DOI:
10.1039/D1PY00312G
(Paper)
Polym. Chem., 2021,
12, 2825-2831
Fabrication of reversible pH-responsive aggregation-induced emission luminogens assisted by a block copolymer via a dynamic covalent bond†
Received
8th March 2021
, Accepted 12th April 2021
First published on 28th April 2021
Abstract
Aggregated induced emission (AIE) molecules with stimuli-responsive properties have attracted increasing attention for many applications. Herein, we synthesized a type of block copolymer poly(ethylene glycol)-block-poly(L-lysine) (PEG-b-PLys) modified with tetraphenylethylene (TPE) via imine bonds (PEG-b-P(Lys-TPE)), which shows reversible pH-responsive AIE characteristics. The presence of pH-sensitive imine bonds enables the reversible detachment of TPE residues from polypeptides with pH variation in the presence of organic solvents, which results in the reversible variation of the fluorescence intensity. We demonstrate that the increase in fluorescence properties is mainly due to the restriction of the intramolecular motion of the TPE moieties during the assembly process of the block copolymer and vice versa, confirming the AIE mechanism. This work offers a convenient and universal strategy to efficiently construct AIEgens with reversible stimuli-responsive properties, which serve as potential candidates for new generations of smart fluorescent nanomaterials.
Introduction
Fluorescent materials show great advantages for biosensing and bioimaging, such as high sensitivity and convenience.1 Typically, traditional organic light-emitting materials with the aggregation caused quenching (ACQ) feature merely emit light in low-concentration solutions, while the luminous intensity reduces or completely disappears as the concentration increases due to the molecular aggregation.2 Inorganic quantum dots (QDs) exhibit excellent fluorescence properties but with limitations such as synthetic difficulties, non-ideal luminous effects and toxicity.3 In contrast, fluorogens with AIE performance show an increased fluorescence intensity upon aggregation.4–6 The main mechanism of the AIE effect is attributed to the restricted intramolecular rotation (RIR) in the aggregated state.7–9 Current research shows that nearly all AIE substances have a common feature in their molecular structure with many benzene rings connected by single bonds. Particularly, tetraphenylethylene (TPE) is one of the widely studied AIE materials.10–12 Tang's group reported TPE as a promising group for determining the critical micelle concentration (CMC), critical micellization temperature (CMT), and the assembly/decomposition of colored amphiphilic molecules.13 They further prepared TPE-based materials with continuously tunable fluorescence emissions facilitated by the self-assembly of block copolymers.14 In addition, a facile method to synthesize stimuli-responsive fluorescent elastomers by coupling TPE to polydimethylsiloxane was reported.15
Stimuli-responsive polymers, a type of smart polymer, can undergo changes in their properties in response to environmental stimuli such as temperature, light, pH, etc.1,2 Many of the stimuli-reactive polymers have been reported, e.g. polypeptides that are particularly striking for biomedical applications due to their biocompatibility, biodegradability and versatility.16–19 However, merely a few studies have been reported on the synthesis of stimuli-responsive AIEgens by introducing AIE molecules into stimuli-responsive polymers.20,21 A couple of approaches have been reported to prepare stimuli-responsive polypeptides, of which the one based on dynamic covalent bond (DCB) construction is of particular interest.22–26 The imine bond is the most widely studied DCB because of its superior pH sensitivity that is suitable for biomedical applications.27,28 Typically, the imine bond is unstable in an acidic solution, but shows reversible re-conjugation at alkaline pH.29 Herein, we incorporated the imine bond to combine the polypeptide copolymers PEG-b-PLys with TPE by a Schiff base reaction to obtain PEG-b-P(Lys-TPE). The as-prepared copolymers exhibit a pH-responsive AIE feature induced by the self-assembly of the block copolymer. Furthermore, the pH effect on the fluorescence intensity of the system was systematically studied. We demonstrated that the presence of an organic solvent enables a reversible behavior by increasing the mobility of the molecules.
Experimental
Materials and methods
ε-Benzyloxycarbonyl-L-lysine was purchased from GL Biochem (Shanghai) Ltd. α-Methoxy-ω-amino poly(ethylene glycol) (mPEG-NH2, Mn = 2000, 5000 g mol−1) was purchased from JenKem Technology Co., Ltd. mPEG-NH2 was stored under a nitrogen atmosphere and placed in a refrigerator at −20 °C. 2-Bromo-1,1,2-tristyrene, 4-formylphenylboronic acid and tetrabutylammonium bromide (TBAB) were purchased from Aladdin. Triphosgene, anhydrous tetrahydrofuran (THF), anhydrous N,N′-dimethylformamide (DMF) and tetrakis (triphenylphosphonium) palladium (Pd(PPh3)4) were purchased from Tansoole (Adamas). Pd(PPh3)4 was stored under a nitrogen atmosphere and placed in a refrigerator at 4 °C. Tetrahydrofuran (THF) and n-hexane were freshly distilled from calcium hydride (CaH2) and purified by passing through activated alumina columns under N2 prior to use. Unless otherwise specified, all chemicals were purchased from commercial sources and used without further drying or purification. Reaction temperatures were controlled using an IKA temperature modulator.
Characterization
Fourier transform infrared (FTIR) spectra were recorded from 2000 to 1200 cm−1 on a JASCO FT/IR 4700 spectrofluorometer. The solution samples were cast on potassium bromide (KBr) plates and dried before measurements. All of the measurements were performed at 20 °C with 32 scans. Tandem gel permeation chromatography (GPC) analysis was conducted using an SSI pump connected to Wyatt Optilab DSP with 0.05 M lithium bromide (LiBr) in DMF as the eluent at a flow rate of 1.0 mL min−1 at 50 °C. Calibration was done using polystyrene standards. All GPC samples were prepared at concentrations of approximately 6 mg mL−1. Nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker AV500 FT-NMR spectrometer at 25 °C. CDCl3 and D2O were used as solvents. UV-vis spectra were obtained using a Shimadzu UV-2910 spectrometer with quartz sample cells with a path length of 2 nm. Fluorescence results were recorded using a Hitachi F-2700 FL spectrophotometer. A Mettler Toledo FE28 pH meter was used to determine the solution pH. Transmission electron microscopy (TEM) experiments were conducted on an FEI Tecnai 20. The polymer solution (7 μL) was pipetted onto a carbon-coated copper grid which was pretreated in a plasma cleaner. The grid was blotted to remove any excess solution and then dyed using 2% uranyl acetate. The samples were dried and stored under ambient conditions before TEM testing. Atomic force microscopy (AFM) studies were conducted using a tapping mode AFM (Bruker Multimode 8 AFM/SPM system) in ambient air with Nanoscope software. Five microliters of polymer solution were placed on mica using a spin-coater, and dried on freshly cleaved mica under ambient conditions before AFM imaging. Minimal processing of the images was carried out using Nanoscope Analysis software from Bruker.
Synthesis of Nε-benzyloxycarbonyl lysine N-carboxyanhydride (ZLys-NCA)
ε-Benzyloxycarbonyl-L-lysine (10 g, 35.71 mmol) (ZLys) and triphosgene (3.71 g, 12.5 mmol, 1.05 equiv.) were added into a 250 mL round-bottom Schlenk flask and dispersed in dry THF (100 mL) under a nitrogen atmosphere. The mixture was stirred at 50 °C under N2 for 2 hours, and the solution was precipitated into petroleum ether (500 mL). The mixture was filtered to obtain a white solid. Then, the solid was dissolved in ethyl acetate (100 mL) and filtered again. Ethyl acetate was removed by rotary evaporation to give a light-yellow solid. Afterwards, the solid was transferred to a glovebox under vacuum or a N2 atmosphere. In the glovebox, the solid was recrystallized in a THF/n-hexane mixture (volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) three times to obtain 6.5 g of a light-yellow solid ZLys-NCA. Yield: 59.4%.
:3) three times to obtain 6.5 g of a light-yellow solid ZLys-NCA. Yield: 59.4%.
Synthesis of the poly(ethylene glycol)-block-poly(Nε-benzyloxycarbonyl-lysine) (PEG-b-PZLys) block copolymer
mPEG-NH2 (Mn = 2000 or 5000 g mol−1) as a macromolecular initiator was used to initiate the ring-opening polymerization (ROP) of ZLys-NCA. mPEG-NH2 and ZLys-NCA (2 g, 6.64 mmol) were dissolved in dry DMF (100 mg mL−1). The mixture was stirred at room temperature under a nitrogen atmosphere. The reaction progress was monitored by FTIR spectroscopy until the characteristic peaks at 1790 and 1850 cm−1 disappeared. The solution was precipitated into ether. The solids were collected by centrifugation, washed three times with ether and dried in a vacuum oven to obtain a light-yellow solid PEG-b-PZLys. Yield: 74.5%–82.3%. PEG-b-PZLys and 4 mol equiv. of HBr (C = 33%, in HAc) were dissolved in CF3COOH (35 mg mL−1) at 0 °C under N2 for 2.5 hours. The solutions were precipitated into ether. The solids were collected by centrifugation, and then dissolved in deionized water. The final product poly(ethylene glycol)-block-polylysine (PEG-b-PLys) was obtained after dialysis and lyophilization. Yield: 64.5%–76%.
Synthesis of 1,1,2-triphenyl-2-(p-formyl)ethylene (TPE-CHO)
2-Bromo-1,1,2-tristyrene (3.35 g, 10 mmol), 4-formylphenylboronic acid (2.25 g, 15 mmol) and TBAB (0.32 g, 1.0 mmol) were added to a three-necked flask. Then the condensation device was set up, and the air in the system was changed to a nitrogen atmosphere. After that, the prepared potassium carbonate aqueous solution (2 mol L−1, 18 mL) and toluene (60 mL) were added to the reaction system. The mixture was stirred at room temperature for 0.5 hours, and then Pd(PPh3)4 (0.01 g, 8.70 × 10–3 mmol) was added immediately, followed by heating to 90 °C for 24 hours. After the reaction was completed, the mixture was poured into water and extracted three times with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate. The solvent was removed by rotary evaporation and the residue was chromatographed on a silica gel column using CH2Cl2/n-hexane (volume ratio of 1:2) as an eluent to obtain 3.45 g of a yellow solid (TPE-CHO). Yield: 95.7%.
Synthesis of the PEG-b-P(Lys-TPE) block copolymer
PEG-b-PLys, TPE-CHO (molar ratio of –NH2/TPE-CHO = 1:2) and a trace amount of triethylamine (TEA) were dissolved in dry DMF (100 mg mL−1) at 25 °C under N2 for 3 days. The solution was precipitated into ether. Afterwards, the solids were collected by centrifugation, washed three times with ether and dried in a vacuum oven to obtain a brown solid PEG-b-P(Lys-TPE). Yield: 83.7%–85.1%.
Reduction of the PEG-b-P(Lys-TPE) block copolymer
PEG-b-P(Lys-TPE), NaBH4 (molar ratio of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–/NaBH4 = 1:8), and a trace amount of CH3OH were dissolved in dry DMF (10 mg mL−1) at 0 °C under N2. The reaction was continued for three days at room temperature, followed by precipitation into ether. Afterwards, the solids were washed three times with deionized water and freeze-dried to obtain a yellow solid H-PEG-b-P(Lys-TPE). Yield: 74.6%–79.8%.
N–/NaBH4 = 1:8), and a trace amount of CH3OH were dissolved in dry DMF (10 mg mL−1) at 0 °C under N2. The reaction was continued for three days at room temperature, followed by precipitation into ether. Afterwards, the solids were washed three times with deionized water and freeze-dried to obtain a yellow solid H-PEG-b-P(Lys-TPE). Yield: 74.6%–79.8%.
Preparation of the sample solution
The stock solution at a concentration of 1 mg mL−1 was first prepared by dissolving the given amount of samples in DMF/THF. Deionized water was then added to prepare the sample solution at a concentration of 0.1 mg mL−1 with the water content varying from 0% to 90% under stirring. Similarly, the polymer mixed solution containing THF and deionized water was prepared as described above. The aqueous solutions were prepared by dialysis against deionized water to remove the organic solvents. DMF/H2O (v/v = 9:1) mixture solutions with different pH values were prepared by adding 10% H2O into the stock DMF solution. Similarly, THF/H2O (v/v = 9:1) mixture solutions with different pH values were prepared by adding 10% H2O into the stock THF solution, followed by evaporating THF to yield a realistic pH value pHreal.
Results and discussion
The TPE-CHO monomer was first synthesized following the reported methods30 (Fig. S1 and Scheme S1†). The diblock copolymer poly(ethylene glycol)-block-poly(Nε-benzyloxycarbonyl-lysine) (PEG-b-PZLys) was synthesized by a ring-opening polymerization of Nε-benzyloxycarbonyl lysine N-carboxyanhydride (ZLys-NCA) using mPEG-NH2 (Mn = 5000 g mol−1) as a macroinitiator, as shown in Scheme 1.31 The 1H NMR spectra show that all peaks of the synthesized block copolymers are well assigned, confirming their chemical structures (Fig. S2a†). The DP of the polypeptide was determined to be 21 from the proton integral ratios of the benzyloxy groups to the methylene groups of PEG. Table S1† summarizes the molecular characteristics of the diblock copolymer PEG112-b-PZLys21. GPC shows a narrow molecular weight distribution with a dispersity (Đ) of 1.13 (Fig. S3†).
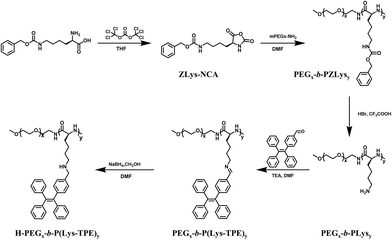 |
| | Scheme 1 Synthetic route to PEGx-b-PLysy and the post-modification procedures. The subscripts x and y represent the average DP of PEG and PLys, respectively. | |
The benzyloxycarbonyl protective groups of the diblock copolymers were removed to yield PEG-b-PLys. The chemical structure of the deprotected diblock copolymer PEG-b-PLys was confirmed by 1H NMR (Fig. S2b†). The disappearance of peaks at 7.23 and 5.12 ppm suggests complete debenzyloxycarbonylation. PEG-b-PLys was then post-modified with TPE-CHO to yield PEG-b-P(Lys-TPE). The 1H NMR spectra show the appearance of peaks at 7–8 ppm, suggesting the successful grafting with TPE (Fig. S2c†). A grafting rate of nearly 100% was determined by the proton integration of TPE to PEG. In addition, the disappearance of peaks at 8.13 ppm suggests quantitative reduction of the Schiff base groups to generate H-PEG-b-P(Lys-TPE) (Fig S2d†).
Self-assembly of PEG112-b-P(Lys-TPE)21 in DMF/H2O
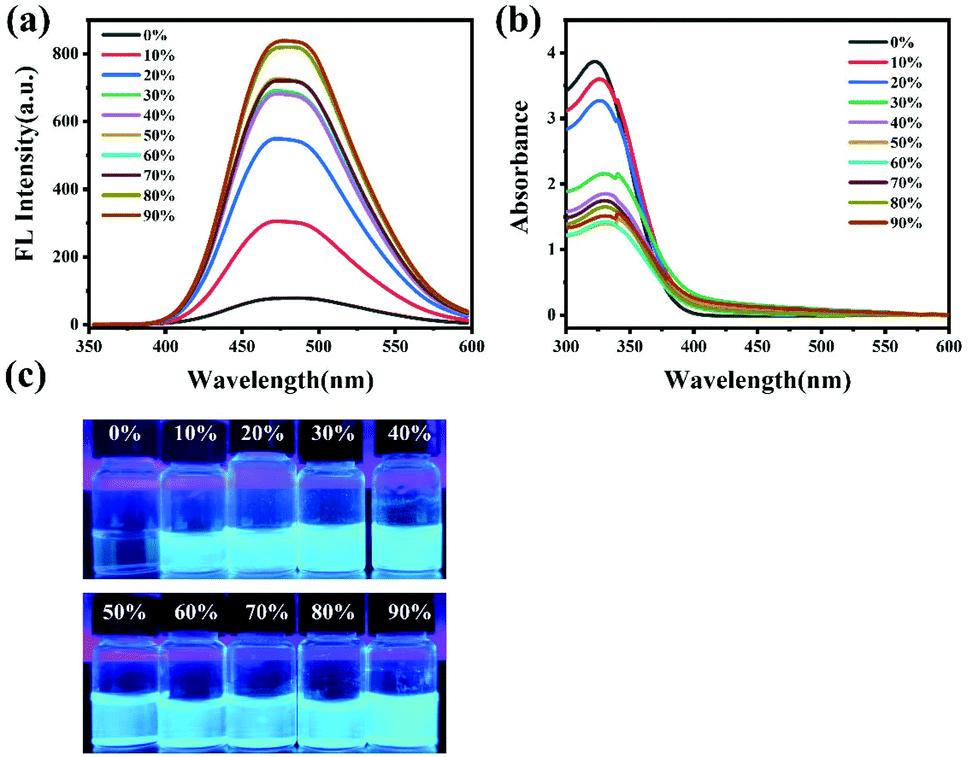
We first explored the self-assembly behavior of the copolymers by adding water into the DMF solution. As the volume ratio of water in the DMF solution of PEG112-b-P(Lys-TPE)21 increases from 0% to 90%, the solution appears opaque. This is because water is a good solvent for PEG and a poor solvent for the TPE residue, which results in the microphase separation of the amphiphiles as the water ratio increases. Simultaneously, the color of fluorescence emission changes from hyaline to blue, which is visible under UV-light (Fig. 1c), indicative of a typical AIE phenomenon. This is further confirmed from the fluorescence spectra (Fig. 1a). The fluorescence of PEG112-b-P(Lys-TPE)21 in DMF solution appears weak due to the quenching effect of aromatic compounds at high concentrations because of the free molecular motion. Upon addition of water to 10%, the fluorescence intensity of the solution increases obviously. Further increasing the water fraction results in a continuous increase of fluorescence intensity. This can be ascribed to the aggregation of hydrophobic TPE moieties during the process of assembly that restricts the dominant nonradiative relaxation, resulting in a fluorescence enhancement of AIE (Scheme 2). In addition, a visible emission peak at ∼480 nm is observed in the fluorescence spectra with the addition of water, which suggests the π–π interaction among TPE during the formation of PEG self-assemblies.32 UV-vis spectroscopy was also conducted to investigate the self-assembly (Fig. 1b). A peak at ∼330 nm decreases with increasing water content. We ascribe this to the assembly of the block copolymers. As expected, the mixed solution of H-PEG112-b-P(Lys-TPE)21 in DMF/H2O shows quite similar results, suggesting the AIE feature of the system (Fig. S4†). In this case, a visible increase in fluorescence intensity is observed as the water fraction reaches 30%, which is possibly because the reduced block copolymer shows enhanced solubility in DMF.
 |
| | Fig. 1 The fluorescence spectra (λex = 330 nm) (a), UV absorption spectra (b) and digital photos (c) under a hand-held UV-lamp (λmax = 365 nm) for PEG112-b-P(Lys-TPE)21 in H2O/DMF mixtures at a concentration of 0.1 mg mL−1 by changing the water fraction from 0% to 90%. | |
 |
| | Scheme 2 The pH-responsive AIE process assisted by the self-assembly of block copolymers. | |
Self-assembly of PEG112-b-P(Lys-TPE)21 in THF/H2O
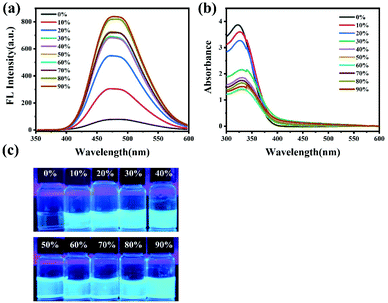
To further confirm the fluorescence mechanism, the self-assembly of PEG112-b-P(Lys-TPE)21 molecules in THF was also conducted. PEG112-b-P(Lys-TPE)21 in the THF/H2O mixed solution displays a similar variation in fluorescence properties with the addition of water. Upon addition of water to 10%, the fluorescence intensity of the solution increases obviously (Fig. S5a and S5c†). The photoluminescence further increases with the continuous addition of water, suggesting the AIE characteristics of PEG112-b-P(Lys-TPE)21.33 The UV-vis results show that a peak appears around 330 nm and decreases with increasing water content because of the formation of the assembly (Fig. S5b†). In spite of the similar tendency in fluorescence variation, the fluorescence intensity value of DMF/H2O (v/v = 1:9) mixed solution is nearly twice that of THF/H2O (v/v = 1:9) mixed solution under the same experimental conditions. This is possibly because the TPE molecules show an increased solubility in THF compared to DMF, considering the lower solubility of THF (δTHF = 18.6) than DMF (δDMF = 24.8). Thus, the presence of DMF may enable increased RIR influence on the assembly, resulting in an enhanced AIE-active fluorescence intensity.
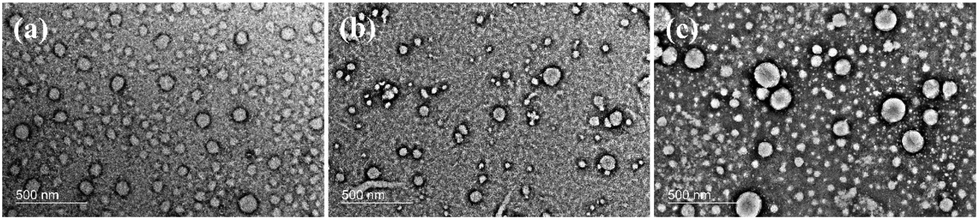
In addition, the difference in the fluorescence phenomena of copolymers in DMF/H2O and THF/H2O also possibly resulted from the different self-assembly behaviors of copolymers in two systems. To investigate the morphology of PEG112-b-P(Lys-TPE)21 self-assemblies, TEM and AFM were performed on the samples. After dialysis, the morphology of PEG112-b-P(Lys-TPE)21 from the DMF/H2O (v/v = 9:1) mixture solvent is observed to be spherical with a diameter of 42.4 ± 6.5 nm (Fig. 2a). AFM images further show a similar morphology with ∼52.5 nm diameter and ∼14.8 nm height. The comparable diameter and height suggest a micelle structure instead of a vesicle (Fig. S6†). With the increase of water fraction, the micellar morphology remains with an increased size (Fig. 2b and c). We further used dynamic light scattering (DLS) to study the average hydrodynamic diameter (Dh) of the micelles (Fig. S7a†). It is observed that Dh increases with increasing water fraction, consistent with the results of TEM. We demonstrate that this is because the Flory–Huggins parameter between the polypeptide and water increases as the water fraction increases, which results in an increase in the aggregation number of the micelles.34 Considering that the diblock copolymer is composed of hydrophilic PEG and hydrophobic P(Lys-TPE) segments, it is expected to form a core–shell structure in aqueous solution. However, in the case of the THF/H2O mixture solution, a hollow vesicle is observed with an apparent contrast between the central and edge parts by TEM (Fig. 3). The vesicular morphology with a diameter of ∼96.2 nm and a much smaller height of ∼6.1 nm is confirmed by AFM (Fig. S8†). The collapsed vesicular structure appears during the sample preparation process. Increasing the water fraction results in a larger size, similar to the results in the DMF/H2O mixture solution (Fig. 3b and c). We ascribe the formation of the spheres to the reduced solubility of TPE molecules in DMF, where the smaller dimension of the P(Lys-TPE) core enables the formation of spheres. In contrast, the stretching of the core-forming block leads to vesicles in THF.35 It is thus conceivable that the restricted chain mobility of P(Lys-TPE) in the spheres in DMF also results in an enhanced fluorescence intensity. Furthermore, the morphology of H-PEG112-b-P(Lys-TPE)21 is similar to that of PEG112-b-P(Lys-TPE)21. A spherical micellar morphology with a diameter of 28.2 ± 2.4 nm is observed from the DMF/H2O (v:v = 9:1) mixture solution. However, as the water content increases to 90%, the self-assembled morphology changes into connected short rods (Fig. S9†). This is possibly because of the increased hydrophobicity of the reduced copolymers, consistent with previous results.36
 |
| | Fig. 2 TEM images of PEG112-b-P(Lys-TPE)21 (0.1 mg mL−1) with a water fraction of 10% (a), 20% (b) and 90% (c) in DMF/H2O solution stained with uranyl acetate. | |
 |
| | Fig. 3 TEM images of PEG112-b-P(Lys-TPE)21 (0.1 mg mL−1) with a water fraction of 10% (a), 20% (b) and 90% (c) in THF/H2O solution stained with uranyl acetate. | |
pH responsiveness of PEG112-b-P(Lys-TPE)21 in DMF/THF and H2O mixtures
Considering the presence of the pH-sensitive imine group, the obtained assemblies are expected to show a pH-triggered transition. To investigate the pH-responsive behavior, the fluorescence intensity of PEG112-b-P(Lys-TPE)21 solution after the addition of aqueous solutions at different pH (vDMF:vH2O = 9:1, 0.1 mg mL−1) was determined by fluorescence spectroscopy. Fig. 4a shows that the fluorescence intensity decreases slightly as the pH reduces from 10.7 to 7.0. The addition of the solution at pH 1.4 significantly reduces the fluorescence intensity, which is because of the detachment of TPE groups on the side chain in acidic solution. To confirm this, the samples were collected and the chemical structure was investigated by 1H NMR (Fig. S10†). The disappearance of peaks at ∼6–8 ppm suggests that the TPE group on the side chain is completely removed. Remarkably, the fluorescence intensity of the mixed solution is recovered after adding an aqueous solution at a pH of 12.6 with stirring (Fig. 4b). The appearance of the typical TPE peaks shows ∼100% re-grafting efficiency, as indicated by 1H NMR (Fig. S11†). Similar behavior was observed in the THF/H2O mixed solution as well (Fig. S12†). It is known that the imine bond combining the polypeptide and TPE groups has a dynamic reversible character upon switching pH values.29 Thus, this can be explained by the fact that the detached TPE groups reconnect with the polypeptide backbone once the external environment is back to the initial state, which results in the recovery of the fluorescence intensity (Fig. 4b and S12b†).
 |
| | Fig. 4 The fluorescence spectra (λex = 330 nm) of PEG112-b-P(Lys-TPE)21 and TPE-CHO solutions with the addition of aqueous solution at different pH (vDMF:vH2O = 9:1, 0.1 mg mL−1) (a), the mixture with the addition of a solution at pH 1.4, followed by the addition of a solution at pH 12.6 with stirring (b), and digital photos under a hand-held UV-lamp (λmax = 365 nm) for PEG112-b-P(Lys-TPE)21 solutions with the addition of aqueous solution at different pH (vDMF:vH2O = 9:1, 0.1 mg mL−1) (c). | |
To confirm the pH-reversible properties, TEM was further performed to investigate the morphology of the assemblies. The TEM images of PEG112-b-P(Lys-TPE)21 solution after adding the solution at pH 1.4 show the absence of the assembly (Fig. 5a). As the alkaline solution was further added, a large amount of small spherical assemblies with a diameter of ∼40.4 nm was observed (Fig. 5b). This is similar to the sample of DMF/H2O (v:v = 9:1) solution prior to pH variation. The restored morphology confirms the reversible behavior of the fluorescence intensity. The AFM images further confirm the spherical morphology with a diameter of ∼63.6 nm and a comparable height of ∼31.3 nm (Fig. S13†). Similarly, in the THF/H2O mixture solution, no particular morphology is observed in the acidic environment (Fig. 6a). Neutralizing the acidic environment with an alkaline solution results in a vesicular morphology, confirmed by both TEM and AFM (Fig. 6b and S14†). This is also similar to the results of the THF/H2O (v:v = 9:1) mixture. Note that the pH value is determined to be ∼8.1 after THF evaporation, near the neutral pH range. We further used DLS to study the average Dh of the vesicles and micelles. In both cases, the DLS analysis of the assembly after successive acid and base treatments shows a similar Dh, confirming the reversible pH-responsiveness (Fig. S15†). In addition, the TPE-CHO molecules in the mixture after acid and base treatments show the absence of fluorescence signal (Fig. 4c). This result further confirms that the reversible pH-responsive behavior of the as-prepared polymers is assisted by the self-assembly of the polypeptide-based copolymers (Scheme 2). We also added a solution of pH 3.3 into the PEG112-b-P(Lys-TPE)21 solutions in DMF/THF and water mixtures (vDMF/THF:vH2O = 9:1, 0.1 mg mL−1), respectively. Similarly, both fluorescence intensities significantly decrease (Fig. S16†). Furthermore, the addition of a basic solution restores the fluorescence in both cases. Such reversible variation in fluorescence intensity is persuasive evidence for the AIE mechanism, where the changes of fluorescence properties are mainly due to the restriction of intramolecular motion during the assembly process.
 |
| | Fig. 5 TEM images for PEG112-b-P(Lys-TPE)21 solutions with the addition of an aqueous solution at pH 1.4 (vDMF:vH2O = 9:1, 0.1 mg mL−1) (a), followed by the addition of a solution at pH 12.6 with stirring (b). Both are stained with uranyl acetate. | |
 |
| | Fig. 6 TEM images for PEG112-b-P(Lys-TPE)21 solutions with the addition of an aqueous solution at pH 2.3 (vTHF:vH2O = 9:1, 0.1 mg mL−1) (a), followed by the addition of a solution at pH 11.7 with stirring (the solution pH value is determined to be ∼8.1 after THF evaporation) (b). Both are stained with uranyl acetate. | |
pH responsiveness of PEG112-b-P(Lys-TPE)21 in H2O
We further examined the reversible pH-responsive behavior in aqueous solution by evaporating THF. The addition of an acidic solution resulted in the absence of assemblies and significantly reduced the fluorescence intensity, similar to that in the presence of organic solvents. This indicates the removal of the TPE group from the polypeptides (Fig. 7). However, as the alkaline solution is continuously added to the acidic solution to achieve a pH of 11.4, the fluorescence intensity is not restored in the entire experimental range (Fig. 7b). This is in sharp contrast to the basic solution without the acid treatment (Fig. 7a). The TEM images also confirm this result by showing the absence of assembly under both acidic and basic pH conditions (Fig. S17†). We thus ascribe this to the lack of organic solvents. The detached TPE residues precipitate from the aqueous solution and can barely reconnect onto the polymer, which results in the irreversible pH-responsive behavior.
 |
| | Fig. 7 FL spectra (λex = 330 nm) for PEG112-b-P(Lys-TPE)21 aqueous solutions (0.1 mg mL−1) at different pH (a) and with pH adjusted from 7 to 3.5 and then to 11.4 (b). | |
Conclusions
In summary, a novel type of poly(ethylene glycol)-block-poly (L-lysine) block copolymer conjugated with TPE (PEG-b-P(Lys-TPE)) by a Schiff base reaction was prepared. Due to the presence of a pH-sensitive imine bond as a linker to combine the polypeptide and the TPE moiety, the obtained system shows reversible pH-responsive AIE characteristics in the presence of an organic solvent. We demonstrate that the variation in fluorescence intensity is generally dominated by the intramolecular motion of the TPE moieties assisted by the self-assembly of the block copolymer. This work provides a convenient and efficient method to construct AIEgens with stimuli-responsive properties, which show great potential for new generations of smart fluorescent nanomaterials.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (52073153 and 51722302) and the Natural Science Foundation of Shandong Province (no. ZR2019JQ17).
Notes and references
- R. Zhang, Y. Yuan, J. Liang, R. T. Kwok, Q. Zhu, G. Feng, J. Geng, B. Z. Tang and B. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 14302–14310 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- M. Noh, T. Kim, H. Lee, C.-K. Kim, S.-W. Joo and K. Lee, Colloids Surf., A, 2010, 359, 39–44 CrossRef CAS.
- J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- B. Z. Tang, X. Zhan, G. Yu, P. P. S. Lee, Y. Liu and D. Zhu, J. Mater. Chem., 2001, 11, 2974–2978 RSC.
- J. Chen, B. Xu, X. Ouyang, B. Z. Tang and Y. Cao, J. Phys. Chem. A, 2004, 108, 7522–7526 CrossRef CAS.
- Q. Zeng, Z. Li, Y. Dong, C. Di, A. Qin, Y. Hong, L. Ji, Z. Zhu, C. K. Jim, G. Yu, Q. Li, Z. Li, Y. Liu, J. Qin and B. Z. Tang, Chem. Commun., 2007, 70–72 RSC.
- M. Wang, G. Zhang, D. Zhang, D. Zhu and B. Z. Tang, J. Mater. Chem., 2010, 20, 1858–1867 RSC.
- Y. Hong, J. W. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- Y. Jiang and N. Hadjichristidis, Chin. J. Polym. Sci., 2019, 37, 930–935 CrossRef CAS.
- Y. Zhao, Y. Wu, S. Chen, H. Deng and X. Zhu, Macromolecules, 2018, 51, 5234–5244 CrossRef CAS.
- X.-F. Duan, J. Zeng, J.-W. Lü and Z.-B. Zhang, J. Org. Chem, 2006, 71, 9873–9876 CrossRef CAS PubMed.
- C. Zhu, S. Pang, J. Xu, L. Jia, F. Xu, J. Mei, A. Qin, J. Sun, J. Ji and B. Tang, Analyst, 2011, 136, 3343–3348 RSC.
- G. Liang, F. Ren, H. Gao, Q. Wu, F. Zhu and B. Z. Tang, Polym. Chem, 2016, 7, 5181–5187 RSC.
- W. Li, D. Huang, J. Wang, W. Shen, L. Chen, S. Yang, M. Zhu, B. Tang, G. Liang and Z. Xu, Polym. Chem, 2015, 6, 8194–8202 RSC.
- T. J. Deming, Prog. Polym. Sci., 2007, 32, 858–875 CrossRef CAS.
- F. Checot, J. Rodriguez-Hernandez, Y. Gnanou and S. Lecommandoux, Biomol. Eng, 2007, 24, 81–85 CrossRef CAS PubMed.
- H. Schlaad and M. Antonietti, Eur. Phys. J. E, 2003, 10, 17–23 CrossRef CAS PubMed.
- T. Koga, M. Matsuoka and N. Higashi, J. Am. Chem. Soc., 2005, 127, 17596–17597 CrossRef CAS PubMed.
- L. Fu, P. Yuan, Z. Ruan, L. Liu, T. Li and L. Yan, Polym. Chem., 2017, 8, 1028–1038 RSC.
- Z. Chen, Z. Ding, G. Zhang, L. Tian and X. Zhang, Molecules, 2018, 23, 1725–1736 CrossRef PubMed.
- H. Lu, J. Wang, Z. Song, L. Yin, Y. Zhang, H. Tang, C. Tu, Y. Lin and J. Cheng, Chem. Commun., 2014, 50, 139–155 RSC.
- W. Zhao, Y. Gnanou and N. Hadjichristidis, Biomacromolecules, 2015, 16, 1352–1357 CrossRef CAS PubMed.
- C. Ge, S. Liu, C. Liang, Y. Ling and H. Tang, Polym. Chem., 2016, 7, 5978–5987 RSC.
- G. Liu and C. M. Dong, Biomacromolecules, 2012, 13, 1573–1583 CrossRef CAS PubMed.
- P. Chen, M. Qiu, C. Deng, F. Meng, J. Zhang, R. Cheng and Z. Zhong, Biomacromolecules, 2015, 16, 1322–1330 CrossRef CAS PubMed.
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., 2003, 115, 4788–4791 CrossRef.
- C. Wang, G. Wang, Z. Wang and X. Zhang, Chem. – Eur. J., 2011, 17, 3322–3325 CrossRef CAS PubMed.
- M. T. Popescu, G. Liontos, A. Avgeropoulos, E. Voulgari, K. Avgoustakis and C. Tsitsilianis, ACS Appl. Mater. Interfaces, 2016, 8, 17539–17548 CrossRef CAS PubMed.
- Q. Wan, M. Liu, D. Xu, H. Huang, L. Mao, G. Zeng, F. Deng, X. Zhang and Y. Wei, J. Mater. Chem. B, 2016, 4, 4033–4039 RSC.
- J. Sun, P. Černoch, A. Völkel, Y. Wei, J. Ruokolainen and H. Schlaad, Macromolecules, 2016, 49, 5494–5501 CrossRef CAS.
- Y. Zhang, H. Xu, X. Ma, Z. Shi, J. Yin and X. Jiang, Macromol. Rapid Commun., 2016, 37, 998–1004 CrossRef CAS PubMed.
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- L. Zhang and A. Eisenberg, Macromolecules, 1999, 32, 2239–2249 CrossRef CAS.
- Y. Yu, L. Zhang and A. Eisenberg, Macromolecules, 1998, 31, 1144–1154 CrossRef CAS.
- Y. Ni, J. Sun, Y. Wei, X. Fu, C. Zhu and Z. Li, Biomacromolecules, 2017, 18, 3367–3374 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional test methods section, synthetic route, 1H NMR spectral data, DLS results, TEM images, AFM images, experimental photos and GPC data. See DOI: 10.1039/d1py00312g |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2021 |

 *ab and
Jing
Sun
*ab and
Jing
Sun