
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diels–Alder cycloaddition polymerization of highly aromatic polyimides and their multiblock copolymers†
Doris
Cristurean‡
 a,
Stephan
Schaumüller‡
a,
Paul
Strasser
a,
Stephan
Haudum
a,
Markus
Himmelsbach
b,
Matthias
Bechmann
c,
Oliver
Brüggemann
a and
Ian
Teasdale
*a
a,
Stephan
Schaumüller‡
a,
Paul
Strasser
a,
Stephan
Haudum
a,
Markus
Himmelsbach
b,
Matthias
Bechmann
c,
Oliver
Brüggemann
a and
Ian
Teasdale
*a
aInstitute of Polymer Chemistry, Johannes Kepler University Linz, Altenbergerstraße 69, 4040 Linz, Austria. E-mail: ian.teasdale@jku.at
bInstitute of Analytical Chemistry, Johannes Kepler University Linz, Altenbergerstraße 69, 4040 Linz, Austria
cInstitute of Organic Chemistry, Johannes Kepler University Linz, Altenbergerstraße 69, 4040 Linz, Austria
First published on 3rd May 2021
Abstract
The ability to prepare block, multiblock and segmented polymers is an essential and established tool in polymer chemistry to tailor the properties of materials and steer the formation of complex nanostructures. The preparation of segmented or block copolymers with pre-defined block lengths is, however, inherently difficult for polyimides, one of the most important and versatile high-performance polymers. The most accessible route to polyimides, a step-growth polyamic acid formation between diamines and dianhydrides, is in dynamic equilibrium, which leads to chain scrambling of attempted block copolymers. We provide herein a solution to this by utilizing a Diels–Alder reaction on phenylethynyl end-functionalized oligomers containing pre-formed, ring-closed imides. The reaction of the alkynes with a bistetraphenylcyclopentadienone chain extender undergoes a chelotropic evolution of CO gas at high temperatures forming phenylene segments and polymerizing the chains in the process. Furthermore, we could use this reaction for the chain extension of different phenylethynyl functionalized telechelic oligoimides and thus produce random multiblock copolymers. Importantly the reaction is also demonstrated to enable chain extension reactions with insoluble oligoimides, considerably expanding the scope of potential as many important polyimides are either insoluble, or poorly soluble, in common organic solvents. This Diels–Alder polymerization is thus demonstrated to be a highly versatile route to prepare novel polyimides with wide-ranging possibilities and considerable potential to prepare advanced materials ranging from electronic applications to high-performance materials.
Introduction
The Diels–Alder (D–A) reaction1 is one of the most applied reactions in organic synthesis2 and has found multifaceted use in macromolecular chemistry.3 This atom efficient [4 + 2] cycloaddition gives the possibility of synthesizing six-membered-rings without an additional catalyst, shows a high functional group tolerance2,4 and thus can be utilized, for example, to prepare block copolymers, for polymer functionalization, cross-linking,3 as well as thermally reversible and self-healing polymers.5,6 Furthermore, polycyclic aromatic compounds can be prepared via the domino [4 + 2] cycloaddition reaction of tetraphenyl-substituted cyclopentadienones with dienophiles at high temperatures, for which the retro-D–A reaction is suppressed by the irreversible chelotropic evolution of CO gas from an intermediate bridged bicyclic cycloadduct.7 This represents a versatile synthesis method towards highly aromatic polymers such as polyphenylenes,8–10 ladder polymers11,12 or graphene nanoribbons.13,14 In particular, the variation of the cyclopentadienone derivatives and the bisacetylene moieties allows the design of a wide range of aromatic polymers15–17 and hyperbranched systems.18,19Aromatic polyimides belong to the group of high-performance polymers and generally exhibit high chain stiffness and rigidity, resulting in excellent stability towards heat and radiation and typically very high glass transition temperatures (Tg) in the range of 200–400 °C.20 Therefore, they are of high importance in wide variety of high demand applications such as the aircraft and space industry,21,22 where stability towards heat and radiation paired with light weight compared to metals are beneficial properties. A second considerable application is in electronics, for which their thermal stability is a valuable asset, as are their excellent properties as dielectric materials.23 The most common and accessible synthesis route is a step-growth polymerization of a diamine and a dianhydride at ambient temperatures, forming high molecular weight polyamic acid as a precursor, followed by a thermally or chemically driven dehydration, yielding the imide.24,25
Block copolymers, combining two or more chemically distinct polymer blocks, are ubiquitous in modern polymer chemistry and soft materials.26,27 Indeed, block copolymers with polyimide components have been realized with several other classes of polymers, for example with polybenzophenone blocks28 or poly(arylene ethersulfone) blocks.29 In these works, reactive end groups were either placed on the ends of both blocks28 or deliberately imbalanced PI blocks were used to ensure reactive anhydride moieties on both ends of a block.29 McGrath and coworkers have utilized the concept of transimidization to create an alternating PI-PDMS block copolymer.30 Another approach to achieving PI copolymers is grafting a macromolecular side chain, for example, PMMA31 or polystyrene,32 onto a PI backbone. However, the preparation of segmented or block copolymers comprising two types of polyimide blocks is more difficult to achieve in a defined manner due to the dynamic equilibrium reaction of the predominant polyamic acid polymerization route and the inherent chain scrambling it causes.25,33,34 In this contribution, we demonstrate a polymerization route to highly aromatic polyimide polymers via a Diels–Alder reaction. Telechelic polyimide oligomers are chain extended via an aromatic bifunctional diene, a synthetic pathway that can be used to prepare multiblock copolymers comprising solely of different polyimide blocks.
Experimental
Materials and methods
If not otherwise mentioned, all starting materials and solvents were purchased in reagent grade. 2,2′-(1,4-Phenylene)bis(1-phenylethane-1,2-dione), compound 8, 9 and the diamines 1 and 10 were purchased from TCI. Compound 4 and 1,2-(diphenyl)propan-2-one were obtained from Alfa Aesar. Compounds 2 and 7 were kindly donated by Evonik Fibres. Compound 7 was dried at 200 °C overnight before use. Diphenyl ether, from Acros Organics, was dried over molecular sieve 3 Å.The 1H nuclear magnetic resonance (NMR) spectra and 13C-NMR spectra were recorded on a Bruker Avance III 300 MHz spectrometer. 19F-NMR spectra were recorded on a Bruker Avance 500 MHz spectrometer. Thermogravimetric analysis (TGA) was carried out on a TA instruments Q5000 under nitrogen. Differential scanning calorimetry measurements (DSC) were performed on a TA instruments Q2000. IR (infrared) spectra were measured with a PerkinElmer 100 Series FTIR spectrometer equipped with ATR using a scan number of 128. For the high-resolution mass spectra, an Agilent 6520 ESI-QTOF (Agilent Technologies, Waldbronn, Germany) was used in positive mode. Methanol with 10 mM ammonium formate was used as an eluent. Size exclusion chromatography (SEC) was performed on a Viscotek GPCmax VE 2001 Solvent/Sample Module equipped with a Viscotek TDA 305 Triple Detector Array. It was run with dimethylformamide with 10 mM LiBr as eluent phase at a flow rate of 0.75 mL min−1. Raman spectra were measured on a BrukerMultiRAM Raman Microscope with an excitation wavelength of 1064 nm (300 mW) in a spectral shift range between 400 and 3600 cm−1.
Synthesis of model compounds and monomers
1H-NMR (300 MHz, DMSO d6) δ/ppm: 8.08, 8.01, 7.99, 7.96, 7.65, 7.64, 7.62, 7.61, 7.58, 7.48, 7.47, 7.46, 7.44, 7.41, 7.39, 7.36, 7.33, 7.30, 3.34.
13C-NMR (75 MHz, DMSO d6) δ/ppm: 166.28, 166.20, 149.94, 145.21, 139.58, 137.20, 132.15, 131.72, 130.75, 130.48, 129.60, 128.86, 128.43, 128.09, 127.21, 126.17, 125.79, 123.81, 121.39, 120.74, 93.41, 87.99, 64.57.
ATR-FTIR (neat): ν 3062 (C–Haromatic), 2210 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 1777 (C
C), 1777 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Oasymm. stretching), 1724 (COsymm stretching), 1613, 1510, 1429, 1368 (C–N), 1221, 1085, 1018, 916, 850, 812, 751, 741, 691, 646.
Oasymm. stretching), 1724 (COsymm stretching), 1613, 1510, 1429, 1368 (C–N), 1221, 1085, 1018, 916, 850, 812, 751, 741, 691, 646.
1H-NMR (300 MHz, CDCl3) δ/ppm: 7.77, 7.75, 7.41, 7.38, 7.36, 7.34, 7.28, 7.22, 7.19, 6.87–6.85.
13C-NMR (75 MHz, CDCl3) δ/ppm: 167.31, 167.21, 150.54, 148.47, 145.26, 141.59, 140.87, 140.26, 140.15, 140.10, 139.77, 138.29, 137.35, 131.39, 131.30, 131.24, 130.47, 130.33, 128.84, 128.52, 128.03, 127.88, 127.38, 127.13, 126.87, 126.81, 126.56, 126.47, 126.12, 126.04, 125.60, 122.21, 120.36, 65.10, 61.56, 60.90.
ATR-FTIR (neat): ν 3055 (C–Haromatic), 3023, 1776 (COasymm. stretching), 1720 (COsymm. stretching), 1599, 1510, 1440, 1363 (C–N), 1201, 1073, 1028, 911, 856, 818, 782, 753, 731, 695.
1H-NMR (300 MHz, CD2Cl2) δ/ppm: 7.26–7.21 (m, 26H), 6.94 (d, 4H, J = 7.3 Hz), 6.78 (s, 4H).
13C-NMR (75 MHz, CD2Cl2) δ/ppm: 200.46, 154.78, 154.52, 133.96, 133.35, 131.26, 131.10, 130.50, 130.44, 129.62, 129.35, 128.89, 128.38, 127.96, 126.04, 125.77.
ATR-FTIR (neat): ν 3053 (C–Haromatic), 3051, 1706 (CO), 1597, 1488, 1442, 1349, 1300, 1121, 1109, 1088, 1070, 1024, 1014, 911, 854 (C–Haromatic), 798, 784, 757, 739, 719, 685.
HRMS (m/z): calc. for C52H34O2 690.2559, found, M + H 691.2632.
Synthesis of telechelic oligomers
The alkyne capped oligomers were prepared according to literature procedure.36 A typical example for the synthesis of a PI-oligomer is described in the following: to 2.79 g (8.00 mmol) of 1 dissolved in 36 mL of DMF in a 3-necked round bottom flask equipped with a mechanical stirrer were added 3.13 g (7.04 mmol) of 8 and 0.48 g (1.92 mmol) of 2. (This amounts to a total solids concentration of 15 wt%.) An N2 stream was applied, and the mixture was stirred for 24 h at room temperature. Then, 2.19 g (21.5 mmol) of acetic anhydride and 3.40 g (43.0 mmol) pyridine were added, and the reaction mixture was again stirred under nitrogen at room temperature for 24 h. The polyimide oligomer PI-5 was collected by precipitation in water and subsequent filtration. The polymer was washed with copious amounts of water and ethanol, then dried in vacuo at 150 °C.PI-2 1H-NMR (300 MHz, CDCl3) δ/ppm: 8.25, 8.21, 8.12, 8.09, 7.80–7.78, 7.46–7.28.
13C-NMR (75 MHz, CDCl3) δ/ppm: 192.83, 166.67, 166.63, 166.03, 150.27, 150.19, 145.94, 145.84, 145.43, 141.86, 140.18 137.22, 135.78, 135.00, 132.13, 132.00, 131.89, 130.32, 130.28, 130.08, 129.97, 129.31, 129.02, 128.93, 128.57, 128.03, 127.98, 126.58, 126.33, 126.17, 124.74, 124.35, 123.77, 122.06, 120.40, 94.27, 87.74, 65.04.
ATR-FTIR (neat): ν 3059 (C–Haromatic), 1777 (COasymm. stretching), 1718 (COsymm. stretching), 1615, 1510, 1448, 1365 (C–N), 1292, 1209, 1161, 1086, 1018, 979, 916, 850, 818, 751, 719, 707, 677.
SEC: Mn = 5.6 kDa, Mw = 9.0 kDa, Đ = 1.60.
PI-4 1H-NMR (300 MHz, CDCl3) δ/ppm: 8.26–8.20, 8.09–8.06, 7.86–7.84, 7.52–7.49, 7.43–7.35.
13C-NMR (75 MHz, CDCl3) δ/ppm: 193.27, 166.41, 150.58, 146.18, 142.24, 140.49, 136.21, 135.26, 132.43, 132.16, 130.62, 129.13, 128.42, 126.79, 126.44, 124.94, 124.47, 120.84, 65.40.
ATR-FTIR (neat): ν 3062 (C–Haromatic), 1777 (COasymm. stretching), 1720 (COsymm. stretching), 1621, 1509, 1448, 1366, 1295, 1249, 1209, 1163, 1092, 1020, 978, 915, 853, 819, 753, 720, 681.
SEC: Mn = 13.7 kDa, Mw = 20.8 kDa, Đ = 1.52.
PI-5 1H-NMR (300 MHz, CD2Cl2) δ/ppm: 8.04–8.00, 7.91, 7.87, 7.84, 7.61, 7.61–7.58, 7.53–7.47, 7.44–7.29.
13C-NMR (75 MHz, CD2Cl2) δ/ppm: 166.95, 166.92, 166.43, 166.31, 150.64, 146.24, 140.56, 139.35, 137.64, 136.37, 133.05 132.75, 132.47, 132.22, 130.96, 130.84, 130.65, 130.32, 129.69, 129.19, 129.11, 128.97, 128.47, 128.42, 126.90, 126.69, 126.48, 125.60, 124.44, 124.03, 122.47, 121.97, 120.88, 94.31, 88.06, 65.45.
19F-NMR (471 MHz, CDCl3) δ/ppm: −63.21.
ATR-FTIR (neat): ν 3063 (C–Haromatic), 1781 (COasymm. stretching), 1720 (COsymm. stretching), 1616, 1510, 1446, 1369 (C–N), 1296, 1254 (C–F), 1209, 1191, 1143, 1100, 1020, 985, 852, 814, 753, 721, 679.
SEC: Mn = 6.0 kDa, Mw = 9.2 kDa, Đ = 1.53.
PI-6 13C-NMR (126 MHz) δ/ppm: 165.93, 158.84, 128.74, 117.03, 94.48, 87.58, 35.59, 30.78.
ATR-FTIR (neat): ν 3068 (C–Haromatic), 2212 (CC), 1760, 1692 (CO), 1378 (C–N), 1261, 1226 (C–Oaromatic), 832, 887, 782; 744, 691.
Diels–Alder polymerization
A typical example for a chain extension reaction with 6 is described in the following: to 1.00 g of oligomer PI-2 (which theoretically contains 0.37 mmol of triple bonds) were added 0.13 g (0.19 mmol) of 6 and 5.6 mL of degassed diphenyl ether in a round bottom flask. (This amounts to a total solids concentration of 200 g L−1.) The mixture was stirred with a magnetic stirrer and refluxed under argon at 260 °C for 72 h. Then the mixture was cooled to room temperature and precipitated in water. The resulting polymer PI-3 was washed with copious amounts of water and ethanol, then dried in vacuo at 150 °C.PI-1 1H-NMR (300 MHz, CD2Cl2) δ/ppm: 7.82–7.80, 7.41, 7.34–7.27, 7.19, 6.90–6.79, 6.68, 6.36.
13C-NMR (75 MHz, CD2Cl2) δ/ppm: 167.36, 150.78, 148.85, 148.73, 145.65, 141.73, 141.20, 141.01, 140.8003, 140.53, 140.38, 140.26, 140.08, 138.85, 138.55, 137.74, 137.34, 137.21, 137.15, 131.68, 131.62, 131.02, 130.63, 128.94, 128.82, 128.41, 128.29, 127.36, 127.25, 127.11, 126.76, 126.50, 126.03, 125.77, 122.08, 120.79, 65.39.
ATR-FTIR (neat): ν 3051 (C–Haromatic), 3018, 2923, 1776 (COasymm. stretching), 1718 (COsymm. stretching), 1599, 1510, 1442, 1362 (C–N), 1209, 1072, 1021, 911, 844, 815, 774, 751, 732, 695, 562.
SEC: Mn = 12.0 kDa, Mw = 15.3 kDa, Đ = 1.28.
PI-3 1H-NMR (300 MHz, CD2Cl2) δ/ppm: 8.27–8.21, 8.10–8.07, 7.87–7.85, 7.53–7.36, 7.26, 6.94–6.86, 6.73, 6.41.
13C-NMR (75 MHz, CD2Cl2) δ/ppm: 206.85, 193.30, 167.34, 166.44, 150.62, 146.29, 146.20, 142.27, 140.99, 140.52, 137.73, 136.24, 135.29, 132.46, 132.19, 131.60, 131.05, 130.66, 130.36, 129.65, 129.16, 128.93, 128.45, 128.40, 126.83, 126.47, 124.96, 124.49, 120.86, 65.43, 30.96.
ATR-FTIR (neat): ν 3060 (C–Haromatic), 1778 (COasymm. stretching), 1722 (COsymm. stretching), 1508, 1446, 1369 (C–N), 1296, 1211, 1091, 1021, 915, 853, 817, 751, 734, 720, 703, 678, 647, 631, 566, 542.
SEC: Mn = 29.7 kDa, Mw = 57.5 kDa, Đ = 1.94.
PI-7 1H-NMR (300 MHz, CD2Cl2) δ/ppm: 8.26–8.20, 8.09–8.07, 7.86–7.84, 7.52–7.49, 7.43–7.35, 7.20–7.08, 7.02–6.99.
13C-NMR (75 MHz, CD2Cl2) δ/ppm: 193.28, 166.41, 150.60, 146.19, 142.26, 140.51, 136.21, 135.28, 132.45, 130.64, 130.07, 129.14, 128.38, 126.81, 126.45, 124.94, 124.48, 123.56, 120.84, 119.13, 65.41.
ATR-FTIR (neat): ν 3061 (C–Haromatic), 1779 (COasymm. stretching), 1720 (COsymm. stretching), 1619, 1610, 1510, 1449, 1369 (C–N), 1297, 1249, 1209, 1164, 1093, 1020, 981, 917, 856, 820, 752, 720, 682.
SEC: Mn = 46.5 kDa, Mw = 178.9 kDa, Đ = 3.85.
PI-8 1H-NMR (300 MHz, CD2Cl2) δ/ppm: 8.01, 7.92, 7.87, 7.40, 7.35, 6.92, 6.85.
13C-NMR (75 MHz, CD2Cl2) δ/ppm: 166.43, 166.31, 150.64, 146.24, 140.56, 139.35, 136.38, 133.06, 132.76, 130.66, 129.19, 128.96, 128.48, 128.43, 127.36, 127.11, 126.91, 126.80, 126.48, 125.60, 124.44, 120.89, 65.46, 53.84.
19F-NMR (471 MHz, CDCl3) δ/ppm: −63.22.
ATR-FTIR (neat): ν 3060 (C–Haromatic), 1784 (COasymm. stretching), 1723 (COsymm. stretching), 1675, 1616, 1510, 1448, 1366 (C–N), 1296, 1255 (C–F), 1209, 1192, 1144, 1089, 1021, 983, 963, 913, 851, 818, 754, 721.
SEC: Mn = 53.3 kDa, Mw = 88.1 kDa, Đ = 1.65.
PI-9 13C-NMR (126 MHz) δ/ppm: 165.04, 158.09, 127.32, 121.70–116.50.
ATR-FTIR (neat): ν 3065 (C–Haromatic), 1777 (COasymm. stretching), 1714 (COsymm. stretching), 1591, 1506, 1476, 1437, 1367 (C–N), 1261, 1226 (C–Oaromatic), 1170, 1114, 1089, 1016, 961, 838, 818, 783, 744, 696, 670.
PI-10 1H-NMR (300 MHz, CD2Cl2) δ/ppm: 8.26–8.21, 8.10–8.07, 8.02–7.99, 7.91–7.84, 7.49, 7.34, 7.22, 7.13–7.08, 7.02–6.99, 6.84, 6.70, 6.38.
13C-NMR (75 MHz, CD2Cl2) δ/ppm: 193.33, 166.47, 150.65, 146.24, 142.34, 140.56, 139.36, 135.35, 133.06, 132.76, 132.53, 131.68, 130.70, 130.66, 130.13, 129.19, 128.49, 128.43, 126.88, 126.49, 125.61, 125.00, 124.54, 124.44, 123.61, 120.89, 119.19, 65.46.
19F-NMR (471 MHz, CDCl3) δ/ppm: −63.22.
ATR-FTIR (neat): ν 3059 (C–Haromatic), 1781 (COasymm. stretching), 1722 (COsymm. stretching), 1617, 1510, 1446, 1367 (C–N), 1293, 1254 (C–F), 1209, 1145, 1092, 1020, 981, 965, 915, 850, 816, 753, 733, 719.
SEC: Mn = 20.5 kDa, Mw = 39.6 kDa, Đ = 1.93.
PI-11 13C-NMR (126 MHz) δ/ppm: 165.35, 157.69, 149.72, 139.62, 127.09, 64.86.
ATR-FTIR (neat): ν 3060 (C–Haromatic), 1780 (COasymm. stretching), 1719 (COsymm. stretching), 1591, 1508, 1476, 1447, 1367 (C–N), 1296, 1257 (C–F), 1229, 1209, 1144, 1088, 1020, 962, 916, 818, 777, 721, 700.
Results and discussion
To develop a D–A chain extension method for polyimides, we first prepared a model compound from the diamine 9,9-bis(4-aminophenyl)fluorene (BAPF) 1 and two equivalents of 4-phenylethynylphthalic anhydride (PEPA) 2 in NMP at room temperature with subsequent chemical dehydration with acetic anhydride and pyridine to form the imide diyne 3 (Scheme 1). Upon adding a 4-fold excess of tetraphenylcyclopentadienone 4 to the imide diyne 3, a full conversion of the terminal alkyne to give compound 5 was observed by 13C-NMR spectroscopy (see ESI86†). The conditions required for this reaction were found to be 260 °C in diphenyl ether for 72 h.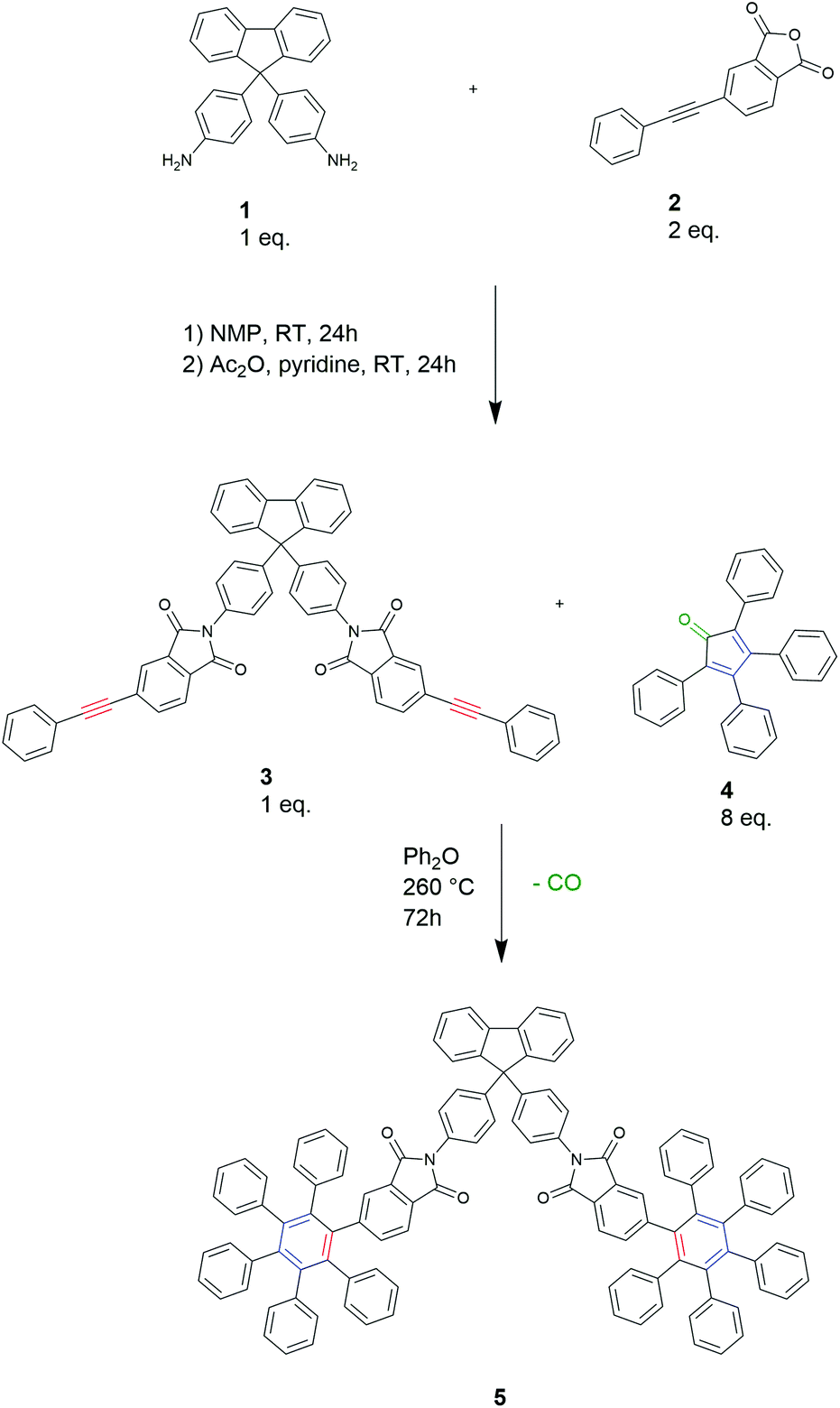 | ||
| Scheme 1 Model reaction for a D–A polyimide formation reaction. | ||
Buoyed by this observation, we then prepared a bifunctional diene 6 in Scheme 2 according to literature procedures.18,35 Compound 6 underwent a Diels–Alder polymerization with diyne 3 to yield the polymer PI-1, a highly aromatic soluble polyimide. The polymerization was carried out under the same conditions as the synthesis of model compound 5 (260 °C, 72 h). Size exclusion chromatography (SEC) of the product PI-1 revealed a polymer with Mn 107.4 kDa and Mw 211.6 kDa. The 13C-NMR spectrum shows the appearance of new signals in the aromatic region, indicating the formation of new aromatic structures. Furthermore, the signals at 167 ppm and 60 ppm resulting from the carbonyl groups and the spirocarbon of 1, respectively, indicate that 1 was introduced successfully into the polymer backbone of PI-1. The signal of the triple bond from the starting material 3 is not detectable by 13C-NMR spectroscopy (see ESI87†) suggesting polymerization is complete. PI-1 was found to have a high decomposition temperature around 561 °C (5% mass loss by TGA) and a high Tg of 396 °C. Furthermore, it was highly soluble in common organic solvents (chloroform, DMSO, DMF and NMP). While our primary motivation for preparing PI-1 was to learn about the polymerization process for the subsequent polymers, PI-1 is in itself an interesting novel polyimide due to its high aromaticity but excellent solubility and high Tg.
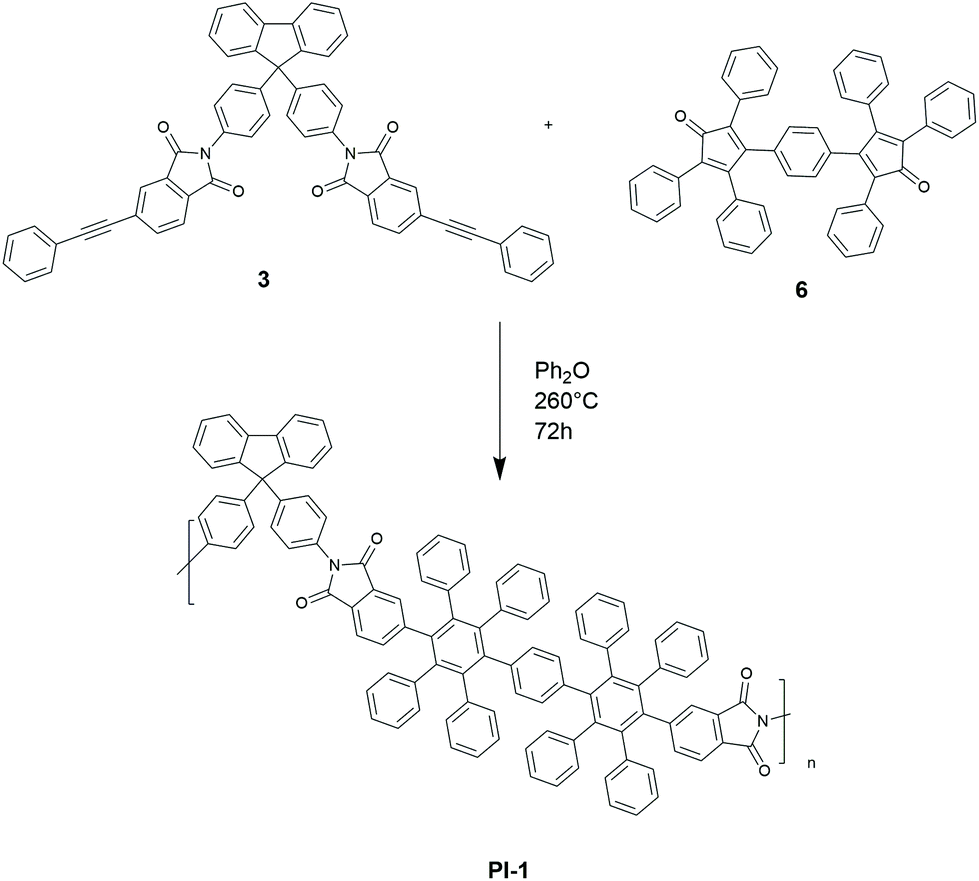 | ||
| Scheme 2 Synthesis of PI-1via D–A polymerization. | ||
Recently, Budy et al. prepared a polyimide with phenylated polyphenylene segments via phenylenediamine monomers (prepared first by D–A) and then copolymerized in the classical two-step polymerisation37 and report increased solubility and processability. However, we aimed to conduct the actual step-growth polymerization by D–A, as this gives the possibility of preparing block copolymers with such segments. To this end we then prepared telechelic, alkyne terminated polyimide oligomers PI-2, PI-4, PI-5 and PI-6via the method reported by Hergenrother38 as shown in Scheme 3. In this conventional two-step route to obtain polyimides, the diamine and the dianhydride were brought to reaction in an aprotic polar solvent (in this case DMF). The addition of PEPA 2 gave end-capped polyamic acid oligomers. In a second step, chemical imidization was performed with acetic anhydride and pyridine. The end-capped oligomers had a calculated Mn of 5.8 kDa for PI-2 and 7.0 kDa for PI-5. The measured Mn values found by SEC were 5.6 kDa for PI-2 and 6.0 kDa for PI-5, thus in good conjunction with the calculated values (Table 1).
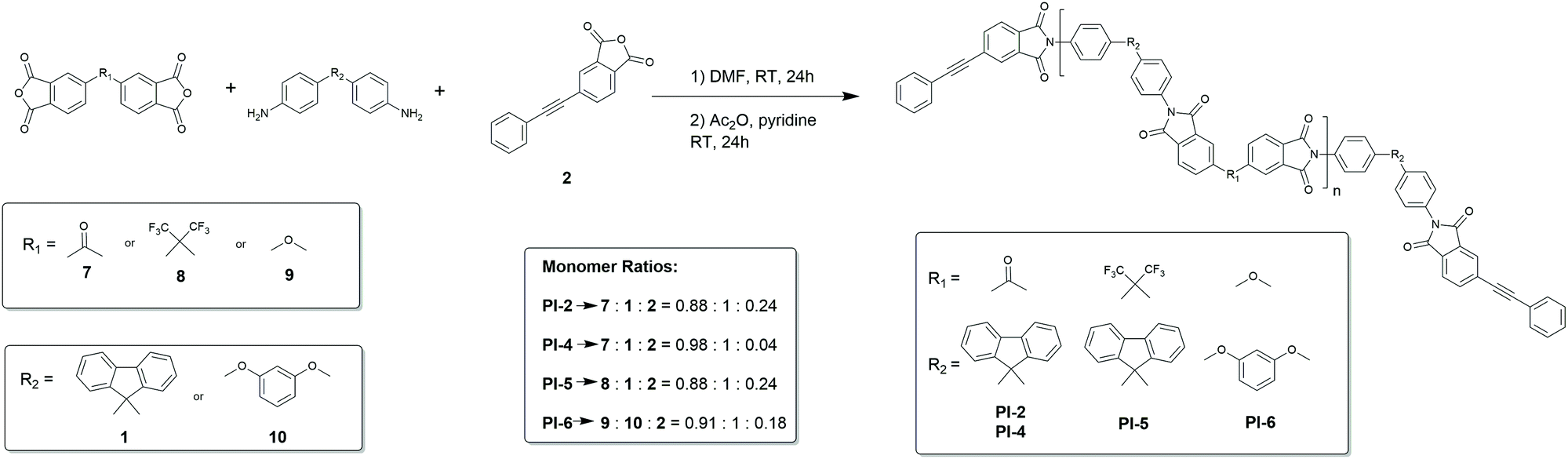 | ||
| Scheme 3 Synthesis route and molar ratios used for phenylethynyl-terminated polyimide oligomers PI-2, PI-4, PI-5 and PI-6. | ||
| Product | M1 | M2 | M n before chain extension/kDa | M w before chain extension/kDA | M n after chain extension/kDa | M w after chain extension/kDa | Đ after chain extension | T g/°C |
|---|---|---|---|---|---|---|---|---|
Molecular weights measured by SEC in DMF/LiBr with multi detection. The oligomers are used in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with compound 6. The two oligomers for PI-10 are used in equimolar amount in relation to triple bond content.a Measured by ESI-Q-TOF. Chromatograms and spectra are provided in the ESI.† :1 ratio with compound 6. The two oligomers for PI-10 are used in equimolar amount in relation to triple bond content.a Measured by ESI-Q-TOF. Chromatograms and spectra are provided in the ESI.† |
||||||||
| PI-1 | 1 | 0.8089a | — | 107.4 | 211.6 | 1.97 | 396 | |
| PI-3 | PI-2 | 5.6 | 9.0 | 29.7 | 57.5 | 1.94 | 381 | |
| PI-7 | PI-4 | 13.7 | 20.8 | 46.5 | 178.9 | 3.85 | 386 | |
| PI-8 | PI-5 | 6.0 | 9.2 | 53.3 | 88.1 | 1.65 | 385 | |
| PI-10 | PI-2 | PI-5 | 5.6/6.0 | 9.0/9.2 | 20.5 | 39.6 | 1.93 | 368 |
Upon reducing the amount of the endcapper 2, longer oligomers are obtained as can be observed from PI-4 which shows an Mn of 13.7 kDa. PI-6 was insoluble in DMF and hence could not be measured via SEC (solubility of all polymers is summarized in ESI†). All alkyne-capped oligomers were characterized by FTIR and NMR spectroscopy showing the characteristic signals of the triple bond at 94 ppm and 87 ppm in the 13C-NMR spectrum. Chain extension of the polyimide oligomer PI-2 with the bifunctional diene 6 was then carried out at 260 °C in diphenyl ether for 72 h (Scheme 4). The successful extension of the polymer chains was monitored via SEC. Fig. 1 shows the peak of PI-3 at a lower retention volume indicating its increased molecular weight compared to PI-2. Moreover, the short-chain oligomers observable between 20 and 22.5 mL of retention volume in PI-2 are no longer present after the chain-extension reaction (Fig. 1). PI-3 was further characterized by 13C-NMR spectroscopy (Fig. 2), indicating the complete transformation of the alkyne chain ends. Based on the results of PI-3, the oligomers PI-4, PI-5 and PI-6 were chain extended using the same method. The extension of PI-4 and PI-5 yielded PI-7 and PI-8, respectively, which show an increased molecular weight as can be observed in the SEC and is listed in Table 1. Additionally, no signals from the alkyne chain ends were observed in the 13C-NMR spectra (see ESI55 and ESI62†) indicating the full conversion of the starting material. The comparison of retention volumes in Fig. 1 indicates that the Mn of the used oligomer carries over directly into the polymer: the oligomer with lower Mn (PI-2) yields the chain extended polymer with lower Mn (PI-3). Consequently, the oligomer with higher molecular mass (PI-4) yields the polymer with higher molecular mass (PI-7) and hence the reaction works well for both chosen oligomer lengths. The extension of PI-6, which is insoluble in diphenylether and other common solvents, was performed in suspension and PI-9 was isolated as an insoluble product. Analysis by FT-Raman spectroscopy (normalized to the signal at 1777 cm−1) revealed a 75% decrease of the signal at 2213 cm−1, originating from the alkyne end groups, compared to the corresponding signal in PI-6 (see Fig. 3a), indicating significant chain extension. A solid-state NMR spectrum (see ESI69†) was recorded where no signals corresponding to the alkyne groups could be detected, further suggesting substantial polymerization. Furthermore, DSC measurements showed an increase in Tg from 207 °C to 218 °C (Fig. 3b) which can be attributed to the chain extension reaction alongside an increase in chain rigidity caused by the newly formed aromatic bridging groups between the oligomers. This observation significantly expands the range of polyimides to which this method can potentially be applied. Since no solubility limitations are observed, a great variety of polyimide oligomers, most of which are poorly soluble in organic solvents, can be considered for the preparation of block copolymers via D–A using chain extension.
 | ||
| Scheme 4 Overview of the synthesized polymers and random block copolymers obtained by chain extension. | ||
 | ||
| Fig. 1 SEC chromatograms of PI-2 (red) and PI-3 (black) as well as PI-4 (blue) and PI-7 (green) measured with a refractive index detector. | ||
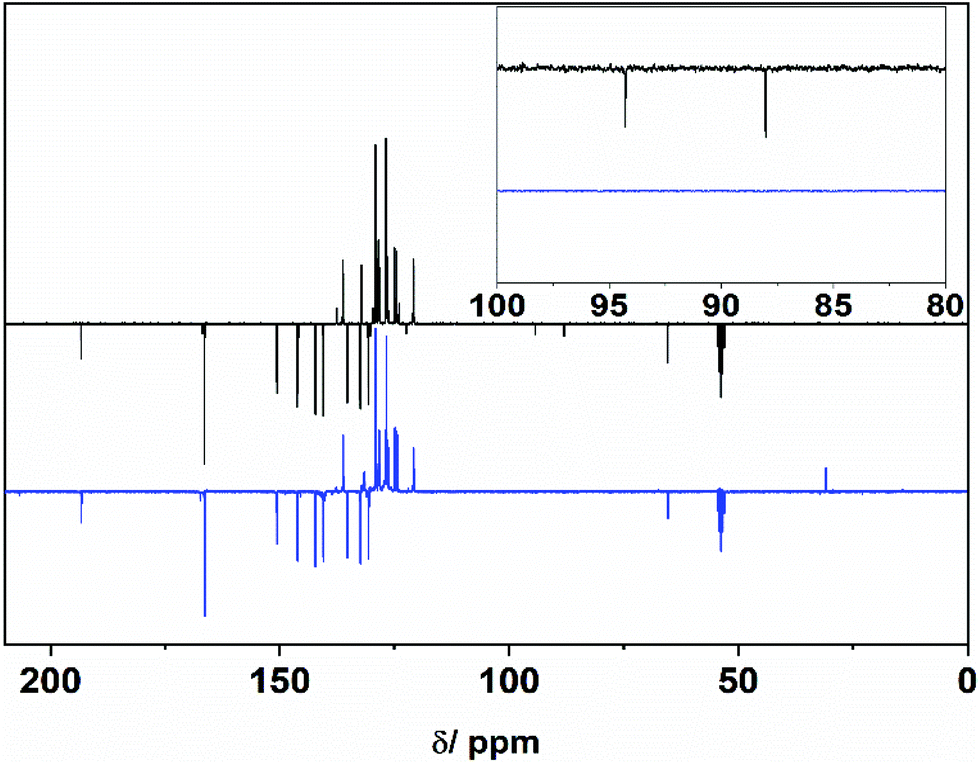 | ||
| Fig. 2 13C-NMR of PI-2 (top) and PI-3 (bottom). Inset zoom region shows the transformation of the alkyne chain ends. | ||
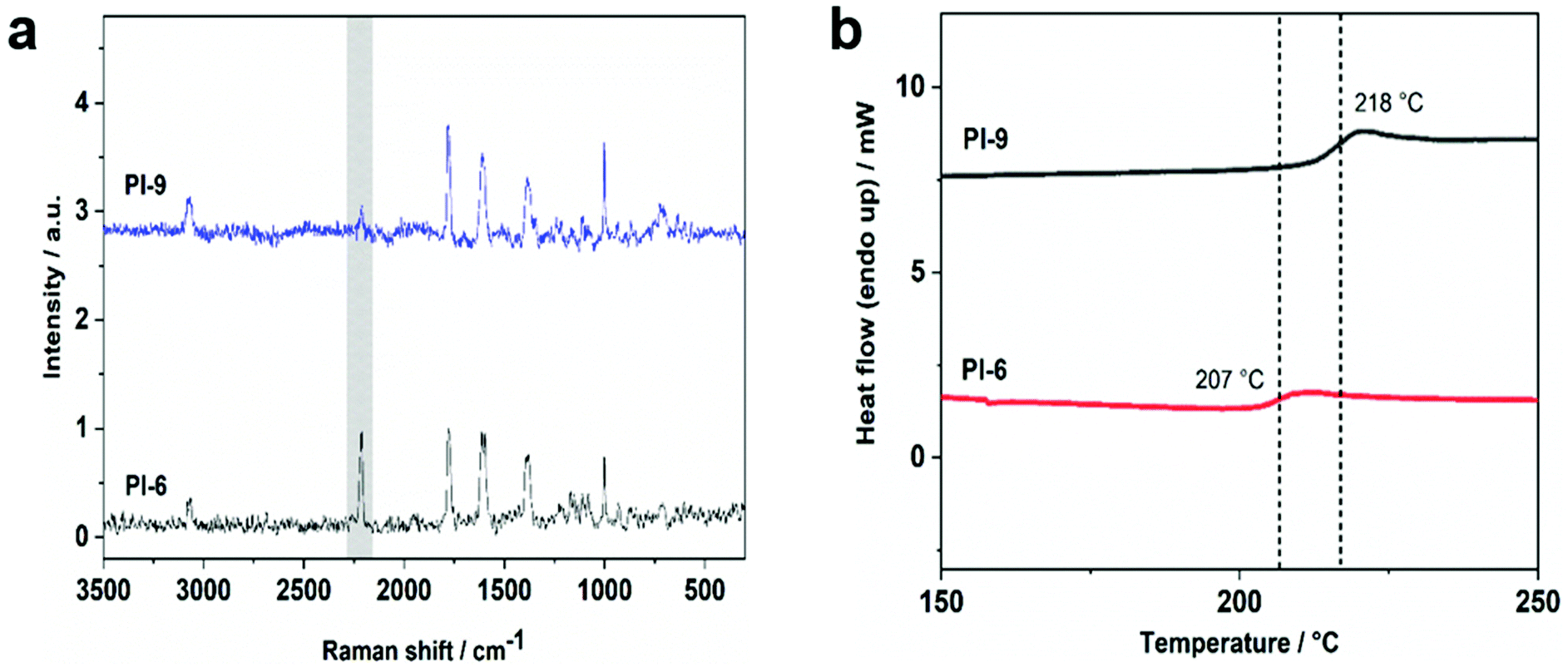 | ||
| Fig. 3 (a) FT-Raman spectrum of PI-6 (bottom) and PI-9 (top) showing the decrease of the triple bond signal at 2213 cm−1 indicating chain extension. (b) DSC curves of PI-6 (bottom) and PI-9 (top) showing an increase of 11 °C in the Tg suggesting that chain extension reaction occurred. | ||
The ability to chain extend polyimides provides the possibility to combine a wide range of monomers and oligomers and thus prepare polyimides with tailored molecular compositions. It is important to note that this is usually not possible during the polyamic acid polymerization step due to chain scrambling during this equilibrium reaction.25,33,34 To this end, we prepared random block copolymers with the oligomers PI-2 and PI-5 and the oligomers PI-5 and PI-6 yielding the block copolymer PI-10 and the block copolymer PI-11 respectively as shown in Scheme 4. PI-11 contains the oligomer PI-6, which causes insolubility of PI-11. Analysis of PI-11 by solid-state NMR spectroscopy showed the conversion of the alkyne groups (see ESI82†). Furthermore, the signal around 60 ppm corresponding to the spiro carbon of the diamine is an indication that the oligomer PI-5 is indeed concurrently present in PI-11. For the soluble PI-10 a significant increase of the molecular weight is detectable by SEC (Table 1). In the 13C-NMR spectrum (see ESI75†) signals resulting from the alkyne groups are not visible anymore indicating complete conversion. The presence of both blocks in the block copolymer PI-10 was detected by NMR spectroscopy in which the fluorine groups of PI-5 and the carbonyl groups of PI-2 are observed (see ESI76 and ESI75† respectively). This observation, along with the monomodal peak in the SEC, evidence the copolymerization of the different blocks and incorporation into the same polymer chains.
Conclusions
A novel route for the preparation of highly aromatic polyimides was demonstrated. First, a model reaction demonstrated the feasibility of preparing aromatic polyimides with polyphenylene segments via the Diels–Alder reaction of phenylethynyl capped imides with tetraphenylcyclopentadienone. A bistetraphenylcyclopentadienone was then prepared according to literature procedures and used to chain extend phenylethynyl functionalized α-ω-telechelic oligoimides. Furthermore, random multiblock copolymers with pre-defined block lengths could be prepared by combining two different PIs. We were also able to show the applicability of this reaction for the chain extension of insoluble oligoimides expanding the potential of the presented approach significantly, since many of the industrially important polyimides are insoluble, due to their superior chemical and mechanical stability. Chain extension via Diels–Alder cycloaddition is thus demonstrated to be a highly versatile route to prepare novel highly aromatic polyimides and their multiblock copolyimides. The D–A chain extension of alkyne-capped oligoimides thus significantly expands the range of polymers possible and could be used to prepare advanced materials with novel properties for applications in high-performance materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Austrian Research Promotion Agency (FFG) grant number 864711 in cooperation with Evonik Fibres GmbH. NMR experiments were performed at the Upper Austrian–South Bohemian Research Infrastructure Center in Linz, co-financed by “RERI-uasb”, EFRE RU2-EU-124/100-2010 (ETC Austria–Czech Republic 2007–2013, Project M00146). We thank Prof. Markus Scharber for FT-Raman spectroscopy measurements.References
- O. Diels and K. Alder, Justus Liebigs Ann. Chem., 1928, 460, 98–122 CrossRef CAS.
- I. C.-Y. Hou, Y. Hu, A. Narita and K. Müllen, Polym. J., 2018, 50, 3–20 CrossRef CAS.
- M. A. Tasdelen, Polym. Chem., 2011, 2, 2133–2145 RSC.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS PubMed.
- Y. L. Liu and T. W. Chuo, Polym. Chem., 2013, 4, 2194–2205 RSC.
- J. Steinkoenig, M. M. Zieger, H. Mutlu and C. Barner-Kowollik, Macromolecules, 2017, 50, 5385–5391 CrossRef CAS.
- M. Hapke, Tetrahedron Lett., 2016, 57, 5719–5729 CrossRef CAS.
- A. J. Berresheim, M. Müller and K. Müllen, Chem. Rev., 1999, 99, 1747–1785 CrossRef CAS PubMed.
- J. K. Stille and G. K. Noren, Macromolecules, 1972, 5, 49–55 CrossRef CAS.
- B. A. G. Hammer and K. Müllen, Chem. Rev., 2016, 116, 2103–21040 CrossRef CAS PubMed.
- U. Scherf, J. Mater. Chem., 1999, 9, 1853–1864 RSC.
- Y. C. Teo, H. W. H. Lai and Y. Xia, Chem. – Eur. J., 2017, 23, 14101–14112 CrossRef CAS PubMed.
- L. Chen, Y. Hernandez, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2012, 51, 7640–7654 CrossRef CAS PubMed.
- A. Narita, X. Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC.
- Z. B. Lim, K. S. Tan, A. V. Lunchev, H. Li, S. J. Cho, A. C. Grimsdale and R. Yazami, Synth. Met., 2015, 200, 85–90 CrossRef CAS.
- S. Grätz, H. Komber, M. Bauer and B. Voit, Polym. Chem., 2019, 10, 698–704 RSC.
- A. L. Rusanov, D. Y. Likhachev, O. V. Kozlova and F. W. Harris, Prog. Polym. Sci., 2006, 31, 749–810 CrossRef CAS.
- K. Stumpe, H. Komber and B. I. Voit, Macromol. Chem. Phys., 2006, 207, 1825–1833 CrossRef CAS.
- F. Morgenroth and K. Müllen, Tetrahedron, 1997, 53, 15349–15366 CrossRef CAS.
- P. M. Hergenrother, High Perform. Polym., 2003, 15, 3–45 CrossRef CAS.
- P. M. Hergenrother, Angew. Chem., Int. Ed. Engl., 1990, 29, 1262–1268 CrossRef.
- R. H. Pater and P. A. Curto, Acta Astronaut., 2007, 61, 1121–1129 CrossRef.
- A. Gumyusenge, X. Luo, Z. Ke, D. T. Tran and J. Mei, ACS Mater. Lett., 2019, 1, 154–157 CrossRef CAS.
- M. Gosh, Polyimides: Fundamentals and Applications, Marcel Dekker Inc., New York, 1996 Search PubMed.
- R. G. Bryant, in Encyclopedia of Polymer Science and Technology, ed. R. G. Bryant, John Wiley & Sons, Inc, Hoboken, N.J., 2002 Search PubMed.
- F. S. Bates, M. A. Hillmyer, T. P. Lodge, C. M. Bates, K. T. Delaney and G. H. Fredrickson, Science, 2012, 336, 434–440 CrossRef CAS PubMed.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS.
- T. Miyahara, J. Miyake, S. Matsuno, M. Watanabe and K. Miyatake, RSC Adv., 2015, 5, 50082–50086 RSC.
- A. S. Badami, A. Roy, H. S. Lee, Y. Li and J. E. McGrath, J. Membr. Sci., 2009, 328, 156–164 CrossRef CAS.
- M. E. Rogers, T. E. Glass, S. J. Mecham, D. Rodrigues, G. L. Wilkes and J. E. McGrath, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 2663–2675 CrossRef CAS.
- A. P. Filippov, E. V. Belyaeva, A. S. Krasova, M. A. Simonova, E. B. Tarabukina, T. K. Meleshko, D. M. Ilgach, N. N. Bogorad and A. V. Yakimansky, Polym. Sci., Ser. A, 2014, 56, 1–9 CrossRef CAS.
- G. D. Fu, E. T. Kang, K. G. Neoh, C. C. Lin and D. J. Liaw, Macromolecules, 2005, 38, 7593–7600 CrossRef CAS.
- D. Wilson, H. D. Stenzenberger and P. M. Hergenrother, Polyimides, Springer Science+Business Media, New York, 1990 Search PubMed.
- A. A. Kuznetsov and A. Y. Tsegelskaya, in Solvents, Ionic Liquids and Solvent Effects, ed. D. Glossman-Mitnik and M. Maciejewska, IntechOpen, 2020, pp. 1–22 Search PubMed.
- S. Xu, M. Adamski, M. Killer, E. M. Schibli, B. J. Frisken and S. Holdcroft, Macromolecules, 2019, 52, 2548–2559 CrossRef CAS.
- C. M. Thompson and P. M. Hergenrother, Macromolecules, 2002, 35, 5835–5839 CrossRef CAS.
- S. M. Budy, J. D. Hall and D. Y. Son, Polym. Chem., 2020, 11, 6273–6280 RSC.
- P. M. Hergenrother, J. W. Connell and J. G. Smith, Polymer, 2000, 41, 5073–5081 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00314c |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |