohols, thiols, carboxylates, and amines. The reaction proceeds via nucleophilic attack at the chloride-bearing carbon and aromaticity is re-established via chloride elimination.25 A widely used N-heteroaromatic for SNAr coupling is 2,4,6-trichloro-1,3,5-triazine (Trz or cyanuric chloride) (Fig. 2).26 Trz is synthesized on industrial scales from the high-temperature trimerization of cyanogen chloride.27 The bulk of global output is devoted to herbicide synthesis e.g. atrazine, and its own synthesis proceeds via step-wise SNAr couplings with isopropylamine and ethylamine. Atrazine's biological mechanism of action, degradation, and associated toxicity are also driven by successive chloride eliminations.28,29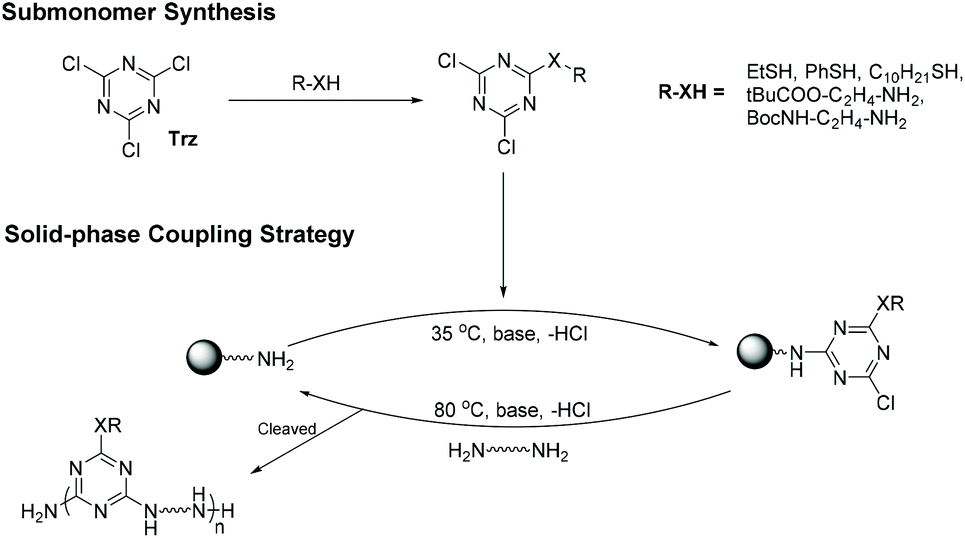 | ||
| Fig. 2 Employing triazine to generate sequence-defined macromolecules on amine-functionalized sold-phase. | ||
Given the efficacy of N-heteroaromatic SNAr couplings this reaction has been used both in-solution and on SP for a variety of applications.30 For example, Sumerlin et al. reported the use of Trz as a BB for the post-polymerization homo- or hetero-functionalization of acrylamide backbones using amines and thiols.31 Wenschuh et al. realized on-resin peptide macrocyclization using N-heteroaromatics such as Trz.32 Trz aside, a range of halogenated N-heteroaromatics have been exploited for SP SNAr couplings including: chloropyrimidines reacting with resin-bound amines;33 resin-bound thiols coupled with 3,6-dichloropyridazine afforded aminopyrazidines after aminolysis cleavage from SP;34 and 2,4,6-triaminopyrimidines were synthesized using SP techniques.35,36
Given the advantages of both SP and SNAr methodology Grate et al. published their iterative strategy for producing SDMs with diverse side-groups using Trz-based BBs (Fig. 2).37 Prior to SP incorporation, Trz-based monomers were generated using either amines or thiols, allowing a sub-monomer's side chain to be set prior to coupling. In the first SDM coupling step, the second chloride is displaced via coupling to a resin-tethered amine. The final chloride displacement is conducted at high temperature with a diamine, affording a new primary amine for future iterations. The initial examples reported consisted mainly of hexameric oligomers with molecular weights up to 1400 g mol−1 with a set of diverse side chains (alkyl, aromatic and charged residues) which led the authors to explore side-chain interaction patterns via molecular dynamics simulations.37 All reported hexamers could be facilely purified by HPLC to provide monodisperse, sequence-defined oligomers. Recently, it has been shown that careful side-chain tailoring allows foldamer structure formation via tailored hydrogen bonding and π–π interactions, akin to secondary structure formation in Nature.38
Active or latent categorization of Trz is challenging due to the multiple, equivalent reactive sites within the ring. The first SNAr coupling to SP could be classified as “active”, however an electrophilic site still exists at the chain end. This remaining ring-bound chloride is therefore a “latent” electrophile in subsequent iterations, therefore N-heteroaromatics like Trz can be considered to be simultaneously active and latent.
Besides interest in N-heteroaromatics for their SNAr reactivity on SP and in-solution,39 there is, to the best of our knowledge, only one SDM methodology exploiting N-heteroaromatics and photochemistry. Barner-Kowollik et al. have previously exploited photo-techniques to produce SDMs,40 and this was further developed to encompass N-heteroaromatics. In 2019, the solution-phase application of photo-responsive tetrazoles for SDM synthesis was disclosed (Fig. 3).41 Using orthogonal photo-responsive ligations, a [4 + 2] Diels–Alder cycloaddition of α-methyl benzaldehydes with fumarates under 365 nm irradiation was conducted. The second coupling joins a carboxylic acid to a pyrene-pended tetrazole via activation under visible light (410 nm), forming a nitrile imine which then couples to a carboxylic acid. The authors demonstrated the formation of palindromic sequences with the highest molecular weights reported reaching 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, as analysed by size-exclusion chromatography after each iterative, precise chain elongation. Aside from this example, photochemical ligations of N-heteroaromatics are a new methodology to accessing SDMs and are likely to be further developed in future.
000 g mol−1, as analysed by size-exclusion chromatography after each iterative, precise chain elongation. Aside from this example, photochemical ligations of N-heteroaromatics are a new methodology to accessing SDMs and are likely to be further developed in future.
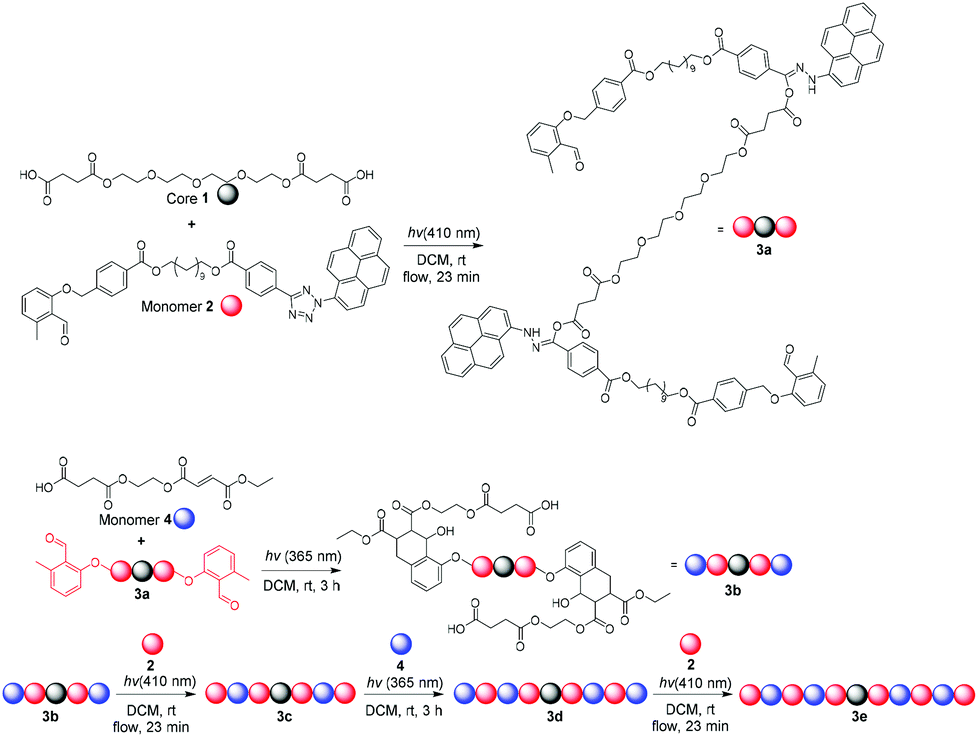 | ||
| Fig. 3 Combination of N-heteroaromatic and photochemistry to generate sequence-defined macromolecules by Barner-Kowollik et al. Reproduced from ref. 41 with permission from Wiley, copyright 2019. | ||
2.2. Maleimides
The maleimide functional group refers to an unsaturated imide housed in a 5 membered-ring. Standard maleimide synthesis proceeds by the condensation of an amine (alkyl or aromatic) with maleic anhydride. BBs containing multiple maleimide units are known, in particular bismaleimides, and take advantage of maleimide's manifold reactivity.42–44 Within polymer chemistry three maleimide reaction modalities (Michael additions, Diels–Alder (DA) cycloadditions, and double-bond polymerizations) have been reported for both backbone and side-chain formation.45,46 Maleimides may undergo Michael addition with carbon, oxygen, nitrogen, and sulfur-based nucleophiles.47 Reactions with thiols, the so-called thiol-maleimide click (a sub-set of the thiol–ene conjugation) has become a powerful coupling technique across the physical sciences, especially in the biological,48 and polymer sciences.47 The thiol-maleimide coupling employs the maleimide as the Michael acceptor and amongst the common double-bond acceptor systems, the maleimide ring is the most reactive due its electron deficiency. The widespread use of the thiol-maleimide click is due to its high selectivity, rapid reaction rates, and relative stability of the conjugate product (high temperature retro-Michael reactions are known).49Thiol–ene click chemistry techniques have been applied for sequence control,50 and maleimide-based couplings have been used for controlling sequence precision.51 Within the realm of iterative couplings, Zhang et al. reported using an iterative exponential growth (IEG) protocol, akin to epoxides (see section 2.5), to produce linear SDMs using the thiol-maleimide click (Fig. 4).52 Bis-functionalized BBs were produced with orthogonal PGs at both termini: (1) a maleimide protected as its furan DA adduct (quantitatively deprotected via retro-DA at high temperatures ca. 110 °C); and (2) an acetyl-protected thiol which is robust to high temperatures and deprotected selectively using either acidic or basic conditions. Splitting a given BB in two batches allows parallel, selective deprotections (Fig. 4). The two orthogonally deprotected batches are then merged, in a second step, and coupled via thiol-maleimide click, with the maleimide being actively consumed. Iterated splitting, orthogonal deprotection, and active coupling afforded a SDM on multi-gram scales. The use of multiple customized BBs with a considered deprotection strategy allows information to be encoded into a primary sequence, which was subsequently decoded using MS/MS techniques.52 The authors reported that IEG-based polymers could be rapidly produce e.g. at the extreme end, 7.9 g of a 128-mer with molecular weights of 27.4 kDa, as determined by size-exclusion chromatography. Zhang et al. have recently published a modified protocol allowing sequence-defined, “digital” dendrimers to be constructed using this methodology,53 expanding the scope of SDM architectures, albeit dendrimers are a specific sub-set of SDMs.54
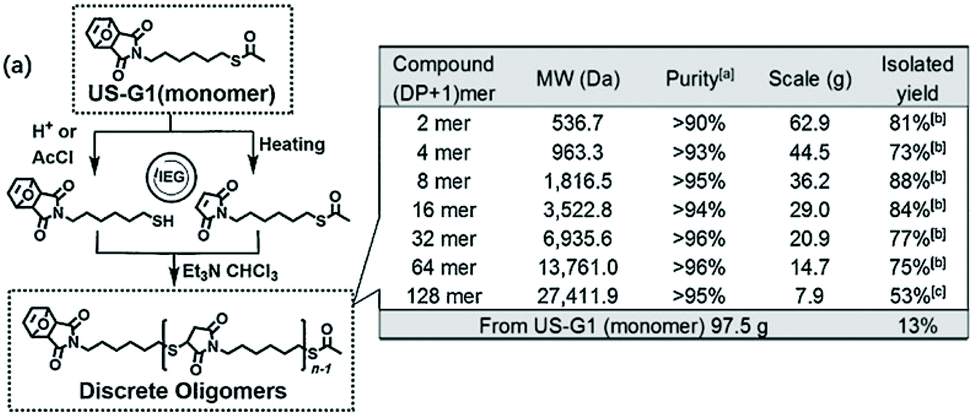 | ||
| Fig. 4 Doubly protected, bis-functionalized monomers containing thiols and maleimides can be converted using iterative exponential growth methodology into sequence-defined macromolecules. Reproduced from ref. 52 with permission from Wiley, copyright 2017. | ||
Due to the success of this IEG approach and the robustness of the chemistry, it is possible to see alternative, iterative syntheses of maleimides and the thiol-maleimide click. No feasible limitation on sequence length, architecture or side-chain formation is foreseeable, except that provided by the given synthetic method e.g., standard limitations of a SP strategy. Alternatively, employing maleimides as dienophiles in combination with dienes like furan,55 butadiene derivatives,56 and anthracene has also produced macromolecules.57 To the best of our knowledge, DA cycloadditions to form SDMs has not been reported, except for sequence-controlled macromolecules which has recently been reviewed.51 Additionally, thermally-induced, retro-DA reactions are useful for thermoreversible materials.58
2.3. Cyclic anhydrides
Organic acid anhydrides are compounds in which two acyl groups share the same bridging oxygen atom e.g., acetic anhydride. Cyclic anhydrides (CAs) are also known with maleic, succinic and glutaric anhydrides perhaps representing the most widely recognized examples of 5- or 6-membered CAs. The ring opening reaction of CAs with nucleophiles is also well known,59 and has been readily applied for a wide range of transformations e.g. reactions of amines and maleic acid derivatives afford the previously introduced maleimides,60 or the production of polypeptides through the ring-opening polymerization of α-amino acid N-carboxyanhydrides.61 Structurally-simple CAs like succinic acid may be viewed as a masked, linear, symmetric dicarboxylic acid, and upon ring-opening CAs reveal a carboxylic acid handle. The aminolysis of succinic acid has been widely applied, for example, for labelling SP, N-linked glycopeptides;62 as a SP anchor for the synthesis of lavendustin A and derivatives;63 or generating combinatorial libraries.64 Masked di-acids CAs can participate in step-wise AA + BB polymerizations and have been demonstrated as ideal compounds for SDM production on SP via an active strategy.Hartmann et al. reported in 2006 using CAs for the synthesis of poly(amidoamines), via treating amine-bearing resins with succinic anhydride. Basic conditions promoted ring-opening, whilst simultaneously generating an amide bond and unmasking a carboxylic acid. The newly revealed carboxylic acid was further reacted with a diamine via active ester formation to regenerate an active amine terminus (Fig. 5).65 The reported polymers could be produced with 10 repeating units, after 20 iterations, and when coupled with a poly(ethylene glycol) chain polymers with molecular weights up to 5000 g mol−1 could be achieved after dialysis in 32% yield.
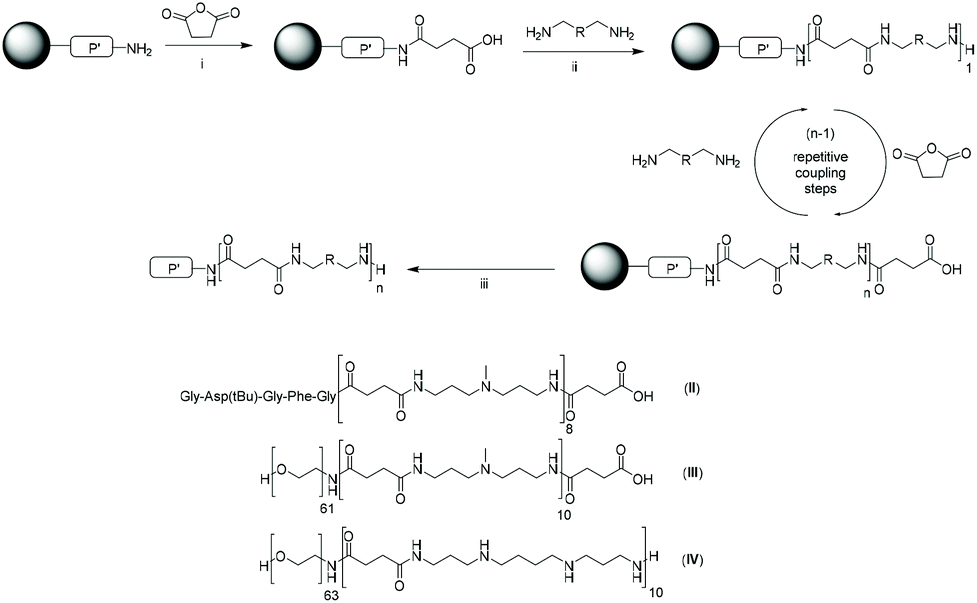 | ||
| Fig. 5 Iterative coupling of succinic anhydride to an amine-functionalized resin and subsequent amidation with diamines generates sequence-defined poly(amidoamines). Reproduced from ref. 65 with permission from ACS, copyright 2006. | ||
From this sub-monomer, two-step, iterative SP protocol many variants have been reported. Hartmann et al. reported the synthesis of sequence-defined poly(amidoamines) for reduction-sensitive, degradable DNA-delivery macromolecules.66 Alternatively, in a similar approach Wagner et al. too generated precision poly(amidoamines), but this method actively employed the CA ring-opening step prior to sequential SP addition. As such a variety of alkyl and aromatic-containing anhydrides were ring-opened with Fmoc-protected amines to afford tailor-made Fmoc-amino acid, which were sequentially added to resin via Fmoc-based peptide coupling strategies.67
Overall, the iterative aminolysis of CA has been a useful methodology to precisely grow SDMs across a range of fields. Iterative CA-diamine couplings have been used for the design of precision polymer medicinal constructs,68 for example monodisperse, cationic structures were used as non-viral polynucleotide delivery vectors.69 For further information on this topic, Wagner et al. have thoroughly reviewed the current state of the art for anti-tumour siRNA delivery.70
2.4. Thiolactones
The recognized versatility of thiols for polymer synthesis has been exploited to access multiple polymeric architectures and functionalization strategies e.g. thiol–ene chemistry (for maleimides see section 2.2).71,72 For example, within our own research group to circumvent the molecular weight limitations of SP, we exploited photo-induced thiol–ene couplings to produce high molecular weight, sequence-defined glycopolymers from lower molecular weight, sequence-defined oligomers.73 The benefits of thiol-based chemistry for iteratively creating SDMs has also been demonstrated, particularly by Du Prez and co-workers, through the use of thiolactones.Thiolactones are akin to their lactone cousins, except the endocyclic O is replaced by S (cyclic esters of mercapto-acids) and 4, 5, and 6-membered rings (denoted as α, β, or γ) are most prevalent (Fig. 6).74 The chemical reactivity of thiolactones also mirrors that of lactones and ring-opening can be induced using nucleophiles, particularly amines. Thiolactone aminolysis simultaneously generates an amide bond and reveals a thiol, which may be further functionalized in either a one-pot fashion or an adjunct step (Fig. 6). Advantageously, thiolactone aminolysis allows the introduction of two separate residual groups with high atom efficiency, in some cases with click-like 100% retention of all atoms.74 In 2011, Espeel et al. reported the one-pot aminolysis and photo-induced, thiol–ene double transformation of thiolactones.75 Further development by multiple research groups expanded upon this initial report and are beyond the scope of this review, but was thoroughly catalogued by Espeel and Du Prez.4,75
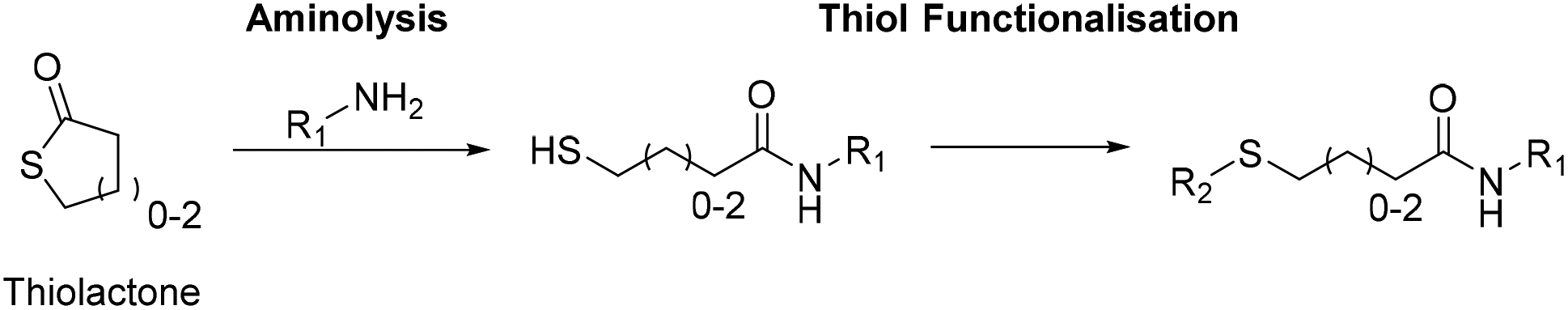 | ||
| Fig. 6 The sequential aminolysis of thiolactones and functionalization of the revealed thiol is the foundation for a range of sequence-defined protocols. Redrawn from ref. 74 with permission from Elsevier, copyright 2014. | ||
With a focus on sequence-definition, thiolactone strategies to SDMs have been developed both on SP and in solution, generally using the latent reactivity of thiolactones. Thiolactone 1, which is commercially-available, has been the foundation of several methodologies given the amine can be easily functionalized e.g., via amide formation. This is best illustrated by the formation of sequence-defined oligomers on SP using 2 developed by Espeel et al.76 Firstly, 1 was modified by introduction of an acrylate side-arm to give 2, which was introduced on to an already thiol-presenting resin via thiol-Michael addition (Fig. 7). Having installed a latent thiolactone on SP, aminolysis allowed the introduction of a range of side chains through amide formation. Any synthetic strategy development is not without challenges and in this case, during the aminolysis step, it was found that liberated thiols could dimerize on the resin to form stable disulfides (Fig. 7). The authors solved the loss of reactive thiol end groups via using dimethylphenylphosphine (PhPMe2). PhPMe2 had the dual function of reducing disulfide-linked dimers in situ and catalyzing the thiol-Michael addition of 2. Overall, the authors in this initial report demonstrated tri-, tetra- and pentameric structures with molecular weights up to 1700 g mol−1 as analysed by NMR, liquid-chromatography mass-spectrometry, and high-resolution mass-spectrometry. Moreover, a range of aliphatic, aromatic and heteroatom-rich functionalities were installed in the amide side-chain. This methodology illustrates the flexibility a latent heterocycle strategy offers, especially using a single BB to rapidly generate diverse SDMs.
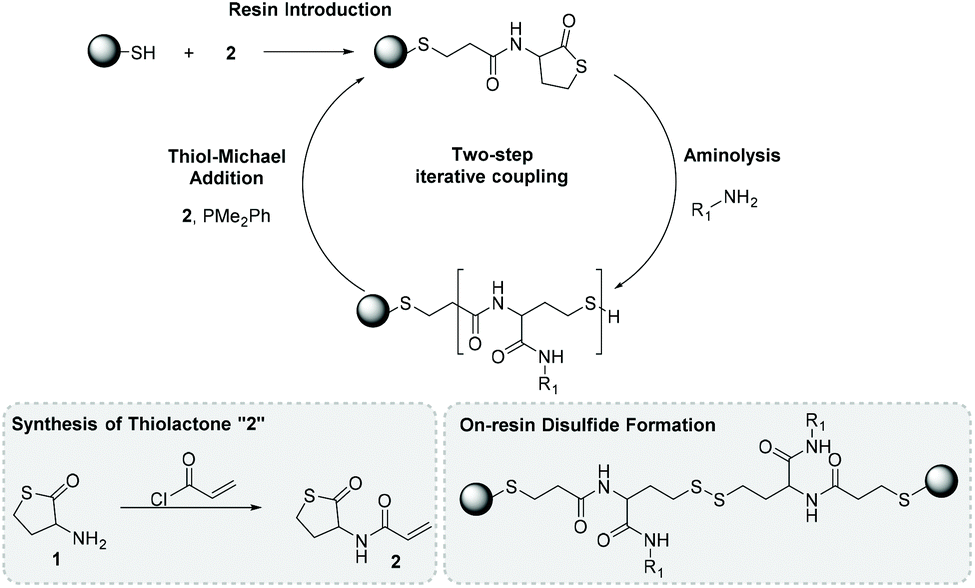 | ||
| Fig. 7 Installation of a latent thiolactone is achieved via thiol-Michael addition and subsequent ring aminolysis generates an amide side-chain and a thiol end-group allowing further iterations. Redrawn from ref. 76 with permission from Wiley, copyright 2013. | ||
Variants of this strategy have now been reported including: automated protocols utilising isocyanate-functionalized thiolactones (in this instance the thiol–ene reaction allowed side-chain installation);77,78 combining thiolactone chemistry in-solution with the Passerini three-component reaction (widely used in its own right to form SDMs)79–81 to convergently form SDMs on gram-scale with molecular weights up to 4 kDa,82 introduction of stereocontrol when defining side-chain identity with an automated protocol,83 and the formation of multi-functional dendrimers.84 These four examples are only selected examples of the extent of thiolactone development for the generation of SDMs with diverse architectures, and the likelihood of further advanced applications and combinations of thiolactone chemistry is high, given the rich modularity available via either active or latent modalities.
2.5. Epoxides
Epoxides are 3-membering ring cyclic ethers with their reactivity largely determined by the high degree of ring strain, which leads to high reactivity with nucleophiles (Fig. 8A). Their characteristic electrophilicity has been widely applied to many areas of synthetic chemistry e.g. polymerization,85 and biosynthesis.86 Successful syntheses have also been transferred to SP synthesis, although not, to the best of our knowledge, for SDM construction. In particular, resin-bound epoxide ring-openings have found utility in: oxazolidinone generation through isocyanate ring-opening and subsequent intramolecular cyclization;87,88 the formation of Shikimic acid-derived substrates for drug discovery via amine-induced ring-opening,89 generation of glycerol derivatives,90 the easy derivatization of phenoxypropanolamines91 and lactones,92 and finally in natural product synthesis by Jacobsen et al.93 and Nicolaou et al.94 Johnson and co-workers reported in 2015 the in-solution use of epoxides in a so-called “iterative exponential growth plus side-functionalization” strategy (IEG+), akin to IEG described in section 2.2, to generate sequence-defined polymers of up to 7 kDa with the controlled installation of stereo-defined side- chains.95 The synthesis hinges on an enantiopure, asymmetric epoxide bearing a silyl-protected alkyne e.g., 3-R or 3-S (Fig. 8B), whereby both terminal end-groups may be manipulated independently. Firstly, the epoxy group can be actively ring-opened at the unhindered endocyclic C atom by azide ion to reveal an alcohol with controlled stereochemistry. Subsequent functionalization via either acetylation or benzylation installs the desired side-chain. In a separate synthetic stream, 3-R or 3-S is treated with a fluoride source e.g., tetra-n-butylammonium fluoride to remove the silyl protecting group, revealing a terminal alkyne. In a subsequent step, the two streams can be combined via copper(I)-catalysed azide–alkyne cycloaddition (CuAAC) to generate a bridging triazole and the first dimer (4) bearing a latent epoxide. The iterative repetition of these two transformations (constituting the IEG+ process) allows for the generation of sequence-defined polymers. The authors found starting materials based upon epichlorohydrin to be ideal as: (1) 3-R/S could be generated on multi-gram scales, (2) azide ring-opening proceeds in a stereospecific and regioselective manner, and (3) the revealing of a secondary alcohol allowed facile, modular side-chain modification.95 Moreover, the easy installation of side-chain stereochemistry allowed Johnson et al. to produce polymers with varying tacticities. In the initial report, SDMs with up to 32 repeat units were produced, this was improved in a subsequent report with masses up to 12.1 kDa reported for SDMs, produced via IEG+, up to 1 g in scale.96 In this report the IEG+ strategy was extended to produce monodisperse diblock copolymers too via introducing allyl side-chain functionality. Using thiol–ene chemistry hydrophobic properties were introduced using n-decanethiol and hydrophilic properties via 1-mercapto-triethyleneglycol monomethyl ether or thioglycerol. The synthesis of a 32-mer with side-chain functionality divided equally between hydrophobic and hydrophilic blocks allowed effective phase separation and the formation of well-ordered morphologies.96 Finally, further development by Johnson and co-workers has shown that IEG+ methodologies can be automated to produce monodisperse SDMs.97 These reports by Johnson et al. demonstrate that epoxide chemistry can be developed in-solution to produce SDMs with high molecular weights and when coupled with click chemistries e.g. thiol–ene or CuAAC can produce a diverse range of SDM structures with varied applications based on either active or latent use of the ring.95–97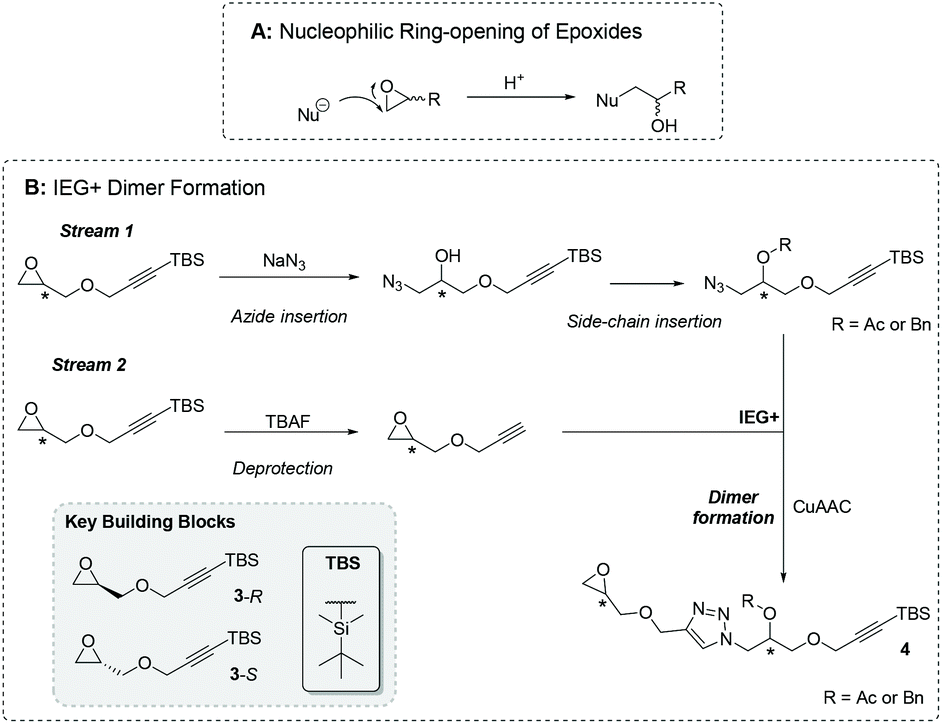 | ||
| Fig. 8 (A) General scheme for epoxide nucleophilic ring-opening; and (B) iterative exponential growth of epoxide-based, sequence-defined macromolecules. | ||
2.6 Cyclic sulfamidate
Cyclic sulfamidates (CSs) are a class of heterocycle which combine both electrophilic and nucleophilic reactivity, and are usually produced as either 5- or 6-membered ring (seven-membered rings are known).98 Saturated, sterically-hindered,99,100 and unsaturated CSs are known in the literature,101,102 but for the purposes of this review only saturated, unhindered rings shall be discussed. As aziridines and azetidine synthons, CSs are employed for their electrophilicity via the regioselective and stereospecific (SN2) ring opening of the endocyclic C–O bond (Fig. 9A). CSs have on occasion been applied as amine nucleophiles.103 CS ring opening reveals a N-sulfate intermediate which after cleavage affords a N–H bond (Fig. 9A). Through a SDM lens, the N-sulfate intermediate may function either as a PG or an end-point itself. Unlike aziridines or azetidines, CS reactivity is not predominantly driven by ring strain meaning 5- and 6-membered CSs retain similar reactivity. CS ring opening has been demonstrated with a diverse set of nucleophiles including thiols,104,105 amines,106,107 halides including fluoride,107,108 and iodide,109 azide,106,107,110 thiocyanate,107 and carbon-centred nucleophiles like organometallics,111–114 cyanide,106 and enolates.107,115 Trifluorothiomethylation has also been demonstrated.116 A recent, comprehensive review of CS synthetic routes by Saeidian et al. details the multiple routes and strategies for accessing CSs.117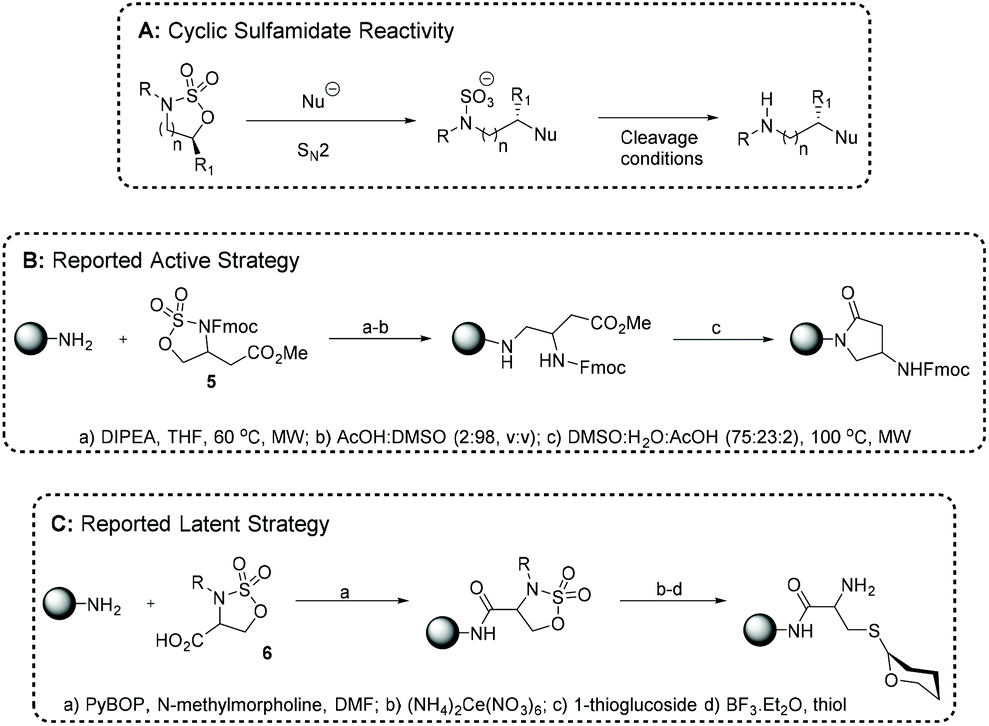 | ||
| Fig. 9 (A) General cyclic sulfamidate reactivity; and literature reports of cyclic sulfamidates being employed on solid-phase in (B) active and (C) latent modalities. | ||
CSs have been widely applied in organic synthesis especially for: heterocyclic synthesis,118 enantiopure amino acid synthesis,107 and carbohydrate synthesis.119,120 Given their properties and advantages CSs are ideal candidates to produce SDMs on SP. In particular, we were attracted to CSs as we have recently reported on the synthesis of sulfated/sulfonated oligo(amidoamines) and identified CSs as versatile building blocks to generate N-sulfated SDMs.
We envisaged iteratively applying cyclic sulfamidate heterocycles in both active and latent modes (akin to thiolactone reactivity – please see section 2.4).121 CSs had been coupled on SP in both active and latent modalities but never in an iterative fashion. For example, Lubell et al. have reported using CSs as active electrophiles to precisely install lactam rings into a growing polypeptide chain (Fig. 9B).122–125 In a typical exemplar, the methodology proceeds via direct ring opening of an ester-functionalized CS e.g. 5 on an amine-bearing resin. CS ring opening generates a secondary amine, extends the main chain's backbone and reveals a terminal N-sulfate functionality. N-Sulfate removal was affected at low pH and further heating forms a lactam ring from the condensation of the secondary amine and pendant ester (Fig. 9B). Alternatively, Cohen et al. reported glycopeptide synthesis using a carboxylate-bearing 6 as a latent electrophile. Amide coupling of 6 to an amine-functionalized resin afforded a latent CS which was attacked by thioglycosides to generate glycoconjugates (Fig. 9C).126,127
We proposed a latent strategy using novel CS 7, whereby an amide coupling would afford a latent electrophile bound to a growing macromolecular chain.121 Resultantly, a range of nucleophiles could be employed to generate ring-opening. For example, using a bis-functionalized building block, in combination with 7, realizes an iterative, step-wise AB + CD chain elongation strategy (Fig. 10A). Alternatively, we envisaged coupling an amine-bearing resin with CS BBs in an active fashion to reveal a N-sulfate serving the role of an amine protecting group. Primary amine bis-alkylation and secondary amine mono-alkylation would yield branching and linear chain growth, respectively. To allow iterative chain elongation N-sulfate cleavage would regenerate a nucleophilic amine end-group, primed for conjugation with another CS (Fig. 10B).121
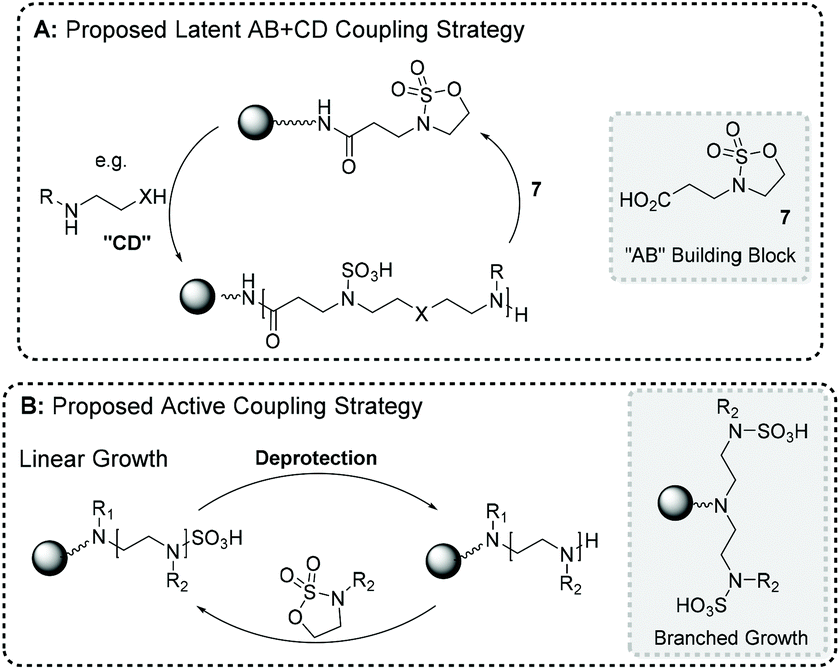 | ||
| Fig. 10 Illustrated proposals for utilising cyclic sulfamidate building blocks in (A) a latent AB + CD and (B) an active iterative manner to generate sequence-defined macromolecules on solid-phase. | ||
Exploring a latent strategy, a range of amide activating agents were employed to couple 7 to an oligo(amidoamine)-presenting resin. PyBOP and Oxyma-based reagents afforded amide bond formation, but nucleophilic components were released during their activation mechanisms e.g. PyBOP expels deprotonated HOBt after initial active ester formation.121 These nucleophilic components then reacted with the latent CS generating undesired side-products (8 – PyBOP-derived and 9 – Oxyma reagent-derived) in significant quantities (Fig. 11). Despite extensive experimentation no conditions were identified which circumvented side product formation, therefore, active coupling strategies were instead investigated. To this end, amine-functionalized resins were successfully alkylated with a range of CS BBs bearing sp3 and sp2-hybridized residual groups e.g., Boc, ethyl, benzyl etc. Primary amine groups could undergo bis-alkylation to afford branched architectures, whereas secondary amines yielded linear chain growth as outlined in Fig. 10. Attempts were then undertaken to find a universal N-sulfate cleavage methodology using mildly acidic solutions and elevated temperatures. Akin to literature, cleavage was observed using pH 5.5 and 50 °C for Boc-functionalized N-sulfates. All examples with sp3-carrying residual groups could not be cleaved, despite combining pH 1 and 100 °C. This synthetic limitation prevented a wide range of CS BBs being employed when building macromolecular chains.121
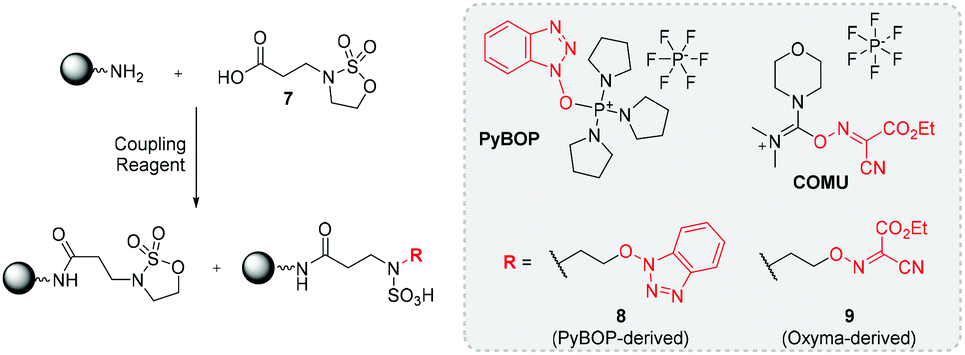 | ||
| Fig. 11 Side product formation from ring-opening during amide coupling of 7 to amine-functionalized resin following a latent strategy. | ||
These challenges highlighted the issues surrounded employing heterocycles for SDM synthesis, but despite the challenges faced in realising both latent and active iterative strategies, we re-deployed the ring-opening of CSs to produce a diverse set of N-sulfated oligo(amidoamine)s using the BB actively. Given our interest in biomimicry and sulfated SDMs, the resultant N-sulfated oligomers were identified as potential glycosaminoglycan mimetics.
2.7. Cyclic boronic esters
Looking to the future of heterocycles in the SDM field, to the best of our knowledge, no reports exist demonstrating the use of cyclic boronic esters to produce SDMs. Cyclic boronic esters are ideal candidates for future investigations given their: long term stability, chemical orthogonality,128 ease of synthesis,129 commercial availability, and their facile transformation into amines, alcohols, thiols and halides.130 Hence, in this section we will briefly outline their reactivity, and sketch potential future directions with illustrative examples focusing on pinacol (pin) and N-methyliminoacetic acid (MIDA) variants. Furthermore, all heterocycles so far described are chemically transformed as either active or latent BBs, however the following boron-mediated reactions are importantly facilitated by the heterocycle itself. After a given coupling a cyclic boronic ester remains chemically unaltered, therefore in this review cyclic boronic esters are classified as latent BBs given their potential later conversion to other functionality.Boronic acids (R-B(OH)2) and esters (R-B(OR1)(OR2)) are organoboranes with two pendant oxygen-bound groups and a carbon group (sp2 or sp3 hybridised), which given an unoccupied pz orbital at boron are Lewic acids, allowing reversible formation of tetrahedral complexes (Fig. 12). This reversibility has been used for sensing,131 separation technologies,132 and cross couplings e.g. the Suzuki–Miyuara reaction.129,133 Pin- and MIDA-boronic esters are structurally diverse at boron: B(pin)'s conformation is planar trigonal whereas B(MIDA)'s is tetrahedral (Fig. 12). Most boronic ester-mediated proceed from a nucleophilic, tetra-coordinated boron species, hence B(pin) is typically more reactive than B(MIDA).134 This reactivity difference has been utilized, in a procedure akin to Fmoc-peptide synthesis, for the natural product synthesis by Burke et al. Using bifunctionalised halo-MIDA boronic esters (AB-type building blocks) a given BB can be coupled via Suzuki–Miyuara methodology to a terminal boronic acid. Pendant B(MIDA) heterocycles are deprotected to the corresponding boronic acid, allowing further iterations. Burke et al. produced pentacyclic secodaphnane alkaloids from three key BBs (Fig. 13),135–137 and have subsequently automated the process, attractive for future SDM syntheses.138
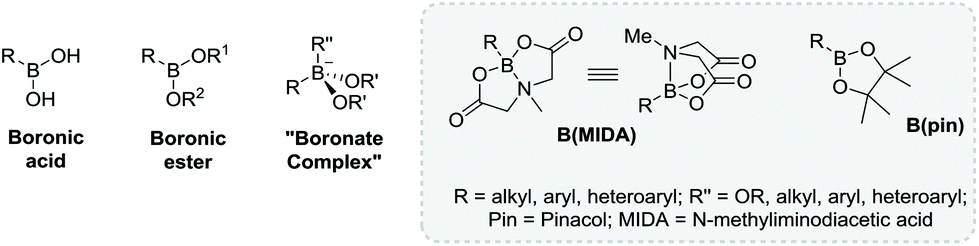 | ||
| Fig. 12 Generic structures of boronic acids, esters and boronate complexes. | ||
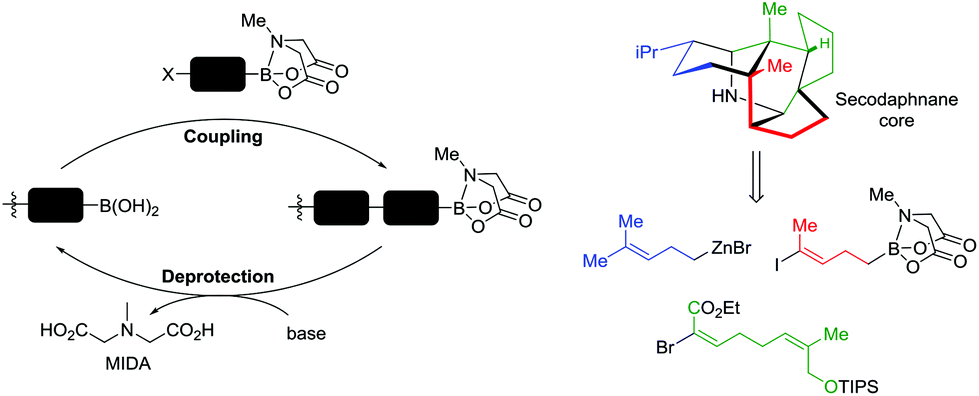 | ||
| Fig. 13 MIDA-boronate iterative coupling strategy and its applications to generating the secodaphnane core via iterative coupling of three tailored building blocks. Redrawn from ref. 135 with permission from AAAS, copyright 2015. | ||
Alternatively, Aggarwal et al. have broadly developed the lithiation–borylation iterative coupling strategy, the full details of which are beyond the scope of this review.139 Lithiation–borylation couplings proceed in three distinct steps: (1) organolithium formation from α-lithiation of a carbamate (Cb) or ester e.g. 2,4,6-triisopropyl benzoates (TIB esters) (Fig. 14); (2) electrophilic trapping of an electrophilic B(pin) BB forms a transient boronate or “ate” complex (Fig. 14A); and finally (3) the “ate” complex undergoes eliminative 1,2-metalate rearrangement to form a new C–C bond with a latent B(pin) being retained (Fig. 14A).139 One pertinent example illustrates its SDM potential as in 2014, Aggarwal et al. reported the “assembly line” synthesis of linear scaffolds containing 10 contiguous stereocentres.140 Using Li-1 alone or mixed with Li-2, chiral information was precisely installed into a growing B(pin)-capped backbone. It was shown that the methyl substitution pattern (primary structure) influenced a chain's helical propensity (secondary structure) (Fig. 14B).140 Despite its exquisite control over stereochemical transfer, lithiation-borylation requires both extremely low-temperatures and expert reagent handling to achieve the high-fidelity iterative coupling results reported. In cases where a SDM's programmed stereochemical fidelity was crucial, these synthetic limitations may also limit the achievable macromolecular weights.
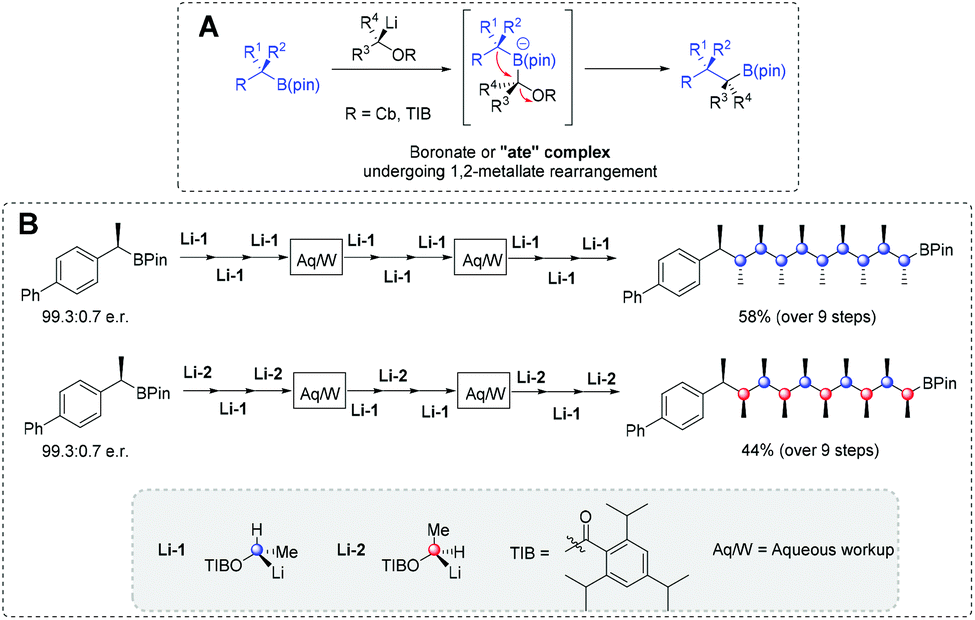 | ||
| Fig. 14 (A) General scheme for C–C bond formation via lithiation–borylation methodology; and (B) the iterative coupling of stereochemically-rich chains. Reproduced from ref. 139 with permission from ACS, copyright 2014. | ||
Both of these examples demonstrate that MIDA boronic ester coupling and lithiation–borylation methodologies are ideal future candidates for producing new SDMs.
3. Future perspectives
The future of the sequence-defined macromolecule field could be developed in several distinct directions e.g.: (1) developing completely new systems and methodologies; (2) redeploying systems exclusively developed for either in-solution or SP methodologies into the other; and (3) transferring developments from other fields into the SDM field.Given the breadth of heterocyclic systems discussed in this review, the development of new iterative coupling strategies utilising hetero- and carbocyclic rings could be a rich vein of development e.g., lactams, aziridines, azetidines etc. Additionally, systems exclusively applied either in solution or on SP could be transferred to the other e.g., epoxides have been well-developed in solution, therefore could be re-applied for iterative SDM assembly on SP. Finally, buoyed by the example of Barner-Kowollik et al. who applied photochemistry to generate SDMs – please see section 2.1,41 advances from the organic and organometallic chemistry communities may provide inspiration and start to percolate through into the sequence-defined polymer community e.g., organo- and metal catalysis, electrochemistry, etc.
Due to the relative youth of this field, we speculate many advances in synthesis, architecture, elemental composition, and ultimately the ways in which sequence-defined polymers will be applied.
4. Conclusions
The wide application of heterocyclic systems for generating monodisperse, sequence-defined macromolecules via iterative couplings is facilitating the thorough investigation of sequence-definition in many varied fields e.g., novel biomimetics, therapeutics, and data storage systems, to name but a few. In this review, we have outlined the current state of the art of solid-phase and solution-phase strategies for a range of 3–6 membered-ring systems (saturated and unsaturated rings) which give access to a diverse range of backbone and side-chain compositions, and polymeric architectures using sequential couplings. Further development of established methodologies is ongoing to advance the application of sequence-defined polymers. The creation of novel strategies based on heterocyclic systems will add to the already-reported, extensive arsenal available to the chemist. The further utilization of techniques compatible with automation in both solution-phase and on solid-phase is also leading to forward strides in how easily sequence-defined systems transition from the academic lab to real-world applications. On all fronts the iterative utilization of heterocyclic systems to construct SDMs is promising as it pushes into further new uncharted territory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SAH would like to thank Dr Christopher Sandford for enlightening conversations and insights into boron chemistry. LH thanks the DFG for financial support through the FOR2327 and CRC1208.Notes and references
- M. A. R. Meier and C. Barner-Kowollik, Adv. Mater., 2019, 31, 1806027 CrossRef PubMed.
- J.-F. Lutz, Macromol. Rapid Commun., 2017, 38, 1700582 CrossRef PubMed.
- W. M. Dawson, G. G. Rhys and D. N. Woolfson, Curr. Opin. Chem. Biol., 2019, 52, 102–111 CrossRef CAS PubMed.
- R. Aksakal, C. Mertens, M. Soete, N. Badi and F. Du Prez, Adv. Sci., 2021, 8, 2004038 CrossRef CAS PubMed.
- J. De Neve, J. J. Haven, L. Maes and T. Junkers, Polym. Chem., 2018, 9, 4692–4705 RSC.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- O. Calin, S. Eller and P. H. Seeberger, Angew. Chem., Int. Ed., 2013, 52, 5862–5865 CrossRef CAS PubMed.
- K. Naresh, F. Schumacher, H. S. Hahm and P. H. Seeberger, Chem. Commun., 2017, 53, 9085–9088 RSC.
- R. Behrendt, P. White and J. Offer, J. Pept. Sci., 2016, 22, 4–27 CrossRef CAS PubMed.
- B. Roy, A. Depaix, C. Périgaud and S. Peyrottes, Chem. Rev., 2016, 116, 7854–7897 CrossRef CAS PubMed.
- B. Sauvagnat, F. Lamaty, R. Lazaro and J. Martinez, Tetrahedron Lett., 1998, 39, 821–824 CrossRef CAS.
- S. A. Hill, C. Gerke and L. Hartmann, Chem. – Asian J., 2018, 13, 3611–3622 CrossRef CAS PubMed.
- S. C. Solleder, R. V. Schneider, K. S. Wetzel, A. C. Boukis and M. A. R. Meier, Macromol. Rapid Commun., 2017, 38, 1600711 CrossRef PubMed.
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC.
- F. Shamout, L. Fischer, N. L. Snyder and L. Hartmann, Macromol. Rapid Commun., 2020, 41, 1900473 CrossRef CAS PubMed.
- R. Faust, Angew. Chem., Int. Ed., 2001, 40, 2251–2253 CrossRef CAS PubMed.
- H. K. Hall Jr. and A. B. Padias, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 625–635 CrossRef.
- R. C. Hartley and S. T. Caldwell, J. Chem. Soc., Perkin Trans. 1, 2000, 477–501, 10.1039/A804421J.
- R. B. Merrifield and J. M. Stewart, Nature, 1965, 207, 522–523 CrossRef CAS PubMed.
- W. R. Gutekunst and C. J. Hawker, J. Am. Chem. Soc., 2015, 137, 8038–8041 CrossRef CAS PubMed.
- Y. Liang, J.-L. Pan, L.-H. Sun, J.-M. Ma, H. Jiang and Z.-L. Li, Macromol. Rapid Commun., 2019, 40, 1900435 CrossRef CAS PubMed.
- J. Zhang, M. E. Matta and M. A. Hillmyer, ACS Macro Lett., 2012, 1, 1383–1387 CrossRef CAS.
- J. A. Nowalk, C. Fang, A. L. Short, R. M. Weiss, J. H. Swisher, P. Liu and T. Y. Meyer, J. Am. Chem. Soc., 2019, 141, 5741–5752 CrossRef CAS PubMed.
- B. R. Elling, J. K. Su, J. D. Feist and Y. Xia, Chem, 2019, 5, 2691–2701 CAS.
- A. Sharma, A. El-Faham, B. G. de la Torre and F. Albericio, Front. Chem., 2018, 6, 516 CrossRef PubMed.
- Encyclopedia of Reagents for Organic Synthesis, DOI:10.1002/047084289X.rn00320.
- Ullmann's Encyclopedia of Industrial Chemistry, DOI:10.1002/14356007.a08_191.
- G. P. Dooley, K. F. Reardon, J. E. Prenni, R. B. Tjalkens, M. E. Legare, C. D. Foradori, J. E. Tessari and W. H. Hanneman, Chem. Res. Toxicol., 2008, 21, 844–851 Search PubMed.
- H. Zhao, Y. Liu, Z. Cui, D. Beattie, Y. Gu and Q. Wang, J. Agric. Food Chem., 2011, 59, 11711–11717 CrossRef CAS PubMed.
- E. R. Felder, W. K. D. Brill and K. Martina, Methods in Enzymology, Academic Press, 2003, vol. 369, pp. 435–469 Search PubMed.
- T. Kubo, C. A. Figg, J. L. Swartz, W. L. A. Brooks and B. S. Sumerlin, Macromolecules, 2016, 49, 2077–2084 CrossRef CAS.
- D. Scharn, L. Germeroth, J. Schneider-Mergener and H. Wenschuh, J. Org. Chem., 2001, 66, 507–513 CrossRef CAS PubMed.
- E. A. Arvanitis, N. Chadha, R. S. Pottorf and M. R. Player, J. Comb. Chem., 2004, 6, 414–419 CrossRef CAS PubMed.
- I. Parrot, C.-G. Wermuth and M. Hibert, Tetrahedron Lett., 1999, 40, 7975–7978 CrossRef CAS.
- F. Guillier, P. Roussel, H. Moser, P. Kane and M. Bradley, Chem. – Eur. J., 1999, 5, 3450–3458 CrossRef CAS.
- C. Wéber, Á. Demeter and I. Greiner, Tetrahedron, 2006, 62, 2304–2312 CrossRef.
- J. W. Grate, K.-F. Mo and M. D. Daily, Angew. Chem., Int. Ed., 2016, 55, 3925–3930 CrossRef CAS PubMed.
- S.-H. Ahn and J. W. Grate, J. Phys. Chem. B, 2019, 123, 9364–9377 CrossRef CAS PubMed.
- S. M. Osman, S. N. Khattab, E.-S. A. Aly, E.-R. Kenawy and A. El-Faham, J. Polym. Res., 2017, 24, 231 CrossRef.
- W. Konrad, F. R. Bloesser, K. S. Wetzel, A. C. Boukis, M. A. R. Meier and C. Barner-Kowollik, Chem. – Eur. J., 2018, 24, 3413–3419 CrossRef CAS PubMed.
- W. Konrad, C. Fengler, S. Putwa and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2019, 58, 7133–7137 CrossRef CAS PubMed.
- L. M. Polgar, E. Hagting, W.-J. Koek, F. Picchioni and M. Van Duin, Polymers, 2017, 9, 81 CrossRef PubMed.
- M. Sava, Des. Monomers Polym., 2013, 16, 14–24 CrossRef CAS.
- M. Sava, I. Sava, V. Cozan and F. Tanasa, J. Appl. Polym. Sci., 2007, 106, 2185–2191 CrossRef CAS.
- E. Dolci, V. Froidevaux, C. Joly-Duhamel, R. Auvergne, B. Boutevin and S. Caillol, Polym. Rev., 2016, 56, 512–556 CrossRef CAS.
- D. J. Hall, H. M. Van Den Berghe and A. P. Dove, Polym. Int., 2011, 60, 1149–1157 CrossRef CAS.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- J. M. J. M. Ravasco, H. Faustino, A. Trindade and P. M. P. Gois, Chem. – Eur. J., 2019, 25, 43–59 CrossRef CAS PubMed.
- A. D. Baldwin and K. L. Kiick, Polym. Chem., 2013, 4, 133–143 RSC.
- S. Martens, J. O. Holloway and F. E. Du Prez, Macromol. Rapid Commun., 2017, 38, 1700469 CrossRef PubMed.
- Q. Shi, Y. Zhang, Z. Huang, N. Zhou, Z. Zhang and X. Zhu, Polym. J., 2020, 52, 21–31 CrossRef CAS.
- Z. Huang, J. Zhao, Z. Wang, F. Meng, K. Ding, X. Pan, N. Zhou, X. Li, Z. Zhang and X. Zhu, Angew. Chem., Int. Ed., 2017, 56, 13612–13617 CrossRef CAS PubMed.
- Z. Huang, Q. Shi, J. Guo, F. Meng, Y. Zhang, Y. Lu, Z. Qian, X. Li, N. Zhou, Z. Zhang and X. Zhu, Nat. Commun., 2019, 10, 1918 CrossRef PubMed.
- E. Abbasi, S. F. Aval, A. Akbarzadeh, M. Milani, H. T. Nasrabadi, S. W. Joo, Y. Hanifehpour, K. Nejati-Koshki and R. Pashaei-Asl, Nanoscale Res. Lett., 2014, 9, 247 CrossRef PubMed.
- A. Gandini, Prog. Polym. Sci., 2013, 38, 1–29 CrossRef CAS.
- V. Marchán, S. Ortega, D. Pulido, E. Pedroso and A. Grandas, Nucleic Acids Res., 2006, 34, e24 CrossRef PubMed.
- B. Gacal, H. Durmaz, M. A. Tasdelen, G. Hizal, U. Tunca, Y. Yagci and A. L. Demirel, Macromolecules, 2006, 39, 5330–5336 CrossRef CAS.
- A. Sanyal, Macromol. Chem. Phys., 2010, 211, 1417–1425 CrossRef CAS.
- I. Atodiresei, I. Schiffers and C. Bolm, Chem. Rev., 2007, 107, 5683–5712 CrossRef CAS PubMed.
- N. Salewska and M. J. Milewska, J. Heterocycl. Chem., 2014, 51, 999–1003 CrossRef CAS.
- H. R. Kricheldorf, Angew. Chem., Int. Ed., 2006, 45, 5752–5784 CrossRef CAS PubMed.
- Y. Tian, Y. Zhou, S. Elliott, R. Aebersold and H. Zhang, Nat. Protoc., 2007, 2, 334–339 CrossRef CAS PubMed.
- R. Devraj and M. Cushman, J. Org. Chem., 1996, 61, 9368–9373 CrossRef CAS.
- J. Perumattam, S. Chakravarty, G. A. McEnroe, R. R. Goehring, B. Mavunkel, S. Suravajjala, W. W. Smith and B. Chen, Mol. Diversity, 1997, 3, 121–128 CrossRef CAS PubMed.
- L. Hartmann, E. Krause, M. Antonietti and H. G. Börner, Biomacromolecules, 2006, 7, 1239–1244 CrossRef CAS PubMed.
- L. Hartmann, S. Häfele, R. Peschka-Süss, M. Antonietti and H. G. Börner, Macromolecules, 2007, 40, 7771–7776 CrossRef CAS.
- D. Schaffert, N. Badgujar and E. Wagner, Org. Lett., 2011, 13, 1586–1589 CrossRef CAS PubMed.
- L. Hartmann and H. G. Börner, Adv. Mater., 2009, 21, 3425–3431 CrossRef CAS PubMed.
- L. Hartmann, S. Häfele, R. Peschka-Süss, M. Antonietti and H. G. Börner, Chem. – Eur. J., 2008, 14, 2025–2033 CrossRef CAS PubMed.
- J. Luo, E. Wagner and Y. Wang, J. Mater. Chem. B, 2020, 8, 2020–2031 RSC.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- A. B. Lowe, Polym. Chem., 2014, 5, 4820–4870 RSC.
- C. Gerke, M. F. Ebbesen, D. Jansen, S. Boden, T. Freichel and L. Hartmann, Biomacromolecules, 2017, 18, 787–796 CrossRef CAS PubMed.
- P. Espeel and F. E. Du Prez, Eur. Polym. J., 2015, 62, 247–272 CrossRef CAS.
- P. Espeel, F. Goethals and F. E. Du Prez, J. Am. Chem. Soc., 2011, 133, 1678–1681 CrossRef CAS PubMed.
- P. Espeel, L. L. G. Carrette, K. Bury, S. Capenberghs, J. C. Martins, F. E. Du Prez and A. Madder, Angew. Chem., Int. Ed., 2013, 52, 13261–13264 CrossRef CAS PubMed.
- S. Martens, J. Van den Begin, A. Madder, F. E. Du Prez and P. Espeel, J. Am. Chem. Soc., 2016, 138, 14182–14185 CrossRef CAS PubMed.
- J. O. Holloway, C. Mertens, F. E. Du Prez and N. Badi, Macromol. Rapid Commun., 2019, 40, 1800685 CrossRef PubMed.
- O. Kreye, T. Tóth and M. A. R. Meier, J. Am. Chem. Soc., 2011, 133, 1790–1792 CrossRef CAS PubMed.
- R. Kakuchi, Polym. J., 2019, 51, 945–953 CrossRef CAS.
- A. Llevot, A. C. Boukis, S. Oelmann, K. Wetzel and M. A. R. Meier, Top. Curr. Chem., 2017, 375, 66 CrossRef PubMed.
- S. C. Solleder, S. Martens, P. Espeel, F. Du Prez and M. A. R. Meier, Chem. – Eur. J., 2017, 23, 13906–13909 CrossRef CAS PubMed.
- C. Mertens, M. Soete, M. L. Ślęczkowski, A. R. A. Palmans, E. W. Meijer, N. Badi and F. E. Du Prez, Polym. Chem., 2020, 11, 4271–4280 RSC.
- N. U. Kaya, F. E. Du Prez and N. Badi, Macromol. Chem. Phys., 2017, 218, 1600575 CrossRef.
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef CAS PubMed.
- I. Vilotijevic and T. F. Jamison, Angew. Chem., Int. Ed., 2009, 48, 5250–5281 CrossRef CAS PubMed.
- H.-P. Buchstaller, J. Comb. Chem., 2003, 5, 789–793 CrossRef CAS PubMed.
- H.-P. Buchstaller, Tetrahedron, 1998, 54, 3465–3470 CrossRef CAS.
- H. Miao, J. A. Tallarico, H. Hayakawa, K. Münger, J. L. Duffner, A. N. Koehler, S. L. Schreiber and T. A. Lewis, J. Comb. Chem., 2007, 9, 245–253 CrossRef CAS PubMed.
- J.-S. Seo, C. M. Yoon and Y.-D. Gong, J. Comb. Chem., 2007, 9, 366–369 CrossRef CAS PubMed.
- W. M. Bryan, W. F. Huffman and P. K. Bhatnagar, Tetrahedron Lett., 2000, 41, 6997–7000 CrossRef CAS.
- N. Gouault, J.-F. Cupif, A. Sauleau and M. David, Tetrahedron Lett., 2000, 41, 7293–7297 CrossRef CAS.
- D. A. Annis, O. Helluin and E. N. Jacobsen, Angew. Chem., Int. Ed., 1998, 37, 1907–1909 CrossRef CAS.
- K. C. Nicolaou, S. A. Snyder, A. Bigot and J. A. Pfefferkorn, Angew. Chem., Int. Ed., 2000, 39, 1093–1096 CrossRef CAS PubMed.
- J. C. Barnes, D. J. C. Ehrlich, A. X. Gao, F. A. Leibfarth, Y. Jiang, E. Zhou, T. F. Jamison and J. A. Johnson, Nat. Chem., 2015, 7, 810–815 CrossRef CAS PubMed.
- Y. Jiang, M. R. Golder, H. V. T. Nguyen, Y. Wang, M. Zhong, J. C. Barnes, D. J. C. Ehrlich and J. A. Johnson, J. Am. Chem. Soc., 2016, 138, 9369–9372 CrossRef CAS PubMed.
- F. A. Leibfarth, J. A. Johnson and T. F. Jamison, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10617 CrossRef CAS PubMed.
- F. Duran, L. Leman, A. Ghini, G. Burton, P. Dauban and R. H. Dodd, Org. Lett., 2002, 4, 2481–2483 CrossRef CAS PubMed.
- G. Jiménez-Osés, A. Avenoza, J. H. Busto and J. M. Peregrina, Tetrahedron: Asymmetry, 2008, 19, 443–449 CrossRef.
- C. D. Navo, N. Mazo, A. Avenoza, J. H. Busto, J. M. Peregrina and G. Jiménez-Osés, J. Org. Chem., 2017, 82, 13250–13255 CrossRef CAS PubMed.
- Y.-J. Chen, Y.-H. Chen, C.-G. Feng and G.-Q. Lin, Org. Lett., 2014, 16, 3400–3403 CrossRef CAS PubMed.
- Y. Liu, Y. Huang, Z. Yi, G. Liu, X.-Q. Dong and X. Zhang, Adv. Synth. Catal., 2019, 361, 1582–1586 CrossRef CAS.
- J. J. Posakony, J. R. Grierson and T. J. Tewson, J. Org. Chem., 2002, 67, 5164–5169 CrossRef CAS PubMed.
- S. De Luca, G. Digilio, V. Verdoliva, P. Tovillas, G. Jiménez-Osés and J. M. Peregrina, J. Org. Chem., 2019, 84, 14957–14964 CrossRef CAS PubMed.
- P. Tovillas, I. García, P. Oroz, N. Mazo, A. Avenoza, F. Corzana, G. Jiménez-Osés, J. H. Busto and J. M. Peregrina, J. Org. Chem., 2018, 83, 4973–4980 CrossRef CAS PubMed.
- J. J. Posakony and T. J. Tewson, Synthesis, 2002, 0859–0864 CrossRef CAS.
- L. Wei and W. D. Lubell, Can. J. Chem., 2001, 79, 94–104 CrossRef CAS.
- C. Huang, L. Yuan, K. M. Rich and J. McConathy, Nucl. Med. Biol., 2013, 40, 498–506 CrossRef CAS PubMed.
- N. Ursinyova, R. B. Bedford and T. Gallagher, Eur. J. Org. Chem., 2016, 2016, 673–677 CrossRef CAS.
- A. Megia-Fernandez, M. Ortega-Muñoz, F. Hernandez-Mateo and F. Santoyo-Gonzalez, Adv. Synth. Catal., 2012, 354, 1797–1803 CrossRef CAS.
- P. Arigala, V. S. Sadu, I.-T. Hwang, J.-S. Hwang, C.-U. Kim and K.-I. Lee, Adv. Synth. Catal., 2015, 357, 2027–2032 CrossRef CAS.
- S. Chang and E. E. Lee, Synthesis, 2010, 2361–2366 CAS.
- M. Eskici, A. Karanfil, M. S. Özer and C. Sarıkürkcü, Tetrahedron Lett., 2011, 52, 6336–6341 CrossRef CAS.
- P. Hebeisen, U. Weiss, A. Alker and A. Staempfli, Tetrahedron Lett., 2011, 52, 5229–5233 CrossRef CAS.
- J. F. Bower, J. Švenda, A. J. Williams, J. P. H. Charmant, R. M. Lawrence, P. Szeto and T. Gallagher, Org. Lett., 2004, 6, 4727–4730 CrossRef CAS PubMed.
- J.-L. Zeng, H. Chachignon, J.-A. Ma and D. Cahard, Org. Lett., 2017, 19, 1974–1977 CrossRef CAS PubMed.
- H. Saeidian, M. Abdoli and Z. Mirjafary, Synthesis, 2015, 47, 1057–1075 CrossRef CAS.
- J. F. Bower, J. Rujirawanich and T. Gallagher, Org. Biomol. Chem., 2010, 8, 1505–1519 RSC.
- M.-F. Alicia, M.-S. Julia, H.-M. Fernando and S.-G. Francisco, Curr. Org. Chem., 2011, 15, 401–432 CrossRef.
- R. E. Meléndez and W. D. Lubell, Tetrahedron, 2003, 59, 2581–2616 CrossRef.
- S. A. Hill, R. Steinfort, S. Mücke, J. Reifenberger, T. Sengpiel and L. Hartmann, Macromol. Rapid Commun., 2021, 2100193 CrossRef PubMed.
- N. Boutard, A. G. Jamieson, H. Ong and W. D. Lubell, Chem. Biol. Drug Des., 2010, 75, 40–50 CrossRef CAS PubMed.
- N. Boutard, S. Turcotte, K. Beauregard, C. Quiniou, S. Chemtob and W. D. Lubell, J. Pept. Sci., 2011, 17, 288–296 CrossRef CAS PubMed.
- A. Geranurimi, C. W. H. Cheng, C. Quiniou, T. Zhu, X. Hou, J. C. Rivera, D. J. St-Cyr, K. Beauregard, V. Bernard-Gauthier, S. Chemtob and W. D. Lubell, Front. Chem., 2019, 7, 23 CrossRef CAS PubMed.
- A. G. Jamieson, N. Boutard, D. Sabatino and W. D. Lubell, Chem. Biol. Drug Des., 2013, 81, 148–165 CrossRef CAS PubMed.
- S. B. Cohen and R. L. Halcomb, Org. Lett., 2001, 3, 405–407 CrossRef CAS PubMed.
- S. B. Cohen and R. L. Halcomb, J. Am. Chem. Soc., 2002, 124, 2534–2543 CrossRef CAS PubMed.
- A. M. Kelly, P.-J. Chen, J. Klubnick, D. J. Blair and M. D. Burke, Org. Lett., 2020, 22, 9408–9414 CrossRef CAS PubMed.
- A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC.
- C. Sandford and V. K. Aggarwal, Chem. Commun., 2017, 53, 5481–5494 RSC.
- T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Angew. Chem., Int. Ed. Engl., 1996, 35, 1910–1922 CrossRef.
- X. Wang, N. Xia and L. Liu, Int. J. Mol. Sci., 2013, 14, 20890–20912 CrossRef PubMed.
- A. J. J. Lennox and G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2013, 52, 7362–7370 CrossRef CAS PubMed.
- G. Berionni, B. Maji, P. Knochel and H. Mayr, Chem. Sci., 2012, 3, 878–882 RSC.
- J. Li, S. G. Ballmer, E. P. Gillis, S. Fujii, M. J. Schmidt, A. M. E. Palazzolo, J. W. Lehmann, G. F. Morehouse and M. D. Burke, Science, 2015, 347, 1221 CrossRef CAS PubMed.
- J. Li, A. S. Grillo and M. D. Burke, Acc. Chem. Res., 2015, 48, 2297–2307 CrossRef CAS PubMed.
- E. M. Woerly, J. Roy and M. D. Burke, Nat. Chem., 2014, 6, 484–491 CrossRef CAS PubMed.
- M. Trobe and M. D. Burke, Angew. Chem., Int. Ed., 2018, 57, 4192–4214 CrossRef CAS PubMed.
- D. Leonori and V. K. Aggarwal, Acc. Chem. Res., 2014, 47, 3174–3183 CrossRef CAS PubMed.
- M. Burns, S. Essafi, J. R. Bame, S. P. Bull, M. P. Webster, S. Balieu, J. W. Dale, C. P. Butts, J. N. Harvey and V. K. Aggarwal, Nature, 2014, 513, 183–188 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |