
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigher-order interfiber interactions in the self-assembly of benzene-1,3,5-tricarboxamide-based peptides in water†
Oleksandr
Zagorodko
 a,
Tetiana
Melnyk
a,
Olivier
Rogier
a,
Vicent J.
Nebot
*ab and
María J.
Vicent
*a
a,
Tetiana
Melnyk
a,
Olivier
Rogier
a,
Vicent J.
Nebot
*ab and
María J.
Vicent
*a
aPolymer Therapeutics Lab. Prince Felipe Research Center, Valencia, Spain. E-mail: vnebot@pts-polypeptides.com; mjvicent@cipf.es
bPTS SL, Valencia, Spain
First published on 28th May 2021
Abstract
Mimicking the complexity of biological systems with synthetic supramolecular materials requires a deep understanding of the relationship between the structure of the molecule and its self-assembly pattern. Herein, we report a series of water-soluble benzene-1,3,5-tricarboxamide-based di- and tripeptide derivatives modified with small non-bulky terminal amine salt to induce self-assembly into twisted one-dimensional higher-order nanofibers. The morphology of nanofibers strongly depends on the nature, order, and quantity of amino acids in the short peptide fragments and vary from simple cylindrical to complex helical. From observations of several fiber-splitting events, we detected interfiber interactions that always occur in a pairwise manner, which implies that the C3 symmetry of benzene-1,3,5-tricarboxamide-based molecules in higher-order fibers becomes gradually distorted, thus facilitating hydrophobic contact interactions between fibrils. The proposed mechanism of self-assembly through hydrophobic contact allowed the successful design of a compound with pH-responsive morphology, and may find use in the future development of complex hierarchical architectures with controlled functionality.
Introduction
The self-assembly of natural compounds is often a complex hierarchical process involving multiple levels of organization and accompanied by assembly-induced functionality.1 This complexity results from an intricate system of directional non-covalent interactions, either intermolecular, intramolecular, or with water molecules. Supramolecular interactions of higher orders, such as assembly of collagen fibrils, amyloids, and nucleic acids, are especially interesting and involve changes in the material's bulk properties.2–5 Mimicking such interactions in aqueous media with synthetic supramolecular systems remains a formidable task due to the complex and frequently counterintuitive self-assembly mechanisms.6In the last few decades multiple supramolecular systems with hierarchical self-assembly were reported, including coordination complexes,7–9 porphyrins,10 different peptides11–14 including di-and tri-peptides with different motifs;15,16 as well as synthetic17,18 and natural19,20 polymers. Multi-level control over self-assembly has paved the way for the development of novel advanced materials in medicine and industry;21–23 however, their complexity remains distant from that observed in nature. Therefore, a deeper understanding of the precise mechanisms and the subtle structural effects in self-assembly represent critical requirements in the design of said materials.
1,3,5-benzenetricarboxamide (BTA), one of the most well-known supramolecular motifs, can be derivatized with a range of substituents to tune self-assembly behavior under a broad range of conditions.24 BTA-based molecules that can assemble in an aqueous environment usually contain short hydrophobic peptide sequences or alkyl chains in the core and water-soluble residues at the termini.25–27 Studies have long-since reported the assembly of symmetric BTA-based compounds in aqueous media into defined nanofibers or nanorods due to the directional network of C3-symmetric hydrogen bonds. Recent studies have reported that, upon closer examination, at least some nanofibers in aqueous solutions comprise two smaller fibers, either twisted or parallel.28,29 Furthermore, several studies have reported the formation of more complicated structures (e.g., nanoribbons or triple helices) during the self-assembly of BTA-based molecules in organic solvents or their mixtures with water.30–32 In water, stepwise self-assembly into clearly defined double helical nanorods was reported only for BTA-based Au-containing metalloamphiphiles.33 Additionally, bundles with an order higher than double helices only formed from BTA-based molecules in aqueous solution as metastable intermediates.34 Discoveries of such higher-order interactions were mainly serendipitous and neither the precise mechanisms nor the approaches to specifically design analogous systems are known.
This study reports the hierarchical supramolecular self-assembly of a family of C3-symmetrical BTA derivatives of short di- and tripeptides in water. BTA-based molecules self-assembling in water are normally modified with bulky terminal hydrophilic substituents that increases the solubility of compounds yet limits the formation of higher-order fibers due to steric constraints. Replacing bulky substituents with sterically non-demanding terminal amines (in the form of hydrochloride salts) described herein has been shown to be sufficient for both solubilization and facilitated interfiber interactions thus leading to architecturally complex materials. Furthermore, direct observation of fiber splitting events sheds light on the mechanism of higher-order interactions and underscores the potential for the directional design of future BTA-based systems with complex molecular architecture specifically tailored for various applications.
Results and discussion
Terminology
In this study, one-dimensional (1D) fibers consisting of 2N fibrils will be classified according to their order N (e.g., zero-order fibers or fibrils [N = 0, a single stack of BTA-derivative molecules], first-order fibers [N = 1, two intertwined fibrils], second-order fibers [N = 2, two intertwined fibers each of which consists of two smaller fibrils], etc.). Other higher-order structures are named according to their morphology without defining the number of interacting fibrils (e.g., nanoribbons or nanosheets, etc.).Design and critical aggregation concentration screening
To study the structure-dependent appearance of higher-order interfiber interactions during self-assembly, we synthesized a family of compounds containing BTA cores with attached short sequences of hydrophobic amino acids and hydrophilic ethylenediamine (EDA) termini (Fig. 1A). EDA termini played a crucial role in the design of the molecule because terminal amine hydrochloride salts promote the solubility of the compounds but could not sufficiently shield the large hydrophobic part of the molecule, thus forcing interactions between 1D supramolecular stacks.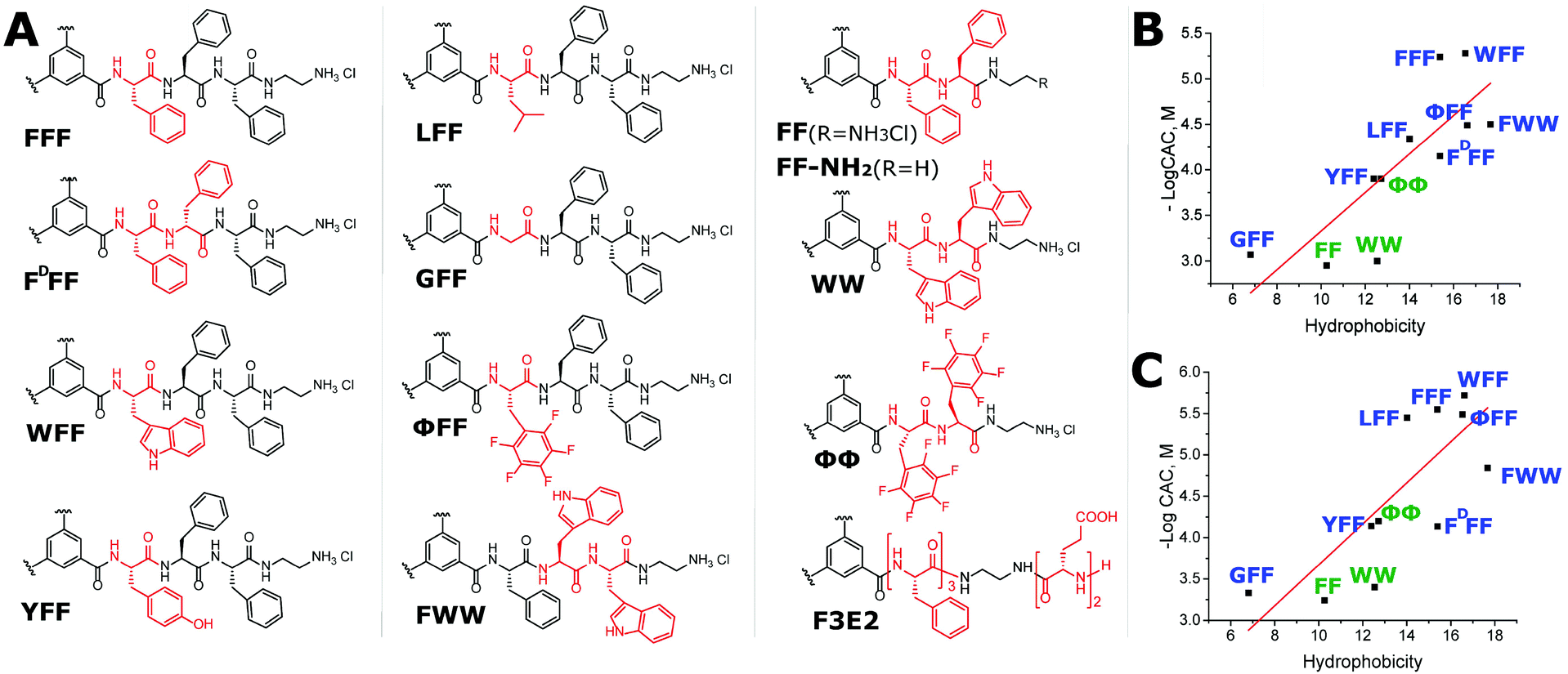 | ||
| Fig. 1 (A) General structure of BTA derivatives. Nomenclature of each compound represents the corresponding peptide sequence with one-letter coded L-amino acids. Pentafluorophenylalanine is assigned as Φ and D-Phenylalanine as DF. (B and C) Dependence of peptide hydrophobicity on critical aggregation concentration of the compounds in water (B) and 0.15 M NaCl (C). | ||
Compounds are named according to their amino acid sequences, represented as tripeptides XFF, (X = L-phenylalanine (F), D-phenylalanine (DF) tryptophan (W), leucine (L), glycine (G), tyrosine (Y), or pentafluorophenylalanine (Φ)), or FWW, and dipeptides FF, WW, and ΦΦ (Fig. 1A). Additionally, we prepared compound F3E2 (E = glutamic acid) with a pH-dependent number of terminal charges and higher chain flexibility, which will be discussed separately.
We determined the critical aggregation concentration (CAC) for each compound in pure water and 0.15 M NaCl solution mimicking physiological salt concentration. The self-assembly of BTA-based compounds in aqueous solutions is coupled with the formation of hydrophobic domains, which can be probed with solvatochromic dyes such as Nile Red (NR).24 For most compounds, a blue shift of the NR peak maximum from 647 nm to around 630 nm was observed after reaching CAC (see Table 1 for exact values). Stronger blue shifts were detected for WW, FWW, WFF, and YFF (607, 615, 626, and 605 nm, respectively), which signifies the formation of more hydrophobic domains.35
| Compounds | CAC in watera, mM | CAC in 150 mM NaCla, mM | λ max , nm | h , kcal mol−1 | Power lawc |
R
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
|
Amide I maxd | Amide II maxd |
|---|---|---|---|---|---|---|---|---|
| a Wavelength of the NR peak maximum after reaching CAC, as determined by fluorimetry. b Hydrophobicity calculated as h = − ΣiΔGi, where ΔGi is a free energy of transfer from water to n-octanol of an individual amino acid residue. c As determined by SAXS. d As determined by FTIR. e Calculated for 150 mM NaCl solutions. | ||||||||
| FF | 1.11 | 0.48 | 630 | 10.26 | −1e | 4.27e | — | — |
| WW | 1.02 | 0.31 | 607 | 12.54 | −1e | 3.92e | — | — |
| ΦΦ | 0.125 | 0.047 | 630 | 12.72 | −1e | 3.61e | — | — |
| FFF | 0.0058 | 0.0028 | 630 | 15.39 | −1 | 4.50 | 1637 | 1534 |
| LFF | 0.0451 | 0.0035 | 630 | 14.01 | −1 | 4.42 | 1638 | 1534 |
| GFF | 0.835 | 0.4671 | 630 | 6.81 | −2 | 7.84e | 1635 | 1532 |
| YFF | 0.125 | 0.071 | 605 | 12.39 | — | — | 1631 | 1544 |
| ΦFF | 0.032 | 0.0019 | 632 | 16.62 | −2 | 6.02 | 1639 | 1540 |
| WFF | 0.0052 | 0.0032 | 626 | 16.53 | −1 | 3.99 | 1636 | 1535 |
| FWW | 0.031 | 0.014 | 615 | 17.67 | −1.5 | 5.18 | 1638 | 1534 |
| FDFF | 0.087 | 0.073 | 630 | 15.39 | — | — | 1645 | 1546 |
Fluorimetry studies in water revealed that the CAC values of the compounds increased in the order WFF < FFF < FWW ∼ ΦFF < LFF < FDFF < YFF < GFF for the tripeptides and ΦΦ < WW < FF for the dipeptide derivatives. In 150 mM NaCl solution, the order was similar for the BTA-dipeptides and changed slightly for the BTA-tripeptides (ΦFF < FFF < WFF < LFF < FWW < YFF∼FDFF ≪ GFF). CAC values decreased in 150 mM NaCl solution for all compounds due to charge screening (see exact values in Table 1).
As BTA core and EDA termini represent conserved structural elements, differences in self-assembly arise exclusively from the interactions between amino acids. When the molecule assembles in a 1D stack, two sets of non-covalent interactions are expected to form between the short peptides, namely hydrogen bonds between amide groups (of BTA and peptides) and hydrophobic interactions between the amino acid residues. If this self-assembly mechanism occurs for all the compounds in a similar manner, CAC values should directly correlate only with residue hydrophobicity, as the number of amide interactions is conserved. We employed the established Wimley-White whole residue octanol scale to calculate hydrophobicity (h) (see exact values in Table 1).36 When plotting h vs. −logCAC, the −logCAC decreases in a manner close to linear upon an increase in total hydrophobicity both in water and in the presence of salt (Fig. 1B and C). Tryptophan-rich compounds deviate from the linear trend; however, it remains unclear if this deviation occurs due to the overestimation of the hydrophobicity values for using the octanol scale (for FWW).37
For most compounds, the CAC in the presence of salt was two to three times lower than in pure water except for LFF and ΦFF (more than ten times lower) and FDFF (1.2 times lower). In the case of FDFF, the hydrophobicity appears similar to FFF; however, three phenyl rings in FDFF cannot achieve co-planarity due to steric complications as a result of a chiral mismatch.38 Screening of Coulomb repulsions does not affect sterical barrier resulting in almost no effect of salt addition on CAC value of FDFF.
In the case of ΦFF, existing models of pentafluorophenyl and benzyl ring interaction suggest more efficient π–π stacking between a fluorinated and a non-fluorinated ring than between the corresponding couples.39 These preferential interactions result in a higher energy barrier for coplanar ring orientation and thereby inhibit supramolecular polymers. In the presence of salt, ΦFF possessed the lowest CAC value within the studied compounds; however, the slope of the curve used for CAC calculations was also the lowest for ΦFF (see ESI Fig. S1†), suggesting that the compound initially aggregated into clusters and supramolecular polymerization started at higher concentration.
The observed difference in CACs in water and salt solution of LFF is harder to explain. A study demonstrated that a Leu–Phe–Phe tripeptide tended to form more disordered aggregates due to steric hindrance,40 which may cause the high CAC of LFF in water. Coulomb screening apparently forces the molecule to planarity, although the mechanism remains unclear.
Self-assembly of BTA-tripeptide derivatives
As some BTA-tripeptide derivatives form small clusters on early stages of self-assembly, we performed measurements at concentrations 5–10 times higher than the CAC. TEM with phosphotungstic acid staining was employed to study the morphology of the nanosystems (Fig. 2A–H). FFF, WFF, LFF, and FWW assembled into thin, defined separated nanorods 30–500 nm long, and ΦFF formed fibers of various thicknesses (up to order 2 and 3). YFF displayed a limited ability to form fibers, and FDFF failed to form any defined structure. GFF formed very thick fibers with a diameter of 15 to 30 nm with several different morphologies simultaneously present (e.g., simple long and short fibers and intertwined higher-order fibers) (Fig. 2G). On several observed fiber-splitting events, fibers always split into two smaller ones. In one such event, double paired fiber splitting was detected (Fig. 2J). Detailed analysis of the images obtained for ΦFF and GFF revealed the presence of fibers with three distinct thicknesses (Fig. 2K and L) suggesting that the thickest fibers are of second or possibly third order (consists of 4 or 8 smaller fibers). | ||
| Fig. 2 TEM images of BTA-derivatives of tripeptides: FFF (A), WFF (B), LFF (C), YFF (D), ΦFF (E), FWW (F), GFF (G), and FDFF (H). Staining – phosphotungstic acid. Scale bars: 200 nm (white), 50 nm (black); SAXS profiles (I), sample concentration for all the measurements: 10× CAC. Example of GFF fibre splitting (J) with splitting points shown by white arrows. Scale bar 200 nm. Different fiber morphologies observed in GFF (K) and ΦFF (L) samples. Scale bar 50 nm. expansion of amide A, amide I and amide II regions on FTIR spectra of the compounds (M). | ||
Small-angle X-ray scattering (SAXS) studies (Fig. 2I) confirmed that all compounds (except YFF and FDFF) assembled into defined structures in solution, and corresponding Kratky plots (see ESI Fig. S2A†) confirmed the formation of defined aggregates, as seen from the bell-like shape of the curve at the low and intermediate q-range. Power law dependencies in medium q-range (Table 1 and Fig. 2I) for FFF, WFF, and LFF are close to −1, a value typical for cylindrical objects. P(r) pair distribution functions are rarely of practical use for supramolecular polymers as fiber lengths often exceed the maximum resolution of the machine; however, they can be used to extract Rc values. P(r) functions for FFF, WFF, and LFF had an appearance typical for cylindrical objects, with one defined maximum that matched the radius of cross-section (Rc) calculated using modified Guinier plot (see ESI Fig. S2B†). For FWW, GFF, and ΦFF, observed power law dependencies of −1.5, −2, and −2, respectively, suggest that these systems comprise fibers and/or fractal structures. The average Rc values for ΦFF and GFF were the highest among the whole group (6.02 and 7.84 nm, respectively). The P(r) function of FWW displayed one defined peak corresponding to the Rc; however, we failed to identify similar peaks for GFF and ΦFF (see ESI Fig. S2C†) due to high polydispersity in length and the presence of fibers with various diameters (as evident from TEM images). Overall, the Rc values obtained for all BTA-tripeptides derivatives exceeded the theoretically calculated radii of zero-order fibers (∼1.5 nm), suggesting first or second-order structure for observed fibers except for ΦFF and GFF fibers, which may attain third-order structure.
FTIR analyses was used to study hydrogen bonding between the molecules. We measured spectra for thin films formed from dried compound solutions in hexafluoroisopropanol/water mixture. The spectra for the BTA-tripeptide derivatives in amide A, amide I, and amide II regions represent a complex overlap of self-assembly sensitive or non-sensitive signals from distinct amides (from BTA, the peptide, and the ethylenediamine spacer) and characteristic peaks of terminal amine hydrochloride. Amides from non-aggregated BTA display peaks at 3306 cm−1, 1655 cm−1 (in the amide I region), and 1530 cm−1 (in amide II region), and the peaks attributed to hydrogen bonding between BTA molecules shift to 3240 cm−1, 1640 cm−1, and 1560 cm−1.41 Amide vibrations of peptides in beta-sheet conformations correspond to peaks at around 3220 cm−1, 1640 cm−1, and 1537 cm−1, while characteristic peaks corresponding to random coil conformation, while still debatable, have been reported at 1645–1660 cm−1.42 In amide I and amide II regions, FFF, WFF, LFF, and FWW displayed peak maxima at 1637 cm−1 and 1534–1535 cm−1. For ΦFF, GFF, and FDFF, both peaks became blue-shifted, while the amide I peak red-shifted and amide II blue-shifted for YFF (see Table 1 for exact values). Vibrations in the amide A and amide B regions are less sensitive to conformation and hydrogen bonding, and all spectra appear similar except for residue-specific peaks (e.g., tryptophan-containing compounds).
FTIR peak deconvolution was used to extract information regarding the ratio of amides participating in hydrogen bonding (ESI Fig. S3†). The peaks present at 1636–1638 cm−1 and 1534–1535 cm−1 suggest a beta-sheet conformation. A peak at 1660 cm−1, indicative of amides not involved in hydrogen bonding, represents the most pronounced difference between the spectra of all studied compounds. FDFF, GFF, and YFF display the highest peak intensity, emphasizing a higher amount of disordered aggregates or unimers, while FFF, WFF, and LFF display the lowest peak intensity, emphasizing the prevailing ordered structures. The almost identical vibration frequencies of BTA and peptide amides in both aggregated and disaggregated states, and a lower amount of BTA amides, prevents the unambiguous assignment of peaks belonging to BTA.
Circular dichroism studies for the BTA-tripeptide compounds were performed in water and 150 mM NaCl in small-volume cuvettes (approx. 35 μL) to minimize the influence of NaCl absorption. A previous study reported that upon BTA self-assembly, one or two peaks at around 215–220 nm and a lower intensity peak at 250 nm appear on the CD spectrum.43 The signal of beta-sheet conformation is also present in the 215–220 nm region. As each of the analyzed compounds possessed a distinct CD spectrum, we compared the spectra in water and 150 mM NaCl, as salt will likely induce self-assembly, and corresponding peaks will increase in intensity (Fig. 3). Of note, we observed a moderate decrease in intensity due to stronger absorption of light by the formed fibers (see UV spectra on ESI Fig. S4†). Peak positions, however, remained unchanged. Spectra of FFF, FDFF, LFF, ΦFF, GFF, and YFF exhibited four troughs at 195–196 nm (disorganized unimers), 204–206 nm (intrinsic signal of peptides containing FF dipeptide sequence overlapping with peaks of amide bonds between peptides), 214–220 nm (signal of BTA stacking overlapped with beta-sheet signals), and at around 250 nm (very weak, probably from BTA stacking). In the presence of salt, significant changes were observed only for LFF, ΦFF and GFF. The peak at 195 nm was less pronounced and almost disappeared in salt-containing solutions, which coincided with the appearance of a trough at 214–220 nm.
 | ||
| Fig. 3 CD spectra of BTA-derivatives of tripeptides in water (black solid line) and 0.15 M NaCl (red dotted line). Scaling value is shown in brackets. Sample concentration for all the measurements: 5 × CAC. | ||
The CD spectra for tryptophan-containing compounds WFF and FWW were substantially different from other compounds. The CD spectrum of FWW displayed one strong maximum and minimum at 231 and 213 nm, respectively, small minima at 195 nm and 275 nm, and a weakly defined peak at 205 nm. Strong intensity signals 231 and 213 nm are intrinsic to the molecule and remained constant upon the heating of diluted solutions of the compound (see ESI Fig. S5†), thereby confirming the origin of the peaks from exciton coupling between two tryptophans.44 The WFF spectrum exhibited five distinct minima and four maxima both in water and 150 mM NaCl due to the presence of tryptophan residues.45 Peaks corresponding to BTA stacking and beta-sheet conformation overlap with those peaks corresponding to electronic transitions in tryptophan and phenylalanine and cannot be directly identified, except for the conformationally-independent peak of tryptophan at 293 nm. Interestingly, we also observed a trough at 293 nm for FWW, but a peak for WFF, findings that provide evidence for the different orientation and solvent exposure of tryptophan chromophores in FWW and WFF.46
Strong CD signals observed for WFF and ΦFF and especially for FWW and FFF originate from exciton coupling47 between the aromatic residues; however, it is not exactly clear if coupling occurs between the residues on the same tripeptide or on different ones.
Finally, for FDFF, the presence of salt led to the disappearance of a low-intensity trough at 204 nm and induced the presence of a negative signal at 195 nm, confirming that the compound does not self-assemble into defined 1D supramolecular polymers. The spectrum of YFF remained almost unaltered in the presence of salt, demonstrating a strong trough at 195 nm and a small trough at 214 nm, confirming that this compound also remains predominantly disassembled.
While multiple peaks in different compounds have opposing signs (suggesting different interactions between residues), CD measurements support the formation of directional networks of non-covalent interactions from BTA and peptides in a beta-sheet-like conformation for FFF, WFF and FWW in both water and salt solution and for LFF, ΦFF and GFF in the presence of salt.
Self-assembly of BTA-dipeptide derivatives
Given the relatively high CAC values of FF and WW in water, we performed an analysis of BTA-dipeptide derivatives in 150 mM NaCl solution at a concentration four times exceeding the CAC. TEM revealed the formation of straight fibers with a uniform thickness of 8.2 ± 1 nm for FF (Fig. 4A) and fibers of varying thickness (11 ± 4 nm) with occasional helical twisting for WW (Fig. 4B). ΦΦ reproducibly formed a distinct type of fiber with a thickness of 9.1 ± 0.6 nm and a length of 50–500 nm that displayed a defined helicity with a full-twist length of around 50 nm (Fig. 4C). As in the case with BTA-tripeptide derivatives, the thickness of all the generated fibers far exceeded the theoretically calculated diameter of the molecules (approximately 3 nm). No splitting events were detected for any of the compounds. | ||
| Fig. 4 TEM images of BTA-derivatives of dipeptides FF (A), WW (B), and ΦΦ (C). Scale bars: 200 nm (white), 50 nm (black). SAXS profiles of BTA-derivatives of dipeptides in water (grey) and 150 mM NaCl (black) (D). Cryo-TEM images of FF-NH2 (E) and the magnification on selected regions (F). Scale bars: 500 nm (white) and 50 nm (black). | ||
SAXS analyses in water revealed almost complete disassembly of FF, the partial organization of WW with a power law dependence around −0.4, and the full assembly of ΦΦ with a power law dependence of around −1 (Fig. 4D). In 0.15 M NaCl, curves of all the compounds displayed power law dependence of −1, typical for cylindrical objects. Kratky plots confirmed the appearance of organized structures (see ESI Fig. S6†). P(r) functions for ΦΦ displayed oscillations with a 50 nm periodicity, similar to the results obtained by TEM (see ESI Fig. S7†). Nevertheless, oscillations in P(r) functions cannot be considered entirely conclusive for this polydisperse system. For WW, the P(r) function appeared similar to FWW and typical for rod-like objects. The Rc values obtained from modified Guinier plot decrease from 4.3 nm for FF to 3.9 nm for WW and 3.6 nm for ΦΦ following the increase in hydrophobicity of the amino acid residues. Upon increasing hydrophobicity, fibers become more tightly packed, which implies a minimal exposure of hydrophobic amino acid residues to the solvent in higher-order fibers.
Self-assembly of BTA-dipeptide derivative with a terminal amine group
While all the synthesized BTA di- and tri-peptide derivatives are insoluble in basic form, we serendipitously discovered that FF-NH2 with terminal amines (rather than hydrochlorides) formed gels after keeping the precipitate in water over two to four weeks. During this period, the liquid transformed into a transparent self-supporting gel (see ESI Fig. S8†) without a noticeable change in the quantity of the precipitate. Other compounds failed to form gels under similar conditions. Apparently, FF-NH2 has very low solubility in water and reaches the gelation point after an extended time via a process of slow solution saturation. This sample was used to study self-assembly in the absence of terminal charges.Cryo-TEM analysis demonstrated that the gelated FF-NH2 sample contained multiple helical nanofibers with a small portion of thinner fibers of two different thicknesses (Fig. 4E), and we detected multiple examples of large fibers splitting into thinner fibers (Fig. 4F). As in the case of GFF, only paired interactions were detected. Considering the presence of fibers with three different diameters, we suggest that the thickest FF-NH2 fibers have a third-order structure, similar to GFF and one order higher than FF with charged terminal amines.
Proposed mechanism of higher-order interfiber interactions
If we consider that all the studied derivatives described are mainly composed of hydrophobic amino acids with small hydrophilic termini, then applying the generally accepted mechanism of BTA self-assembly with the formation of a single stack would require the exposure of the majority of the hydrophobic residues to water that is a thermodynamically unfavorable. The formation of first or higher-order helical fibers through molecule distortion will significantly decrease the solvent exposure of hydrophobic residues (Fig. 5A). The high values of the fiber radius for the studied compounds, far exceeding those theoretically predicted for a single BTA stack, confirm this mechanism. Considering a report of the formation of first-order fibers (twisted and parallel) for BTA modified with alkyl chains and short PEG,28 solvent exposure minimization might play a pivotal role in the aqueous self-assembly of a broad range of BTA-based molecules. | ||
| Fig. 5 Schematic representation of the proposed mechanism of higher-order fiber formation. (A) Illustration of how a decrease in solvent-accessible area favors the formation of higher-order fibers with representative structures of the primary fibril and first and second-order fibrils. (B) Illustration of hydrophobic contact interactions responsible for stabilization of higher-order fibers. | ||
BTA-based molecules are traditionally depicted as perfectly C3-symmetrical units; however, analysis of the available crystalline BTA-based molecule structure provides evidence that even the amide groups of BTA do not necessarily retain C3 symmetry in three-dimensional space (relative to the BTA ring).48–52 The molecule can be distorted from flat and C3 symmetrical, which can be further enhanced by substituent flexibility; however, the hydrogen bond network between the BTA amide groups remains unperturbed.
Peptide chains themselves possess high levels of flexibility, with only three amino acids needed for a 180° turn in proteins (the so-called protein β-turn).53 Hydrophobic tripeptides are quite bulky for β-turn, and FTIR and CD data confirm that tripeptides in BTA derivatives are in a beta-sheet conformation (except FDFF and YFF). CD data and the different behavior of BTA-tripeptide derivatives in water and 0.15 M NaCl suggest that the mechanism of self-assembly and orientation of the residues in assembled state varies between compounds. We detected higher-order fibers in all the systems except for FDFF, suggesting that their formation mainly correlates to peptide flexibility and the formation of an unperturbed hydrogen bond network.
To gain insight into the mechanism of molecule organization in higher-order fibers, we considered that detected fiber splitting always occurs in a pairwise manner in FF-NH2 and GFF. The glycine spacer in GFF is non-bulky and provides higher flexibility to diphenylalanines, thus allowing a more efficient distribution of surface charge, which leads to the formation of very thick twisted fibers. Compared to FF, FF-NH2 possesses no surface charges, and Coulomb repulsions facilitate interactions between the fibers. It would be logical to conclude (i) that the fibrils in the bundle are held together by hydrophobic interactions between the outer phenylalanine/tryptophan residues and (ii) that charged amine hydrochloride groups remain on the surface of the fiber and do not interact with deeper layers of the hydrophobic core, thus excluding a hexagonal packing mechanism previously described for crystalline BTA derivatives.34
Beta-sheets in proteins can interact in various manners to form higher order supramolecular superstructures.54 While it remains challenging to identify the exact mechanism involved, we believe that hydrophobic contact interactions provide the most plausible explanation (see Fig. 5B). In proteins, superstructures formed through hydrophobic contact may have either C2 or C3 symmetry.54 We only detected C2 symmetric interactions for the systems described herein, which is probably caused by the limited redistribution of the surface charges. Indeed, every increase in fiber order would require the denser organization of surface charges (as in the case of ΦFF and GFF) or cause more steric constraints (as in the case of FF-NH2) (Fig. 5A). For the described systems, the highest fiber order is apparently limited to 3 or 4.
Presuming the validity of the described self-assembly mechanism, a further increase in hydrophobic chain size and/or the shifting of the charged group further from the core should facilitate molecular packing and lead to the formation of 2D supramolecular structures such as nanoribbons.
Nanoribbon formation
Hierarchical pairwise interfiber interaction implies that formation of a hydrophobic region during fiber formation occurs only on one side of the fiber rather than in several places simultaneously. A fiber with two interaction sites can form two-dimensional objects; however, that process would require much stronger distortion of the molecule to keep all the charges on the surface. In this case, an increase in the hydrophobic core diameter and distancing the charged group further from the core should prompt the formation of nanoribbon- or membrane-like objects. Initially, we prepared BTA-based tetraphenylalanine derivatives, but self-assembly studies failed due to compounds’ insolubility in water. To avoid this problem, we designed F3E2 bearing a FFF core with diglutamate termini.At basic pH, the ionization of both carboxylic groups should prevent higher order interfiber interactions due to strong Coulomb repulsions. At acidic pH, protonation only affects the terminal amine, thereby facilitating interfiber interactions. F3E2 remained soluble at both pH values, and NR fluorimetry demonstrated stable assembly with CAC values of 32 μM at pH 8.0 and 0.3 μM at pH 3.0.
TEM analysis of F3E2 at pH 8.0 demonstrated the formation of thin fibers (Fig. 6A), most probably consisting of double-helical fibers, similar to a previously reported compound with an FFF core and ten glutamic residues per arm.29 At pH 3.0, we observed various distinct morphologies, including separate fibers, parallel fiber doublets, short flat objects, and twisted nanoribbons consisting of multiple interacting fibers (Fig. 6B). We also observed multiple nanoribbon splitting events (the most representative example is depicted in Fig. 6C). Contrary to the pairwise splitting observed for GFF and FF-NH2, F3E2 surface split into at least six recognizable smaller fibers. These findings confirm that fibers interact with two neighboring molecules as shown in Fig. 6D, thus supporting the hypothesis that terminal charges remain on the fibers’ surface and, together with substituent flexibility and hydrophobicity, determine higher-order structure formation.
 | ||
| Fig. 6 TEM images of F3E2 at pH 8.0 (A) and pH 3.0 (B) with the magnification of a selected region (C). Scale bars: 200 nm (white), 50 nm (black). Proposed model of BTA molecule packing in F3E2 at different pH values (D). | ||
While confirmation or correction of the proposed mechanism of self-assembly requires additional studies on related compounds, we believe that our findings can already aid the design of BTA-based architectures with a complex hierarchical organization and tailored functionalities.
Materials and methods
Materials
All reagent grade chemicals were used without purification. Anhydrous N,N-dimethylformamide (DMF, ≥99.8% anhydrous), methanol (HPLC grade), and tetrahydrofuran anhydrous (99.8%) were purchased from Scharlab SL (Sentmenat, Spain). All protected amino acids were purchased from Iris Biotech.Synthesis of compounds
Detailed synthetic procedures for all the compounds are provided in ESI.†Solution preparation
A fixed amount of compound was weighed and transferred quantitatively to a 2 mL volumetric flask. Compounds were dissolved in water or 150 mM NaCl solution under mild shaking or sonication, and the volume of the solution was adjusted with the same solvent. Stock solutions were equilibrated at room temperature overnight. After dilution, samples were equilibrated for at least 3 h at room temperature. pH values of all solutions were in a range of pH 4.0 to 5.5.Nuclear magnetic resonance spectroscopy
NMR measurements were performed on a Bruker 300 UltrashieldTM (Bruker, Billerica, United States) or on Avance III 500 MHz Bruker (Bruker, Billerica, United States). All measurements were performed at room temperature, and spectra were processed and analyzed using TopSpin software.Circular dichroism spectroscopy
To measure the absorption of polarized light, the prepared solution was transferred to a quartz cuvette with a light path length of 1 cm or 0.1 mm and measured five times at 25 °C with a J-1500 spectrometer (JASCO corporation, Easton, United States). A nitrogen flow of 2.7 L min−1 was led through the spectrometer. CD spectra were measured in 0.1 mm cuvettes in water and 150 mM NaCl solution at concentration 5× CAC. Temperature variation spectra of FWW were measured in a cuvette with a light path length of 1 cm at concentration 0.03 mM in a temperature range from 10 to 80 °C and equilibrated for 15 min at each temperature.Fourier transform infrared spectroscopy
ATR-FTIR measurements were performed on an FT/IR-4700 (JASCO, Easton, United States) using a 400–4000 cm−1 range. In a typical experiment, one drop of a 5 mg mL−1 solution of the compound in water:hexafluoroisopropanol (1:1) was placed on the diamond crystal and allowed to dry for 20 min.
Fluorescent spectroscopy
Fluorescence spectroscopy measurements were performed on an FP-6500 spectrofluorimeter (JASCO, Easton, United States) with an excitation wavelength of 550 nm and measured emission spectrum in a range from 575 to 740 nm. Samples were dissolved in water or 0.15 M sodium chloride solution and allowed to equilibrate for 3 h. To 800 μL of each solution, 1 μL of 1 mg ml−1 NR solution in acetone was added, and the samples were additionally equilibrated for 30 min.Transmission electron microscopy and cryo-TEM
The TEM and cryo-TEM experiments were performed on the FEI Tecnai Spirit G2 transmission electron microscopy with a digital camera (Soft Image System, Morada, United States) and ITEMJ image capture software (Osaka, Japan). Samples for TEM were applied directly onto carbon film on 200 mesh copper grids. Excess of the sample was carefully removed by capillarity, and the grids were immediately stained with one drop of 0.1% phosphotungstic acid for 30 s. Excess stain was removed by capillary action. Samples for cryo-TEM were vitrified with a Vitrobot (ThermoFisher Scientific).Small-angle X-ray scattering
The SAXS measurements were performed at the BL11 beamline of the Alba synchrotron (Barcelona, Spain). The observed q range was 4.24 × 10–3 Å−1 ≤ q ≤ 3.52 Å−1, where q is the magnitude of the scattering vector q = (4π/λ)sin(θ/2). Measurements were performed in Hilgenberg Mark-tubes made of glass N50 packed with green perforated plastic disk (length 80 mm, outer diameter 1.5 mm, wall thickness 0.01 mm). All solutions were equilibrated for 24 h before measurement. Experiments were performed with the use of in-house cells and cell holders. Analysis and fitting were performed with ATSAS software packages. P(r) functions were extracted with GNOM.Mass spectrometry
MS analysis was performed using an AB Sciex QTRAP 4500 mass spectrometer (Sciex, Singapore) with electrospray ionization source. Sample was introduced via direct injection of 20 μL of 100 μg mL−1 solution. Analysis parameters: mobile phase 0.1% TFA in water:0.1% TFA in acetonitrile (1:1); flow: 0.4 mL min−1; capillary voltage: 3.5 kV; fragmentation voltage: 70 V; gas flow: 11 L min−1, nebulizer gas pressure: 15 psi; mode – positive.
Molecular radius calculation
Radius of fully stretched molecules was calculated using Gaussian software package.Conclusions
We studied the self-assembly of a family of soluble BTA derivatives of di- and tri-peptides with solubility provided by a single terminal protonated amino group. Amino groups are non-bulky and cannot entirely shield hydrophobic residues from water, and thus force the formation of higher-order fibers. In addition to the well-described formation of BTA-based nanofibers and recently reported double-helical fibers, we discovered that more complicated structures (higher-order fibers and nanoribbons) could form in an aqueous solution. While fiber diameter for most systems strongly exceeded the theoretically estimated size of molecules, we observed the formation of mixtures of fibers of different order for WW, ΦFF, GFF, and FF-NH2.The proposed mechanism of higher-order fiber formation suggests that: (1) fibers interact to minimize the exposure of hydrophobic groups to water, (2) molecules in bundles become distorted away from C3 symmetry to facilitate hydrophobic contact interactions, and (3) an increase in fiber order will lead to an increase in surface charges concentration and steric constraints until a critical value is reached that limits further interactions of the higher-order fibers.
Applying this mechanism to the design of BTA-based systems with controlled morphology, we successfully prepared a pH-responsive F3E2 that assembled into thin fibers at pH 8.0 and mixed complex morphologies such as nanoribbons at pH 3.0. Splitting of nanoribbons into smaller fibers did not occur in a pair-like manner as observed for BTA-tripeptides due to facilitated hydrophobic contact interactions.
Nanoribbons and higher-order helical fibers occurred only in a mixture of various morphologies, except compound ΦΦ, which only formed helical nanofibers. Due to the highly sensitive nature of the supramolecular polymerization, the possible attainment of a single morphology in a controlled manner will require more detailed studies regarding the role of subtle changes in polymerization conditions, such as solution preparation, temperature, time, and aging. Nevertheless, we anticipate that this study will facilitate and accelerate the future development of BTA-based advanced dynamic supramolecular materials with controlled hierarchical architecture and functionality.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Prof. Beatriu Escuder for fruitful discussions. The authors acknowledge Stuart P. Atkinson for English editing and E. Solano and M. Malfois from BL-11 NCD-SWEET beamline at ALBA Synchrotron for their support with SAXS experiments. Authors are especially grateful to Silvia Stifano for her valuable help with mass spectrometry experiments. The authors would like to thank the Spanish Ministerio de Economía y Competitividad (SAF2016-80427-R, PTQ-13-06465, and PID2019-108806RB-I00) and the H2020 European Research Council (grant ERC-CoG-2014-648831 MyNano) for financial support. TM is supported by an AECC Valencia Ph.D. grant. Part of the equipment employed in this work has been funded by Generalitat Valenciana and co-financed with FEDER funds (PO FEDER of Comunitat Valenciana, 2014–2020).Notes and references
- A. C. Mendes, E. T. Baran, R. L. Reis and H. S. Azevedo, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 582–612 CAS.
- J. Daban, FEBS Lett., 2020, 594, 395–411 CrossRef CAS PubMed.
- D. O. Sohutskay, T. J. Puls and S. L. Voytik-Harbin, Studies in Mechanobiology, Tissue Engineering and Biomaterials, in Multi-scale Extracellular Matrix Mechanics and Mechanobiology, ed. Y. Zhang, Springer International Publishing, Springer, Cham, 2020, vol. 23, pp. 203–245 Search PubMed.
- S. Zhu, Q. Yuan, T. Yin, J. You and Z. Gu, J. Mater. Chem. B, 2018, 6, 2650–2676 RSC.
- G. Wei, Z. Su, N. P. Reynolds, P. Arosio, I. W. Hamley and R. Mezzenga, Chem. Soc. Rev., 2017, 46, 4661–4708 RSC.
- A. A. Kornyshev, D. J. Lee, S. Leikin and A. Wynveen, Rev. Mod. Phys., 2007, 79, 943–996 CrossRef CAS.
- G. Huo, X. Shi, Q. Tu, Y. Hu, G. Wu, G. Yin, X. Li, L. Xu, H. Ding and H. Yang, J. Am. Chem. Soc., 2019, 141, 16014–16023 CrossRef CAS PubMed.
- C. Zhang, F. Wang, R. S. Patil, C. L. Barnes, T. Li and J. L. Atwood, Chem. – Eur. J., 2018, 24, 14335–14340 CrossRef CAS PubMed.
- B. Li, T. He, Y. Fan, X. Yuan, H. Qiu and S. Yin, ChemComm, 2019, 55, 8036–8059 RSC.
- S. Wang, W. Lin, X. Wang, T. Cen, H. Xie, J. Huang, B. Zhu, Z. Zhang, A. Song, J. Hao, J. Wu and S. Li, Nat. Commun., 2019, 1399 CrossRef PubMed.
- B. O. Okesola, Y. Wu, B. Derkus, S. Gani, D. Wu, D. Knani, D. K. Smith, D. J. Adams and A. Mata, Chem. Mater., 2019, 31, 7883–7897 CrossRef CAS PubMed.
- R. Martí-Centelles and B. Escuder, ChemNanoMat, 2018, 4, 796–800 CrossRef.
- J. Yang, J. Song, Q. Song, J. Y. Rho, D. H. Mansfield, S. C. L. Hall, M. Sambrook, F. Huang and S. Perrier, Angew. Chem., 2020, 59, 8860–8863 CrossRef CAS PubMed.
- L. E. R. O’Leary, J. A. Fallas, E. L. Bakota, M. K. Kang and J. D. Hartgerink, Nat. Chem., 2011, 3, 821–828 CrossRef PubMed.
- W. Liyanage and B. L. Nilsson, Langmuir, 2016, 32, 787–799 CrossRef CAS PubMed.
- S. Hsu and R. D. Chakravarthy, New J. Chem., 2018, 42, 4443–4449 RSC.
- K. Deepthi, R. R. B. Amal, V. R. Rajeev, K. N. N. Unni and E. B. Gowd, Macromolecules, 2019, 52, 2889–2899 CrossRef CAS.
- Y. Lu, J. Lin, L. Wang, L. Zhang and C. Cai, Chem. Rev., 2020, 120, 4111–4140 CrossRef CAS PubMed.
- H. Zhang, Y. Wang, H. Zhang, X. Liu, A. Lee, Q. Huang, F. Wang, J. Chao, H. Liu, J. Li, J. Shi, X. Zuo, L. Wang, L. Wang, X. Cao, C. Bustamante, Z. Tian and C. Fan, Nat. Commun., 2019, 10, 1006 CrossRef CAS PubMed.
- R. Freeman, M. Han, Z. Álvarez, J. A. Lewis, J. R. Wester, N. Stephanopoulos, M. T. McClendon, C. Lynsky, J. M. Godbe, H. Sangji, E. Luijten and S. I. Stupp, Science, 2018, 362, 808–813 CrossRef CAS PubMed.
- T. Shimizu, W. Ding and N. Kameta, Chem. Rev., 2020, 120, 2347–2407 CrossRef CAS PubMed.
- L. Chen and H. Yang, Acc. Chem. Res., 2018, 51, 2699–2710 CrossRef CAS PubMed.
- A. Restuccia, D. T. Seroski, K. L. Kelley, C. S. O. Bryan, J. J. Kurian, K. R. Knox, S. A. Farhadi, T. E. Angelini and G. A. Hudalla, Commun. Chem., 2019, 2, 53 CrossRef.
- S. Cantekin, F. A. de Greef and A. R. Palmans, Chem. Soc. Rev., 2012, 6125–6137 RSC.
- M. H. Bakker, C. C. Lee, E. W. Meijer, P. Y. W. Dankers and L. Albertazzi, ACS Nano, 2016, 1845–1852 CrossRef CAS PubMed.
- R. Appel, J. Fuchs, S. M. Tyrrell, P. A. Korevaar, M. C. A. Stuart, I. K. Voets, M. Schönhoff and P. Besenius, Chem. – Eur. J., 2015, 21, 19257–19264 CrossRef CAS PubMed.
- C. M. Berac, L. Zengerling, D. Straβburger, R. Otter, M. Urschbach and P. Besenius, Macromol. Rapid Commun., 2020, 41, 1900476 CrossRef CAS PubMed.
- R. P. M. Lafleur, S. Herziger, S. M. C. Schoenmakers, A. D. A. Keizer, J. Jahzerah, B. N. S. Thota, L. Su, P. H. H. Bomans, N. A. J. M. Sommerdijk, A. R. A. Palmans, R. Haag, H. Friedrich, C. Böttcher and E. W. Meijer, J. Am. Chem. Soc., 2020, 142, 17644–17652 CrossRef CAS PubMed.
- O. Zagorodko, V. J. Nebot and M. J. Vicent, Polym. Chem., 2020, 11, 1220–1229 RSC.
- Z. Shen, Y. Sang, T. Wang, J. Jiang, Y. Meng, Y. Jiang, K. Okuro, T. Aida and M. Liu, Nat. Commun., 2019, 10, 3976 CrossRef PubMed.
- Z. Shen, T. Wang and M. Liu, Angew. Chem., Int. Ed., 2014, 126, 13642–13646 CrossRef.
- A. R. A. Palmans, I. K. Voets and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 336–343 CrossRef PubMed.
- B. Kemper, L. Zengerling, D. Spitzer, R. Otter, T. Bauer and P. Besenius, J. Am. Chem. Soc., 2017, 140, 534–537 CrossRef PubMed.
- M. A. Vandenberg, J. K. Sahoo, L. Zou, W. Mccarthy and M. J. Webber, ACS Nano, 2020, 14, 5491–5505 CrossRef CAS PubMed.
- D. L. Sackett and J. Wolff, Anal. Biochem., 1987, 167, 228–234 CrossRef CAS PubMed.
- C. J. Pace and J. Gao, Acc. Chem. Res., 2013, 46, 907–915 CrossRef CAS PubMed.
- S. Marchesan, L. Waddington, C. D. Easton, D. A. Winkler, L. Goodall, J. S. Forsythe and P. G. Hartley, Nanoscale, 2012, 4, 6752–6760 RSC.
- A. M. Garcia, M. Melchionna, O. Bellotto, S. Kralj, S. Semeraro, E. Parisi, D. Iglesias, P. D'Andrea, R. De Zorzi, A. V. Vargiu and S. Marchesan, ACS Nano, 2021, 15, 3015–3025 CrossRef CAS PubMed.
- W. C. Wimley, T. P. Creamer and S. H. White, Biochemistry, 1996, 35, 5109–5124 CrossRef CAS PubMed.
- R. Wolfenden, J. Gen. Physiol., 2007, 127, 357–362 CrossRef PubMed.
- P. J. M. Stals, M. M. J. Smulders, R. Martín-Rapún, A. R. A. Palmans and E. W. Meijer, Eur. J. Chem., 2009, 15, 2071–2080 CrossRef CAS PubMed.
- D. Usoltsev, V. Sitnikova, A. Kajava and M. Uspenskaya, Biomolecules, 2019, 9, 1–17 CrossRef PubMed.
- X. Lou, R. P. M. Lafleur, C. M. A. Leenders, S. M. C. Schoenmakers, N. M. Matsumoto, M. B. Baker, J. L. J. van Dongen, A. R. A. Palmans and E. W. Meijer, Nat. Commun., 2017, 8, 15420 CrossRef PubMed.
- A. Roy, P. Bour and T. A. Keiderling, Chirality, 2009, 171, 163–171 CrossRef PubMed.
- E. Longo, R. Hussain and G. Siligardi, Int. J. Pharm., 2015, 480, 84–91 CrossRef CAS PubMed.
- T. Izumi and H. Inoue, J. Biochem., 1976, 79, 1309–1321 CrossRef CAS PubMed.
- C. Bortolini, L. Liu, S. V. Hoffmann, N. C. Jones, T. P. J. Knowles and M. Dong, ChemistrySelect, 2017, 2, 2476–2479 CrossRef CAS.
- L. Rajput and K. Biradha, J. Mol. Struct., 2008, 876, 339–343 CrossRef CAS.
- X. Hou, Z. Wang, M. Overby, A. Ugrinov, C. Oian, R. Singh and Q. R. Chu, Chem. Commun., 2014, 50, 5209–5211 RSC.
- A. D. Lynes, C. S. Hawes, K. Byrne, W. Schmitt and T. Gunnlaugsson, Dalton Trans., 2018, 47, 5259–5268 RSC.
- A. D. Lynes, C. S. Hawes, E. N. Ward, B. Haffner, M. Mobius, K. Byrne, W. Schmitt, R. Palb and T. Gunnlaugsson, CrystEngComm, 2017, 19, 1427–1438 RSC.
- G. Basuyaux, A. Desmarchelier, G. Gontard, N. Vanthuyne, J. Moussa, H. Amouri, M. Raynal and L. Bouteiller, Chem. Commun., 2019, 55, 8548–8551 RSC.
- A. M. C. Marcelino and L. M. Gierasch, Biopolymers, 2008, 89, 380–391 CrossRef CAS PubMed.
- P. Cheng, J. D. Pham and J. S. Nowick, J. Am. Chem. Soc., 2012, 135, 5477–5492 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00304f |
| This journal is © The Royal Society of Chemistry 2021 |