DOI:
10.1039/D1PY00290B
(Paper)
Polym. Chem., 2021,
12, 2832-2839
Physiologically relevant pH- and temperature-responsive polypeptide hydrogels with adhesive properties†
Received
3rd March 2021
, Accepted 12th April 2021
First published on 14th April 2021
Abstract
Physiologically relevant pH- and temperature-responsive polypeptide biomaterials have attracted increasing attention due to their versatile morphologies and functions responding to ambient changes. Herein, we prepare a series of polypeptide hydrogels via a ring-opening polymerization and post-modification strategy, which show tunable gelation temperature and pH, reversible sol–gel phase transition and adhesive properties. This approach is mainly based on the self-assembly of an amphiphilic polymer employing poly(ethylene glycol) and poly(L-glutamic acid) derivatives as temperature and pH responsive segments, respectively. We experimentally demonstrate that the temperature and pH responsiveness is a result of comprehensive effects of the variation of intermolecular interactions, electrostatic repulsion and secondary structures. The hydrogel is degradable and has good biocompatibility in vivo. Moreover, the hydrogel showed adhesive properties to a wide range of materials. This study provides a new class of stimuli-responsive hydrogels that respond to physiologically relevant pH and temperature and exhibit unique adhesive properties, as well as the underlying gelation mechanism.
Introduction
Hydrogel is a three-dimensional polymer network containing water, which has been widely explored as biomaterials owing to its soft property and hydrated form, similar to that of the native extracellular matrix.1–3 Stimuli responsive hydrogels have been extensively developed in order to realize different functions such as acting as sustained or triggered drug release platforms, facile cell collection systems and sensors.4–6 Smart hydrogels can respond to chemical and physical signals, including molecules, ions, pH, temperature, light, and magnetic/electric field, to name a few.7,8 Classic temperature-responsive hydrogels are considered excellent candidates for localized delivery in vivo.9,10 Specifically, injectable hydrogels based on temperature responsiveness have been developed as minimally invasive drug/cell delivery systems to decrease the hurt and pain to patients, as well as damage from shear to cells.11,12 Since injected polymer solutions rapidly undergo sol–gel transition at physiology temperature, the formed hydrogels can retain drugs or cells in situ and act as delivery depots, while their drug/cell release behavior can be adjusted by strength, mesh size, swelling or degradability of the hydrogels.13 Additionally, pH is an important stimulus that has received considerable attention in terms of stimuli-responsive systems for biomedical applications, especially in the physiologically relevant range (pH 5.5–7.4).14,15 Moreover, dual or multiple stimuli-responsive systems are advantageous in precise control of the functions of intelligent systems.16
In the last few decades, a series of temperature and pH dual stimuli-responsive hydrogels with different functions have been designed and prepared successively.17,18 Poly(N-isopropylacrylamide) (PNIPAAm), poly(N,N-diethylacrylamide (PDEAAm), poly(ethylene oxide) (PEO) and poly(propylene oxide) (PPO) are typical systems that allow for the introduction of ionizable group containing segments to prepare temperature and pH dual stimuli-responsive hydrogels.19–22 The block copolymers of poly(urethane amino sulfamethazine), poly(β-amino ester urethane) or polypeptide are also used for the preparation of dual responsive hydrogels.23–25 Compared to other synthetic hydrogels, polypeptide hydrogels utilize amino acid or amino acid derivates as building blocks, which endow these materials with enhanced biocompatibility, biodegradability, and unique secondary structures analogous to natural proteins.26–33 These unique properties and potential in the area of biomedical applications have triggered the development of polypeptide hydrogels. Deming and co-workers prepared a series of amphiphilic block copolypeptide hydrogels from lysine, leucine, valine and glutamic acid via the ring-opening polymerization (ROP) of α-amino acid-N-carboxyanhydride (NCA) using Co(PMe3)4 as the initiator.34 Additionally, gelation of the artificial protein hydrogel mainly depends on the amphiphilic nature and chain conformations of the polypeptides. NCA-derived polypeptide-based hydrogels were further expanded to multiple stimuli-responsive hydrogels. Li and co-workers developed thermal- and pH-responsive hydrogels, which employed poly(ethylene glycol)-b-poly(L-glutamate)s as the backbone and glutamic acid as a pH responsive group.35 Oligo(ethylene glycol) (OEG) was introduced into the side chain to provide temperature responsive characteristics. Both pH- and temperature-responsive properties could be adjusted by the degree of polymerization of the polypeptide blocks. Due to the low pKa (4.1) of glutamic acid residues and the insufficient hydrophobicity of OEG side chains, the pH of sol–gel transition was below 4.4, and the sol–gel transition temperature was above 40 °C, which limits their applications under physiological conditions.14 Lee and co-workers designed pH- and temperature-responsive hydrogels composed of poly(ethylene glycol) (PEG), poly(γ-benzyl-L-glutamate) (PBLG) and oligo(sulfamethazine) (OSM).36 The polypeptide block copolymers underwent sol–gel transition at physiologically relevant temperature. The OSM segment responds to pH over 7.6, owing to its relatively high pKa (8.3). At pH 8.3, sulfonamide groups underwent ionization, which further caused gel–sol transition. Hao and co-workers synthesized polypeptide hydrogels based on triblock copolymers methoxy-poly(ethylene glycol)-b-poly(L-lysine)-b-poly(L-valine).37 Less pH-dependence on sol–gel transition was observed between pH 5.5 and 7.5, due to the higher pKa (10.5) of lysine residues.14 Physiologically relevant pH was critical to maintaining the bioactivity of bioactive molecules and cells. Therefore, the development of physiologically relevant pH- and temperature-responsive polypeptide hydrogels remains a challenge. Thus, we propose the synthesis of stimuli-responsive hydrogels utilizing copolymers of PEG and poly(L-glutamate) containing hydrophobic and ionizable groups that respond to stimuli under physiological conditions.
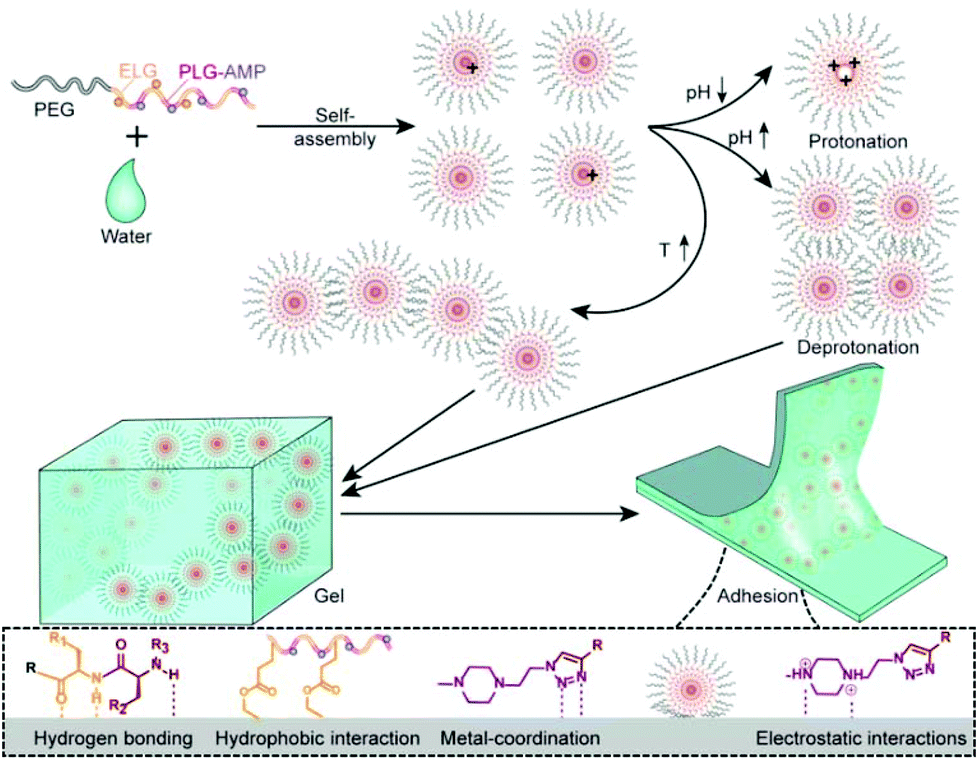
In this study, we developed a new series of physiologically relevant pH- and temperature-responsive polypeptide hydrogels with rapid sol–gel transition behaviors and adhesive properties via a ROP and post-modification strategy (Scheme 1). It is known that the 1,2,3-triazole ring and N,N′-substituted piperazine are generally protonation and pH-sensitive sites.38 Thus, we modified the polypeptide with 1-(2-azidoethyl)-4-methylpiperazine (AMP) via “click” chemistry of alkynyl and azide, generating two kinds of pH-responsive groups. Different experiments were conducted in which the composition, degree of polymerization (DP) and block structure of polypeptide segments were varied to synthesize a series of polypeptide-containing amphiphiles (PPCAs). As expected, PPCAs possess excellent gelation capability. The gelation temperature ranged from 10 to 70 °C, and gelation concentration reached as low as 1.5% (w/v). Meanwhile, the PPCA hydrogels underwent reversible sol–gel transition in response to the pH change. Moreover, the prepared hydrogels exhibit good biocompatibility and biodegradation, and could adhere to various materials.
 |
| | Scheme 1 Synthesis routes of EG45(ExPk)m (A) and EG45(ExPAy)m (B). | |
Results and discussion
Design and synthesis of polypeptide-containing amphiphiles
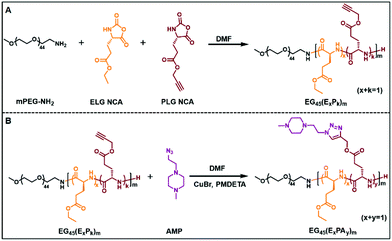
PPCAs were synthesized via a ROP and post-modification strategy as described in Schemes 1, S1 and S2.† In order to obtain PPCAs, methoxy poly(ethylene glycol)-block-poly((γ-ethyl-L-glutamate)-co-(γ-propargyl-L-glutamate)) (mPEG-b-P(ELG-co-PLG)) and P(ELG-co-PLG)-b-PEG-b-P(ELG-co-PLG) were first prepared via the ROP of ELG NCA and PLG NCA using mPEG-NH2 or NH2-PEG-NH2 as the macro-initiator, according to previously reported procedures with some modifications.39–41 Then, piperazine-containing AMP was introduced into the polymer by “click” chemistry between the azido group of AMP and the alkynyl group of PLG, providing PPCAs with a pH responsive capacity. The prepared diblock and triblock copolymers were abbreviated as EGn(ExPAy)m and (ExPAy)mEGn(ExPAy)m, respectively, where E and PA represent ELG and AMP-grafted PLG residues, as well as n and m are DPs of PEG and polypeptide blocks, and x and y are the molar fractions of the amino acid residues in the polypeptide block. ELG, PLG, ELG NCA and PLG NCA were proved by 1H NMR (Fig. S1 to S4†). The structure of AMP was confirmed by 13C NMR and ESI-MS (ESI+) as shown in Fig. S5 and S6.† In order to explore the effects of the PPCA block structure on gelation, mPEG-NH2 and NH2-PEG-NH2 were used as initiators producing a series of diblock and triblock copolymers. The 1H NMR spectrum of PEG and polypeptide segments clearly showed the appropriate peaks (Fig. S7†). The DP of polypeptide segments and contents of ELG and PLG residues were calculated through comparison of the integration ratio of PEG (δ, 3.64–3.92 ppm, –CH2–CH2–) to ELG (δ, 1.18 ppm, –CH3) and PLG (δ, 2.36 ppm, –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), respectively. After the “click” reaction, the grafting ratio of AMP was quantified by comparing the integration ratio of ELG (δ, 1.18 ppm, –CH3) or mPEG (δ, 3.44 ppm, –O–CH3) to AMP (δ, 5.13–5.32 ppm, two –CH2– adjacent the triazole ring) (Fig. S8†). The details of various polymers are listed in Tables S1 and S2.†
CH), respectively. After the “click” reaction, the grafting ratio of AMP was quantified by comparing the integration ratio of ELG (δ, 1.18 ppm, –CH3) or mPEG (δ, 3.44 ppm, –O–CH3) to AMP (δ, 5.13–5.32 ppm, two –CH2– adjacent the triazole ring) (Fig. S8†). The details of various polymers are listed in Tables S1 and S2.†
Thermal and pH response
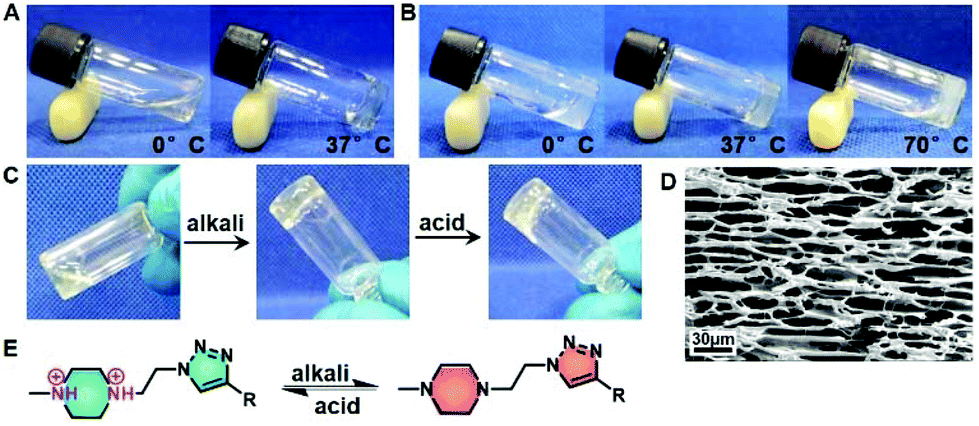
Amphiphilic block polymers tend to self-assemble into various nanostructures when dissolved in thermodynamically good solvents for one or more blocks but not for others.42,43 Once reaching the critical concentration, they show phase transition in macrography.44 Previous studies have characterized the gelation of amphiphilic block polymers containing PEG and hydrophobic blocks.45,46 Our study found that PPCAs possessed superior gelation capability. EG45(E1/2PA1/2)20 and EG45(E3/4PA1/4)20 solutions in PBS at relatively low concentrations (3% (w/v) and 1.8% (w/v), respectively) both gelled within 5 min at 37 °C. According to the photos shown in Fig. 1A and B, EG45(E1/2PA1/2)20 solution is limpid and has better solubility in water than EG45(E3/4PA1/4)20 at 0 °C and pH 7.4. We also found the solubility of EG45(E1/2PA1/2)16 and EG45(E1/2PA1/2)12 is better than EG45(E3/4PA1/4)16 and EG45(E2/3PA1/3)12. This phenomenon can be attributed to the protonated PA's stronger hydrophilicity compared to ELG. The results of the acid–base titration also verified the stronger buffering ability of EG45(E1/2PA1/2)20 than EG45(E3/4PA1/4)20 (Fig. S9†). During the heating process, EG45(E3/4PA1/4)20 dehydrated easily, while EG45(E1/2PA1/2)20 showed no syneresis. In addition to hydrophilicity, this may be due to the triazole ring which enhanced self-assembly interactions. Furthermore, the SEM image shown in Fig. 1D indicates that the hydrogel has a porous structure.
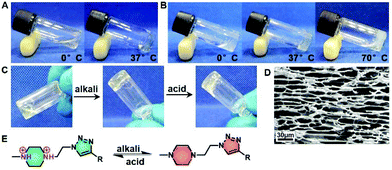 |
| | Fig. 1 Thermal responsive sol–gel behavior of EG45(E1/2PA1/2)20 (3% (w/v)) (A) and EG45(E3/4PA1/4)20 (1.8% (w/v)) (B). (C) Rapid reversible sol–gel behavior of 300 μL of 3% (w/v) EG45(E1/2PA1/2)20 in PBS, after addition of a drop of 5 M NaOH or HCl. (D) SEM image of 3% (w/v) EG45(E1/2PA1/2)20 hydrogels. Scale bar: 30 μm. (E) Deprotonation and protonation of 1-methyl-4-ethyl-piperazine (MEPP) in response to pH change. | |
The N,N′-substituted piperazine and triazole ring are common H-bond acceptors in a neutral environment, and can adjust the polymer's hydrophilicity via protonation and deprotonation.47,48 Hydrophilic/hydrophobic balance is a key factor in the gelation of amphiphilic block copolymers, associated with gelation concentration, temperature and hydrogel strength. As shown in Fig. 1C, EG45(E1/2PA1/2)20 solution at approx. pH 6 rapidly converts to a hydrogel after the addition of a small amount of alkali liquor. Upon addition of acid, the hydrogel quickly reverted to a sol status after shaking slightly. The fast pH-responsive reversible sol–gel transition of PPCAs may be associated with the protonation and deprotonation sequence (Fig. 1E).
Sol–gel transition
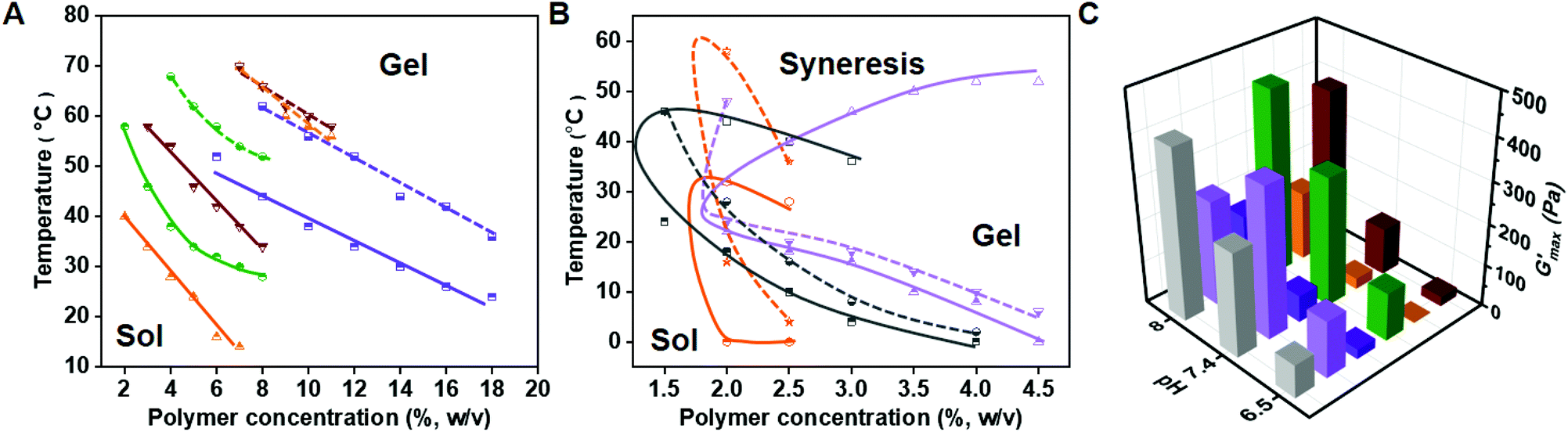
To investigate the gelation behavior of PPCA solutions in PBS, its sol–gel transition at different pH values was examined using the inversion tube method (Fig. 2A and B). As the critical gelation concentrations (CGCs) of EG45(E1/3PA2/3)12, EG45(E1/4PA3/4)16 and EG45(E1/4PA3/4)20 were above 20% (w/v), we focused on the other samples, (EG45(E1/2PA1/2)12, EG45(E2/3PA1/3)12, EG45(E1/2PA1/2)16, EG45(E3/4PA1/4)16, EG45(E1/2PA1/2)20, EG45(E3/4PA1/4)20 and (E1/2PA1/2)10EG45(E1/2PA1/2)10). By comparing the CGC and critical gelation temperature (CGT) of PPACs with the same polypeptide length in Fig. 2A and B, we observed that the CGC and CGT increase with increasing the PA molar content in the polypeptide segments, which is probably due to an increase in hydrophilicity. As shown in Fig. 2B, the hydrogels of PPCAs with a 25% or 33.3% molar PA residue content in the polypeptide block undergoes syneresis at pH 7.4 during the programmed temperature increase, while the syneresis is relieved at pH 6.5, and even the EG45(E2/3PA1/3)12 hydrogel shows no syneresis at pH 6.5 at elevated temperatures up to 70 °C. In contrast, no syneresis is observed for all PPCA hydrogels with a 50% molar PA residue content, upon increasing the temperature to 70 °C at both pH 7.4 or 6.5 (Fig. 2A). Obviously, PPCA hydrogels with higher PA contents have better water-retaining capability at elevated temperatures. Additionally, the CGC and CGT of PPCAs decrease with an increase in pH. For PPCAs with a 50% molar PA residue content in the polypeptide block, the CGC and CGT decrease upon increasing the DP of the polypeptide block. For PPCAs with a 25% molar PA residue content in the polypeptide block, the CGT decreases with an increase in the DP of the polypeptide block. The pH effect on gelation increases with an increase in the DP of the polypeptide. By comparing EG45(E1/2PA1/2)20 and (E1/2PA1/2)10EG45(E1/2PA1/2)10, it was discovered that the diblock copolymer showed a lower CGC and CGT than the triblock copolymer at pH 7.4, which may be attributed to the difference in intermolecular interactions between the polypeptide segments. Overall, PPCAs can undergo sol–gel phase transition at the physiologically relevant temperature and at pH 6.5 and 7.4.
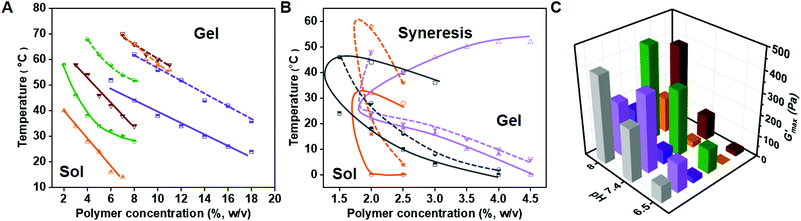 |
| | Fig. 2 Phase diagrams of PPCAs with 50% (A) and 25% or 33.3% (B) PA molar contents in the polypeptide blocks, measured by the test tube inversion method. (A) Purple line, EG45(E1/2PA1/2)12; green line, EG45(E1/2PA1/2)16; orange line, EG45(E1/2PA1/2)20; and red line, (E1/2PA1/2)10EG45(E1/2PA1/2)10. (B) Grey line, EG45(E2/3PA1/3)12; light purple line, EG45(E3/4PA1/4)16; and orange line, EG45(E3/4PA1/4)20. The dotted line and solid line represent the sol–gel transition at pH 6.5 and 7.4, respectively. (C) G′max of PPCA hydrogels, defined as the maximum value in the test from 4 °C to 60 °C; grey, EG45(E2/3PA1/3)12, 3% (w/v); light purple, EG45(E3/4PA1/4)16, 3% (w/v); purple, EG45(E1/2PA1/2)12, 9% (w/v); green, EG45(E1/2PA1/2)16, 9% (w/v); orange, EG45(E1/2PA1/2)20, 9% (w/v); and red, (E1/2PA1/2)10EG45(E1/2PA1/2)10, 9% (w/v). | |
Rheological properties
The mechanical property of hydrogels is a fundamental property for biomedical applications. Due to its thermal and pH responsiveness, we studied the rheological properties of the hydrogel as a function of temperature at different pH values. Based on the obtained results from the sol–gel phase diagram, we tested the rheological properties of 9% (w/v) hydrogels of PPCAs with a 50% PA molar content and 3% (w/v) hydrogels of PPCAs with a 25% or 33.3% PA molar content (Fig. 2C and S10†). Firstly, the max storage moduli (G′max) of PPCA hydrogels were compared between PPCAs with the same polypeptide chain length. Although the concentration of the EG45(E2/3PA1/3)12 hydrogel is a third of the EG45(E1/2PA1/2)12 hydrogel, G′max of the EG45(E2/3PA1/3)12 hydrogel is more than 2-fold that of the EG45(E1/2PA1/2)12 hydrogel. However, G′max of the EG45(E3/4PA1/4)16 hydrogel at pH 8 is weaker than that of EG45(E1/2PA1/2)16 due to syneresis in the EG45(E3/4PA1/4)16 hydrogel at 45 °C. Therefore, the molar fraction of PA in the polypeptide block affects G′max. The higher PA molar content may lead to lower G′max at a given DP of the polypeptide and pH. With an increase in the DP of the polypeptide block containing 50% molar content of PA from 12 to 20, G′max of the PPCA hydrogels firstly increases and then decreases. Additionally, G′max of the (E1/2PA1/2)10EG45(E1/2PA1/2)10 hydrogel is higher than that of EG45(E1/2PA1/2)20. G′max of the PPCA hydrogels gradually increases upon increasing the pH from pH 6.5 to 8 except for the EG45(E3/4PA1/4)16 hydrogel. Overall, G′max values of the tested PPCA hydrogels are in the range of 2–445 Pa, which is dependent on the composition of PPCAs and influenced by environmental stimuli.
Mechanism of hydrogel formation
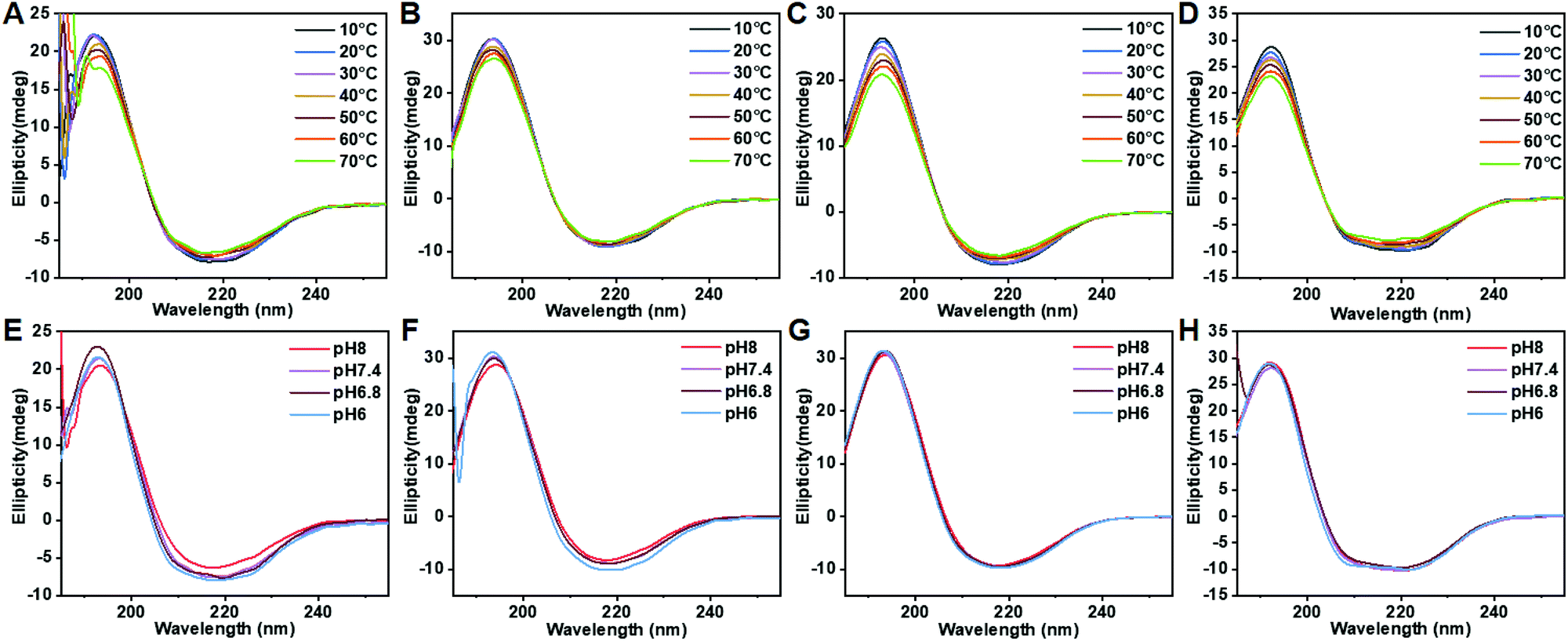
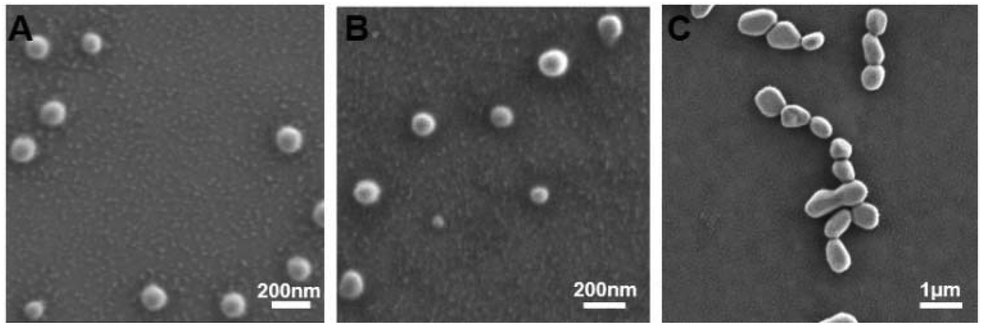
Polypeptide conformation was first studied by circular dichroism (CD) as a function of composition, temperature and pH. As shown in Fig. 3 and S11,† PPCAs mainly adopt the β-sheet conformation. In addition, some typical absorptions for α-helix conformation in (E1/2PA1/2)10EG45(E1/2PA1/2)10 besides the β-sheet were observed. The formation of α-helix conformation is associated with steric hindrance and electric charge of side chains. The secondary structure of PPCAs shows less dependence on pH from pH 6 to pH 8, and the absorption is slightly weakened with an increase in temperature. This reveals that protonation shows only slight influence on the secondary conformation of PPCAs. Additionally, the H-bonding interaction is slightly destroyed with an increase in temperature, which leads to a slight decrease of β-sheets at high temperatures. The formation of β-sheet conformation is beneficial for gelation and this dramatically decreases the CGC of PPCAs as described in the segment of sol–gel transition. The conformation of the polypeptide is associated with the core stability of the micelle in aqueous solution. The micelle structure was determined by the pyrene-probe fluorescence method. The critical micelle concentration (CMC) of EG45(E1/2PA1/2)12 is about 0.0245 mg mL−1 (Fig. S12†). SEM images also show individual micelles with sizes of around 100–150 nm for EG45(E1/2PA1/2)12 and EG45(E1/2PA1/2)16 (Fig. 4A and B). Additionally, the self-assembly of EG45(E1/2PA1/2)16 tends to fuse into larger, irregular particles at the micrometer level as the temperature increases to 55 °C (Fig. 4C). We further employed 13C NMR to explore the gelation process as a function of temperature at pH 6.5 and 7.4. The signals of PEG (about 70 ppm) gradually decrease with an increase in temperature (Fig. S13†). This accounts for the collapse of micelles, which also results in the reduction of signals of the polypeptide backbone and 1-methyl-4-ethyl-piperazine (MEPP) side group. Meanwhile, the signals of MEPP decrease more rapidly at pH 7.4 compared to that at pH 6.5. Thus, increasing the temperature and pH benefited the fusion or interaction of micelles and promoted gelation. We predicted that the PPCA hydrogel was obtained after self-assembly of PPCAs, micelle fusion and entanglement under the stimulation of increasing temperature and pH values (Scheme 2).
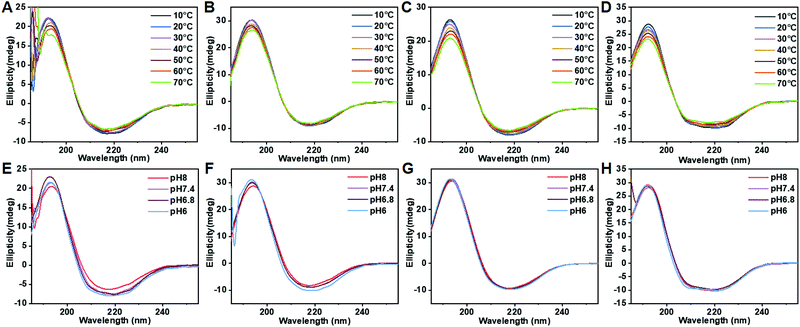 |
| | Fig. 3 Circle dichroism (CD) spectra of PPCAs (0.2 mg mL−1) as a function of temperature (A–D) and pH (E–H). (A) and (E), EG45(E1/2PA1/2)12; (B) and (F), EG45(E1/2PA1/2)16; (C) and (G), EG45(E1/2PA1/2)20; and (D) and (H), (E1/2PA1/2)10EG45(E1/2PA1/2)10. | |
 |
| | Fig. 4 SEM photographs of EG45(E1/2PA1/2)12 at 25 °C (A), and EG45(E1/2PA1/2)16 at 25 °C (B) and 55 °C (C). | |
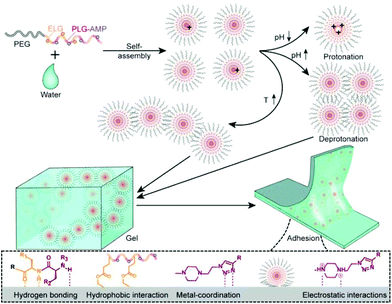 |
| | Scheme 2 Illustration of the gelation and adhesion mechanism of PPCA hydrogels. | |
Biocompatibility and degradation
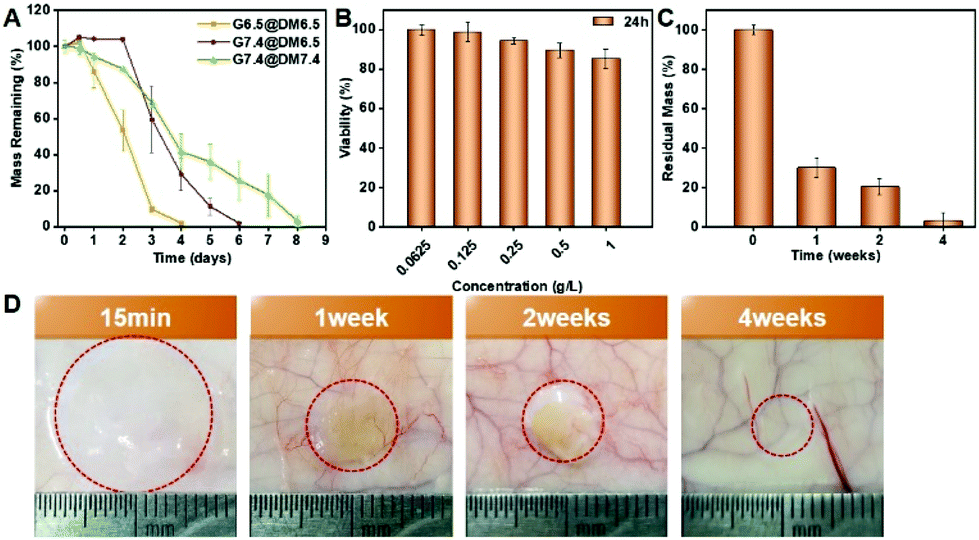
We firstly evaluated the degradation of the PPCA hydrogel in vitro, using 3% (w/v) EG45(E2/3PA1/3)12, 9% (w/v) EG45(E1/2PA1/2)12 and 3% (w/v) EG45(E3/4PA1/4)16 hydrogels as examples. In the degradation medium of PBS (pH 7.4), EG45(E2/3PA1/3)12, EG45(E1/2PA1/2)12 and EG45(E3/4PA1/4)16 hydrogels that were prepared at pH 7.4 completely degraded within 3 days, 13 days and 9 days, respectively (Fig. S14† and Fig. 5A). Compared to the 3% (w/v) EG45(E3/4PA1/4)16 hydrogel, the 3% (w/v) EG45(E2/3PA1/3)12 hydrogel with a shorter polypeptide block and fewer ELG residues shows a faster degradation rate. Additionally, although G′max of the 3% (w/v) EG45(E3/4PA1/4)16 hydrogel is higher than that of the 9% (w/v) EG45(E1/2PA1/2)12 hydrogel (Fig. 2C), the degradation rate of the 3% (w/v) EG45(E3/4PA1/4)16 hydrogel is faster than that of the 9% (w/v) EG45(E1/2PA1/2)12 hydrogel. As the degradation rate of the EG45(E3/4PA1/4)16 hydrogel is moderate, we further investigated the degradation behaviors of EG45(E3/4PA1/4)16 hydrogels that were prepared at various pH values and incubated in different degradation media. As shown in Fig. 5A, the degradation rate of G7.4 in medium at pH 7.4 is slower than that at pH 6.5. This is because the osmotic pressure between the hydrogel and degradation medium causes hydrogel swelling and promotes hydrogel erosion. The weaker strength of the hydrogel at pH 6.5 accelerates hydrogel degradation. Fig. 5B shows that PPCAs have no cytotoxicity toward B16F10 cells at the tested concentrations. We further tested the hydrogel degradation in the subcutaneous layer of rats in vivo, and evaluated their biocompatibility. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Jilin University and approved by the Animal Ethics Committee of Changchun Institute of Applied Chemistry, CAS. The rats were sacrificed at given time intervals (15 min, 1 week, 2 weeks, and 4 weeks) after subcutaneous injection of EG45(E3/4PA1/4)16 solution. The hydrogel is clearly observed at 15 min post-injection, which verifies the sol–gel phase transition in vivo. The hydrogel shows over 60% of mass loss in the first week and almost complete degradation after 4 weeks. The hematoxylin–eosin staining (H&E) images of skin tissues surrounding the hydrogels show mild acute inflammation in the first 2 weeks, which gradually decreases as the hydrogel degrades (Fig. S15†). Overall, there is no obvious or long-term damage to the surrounding tissue and the hydrogel can be tolerated by the tissue. Therefore, the biodegradable PPCA hydrogel exhibits good biocompatibility in vivo.
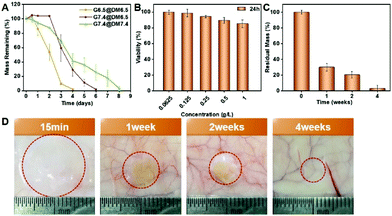 |
| | Fig. 5 (A) Degradation of the EG45(E3/4PA1/4)16 hydrogel in vitro (n = 3). The hydrogels that were prepared at pH 6.5 and 7.4 are defined as G6.5 and G7.4, respectively. The degradation media (PBS) at pH 6.5 and 7.4 are defined as DM6.5 and DM7.4, respectively. (B) Cell cytotoxicity of PPACs (n = 5). (C) Residual hydrogel masses at various time intervals in vivo (n = 3). (D) Representative photograms of hydrogels at various time intervals in vivo. EG45(E3/4PA1/4)16 was used for the tests in (A)–(D), and the polymer concentration was 3% (w/v) for the hydrogel samples in (A), (C) and (D). | |
Adhesion properties
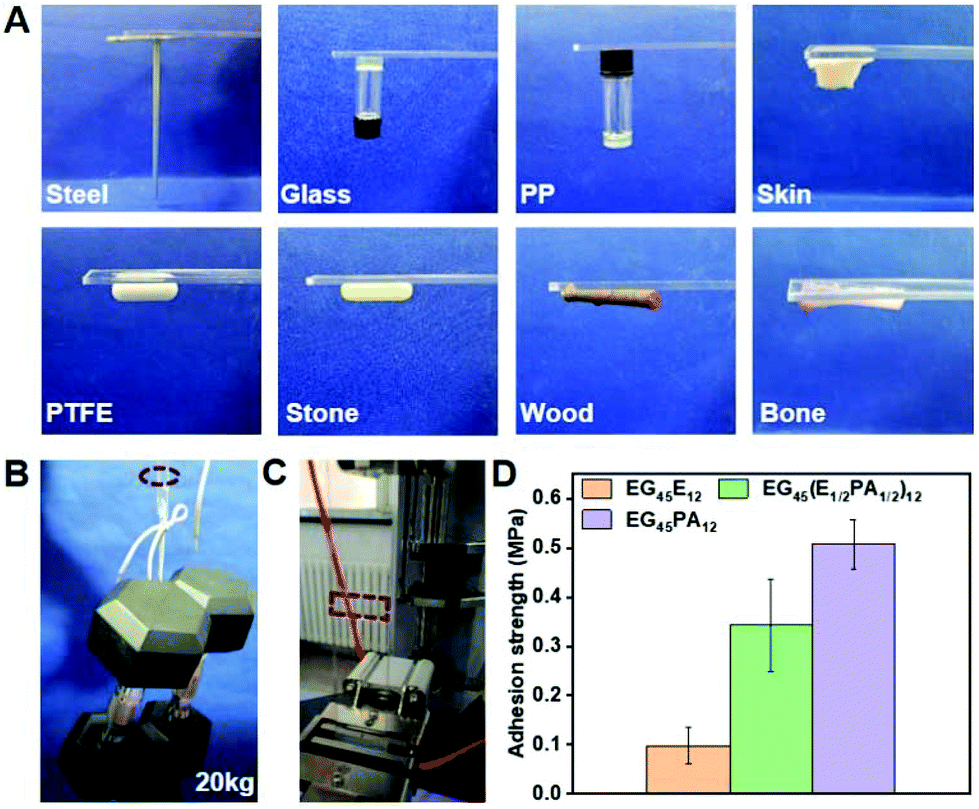
Since the polypeptide and MEPP groups provide the feasibility for forming H-bonding between copolymer chains and adhesive interfaces, we examined the adhesive properties of PPCAs on different materials.49 As shown in Fig. 6A, firm adhesion can be formed between the hydrogels and steel, glass, polypropylene, Teflon, stone, wood, bone and skin. The adhesive property is derived from the special structure of PPCAs. The carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of the polypeptide, the nitrogen–hydrogen group (–NH–) of the polypeptide and protonated MEPP can form hydrogen bonding with the hydroxyl (–OH) or amine (NH2) group in the materials. The ethyl group (–CH2CH3) of ELG, deprotonated MEPP and 1,2,3-triazole ring are easy to form hydrophobic interactions with the hydrophobic materials. The nitrogen–nitrogen double bond (–NN–) of 1,2,3-triazole ring can form metal-coordination with metal. After protonation, MEPP can form electrostatic interactions with the adhesion interface. Thus, the adhesive property is a result of comprehensive effects of hydrogen bonding, hydrophobic interaction, metal-coordination, electrostatic interactions, and so on (Scheme 2). The hydrogels of EG45(E1/2PA1/2)12 and EG45PA12 both firmly adhere to the interfaces of PMMA, which load at least 20 kg, and the adhesion areas are 25 × 20 mm (Fig. 6B and Video S1†). We further explored the adhesive property of PPCAs with comparable polypeptide chain lengths. As shown in Fig. 6D, EG45E12 and EG45PA12 hydrogels have the lowest and strongest adhesion strengths, respectively, with EG45(E1/2PA1/2)12 hydrogels located in the middle. Hence, we propose that both the triazole ring and MEPP groups are favorable factors to create interface adhesion. This may explain why the degradation rate of the 3% (w/v) EG45(E3/4PA1/4)16 hydrogel is faster than that of the 9% (w/v) EG45(E1/2PA1/2)12 hydrogel. The PA content of EG45(E1/2PA1/2)12 is higher than that of EG45(E3/4PA1/4)16. Adhesion is a comprehensive result of the interactions between the adhesive and interface as well as the strength of the adhesive itself.50 The adhesive strength at pH 8 is stronger than that at pH 7.4 (Fig. S16†), which may be caused by the increased strength of the hydrogel (Fig. 2C), and the hydrophobic interaction between the hydrogel and interface. Therefore, the adhesive capability of PPCAs can be adjusted by varying the polypeptide composition and pH. Combined with the advantages of good biocompatibility and degradation, the adhesive PPCA hydrogels show potential in drug delivery systems, wound dressings and adhesive sensors.
O) of the polypeptide, the nitrogen–hydrogen group (–NH–) of the polypeptide and protonated MEPP can form hydrogen bonding with the hydroxyl (–OH) or amine (NH2) group in the materials. The ethyl group (–CH2CH3) of ELG, deprotonated MEPP and 1,2,3-triazole ring are easy to form hydrophobic interactions with the hydrophobic materials. The nitrogen–nitrogen double bond (–NN–) of 1,2,3-triazole ring can form metal-coordination with metal. After protonation, MEPP can form electrostatic interactions with the adhesion interface. Thus, the adhesive property is a result of comprehensive effects of hydrogen bonding, hydrophobic interaction, metal-coordination, electrostatic interactions, and so on (Scheme 2). The hydrogels of EG45(E1/2PA1/2)12 and EG45PA12 both firmly adhere to the interfaces of PMMA, which load at least 20 kg, and the adhesion areas are 25 × 20 mm (Fig. 6B and Video S1†). We further explored the adhesive property of PPCAs with comparable polypeptide chain lengths. As shown in Fig. 6D, EG45E12 and EG45PA12 hydrogels have the lowest and strongest adhesion strengths, respectively, with EG45(E1/2PA1/2)12 hydrogels located in the middle. Hence, we propose that both the triazole ring and MEPP groups are favorable factors to create interface adhesion. This may explain why the degradation rate of the 3% (w/v) EG45(E3/4PA1/4)16 hydrogel is faster than that of the 9% (w/v) EG45(E1/2PA1/2)12 hydrogel. The PA content of EG45(E1/2PA1/2)12 is higher than that of EG45(E3/4PA1/4)16. Adhesion is a comprehensive result of the interactions between the adhesive and interface as well as the strength of the adhesive itself.50 The adhesive strength at pH 8 is stronger than that at pH 7.4 (Fig. S16†), which may be caused by the increased strength of the hydrogel (Fig. 2C), and the hydrophobic interaction between the hydrogel and interface. Therefore, the adhesive capability of PPCAs can be adjusted by varying the polypeptide composition and pH. Combined with the advantages of good biocompatibility and degradation, the adhesive PPCA hydrogels show potential in drug delivery systems, wound dressings and adhesive sensors.
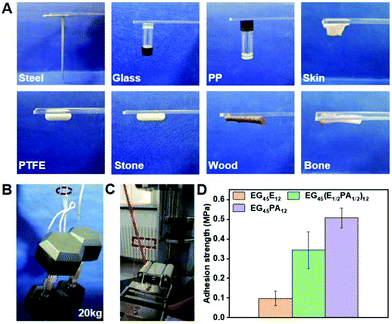 |
| | Fig. 6 Adhesive properties of PPACs. (A) Adhesive of EG45(E1/2PA1/2)12 (9% (w/v)) to various substrates. (B) Macroscopic adhesion tests of EG45(E1/2PA1/2)12 (9% (w/v)) to the PMMA platelet; the adhesion areas were 25 × 20 mm and 20 mg EG45(E1/2PA1/2)12 solution was added. (C) Image of the tensile test. (D) Adhesion strength of PPCAs with the same polypeptide chain length to the PMMA platelet, and the concentration was unified at 9% (w/v). | |
Conclusions
In summary, we successfully prepared a series of pH and temperature dual responsive PEG-polypeptide block copolymer hydrogels with adhesive properties. The sol–gel transition temperatures ranging from 10 °C to 70 °C at pH 7.4 can be adjusted by the concentration and composition of PPCAs. The gelation temperature and storage modulus of the PPCA hydrogels were markedly affected by changing the pH from 6.5 to 7.4. Additionally, the hydrogels showed reversible sol–gel transitions in response to a single stimulus of pH change. Thus, customized hydrogels that respond to physiologically relevant pH and temperature can be realized using our system. Subsequently, we confirmed the good biocompatibility and biodegradability of the PPCA hydrogels in vitro and in vivo. In addition, the polypeptide hydrogels exhibited unique adhesive properties, and the adhesion strength was dependent on the molar fractions of the triazole ring and N,N′-substituted piperazine groups, and pH. Therefore, this study provides a new strategy for the construction of physiologically relevant stimuli-responsive hydrogels with unique functions, such as adhesive properties.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (projects 51973218, 51833010, and 51622307) and the Youth Innovation Promotion Association, CAS.
References
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18–23 CrossRef.
- C. D. Spicer, Polym. Chem., 2020, 11, 184–219 RSC.
- Y. Rong, Z. Zhang, C. He and X. Chen, Sci. China: Chem., 2020, 63, 1100–1111 CrossRef CAS.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS.
- C. D. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- D.-L. Liu, X. Chang and C.-M. Dong, Chem. Commun., 2013, 49, 1229–1231 RSC.
- S. H. Lee and H. Shin, Adv. Drug Delivery Rev., 2007, 59, 339–359 CrossRef CAS PubMed.
- Y. Gao and C. M. Dong, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1067–1077 CrossRef CAS.
- B. Jeong, Y. Bae, D. Lee and S. Kim, Nature, 1997, 388, 860–862 CrossRef CAS PubMed.
- M. Park, M. Joo, B. Choi and B. Jeong, Acc. Chem. Res., 2011, 45, 424–433 CrossRef PubMed.
- C. He, S. W. Kim and D. S. Lee, J. Controlled Release, 2008, 127, 189–207 CrossRef CAS PubMed.
- L. Yu and J. D. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- X. Zhang, L. Cheng, L. Feng, Y. Peng, Z. Zhou, G. Yin, W. Li and A. Zhang, Polym. Chem., 2019, 10, 2305–2315 RSC.
- E. S. Gil and S. M. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
- X. Yin, A. S. Hoffman and P. S. Stayton, Biomacromolecules, 2006, 7, 1381–1385 CrossRef CAS PubMed.
- K. S. Soppimath, D. C. W. Tan and Y. Y. Yang, Adv. Mater., 2005, 17, 318–323 CrossRef CAS.
- N. Singh and D. Lee, J. Controlled Release, 2014, 193, 214–227 CrossRef CAS PubMed.
- W. Shim, J. Yoo, Y. Bae and D. Lee, Biomacromolecules, 2005, 6, 2930–2934 CrossRef CAS PubMed.
- W. Wu, W. Li, L. Q. Wang, K. Tu and W. Sun, Polym. Int., 2010, 55, 513–519 CrossRef.
- J. Kim, S. Lee, S. Kim and Y. Lee, Polymer, 2002, 43, 7549–7558 CrossRef CAS.
- B. Guo and Q. Gao, Carbohydr. Res., 2007, 342, 2416–2422 CrossRef CAS PubMed.
- J. Zhang, L. Y. Chu, Y. K. Li and Y. M. Lee, Polymer, 2007, 48, 1718–1728 CrossRef CAS.
- C. Huynh, M. Nguyen and D. Lee, Acta Biomater., 2011, 7, 3123–3130 CrossRef CAS PubMed.
- T. Le, H. Duong, T. Thambi, V. Phan, J. Jeong and D. Lee, Biomacromolecules, 2018, 19, 3536–3548 CrossRef PubMed.
- Y. Li, H. Yang and D. Lee, J. Controlled Release, 2020, 330, 151–160 CrossRef PubMed.
- F. Gelain, Z. Luo and S. Zhang, Chem. Rev., 2020, 120, 13434–13460 CrossRef CAS PubMed.
- F. Raza, H. Zafar, Y. Zhu, Y. Ren, U. Aftab, A. Khan, X. He, H. Han, M. Aquib, K. BoakyeYiadom and L. Ge, Pharmaceutics, 2018, 10, 16–37 CrossRef PubMed.
- X. Zhou and Z. Li, Adv. Healthcare Mater., 2018, 7, 1800020–1800037 CrossRef PubMed.
- E. Liarou, S. Varlas, D. Skoulas, C. Tsimblouli, E. Sereti, K. Dimas and H. Iatrou, Prog. Polym. Sci., 2018, 83, 28–78 CrossRef CAS.
- T. J. Deming, Prog. Polym. Sci., 2007, 32, 858–875 CrossRef CAS.
- M. Patel, H. J. Lee, S. Park, Y. Kim and B. Jeong, Biomaterials, 2018, 159, 91–107 CrossRef CAS PubMed.
- C. He, X. Zhuang, Z. Tang, H. Tian and X. Chen, Adv. Healthcare Mater., 2012, 1, 48–78 CrossRef CAS PubMed.
- Z. Song, Z. Tan and J. Cheng, Macromolecules, 2019, 52, 8521–8539 CrossRef CAS.
- A. Nowak, V. Breedveld, L. Pakstis, B. Ozbas, D. Pine, D. Pochan and T. Deming, Nature, 2002, 417, 424–428 CrossRef CAS PubMed.
- F. Meng, Y. Ni, S. Ji, X. Fu, Y. Wei, J. Sun and Z. Li, Chin. J. Polym. Sci., 2017, 35, 1243–1252 CrossRef CAS.
- M. Turabee, T. Thambi, H. Duong, J. Jeong and D. Lee, Biomater. Sci., 2018, 6, 661–671 RSC.
- S. Zhao, H. Zhu, Z. Chen, S. Shuai, N. Zhang, Y. Liu, Z. Rao, Y. Li, C. Zhao, K. Zhou, W. Ge and J. Hao, Mater. Res. Express, 2019, 6, 085711 CrossRef CAS.
- Z. Song, R. A. Mansbach, H. He, K.-C. Shih, R. Baumgartner, N. Zheng, X. Ba, Y. Huang, D. Mani, Y. Liu, Y. Lin, M.-P. Nieh, A. L. Ferguson, L. Yin and J. Cheng, Nat. Commun., 2017, 8, 92 CrossRef PubMed.
- Y. Cheng, C. He, C. Xiao, J. Ding, H. Cui, X. Zhuang and X. Chen, Biomacromolecules, 2013, 14, 468–475 CrossRef CAS PubMed.
- Y. Cheng, C. He, C. Xiao, J. Ding, X. Zhuang, Y. Huang and X. Chen, Biomacromolecules, 2012, 13, 2053–2059 CrossRef CAS PubMed.
- A. C. Engler, H. I. Lee and P. T. Hammond, Angew. Chem., Int. Ed., 2009, 48, 9334–9338 CrossRef CAS PubMed.
- L. Li, Q. Wang, R. Lyu, L. Yu, S. Su, F. Du and Z. Li, Polym. Chem., 2018, 9, 4574–4584 RSC.
- A. H. Gröschel, F. H. Schacher, H. Schmalz, O. V. Borisov, E. B. Zhulina, A. Walther and A. H. E. Müller, Nat. Commun., 2012, 3, 710 CrossRef.
- Y. Shen, X. Fu, W. Fu and Z. Li, Chem. Soc. Rev., 2015, 44, 612–622 RSC.
- Y. Cheng, C. He, J. Ding, C. Xiao, X. Zhuang and X. Chen, Biomaterials, 2013, 34, 10338–10347 CrossRef CAS.
- S. Cui, L. Yu and J. Ding, Macromolecules, 2018, 51, 6405–6420 CrossRef CAS.
- S. Chang, X. Wu, Y. Li, D. Niu, Y. Gao, Z. Ma, J. Gu, W. Zhao, W. Zhu, H. Tian and J. Shi, Biomaterials, 2013, 34, 10182–10190 CrossRef CAS PubMed.
- M. Juríček, P. Kouwer and A. Rowan, Chem. Commun., 2011, 47, 8740–8749 RSC.
- A. Faghihnejad, K. Feldman, J. Yu, M. Tirrell, J. Israelachvili, C. Hawker, E. Kramer and H. Zeng, Adv. Funct. Mater., 2014, 24, 2322–2333 CrossRef CAS.
- C. Cui, C. Fan, Y. Wu, M. Xiao, T. Wu, D. Zhang, X. Chen, B. Liu, Z. Xu, B. Qu and W. Liu, Adv. Mater., 2019, 31, 1905761 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00290b |
|
| This journal is © The Royal Society of Chemistry 2021 |

 *ab and
Xuesi
Chen
*ab and
Xuesi
Chen