
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polybenzoxazines: a sustainable platform for the design of fast responsive and catalyst-free vitrimers based on trans-esterification exchanges†
Antoine
Adjaoud
ab,
Acerina
Trejo-Machin
ab,
Laura
Puchot
a and
Pierre
Verge
 *a
*a
aLuxembourg Institute of Science and Technology, Materials Research and Technology Department, 5 Avenue des Hauts-Fourneaux, L-4362 Esch-sur-Alzette, Luxembourg. E-mail: pierre.verge@list.lu
bUniversity of Luxembourg, 2, Avenue de l'Université, L-4365 Esch-sur-Alzette, Luxembourg
First published on 14th May 2021
Abstract
This work explores a new strategy, aiming for the synthesis of catalyst-free vitrimers by taking advantage of the abundant number of tertiary amines covalently bound into a polybenzoxazine network. A bio-based monomer was obtained by reacting 4,4-bis(4-hydroxyphenyl)valeric acid, polyethylene glycol, paraformaldehyde and mono-ethanolamine, via consecutive solvent-free Fischer esterification and Mannich-like ring-closure. The two-step reaction led to the formation of quadri-telechelic benzoxazine-terminated polyethylene glycol monomers, containing ester bonds and aliphatic hydroxyl groups. The structural features of the resulting products were substantiated by 1H NMR, 13C NMR, elemental analysis, and FTIR. The occurrence of the thermally induced ring-opening polymerization was monitored by rheological measurements and DSC. At 140 °C, the monomers show a short gelation time (145 seconds). Once polymerized, the polybenzoxazine exhibits a relatively high temperature of α mechanical relaxation (93 °C). Due to the ability of tertiary amines to catalyze transesterification reactions, and to the abundant number of hydroxyl groups, the material enables exchange reactions without the use of an external catalyst. It possesses all the typical characteristics of a vitrimer, such as recycling, reshaping, and self-healing. Short stress-relaxation times were measured (116 s at 170 °C). Finally, the effect of the structural features of the vitrimer was investigated by tuning the crosslinking density of the network and the number of hydroxyl groups, shedding more light on the mechanism of self-catalysis and the range of properties. Therefore, such a strategy constitutes an efficient and versatile route for an easy elaboration of mono-component, catalyst-free, and fast responsive vitrimers.
1. Introduction
Over the past few decades, the apogee of industrialization has led to a compelling increase in the consumption of thermosets worldwide, resulting in an abundant source of wastes which cannot be recycled or repaired.1 To address this issue, reversible dissociative or associative bonds have been inserted within the chemical structure of thermosets to create covalent adaptable networks (CANs), enabling self-healability or recyclability.2–4 In 2011, Leibler and co-workers successfully introduced dynamic covalent bonds (DCBs) in thermoset polymer networks and developed the first “vitrimer”, combining the antithetical properties of a thermoplastic and a thermoset.5 At service temperatures, vitrimers behave like a traditional thermoset with high mechanical properties, chemical resistance, and insolubility. At higher temperatures, they display the fluidity and the reprocessing ability of a thermoplastic.5–7The different strategies and methodologies conceived to design performant vitrimers can be found in the excellent reviews published by Du Prez et al.,8–11 and Hillmyer and Dichtel.12 Among these strategies, the dynamic transesterification reaction (TER) occurring between ester bonds and hydroxyl (–OH) groups has been reported as the most representative chemistry for driving vitrimer behaviors. In most cases, the TER requires the use of an external catalyst to trigger fast enough exchange reactions within a reasonable range of conditions. Numerous catalysts can be employed to control the transesterification reaction,13 but solid or organic basic catalysts are preferred within a context of vitrimer material elaboration. Among them, zinc acetate (Zn(OAc)2) is the most widely used, owing to its high efficiency.5–7 Tertiary amine (NR3) groups are also well-known sites of TER catalysis.13–15 In one of their pioneering works, Leibler et al. evidenced that triazobicyclodecene (TBD), a bicyclic base containing a NR3 group, gave similar results to Zn(OAc)2.5 Later, Williams et al.,16 and more recently Zhang et al.,17 have both demonstrated in elegant approaches that NR3 covalently bound to the polymer network exert an internal catalysis on the transesterification reactions occurring in epoxy-acid systems. These studies join other recent research works dedicated to the development of internally catalyzed (or catalyst-free) vitrimers.18–22 It is noteworthy that in the absence of a catalyst, the lifetime of a vitrimer relying on a transesterification mechanism could be greatly enhanced, particularly as the risk of ester hydrolysis would be reduced.8 Thus, the elaboration of catalyst-free vitrimers appears of great importance, and the use of polymer structures containing tertiary amines seems to be a reasonable choice.
Polybenzoxazines (PBZs) are mono-component thermoset, polymers constituted of an abundant number of NR3 coming from the auto-catalyzed ring-opening polymerization (ROP) of benzoxazines.23 They are also a promising alternative to phenolic and epoxy resins thanks to their unique mechanical and thermal properties, such as a high glass transition temperature, near-zero shrinkage upon polymerization, and high char yield polymers.24 Benzoxazine monomers are obtained from the condensation reaction of a phenolic compound, a primary amine and paraformaldehyde (PFA). For the past ten years, there has been an explosive growth of research works on polybenzoxazines, dedicated to the use of natural phenolic compounds and amines25–43 as synthons of their monomers. As PFA can be synthesized from bio-ethanol or carbon dioxide, these precursors could be considered as 100% bio-based.44 However, to solve the concern of its carcinogenetic, its substitution by more friendly aldehydes has been under the spotlight of very significant investigations.45,46 It makes no doubts that polybenzoxazines with an excellent life cycle impact will emerge in the coming years.
As they are constituted from a permanent covalent network, PBZs suffer from similar drawbacks to traditional thermoset resins, and they cannot be recycled or reprocessed if they have not been specifically designed with this aim. Few works address this issue, mostly those written by Yagci and Kiskan.47–49 Recently, Verge et al. developed the first vitrimer involving PBZs, showing recyclability, reshaping, and reversible adhesion properties.50 The rearrangement of the permanent network was possible thanks to exchangeable disulfide bonds. However, to the best of our knowledge, none of the self-healable or reprocessable PBZs reported so far demonstrate vitrimer features from TER exchanges. Besides this, and more importantly, the NR3 generated during the ROP of benzoxazines have never been considered as internal catalysts for TER exchanges.
Inspired by a bio-based PBZ we developed in the past,51 and motivated by the outstanding achievements made on vitrimers over the past decade, this work attempts to illustrate how PBZs could be used to design catalyst-free vitrimers based on a transesterification mechanism. 4,4-Bis(4-hydroxyphenyl)valeric acid, more commonly known as diphenolic acid (DPA), is a bio-based diphenol coming from levulinic acid, a chemical extracted from lignocellulosic biomass.52 DPA was used to esterify the end-chains of polyethylene glycol (PEG) to form a quadri-telechelic phenol-terminated PEG (PEG-DPA) containing ester bonds. Thus, the hydroxyphenyl moieties were reacted via a Mannich-like condensation with mono-ethanolamine (mea), an amine derived from L-serine, sorbitol or glycolaldehyde.53,54 This reaction led to the formation of PEG-DPA-mea, a quadri-telechelic benzoxazine-terminated polyethylene glycol monomer with pending aliphatic –OH groups connected to the nitrogen atom (in β-position). These –OH groups were expected to transesterify with the ester bonds. The following paragraphs describe the synthesis of a series of bio-based material and the evidence that their vitrimer behavior does not require the use of an external catalyst. These materials fit many of the Green Chemistry and Engineering principles, such as “Use of renewable feedstocks”, “Safer solvents and auxiliaries”, “Durability rather than immortality”, “Renewable rather than depleting”, and “Design for Commercial after-life”, to cite but a few.
2. Experimental section
2.1 Materials and chemicals
4,4-Bis(4-hydroxyphenyl)valeric acid (95%, diphenolic acid, DPA), polyethylene glycol (average Mn = 200, 400 and 2000 g mol−1; PEG200, PEG400, PEG2000), mono-ethanolamine (≥98%, mea), furfurylamine (≥99%, fa) paraformaldehyde (95%, PFA), and para-toluene sulfonic acid monohydrate (≥98.5%, p-TSA) were purchased from Sigma-Aldrich. All chemicals were reagent grade and used as received without any further purification.2.2 Equipment and characterizations
Nuclear Magnetic Resonance (NMR) spectroscopy was performed on an AVANCE III HD Bruker spectrometer equipped with a 5 mm BBO-probe operating at a proton frequency of 600 MHz. All chemical shifts are given as δ value (ppm) referenced to tetramethylsilane (TMS) as an internal standard. Assignments were performed using a combination of COSY, HSQC, and HMBC spectra. Methyl peak (–CH3) in α-position of the aromatic ring was used as the reference for the integration of all other peaks (6.00H). Peak multiplicity was indicated as follows: singlet (s); doublet (d); triplet (t) or multiplet (m). The coupling constants (J) were reported in Hertz (Hz).Fourier transform infrared spectroscopy (FTIR) was conducted on a Bruker TENSOR 27 instrument in the attenuated total reflection (ATR) mode using a diamond crystal. All spectra were recorded at room temperature in direct absorbance mode across 4000–400 cm−1 frequency range (32 scans, 4 cm−1 spectral resolution).
Elemental analysis (CHNS measurements) was performed on a Vario MACRO cube CHNS/O from Elementar France SARL. Samples were put into an oxygen-enriched furnace at 1150 °C, where a combustion process converted carbon-to-carbon dioxide; hydrogen to water; nitrogen-to-nitrogen gas/oxides of nitrogen and sulfur-to-sulfur dioxide. The combustion products were swept out of the combustion chamber by an inert carrier gas (He, 600 mL min−1) and passed over heated (850 °C) high purity copper. The separation of the measuring components took place as follows: nitrogen (N2) was not adsorbed in the adsorption columns and was the first measuring component to enter the thermal conductivity detector directly. The other components were adsorbed in their respective adsorption column. Each of these columns was then heated separately to the corresponding desorption temperature (Tdesorpt.) in order to release the components in the following order: CO2 (Tdesorpt. = 240 °C), H2O (Tdesorpt. = 150 °C) and SO2 (Tdesorpt. = 100 °C or 230 °C). After desorption, each component was transported by the carrier gas flow into the measuring cell of a thermal conductivity detector (TCD).
Differential scanning calorimetry (DSC) thermograms were recorded on a Netzsch DSC 204 F1 Phoenix device in standard pierced aluminum crucibles (40 μL). A linear heating ramp from −25 to 300 °C at 10 °C min−1 rate was applied under inert atmosphere (N2). The DSC thermogram of the PBZ material was recorded over two consecutive heating–cooling cycles from −25 to 250 °C (heating rate of 10 °C min−1 and cooling rate of 20 °C min−1) to determine the glass transition (Tg) of the polymer and to ensure complete polymerization.
Thermogravimetric analysis (TGA) was completed on the Mettler Toledo TGA 2 device in standard ceramic alumina pan from 25 to 800 °C at 10 °C min−1 rate under air or inert atmosphere (N2). The maximum of the degradation temperature was determined using the derivative of the TGA curve (DTG).
Rheological measurements were recorded using an Anton Paar Physica MCR 302 rheometer equipped with a CTD 450 temperature control device. Stress relaxation experiments were performed on disk-shaped solid geometry with a disposable aluminum plate–plate (ϕ = 25 mm). The relaxation modulus was followed as a function of time for 2000 s between 120 °C and 170 °C with a constant applied strain of 1% and normal force of 20 N. The original relaxation modulus (G0) was extracted from the initial plateau of the stress relaxation curves (onset point of the second derivative curve). For the isothermal rheo-kinetic measurements, small quantities of the samples were loaded in a parallel plate-plate geometry (ϕ = 25 mm). The polymerization measurements were recorded in the oscillation mode at a frequency of 1 Hz and a controlled strain of 0.1%. Heating ramps of 20 °C min−1 were applied to reach a temperature of 140 °C. The sample deformation was ramped linearly from 1% to 0.2% to remain within the instrument's limitation and to maintain a linear viscoelastic behavior as the moduli (G′ storage modulus, G′′ loss modulus) increase by several orders of magnitude upon curing. The gap between the plates was maintained at 0.5 mm during all the experiments. Rheology temperature sweep curves were performed on the bar-shaped solid in rectangular-torsion mode under constant deformation of 0.1% (1 Hz).
Dilatometry thermogram was recorded on the Netzsch DIL 402 C apparatus from 25 to 200 °C (2 °C min−1) under an inert atmosphere (N2) and a constant force to avoid buckling (30 cN). The material was introduced in a hollow cylinder capped at the extremities by two solid cylindrical end pieces to limit the side expansion.
Dynamic mechanical analysis (DMA) was used to evaluate the mechanical properties of the material after reprocessing. The cured and reprocessed materials were analyzed at room temperature (RT) in tension and in single cantilever mode (effective lengths of 10 and 5 mm, respectively, width 5 mm, thickness 1.25 mm). The measurements were performed at 1 Hz using a preload force of 0.05 N and a sinusoidal strain of 10 μm. The storage modulus (E′) was recorded as a function of time. The reported values are an average of three values.
Optical microscopy was used to illustrate the self-healing performance of the PBZ vitrimer. The surface morphology of scratched samples was examined using the Nikon Universal Design Microscope UDM ECLIPSE LV100D-U (optics ×20, gain ×2.40, exposure 6 ms). The healing process was conducted at 150 °C in a convection oven. The width of the crack was measured before and after the healing process in a dynamic contrast mode at different time intervals.
Swelling experiments were conducted in acetone, chloroform and water by immersion at room temperature of 25 mg of the material in 2 mL of solvent at different time intervals for poly(PEG400-DPA-mea). The crosslinking density was measured by swelling tests in water at different time intervals. The swelling ratio (W) is determined according to the eqn (1):
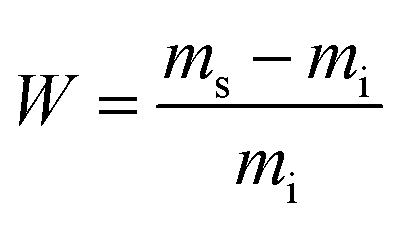 | (1) |
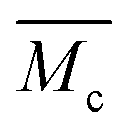 ) was calculated by applying the Flory Rehner equation (2):
) was calculated by applying the Flory Rehner equation (2):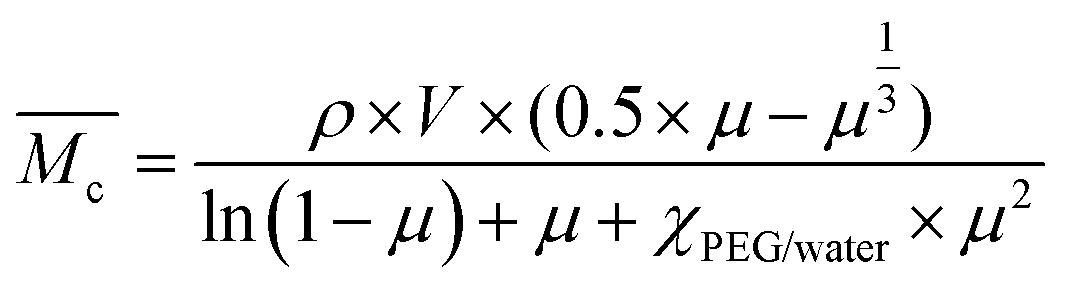 | (2) |
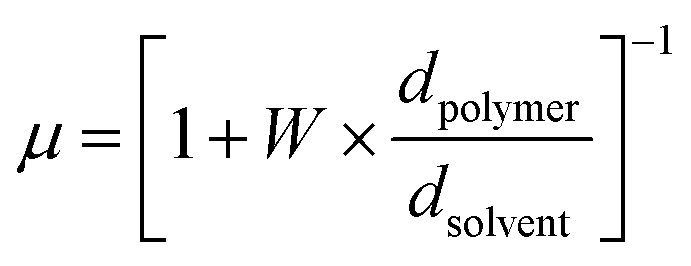 | (3) |
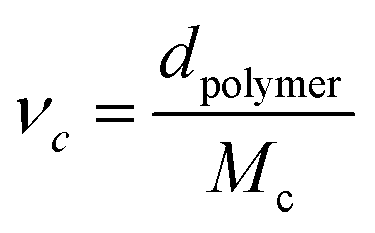 | (4) |
2.3 Synthetic procedure
PEG200-DPA (brown powder). 1H NMR (DMSO-d6, 600 MHz, 298°K): δ (ppm) = (assignment, multiplicity (coupling constant), [attribution], experimental integration, theoretical integration). δ = 1.46 (C–CH3*, s, [c], exp 6.00H, th 6.00H); δ = 2.02 (CH2–CH2*–C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, t (J = 6.91 Hz), [a], exp 3.98H, th 4.00H); δ = 2.24 (C–CH2*–CH2, t (J = 7.37 Hz), [b], exp 3.97H, th 4.00H); δ = 3.48 (O–CH2*, t (J = 3.66 Hz), [3], exp 9.01H); δ = 3.55 (OC–O–CH2-βCH2*, m, [1], exp 4.04H, th 4.00H); δ = 4.06 (OC–O-αCH2*–CH2, t, [2], exp 3.78H, th 4.00H); δ = 6.65 (CCH*–CH, d (J = 8.56 Hz), [e], exp 8.00H, th 8.00H); δ = 6.93 (CH–CH*C–O, d (J = 8.44 Hz), [d], exp 8.00H, th 8.00H); δ = 9.18 (Ar–OH*, s, [f], exp 3.99H, th 4.00H).
O, t (J = 6.91 Hz), [a], exp 3.98H, th 4.00H); δ = 2.24 (C–CH2*–CH2, t (J = 7.37 Hz), [b], exp 3.97H, th 4.00H); δ = 3.48 (O–CH2*, t (J = 3.66 Hz), [3], exp 9.01H); δ = 3.55 (OC–O–CH2-βCH2*, m, [1], exp 4.04H, th 4.00H); δ = 4.06 (OC–O-αCH2*–CH2, t, [2], exp 3.78H, th 4.00H); δ = 6.65 (CCH*–CH, d (J = 8.56 Hz), [e], exp 8.00H, th 8.00H); δ = 6.93 (CH–CH*C–O, d (J = 8.44 Hz), [d], exp 8.00H, th 8.00H); δ = 9.18 (Ar–OH*, s, [f], exp 3.99H, th 4.00H).
13C NMR (DMSO-d6, 600 MHz, 298°K): δ (ppm) = 27.7 [e]; 30.3 [b]; 36.7 [c]; 44.3 [d]; 63.6 [2]; 68.7 [1]; 70.2 [3]; 115.2 [h]; 128.2 [g]; 139.7 [f]; 155.5 [i]; 173.5 [a].
PEG400-DPA (brown viscous substance). 1H NMR (DMSO-d6, 600 MHz, 298°K): δ (ppm) = (assignment, multiplicity (coupling constant), [attribution], experimental integration, theoretical integration). δ = 1.44 (C–CH3*, s, [c], exp 6.00H, th 6.00H); δ = 2.03 (CH2–CH2*–C
O, t (J = 7.90 Hz), [a], exp 4.00H, th 4.00H); δ = 2.24 (C–CH2*–CH2, t (J = 7.93 Hz), [b], exp 3.99H, th 4.00H); δ = 3.48 (O–CH2*, t (J = 2.78 Hz), [3], exp 30.3H); δ = 3.55 (OC–O–CH2-βCH2*, m, [1], exp 4.13H, th 4.00H); δ = 4.06 (OC–O-αCH2*–CH2, t, [2], exp 3.98H, th 4.00H); δ = 6.65 (CCH*–CH, d (J = 8.57 Hz), [e], exp 8.02H, th 8.00H); δ = 6.93 (CH–CH*C–O, d (J = 8.53 Hz), [d], exp 7.98H, th 8.00H); δ = 9.19 (Ar–OH*, s, [f], exp 4.02H, th 4.00H).
13C NMR (DMSO-d6, 600 MHz, 298°K): δ (ppm) = 27.7 [e]; 30.3 [b]; 36.7 [c]; 44.3 [d]; 63.6 [2]; 68.7 [1]; 70.2 [3]; 115.1 [h]; 128.2 [g]; 139.6 [f]; 155.5 [i]; 173.5 [a].
FTIR (cm−1); very strong (vs), strong (s), medium (m), weak (w), broad (br): 3339 (–OH stretching, br), 2869 (C–H stretching, vs), 1730 (CO stretching from the ester, s), 1625–1475 (CC stretching vibrations from the aromatic ring, m), 1175 (phenol C–O stretching and –OH in plane deformation, w), 1083 (C–O stretching from the ester, s).
PEG2000-DPA (brown wax). 1H NMR (DMSO-d6, 600 MHz, 298°K): δ (ppm) = (assignment, multiplicity (coupling constant), [attribution], experimental integration, theoretical integration). δ = 1.46 (C–CH3*, s, [c], exp 6.00H, th 6.00H); δ = 2.03 (CH2–CH2*–C
O, t (J = 8.04 Hz), [a], exp 4.11H, th 4.00H); δ = 2.24 (C–CH2*–CH2, t (J = 8.12 Hz), [b], exp 4.00H, th 4.00H); δ = 3.51 (O–CH2*&OC–O–CH2-βCH2*, [3,1], exp 191.14H); δ = 4.06 (OC–O-αCH2*–CH2, t, [2], exp 4.00H, th 4.00H); δ = 6.65 (CCH*–CH, d (J = 8.61 Hz), [e], exp 7.95H, th 8.00H); δ = 6.93 (CH–CH*C–O, d (J = 8.61 Hz), [d], exp 7.96H, th 8.00H); δ = 9.18 (Ar–OH*, s, [f], exp 4.07H, th 4.00H).
13C NMR (DMSO-d6, 600 MHz, 298°K): δ (ppm) = 27.7 [e]; 30.3 [b]; 36.7 [c]; 44.3 [d]; 63.6 [2]; 68.7 [1]; 70.2 [3]; 115.1 [h]; 128.2 [g]; 139.6 [f]; 155.5 [i]; 173.5 [a].
PEG200-DPA-mea (yellow powder). 1H NMR (DMSO-d6, 600 MHz, 298 K): δ (ppm) = (assignment, multiplicity (coupling constant), [attribution], experimental integration, theoretical integration). δ = 1.47 (C–CH3*, s, [c], exp 6.00H, th 6.00H); δ = 2.03 (CH2–CH2*–C
O, m, [a], exp 3.97H, th 4.00H); δ = 2.25 (C–CH2*–CH2, m, [b], exp 3.87H, th 4.00H); δ = 2.71 (N–CH2*–CH2, m, [i], exp 6.06H, th 8.00H); δ = 3.48 (O–CH2*, m, [3], exp 11.01H); δ = 3.55 (CH2–CH2*–OH & OC–O–CH2-βCH2*, m, [1, j], exp 10.60H, th 12.00H); δ = 3.91 (Ar–CH2*–N, s, [h], exp 5.30H, th 8.00H); δ = 4.05 (OC–O-αCH2*–CH2, m, [2], exp 3.98H, th 4.00H); δ = 4.52 (CH2–OH*, s, [k], exp 3.34H, th 4.00H); δ = 4.78 (O–CH2*–N, s, [g], exp 5.62H, th 8.00H); δ = 6.82 (CCH*–CH & CCH*–C & CH–CH*C–O, m, [d, f, e], exp 11.78, th 12.00H).
13C NMR (DMSO-d6, 600 MHz, 298 K): δ (ppm) = 27.5 [e]; 30.3 [b]; 36.5 [c]; 44.5 [d]; 50.6 [m]; 54.0 [n]; 60.0 [o]; 63.6 [2]; 68.7 [1]; 70.2 [3]; 83.1 [l]; 115.9 [i]; 120.3 [k]; 126.0 [g]; 126.6 [h]; 140.8 [f]; 152.3 [j]; 173.5 [a].
PEG400-DPA-mea (orange viscous substance). 1H NMR (DMSO-d6, 600 MHz, 298 K): δ (ppm) = (assignment, multiplicity (coupling constant), [attribution], experimental integration, theoretical integration). δ = 1.45 (C–CH3*, s, [c], exp 6.00H, th 6.00H); δ = 2.02 (CH2–CH2*–C
O, m, [a], exp 4.03H, th 4.00H); δ = 2.29 (C–CH2*–CH2, m, [b], exp 4.05H, th 4.00H); δ = 2.72 (N–CH2*–CH2, m, [i], exp 5.92H, th 8.00H); δ = 3.48 (O–CH2*, m, [3], exp 30.68H); δ = 3.55 (CH2–CH2*–OH & OC–O–CH2-βCH2*, m, [1, j], exp 10.20H, th 12.00H); δ = 3.88 (Ar–CH2*–N, s, [h], exp 5.96H, th 8.00H); δ = 4.06 (OC–O-αCH2*–CH2, m, [2], exp 3.99H, th 4.00H); δ = 4.51 (CH2–OH*, s, [k], exp 3.88H, th 4.00H); δ = 4.80 (O–CH2*–N, s, [g], exp 5.93H, th 8.00H); δ = 6.81 (CCH*–CH & CCH*–C & CH–CH*C–O, m, [d, f, e], exp 12.07, th 12.00H).
13C NMR (DMSO-d6, 600 MHz, 298 K): δ (ppm) = 27.5 [e]; 30.3 [b]; 36.5 [c]; 44.5 [d]; 50.6 [m]; 54.0 [n]; 60.0 [o]; 63.7 [2]; 68.7 [1]; 70.2 [3]; 83.1 [l]; 116.0 [i]; 120.3 [k]; 126.0 [g]; 126.6 [h]; 140.8 [f]; 152.3 [j]; 173.5 [a].
FTIR (cm−1); very strong (vs), strong (s), medium (m), weak (w), broad (br): 3339 (–OH stretching, br), 2869 (C–H stretching, vs), 1730 (CO stretching from the ester, s), 1625–1475 (CC stretching vibrations from the aromatic ring, m), 1230 (C–O–C oxazine asymmetric stretching, m), 1083 (C–O stretching from the ester, s), 1030 (primary alcohol C–O stretching and –OH in plane deformation, w), 928 (C–H out-of-plane vibration in the trisubstituted benzene ring, m).
Elemental analysis (exp, th): C (67, 68), H (150, 98), N (4, 4), S (<0.05, 0).
PEG2000-DPA-mea (yellowish wax). 1H NMR (DMSO-d6, 600 MHz, 298 K): δ (ppm) = (assignment, multiplicity (coupling constant), [attribution], experimental integration, theoretical integration). δ = 1.47 (C–CH3*, s, [c], exp 6.00H, th 6.00H); δ = 2.04 (CH2–CH2*–C
O, m, [a], exp 3.81H, th 4.00H); δ = 2.26 (C–CH2*–CH2, m, [b], exp 4.00H, th 4.00H); δ = 2.72 (N–CH2*–CH2, m, [i], exp 5.44H, th 8.00H); δ = 3.51 (O–CH2*& CH2–CH2*–OH & OC–O–CH2-βCH2*, m, [3, 1, j], exp 197.19H); δ = 3.92 (Ar–CH2*–N, s, [h], exp 5.61H, th 8.00H); δ = 4.07 (OC–O-αCH2*–CH2, m, [2], exp 4.06H, th 4.00H); δ = 4.52 (CH2–OH*, s, [k], exp 3.86H, th 4.00H); δ = 4.78 (O–CH2*–N, s, [g], exp 5.41H, th 8.00H); δ = 6.83 (CCH*–CH & CCH*–C & CH–CH*C–O, m, [d, f, e], exp 12.97, th 12.00H).
13C NMR (DMSO-d6, 600 MHz, 298 K): δ (ppm) = 27.6 [e]; 30.3 [b]; 36.6 [c]; 44.5 [d]; 50.6 [m]; 54.0 [n]; 60.0 [o]; 63.7 [2]; 68.7 [1]; 70.3 [3]; 83.1 [l]; 115.9 [i]; 120.3 [k]; 126.0 [g]; 126.6 [h]; 140.8 [f]; 152.3 [j]; 173.5 [a].
PEG400-DPA-mea75/fa25. 1H NMR (DMSO-d6, 600 MHz, 298 K): δ (ppm) = (assignment, multiplicity (coupling constant), [attribution], experimental integration, theoretical integration). δ = 1.46 (C–CH3*, s, [c], exp 6.00H, th 6.00H); δ = 2.04 (CH2–CH2*–C
O, m, [a], exp 3.89H, th 4.00H); δ = 2.26 (C–CH2*–CH2, m, [b], exp 3.99H, th 4.00H); δ = 2.72 (N–CH2*–CH2, m, [i], exp 4.73H, th 6.00H); δ = 3.49 (O–CH2*, m, [3], exp 32.49H); δ = 3.55 (CH2–CH2*–OH & OC–O–CH2-βCH2*, m, [1, j], exp 8.88H, th 10.00H); δ = 3.90 (fa-CH2*–N & Ar–CH2*–N, s, [l, h], exp 7.43H, th 10.00H); δ = 4.07 (OC–O-αCH2*-CH2, m, [2], exp 4.01H, th 4.00H); δ = 4.51 (CH2–OH*, s, [k], exp 2.75H, th 3.00H); δ = 4.78 (O–CH2*–N, s, [g], exp 5.77H, th 8.00H); δ = 6.30 (fa-CCH*–CH, s, [m], exp 0.91H, th 1.00H); δ = 6.41 (fa-CH–CH*CH, s, [n], exp 0.92H, th 1.00H); δ = 6.82 (CCH*–CH & CCH*–C & CH–CH*C–O, m, [d, f, e], exp 11.96, th 12.00H); δ = 7.60 (fa-CHCH*–O, s, [o], exp 0.91H, th 1.00H).
13C NMR (DMSO-d6, 600 MHz, 298 K): δ (ppm) = 27.6 [e]; 30.3 [b]; 36.5 [c]; 44.5 [d]; 48.0 [p]; 49.6 [mfa]; 50.6 [mmea]; 54.0 [n]; 60.0 [o]; 63.7 [2]; 68.7 [1]; 70.3 [3]; 81.8 [lfa]; 83.1 [lmea]; 109.1 [r]; 110.9 [s]; 115.9 [i]; 119.6 [kfa]; 120.3 [kmea]; 126.0 [g]; 126.6 [h]; 140.8 [f]; 143.1 [t]; 152.3 [j&q]; 173.5 [a].
PEG400-DPA-mea50/fa50. 1H NMR (DMSO-d6, 600 MHz, 298 K): δ (ppm) = (assignment, multiplicity (coupling constant), [attribution], experimental integration, theoretical integration). δ = 1.48 (C–CH3*, s, [c], exp 6.00H, th 6.00H); δ = 2.05 (CH2–CH2*–C
O, m, [a], exp 3.94H, th 4.00H); δ = 2.27 (C–CH2*–CH2, m, [b], exp 3.96H, th 4.00H); δ = 2.72 (N–CH2*–CH2, m, [i], exp 3.05H, th 4.00H); δ = 3.49 (O–CH2*, m, [3], exp 31.12H); δ = 3.55 (CH2–CH2*–OH & OC–O–CH2-βCH2*, m, [1, j], exp 7.32H, th 8.00H); δ = 3.90 (fa-CH2*–N & Ar–CH2*–N, s, [l, h], exp 9.58H, th 12.00H); δ = 4.07 (OC–O-αCH2*–CH2, m, [2], exp 4.00H, th 4.00H); δ = 4.52 (CH2–OH*, s, [k], exp 1.81H, th 2.00H); δ = 4.78 (O–CH2*–N, s, [g], exp 6.10H, th 8.00H); δ = 6.29 (fa-CCH*–CH, s, [m], exp 1.78H, th 2.00H); δ = 6.40 (fa-CH–CH*CH, s, [n], exp 1.89H, th 2.00H); δ = 6.84 (CCH*–CH & CCH*–C & CH–CH*C–O, m, [d, f, e], exp 11.91, th 12.00H); δ = 7.59 (fa-CHCH*–O, s, [o], exp 1.87H, th 2.00H).
13C NMR (DMSO-d6, 600 MHz, 298 K): δ (ppm) = 27.5 [e]; 30.3 [b]; 36.5 [c]; 44.5 [d]; 48.0 [p]; 49.6 [mfa]; 50.6 [mmea]; 54.0 [n]; 60.0 [o]; 63.7 [2]; 68.7 [1]; 70.2 [3]; 81.8 [lfa]; 83.1 [lmea]; 109.1 [r]; 110.9 [s]; 115.9 [i]; 119.6 [kfa]; 120.3 [kmea]; 126.0 [g]; 126.6 [h]; 140.8 [f]; 143.1 [t]; 152.3 [j&q]; 173.5 [a].
PEG400-DPA-mea25/fa75. 1H NMR (DMSO-d6, 600 MHz, 298 K): δ (ppm) = (assignment, multiplicity (coupling constant), [attribution], experimental integration, theoretical integration). δ = 1.48 (C–CH3*, s, [c], exp 6.00H, th 6.00H); δ = 2.05 (CH2–CH2*–C
O, m, [a], exp 4.00H, th 4.00H); δ = 2.27 (C–CH2*–CH2, m, [b], exp 3.97H, th 4.00H); δ = 2.72 (N–CH2*–CH2, m, [i], exp 1.55H, th 2.00H); δ = 3.49 (O–CH2*, m, [3], exp 31.38H); δ = 3.56 (CH2–CH2*–OH & OC–O–CH2-βCH2*, m, [1, j], exp 6.09H, th 6.00H); δ = 3.89 (fa-CH2*–N & Ar–CH2*–N, s, [l, h], exp 10.79H, th 14.00H); δ = 4.07 (OC–O-αCH2*–CH2, m, [2], exp 3.94H, th 4.00H); δ = 4.51 (CH2–OH*, s, [k], exp 1.01H, th 1.00H); δ = 4.78 (O–CH2*–N, s, [g], exp 5.82H, th 8.00H); δ = 6.29 (fa-CCH*–CH, s, [m], exp 2.71H, th 3.00H); δ = 6.40 (fa-CH–CH*CH, s, [n], exp 2.73H, th 3.00H); δ = 6.84 (CCH*–CH & CCH*–C & CH–CH*C–O, m, [d, f, e], exp 11.99, th 12.00H); δ = 7.69 (fa-CHCH*–O, s, [o], exp 2.80H, th 3.00H).
13C NMR (DMSO-d6, 600 MHz, 298 K): δ (ppm) = 27.5 [e]; 30.3 [b]; 36.5 [c]; 44.5 [d]; 48.0 [p]; 49.6 [mfa]; 50.6 [mmea]; 54.0 [n]; 60.0 [o]; 63.7 [2]; 68.7 [1]; 70.2 [3]; 81.8 [lfa]; 83.1 [lmea]; 109.1 [r]; 110.8 [s]; 116.0 [i]; 119.6 [kfa]; 120.3 [kmea]; 126.2 [g]; 126.8 [h]; 141.1 [f]; 143.1 [t]; 152.3 [j&q]; 173.5 [a].
3. Results and discussion
For the sake of clarity, the first part of the discussion is focusing on the synthesis and the characterization of PEG400-DPA-mea. All the materials discussed later in this manuscript (subsection 3.2.2) have been prepared and characterized following the same procedures, detailed in the ESI.†3.1 Synthesis and characterization of the vitrimer precursors
PEG400-DPA-mea was synthesized solventless in two steps (Scheme 1), following a procedure previously reported by our group.51 The phenolation of polyethylene glycol 400 (PEG400) via a Fischer esterification was realized with diphenolic acid (DPA), with p-toluene sulfonic acid as the catalyst (p-TSA), and led to the formation of a quadri-telechelic phenol terminated PEG (PEG400-DPA). It is noteworthy that PEG400 was specifically considered for the synthesis of the benzoxazine monomers because its industrial production adheres to many of the principles of the Green Chemistry.56 PEG400-DPA was purified by liquid–liquid extractions to remove non-hydrolyzed p-TSA. PEG400-DPA was then reacted with mono-ethanolamine (mea) and paraformaldehyde (PFA) via Mannich-like condensation followed by a ring-closure process. Mea was purposely used to cap the extremities of the monomer with pending –OH groups, which are able to react further by transesterification with the ester bonds of the PEG400-DPA backbone. In fine, a quadri-telechelic benzoxazine-terminated PEG monomer was obtained (PEG400-DPA-mea). It is noteworthy that this second step, i.e. Mannich-like condensation with an amino-alcohol, may compete with various detrimental side reactions when performed in a solvent, leading to the formation of hydroxybenzylaminoethanol, oxazolidine, triazacyclohexane, or diazaheptane derivatives (depending on the initial molar ratio).57,58 To sidestep these competing reactions, the Mannich condensation was performed in various conditions (Table S1†). The highest conversion yields of phenolic counterparts into oxazine rings were obtained when the synthesis was performed in the absence of solvents and below 100 °C.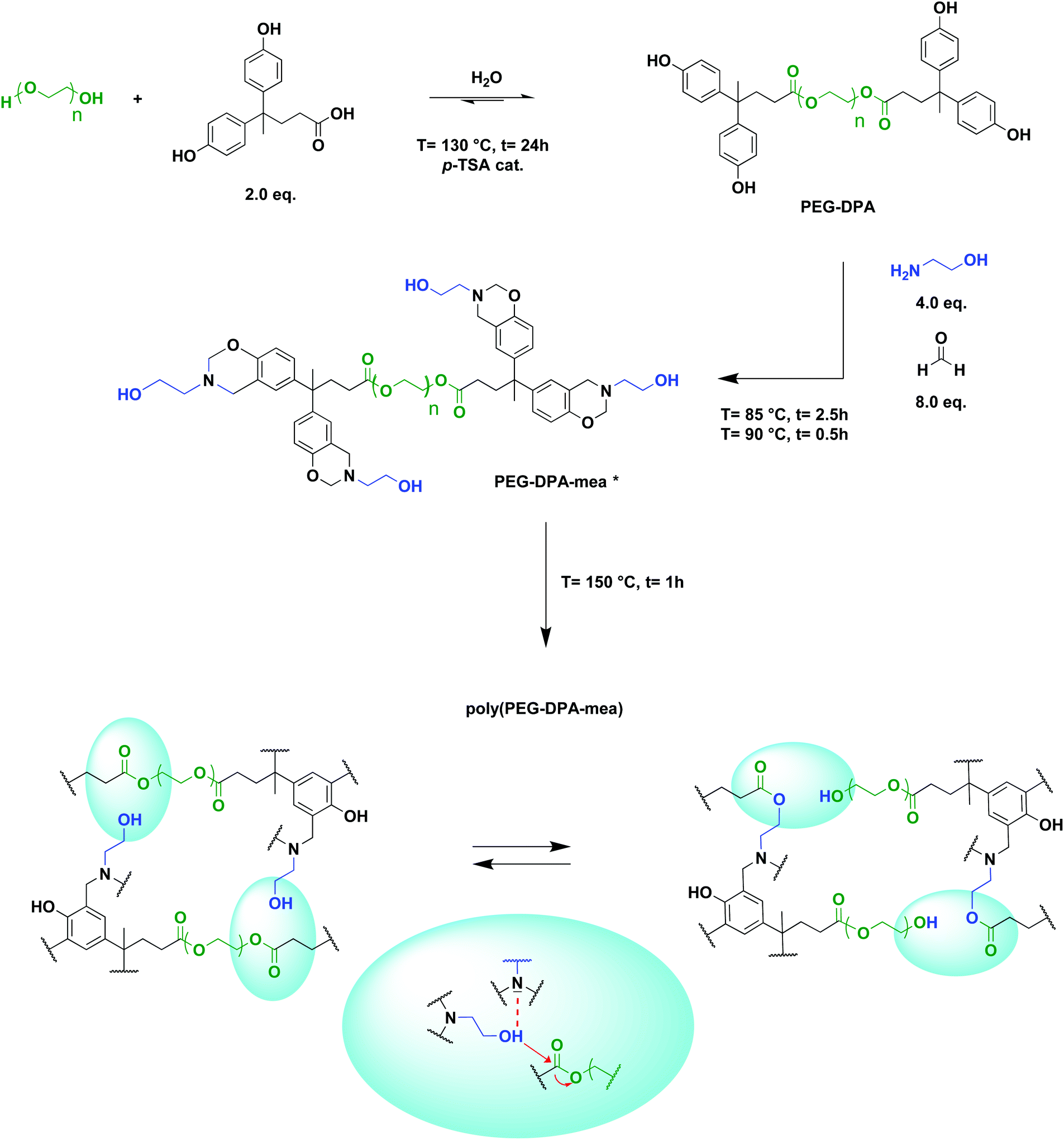 | ||
| Scheme 1 Synthetic pathway to obtain poly(PEG-DPA-mea), and expected structure of the resulting vitrimer through topology rearrangement by transesterification reactions catalyzed by the in situ generated tertiary amines (*25% of unclosed benzoxazine rings). | ||
The structural features of PEG400-DPA and PEG400-DPA-mea were characterized by 1H NMR, 13C NMR, FTIR and elemental analysis (Fig. 1, Fig. S1–3†). The 1H NMR spectrum of PEG400-DPA (Fig. 1a) reveals peaks corresponding to methylene protons [1,2] adjacent to the carbonyl group in β- and α-position (δ = 3.55 and = 4.06 ppm respectively), attesting the successful esterification of PEG400. Furthermore, the characteristic carbonyl peak [a] at δ = 173.5 ppm in 13C NMR (Fig. S1†) and its correlation in 2D NMR with DPA characteristic methylene protons [1,2] confirm the formation of the ester bond. The absence of impurities or by-products peaks suggests that quantitative conversion was nearly reached. From the 1H NMR spectrum of PEG400-DPA-mea (Fig. 1b), the characteristic peaks corresponding to the formation of the benzoxazine rings are revealed at δ = 3.88 and δ = 4.80 ppm, which correspond to Ar–CH2*–N [h] and O–CH2*–N [g], respectively. The experimental integrations of the methylene protons of the oxazine rings are 5.81H and 5.95H, respectively, corresponding to roughly 75% of the closed benzoxazine rings (theoretical value of 8.00H each). The remaining 25% of end-capping is attributed to opened benzoxazine rings labelled with * in Fig. 1b. In such opened structures, the primary amine has reacted with the phenolic group, but the ring is not closed between the –OH and the nitrogen. The non-closed structures can still be involved in the crosslinking process and affect the rate of transesterification reactions similarly to closed structures. The disappearance of the peak at δ = 9.2 ppm suggests that 100% of the phenolic groups reacted, and this is confirmed by elemental analysis as the experimental ratio of carbon and nitrogen atoms C/N is equal to 67/4 (theoretical C/N = 68/4). The wide signal centered at δ = 4.51 ppm is attributed to the aliphatic hydroxyl groups –OH [k] (ex. 3.88H, th 4.00H). In the 13C NMR spectrum (Fig. S2†), the characteristic oxazine ring carbons are found at δ = 50.6 and δ = 83.1 ppm (for Ar–*CH2–N [m] and O–*CH2–N [l], respectively). The formation of PEG400-DPA-mea was also confirmed by FTIR analysis (Fig. S3†). The strong absorption peak at 1730 cm−1 is typical of the ester CO stretching. The presence of the benzoxazine rings, confirmed by the characteristic bands at 1230 and 932 cm−1 (attributed to the C–O–C asymmetric stretching and the C–H out-of-plane vibration in the trisubstituted benzene ring, respectively), and by the decrease of the intensity of the –OH stretching vibration centered at 3339 cm−1. Finally, the peak at 1030 cm−1 corresponds to the absorption of the primary alcohol C–O stretching and –OH in-plane deformation.
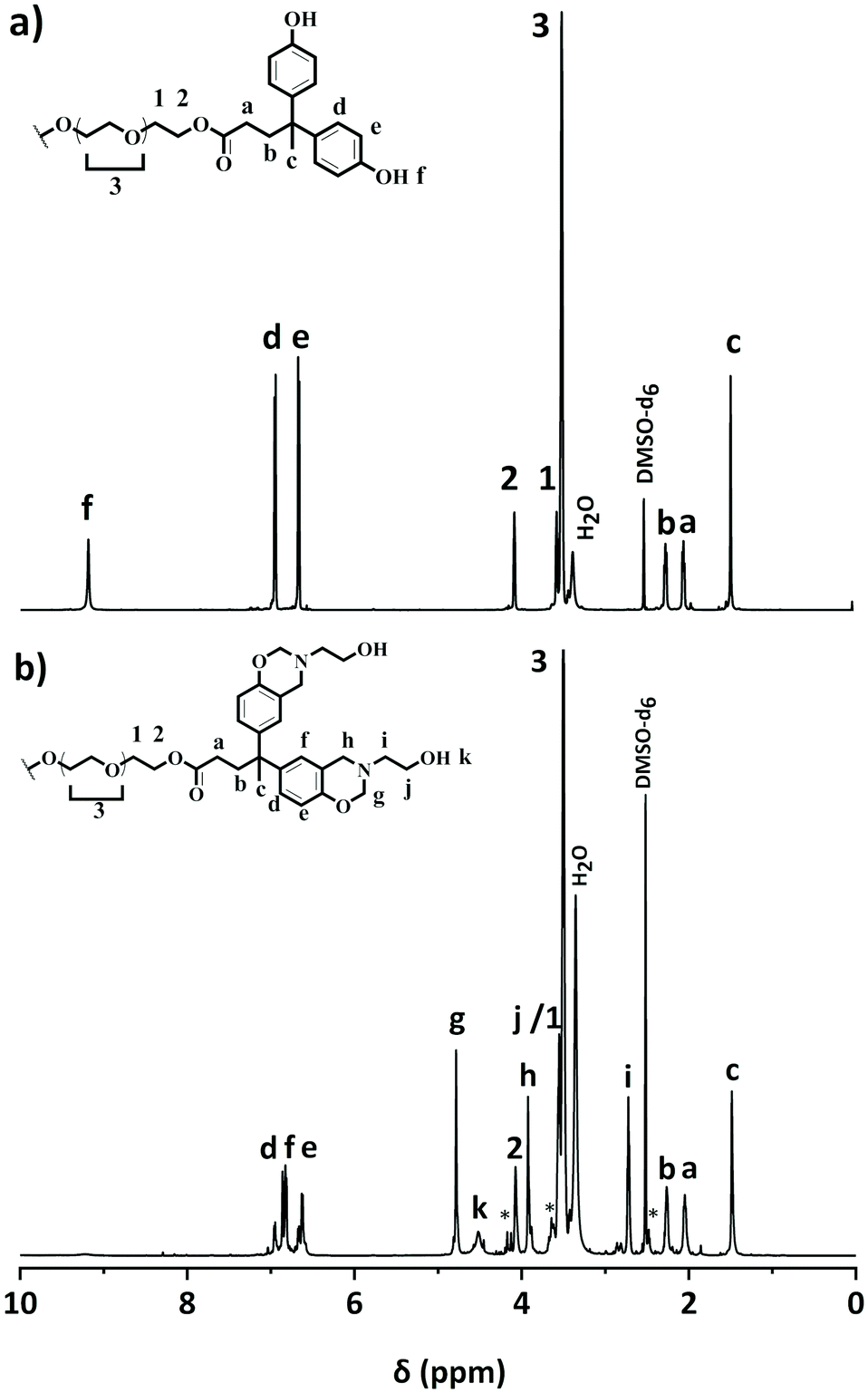 | ||
| Fig. 1 1H NMR spectrum of (a) PEG400-DPA and (b) PEG400-DPA-mea (DMSO-d6). | ||
DSC measurements revealed that PEG400-DPA and PEG400-DPA-mea have a similar softening temperature (5.6 and 7.2 °C respectively, Fig. S4†). An exothermic peak (Texo,1 = 110–220 °C) was observed only in the case of PEG400-DPA-mea, attributed to the ring-opening polymerization (ROP) of benzoxazine moieties. It is worth indicating the benzoxazine ROP is triggered at a very low temperature compared to traditional benzoxazines, generally in the range of 150–200 °C.59 The apparent low ROP temperature is related to the activating effect of alcohol functionality through hydrogen bonding, as previously reported in the literature.60,61 A second exothermic peak centered around 250 °C (Texo,2) is attributed to a thermal degradation, also confirmed by TGA (Fig. S5†). PEG400-DPA-mea was subjected to a curing treatment monitored by rheology at 140 °C (Fig. 2a). The recorded rheograms present the evolution of the storage modulus (G′) and the loss modulus (G′′) during the polymerization of the monomer. The behavior of PEG400-DPA-mea when heated is typical to thermosets with a significant increase in both G′ and G′′. The approximate gelation (tgel), defined as the crossover point of G′ and G′′, was reached quickly after 145 s of isothermal curing. This high reactivity could be explained by the high functionality of the system. Rheological characterization of polymerized PEG400-DPA-mea, so-called poly(PEG400-DPA-mea), is reported in Fig. 2b. The measurement was done in torsion mode on a bar sample cured in a bar-shape mold as described in the Experimental section (one hour at T = 150 °C). The DSC thermogram of poly(PEG400-DPA-mea) did not reveal an exothermic peak, indicating a complete polymerization (Fig. S6†). The moduli were measured to 0.6 GPa and 10 MPa in the glassy and rubbery states, respectively. The α mechanical relaxation (Tα) was measured at 93 °C according to the maximum of the tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ. The significant broad tan delta is characteristic of networks constituted of a wide distribution of chemical structures, assigned to the transesterification reactions occurring during the curing. The Tg of poly(PEG400-DPA-mea) was also measured by dilatometry (Fig. S7†). The transition was measured at the onset of the inflection (79 °C) and is more representative of the service temperature of the vitrimer than the Tα. It is worth indicating that the low thermal expansion associated to the Tg is consistent with the near-zero volumetric expansion characteristic of polybenzoxazines.24
δ. The significant broad tan delta is characteristic of networks constituted of a wide distribution of chemical structures, assigned to the transesterification reactions occurring during the curing. The Tg of poly(PEG400-DPA-mea) was also measured by dilatometry (Fig. S7†). The transition was measured at the onset of the inflection (79 °C) and is more representative of the service temperature of the vitrimer than the Tα. It is worth indicating that the low thermal expansion associated to the Tg is consistent with the near-zero volumetric expansion characteristic of polybenzoxazines.24
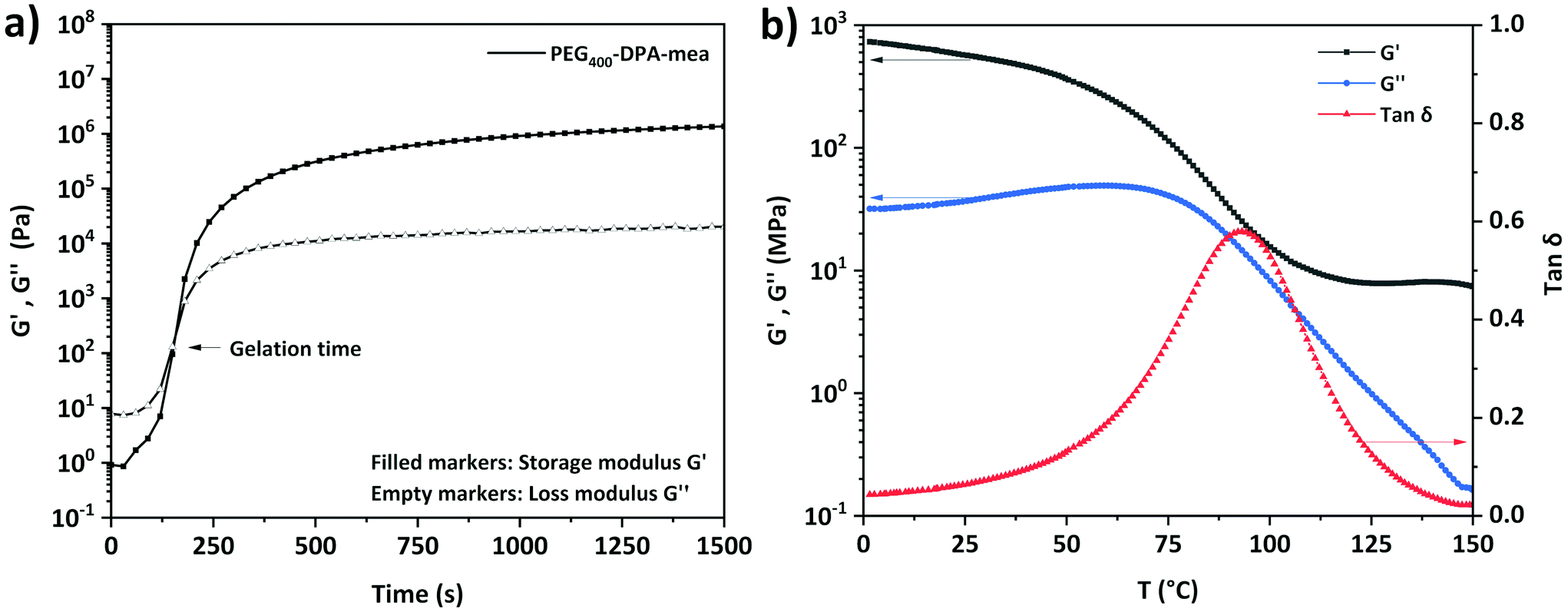 | ||
| Fig. 2 (a) Isothermal rheology monitoring of PEG400-DPA-mea at 140 °C (b) Rheology temperature sweep curves in torsion mode of poly(PEG400-DPA-mea). | ||
3.2 Characterization of the polybenzoxazine vitrimer
 | (5) |
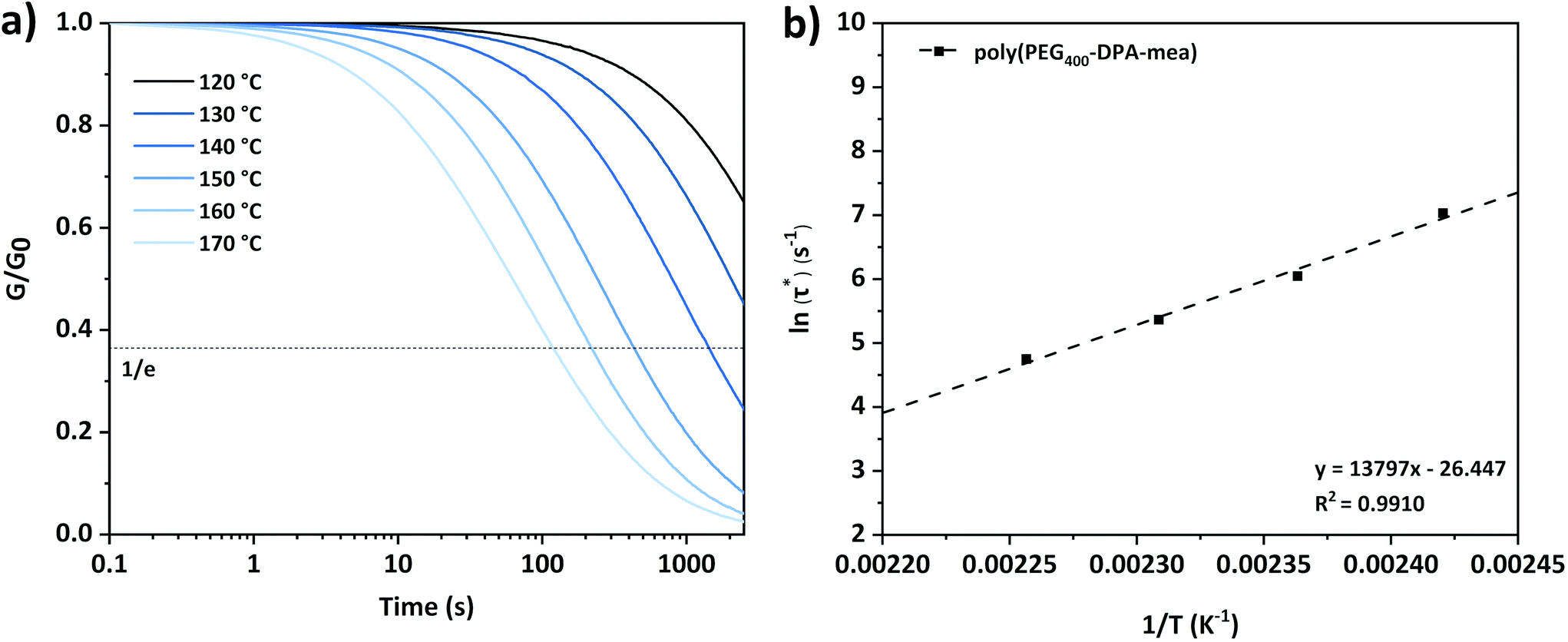 | ||
| Fig. 3 (a) Stress relaxation curves of poly(PEG400-DPA-mea) at different temperatures and (b) Arrhenius plot obtained from stress relaxation experiment of poly(PEG400-DPA-mea). | ||
The linear fitting of ln (τ*) versus 1/T plot (correlation coefficient of 0.991) suggests that the network follows an Arrhenius-like flow characteristic. The activation energy (Ea) of transesterification reactions was obtained from the slope of the Arrhenius equation and yielded 115 kJ mol−1 (Fig. S7;† the detail of the calculation is reported in the ESI†). The theoretical Tv was found at 88 °C for poly(PEG400-DPA-mea), in the range of the vitrimers developed by Leibler et al. for epoxy/acid systems.5,6 In the present work, the catalytic effect originates from the neighboring group participation (NGP) of the NR3 groups in the mechanism of the TER (Scheme 1), in a similar fashion than described by Du Prez et al. in a recent review.11 The activation of the –OH groups is promoted by intramolecular hydrogen bonding with the lone electron pair carried by the nitrogen atoms. While the energy of the H–O bonds decreased owing to the N–H interaction, the atom of oxygen became nucleophilic enough to attack the electrophilic center of the carbonyl bond, allowing the TER to occur.
| Network | Type of internal catalysis | 103*νca (mol cm)−3 |
N/COOb | OH/COOd |
T
g
(°C) |
T
α
(°C) |
E a KJ mol−1) | τ*i (S) [T] | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|
|
a Determined from the Flory Rehner equation (2).
b Number of NR3 functionalities per ester bonds (*confirmed by elemental analysis).
c From the literature.
d Number of –OH per ester bonds (* determined from 1H NMR).
e Determined by dilatometry experiment.
f Determined by DSC experiment.
g Measured from the maximum peak of tanδ curve.
h Extrapolated from the Arrhenius equation.
i Determined by stress relaxation experiment.
|
||||||||||
| Epoxy resin | Excess of –COOH groups/β-hydroxy ester | — | — | 1–2c | — | 8 | 104 | ∼104 [150 °C] | 18 | |
| Poly(hydroxyethylmethacrylate) | β-Keto ester | 2.4 ± 0.3c | — | 1.5c | — | 130 | 111 | ∼1000 [150 °C] | 21 | |
| Epoxy resin | Excess of –OH groups | — | — | >8c | 63f | 70 | 63 | ∼4500 [180 °C] | 19 | |
| Epoxy resin | Excess of –OH groups | — | — | 0.625c | — | 57 | 64 | ∼5000 [180 °C] | 20 | |
| Epoxy resin | Covalently bonded NR3 groups | — | 0.027 c | 0.9c | — | 69 | 94 | ∼ 1000 [160 °C] | 16 | |
| Epoxy resin | Covalently bonded NR3 groups/β-hydroxy ester | — | 0.5 c | 1c | −1f | 22 | — | >104 [170 °C] | 17 | |
| Epoxy resin | Covalently bonded NR3 groups/excess of –OH groups | — | <0.1c | 1c | 120 f | 135 | 125 | >104 [170 °C] | 22 | |
| Polybenzoxazine | Poly(PEG200-DPA-mea) | Covalently bonded NR3 groups/excess of –OH groups | 81 ± 5 | 2 | 1.67* | 125 | — | 106 | 814 [150 °C] | This work |
| Poly(PEG400-DPA-mea) | 24 ± 1 | 2* | 1.95* | 79 | 93 | 115 | 422 [150 °C] | |||
| Poly(PEG400-DPA-mea75/fa25) | 69 ± 1 | 2 | 1.38* | 113 | — | 126 | 2012 [150 °C] | |||
| Poly(PEG400-DPA-mea50/fa50) | 118 ± 17 | 2 | 0.91* | 121 | — | 131 | 3172 [150 °C] | |||
| Poly(PEG400-DPA-mea25/fa75) | 117 ± 19 | 2 | 0.50* | 125 | — | 136 | 3410 [150 °C] | |||
| Poly(PEG2000-DPA-mea) | 1.5 ± 0.1 | 2 | 1.93* | — | −34 | 154 | 36 [150 °C] | |||
The effect of the structural features of the benzoxazine-based vitrimer was investigated by tuning the crosslinking density of the material and the number of –OH groups. The effect of the crosslinking density was assessed by preparing two materials with a PEG of molecular weight of either 200 or 2000 g mol−1. In these materials, the ratio NR3/ester/–OH groups (N/COO/OH) was similar (2/1/2), but the concentration of the dynamic exchangeable sites was decreasing along with the molecular weight of the PEG. Once polymerized, these materials were annotated as poly(PEG200-DPA-mea) and poly(PEG2000-DPA-mea) for the sake of clarity (Table 1, rows 8 and 13). The effect of the number of –OH groups was investigated by preparing three additional materials, where a part of mono-ethanolamine was substituted by furfurylamine (fa). They were called poly(PEG-DPA-mea75/fa25), poly(PEG-DPA-mea50/fa50), poly(PEG-DPA-mea25/fa75), corresponding to materials where 75%, 50% and 25% of mea was substituted by 75%, 50% and 25% of fa, respectively. NMR revealed that they correspond to materials containing a ratio of N/COO/OH equal to 2/1/1.38, 2/1/0.91 or 2/1/0.5, in respect to the order given in the sentence above (Table 1, rows 10–12). The details of the preparation and characterization of all the materials can be found in the ESI (Fig. S8–14 and S18–25†).
As expected, the crosslinking density was decreasing when the PEG molecular weight was increasing, reaching (81 ± 5) × 103 mol cm−3, (24 ± 1) × 103 mol cm−3 and (1.5 ± 0.1) × 103 mol cm−3 for poly(PEG200-DPA-mea), poly(PEG400-DPA-mea) and poly(PEG2000-DPA-mea) respectively. The crosslinking density of poly(PEG400-DPA-meax/fa100−x) were found to be similar to poly(PEG200-DPA-mea), close to 100 × 103 mol cm−3. They are higher than νc of (polyPEG400-DPA-mea) in reason of the involvement of the furan ring in the network formation.30,41,59
According to the stress relaxation curves, all these materials behaved like vitrimers (Tables S2–3, Fig. S15–17 and S26–29†). Their Ea and τ* at 150 °C were determined from isothermal relaxation measurements (Table 1, column 8 and 9) and plotted in Fig. 4a and Fig. 4b as a function of their νc. A clear dependence of the Ea on the number of –OH groups is depicted on Fig. 4a (values encircled by a dashed line). Despite their similar crosslinking density, the Ea of poly(PEG-DPA-mea75/fa25), poly(PEG-DPA-mea50/fa50), and poly(PEG-DPA-mea25/fa75) are increasing while the amount of –OH groups is decreasing (Ea = 126, 131, 136 kJ mol−1 respectively). In the case of poly(PEG2000-DPA-mea), which crosslinking density is the lowest, Ea reached the value of 156 kJ mol−1. In the more crosslinked network (poly(PEG200-DPA-mea)), Ea decreased to 106 kJ mol−1. Lowering the concentration of –OH groups, either by decreasing the νc or the total amount of –OH groups leads to an increase of the Ea. These experiments confirm that the energy needed to activate the dynamic exchanges is clearly governed both by the excess of –OH groups and the concentration of dynamic sites in the network. It is worth indicating the Ea related to the relaxation of theses PBZs vitrimer are in the same range than other reported vitrimers catalyzed either by external or internal tertiary amine.5,6,16,22 However it cannot be excluded that aromatic –OH groups may also be involved in the transesterification mechanism and contribute to the high value of Ea. Indeed, the transesterification with phenoxy is well documented.64–66 Nevertheless, it is worthy to mention that even after a couple of hours at 170 °C, the relaxation of PEG400-DPA-fa, i.e. in the absence of aliphatic –OH was not observed. The results do not exclude the possible involvement of the phenoxy groups in the exchanges, but it is not observed in the conditions of the experiments described here.
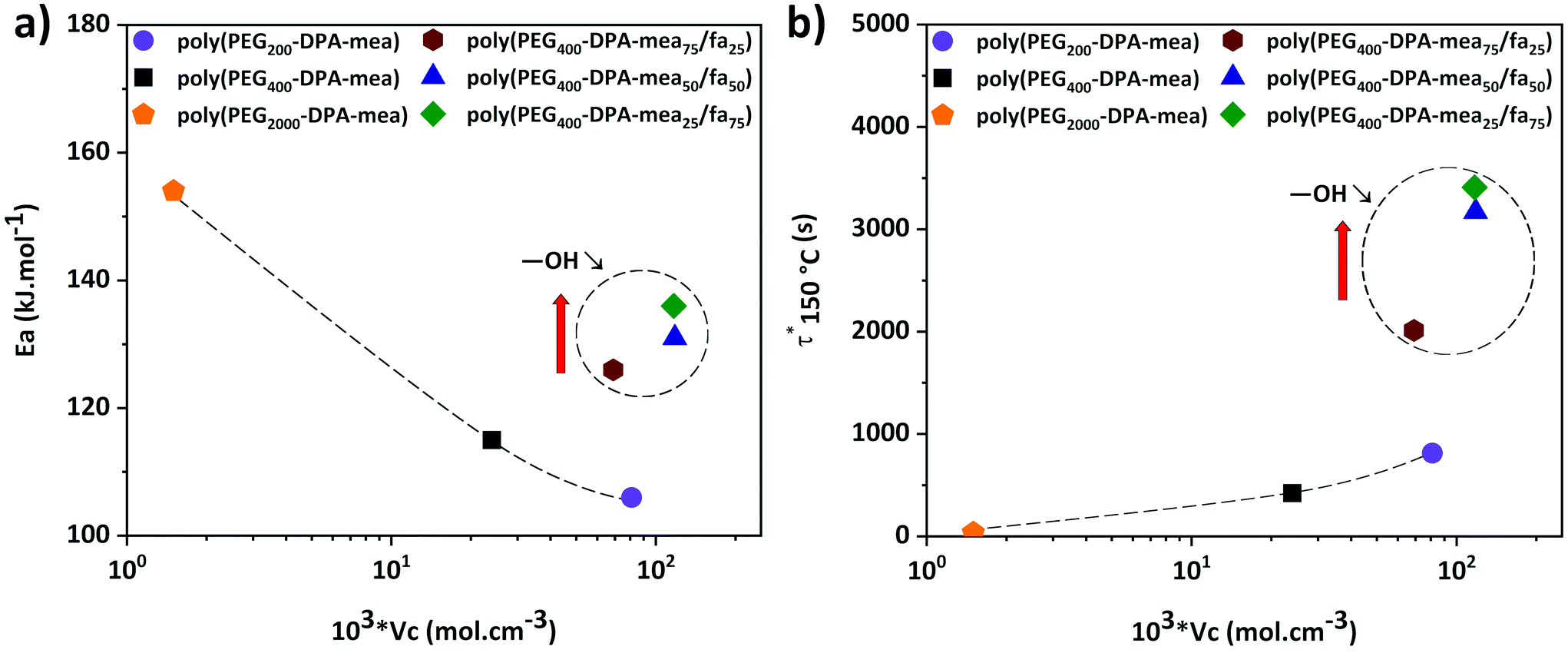 | ||
| Fig. 4 Evolution of τ*150 °C and Ea as a function of the crosslinking density of the different polybenzoxazine vitrimer. | ||
The evolution of τ* follows another trend (Fig. 4b). A clear dependence exists with the concentration of –OH groups, as attested by the significant increase of τ* when decreasing the amount of –OH groups for materials of similar νc (poly(PEG400-DPA-meax/fa100−x)). τ* remains reasonable (lower than 1 hour at 150 °C) even for the material displaying the lower amount of –OH groups (Table 1, row 12). However, despite their lowest concentration of dynamic sites, the fastest behaviors were reached for the materials with the lowest νc (Table 1, rows 9 and 13). This trend indicates that the speed of the dynamic exchange is also driven by the mobility of the chains.62,63
The very short τ* compared to similar catalyst-free vitrimers can be explained by the abundant number of NR3 groups (coming from the benzoxazine ROP). Indeed, the polymerization of PEGn-DPA-meax/fa100−x follows a ROP, which leads to the formation of a network with an abundant number of tertiary amines, significantly higher in comparison to the previously reported catalyst-free vitrimers (Table 1, column 4). Therefore, the nucleophile substitution of the ester bonds by the –OH groups is highly promoted. This behavior is in adequation with one of the first observations made by Leibler et al. about the relationship between the amount of catalyst and the short τ* of vitrimers.6 Moreover, the catalytic effect is more pronounced thanks to the close position of the NR3 groups regarding the –OH groups, in agreement with the NGP theory recently disclosed by Du Prez et al.11 and illustrated in Scheme 1.
3.3 Self-healing, recycling, reprocessing and reshaping
Additional swelling experiments were performed in different solvents for poly(PEG400-DPA-mea). The material is insoluble in acetone, water, and chloroform, in which it swells by 14%, 38%, and 103% respectively after 24 hours (Fig. S30†). Later, the samples were dried and weighted to determine the soluble part, measured to be lower than 5 wt%. The chemical recyclability of poly(PEG400-DPA-mea) was illustrated by its chemical decomposition in acetic acid at RT (Fig. S31†). The self-healability of poly(PEG400-DPA-mea) was demonstrated by surface morphology analysis recorded by optical microscopy (Fig. 5a). The cured material was notched with a scalpel blade, forming a scratch of 17.7 μm initial width. When exposed at 150 °C in pressure-free conditions, the width of the damaged area gradually decreased with time, reaching 4.6 μm (i.e. less than 25% of the initial scratch width) after 120 min. The reshaping, twisting and bending of materials made of poly(PEG400-DPA-mea) were straightforward, thanks to the fast relaxation process (Fig. 5b). The reprocessability was investigated by grinding the material into a fine powder, after which it was compressed to the shape of either a disk or a bar. The reprocessed bar was then subjected to dynamic mechanical tests in tensile and single cantilever modes to determine the impact of the reprocessing on their mechanical properties (Fig. 6). The storage modulus (E′) of poly(PEG400-DPA-mea) (1307 ± 86 MPa) was fully recovered even after being ground and reshaped five times (1306 ± 81 MPa). Besides storing the mechanical properties, the overlap of the FTIR spectra of both cured and reprocessed materials provides evidence that poly(PEG400-DPA-mea) did not show any deterioration of its chemical structure after 5 recycling steps (Fig. S32†).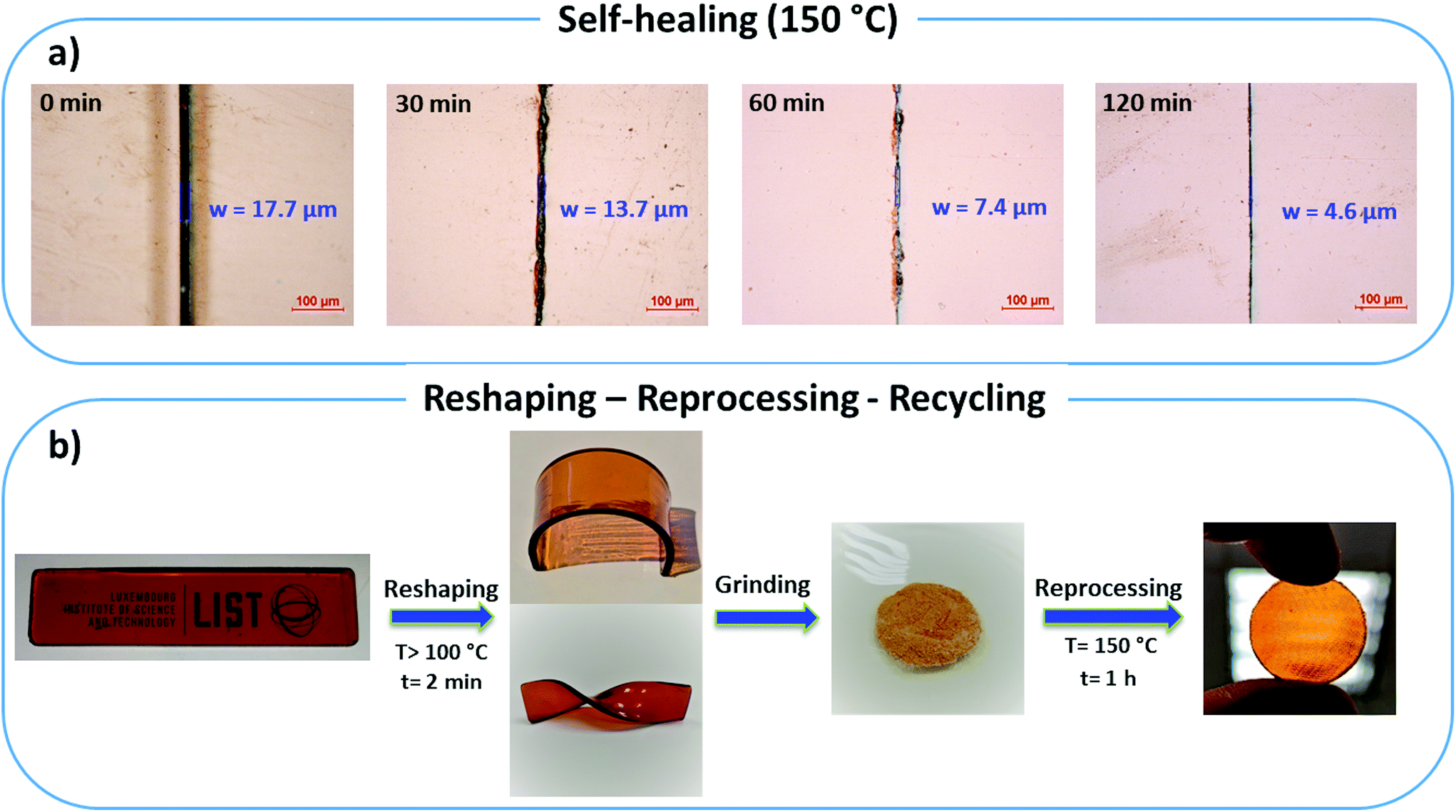 | ||
| Fig. 5 (a) Self-healing behavior of poly(PEG400-DPA-mea) (b) Life cycle of the bio-based polybenzoxazine vitrimer by reshaping, reprocessing and recycling. | ||
 | ||
| Fig. 6 Storage modulus of poly(PEG400-DPA-mea) determined in tensile and single cantilever mode after successive recycling (one, two and five times respectively). | ||
Conclusions
This study relates the synthesis and the properties of a catalyst-free and bio-based polybenzoxazine vitrimer. The synthesis pathway consisted of a solventless two-step reaction, including the Fischer esterification of 4,4-bis(4-hydroxyphenyl) valeric acid and polyethylene glycol, which was then reacted with mono-ethanolamine and paraformaldehyde via a Mannich-like condensation. Quadri-telechelic benzoxazine-terminated polyethylene glycol monomers containing ester bonds and pendant aliphatic hydroxyl groups (PEG400-DPA-mea) were obtained. The chemical structure of the monomers was confirmed by 1H NMR, 13C NMR, elemental analysis, and FTIR. The vitrimer (poly(PEG400-DPA-mea)) was formed by triggering the ring-opening polymerization of the benzoxazine moieties at 150 °C for one hour, leading to a covalently bonded network with an abundant number of tertiary amines per ester bond. The mechanical properties of the resulting PBz were assessed by rheological characterization. The achievement of a cross-linked structure was attested by the ability of the material to swell in common organic solvents, with a soluble fraction lower than 5 wt%. Due to the ability of tertiary nitrogen groups to catalyze transesterification reactions, the material enabled fast exchange reactions without the use of an external catalyst, as attested by short stress-relaxation times (116 s at 170 °C). The activation energy of the ester bond exchange was found to be 115 kJ mol−1, in the range of transesterification reactions catalyzed by tertiary amines. The effect of the structural features of the vitrimer was also investigated by tuning the crosslinking density of the network and the amount of –OH groups. It showed that the energy needed to activate the dynamic exchanges is clearly governed both by the excess of –OH groups and the concentration of dynamic sites in the network. The very short relaxation times were explained by the neighboring group participation theory, the abundant number of tertiary N groups and the flexibility of the network.The self-healing, recycling, reshaping, and reprocessing of poly(PEG400-DPA-mea) without any loss of properties were successfully demonstrated. Therefore, this research reflects the suitability of polybenzoxazines to design easily synthesizable mono-component, catalyst-free, and fast responsive vitrimers.
Abbreviations
| CAN | Covalent adaptable network |
| DCB | Dynamic covalent bond |
| TER | Transesterification reaction |
| –OH | Aliphatic hydroxyl |
| Zn(OAc)2 | Zinc acetate |
| NR3 | Tertiary amine |
| TBD | Triazobicyclodecene |
| PBZs | Polybenzoxazines |
| ROP | Ring-opening polymerization |
| PFA | Paraformaldehyde |
| DPA | Diphenolic acid |
| PEG | Polyethylene glycol |
| PEG-DPA | Quadri-telechelic phenol-terminated PEG |
| mea | Mono-ethanolamine |
| PEG-DPA-mea | Quadri-telechelic benzoxazine-terminated polyethylene glycol monomer |
| fa | Furfurylamine |
| p-TSA | para-Toluene sulfonic acid |
| NMR | Nuclear magnetic resonance |
| TMS | Tetramethylsilane |
| FTIR | Fourier transform infrared spectroscopy |
| ATR | Attenuated total reflection |
| N2 | Nitrogen |
| T desorpt. | Desorption temperature |
| TCD | Thermal conductivity detector |
| DSC | Differential scanning calorimetry |
| T g | Glass transition |
| TGA | Thermogravimetric analysis |
| DTG | Derivative tthermogravimetric Analysis |
| DMA | Dynamic mechanical analysis |
| RT | Room temperature |
| G 0 | Original relaxation modulus |
| G′/E′ | Storage modulus |
| G′′ | Loss modulus |
| W | Swelling ratio |
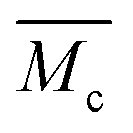
| Molecular weight between crosslinks |
| ν c | Crosslinking density |
| DMSO-d6 | Deuterated dimethylsulfoxide |
| Poly(PEG-DPA-mea) | Polymerized PEG-DPA-mea |
| PEGn | Polyethylene glycol of molecular weight n (g mol−1) |
| Tan δ | Loss factor |
| τ* | Relaxation time |
| E a | Activation energy |
| NGP | Neighboring group participation |
Author contributions
Antoine Adjaoud: validation, investigation, writing – original draft, writing – review and editing. Acerina Trejo-Machin: validation, investigation, writing – review and editing. Laura Puchot: validation, investigation, writing – original draft, writing – review and editing. Pierre Verge: conceptualization, validation, investigation, resources, writing – original draft, writing – review and editing, supervision, project administration.Funding sources
This research was supported by the Luxembourg National Research Fund (FNR), Grant number: C18/MS/12538602.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank the FNR for the funding of the project LIGNOBENZ (C18/MS/12538602). The authors are also extremely thankful to Benoit Marcolini, Régis Vaudemont, Lindsey Auguin, and Denis Pittois. The authors would also like to thank Laurent Sabatie and Dr Daniel Schmidt for the intellectual and technical discussions related to vitrimers.Notes and references
- K. L. Forsdyke and T. F. Starr, Thermoset Resins, iSmithers Rapra Publishing, 2002 Search PubMed.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- S. D. Bergman and F. Wudl, J. Mater. Chem., 2008, 18, 41–62 RSC.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- W. Denissen, J. Winne and F. Du Prez, Chem. Sci., 2015, 7, 30–38 RSC.
- J. Winne, L. Leibler and F. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- M. Guerre, C. Taplan, J. M. Winne and F. E. Du Prez, Chem. Sci., 2020, 11, 4855–4870 RSC.
- F. Van Lijsebetten, J. O. Holloway, J. M. Winne and F. E. Du Prez, Chem. Soc. Rev., 2020, 49, 8425–8438 RSC.
- D. J. Fortman, J. P. Brutman, G. X. De Hoe, R. L. Snyder, W. R. Dichtel and M. A. Hillmyer, ACS Sustainable Chem. Eng., 2018, 6, 11145–11159 CrossRef CAS.
- J. Otera, Chem. Rev., 1993, 93, 1449–1470 CrossRef CAS.
- U. Schuchardt, R. Sercheli and V. Matheus, J. Braz. Chem. Soc., 1998, 9, 199–210 CrossRef CAS.
- M. K. Kiesewetter, M. D. Scholten, N. Kirn, R. L. Weber, J. L. Hedrick and R. M. Waymouth, J. Org. Chem., 2009, 74, 9490–9496 CrossRef CAS PubMed.
- F. I. Altuna, C. E. Hoppe and R. J. J. Williams, Eur. Polym. J., 2019, 113, 297–304 CrossRef CAS.
- Y. Li, T. Liu, S. Zhang, L. Shao, M. Fei, H. Yu and J. Zhang, Green Chem., 2020, 22, 870–881 RSC.
- F. I. Altuna, V. Pettarin and R. J. J. Williams, Green Chem., 2013, 15, 3360–3366 RSC.
- J. Han, T. Liu, C. Hao, S. Zhang, B. Guo and J. Zhang, Macromolecules, 2018, 51, 6789–6799 CrossRef CAS.
- T. Liu, S. Zhang, C. Hao, C. Verdi, W. Liu, H. Liu and J. Zhang, Macromol. Rapid Commun., 2019, 40, 1800889 CrossRef PubMed.
- S. Debnath, S. Kaushal and U. Ojha, ACS Appl. Polym. Mater., 2020, 2, 1006–1013 CrossRef CAS.
- C. Hao, T. Liu, S. Zhang, W. Liu, Y. Shan and J. Zhang, Macromolecules, 2020, 53, 3110–3118 CrossRef CAS.
- X. Ning and H. Ishida, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 1121–1129 CrossRef CAS.
- H. Ishida, in Handbook of Benzoxazine Resins, ed. H. Ishida and T. Agag, Elsevier, Amsterdam, 2011, pp. 3–81 Search PubMed.
- E. Calò, A. Maffezzoli, G. Mele, F. Martina, S. E. Mazzetto, A. Tarzia and C. Stifani, Green Chem., 2007, 9, 754–759 RSC.
- N. N. Ghosh, B. Kiskan and Y. Yagci, Prog. Polym. Sci., 2007, 32, 1344–1391 CrossRef CAS.
- M. Comí, G. Lligadas, J. C. Ronda, M. Galià and V. Cádiz, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4894–4903 CrossRef.
- L. Dumas, L. Bonnaud, M. Olivier, M. Poorteman and P. Dubois, J. Mater. Chem. A, 2015, 3, 6012–6018 RSC.
- L. Puchot, P. Verge, T. Fouquet, C. Vancaeyzeele, F. Vidal and Y. Habibi, Green Chem., 2016, 18, 3346–3353 RSC.
- C. Wang, J. Sun, X. Liu, A. Sudo and T. Endo, Green Chem., 2012, 14, 2799–2806 RSC.
- N. K. Sini, J. Bijwe and I. K. Varma, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 7–11 CrossRef CAS.
- P. Thirukumaran, A. Shakila Parveen and M. Sarojadevi, ACS Sustainable Chem. Eng., 2014, 2, 2790–2801 CrossRef CAS.
- P. Froimowicz, C. R. Arza, L. Han and H. Ishida, ChemSusChem, 2016, 9, 1921–1928 CrossRef CAS PubMed.
- L. Dumas, L. Bonnaud, M. Olivier, M. Poorteman and P. Dubois, Green Chem., 2016, 18, 4954–4960 RSC.
- X. Liu, R. Zhang, T. Li, P. Zhu and Q. Zhuang, ACS Sustainable Chem. Eng., 2017, 5, 10682–10692 CrossRef CAS.
- B. Kiskan, React. Funct. Polym., 2018, 129, 76–88 CrossRef CAS.
- M. L. Salum, D. Iguchi, C. R. Arza, L. Han, H. Ishida and P. Froimowicz, ACS Sustainable Chem. Eng., 2018, 6, 13096–13106 CrossRef CAS.
- L. Bonnaud, B. Chollet, L. Dumas, A. A. M. Peru, A. L. Flourat, F. Allais and P. Dubois, Macromol. Chem. Phys., 2019, 220, 1800312 CrossRef.
- N. Amarnath, S. Shukla and B. Lochab, ACS Sustainable Chem. Eng., 2018, 6, 15151–15161 CrossRef CAS.
- M. Monisha, N. Amarnath, S. Mukherjee and B. Lochab, Macromol. Chem. Phys., 2019, 220, 1970005 CrossRef.
- S. Ren, X. Miao, W. Zhao, S. Zhang and W. Wang, Mater. Today Commun., 2019, 20, 100568 CrossRef CAS.
- Y. Lyu and H. Ishida, Prog. Polym. Sci., 2019, 99, 101168 CrossRef CAS.
- N. Amarnath, S. Shukla and B. Lochab, ACS Sustainable Chem. Eng., 2019, 7, 18700–18710 CrossRef CAS.
- L. E. Heim, H. Konnerth and M. H. G. Prechtl, Green Chem., 2017, 19, 2347–2355 RSC.
- R. Tavernier, L. Granado, G. Foyer, G. David and S. Caillol, Macromolecules, 2020, 53, 2557–2567 CrossRef CAS.
- R. Tavernier, L. Granado, G. Foyer, G. David and S. Caillol, Polymer, 2020, 123270 Search PubMed.
- O. S. Taskin, B. Kiskan and Y. Yagci, Macromolecules, 2013, 46, 8773–8778 CrossRef CAS.
- M. Arslan, B. Kiskan and Y. Yagci, Macromolecules, 2015, 48, 1329–1334 CrossRef CAS.
- M. Arslan, B. Kiskan and Y. Yagci, Macromolecules, 2018, 51, 10095–10103 CrossRef CAS.
- A. Trejo-Machin, L. Puchot and P. Verge, Polym. Chem., 2020, 11, 7026–7034 RSC.
- A. Trejo-Machin, P. Verge, L. Puchot and R. Quintana, Green Chem., 2017, 19, 5065–5073 RSC.
- J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang, G. G. Neuenscwander, S. W. Fitzpatrick, R. J. Bilski and J. L. Jarnefeld, Resour., Conserv. Recycl., 2000, 28, 227–239 CrossRef.
- V. Froidevaux, C. Negrell, S. Caillol, J.-P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed.
- M. Pelckmans, T. Renders, S. Van de Vyver and B. F. Sels, Green Chem., 2017, 19, 5303–5331 RSC.
- C. Özdemir and A. Güner, J. Appl. Polym. Sci., 2006, 101, 203–216 CrossRef.
- J. Chen, S. K. Spear, J. G. Huddleston and R. D. Rogers, Green Chem., 2005, 7, 64–82 RSC.
- H. A. Bruson, J. Am. Chem. Soc., 1936, 58, 1741–1744 CrossRef CAS.
- N. G. Kandile, T. M. A. Razek, A. M. Al-Sabagh and M. M. T. Khattab, Egypt. J. Pet., 2014, 23, 323–329 CrossRef.
- A. Trejo-Machin, A. Adjaoud, L. Puchot, R. Dieden and P. Verge, Eur. Polym. J., 2020, 124, 109468 CrossRef CAS.
- B. Kiskan, B. Koz and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6955–6961 CrossRef CAS.
- R. Kudoh, A. Sudo and T. Endo, Macromolecules, 2010, 43, 1185–1187 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, M. A. Hillmyer and W. R. Dichtel, J. Appl. Polym. Sci., 2017, 134, 44984 CrossRef.
- S. Zhao and M. M. Abu-Omar, Macromolecules, 2019, 52, 3646–3654 CrossRef CAS.
- R. S. Porter and L.-H. Wang, Polymer, 1992, 33, 2019–2030 CrossRef CAS.
- Y. Yuan and E. Ruckenstein, Polymer, 1998, 39, 1893–1897 CrossRef CAS.
- P. Baumhof, R. Mazitschek and A. Giannis, Angew. Chem., Int. Ed., 2001, 40, 3672–3674 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00324k |
| This journal is © The Royal Society of Chemistry 2021 |