
Substituted polynorbornene membranes: a modular template for targeted gas separations†
Xinyi
Wang‡
a,
Trevor J.
Wilson‡
a,
Dmitry
Alentiev
 b,
Maria
Gringolts
b,
Eugene
Finkelshtein
b,
Maxim
Bermeshev
*b and
Brian K.
Long
*a
b,
Maria
Gringolts
b,
Eugene
Finkelshtein
b,
Maxim
Bermeshev
*b and
Brian K.
Long
*a
aDepartment of Chemistry, University of Tennessee, Knoxville, Knoxville, TN 37996, USA. E-mail: Long@utk.edu
bA.V. Topchiev Institute of Petrochemical Synthesis RAS, Moscow, 119991, Russia. E-mail: bmv@ips.ac.ru
First published on 26th April 2021
Abstract
Polynorbornenes are ideal materials for systematic structure–property investigations designed to correlate gas-transport properties to polymer structure. The modular nature of norbornene-based systems provides a facile route towards the synthesis of diverse polymeric materials. Though many valuable correlations between gas-permeability and polynorbornene structure have been summarized in a prior review (2017), many of these efforts focused heavily on the design of materials with high permeabilities. More recently, the design of next-generation polynorbornene membranes has shifted toward the development of materials that are highly selective for multiple targeted gas separations. Since 2017, tremendous progress has been realized in the preparation of stable and highly selective polynorbornenes for membrane-based gas separations. Examples include polynorbornene derivatives that exhibit very high selectivity for CO2/N2 and light hydrocarbon (e.g., C3+/C1) separations, some of which exhibit gas separation performances that approach or exceed the 2008 upper bound. Herein, we summarize prior findings, present recent developments in the field of advanced polynorbornene membrane materials for targeted gas separations, and provide an outlook for the future of this field of research.
1. Introduction
1.1. Gas separation membranesThe development of membrane mediated gas separation technology has been the focus of extensive academic and industrial research efforts for many decades. As shown in Fig. 1, these systems utilize thin polymer films that preferentially permeate one gaseous species over another, enriching the downstream in the more permeable species. Membrane systems are an attractive alternative to conventional separations due to their low energy requirements, small footprint, and mechanical simplicity.1 This technology has matured rapidly and has been successfully applied to several notable industrial separations such as: hydrogen recovery, nitrogen production, natural gas processing, and industrial vapor recovery.2
 | ||
| Fig. 1 Schematic representation of a membrane driven gas separations. | ||
Within the membrane community, the solution-diffusion model is frequently used to describe mass transport in dense polymeric membranes.2–10 According to this model; as a pressure differential is applied across a membrane, penetrant molecules sorb into the upstream side of the membrane, diffuse through the polymer matrix down a chemical potential gradient, then desorb into the downstream side of the membrane. Thus, for a given penetrant molecule “A”, the overall permeability (PA) can be expressed as the product of its solubility coefficient (SA) and its diffusivity coefficient (DA) according to:
| PA = SA·DA |
The ratio of permeabilities of gas A over gas B is defined as the ideal selectivity (αA/B), where by convention, gas A is the faster permeating gas, as shown below:
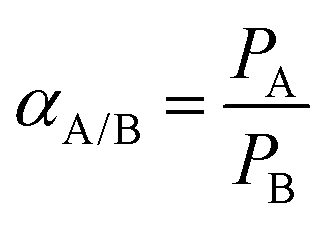
Permeability and selectivity are frequently used to benchmark membrane performance and can be plotted on a log–log plot of ideal selectivity versus permeability, as is shown in Fig. 2 and 3. Generally, attempts to increase membrane permeability via chemical modification of the membrane material are met with a proportional decrease in selectivity, and vice versa. This problematic behavior is referred to as the permeability–selectivity tradeoff and has made the realization of membranes that are both high permeability and high selectivity a challenging endeavor.
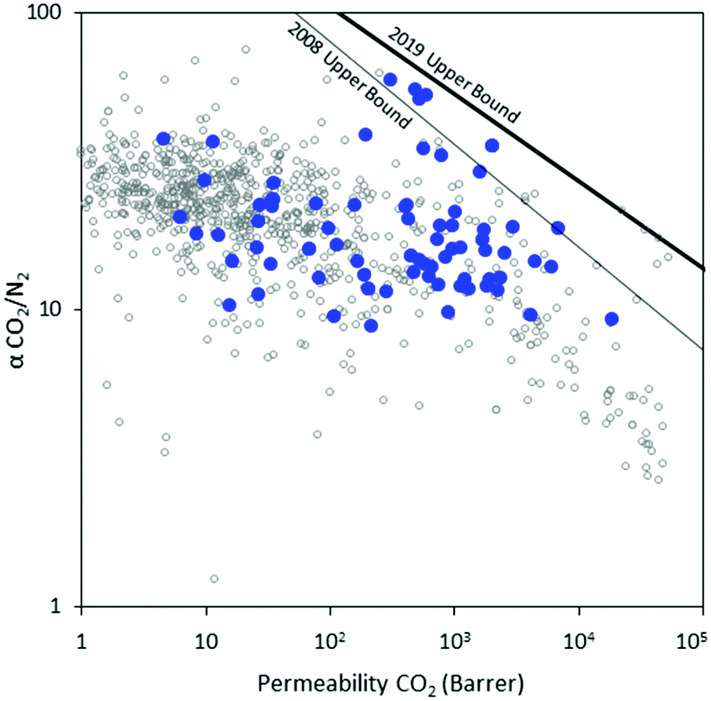 | ||
| Fig. 2 Robeson plot11,12 of CO2/N2 separation performance for a breadth of previously reported non-norbornene-based materials (open gray), and norbornene-based materials described herein (blue). | ||
 | ||
| Fig. 3 Robeson plot11–13 showing CO2/CH4 separation performance for a breadth of previously reported non-norbornene-based materials (open gray), and norbornene-based materials described herein (blue). | ||
Among the myriad materials developed in the pursuit of gas separation membrane materials, substituted polynorbornenes (PNBs) have emerged as a promising material class. The inherently modular nature of norbornyl-based monomers and diverse polymerization methods available lend themselves to the vast diversity of PNB-derived membrane materials reported in the literature.14,15 More specifically, the ability to easily alter monomeric substituents enables researchers to design materials that take advantage of noncovalent polymer–gas interactions without changing the commonality of the polymer backbone. In addition to the tunability of substituent chemistry, careful catalyst selection can afford structurally distinct backbone architectures and tacticity by controlling the mechanism through which the polymerization propagates.16–20 In this way, PNBs can be viewed as a structural scaffold upon which various substituents can be affixed, and multiple polymeric backbone structures accessed.
Though prior reviews of PNB-based gas separation membranes exist,14,21 there has been a veritable explosion of reports investigating these materials in recent literature. As such, we believe that a general overview of the state of the field is warranted. In this perspective, we will discuss recent developments in the field of PNB-based membrane materials, present some of their limitations and challenges, and pose an outlook for the future of this exciting field. We will begin with a brief introduction to the synthetic approaches used in the synthesis of norbornyl monomers, as well as some of the common polymerization techniques applied to these monomers. We will then describe many of the most recent advances in membrane mediated gas separations that utilize PNBs. These discussions include: PNBs bearing organosilicon pendant groups, advances in fluorine containing PNBs, PNBs with hydrocarbon sidechains, PNBs bearing rigid ladder type sidechains, PNBs with aromatic biphenyl fragments as their sidechain, and a new class of ladder type polymers synthesized through annulation reactions between norbornadiene and haloarenes. We will also discuss related work on post polymerization modification (PPM), and NB-based composites.
1.2. Monomer and polymer synthesis
As the unifying structural motif present in all the materials discussed herein, the norbornyl moiety offers several key advantages: (1) the relative ease of monomer synthesis via cycloaddition facilitates the synthesis of norbornyl monomers with a wide range of functionalities, (2) the strained bicyclic olefin can be polymerized through several mechanisms, and (3) by employing these various polymerization mechanisms, a variety of thermomechanical properties may be obtained, which is heavily dictated by polymer backbone structure.
The most convenient and straight-forward synthetic route to substituted norbornyl monomers involves the prototypical [4π + 2π] cycloaddition between a diene and a dienophile, more commonly known as a Diels–Alder reaction (Scheme 1a). These reactions provide the unique [2.2.1]bicyclic structure of norbornene-derivatives in a single step, and can often be performed on a multi-gram-scale in moderate to excellent yields. However, the synthesis of substituted norbornenes using Diels–Alder reactions typically provides a mixture of exo- and endo-substituted isomers that have differential polymerization reactivity, with the former often showing higher reactivity, regardless of polymerization mechanism.
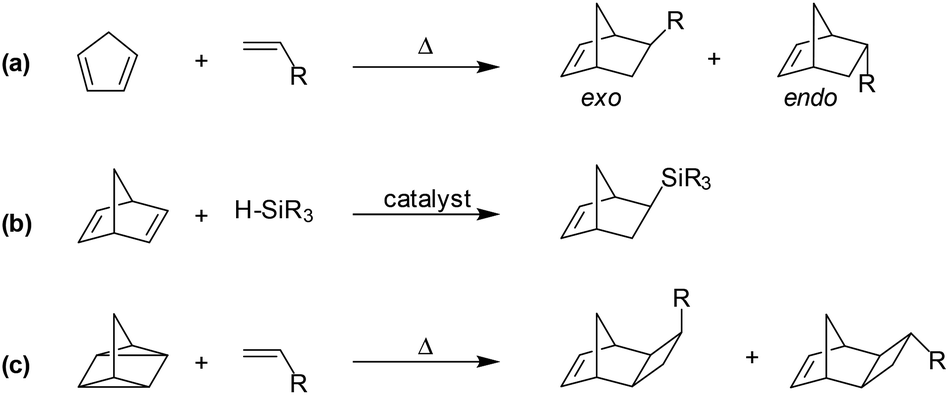 | ||
| Scheme 1 Representative synthetic routes to various substituted norbornene and tricyclononene monomers: (a) Diels–Alder, (b) metal-catalyzed hydrosilation, and (c) [2σ + 2σ + 2π] cycloaddition. | ||
To obviate the reactivity differences of exo- and endo-substituted isomers (Scheme 1a), pure exo-substituted norbornenes have been synthesized via stereoselective catalytic transformation of norbornadiene (Scheme 1b).16,22,23 Furthermore, closely related monomers bearing a third fused ring, commonly referred to as tricyclononenes (TCNs), may be synthesized using thermal [2σ + 2σ + 2π] cycloaddition reaction between quadricyclane and various vinyl reagents (Scheme 1c). This stereospecific route affords a monomer in which the fused cyclobutyl ring is exclusively in its exo-isomer form (Scheme 1c).24
Norbornene based monomers may be polymerized through a variety of mechanisms. These include radical, ionic, ring opening metathesis polymerization (ROMP), addition polymerization (AP), and catalytic arene-norbornene annulation (CANAL). While radical and ionic polymerizations of norbornene derivatives have been reported, these mechanisms tend to produce ill-defined oligomers that are poorly suited for membrane fabrication.25–29 As such, most PNB membrane literature is focused on materials synthesized through AP, and ROMP (Scheme 2a & b, respectively). An additional and more recently developed polymerization technique in norbornene inspired membrane materials has been the advent of CANAL, which yields ladder type structures that demonstrate high thermal stability, high BET surface areas, and promising gas transport properties (Scheme 2c).30–32
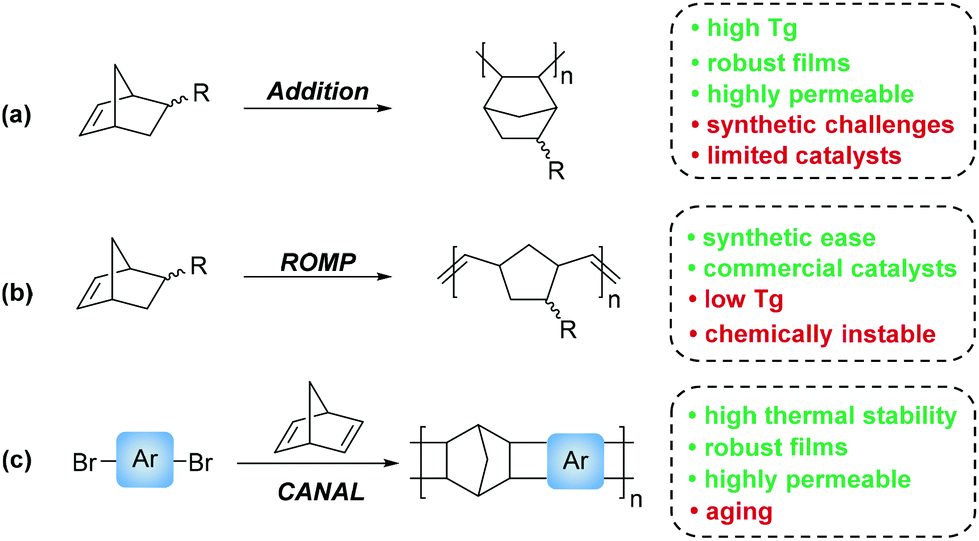 | ||
| Scheme 2 Polymerization methods that are applicable to norbornene-based polymers for gas separation membranes: (a) addition polymerization (AP), (b) ring opening metathesis polymerization (ROMP), and (c) catalytic arene-norbornene annulation (CANAL). | ||
While all PNB materials are derived from related norbornyl monomers, the final polymers display markedly different bulk properties. For example, PNBs synthesized through ROMP, which we will refer to as metathesis PNBs (MPNBs), tend to have significantly lower Tg's, lower Td's, lower gas permeabilities, exhibit greater main chain flexibility, and are prone to oxidation along the residual backbone olefin making them less chemically stable. In many cases, the unsaturated backbone is hydrogenated to impart greater chemical stability. However, these main chain olefins have also been used as reactive handles for intentional post-polymerization modifications (PPM). While the material properties of MPNBs are less attractive than their AP counterparts, their synthesis is well-established with many commercial ROMP catalysts available. Most notable of which are the ruthenium centered Grubbs, and molybdenum-based Schrock catalysts that exhibit rapid polymerization kinetics, and are less prone to substrate stereochemistry and substituent incompatibilities relative to most AP catalysts (Fig. 4a–c).
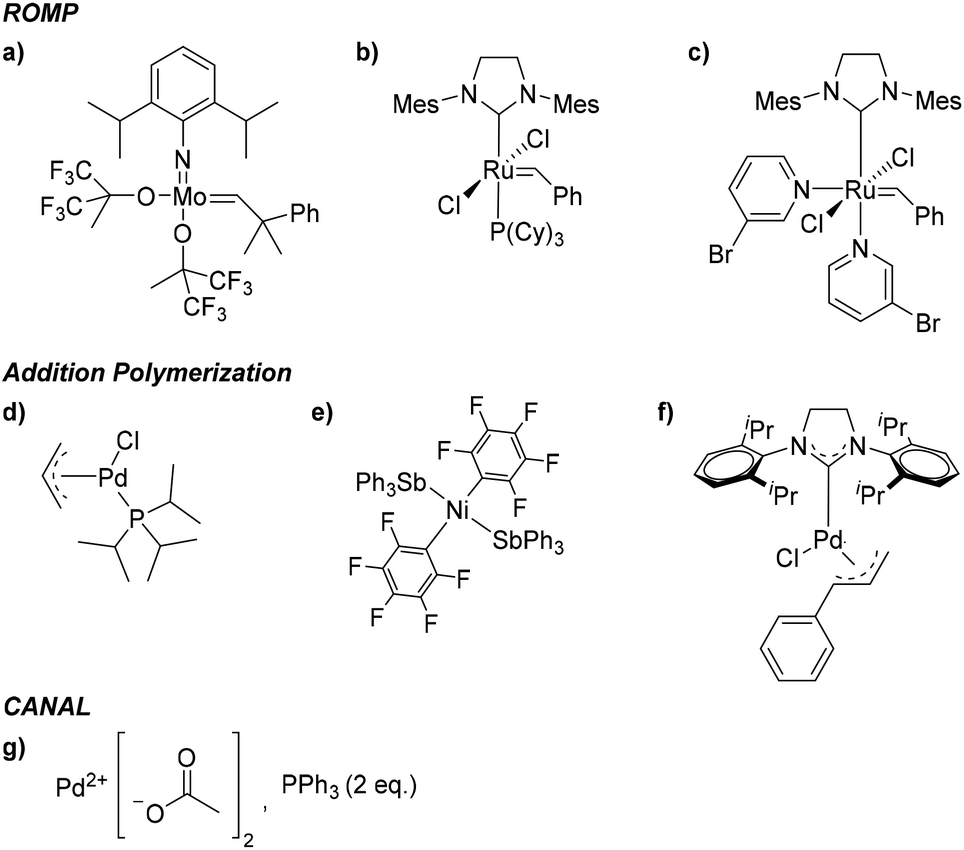 | ||
| Fig. 4 Representative catalyst examples used in polynorbornene synthesis via ROMP (a–c), AP (d–f), and CANAL (g). | ||
In contrast to the material limitations of MPNBs, addition polynorbornenes (APNBs) tend to display high thermal stability, higher gas permeabilities, better mechanical integrity, and are more chemically inert than their MPNB counterparts due to their rigid, saturated polymer backbone. Additionally, APNBs frequently display higher Tg's due to the retention of the rigid bicyclic norbornane motif along the polymer backbone. While APNBs have more attractive thermal and mechanical properties, they are synthetically more challenging to access. Many AP catalysts capable of polymerizing unsubstituted norbornene exhibit poor tolerance to polar and/or heteroatom-functionalized monomers. Recent successes in catalyst screening and development have enabled access to a wide range of polar and heteroatom functionalized APNBs.33–40 A select few of these catalysts are shown in (Fig. 4d–f). These advances have contributed to the growing library of substituted APNBs applied to the separation of gaseous mixtures.
Polymers synthesized through CANAL are among the newest norbornene derived materials to be applied to gaseous separations. Since the seminal report by Xia and coworkers in 2014,31 a variety of CANAL derived materials have been synthesized and their gas transport properties studied.30,32,41,42 These materials exhibit robust mechanical properties, high thermal stabilities, and exceptional gas permeabilities. These properties are likely due to the highly rigid backbone structure provided by the retention of the bicyclic norbornane motif and the planar arene fragments that constitute the backbone repeat unit. In the initial synthetic report of CANAL polymers, Xia and coworkers demonstrated that the highest conversions were achieved using Pd(OAc)2 in the presence of two equivalents of triphenyl phosphine (Fig. 4g). While other ligands were investigated, no other system was capable of granting comparable yields as those seen with the triphenyl phosphine system (>98%).31
2. Metathesis and addition polynorbornenes bearing organosilicon substituents
The incorporation of pendant silicon containing substituents onto PNBs has become an increasingly common synthetic trope in recent literature.6,16,18,20,22,34,43,44 This is particularly true for membranes designed to target CO2/N2 separation and hydrocarbon separations where the gases of interest are the more condensable component. This strategy was initially inspired by silicone rubbers, such as polydimethylsiloxane (PDMS), whose gas transport properties obey solubility-controlled selectivity, or “reverse selectivity”, for hydrocarbon gases. For these materials, the higher molecular weight hydrocarbons, such as propane and butane, are more permeable than lighter hydrocarbons (e.g. methane), and thus valuable heavy hydrocarbon petrochemical precursors can be separated in higher flux.Despite their desirable transport properties, silicone rubbers are often crosslinked to achieve good film forming properties, which limits their processability for practical membrane applications. It was found that affixing organosilicon pendant chains to a glassy polymer backbone, such as APNB, not only imbues the material with drastically increased gas permeability, but in some cases retains the solubility-controlled selectivity that is observed when using rubbery PDMS, which is rarely observed for other high Tg glassy polymers.6,17,18,20,45–47 In the last five years, a host of materials bearing various silicon containing substituents have been synthesized and their gas transport properties studied in-depth. This section is not intended to exhaustively review these materials, but rather to provide a select perspective regarding recent developments in the field of silicon containing MPNB and APNB materials.
2.1. Metathesis polynorbornenes bearing organosilicon groups
The synthetic ease of ROMP chemistry has facilitated the synthesis of a wide variety of organosilicon functionalized MPNBs. Among the earliest materials of this type to be applied to gas separation membranes were trimethylsilyl functionalized MPNBs. It was found that nearly an order of magnitude increase in gas permeability was achieved by introducing a single trimethylsilyl substituent per repeat unit relative to unsubstituted MPNB (2.2vs.2.1).48–52 Subsequent studies replacing these methyl groups with other aryl, alkyl or alkylsilyl groups, such as Ph (2.3), i-Pr (2.4), or CH2SiMe3 (2.5), typically resulted in reduced gas permeabilities.14,52,53Introduction of additional trimethylsilyl groups, as well as varying the relative position of the trimethylsilyl substituents, has been studied for both MPNBs (2.11, 2.12) and metathesis-polytricyclononenes (MPTCNs) (2.19, 2.20). Therein, a marked increase in gas permeability coefficients was observed, with a 3–4-fold increase for MPNB and a 2-fold increase for MPTCN.48,51,52 The most pronounced effects were observed when these groups were trans-vicinal to one another (2.11, 2.19) while geminal-substituted analogues experienced this to a lesser effect (2.12, 2.20).14,48,51,52 A recent study of MPTCNs bearing three SiMe3 groups (2.21) revealed even higher permeability compared to the analogous MPTCN with one or two SiMe3 groups per repeat unit.43 Many of these studies, such as those describing polymers (2.3–2.7) and (2.11–2.12 and 2.19–2.20), were discussed in a previous review and will not be discussed in detail in this perspective.14,54
The exo-/endo-orientation of the monomers used also strongly influences the gas permeability of MPNB bearing SiMe3 groups (2.2). The permeability of exo-2.2, which was synthesized from the pure exo-monomer isomer, turned out 1.5–2 fold higher than that of 2.2 that contains a mixture of exo- and endo-isomers (Table 1).16 This effect is explained by the difference in microstructure between the polymer chains forming from exo- and endo-isomers of the initial monomer. The chains formed from the endo-isomer are believed to pack with less free volume than the chains formed from exo-isomer. It is also interesting that the gas transport properties of exo-2.2 are close to those of the trimethylsilyl-substituted MPTCN 2.13. Taking into account the exo orientation of cyclobutane ring, it could be concluded that the orientation of substituent more strongly contributes to the gas transport properties than the presence of the fused cyclobutane ring.16
| Polymer | P (Barrer) | α ij = Pi/Pj | C4H10/CH4 | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N2 | O2 | CO2 | CH4 | C2H6 | C3H8 | C4H10 | O2/N2 | CO2/CH4 | CO2/N2 | |||
| a Gas permeability was measured through gas chromatographic method. b Gas permeability was measured via gas time-lag method. c Polymer film was crosslinked using 0.1 mass% of a Sn-catalyst (“x” before a number of a polymer refers that this polymer was crosslinked). | ||||||||||||
| 2.1 | 1.5 | 2.8 | 15.4 | 2.5 | — | — | — | 1.9 | 6.3 | 10.3 | — | 49 |
| 2.2 | 4.2 | 16.9 | 63.4 | 7.3 | — | — | — | 4.0 | 8.7 | — | 48 | |
| 2.2 | — | — | — | — | 11.1 | 3.2 | — | — | — | — | — | 20 |
| exo-2.2 | 7.1 | 28 | 118 | 17 | — | — | — | — | 6.9 | 16.6 | — | 16 |
| 2.8 | — | — | — | 1.4 | — | 1.0 | 10.2 | — | — | — | 7.4 | 20 |
| 2.9 | — | — | — | 13.3 | — | 12.8 | 75.1 | — | — | — | 5.7 | 20 |
| 2.10 | — | — | — | 97.7 | — | 241.8 | 2099.8 | — | — | — | 21.5 | 20 |
| 2.13 | 6.8 | 28 | 112 | 14.3 | — | — | — | 4.1 | 7.8 | — | 52 | |
| 2.14 | 55 | 170 | 830 | 180 | 270 | 290 | 1940 | 3.1 | 4.6 | 15.1 | 10.8 | 57 |
| 2.14 | 90 | 235 | 880 | 260 | — | — | — | 2.6 | 3.4 | 9.8 | — | 43 |
| 2.15 | 4.9 | 16 | 190 | 11 | 13 | 10 | 27 | 3.3 | 17.3 | 38.8 | 2.5 | 17 |
| 2.16 | 18 | 55 | 400 | 50 | 93 | 122 | 1260 | 3.1 | 8.0 | 22.2 | 25.2 | 17 |
| x2.16 | 18 | 54 | 407 | 51 | 100 | 116 | 1100 | 3.0 | 8.0 | 22.6 | 21.6 | 17 |
| x2.17 | 50 | 150 | 960 | 170 | 380 | 615 | 4100 | 3.0 | 5.6 | 19.2 | 24.1 | 17 |
| x2.18 | 68 | 188 | 1100 | 250 | 700 | 1300 | 8100 | 2.8 | 4.4 | 16.2 | 32.4 | 17 |
| 2.21 | 35 | 110 | 515 | 70 | — | — | — | 3.1 | 7.4 | 14.7 | — | 43 |
| 2.21 | 40 | 130 | 570 | 70 | — | — | — | 3.3 | 8.1 | 14.3 | — | 43 |
Another structure–property relationship has been demonstrated by probing the influence of substituent central atom size on the gas permeability of substituted PTCNs. The study of a series of MPTCNs bearing EMe3 (where E = Si, Ge, C) substituents (2.13, 2.13a, and 2.13b, respectively) demonstrates that the highest gas permeability was observed for Si-substituted MPTCN 2.13.55 Replacement of the Si atom by larger Ge (2.13a) or smaller C (2.13b) atom resulted in reduced gas permeability. The authors hypothesized that this effect is due to the trimethylsilyl group possessing the most optimal combination of rigidity and volume to form the largest free volume when chain packing. The tert-butyl group occupies less volume, whereas the –GeMe3 group is not as rigid as –SiMe3 group despite its larger volume. The increase in EMe3 substituent flexibility in the row of E = C < Si < Ge was confirmed by the decrease in glass transition temperatures of polymers bearing EMe3 substituents in the same row (Fig. 5).55
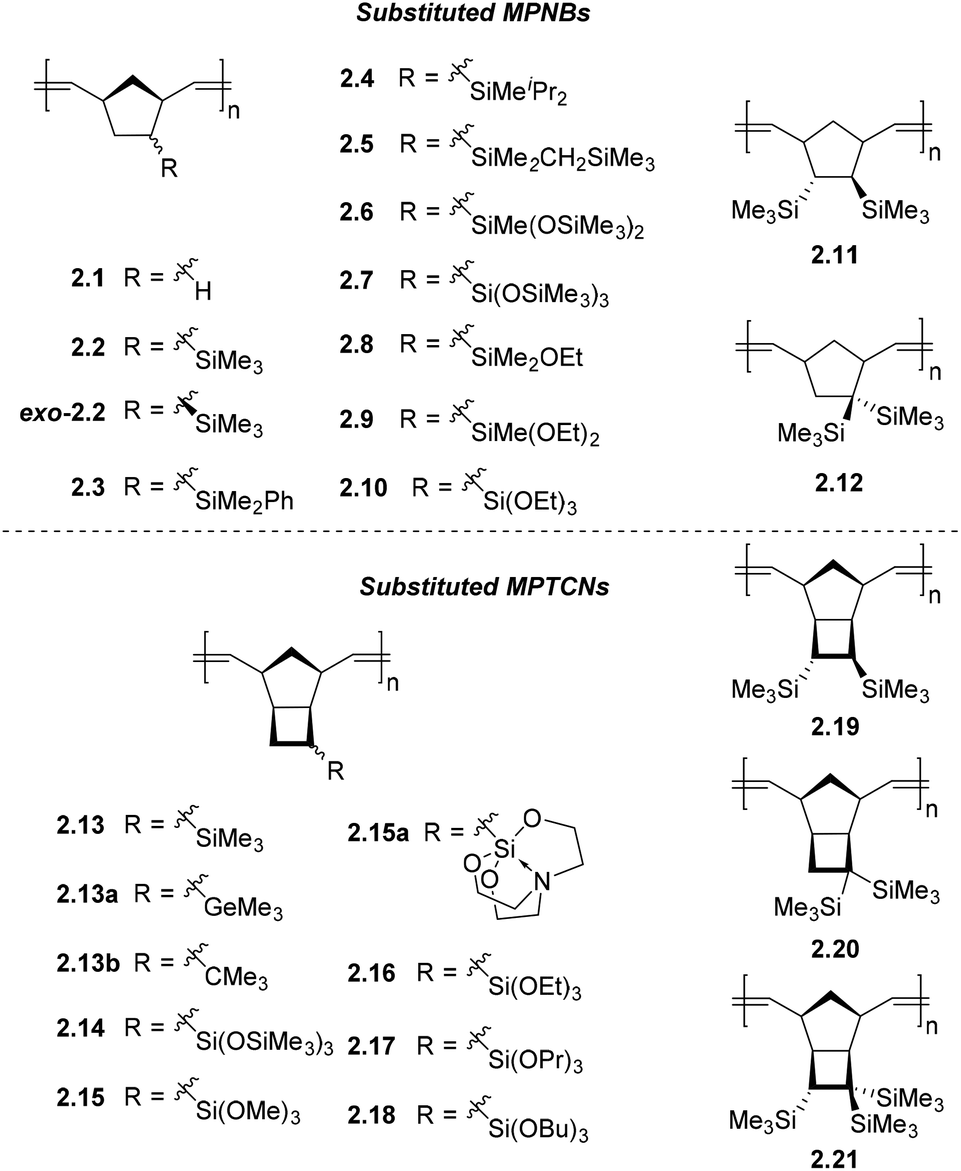 | ||
| Fig. 5 Metathesis polynorbornenes (MPNBs) and polytricyclononenes (MPTCNs) bearing organosilicon substituents. | ||
The highly flexible siloxane (–Si–O–Si–) linkage is also hypothesized to be a key structural feature responsible for the high gas diffusion seen in PDMS, as it potentially contributes to larger free volumes combined with rapid segmental chain dynamics.21,56 This segmental chain motion is of key importance in the formation of transient inter-chain gaps believed to govern gaseous diffusion through dense polymer matrices. Inspired by this structure–property relationship, a variety of siloxane functionalized MPNBs, such as 2.6 and 2.7, have been developed and their gas transport properties studied in-depth. We will intentionally exclude some earlier works on siloxane-containing MPNBs with improved permeability as they were discussed in a recent review.21 However, a recent example is MPTCN 2.14 which bears trimethylsilyl moieties linked to the backbone via Si–O–Si units, and the gas permeability generally increased more than 4-fold for all tested gases without significant decrease in ideal selectivity, as compared to 2.13.57 MPTCN 2.14 also showed solubility controlled permeation for hydrocarbons, as the permeability increased from 180 Barrer for CH4 to 1940 Barrer for C4H10, probably as a result of their flexible siloxane substituents.
Inspired by the exceptional permeabilities and selectivities seen in polymers bearing siloxane moieties for a variety of gas pairs, it was hypothesized that incorporation of alkoxysilane (Si–O–C) fragments may also improve membrane performance relative to polymers bearing trimethylsilyl substituents. One report by Sundell, Lawrence and coworkers revealed such improvements in a series of MPNBs (2.2, 2.8–2.10).20 This systematic investigation incrementally replaced the methyl groups in the MPNB (2.2) with one, two, and three ethoxy groups (2.8, 2.9, and 2.10 respectively). This structural perturbation increased C4H10/CH4 selectivity from α = 5.7 to α = 21.5 without significantly compromising permeability. In the case of 2.10, CH4 permeability increased by 5-fold compared to MPNB 2.2.20 This initial study revealed that MPNBs 2.8–2.10 exhibit solubility-controlled separation for light hydrocarbon gases,20 which is believed to originate from their flexible alkoxysilyl substituents. These characteristics make them particularly well suited for the separation of gaseous hydrocarbons, while the presence of the ethoxysilyl moieties enables them to be crosslinked through sol–gel chemistry to improve their stability over time, as well as to potentially further enhance diffusivity selectivity (see section 5 on PPM).
Similarly, a series of MPTCNs bearing increasingly bulky trialkoxysilyl-substituents from trimethoxysilyl to tributoxysilyl (2.15–2.18) were studied and compared to 2.11. The glass transition temperature of these polymers decreased with increasing alkoxysilyl length (113 °C for 2.13 to −44 °C for 2.18). For the two MPTCNs 2.13 and 2.15, which are in their glassy state at room temperature, the permeability coefficient of 2.15 decreased in all gases tested compared to 2.13, except for CO2 (P = 190 Barrer for 2.15, compared to P = 112 Barrer for 2.13), which is attributed to the specific interaction between CO2 and the methoxysilyl units. In contrast, MPTCNs in their rubbery state at room temperature (x2.16–x2.18, where the “x” denotation refers to the fact that a polymer is crosslinked, also see section 5.3 for more discussion about crosslinking), it was found that increasing alkoxysilyl substituent length increases polymer chain flexibility and thus stronger self-plasticization effects, which facilitates gas diffusivity in rubbery polymers. The presence of longer and flexible substituents also enhanced the solubility of gases (especially for hydrocarbons), leading to the overall increase in permeability coefficients for all gases tested.17 Polymers x2.16–x2.18 are each more permeable than 2.13 for all gases tested, and notably, MPTCN x2.17 with propoxysilyl-substituents and x2.18 with butoxysilyl-substituents are the first reported metathesis-polymers that possess higher permeability than their AP analogs.17 MPTCNs x2.16–x2.18 also showed solubility-controlled permeation of hydrocarbons, which is likely due to the combined effect of having relatively rigid main chains and flexible rubbery-like alkoxysilyl substituents.
A MPTCN bearing a silatrane substituent (2.15a) proved less permeable in comparison to other Si-substituted ROMP polynorbornenes. The silatrane group is notable due to the presence of its rigidly fixed (–Si–O–C–) fragments. It was hypothesized that the low permeability of 2.15a could be caused by two factors. First, it could be hypothesized that silicon-rich domains may not be formed due to the rigidly fixed (Si–O–C) fragments of 2.15a, as compared to domains formed in analogous polymers bearing flexible trialkoxysilyl groups. Alternatively, the presence of nitrogen atoms within the silatrane group may increase the net molecular dipole through induction, which may promote denser chain packing caused by strengthened dipole–dipole interactions between chains.58
Lastly, it should be noted that the double bond embedded within the backbone of MPNBs may also be subjected to PPM, which may improve their stability over time or improve their performance in some cases. A detailed discussion regarding PPM of MPNBs will be discussed in detail in section 5.
2.2. Addition polynorbornenes bearing organosilicon substituents
The synthesis of homo-APNBs bearing organosilicon substituents has proven a greater synthetic challenge than those synthesized via ROMP. This is primarily due to the poor tolerance of many AP catalysts to polar and/or heteroatom functionalized monomers. In recent literature, considerable progress has been made addressing this synthetic limitation, and several catalysts are now known to polymerize polar and/or heteroatom functionalized monomers through AP.6,17,18,20,34,36,44,47,59 Alternatively, copolymerization with unsubstituted NB has been used to achieve polymers of sufficient molecular weight for film formation when homopolymerization has proven challenging.60 However, this approach is becoming less common as the availability of tolerant catalysts continues to increase.In other cases, addition polytricyclononenes (APTCNs) (i.e.2.22–2.27) were designed such that the substituent is placed further from the polymerizable double bond, preventing deleterious interactions between the propagating catalyst center and the monomer substituent. Many of these monomers have been successfully polymerized using a Pd-based catalyst system to high molecular weight.57 Though these synthetic challenges have limited studies of APNB/APTCN in the past, the permeability of these polymers is typically several folds higher than their ROMP analogues without compromising selectivity, which makes them promising candidates for commercial membrane applications (Fig. 6).
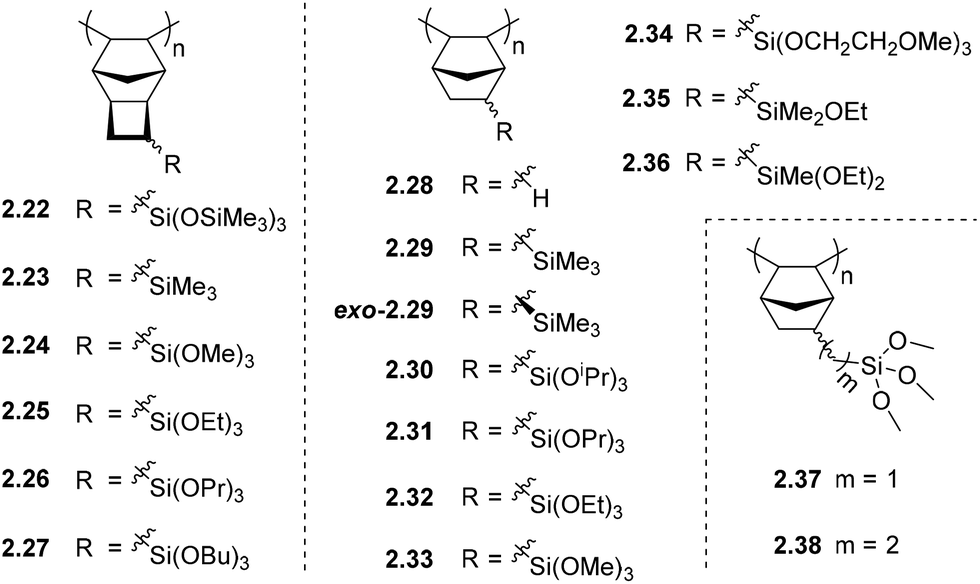 | ||
| Fig. 6 Addition polynorbornenes (APNBs) and polytricyclononenes (APTCNs) bearing organosilicon substituents. | ||
Initial studies on APNBs revealed that the permeability coefficients of trimethylsilyl-substituted APNB (2.29) increased several-fold as compared to unfunctionalized PNB (2.28). Interestingly, the difference in permeability between exo-2.29 (Table 2) and 2.29, which is produced from a mixture of exo-/endo-monomer isomers, is significantly less than the analogous difference in the case of metathesis polymers (2.2vs.exo-2.2, Table 1). This is hypothesized to arise as the APNB 2.29 is enriched in the exo-monomer isomer due to the low addition polymerization reactivity of the endo-isomer, whereas the metathesis polymerization of exo-/endo-monomers mixtures frequently contain endo-isomer contents that are more similar to that of the initial monomer feed.16 Nonetheless, the resultant gas selectivities of trimethylsilyl-substituted APNBs 2.29 and exo-2.29 were not high enough to draw near the upper bound for a variety gas pairs, and improvements are required before reaching industrial relevance.46,52,61
| Polymer | P (Barrer) | α ij = Pi/Pj | Ref. | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N2 | O2 | CO2 | CH4 | C2H6 | C3H8 | C4H10 | O2/N2 | CO2/CH4 | CO2/N2 | C4H10/CH4 | ||
| 2.22 | 420 | 960 | 4020 | 1250 | 2370 | 3490 | 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 200 |
2.3 | 8.0 | 9.6 | 17.8 | 57 |
| 2.23 | 430 | 1080 | 5980 | 1130 | 1720 | 1880 | 14700 |
2.5 | 5.3 | 13.9 | 13.0 | 17 |
| 2.24 | 56 | 141 | 2000 | 130 | 240 | 335 | 4050 | 2.5 | 15.4 | 35.7 | 31.2 | 17 |
| 2.25 | 47 | 131 | 1000 | 153 | 305 | 510 | 5000 | 2.8 | 6.5 | 21.3 | 32.7 | 17 |
| 2.26 | 35 | 93 | 470 | 84 | 220 | 440 | 4100 | 2.7 | 5.6 | 13.4 | 48.8 | 17 |
| 2.27 | 29 | 78 | 440 | 100 | 250 | 440 | 3600 | 2.7 | 4.4 | 15.2 | 36.0 | 17 |
| 2.28 | 1.5 | 6.9 | 33.6 | 2.6 | — | — | — | 4.6 | 12.9 | 22.4 | — | 46 |
| 2.29 | 297 | 780 | 4350 | 790 | 1430 | 1740 | 17500 |
2.6 | 5.5 | 14.6 | — | 46 |
| 2.29 | — | — | — | 678.9 | — | 856.4 | 7040.7 | — | — | — | 8.22 | 20 |
| exo-2.29 | 430 | 1100 | 5500 | 1100 | 1600 | 1800 | 15200 |
2.5 | 5.0 | 12.7 | 14.2 | 16 |
| 2.30 | 90 | — | 1120 | — | — | — | — | — | — | 12 | — | 6 |
| 2.31 | 47 | — | 650 | — | — | — | — | — | — | 14 | — | 6 |
| 2.32 | 57 | — | 940 | — | — | — | — | — | — | 16 | — | 6 |
| 2.32 | — | — | — | 176.2 | — | 499.1 | 3837.6 | — | — | — | 21.8 | 20 |
| 2.33 | 56 | — | 1600 | — | — | — | — | — | — | 29 | — | 6 |
| 2.37 | 23 | — | 770 | — | — | — | — | — | — | 33 | — | 6 |
| 2.38 | 16 | — | 560 | — | — | — | — | — | — | 35 | — | 6 |
| 2.35 | — | — | — | 57.7 | — | 112.7 | 1620.9 | — | — | — | 28.1 | 20 |
| 2.36 | — | — | — | 49.0 | — | 100.1 | 1104.3 | — | — | — | 22.5 | 20 |
It has been proposed that incorporation of siloxane fragments could also improve the APNB/APTCN membrane selectivity, as was previously observed in MPNBs. Surprisingly, the siloxane substituted APTCN 2.22 did not markedly change light gas permeability as compared to trimethylsilyl-substituted 2.23, which contradicts the trend observed for the analogous metathesis polymers. However, a near 50% increase in permeability is observed for gaseous higher hydrocarbons (C2+), while their solubility-controlled selectivity leads to a nearly 50% increase in the overall selectivity for C4/C1.57 The flexible siloxane linkage can be also applied as a crosslinker in a polymer matrix, which will be discussed in section 5.3.45
Slight perturbations in the structure of alkoxysilyl substituents in various APNBs and APTCNs have been shown to have a strong influence on the CO2/N2 and C4H10/CH4 transport properties of the resulting membranes (Table 2). For example, changes in alkoxysilane branching (2.30 and 2.31) and tether length (2.33, 2.37 and 2.38) both led to traditional permeability–selectivity tradeoffs with CO2/N2 separation (i.e., from 2.30 to 2.31, CO2 permeability decreased by 40% while CO2/N2 selectivity increased by 16%; from 2.33 to 2.38, CO2 permeability decreased by 52% while CO2/N2 selectivity increased by 81%). In contrast, changes in alkoxysilane chain-length (2.31–2.33) led to a perpendicular approach towards the 2008 upper bound for CO2/N2 (for polymers 2.31–2.33, CO2 permeability increased by 146% and CO2/N2 selectivity increased by 107%).6
In another study of APNBs utilized for heavy gas separation, the methyl groups in 2.29 were replaced with one, two, or three ethoxy groups (2.35, 2.36, and 2.32 respectively). This led to an initial increase in C4H10/CH4 selectivity but decrease in permeability as compared to 2.29. Triethoxysilyl-substituted polymer 2.32 showed that some degree of permeability was restored (C4H10 permeability of 3837 Barrer), albeit with slightly decreased selectivity (α(C4H10/CH4) = 21.8).18,20,47 These materials can also be treated with non-solvents, such as lower alcohols, to swell the polymer matrix, increase free volume size, and thus improve membrane performance.18
A series of APTCNs bearing increasing alkoxysilane chain lengths (2.24–2.27) has also been investigated and compared to APTCNs bearing simple trimethylsilyl-substituents (2.23).17 Similar to the APNBs discussed above (2.30–2.33 and 2.37–2.38), increasing alkoxy chain length in APTCNs generally led to decreased permeability of light gases due to a denser chain packing and lower free volume. These APTCNs also exhibited high ideal CO2/N2 selectivities. The highest selectivity was observed in the case of 2.24, which had a CO2/N2 selectivity of 35.7 and CO2 permeability of 2000 Barrer, locating this polymers performance above the 2008 CO2/N2 upper bound. Similar to APNBs, alkoxysilyl substituted APTCNs also exhibit solubility-controlled permeation of hydrocarbons, and surprisingly, the permeability of C2–C4 hydrocarbons is weakly dependent on the size of the alkoxy groups. Ideal C4/C1 selectivities of trialkoxysilyl substituted APTCNs (2.24–2.27) were in the range of 30–50 (Table 2), with the highest being observed for polymer 2.26. It was also the highest C4/C1 selectivity ever found for PNBs/PTCNs. It is worth noting that alkoxysilyl substituted APTCNs possess relatively stable gas transport properties over time. For example, polymer 2.27 showed no change in permeability over a few months, suggesting that it is not prone to aging. This was hypothesized to be due to the low free volume arising from the presence of flexible alkoxy fragments.17
To optimize the CO2/N2 separation performance of alkoxysilyl APNBs, Long and coworkers proposed that the incorporation of CO2-philic groups may enhance CO2 solubility, and therefore overall CO2 selectivity of these membrane materials. Toward this goal, tris(2-methoxyethoxy)silyl moieties were introduced onto the rigid APNB backbone (2.34), with the hypothesis that this combination would achieve both the advantages of larger free volume induced by frustrated interchain packing, and thus high permeability, as well as favorable interactions between CO2 and the Lewis basic ether functionalities.6 Among a series of homo- and copolymers containing this tris(2-methoxyethoxy)silyl-substituent, homopolymer 2.34 displayed the highest performance with a CO2/N2 selectivity of 36.7 and CO2 permeability of 755 Barrer. This work was discussed in a recent review and is not discussed in detail here.62 Interestingly, a subsequent study using 2.34 revealed that its H2S/CH4 separation performance exceeds the 2008 Robeson upper bound for this gas pair, and also possesses moderate CO2/CH4 performance. This observation is noteworthy in that most membranes that have good H2S/CH4 separation performance often exhibit poor CO2/CH4 separation performance, and vice versa. Thus, the high performance of 2.34 makes it a promising candidate for dual-purpose natural gas processing membranes.47
2.3. Comparison of metathesis and addition polynorbornene gas separation performance
In general, APNBs/APTCNs typically have higher permeabilities than their metathesis analogues. This is due in part as most APNBs/APTCNs are glassy materials at room temperature, and therefore possess excess non-equilibrium free volume that often leads to higher permeabilities and may contribute to diffusivity-controlled selectivity (separating gases primarily based upon individual gas diffusivity and differences in kinetic diameters). In contrast, MPNBs/MPTCNs are often in their rubbery state at room temperature (with some exceptions that include bulky substituents or relatively more rigid backbones, such as 2.2, 2.13, and 2.15), and often possess smaller free volume, lower overall permeabilities, and solubility-controlled selectivity.The disparate physical characteristics between MPNBs and APNBs lead to stark differences in gas separation performance, even when comparing APNB and MPNB products with identical substituents. For example, MPNBs 2.8–2.10 bearing alkylsilyl and alkoxysilyl substituents generally show enhanced diffusivity due to self-plasticization and enhancement of gas solubility (especially for hydrocarbons) for polymers containing greater alkoxysilyl content. In contrast, APNBs 2.35–2.36 and 2.32 have the same change in substituents, but display a slightly different trend in performance wherein the more flexible substituents are shown to decrease excess non-equilibrium free volume in the membranes, leading to decreased diffusivity. However, these substituents also enhance gas solubility (especially for hydrocarbons).20 Similar trends are seen for polymers 2.15–2.18 and 2.24–2.27.17 One of the notable highlights of these organosilicon containing APNB/APTCNs is that many of them exhibit solubility-controlled selectivity.17,20,57 This desirable separation mechanism, combined with their high gas permeability, makes these membranes promising candidates for commercial gas separation applications.
3. Si-Free metathesis and addition polynorbornenes
3.1. Polynorbornenes with fluorinated substituents
Gas transport properties of MPNBs bearing fluorine-containing substituents were first described in the early 1990s.63,64 In these works, it was found that the introduction of fluorine-containing substituents to the side chain of MPNBs (3.1, 3.2, Fig. 7 and Table 3) resulted in increased gas permeability as compared to unfunctionalized MPNB. This observation is in direct contrast to the effect achieved by the incorporation of chlorine- and bromine-containing moieties in MPNBs.65 Since these initial reports, two new classes of MPNBs bearing fluorinated substituents have been synthesized and their gas separation properties have been studied. The first class of these materials are those derived from norbornene-5,6-dicarboximides with fluoroaryl substituents connected to the imide nitrogen atom (NB-DCIs). Various monomers of this type are readily synthesized from cis-5-norbornene-exo-2,3-dicarboxylic anhydride and an appropriate amine or aniline.66 Synthesis of MPNB-DCIs, (Fig. 7, 3.3–3.5) and their transport properties will be discussed in detail in section 3.3.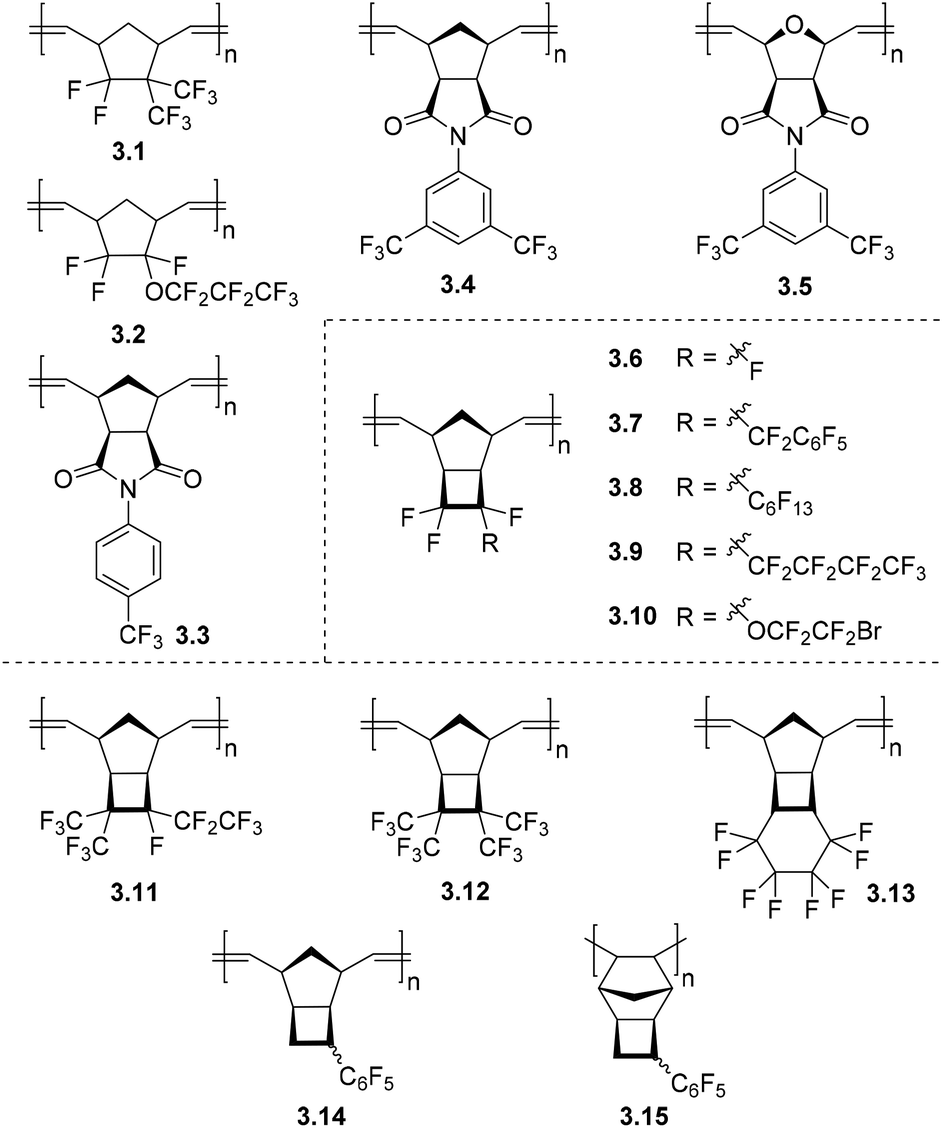 | ||
| Fig. 7 Metathesis polynorbornenes (3.1, 3.2), polynorbornene-5,6-dicarboximides (3.8, 3.9), polyoxanorbornene-5,6-dicarboximide (3.10), polytricyclononenes (3.3–3.7, 3.11–3.14) and addition polytricyclononene (3.15) bearing fluorinated substituents. | ||
| Polymer | P (Barrer) | α ij = Pi/Pj | Ref. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| He | H2 | N2 | O2 | CO2 | CH4 | O2/N2 | He/CH4 | CO2/N2 | H2/CH4 | He/H2 | N2/CH4 | ||
| a The measurements were carried out at 35 °C, in other cases at RT | |||||||||||||
| 3.1 | — | 122 | 11 | 28 | 102 | 9.5 | 2.6 | — | 9.7 | 12.8 | — | 1.1 | 63 |
| 3.1 | — | 166 | 17 | 50 | 200 | 13 | 2.9 | 11.8 | 12.7 | 1.3 | 64 | ||
| 3.2 | — | 130 | 17 | 55 | 200 | 18 | 3.2 | — | 11.8 | 7.2 | — | 0.9 | 64 |
| 3.3 | — | 34 | 1.5 | 6.2 | 34 | 1.6 | 4.2 | — | 23.4 | 14.3 | — | 0.9 | 66 |
| 3.4 | — | 57 | 4.2 | 13.5 | 67 | 4.3 | 3.2 | — | 16.0 | 13.3 | — | 1.0 | 70 |
| 3.5 | 204 | — | 11 | 39.4 | 165 | 11 | 3.5 | 19 | 14.5 | — | — | 1.1 | 71 |
| 3.6 | 29 | 20 | 1.1 | 4.1 | 16 | 0.9 | 3.7 | 32 | 14.5 | 21.2 | 1.5 | 1.3 | 67 |
| 3.7 | 55 | 34 | 2.3 | 7.0 | 26 | 1.9 | 3.0 | 29 | 11.3 | 17.7 | 1.6 | 1.2 | 67 |
| 3.8 | 168 | 92 | 11 | 32 | 105 | 9.0 | 2.9 | 18.9 | 9.5 | 10.3 | 1.8 | 1.2 | 68 |
| 3.9 | 280 | 175 | 24 | 57 | 210 | 16 | 2.4 | 17.5 | 8.8 | 10.9 | 1.6 | 1.5 | 67 |
| 3.10 | 76 | 66 | 5.1 | 17 | 96 | 5.3 | 3.3 | 14.3 | 18.8 | 12.5 | 1.2 | 1.0 | 67 |
| 3.11 | 262 | 203 | 24 | 68 | 277 | 20 | 2.8 | 13.1 | 11.5 | 10.2 | 1.3 | 1.2 | 19 |
| 3.12 | 510 | 700 | 60 | 180 | 730 | 46 | 3.0 | 11.1 | 12.2 | 15.2 | 0.7 | 1.3 | 67 |
| 3.13 | 195 | 137 | 14 | 42 | 184 | 12 | 3.0 | 16.2 | 13.1 | 11.7 | 1.4 | 1.2 | 69 |
| 3.14 | 44 | 41 | 2.3 | 8.2 | 33 | 3.5 | 3.6 | 12.6 | 14.3 | 11.7 | 1.1 | 0.7 | 67 |
| 3.15 | 400 | 300 | 48 | 130 | 620 | 55 | 2.7 | 7.3 | 12.9 | 5.5 | 1.3 | 0.9 | 72 |
A second series of fluoro-substituted polymers are those derived from fluorinated TCNs (3.6–3.10, 3.11–3.15) (Fig. 7), which are prepared by reaction of quadricyclane with various fluorinated olefins (Scheme 1c). The propensity of a fluorinated olefin to undergo cycloaddition with quadricyclane increases as the number of electronegative fluorinated substituents around the olefin increases. However, increased steric bulk of substituents near the olefin also seemingly hinders this reactivity (Fig. 8).67 Metathesis polymerization of fluorine-containing TCNs, NBs, and NB-DCIs affords the corresponding polymers in moderate to high yield.19,66–72 Notably, ROMP of a series of TCNs with high fluorine content was carried out in a mixed solvent system utilizing CH2Cl2 and C6F6 due to the limited solubility of the catalyst and subsequent polymers,67 and a wide variety of fluorinated PTCN with up to four substituents per TCN repeat unit was achieved (Fig. 7).
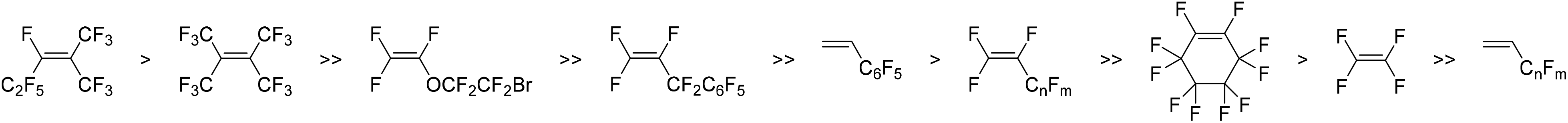 | ||
| Fig. 8 Comparative reactivity of fluorine-containing olefins to undergo cycloaddition with quadricyclane to yield tricyclonone (TCN) monomers. | ||
Gas transport investigations of these fluorinated MTCNs, MPNBs, and MPNB-DCIs demonstrate that the incorporation of perfluoroalkyl substituents has a strong positive influence on the pure gas permeabilities (Table 3) of the studied polymers (3.1, 3.2, 3.8, 3.9, 3.11, 3.12), while the presence of perfluorophenyl substituents in MPNBs (3.7, 3.14) generally lead to lower gas permeability due to π–π stacking interactions and denser polymer packing.19,64,67,68 Among perfluoroalkyl substituents, the influence of trifluoromethyl (–CF3) moieties is more pronounced (Table 3, polymers 3.11, 3.12), and the most permeable polymer was MPTCN 3.12 that bears four trifluoromethyl groups (Table 3).
The effect of fluorine-containing substituents on gas separation properties of MPNBs can be explained in more detail when considering their diffusion and solubility coefficients (Table 4). The incorporation of long perfluoroalkyl or several trifluoromethyl substituents (e.g.3.9, 3.12) results in a noticeable increase in their diffusion coefficients (Table 4).67 This increase was explained by the higher rigidity of the polymer chain in the presence of bulky substituents (3.12). On the other hand, the low cohesion energy, which is characteristic of fluoropolymers, leads to a weak interchain interactions, greater mobility of polymer chains, and less dense packing. The latter is evidenced by an increase in diffusion coefficients of MPNBs with long perfluoroalkyl-substituents (e.g.3.9).
| Polymer | T g (°C) | D × 108 (cm2 s−1) | S × 103 (cm3 (cm3 cmHg)−1) | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| N2 | O2 | CO2 | CH4 | N2 | O2 | CO2 | CH4 | |||
| a Measurements were made at room temperature, unless otherwise noted. b Measurements were made at 35 °C | ||||||||||
| 3.1 | 176 | 33 | 65 | 29 | 13.5 | 20 | 37 | 240 | 39 | 63 |
| 3.1 | 169 | 8.4 | 14 | 8.4 | 3.3 | 7.6 | — | 109 | 19 | 64 |
| 3.2 | 77 | 44 | 57 | 40 | 23 | 3.9 | 9.6 | 50 | 7.8 | 64 |
| 3.3 | 107 | 12 | 23 | 14 | 3.1 | 0.9 | 1.8 | 12 | 3.0 | 66 and 67 |
| 3.4 | 107 | 12 | 24 | 8.2 | 3.6 | 1.9 | 2.9 | 32 | 5.3 | 67 and 70 |
| 3.5 | 73 | 37 | 71 | 35 | 14 | 3.0 | 4.5 | 30 | 6.4 | 68 and 71 |
| 3.6 | 89 | 100 | 160 | 76 | 28 | 2.4 | 3.6 | 28 | 5.7 | 67 |
| 3.7 | 106 | 20 | 46 | 24 | 7.8 | 2.5 | 3.7 | 40 | 6.8 | 67 |
| 3.8 | 222 | 2.8 | 9 | 2.2 | 0.8 | 5.3 | 7.2 | 160 | 21 | 66 and 68 |
| 3.9 | 168 | 8.3 | 19 | 4.8 | 3.3 | 5.1 | 7.2 | 140 | 13 | 67 and 70 |
| 3.10 | 182 | 67 | 161 | 76 | 20 | 1.6 | 2.4 | 22 | 5.3 | 67 and 71 |
| 3.11 | 150 | 37 | 78 | 43 | 12 | 3.6 | 8.6 | 64 | 16 | 19 |
| 3.12 | 199 | 81 | 180 | 87 | 24 | 7.4 | 10 | 84 | 19 | 67 |
| 3.13 | 154 | 25 | 53 | 26 | 7.2 | 5.6 | 7.8 | 69 | 16 | 69 |
| 3.14 | 107 | 10 | 23 | 9.2 | 3.5 | 2.3 | 3.6 | 36 | 10 | 67 |
| 3.15 | >300 | 27 | 60 | 28 | 8.4 | 18 | 22 | 220 | 65 | 72 |
The differences in the solubility coefficients of fluorinated MPNBs were not as significant as those seen in the diffusion coefficients.67 The most permeable polymer 3.12 displayed a moderately high solubility coefficient. More significant differences are noticeable in their solubility coefficients for CO2 (S(CO2)), which are higher than for other gases, and the highest S(CO2) values are observed for MPNB-DCIs 3.3–3.4 (Table 4) that may be explained by their higher affinity for CO2.66,70
An interesting feature of fluorine-containing polymers is their rather high selectivity for gas pairs containing methane (i.e. H2/CH4, He/CH4) (Table 3). Polymer 3.12 is of particular interest in that the trade-off effect between permeability and selectivity is negligible, showing a significant increase in CH4 permeability coefficient, as compared to polymer 3.11, while exhibiting very little change in ideal selectivity. For other gas pairs, traditional trade-off behaviour was observed.67
Very recently, the gas permeability of fluorinated APNB 3.15 was published for the first time.72 In the presence of the three-component catalytic system (Pd(OAc)2, Na[B(3,5-(CF3)2C6H5)4], and PCy3), the perfluorophenyl APTCN was obtained (Table 3). Polymer 3.15 displayed high permeability for all investigated gasses, with only one MPTCN (3.12) exhibiting higher gas permeability (Table 3). Comparison of 3.15 with its metathesis isomer 3.14 (Table 3) shows that the higher permeability of the addition polymer can be explained by an observed increase in diffusion and solubility coefficients, respectively (Table 4). Therein, it was found that 3.15's diffusion coefficients increased ∼2.5 fold, and its solubility coefficients increased ∼7 fold times. The observed increase in diffusion coefficients was explained by the less dense packing of the more rigid APTCN backbone, which is confirmed by WAXD data and thermal analysis.72 The trade-off effect of O2/N2 and CO2/N2 gas permeability and selectivity for polymer 3.15 is negligible in relation to gas pairs compared to 3.14, but is not realized for gas pairs containing CH4. A possible explanation may be the higher sorption capacity of polymer 3.15; however, mass transport in these fluorine containing APNBs is not yet well understood, and further investigation will require new fluorine containing APNBs to be synthesized and studied.
In addition to interesting gas transport properties, fluorine-containing MPNBs and APNBs exhibit good thermal and chemical stability. Their decomposition temperatures are routinely ≥300 °C, with many ≥400 °C. In contrast to other MPNBs, fluorine-containing MPNBs are typically resistant to oxidants. For example, in the presence of K2Cr2O7/H2SO4 the main chain double bonds of MPNB 3.12 were not oxidized.67
Thus, both MPNBs and APNBs bearing fluorine-containing substituents combine desirable thermal and chemical stability with tunable gas permeability and good selectivities for gas pairs containing CH4 or CO2 (e.g. H2/CH4, He/CH4, CO2/CH4 and CO2/N2).67,72 These characteristics make these membrane materials promising candidates for the removal of light gases from multi-component hydrocarbon gas streams. Two approaches, including changing the structure of main chains and introducing CF3 or long perfluoroalkyl substituents, make it possible to deliberately influence the diffusion and solubility of gases in fluoro-PNBs. Among PNBs with fluorinated substituents, APNBs with perfluorophenyl groups and MPNBs bearing four trifluoromethyl (–CF3) moieties per repeat unit show the highest permeability. Further development of PNBs with fluorinated substituents and investigations of their gas transport properties are promising future avenue of research. This is especially true of fluorine containing APNBs, as they tend to exhibit greater permeability than their metathesis analogues.
3.2. Polynorbornenes with hydrocarbon/carbocyclic substituents
Bermeshev and coworkers recently investigated the permeation of C1–C4 hydrocarbons and mixed gases through addition poly(5-n-alkylnorbornenes) (3.17–3.18) (Fig. 9).73 While these poly(5-n-alkylnorbornenes) exhibited overall low gas permeability, they displayed a unique feature for glassy polymers – solubility-controlled permeation of hydrocarbons – and had enhanced C4/C1 separation selectivity. It should be noted that a slight increase in permeability was observed in the transition from 3.17 to 3.18.74 However, the increase in permeability of heavy gaseous hydrocarbons with increasing alkyl fragments length turned out to be stronger (Table 5).
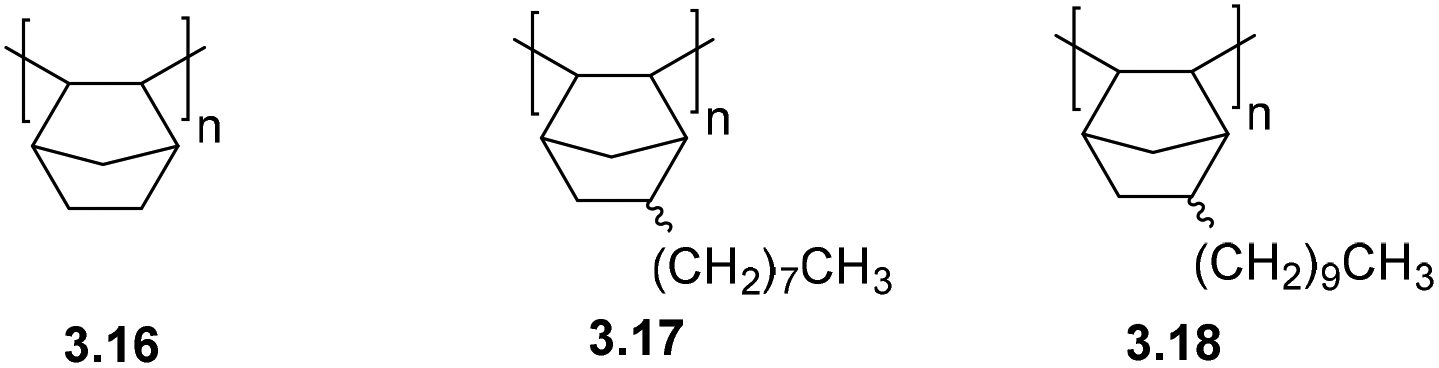 | ||
| Fig. 9 Structures of addition poly(5-n-alkylnorbornenes). | ||
| Polymer | P (Barrer) | α(C4/C1) | D × 108 (cm2 s−1) | α D(C4/C1) |
S × 102a (cm3(STP) cm−3 cmHg−1) |
α S(C4/C1) | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| O2 | CH4 | n-C4H10 | CH4 | n-C4H10 | CH4 | n-C4H10 | |||||
| a S was calculated as P/D. | |||||||||||
| 3.16 | 6.9–11 | 3.1 | 1.0 | 0.3 | — | — | — | — | — | — | 14 and 73 |
| 3.17 | — | 16 | 210 | 13 | 33 | 5.6 | 0.17 | 0.5 | 38 | 78 | 73 |
| 3.18 | 25 | 24 | 340 | 14 | 60 | 13 | 0.22 | 0.4 | 26 | 65 | 73 and 74 |
In contrast to poly(5-n-alkylnorbornenes), unsubstituted APNB 3.16 possesses diffusion-controlled hydrocarbon permeation. This strongly suggests that the presence of alkyl fragments is the principle contributing factor in the solubility-controlled hydrocarbon permeation of poly(n-alkylnorbornenes).73 While the overall C4/C1 permeability selectivities of 3.17 and 3.18 were close, detailed investigations revealed that their performance stems from a different combination of diffusion and solubility coefficients between these two polymers (Table 5).73 The diffusivity of gaseous hydrocarbons through polymers 3.17 and 3.18 increases as a function of increasing alkyl group length, and an opposite trend was observed for hydrocarbon solubility, with these effects being stronger for heavier hydrocarbons (Table 5). The higher diffusion coefficients of 3.18 in comparison to polymer 3.17 were explained by the reduction of energy barriers for gas diffusion due of the presence of longer and more flexible substituents.73 Because of the comparatively high hydrocarbon separation selectivitites (α ≤ 14 for C4/C1) and stable gas permeability over time, these polymers were considered as potential membrane materials for hydrocarbon separation for the first time.
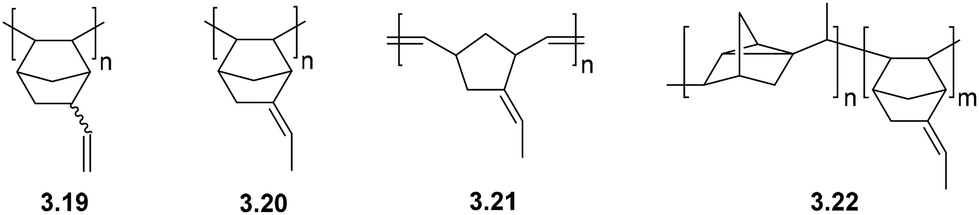 | ||
| Fig. 10 Structures of polynorbornenes bearing unsaturated substituents. | ||
| Polymer | A (nm) | P (Barrer) | D(O2) × 108 (cm2 s−1) | α ij = Pi/Pj | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| He | O2 | CO2 | CH4 | O2/N2 | CO2/N2 | ||||
| a Kuhn segment length. b The polymer film was prepared directly from the polymerization mass | |||||||||
| 3.19 | 6.0 | 120 | 48 | 310 | 24 | 40 | 3.7 | 24 | 15 |
| 3.20 | — | 390 | 100 | 620 | 58 | 94 | 3.1 | 19 | 15 |
| 3.20 | 6.3 | 300 | 80 | 400 | 45 | 73 | 3.2 | 16 | 15 |
| 3.21 | — | 26 | 6.7 | 30 | 4.8 | 29 | 4.2 | 19 | 75 |
| 3.22 | — | 39 | 7.8 | 36 | 3.0 | 22 | 4.3 | 20 | 15 |
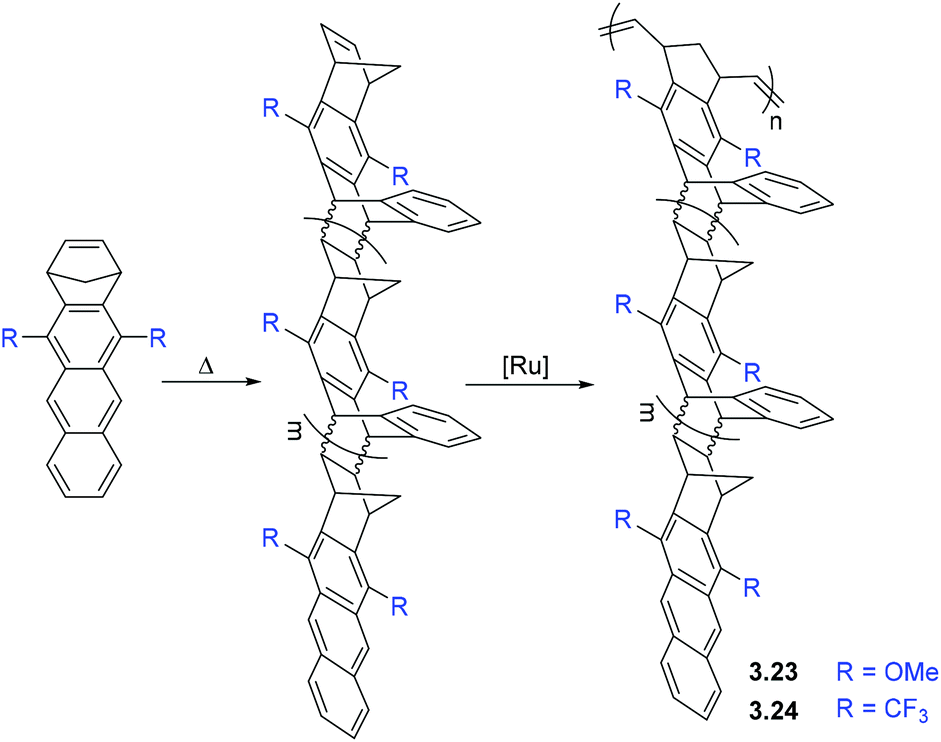 | ||
| Scheme 3 Synthesis of the polynorbornenes with side chain porosity. | ||
BET analysis of polymers 3.23–3.24 revealed that they have high surface areas (≤800 m2 g−1).77 It is worth noting that the polycyclic monomers were not porous, themselves, and the porosity emerged only upon polymerization. Such polymers were considered as materials for gas storage and sorption of organic vapors. Smith and coworkers studied the gas transport properties of polymers 3.23 and 3.24, and found that they display gas permeabilities comparable to those seen in many PIMs (Table 7).77 Permeability coefficients of the polymers increased in the order of P(N2) < P(CH4) < P(O2) ≈ P(He) < P(H2) < P(CO2). The ultrahigh CO2 and H2 permeability coefficients observed for 3.24 (21000 and 8300 Barrer, respectively) demonstrate the remarkable transport properties of this emerging class of materials. The H2/CH4 selectivity of 3.24 surpassed the corresponding 2008 upper bound. Similar to PIMs, there was a large contribution of solubility to overall CO2 permeability for both 3.23 and 3.24. However, a strong dependence of gas transport properties on the nature of substituents (R, Scheme 3) was observed. For example, polymer 3.24 exhibited much higher gas permeability than 3.23, which is consistent with the difference in BET surface areas and can be explained by the difference in free volume of polymers bearing methoxy (–OMe) and trifluoromethyl (–CF3) substituents.
| Polymer | Sample treatmenta | Aging time (h) | P (Barrer) | D × 108 (cm2 s−1) | α ij = Pi/Pj | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| H2 | N2 | O2 | CO2 | O2 | CO2 | O2/N2 | CO2/N2 | |||
| a A: film was soaked in ethanol for 48 h, dried in air for 24 h, and dynamic vacuum applied at 35 °C for 8 h; A1: film was soaked in ethanol for 48 h, dried in air for 24 h, and dynamic vacuum applied at 35 °C for 4 h; B: film was thermally annealed at 120 °C under dynamic vacuum for 24 h, treated by methanol vapor at 180 mbar (partial pressure of methanol) for 12 h, and dynamic vacuum applied at 100 °C for 16 h; C: film was thermally annealed at 120 °C under dynamic vacuum for 24 h, treated by methanol vapor at 160–200 mbar (partial pressure of methanol) for 12 h and dynamic vacuum applied at 70 °C overnight; D: film was thermally annealed at 120 °C for 24 h, and dynamic vacuum applied. | ||||||||||
| 3.23 | A | 1 | 1409 | 153 | 414 | 2900 | 153 | 91.4 | 2.7 | 19 |
| A | 1000 | 1127 | 112 | 313 | 2154 | 134 | 74.3 | 2.8 | 19 | |
| B | 1 | 535 | 46 | 141 | — | 78.4 | — | 3.1 | — | |
| C | 100 | 698 | 58 | 181 | 1357 | 120 | 97.4 | 3.1 | 23 | |
| D | 48 | 587 | 52 | 150 | 1072 | 54.5 | 36.0 | 2.9 | 21 | |
| 3.24 | A1 | 1 | 8303 | 2367 | 4354 | 21266 |
995 | 633 | 1.8 | 9.0 |
| A | 1 | 8327 | 1993 | 4035 | 18490 |
888 | 513 | 2.0 | 9.3 | |
| A | 1000 | 7285 | 1464 | 3326 | 15104 |
632 | 418 | 2.3 | 10 | |
| B | 500 | 4736 | 935 | 2088 | 9919 | — | — | 2.2 | 11 | |
| C | 80 | 4864 | 1195 | 2346 | 11144 |
— | 461 | 2.0 | 9.3 | |
| C | 1100 | 4661 | 1008 | 2187 | 10324 |
— | 443 | 2.2 | 10 | |
| D | 120 | 3048 | 523 | 1244 | 6361 | 429 | 257 | 2.4 | 12 | |
| PIM-1 | C | 1 | 5251 | 777 | 2177 | 12318 |
517 | 268 | 2.8 | 16 |
| C | 1000 | 4437 | 576 | 1736 | 10005 |
450 | 293 | 3.0 | 17 | |
| D | 1 | 3293 | 444 | 1258 | — | — | — | 2.8 | — | |
Similar to PIMs, the gas transport properties of polymers with side chain porosity (3.23–3.24) demonstrated a dramatic dependence on the history of a polymer sample and aging time. The most permeable films were obtained after soaking in ethanol, drying in air and evacuation for 4 h.77 Significantly lower permeability was observed after thermal treatment of polymer films, which was expected as heat treatment is known to accelerate the aging process. This aging phenomenon was more pronounced for the less permeable 3.23, as compared to 3.24, which could be explained by stability of smaller free volume elements created by the trifluoromethyl-substituted polymer.
An important aspect to consider regarding these materials is that plasticization of the polymer matrix (by penetrant molecules) may promote a reduction of selectivity at high feed pressure. CO2 permeation studies of 3.23 and 3.24 at feed pressures up to 51 bar indicate that the point of plasticization for these materials is above 51 bar, which is significantly higher than those measured for other PIM materials.77 This effect was attributed to high polymer chain cohesion energy that is typical of ROMP polymers.
Sundell, Lawrence, and coworkers outlined a new approach to the synthesis of PNBs bearing aromatic substituents.23 This novel approach was sub-divided into two mechanistically distinct synthetic routes (Scheme 4). The first involves CANAL between 2,5-norbornadiene and a brominated biphenyl reagent, resulting in exo-TCN containing biphenyl fragment. The second route utilized Heck coupling between 2,5-norbornadiene and a brominated biphenyl reagent, resulting in exo-biphenyl-substituted NB. In contrast to monomers synthesized via traditional Diels–Alder reactions, both routes selectively yielded exo-substituted NBs that exhibit high polymerization activity.
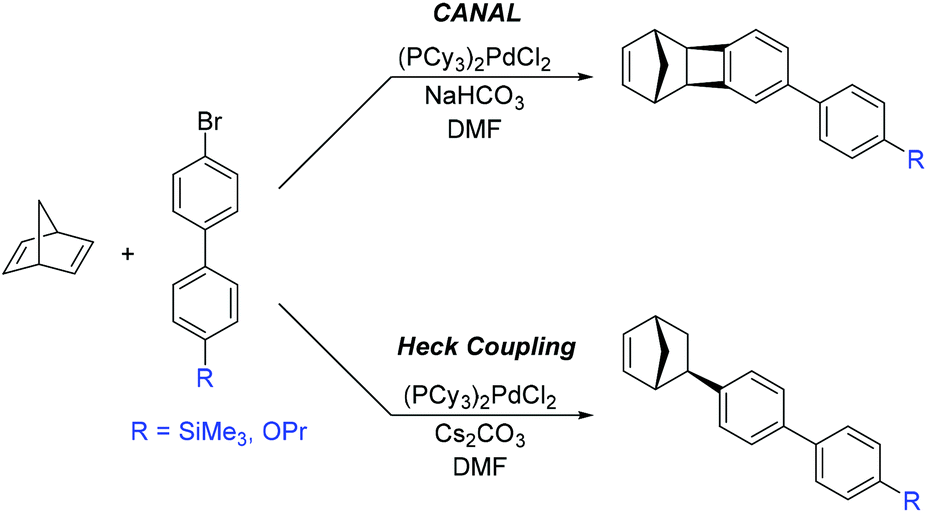 | ||
| Scheme 4 Synthesis of norbornenes bearing biphenyl substituents. | ||
A series of MPNBs and APNBs bearing biphenyl substituents were synthesized using either Grubbs 1st generation catalyst (ROMP) or a palladium-based addition polymerization catalyst (Fig. 11) to yield either propoxy (–O(CH3)2CH3) or trimethylsilyl (–SiMe3) substituted polymers.23 For this polymer series, two correlations between structure and gas transport properties were observed. First, addition polymers primarily displayed increased gas permeability as compared to their metathesis analogues. Second, the overall gas permeability of these polymers depended heavily on the monomer synthetic route chosen (CANAL versus Heck coupling). In general, monomers synthesized via CANAL provided polymers with higher gas permeability and higher free volume than those prepared through the Heck route. This could be explained by the fact that fused cyclobutane ring formed during CANAL reaction contributes to the rigidity of the monomer/polymer structure (e.g. polymers 3.26 and 3.28–3.29), whereas the norbornyl–biphenyl bond formed in the Heck reaction is susceptible to bond rotation (e.g. polymer 3.25 and 3.27).
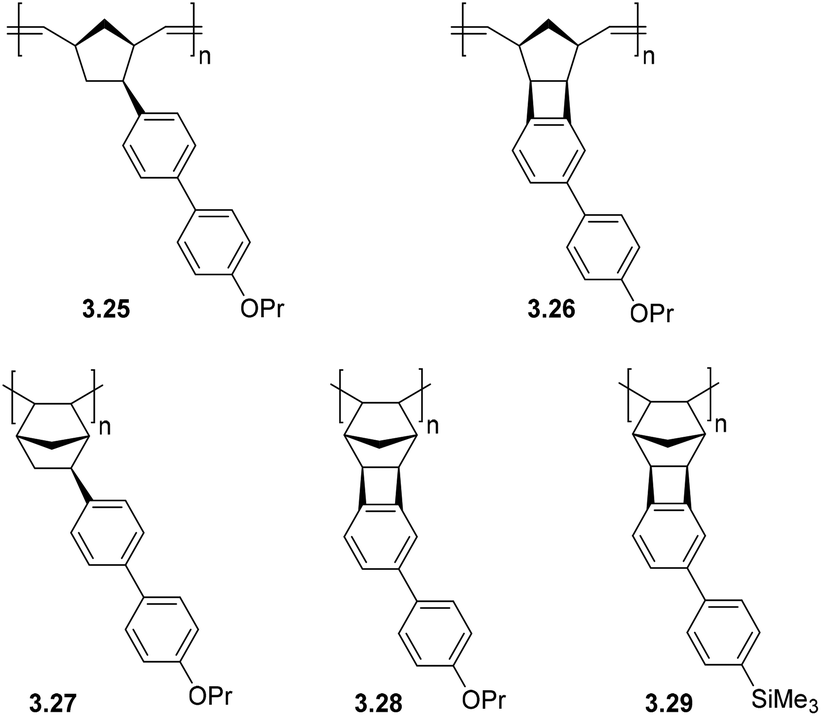 | ||
| Fig. 11 Addition and metathesis polynorbornenes bearing biphenyl substituents. | ||
Metathesis and addition polynorbornenes bearing propoxy (–O(CH2)2CH3) substituted biphenyl moieties (3.25–3.28) displayed low gas permeabilities as compared to trimethylsilyl substituted analogues (e.g.3.29) (Table 8). This was hypothesized to be a consequence of π–π stacking interactions between aromatic rings in polymers 3.25–3.28, which promotes dense packing of the chains and reduced gas diffusion. A significant increase in gas permeability and FFV was observed when replacing propoxy groups (–O(CH2)2CH3) in addition polymers with trimethylsilyl (–SiMe3) groups (3.29, Table 8). Gas separation selectivity of the polymers from this series obeyed traditional permeability–selectivity trade-off behavior; however, a valuable feature of these APTCNs was their high solubility-controlled selectivities for hydrocarbon separations (α ≤ 28 for C4/C1). It is worth noting that for most PNBs, higher C4/C1 gas separation selectivities are observed only in the case of PNBs bearing alkoxysilyl fragments (see section 2.3). Furthermore, PNBs (including PTCNs) containing biphenyl fragments also displayed high mixed hydrocarbon separation selectivities (Table 9). The highest values were obtained for the polymer bearing trimethylsilyl substituents (3.29). These values are comparable to those of PNBs bearing alkoxysilyl substituents.23 Therefore, the introduction of aromatic rings bearing silyl- or alkoxy-substituents in the side chain of polynorbornenes may be a promising approach to enhance hydrocarbons separation selectivity.
| Polymer | FFVa (%) | P (Barrer) | α ij = Pi/Pj | ||||||
|---|---|---|---|---|---|---|---|---|---|
| He | N2 | O2 | CO2 | CH4 | n-C4H10 | He/N2 | C4/C1 | ||
| a FFV was calculated using a computational model. | |||||||||
| 3.25 | — | 7.0 | 0.46 | 1.57 | 8.33 | 0.63 | — | 15.2 | — |
| 3.26 | — | 9.5 | 0.7 | 2.24 | 12.5 | 0.85 | — | 15 | — |
| 3.27 | 26.4 | 11.9 | 1.2 | 3.9 | 27 | 2.17 | — | 10.3 | — |
| 3.28 | 31.4 | 25.8 | 3.39 | 11.1 | 77 | 6.35 | 179 | 7.6 | 28 |
| 3.29 | 33.9 | 150 | 41 | 120 | 710 | 84 | 1800 | 3.8 | 21 |
| Polymer | p (psi) | P (Barrer) | α(C3/C1) | α(C4/C1) | |||
|---|---|---|---|---|---|---|---|
| CO2 | CH4 | C3H8 | n-C4H10 | ||||
| 3.26 | 500 | 144 | 24.0 | 84 | 190 | 3.5 | 7.9 |
| 3.26 | 800 | 194 | 32.7 | 134.4 | 321.6 | 4.1 | 9.8 |
| 3.28 | 500 | 253 | 46 | 210 | 500 | 4.5 | 10.9 |
| 3.28 | 800 | 310 | 58 | 290 | 740 | 5.1 | 12.8 |
| 3.29 | 500 | 1200 | 280 | 1800 | 5000 | 6.5 | 17.8 |
| 3.29 | 800 | 1400 | 330 | 2300 | 6500 | 7.1 | 19.6 |
In summary, introducing hydrocarbon or carbocyclic substituents in the side chains are both fruitful approaches to enhance the gas transport properties of PNBs. Firstly, the introduction of long alkyl fragments (e.g. octyl and decyl groups) was found to increase hydrocarbon separation selectivity. Therefore, it is possible to switch on/off solubility-controlled permeation of hydrocarbons for polymers using this approach. It should be expected that the introduction of longer alkyl groups (e.g. dodecyl groups) might result in a further increase in C3+/C1 hydrocarbon separation selectivity.
Secondly, the introduction of polycyclic fragments in the side chain of MPNBs proved to be a powerful tool to enhance PNB gas permeability and gas separation selectivity (especially, for CO2/N2 separation). Further development of this approach would be interesting as it can be expected that APNBs bearing analogous polycyclic substituents to possess even more attractive combination of gas permeability and separation selectivity. Lastly, the synthesis of PNBs bearing aromatic substituents in the side chain via Heck or CANAL reactions was efficient and simple. Utilizing this approach, it was established that APNBs containing biphenyl fragments possess a high selectivity of hydrocarbons separation (for individual and mixed gases), and further polymer structure tuning may lead to attractive combinations of permeability and selectivity.
3.3. Polynorbornenes bearing imide moieties
A promising class of monomers for polynorbornene-based membranes are 5-norbornene-2,3-dicarboxyimides (NB-DCIs) (Scheme 5). A valuable feature of NB-DCIs is the simplicity in their synthesis. They can be obtained by the reaction of 5-norbornene-2,3-dicarboxyanhydride with an amine or aniline, followed by subsequent dehydration of the resultant amic acid.66,70,81,82 The associated polymers 3.30–3.35 contain imide moieties, drawing similarity to heavily studied polyimides. While traditional polyimides typically display low gas permeability, they exhibit desirable separation selectivity, as well as excellent thermomechanical properties. These characteristics make them attractive candidates for membrane materials. To date, gas transport studies of poly(NB-DCIs) have been limited to metathesis polymers, while addition polymerization of NB-DCIs usually leads to low-molecular-weight products that are poorly suited for membrane fabrication. ROMP polymerization of synthesized NB-DCIs results in MPNBs 3.30–3.35 bearing substituted imide moieties (Scheme 5).66,70,71,81–87 Exhaustive hydrogenation of the polymers in the presence of Wilkinson's catalyst or diimide reduction leads to saturated polymers h3.30–h3.35, which imbues the materials with enhanced chemical stability relative to unsaturated metathesis polymers.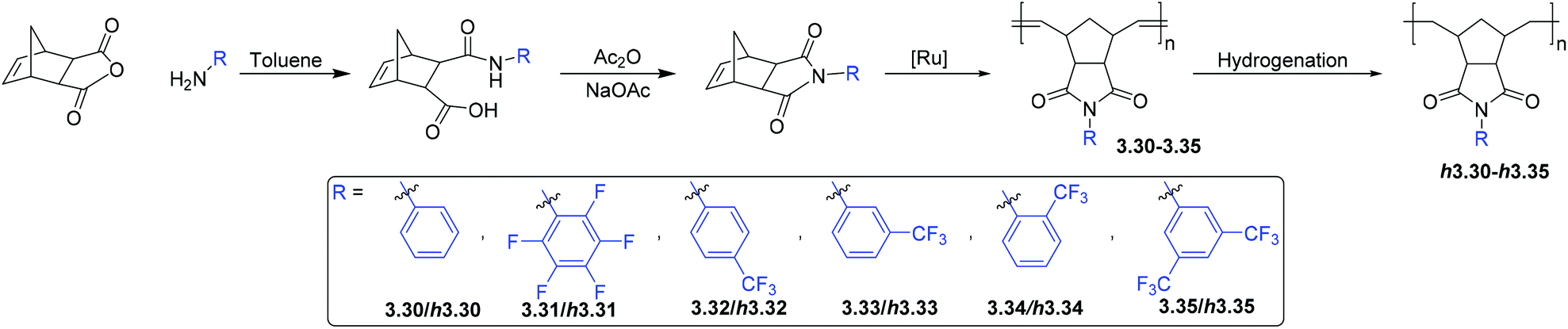 | ||
| Scheme 5 Synthesis and polymerization of NB-DCIs. | ||
Gas transport properties of metathesis poly(NB-DCI)s have been extensively studied from 2000 to 2010,70,71,81,83–87 and were described in a previous review.14 These polymers displayed comparatively low gas permeability that could be explained by strong interchain dipole–dipole interactions between imide moieties. However, they displayed attractive CO2/CH4, H2/C3H6 and CO2/N2 selectivities. It was also observed that the introduction of rigid and bulky carbocyclic substituents (e.g. cyclopentyl, cyclohexyl, adamantyl groups) onto poly(NB-DCI)s did not result in noticeable increase in gas permeability.83,85
A significant increase in gas permeability was observed when fluorine-containing substituents were appended to poly(NB-DCI)s (polymers 3.31–3.35).70,71 The effect was stronger in the case of trifluoromethyl (–CF3) groups bonded via an aromatic ring, and the position of trifluoromethyl group about the aromatic ring was also shown to alter the gas transport properties of poly(NB-DCI)s.66 Therein, polymers bearing trifluoromethyl substituents in para- or ortho-positions (3.32 and 3.34, respectively) displayed higher gas permeabilities than polymer 3.33 that bears a trifluoromethyl substituent in the aromatic ring's meta-position (Table 10). The higher gas permeabilities of polymers 3.32 and 3.34 was accompanied by both higher diffusivity and higher solubility coefficients than was observed for 3.33.
| Polymer | P (Barrer) | D(CO2)a | S(CO2)b | α ij = Pi/Pj | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| CO2 | N2 | CH4 | C3H6 | CO2/CH4 | H2/C3H6 | ||||
| a Diffusion coefficient in 10−8 cm2 s−1. b Solubility coefficient in 10−2 cm3(STP) cm−3 cmHg−1. | |||||||||
| 3.30 | 11.4 | 0.31 | 0.51 | — | 1.81 | 6.32 | 21 | — | 81 |
| 3.31 | 34.6 | 1.30 | 1.48 | 1.08 | 2.62 | 13.2 | 23 | 36 | 66 |
| 3.32 | 9.8 | 0.36 | 0.37 | 0.26 | 0.91 | 10.9 | 27 | 58 | 66 |
| 3.33 | 34.4 | 1.47 | 1.59 | 1.20 | 2.16 | 16.0 | 22 | 29 | 66 |
| 3.34 | 67.3 | 4.20 | 4.28 | 3.79 | 4.81 | 14.0 | 16 | 15 | 70 |
| 3.35 | 25.2 | 1.55 | 1.37 | 1.24 | 1.50 | 17.1 | 18 | 31 | 70 |
| h3.30 | 4.51 | 0.12 | 0.15 | — | 0.72 | 6.26 | 30 | — | 81 |
| h3.34 | 26.1 | 1.32 | 1.24 | 1.20 | 3.14 | 8.32 | 21 | 31 | 82 |
| h3.35 | 6.12 | 0.30 | 0.27 | 0.30 | 0.79 | 7.73 | 23 | 43 | 82 |
Hydrogenation of metathesis poly(NB-DCI)s (h3.30–h3.35) resulted in reduced gas permeability, which was accompanied by a decrease in both diffusion and solubility coefficients (Table 10).82 In general, gas separation selectivities of hydrogenated poly(NB-DCI)s were significantly higher than those of initial ROMP polymers (Table 10). Also, it was found that CO2/CH4 separation selectivity for these polymers weakly depended on the position of trifluoromethyl groups, whereas H2/C3H6 separation selectivity was significantly higher (α = 58) in the case of polymer 3.32 that bears a meta-substituted trifluoromethyl group (Table 10).
As previously mentioned, attempts to synthesize AP(NB-DCI)s have typically resulted in low molecular weight materials with poor film forming properties, and are therefore absent from the literature. However, taking into account the higher gas permeability and selectivity of most glassy addition polymers, as compared to their analogous metathesis polymers, synthesis of AP(NB-DCI)s is an interesting avenue for future research.
4. Polynorbornenes synthesized via catalytic arene-norbornene annulation (CANAL)
APNBs containing norbornene fragments in the main chain possess high chain rigidity, which typically results in large free volumes and enhanced gas permeabilities (see section 2.2). Recent reports have demonstrated that these rigid bicyclic motifs may be incorporated into the main chain of a polymer through the use of catalytic arene-norbornene annulation (CANAL).31 Owing to extreme rigidity of the structures formed by CANAL, the reaction is a powerful tool for designing microporous materials for gas storage and gas separation membrane applications.There are two prevailing synthetic strategies in which CANAL has been applied to the synthesis of new polymeric materials. The first strategy is the direct usage of CANAL for the synthesis of ladder polymers containing alternating norbornyl and aryl fragments (Scheme 6).41 It is worth mentioning that when functionalized norbornene derivatives are applied in the polymerization, polymers with precise sequence control (i.e. polymers that contain perfectly alternating aryl substituents along the main chain) can be achieved, providing a powerful synthetic tool for targeted macromolecular design.
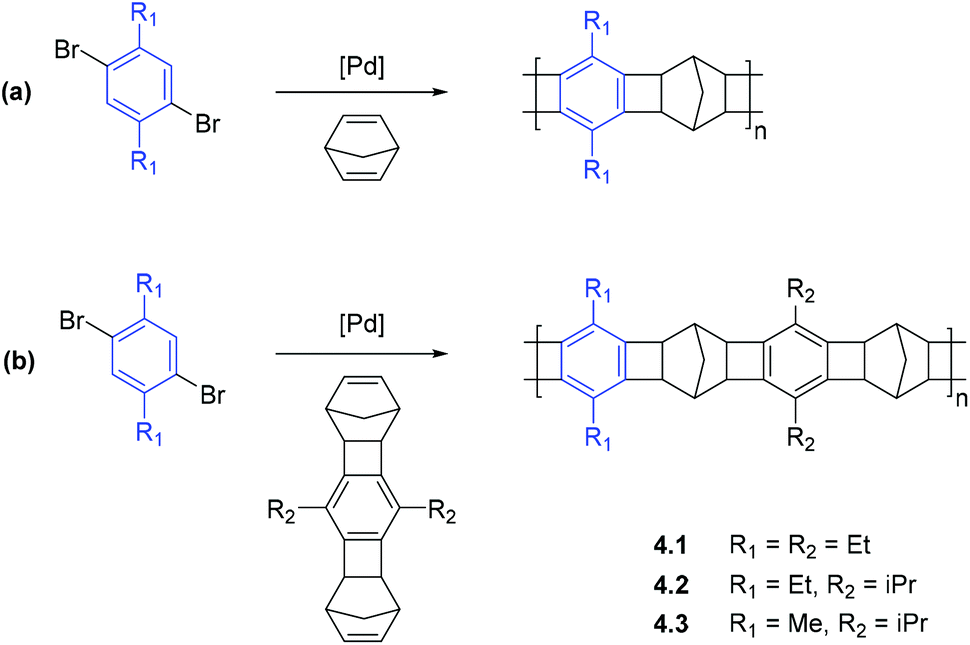 | ||
| Scheme 6 Synthesis of ladder copolymers via CANAL using (a) norbornadiene or (b) functionalized dinorbornene derivatives.41 | ||
Xia, Smith, and coworkers reported the synthesis of a series of glassy ladder polymers bearing alkyl (methyl, ethyl, isopropyl) substituents via CANAL (4.1–4.3, Scheme 6), and provided a detailed analysis of their gas transport properties.41 The synthesized ladder CANAL polymers 4.1–4.3 showed moderate to high gas permeability, yet were less permeable than trimethylsilyl (–Me3Si) substituted APNBs. Nonetheless, their gas transport properties were similar to APNBs with flexible Si-containing side chains. The nature of alkyl groups in CANAL ladder polymers exerted a complex influence on gas permeability, and the most highly permeable polymer was 4.3 that bears both methyl and isopropyl substituents in its monomer units. The replacement of these alkyl groups by ethyl groups led to a decrease in gas permeability. This trend correlates with the change of FFV values (Table 11) and surprisingly, the increase in gas permeability mainly resulted from the increase in solubility coefficients, whereas diffusivity coefficients changed nonlinearly regardless of FFV values (Table 12). Gas separation selectivities for ladder CANAL polymers with different alkyl groups were similar (Table 11), which was attributed to the similarity of diffusivity selectivity and solubility selectivity among these polymers (Table 12).41
| Polymer | S BET (m2 g−1) | FFV (%) | P (Barrer) | α ij = Pi/Pj | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | N2 | O2 | CO2 | CH4 | O2/N2 | CO2/CH4 | H2/N2 | ||||
| a Polymer was heated under vacuum at 120 °C for 24 h, treated with 200—300 mbar of methanol vapor at 35 °C for 8 h, and heated under vacuum at 100 °C for 12 h. b Polymer was heated under vacuum at 120 °C for 24 h. c Polymer was aged for 300 days. | |||||||||||
| 4.1 | — | 23 | 880 | 110 | 310 | 1300 | 270 | 2.8 | 4.8 | 8.0 | 41 |
| 4.1 | — | 23 | 860 | 95 | 270 | 1200 | 200 | 2.8 | 6.0 | 9.1 | 41 |
| 4.2 | — | 26 | 1200 | 150 | 410 | 1800 | 350 | 2.7 | 5.1 | 8.0 | 41 |
| 4.2 | — | 26 | 1200 | 150 | 420 | 1900 | 360 | 2.8 | 5.3 | 8.0 | 41 |
| 4.3 | — | 27 | 1500 | 190 | 490 | 2200 | 380 | 2.6 | 5.8 | 7.9 | 41 |
| 4.3 | — | 27 | 1550 | 180 | 500 | 2300 | 390 | 2.8 | 5.9 | 8.6 | 41 |
| 4.4 | 216 | 26 | 462 | 20.7 | 81 | 419 | 16.3 | 3.9 | 26 | 22 | 30 |
| 4.5 | 533 | 29 | 1154 | 91 | 319 | 1691 | 108 | 3.5 | 15 | 13 | 30 |
| 4.6 | 199 | 25 | 282 | 7.0 | 33 | 157 | 4.8 | 4.7 | 33 | 40 | 30 |
| 4.7 | 881 | 28.6 | 2760 | 97 | 463 | 1678 | 121 | 4.8 | 13.9 | 28.5 | 42 |
| 4.7 | 881 | 28.6 | 1163 | 39 | 204 | 749 | 53 | 5.2 | 14.1 | 29.8 | 42 |
| 4.8 | 987 | 29.1 | 3608 | 162 | 747 | 2520 | 205 | 4.6 | 12.3 | 22.3 | 42 |
| 4.8 | 987 | 29.1 | 2452 | 110 | 528 | 1751 | 129 | 4.8 | 13.6 | 22.4 | 42 |
| Polymer | D × 108 (cm2 s−1) | S × 102 (cm3(STP) cm−3 cmHg−1) | α D | α S | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| O2 | CO2 | O2 | CO2 | O2/N2 | CO2/CH4 | O2/N2 | CO2/CH4 | ||
| a Polymer was heated under vacuum at 120 °C for 24 h, treated with 200—300 mbar of methanol vapor at 35 °C for 8 h, and heated under vacuum at 100 °C for 12 h. b Aged for 300 days. | |||||||||
| 4.1 | 160 | 91 | 1.9 | 14 | 1.9 | 2.3 | 1.4 | 2.15 | 41 |
| 4.2 | 200 | 130 | 2.1 | 14 | 2.3 | 2.5 | 1.2 | 2.2 | 41 |
| 4.3 | 160 | 85 | 3.1 | 26 | 1.9 | 2.1 | 1.35 | 2.6 | 41 |
| 4.4 | 37.6 | 12.9 | 2.16 | 32.5 | 3.6 | 5.2 | 1.1 | 4.8 | 30 |
| 4.5 | 118 | 39 | 2.7 | 43.4 | 3.2 | 3.1 | 1.1 | 5.0 | 30 |
| 4.6 | 31.8 | 10.5 | 1.04 | 15.0 | 4.0 | 5.8 | 1.2 | 5.5 | 30 |
| 4.7 | 94 | 34.7 | 4.9 | 48 | 4.22 | 4.95 | 1.13 | 2.80 | 42 |
| 4.7 | 53 | 20.9 | 3.9 | 36 | 4.78 | 5.09 | 1.09 | 2.79 | 42 |
| 4.8 | 175 | 58.0 | 4.3 | 43 | 3.98 | 5.09 | 1.16 | 2.42 | 42 |
| 4.8 | 130 | 43.0 | 4.1 | 41 | 4.13 | 5.21 | 1.16 | 2.60 | 42 |
The second synthetic strategy employing CANAL is to access rigid monomeric species that can be used in subsequent polycondensation reactions (Scheme 7).30,42 For instance, CANAL reaction between 2,5-norbornadiene and various substituted bromoanilines yields diamine functionalized norbornane fragments that have been used for the synthesis of polyimides and PIM-like materials. For example, diamine functionalized monomers synthesized via CANAL may be reacted with dianhydrides, such as 6FDA, to yield polyimides (4.4–4.6).30 These unique oligocyclic monomers are of further interest in that the angle between the two planes in which terminal aniline fragments are placed was found to be ∼129°.30 In combination with the rigidity of the main chain, this structural characteristic draws close analogy to those used to access PIMs. Indeed, reaction of CANAL diamine monomers with formaldehyde dimethyl acetal yields polymers that feature both CANAL and Tröger's base units along the polymer main chain (4.7–4.8).42
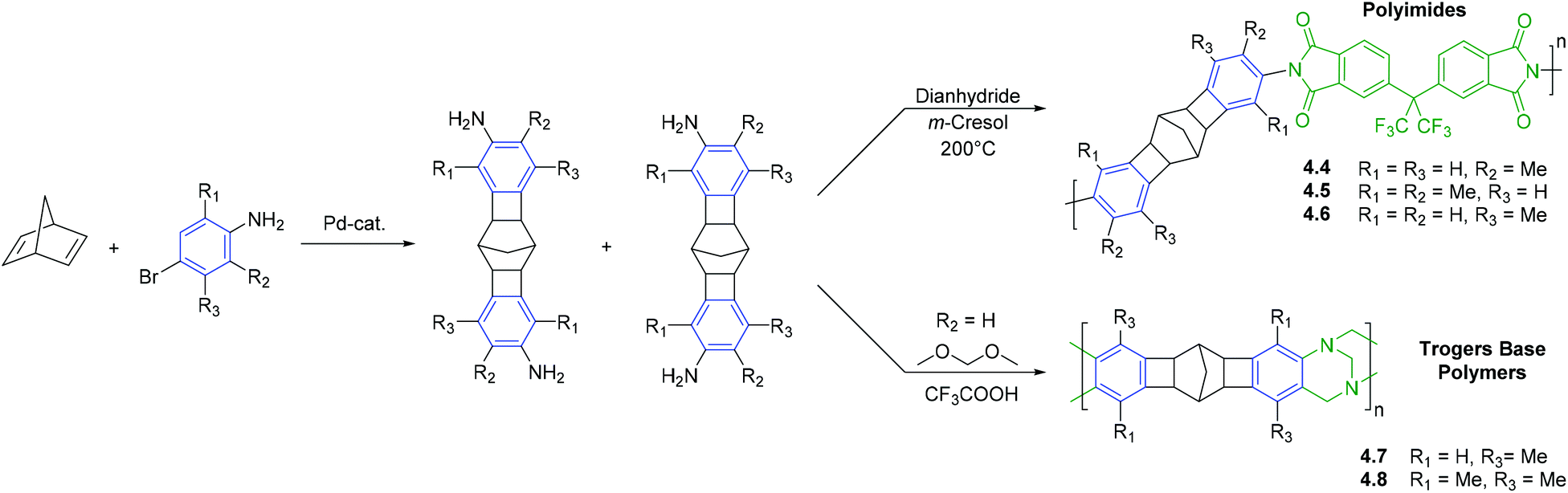 | ||
| Scheme 7 Synthesis of polyimides or Tröger's base polymers that contain subunits prepared via CANAL. | ||
CANAL polymers 4.4, 4.5, and 4.6 displayed moderate to high gas permeability and noticeably increased gas separation selectivities (Table 11). A strong influence of the number and position of the methyl groups on gas permeability was also observed. The transition from one to two methyl groups per aromatic ring (4.4 and 4.5), as well as ortho- or meta-orientation of the methyl groups relative to imide linkage (4.4 and 4.6), resulted in significant increase in gas permeability level. The influence was consistent with difference in BET surface areas, as well as FFV calculated by molecular dynamics simulation and WAXD data. The diffusion and solubility coefficients of CANAL-polyimides were found to be 1–2 orders of magnitude higher than those of non-porous polyimides (Table 12).30 CANAL-polyimide gas separation selectivities were notable: α ≤ 4.7, 33, 40 for O2/N2, CO2/CH4 and H2/N2 gas pairs, respectively. It was emphasized that gas transport characteristics of these polymers are closer to 2008 upper bound for CO2/CH4 gas separations than those of commercial polyimides and a number of PIM-PIs.30
The incorporation of Tröger's base-containing repeat units into the polymer main is another commonly used tool for tuning gas transport properties.78,88–90 Tröger's base polymers featuring CANAL units (4.7 and 4.8) exhibited high gas permeability (P(CO2) = 1600–2500 Barrer), as well as high gas separation selectivities (α ≤ 4.8, 13.9, 28.5 for O2/N2, CO2/CH4 and H2/N2 gas pairs, respectively) (Table 12). The gas separation performance of polymers 4.7 and 4.8 was above the 2008 upper bound for O2/N2 gas separations, making them attractive for O2 separations form air.
The aging of polymers 4.7 and 4.8 was unusual in that though a reduction in gas permeability coefficients were observed as a function of time, they only exhibited a small increase in gas separation selectivity after aging (Table 11), which contrasts what is typically observed for other PIM materials. The authors suggested that the reason for this may be the collapse of large free volume elements without the formation of size-selective ultramicropores (<7 Å). Surprisingly, the more permeable 4.8 was less prone to aging, in which P(O2) decreased by only 29% after 300 days of aging, whereas polymer 4.7 decreased by 56%. This aging resulted in significant reductions in diffusion coefficients and a slight increase in diffusion selectivity (αD) values, whereas there was no notable effect of aging on solubility selectivity (αS) values (Table 12).42
In summary, CANAL chemistry has proven to be a powerful tool in the design of new polymer membrane materials for gas separation. Gas permeability of CANAL polymers derived from norbornene derivatives was found to be lower than APNBs, but higher than MPNBs. The versatility of the CANAL reaction and promising combination of gas permeability and selectivity make CANAL polymers a potential alternative to traditional PIMs and TR-polymers.91,92 The persistence of gas transport properties over time for CANAL ladder polymers is a current practical limitation and will need to be addressed if these materials are to realize their full potential.
We anticipate that future research efforts will focus heavily on the development of new CANAL-derived monomers and polymers. For example, a new series of CANAL ladder polymers were recently synthesized via the annulation of norbornadiene with five-membered heteroaryl halides to yield from dinorbornene monomers.93 These monomers were then polymerized in the presence of dihaloarenes. Based on the structural rigidity and non-parallel orientation of the norbornyl units within these polymers, it may be surmised that these materials may have promising gas transport properties, similar to those of traditional PIMs.
Another potential way to expand the utility of CANAL reactions for gas separation membranes could be to use oxanorbornene moieties rather than simple norbornene-based species.94 It is hypothesized that the ladder polymers containing oxanorbornane fragments may possess enhanced CO2 permeability due to the presence of additional polar groups. For example, if the CO2/N2 separation selectivities of CANAL polymers containing oxanorbornene motifs are estimated from prior reports, it is anticipated that they will be rather high (values of CO2/N2 selectivity calculated from original data ranged from α = 12–22). Given the promising synthetic potential for the introduction of various substituents into the side chain of CANAL polymers, we expect that materials with increased selectivity for CO2/N2 separation based on the CANAL reaction will be a fruitful avenue of research in the future.
5. Post-polymerization modification (PPM) of polynorbornenes
PPM of polymeric materials is an additional method by which thermomechanical and gas transport properties may be tuned.95 PPM obviates the need to design and synthesize new monomers and polymers by harnessing reactive moieties present within polymeric structures. Although this approach possesses several advantages over the traditional approach of new monomer and polymer design, it has only recently been applied to improve the gas permeability of PNBs.5.1. Modification of main chain double bonds in MPNBs
The presence of double bonds in the main chain of MPNBs provides a reactive handle for targeted PPM. There are examples of MPNB backbone double bonds modified by hydrogenation,56,82,96 sulfonation,97,98 bromination,99–101 epoxidation,100,102,103 catalytic dihydroxylation,104 cycloaddition reaction,105 thiol–ene chemistry,106etc. Nevertheless, only hydrogenation,56,82,96 epoxidation, and gem-difluorocyclopropanation107–109 have been used as a means of tuning gas-transport properties of MPNBs (Scheme 8). | ||
| Scheme 8 (a) Epoxidation and gem-difluorocyclopropanation of double bonds along the backbone of MPNBs 5.1 and 5.2108,112 and (b) poly(4,4-dimethyltricyclo[5.2.1.02,6]-4-siladec-8-ene).109 Conditions: (i) m-chloroperbenzoic acid; (ii) sodium chlorodifluoroacetate, Δ. | ||
Most often, MPNBs are hydrogenated to obviate oxidation reactions that compromise the chemical stability of unsaturated MPNBs. Exhaustive hydrogenation of MPNBs is generally performed via diimide reduction, which is generated in situ from p-toluenesulfonylhydrazide,56,82,96,110 or by using hydrogen over Ru- or Pd-catalysts.110,111 The effect of hydrogenation on the gas permeability of MPNBs has been described in previous reviews,14,54 and in most cases it has been found to lead to a slight decrease in permeability coefficients.
The main goal of epoxidation and gem-difluorocyclopropanation of MPNBs was to increase the rigidity of the polymer backbone, which would hypothetically result in higher gas permeability. Epoxidation of MPNBs was carried out using m-chloroperbenzoic acid though some degradation of the MPNBs was observed, which was more prevalent when using chloroform as a solvent than toluene.112 However, careful selection of reaction conditions resulted in the exhaustive epoxidation of double bonds to yield polymers 5.3–5.4. gem-Difluorocyclopropanation of MPNBs was performed by using difluorocarbene, which was generated in situ. The presence of bulky trimethylsilyl (–SiMe3) substituents in MPNBs (e.g.5.2, Scheme 8) significantly reduced the rate of both epoxidation and difluorocyclopropanation of the double bonds. In the case of difluorocyclopropanation of 5.2, it was impossible to modify all double bonds with polymer 5.6 reaching ∼84% conversion to the difluorocyclopropanated form.108 The authors attributed the lower activity of double bonds in MPNB 5.2 to steric shielding by the trimethylsilyl substituents.108 Indeed, it was found that moving the silicon-containing fragment away from the double bond in silacyclopentane-containing MPNB 5.8 yielded complete difluorocyclopropanation of the double bonds to produce polymer 5.9 (Scheme 8b).109 It should be noted that polymer 5.9 is the first reported PNB with both silicon- and fluorine-containing substituents.
Gas permeation testing revealed that epoxidized (5.3) and difluorocyclopropanated (5.5) MPNBs exhibited opposite behaviors in regard to gas permeability, as compared to the native unsaturated MPNB 5.1 (Table 13). For example, the permeability coefficients of epoxidized polymers 5.3 and 5.4 were much lower across all gases tested than 5.1 and 5.2. This decrease in permeability was accompanied by a more significant decrease in diffusion coefficients than was observed for solubility coefficients. The only exception was the S(CO2), which increased after epoxidation. In contrast, gem-difluorocyclopropanation resulted in a significant increase in gas permeability for polymers 5.5, 5.6 and 5.9. This was attributed to the increase in solubility coefficients (especially, in the case of CO2), while the diffusion coefficients did not change significantly.108 Similar to epoxidized MPNBs, the effect of gem-difluorocyclopropanation on gas permeability was less pronounced in the case of the more permeable silicon-substituted MPNBs. Interestingly, this series (5.1, 5.5, 5.6) did not obey typical permeability–selectivity trade-off behavior (Table 13). Moreover, the ideal selectivity for gas pairs containing CH4 increased after difluorocyclopropanation, which is consistent with previous observations that fluorine-containing polymers may have increased ideal selectivity for separations involving CH4 (see section 3.1).113
| Polymer | T g (°C) | P (Barrer) | α ij = Pi/Pj | D CO2/ D N2 | D(O2) × 108 (cm2 s−1) | S CO2/ S N2 | S (cm3 (STP) m−3 atm−1) | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| O2 | N2 | CO2 | CH4 | O2/N2 | CO2/N2 | CO2/CH4 | O2 | CO2 | ||||||
| 5.1 | 41 | 2.3 | 0.43 | 9.3 | 0.78 | 5.2 | 21 | 12 | 1.5 | 12.8 | 14 | 0.14 | 1.3 | 107 |
| 5.2 | 103 | 34 | 8.3 | 140 | 19 | 4.1 | 17 | 7.4 | 1.4 | 57 | 12 | 0.45 | 3.6 | 107 |
| 5.3 | 64 | 0.2 | 0.03 | 1.1 | 0.04 | 6.8 | 35 | 26 | 1.1 | 2.1 | 31 | 0.08 | 2.4 | 107 |
| 5.4 | 118 | 10 | 2.2 | 47 | 4.2 | 4.7 | 22 | 11 | 0.8 | 22 | 23 | 0.36 | 5.2 | 107 |
| 5.5 | 94 | 5.1 | 1.1 | 23.1 | 1.5 | 4.6 | 21 | 15 | 1.1 | 11.5 | 20 | 0.34 | 4.4 | 108 |
| 5.6 | 117 | 44 | 11 | 217 | 21 | 4.0 | 20 | 10 | 1.2 | 56.5 | 17 | 0.60 | 6.6 | 108 |
| 5.7 | 90 | 3.3 | 0.64 | 13.8 | 0.77 | 5.2 | 22 | 18 | 0.8 | 6.7 | 27 | 0.37 | 5.6 | 108 |
| 5.8 | 111 | 5.7 | 1.2 | 24 | 2.5 | 4.8 | 20 | 9.7 | 1.4 | 17.8 | 14 | 0.24 | 2.1 | 109 |
| 5.9 | 171 | 26 | 9.2 | 115 | 13.5 | 2.8 | 13 | 8.5 | 0.7 | 26.2 | 18 | 0.75 | 8.8 | 109 |
An interesting result was observed for a polymer 5.7, which contains both epoxidized and difluorocyclopropanated monomer units (Table 13).108 Dual modification of MPNB led to a simultaneous increase in selectivity and permeability, and the resulting “copolymer” possessed the highest S(CO2) value among its analogues (5.1, 5.3, 5.5). This was hypothesized to result due to the combination of increased gas permeability arising from the presence of CF2 units, as well as increased CO2 solubility arising from the epoxy groups present. These modified polymers demonstrated reasonably high thermal stability, good mechanical properties, and better storage stability under ambient conditions, as compared to films of unmodified MPNB 5.1.108
5.2. Modification of side chain in APNBs
In contrast to MPNBs, the fully saturated APNB backbone provides no reactive entities upon which PPM can be performed. Therefore, the most promising means of PPM of APNBs is through transformations of reactive pendant moieties along the main polymer chain.114–116 It should be noted that the synthesis of high molecular weight APNBs that possess reactive side groups, as well as good film-forming and mechanical properties for measuring gas transport parameters, is a challenging synthetic problem.With an active three-component system featuring a Pd-based catalyst bearing an N-heterocyclic carbene, high-molecular-weight APNB poly(5-vinylnorbornene), which contains vinyl pendant groups, was synthesized.15,33 The appendant vinyl groups provided suitable functional handles for PPMs including hydrogenation (5.11), cyclopropanation (5.12), epoxidation (5.13), and thioacetylation (5.14) (Scheme 9).117 In most cases, an exhaustive conversion of double bonds was achieved with no observable reduction in polymers molecular weights. However, isomerization of the vinyl double bond was observed during radical thioacetylation attempts. As a result, the synthesized polymer contained ∼10% unreacted ethylidene-norbornene units (Scheme 9).
 | ||
| Scheme 9 PPM of side double bonds in addition poly(5-vinylnorbornene).117 | ||
Hydrogenated and cyclopropanated polymers 5.11 and 5.12, respectively, showed increased gas permeabilities, which was primarily due to increases in diffusion coefficients (Table 14). The highest selectivity for these polymers was observed for the gas pairs CO2/CH4, CO2/N2 and H2/CH4, and no traditional permeability/selectivity trade-off relationship was observed for cyclopropanated polymer 5.12 as compared to unmodified 5.10.117 These polymers also exhibited unusual solubility-controlled selectivity for light hydrocarbons (αC4/C1 > 1), which is usually observed for rubbery polymers and highly permeable porous glassy polymers.118
| Polymer | P (Barrer) | α ij = Pi/Pj | D CO2/ D N2 | D × 108 (cm2 s−1) | S CO2/ S N2 | S (cm3 (STP) m−3 atm−1) | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| O2 | N2 | CO2 | C1 | C4a | CO2/N2 | CO2/CH4 | C4/C1a | CO2 | CH4 | CO2 | C4a | ||||
| a S(C4) is from ref. 118. | |||||||||||||||
| 5.10 | 48 | 13 | 310 | 24 | — | 24 | 13.1 | — | 2.1 | 31 | 6.2 | 11 | 13 | — | 15 |
| 5.11 | 72 | 21 | 390 | 37 | 149 | 16 | 9.2 | 4.0 | 2.0 | 45 | 10 | 8 | 10 | 180 | 117 |
| 5.12 | 70 | 22 | 510 | 44 | 380 | 23 | 11.6 | 8.6 | 1.8 | 57 | 14 | 13 | 12 | 250 | 117 |
| 5.13 | 7.6 | 3.8 | 120 | 7.1 | 27.5 | 31 | 16.8 | 3.9 | 0.8 | 9.1 | 3.0 | 37 | 17 | 200 | 117 |
| 5.14 | 1.3 | 0.6 | 33 | 1.5 | 4.7 | 56 | 21.9 | 3.1 | 1.1 | 8.5 | 2.1 | 50 | 5.1 | 68 | 117 |
| 5.15 | — | 0.9 | 25 | — | — | 29 | — | — | 1.6 | 5.0 | — | 19 | 4.1 | — | 59 |
| 5.16 | — | 0.6 | 22 | — | — | 34 | — | — | 1.5 | 2.9 | — | 22 | 5.7 | — | 59 |
| 5.17 | — | 0.7 | 23 | — | — | 32 | — | — | 2.5 | 2.6 | — | 14 | 6.7 | — | 59 |
| 5.18 | — | 0.8 | 24 | — | — | 32 | — | — | 1.1 | 3.5 | — | 31 | 4.9 | — | 59 |
| 5.19 | — | 0.2 | 15 | — | — | 74 | — | — | 1.6 | 2.1 | — | 44 | 5.7 | — | 59 |
Epoxidized and thioacetylated polymers 5.13 and 5.14, respectively, showed a significant decrease in gas permeability as compared to base polymer 5.10 (Table 14).117 This was attributed to the general decrease in both diffusion and solubility coefficients (except S(CO2) of 5.13), and was hypothesized to be due to denser interchain packing arising from dipole–dipole interactions between pendant polar functional groups. The observed increase in S(CO2) for polymer 5.13 (Table 14), as compared to polymers 5.10–5.12 and 5.14, can be explained by specific interactions between CO2 molecules and the epoxy group, which was also observed for other epoxidized MPNBs 5.3 and 5.4 (Table 13). Despite the low permeabilities of epoxidized polymer 5.13 and thioacetylated polymer 5.14, they both exhibited solubility-controlled selectivity (Table 14).
Another approach to PPM of APNBs was the introduction of CO2-philic substituents. Imidazole and amidoxime functionalities have been shown, both experimentally and computationally, to contribute desirable increases in CO2 affinity. This phenomenon has been observed for a variety of polymers, such as PIMs, PTMSP, and others.119–121 However, the direct polymerization of monomers bearing these functional groups has proven problematic due to the incompatibility of commonly employed catalysts with polar and/or heteroatom functionalized monomers. To circumvent this synthetic limitation, the desired polymers were synthesized through PPM of APNBs containing pendant bromomethyl substituents (Scheme 10).59
 | ||
| Scheme 10 Post polymerization modification of addition poly(norbornene-co-5-bromomethyl-2-norbornene) (5.15).59 | ||
Toward this goal, 5-bromomethyl-2-norbornene and unsubstituted norbornene were copolymerized to ensure polymers of sufficient molecular weight to achieve good mechanical properties were obtained (5.15) (Scheme 10). The pendant alkyl bromides atoms were then replaced by the corresponding functional group by nucleophilic substitution reactions (Scheme 10). In the case of benzimidazole, only half of the bromine atoms were converted (polymer 5.16), possibly due to the lower nucleophilicity and greater steric hindrance of benzimidazole.59 Conversely, the imidazole and nitrile functionalized analogs were synthesized with quantitative substitution by imidazole (5.17) and nitrile (5.18), respectively. Amidoxime functionalized polymer (5.19) was insoluble in common NMR solvents, and thus its conversion could not be assessed quantitatively. With the exception of amidoximated polymer 5.19, PPMs to yield polymers 5.16–5.18 did not provide significant changes in overall gas permeability (Table 14). However, incorporation of these CO2-phillic moieties did result in increased CO2/N2 selectivity. The highest value of the CO2/N2 gas pair selectivity was achieved with amidoximated polymer 5.19; however, this PPM also led to a significant decrease in the copolymer permeability (Table 14).
5.3. Crosslinked polynorbornenes
Intentional crosslinking is a common approach to improve swelling and plasticization resistance, as well as mechanical and physical aging properties of polymer membranes.91,122–124 The effect of crosslinking on gas permeability is rather complex and generally remains controversial. In most cases, crosslinking leads to a decrease in permeability, but not always, which may depend on the type of crosslinking agent, degree of crosslinking, and structural features of macromolecules, etc. However, there are examples of crosslinked polynorbornenes with performances that exceed the 2008 Robeson upper bound.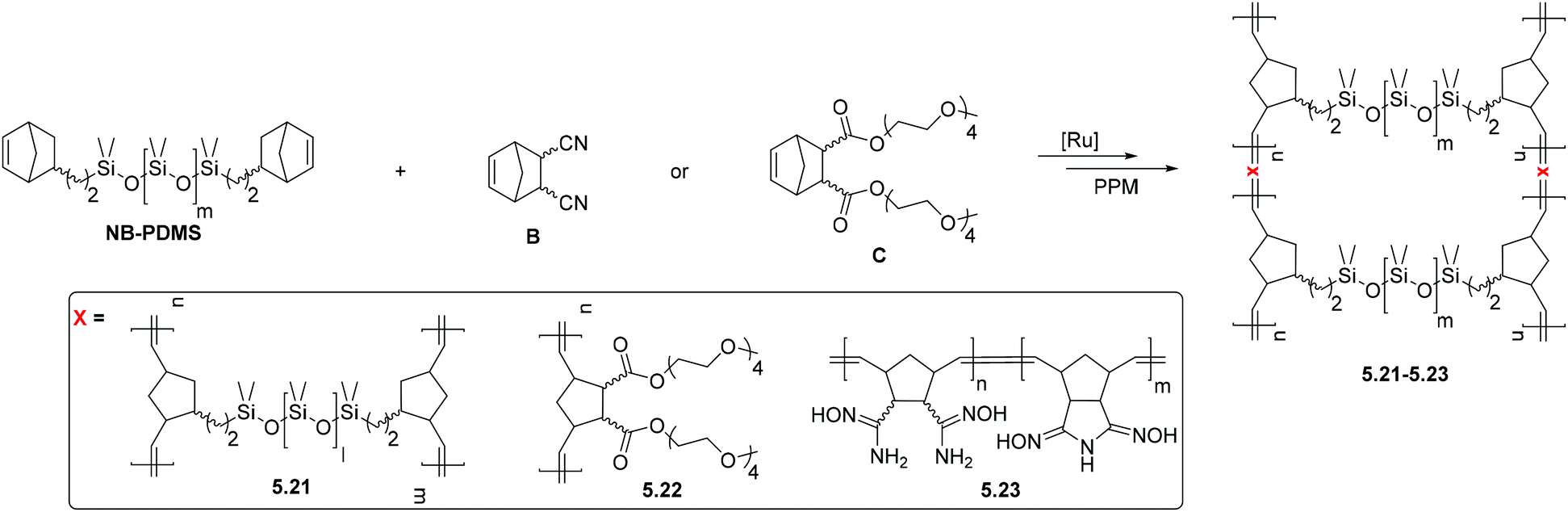 | ||
| Scheme 11 Synthesis of crosslinked norbornene-dimethylsiloxane-based membranes using ROMP. | ||
| Entry | Polymer | T g (°C) | Gel fr./xLda | P (Barrer) | α ij = Pi/Pj | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | O2 | N2 | CO2 | C1 | C4 | CO2 /N2 | C4/C1 | |||||
| a Gel fraction (% mass)/crosslink density (mol cm−3 × 105). b 0.4% mol of Grubbs 2nd generation catalyst to monomer was added. c 0.8% mol of Grubbs 2nd generation catalyst to monomer was added. d 3.7% mol of Grubbs 2nd generation catalyst to monomer was added. e 18 wt% of monomer C (Scheme 11) was added as comonomer. f 34 wt% of monomer C was added as comonomer. g Polymer treated with methanol and hydroxylamine for 39 h. h Polymer treated with methanol and hydroxylamine for 82 h. | ||||||||||||
| 1 | 5.20 | −123 | —/7.81 | — | — | 460 | 3545 | — | — | 7.7 | — | 128 |
| 2 | 5.21 | −125 | —/0.57 | — | — | 275 | 4030 | — | — | 14.7 | — | 128 |
| 3 | 5.21 | −125 | —/1.19 | — | — | 490 | 6700 | — | — | 13.8 | — | 128 |
| 4 | 5.21 | −125 | —/1.47 | — | — | 422 | 5040 | 12.0 | — | 128 | ||
| 5 | 5.22 | −124/−54 | — | — | — | 770 | 6200 | — | — | 8.1 | — | 127 |
| 6 | 5.22 | −124/−57 | — | — | — | 180 | 3400 | — | — | 18.8 | — | 127 |
| 7 | 5.23 | −125 | — | — | — | 360 | 6800 | — | — | 18.8 | — | 126 |
| 8 | 5.23 | −125 | — | — | — | 320 | 4800 | — | — | 15.0 | — | 126 |
| 9 | 5.24 | −53 | 98.5/— | — | — | 5.1 | 301 | 16.8 | — | 59.2 | 17 | 129 |
| 10 | 5.25 | −61 | 98.9/— | — | — | 8.6 | 476 | 28.0 | — | 55.0 | 17 | 129 |
| 11 | 5.26 | −53 | 94.6/— | — | — | 10.0 | 515 | 32.0 | — | 51.0 | 16 | 130 |
| 12 | 5.27 | 176/227 | — | 16.4 | 2.08 | 0.5 | 11 | 0.43 | — | 20.9 | — | 125 |
| 13 | 5.28 | 224 | — | 18.9 | 2.42 | 0.5 | 12 | 0.52 | — | 24.0 | — | 125 |
An effective approach to increase CO2 permeability and selectivity was achieved by the copolymerization of NB-PDMS with functionalized norbornenes comonomers (B and C) via ROMP (Scheme 11). The incorporation of functionalized norbornene comonomers was desired to improve CO2 solubility (polymers 5.22 and 5.23), and the crosslink density of 5.22 was controlled by the amount of NB-PDMS comonomer. Copolymer 5.22 exhibited an enhanced CO2 permeability (Table 15, entries 5 and 6). The synthesis of copolymer 5.23 included ROMP of NB-PDMS and a dicyano-substituted norbornene comonomer (B) followed by the transformation of CN groups into amidoxime substituents via the treatment with methanol and hydroxylamine.126 The degree of CN group conversion to mono-, di-, and cyclic amidoximes depended on the duration of the copolymer's treatment, and their gas separation properties varied depending on the ratio of mono-, di-, and cyclic amidoxime content. The highest values of permeability (P(CO2) = 6800 Barrer) and selectivity were achieved when 39% of the CN groups were converted to amidoximes (Table 15, entry 7), and these results exceeded the 2008 Robeson upper bound. Upon extending amidoximation reaction times, mainly cyclic amidoxime species were observed. These cyclic species have a lower CO2-philicity, and therefore exhibited a significant decrease in CO2 permeability (entry 8). A good correlation between the calculated binding energies of CO2-polymer units and the observed changes in the CO2 transport parameters was observed, which presents an opportunity for CO2-philicity prediction.126
Similar approaches were also implemented using norbornene-5,6-dicarboximide derivatives (Scheme 12). Therein, a NB functionalized telechelic poly(propylene glycol)(PPG)-block-poly(ethylene glycol)(PEG)-block-PPG (PPG-b-PEG-b-PPG) block copolymer (Scheme 12), as well as a similar PDMS-containing macromonomer, were copolymerized via ROMP to form crosslinked membranes.129 PEG is known to be a high CO2-philic material because of the presence of polar C–O bonds, however, their crystallinity has a negative impact on permeability and thus separation performance.132,133
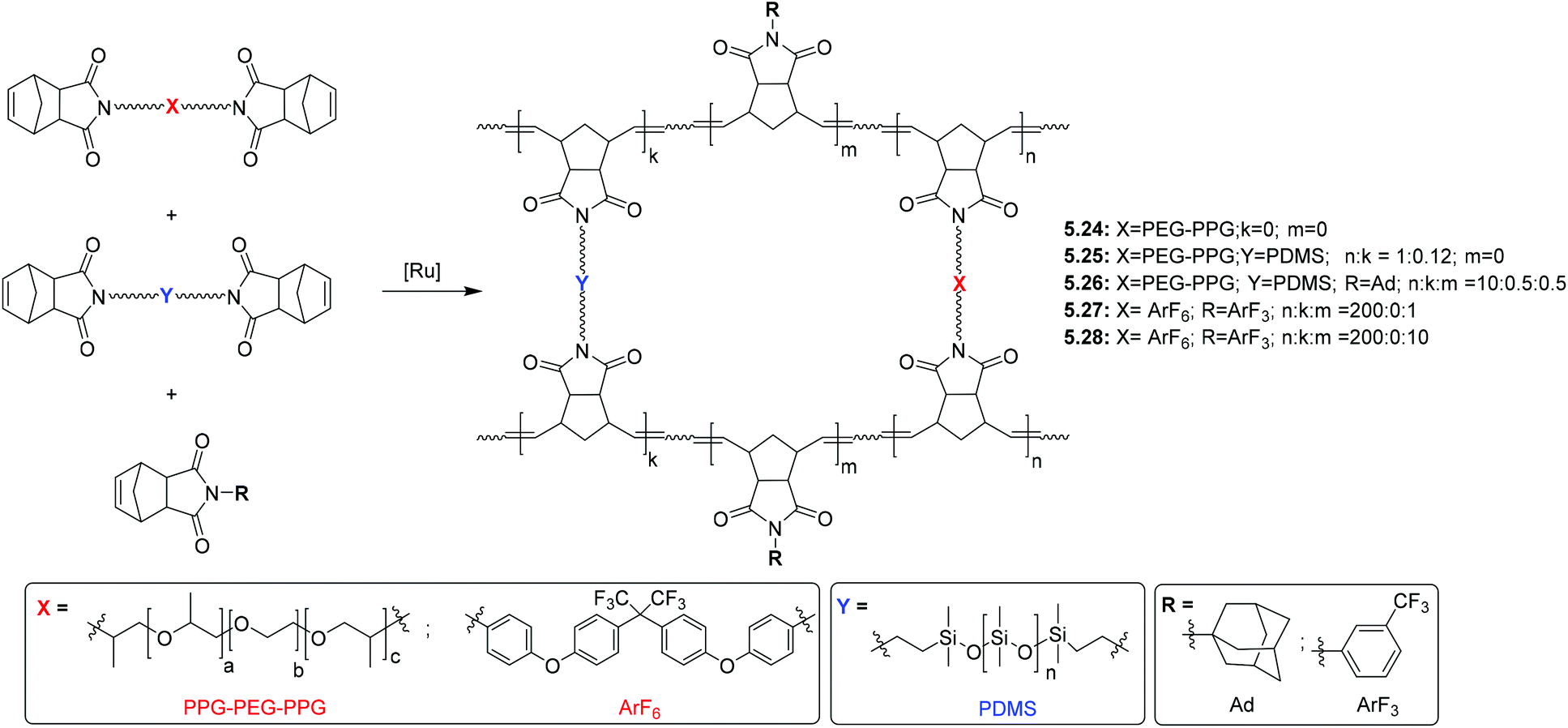 | ||
| Scheme 12 Synthesis of cross-linked norbornene-5,6-dicarboximide – PDMS based membranes using ROMP. | ||
To address this problem, PPG was incorporated as the pendant methyl moieties (–CH3) hinder PEO chain packing and thus promotes gas permeability. Both PPG-PEG-PPG and PDMS membranes are rubbery and possess poor mechanical properties without proper modifications. To address this, the adamantane containing norbornene-dicarboximide comonomers (R = Ad, Scheme 12) was copolymerized via ROMP to control the crosslinking density and size-sieving properties of polymers 5.24–5.26.130 These membranes showed excellent CO2/N2 selectivity and high CO2 permeability (Table 15, entries 9–11), which placed their gas separation performance beyond the 2008 Robeson upper bound.129,130
Interestingly, the study of the film surface morphology revealed the formation of a well-interconnected network structure, which endows the rubbery PPG-PEG-PPG/PDMS membranes (5.24, 5.25) with gas transport properties usually observed in glassy polymers, such as high stability and high mechanical and thermal properties (Table 16). Notably, these membranes displayed excellent anti-aging (up to 8 months) properties and anti-plasticization behavior (up to 25 atm CO2) (Table 16).129
| Entry | Polymer | E (MPa) | ε (%) | σ (MPa) | Aging, plasticization | Ref. |
|---|---|---|---|---|---|---|
| a Crosslinked APTCN obtained by copolymerization of trimethylsilyl TCN with 0.3 mol% DNBD. | ||||||
| Metathesis polynorbornenes | ||||||
| 1 | x2.10 | — | 300 | 5.2 | Selectivity did not change up to 800 psi | 20 |
| 2 | 5.24 | 549 | 7.7 | 11 | — | 129 |
| 3 | 5.25 | 379 | 10 | 10 | Stable for 8 months, no plasticization up to 25 atm CO2 | 129 |
| 4 | 5.27 | 1614 | — | 45 | — | 125 |
| 5 | 5.28 | 1869 | — | 57 | No plasticization up to 14 atm CO2 | 125 |
| Addition polynorbornenes | ||||||
| 6 | x2.32 | — | 23 | 24 | α decreases 27% as P increases from 500 to 800 psi. α constant for 5 d at 800 psi | 136 |
| 7 | 2.23 | 650 | 8 | 35 | — | 135 |
| 8 | 5.29 | 640 | 11 | 33 | — | 135 |
Similarly, crosslinked norbornene-dicarboximide frameworks containing 4,4′-(hexafluoroisopropylidene)bis(p-phenyleneoxy) fragment (ArF6) were synthesized via ROMP with N-3-trifluoromethylphenyl-norbornene-5,6-dicarboximide (X = ArF6 and R = ArF3, Scheme 12) to yield polymers 5.27 and 5.28.125 The chain transfer agent, cis-1,4-diacetoxy-2-butene, was applied to improve the solubility of the crosslinked polymer. Membranes of 5.27 and 5.28 also showed high CO2 anti-plasticization resistance (Table 16) and good mechanical characteristics.
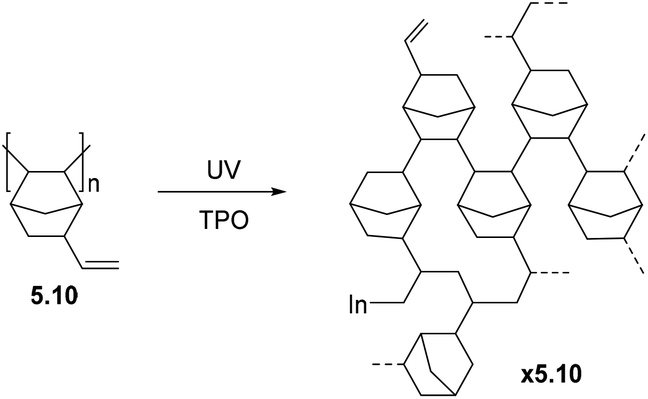 | ||
| Scheme 13 Addition poly(5-vinyl-2-norbornene) cross-linked by UV-irradiation in the presence of the photo-initiator diphenyl-(2,4,6-trimethylbenzoyl)phosphine oxide (TPO). | ||
| Entry | Polymer | Gel fr./xLda | P (Barrer) | α ij = Pi/Pj | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| He | O2 | N2 | CO2 | C1 | C4 | CO2 /N2 | C4/C1 | ||||
| a Gel fraction (%mass)/crosslink density (mol cm−3 × 105). b Properties for CO2/CH4 gas mixture. c PNB 5.10 crosslinked with 1.3 mol% TPO as photo initiator. d PNB 5.10 crosslinked with 33 mol% TPO as photo initiator. e Crosslinked APTCN was obtained by copolymerization of trimethylsilyl TCN with 0.3 mol% DNBD. f Trimethylsilyl TCN copolymerizing with 3 mol% triethoxysilyl TCN. g Polymer 5.30 in entry 5 crosslinked by sol–gel chemistry. h Trimethylsilyl TCN copolymerizing with 10 mol% triethoxysilyl TCN. i Polymer 5.30 in entry 7 crosslinked by sol–gel chemistry. | |||||||||||
| 1 | 5.10 | 0 | — | — | — | 27b | — | — | 18.9b | — | 76 |
| 2 | x5.10 | — | — | — | — | 20b | — | — | 22.0b | — | 76 |
| 3 | x5.10 | — | — | — | — | 10b | — | — | 21.9b | — | 76 |
| 4 | 5.29 | 0 | 1300 | 1000 | 430 | 5400 | 1000 | 11000 |
12.5 | 11 | 135 |
| 5 | 5.30 | 0 | 1180 | 990 | 400 | 5180 | 1030 | 14640 |
13.0 | 14 | 45 |
| 6 | x5.30 | 73/— | 1090 | 940 | 375 | 4710 | 960 | 15460 |
12.6 | 16 | 45 |
| 7 | 5.30 | 0 | 930 | 800 | 310 | 4670 | 830 | 16700 |
15.1 | 20 | 45 |
| 8 | x5.30 | 80/— | 1020 | 730 | 285 | 4130 | 770 | — | 14.5 | — | 45 |
For the crosslinking of saturated and relatively inert APNBs, copolymerization of a primary monomer with a small amount of a bifunctional crosslinking agent was shown to be an efficient approach. The dimer of norbornadiene-2,5 (DNBD) was chosen as a bifunctional crosslinking agent and copolymerized with TCNSiMe3 (Scheme 14).135 DNBD contains two norbornyl fragments, which can be polymerized independently and thus crosslink PNB chains. To maintain the solubility of the resulting copolymer, the percentage of the crosslinking agent is chosen carefully, with no more than 0.3 mol% of DNBD necessary to obtain polymer 5.29, while 1 mol% DNBD results in an insoluble polymer. The permeability and selectivity of the obtained copolymer 5.29 (Table 17) was slightly lower than that of non-crosslinked polymer 2.23 (Table 2, section 2.2), while its plasticization resistance is better than unmodified 2.23. The main advantages of these crosslinked copolymers were their higher molecular weights and better film-forming properties.135 The attempt using 5-ethylidene-2-norbornene containing endo- and exo-cyclic double bonds as a bifunctional crosslinking agent did not result in a crosslinked variant of polymer 2.29 due to the low reactivity of the tri-substituted exo-cyclic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds.137
C bonds.137
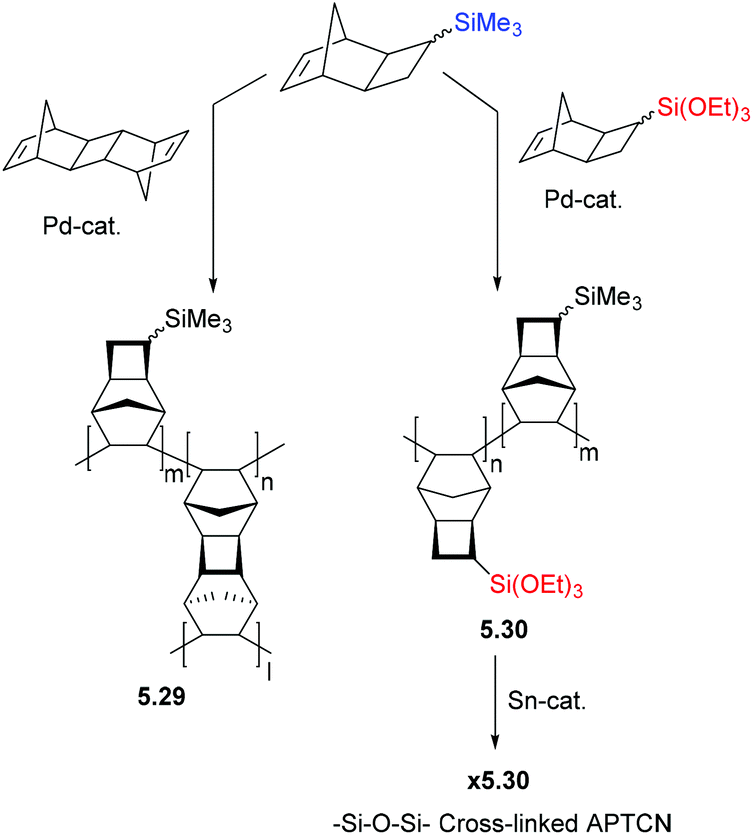 | ||
| Scheme 14 Synthesis of crosslinked addition polytricyclononenes (APTCNs) bearing organosilicon substituents. | ||
The absence of reactive centers in trimethylsilyl-substituted (–SiMe3) APNBs, as well as their rigid structure, makes them chemically stable and prevents the possibility of crosslinking. Addition copolymerization of trimethylsilyl-substituted TCN (TCNSiMe3) with triethoxysilyl-substituted TCN (TCNSi(OEt)3) to yield copolymers 5.30 with varying content of TCNSi(OEt)3 units (Scheme 14). Hydrolysis in the presence of tin(dimethyldineodecanoate) successfully resulted in cross-linked polymer membranes (x5.30) (Scheme 14 and Table 17).45 The introduction of even 3% TCNSi(OEt)3 imparted efficient crosslinking of the copolymer x5.30 (73% yield of the gel fraction) (Table 17, entry 6) with practically unchanged gas permeability parameters compared to non-crosslinked 5.30 of the same composition (Table 17, entry 5), as well as that of APTCNSiMe3 (polymer 2.23, Table 2, section 2.2). When the content of TCNSi(OEt)3 units in the copolymer is increased, the permeability of penetrant gases decreases (Table 17, entries 5–8). Lastly, butane permeability increased upon the introduction of alkoxysilyl-containing units within trimethylsilyl-substituted APTCN 2.23. This led to an increased C4/C1 selectivity for copolymers 5.30 and x5.30 (Table 17, entries 5–7).
In summary, a series of PPMs of MPNBs and APNBs were synthesized and their membrane gas separation performance evaluated. However, most of these PPMs did not lead to simultaneous enhancements in gas permeability and selectivity. Only gem-difluorocyclopropanation of MPNBs (Scheme 8), the introduction of CO2-philic groups into side chains of APNBs (Scheme 10), and the in situ ROMP assembly method (Scheme 12) provided an increase in both gas permeability and selectivity. The in situ ROMP assembly method, as well as the introduction of CO2-philic groups into side chains of APNBs seem to be promising routes to access membranes with gas separation performance near or above the Robeson upper bound. Furthermore, the observed positive effects achieved from the incorporation fluorine-containing groups into PNBs will be a promising direction for future research efforts. For example, the liquid-phase fluorination of APNBs is a promising modification of these polymers.138
6. Mixed matrix membranes
While targeted macromolecular design has proven a powerful tool to tune the gas separation performance of polymeric membranes, this approach often requires sophisticated synthetic expertise. A more experimentally accessible approach is through the incorporation of filler materials, especially nanoparticles, into a polymeric matrix. These mixed matrix membranes (MMMs) have promising transport properties, and are often much simpler to synthesize and fabricate.While the MMM approach has been applied to various polymeric materials,139–141 there are only a select few published examples in which gas transport properties of PNB matrix-based composites are described. The first composites of this kind were prepared by physical blending highly permeable polymer 2.23 with cyclodextrin or calixarene derivatives (Fig. 12).142,143 It was hypothesized that advantageous interactions between the cyclodextrin/calixarene filler materials and penetrant gas molecules would impart the membrane with enhanced separation capacity. Additionally, it was believed that these filler materials would constitute free volume elements that could enhance overall gas permeability.
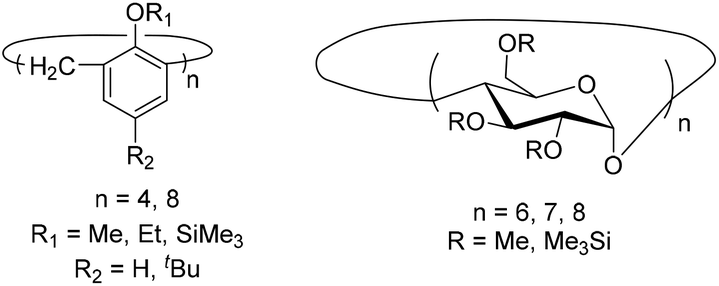 | ||
| Fig. 12 Calixarenes and cyclodextrines used for preparation of MMMs based on polymer 2.23. | ||
The organic composition of these cyclodextrin and calixarene fillers provided good compatibility with the polymeric matrix, and no agglomeration of filler was observed up to loadings of 29 wt% in the PNB matrix (2.23).142,143 The influence of calixarene/cyclodextrin ring size and nature of substituents on gas permeability and selectivity of the composites was studied. Trends found for composites with calixarenes and with cyclodextrins were the same; however, all of the investigated composites displayed decreased permeability and increased selectivity towards various gas pairs, as compared to the pure host matrix material 2.23. The source of decreased permeability within these composites was hypothesized to be due to the reduction of free volume when calixarene or cyclodextrin was incorporated into 2.23.
The lower FFVs observed for calixarene/cyclodextrin-2.23 composites were confirmed by positron annihilating lifetime spectroscopy (PALS) and the results of low temperature nitrogen adsorption/desorption method.142,143 The incorporation of calixarenes or cyclodextrins with smaller ring size or less bulky substituents in the upper and lower rims led to a more dramatic decrease in gas permeability. Interestingly, selectivity for the gas pairs with similar molecular sizes did not change substantially (15–50%) when calixarenes or cyclodextrins were added into 2.23, whereas the selectivity for gas pairs of quite different molecular dimensions (e.g. He/N2 or H2/CH4) increased. For example, the introduction of calix[4]arene with R1 = Me and R2 = tert-Bu (17 wt%) into 2.23 resulted in a 3–7 fold decrease in gas permeability and a 20–80% increase in selectivity. At the same time, a 5–45% decrease in gas permeability and a 12–65% increase in selectivity were observed for the similar composite containing calix[8]arene with R1 = SiMe3 and R2 = tert-Bu (17 wt%).
One of the limitations of MMMs containing inorganics filler is poor compatibility between the inorganic filler and the polymer matrix.140 For example, MMMs from physical blends of polymer 2.1 and metal–organic frameworks (MOFs) were found to be brittle and not suitable for studying gas-transport properties.144 Using NB modified UiO66-NH2 (Scheme 15) as a MOF-type filler and as a co-monomer in ROMP with norbornene, Zhang, Huang, and coworkers prepared MMMs with covalently attached MOF fillers (up to 50 wt%) with markedly improved mechanical properties.144 This approach allowed the authors to enhance both gas permeability and selectivity relative to native MPNB (2.1). These MMMs exhibited excellent gas separation performance for H2/CO2 and H2/N2, surpassing their respective 2008 Robeson upper bounds (PH2 = 91–230 Barrer, while H2/N2 and H2/CO2 selectivities were >1000 and 6–7, respectively). To show the scalability of their facile technique to construct PNB-MOF MMMs, a large area membrane (98 cm × 165 cm) with the thickness of the selective layer about 3–5 μm was successfully prepared.
 | ||
| Scheme 15 Synthesis of NB modified UiO66-NH2. | ||
Later, Kim and coworkers145 used the same filler (NB modified UiO66-NH2) to prepare crosslinked MMMs consisting of covalently attached UiO66-NH2 and a rubbery PEG/PPG-PDMS copolymer matrix. MMMs with up to 5 wt% filler were obtained through ROMP of di-NB-functionalized PEG/PPG and PMDS macromonomers (Scheme 16) with norbornene modified UiO66-NH2 and in situ membrane casting. The obtained MMMs exhibited significantly improved gas permeability in comparison to the pristine copolymer from di-NB-functionalized PEG/PPG and PMDS macromonomers, while CO2/N2 selectivity remained almost unchanged. Gas separation performance of these MMMs for CO2/N2 surpassed the Robeson 2008 upper bound (P(CO2) ≤ 585 Barrer and α(CO2/N2) = 53 vs. P(CO2) = 437 Barrer and α(CO2/N2) = 56 for the pristine copolymer) and these composites were found to resist aging when stored up to 11 months.
 | ||
| Scheme 16 Copolymerization of NB modified UiO66-NH2 with dinorbornene-functionalized PEG/PPG and PMDS macromonomers. | ||
Nishide, Oyaizu and coworkers146,147 descried highly oxygen-scavenging composite films from a polynorbornene matrix, Fe(II)salen-benzylimidazole as an oxidative catalyst, and methyl linoleate, which was used both as a polymer plasticizer and as an oxidation trigger (Scheme 17). Host matrices included polymers 2.1, 3.21, 6.1 and 6.2, with the highest oxygen-scavenging capacity (up to 300 ml of O2 gas at STP/g(film)) being achieved for films based on polymer 3.21 that contains more oxygen-consuming allylic hydrocarbon bonds. Gas permeability of such films were compared to the permeability of pure polymer 2.1 (O2 and N2 gas permeabilities for 6.1/Fe(II)salen films were 7.5 and 3.5 Barrer, respectively, vs. 2.8 and 1.5 Barrer for polymer 2.1 alone).146
 | ||
| Scheme 17 Synthesis of MPNBs bearing imidazolyl-substituents. | ||
Several papers describing gas-transport properties of composites derived from a polymer only partly containing norbornene units have also been published. For instance, physical blends of NB-ethylene copolymer with various surface modified graphitic nano-sheets were reported as high performance membrane materials for H2/CH4 and H2/CO2 separation.148 Nanocomposites based on NB-ethylene copolymer/linear low-density polyethylene blends with different modified organoclays were reported as barrier materials and their oxygen/water permeability was evaluated.149 However, these materials are beyond the PNB focused scope of this perspective.
7. Physical aging and plasticization
Physical aging and plasticization of polymers by penetrants are well-known issues that present considerable obstacles to the commercial adoption of polymeric membrane materials for gas separation applications. These issues are particularly pronounced for large free volume and highly gas permeable glassy polymers (e.g. PIMs, polyacetylenes, etc.),91 which often possess large amounts of excess non-equilibrium free volume. These polymers tend to undergo chain rearrangements and densification, ultimately moving toward equilibrium chain packing and resulting in the instability of their gas transport properties over time. The affinity between polymer and penetrant gas can additionally lead to swelling and plasticization of the polymer matrix, exacerbating these issues further. Research efforts to study and minimize the physical aging and plasticization of glassy polymers have a long history and are still relevant today.91,150,151 However, studies of PNB membrane properties, in general, have been limited to the estimation of gas permeability and diffusivity coefficients for freshly prepared films, and there is a general lack of information regarding PNB aging and plasticization in the literature.The most common method to evaluate physical aging in highly permeable glassy polymers (including APNBs and CANAL polymers) is by monitoring gas permeability and diffusion coefficients as a function of time.30,42,77,152 Reduced physical aging was found for MPNBs with side chain porosity (3.23, 3.24). For example, helium permeability (P(He)) for these polymers decreased by 8–15% within 1000 h. As a comparison, the helium permeability (P(He)) of PIM-1 decreased by roughly 45% for the same time period. Polymers 3.23 and 3.24 also exhibited exceptional resistance to plasticization, wherein CO2 plasticization pressure was measured to be >51 bar.77 Similarly, crosslinked composite materials based on UiO66-NH2 and a metathesis polymer matrix derived from di-NB-functionalized PEG/PPG and PMDS macromonomers (Scheme 16) showed both good plasticization resistance to CO2 and the stability of gas transport properties over more than 11 months.145
Trimethylsilyl (–SiMe3) substituted APNBs are prone to the physical aging because of large free volume. Therein, a 3–5% reduction in O2 and N2 permeability coefficients was observed for APTCNs bearing vicinal-substituted –SiMe3 groups over a time period of three weeks.61 Its more permeable APTCN isomer that bears geminal substituted trimethylsilyl groups exhibited a sharper reduction in P(O2) and P(N2), decreasing by 9–13% in just two weeks.153 Kinetic investigations into the physical aging of APTCNs bearing vicinal-substituted –SiMe3 groups showed that selectivities for the gas pairs H2/N2, O2/N2 and CO2/CH4 increased 1.2–1.4 times over a time period of 400 days.152 Interestingly, the gas transport parameters of this polymer was restored after the immersion of the polymeric films in ethanol.152
In contrast to –SiMe3 substituted APNBs, polymers bearing long alkyl or alkoxysilyl substituents have been shown to exhibit aging resistance, which is believed to be due to their lower free volume content. Similarly, it was found that the O2 and N2 permeability of APTCN polymer 2.27 and APNB polymer 3.18 did not change within a few months.17,73 Alkoxysilyl substituted APNBs (2.30–2.33, 2.37, 2.38) displayed a good resistance to CO2 plasticization,6 which was investigated by measuring the permeability of n-butane and CO2 at various pressures.6,18 Physical aging of APNBs was also studied using ellipsometry and mechanics,154,155 as well as dielectric spectroscopy.156–158
CANAL polymers surprisingly exhibited comparatively good resistance to aging in spite of large free volume.30,42 After 300 days of aging, polymers 4.7 and 4.8 continued to possess promising O2 permeabilities of 200–500 Barrer (initially, P(O2) was 450–750 Barrer) and O2/N2 selectivities equal to their initial values.42
In order to overcome the deleterious effects of physical aging and plasticization within polymers, crosslinking is particularly promising.91 Crosslinking in PNBs seems particularly attractive as (1) the chemistry of norbornenes and the corresponding polymers is versatile, and (2) a number of crosslinked polynorbornenes have been synthesized and that display promising gas transport properties in combination with resistance to aging and plasticization (see discussion in sections 5.3 and 6, as well as Table 16).20,45,76,125–130,135,136,145
8. Conclusion and future directions
Due to their synthetic versatility and amenability to numerous polymerization methods, norbornene-based monomers and polymers are attractive candidates for myriad applications. Though the first reports of PNB gas transport properties appeared in the late 1980s and early 1990s, essential progress has been achieved in recent years. In the current design of NB-based membrane materials, there are five main approaches – metathesis polymerization, addition polymerization, CANAL reaction to form unique monomers or polymers, post-polymerization modification of synthesized polynorbornenes, and incorporation of fillers to form composites. Through these methods, many new relationships between polymer structure and gas transport properties have been established, many of which are difficult or impossible to achieve for other classes of polymers. By changing the main chain structure, as well as the number and types of side-chain substituents, it has been possible to obtain polymers with targeted properties and even inversion of gas separation performance (e.g., solubility selectivity for hydrocarbon separation, or gas permeability of metathesis and addition polymers). Furthermore, the gas separation performance of some of these polymers have approached or even surpassed the Robeson upper bound.Norbornene-based polymers derived from addition polymerization and CANAL reactions tend to provide large free volume polymers with high gas permeability and moderate selectivities. For these polymers, aging and/or plasticization (especially for CANAL PNBs or PNBs with side chain porosity, see section 7), and insufficient gas separation selectivity are sometimes primary drawbacks. In regards to addition polymerizations, there are several groups of norbornene monomers that have not yet been efficiently polymerized due to catalyst-functional group incompatibilities, and therefore their membrane properties have not been possible to measure. Thus, the development of new addition polymerization catalysts is needed. Similarly, the discovery of novel organic reactions that may be used to assemble new types of polymer backbones based on NB monomers, such as the discovery CANAL, are of critical importance. These new synthetic methods would diversify the number of tools available for the macromolecular design of membrane materials based on norbornene derivatives and may ultimately broaden their overall utility.
MPNBs usually display lower gas permeability and often high selectivity as compared to APNBs and CANAL PNBs. The chemical instability of their main chains due to the presence of unsaturated CC double bonds is a key issue that must be circumvented. This has been generally addressed by PPM reactions, such as hydrogenation, oxidation, and fluorination. However, additional routes and techniques for PPM of MPNBs and MPTCNs will assuredly advance this area further.
Another promising direction in the macromolecular design of PNBs and PTCNs includes the introduction of substituents and/or backbone functionalities that are capable of specific interactions with one of the gaseous components to be separated in a mixture. In such cases, it is often possible to improve gas permeability without significant losses in the separation selectivity of such mixtures. This was demonstrated by the examples of fluoro-, amidoxime-, and oligoethylene oxide-substituted PNBs for the separation of CO2-containing gaseous mixtures. Utilizing MPNBs with polysiloxane, polyethylene glycol, and polypropylene glycol as substituents resulted in a new type of hybrid polymers that are effective for the separation of mixtures containing CO2. Calculation of the binding energies of CO2-polymer units is also very promising for polymer/functional group CO2-philicity prediction.
The incorporation of filler materials, such as nanoparticles, has also proven to be an efficient means of tuning PNB gas separation performance. Though there are only a few reports describing these composite materials, they have shown that it possible to enhance both gas permeability and selectivity of polynorbornenes, and even surpassing the Robeson upper bound. However, the lack of the information regarding gas transport properties of PNB composites is still one of the most challenging problems in this field. Moreover, the traditional drawbacks of composite membrane materials are also present, such as compatibility between a polymer matrix and a filler, mechanical properties, and aggregation of filler's particles.
In closing, it should be noted that the majority of gas separation data reported herein was conducted using pure gases. This experimental design limitation does not reflect the demands of real-world industrial gas streams, which involve gaseous mixtures that are often at elevated temperatures and pressures. Though these pure gas measurements provide tremendous insight and basis for comparing polymeric species, the true utility of these PNB-based materials may not be fully understood until more sophisticated gas transport studies using mixed gas streams (at variable temperature and pressures) can be conducted. Despite these limitations, the development of PNB-derived membrane materials is an extremely promising avenue of research that hold promise to yield next-generation gas separation membranes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
XW, TJW, and BKL gratefully acknowledge financial support from the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division, under Award DE-SC0018179. A portion of this manuscript was prepared within the State Program of TIPS RAS, and the preparation of section 3.3 was supported by the Russian Science Foundation (project no. 20-13-00428). EF, MG, DA and MB are very grateful to Prof. Yuri Yampolskii for useful discussions of gas-transport properties within polynorbornenes.Notes and references
- H. B. Park, J. Kamcev, L. M. Robeson, M. Elimelech and B. D. Freeman, Science, 2017, 356, eaab0530 CrossRef PubMed
.
- M. Galizia, W. S. Chi, Z. P. Smith, T. C. Merkel, R. W. Baker and B. D. Freeman, Macromolecules, 2017, 50, 7809–7843 CrossRef CAS
- A. Bos, I. G. M. Pünt, M. Wessling and H. Strathmann, J. Membr. Sci., 1999, 155, 67–78 CrossRef CAS
- B. D. Freeman, Macromolecules, 1999, 32, 375–380 CrossRef CAS
- S. Kanehashi and K. Nagai, J. Membr. Sci., 2005, 253, 117–138 CrossRef CAS
- C. R. Maroon, J. Townsend, M. A. Higgins, D. J. Harrigan, B. J. Sundell, J. A. Lawrence III, J. T. O'Brien, D. O'Neal, K. D. Vogiatzis and B. K. Long, J. Membr. Sci., 2020, 595, 117532 CrossRef CAS
- C. E. Powell and G. G. Qiao, J. Membr. Sci., 2006, 279, 1–49 CrossRef CAS
- R. Swaidan, B. Ghanem, E. Litwiller and I. Pinnau, Macromolecules, 2015, 48, 6553–6561 CrossRef CAS
- J. G. Wijmans and R. W. Baker, J. Membr. Sci., 1995, 107, 1–21 CrossRef CAS
- Y. Yampolskii, Macromolecules, 2012, 45, 3298–3311 CrossRef CAS
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS
- B. Comesaña-Gándara, J. Chen, C. G. Bezzu, M. Carta, I. Rose, M.-C. Ferrari, E. Esposito, A. Fuoco, J. C. Jansen and N. B. McKeown, Energy Environ. Sci., 2019, 12, 2733–2740 RSC
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165–185 CrossRef CAS
- E. S. Finkelshtein, M. V. Bermeshev, M. L. Gringolts, L. E. Starannikova and Y. P. Yampolskii, Russ. Chem. Rev., 2011, 80, 341–361 CrossRef CAS
- E. V. Bermesheva, A. I. Wozniak, I. L. Borisov, N. P. Yevlampieva, O. S. Vezo, G. O. Karpov, M. V. Bermeshev, A. F. Asachenko, M. A. Topchiy, P. S. Gribanov, M. S. Nechaev, V. V. Volkov and E. S. Finkelshtein, Polym. Sci., Ser. C, 2019, 61, 86–101 CrossRef CAS
- D. A. Alentiev, M. V. Bermeshev, L. E. Starannikova, E. V. Bermesheva, V. P. Shantarovich, V. G. Bekeshev, Y. P. Yampolskii and E. S. Finkelshtein, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1234–1248 CrossRef CAS
- D. A. Alentiev, E. S. Egorova, M. V. Bermeshev, L. E. Starannikova, M. A. Topchiy, A. F. Asachenko, P. S. Gribanov, M. S. Nechaev, Y. P. Yampolskii and E. S. Finkelshtein, J. Mater. Chem. A, 2018, 6, 19393–19408 RSC
- N. Belov, R. Nikiforov, L. Starannikova, K. R. Gmernicki, C. R. Maroon, B. K. Long, V. Shantarovich and Y. Yampolskii, Eur. Polym. J., 2017, 93, 602–611 CrossRef CAS
- M. V. Bermeshev, L. E. Starannikova, S. R. Sterlin, A. A. Tyutyunov, A. N. Tavtorkin, Y. P. Yampolskii and E. S. Finkelshtein, Pet. Chem., 2015, 55, 753–758 CrossRef CAS
- B. J. Sundell, J. A. Lawrence III, D. J. Harrigan, J. T. Vaughn, T. S. Pilyugina and D. R. Smith, RSC Adv., 2016, 6, 51619–51628 RSC
- M. Bermeshev and E. S. Finkelshteina, INEOS Open, 2018, 1, 39–54 CrossRef CAS
- M. A. Guseva, D. A. Alentiev, E. V. Bermesheva, I. A. Zamilatskov and M. V. Bermeshev, RSC Adv., 2019, 9, 33029–33037 RSC
- B. J. Sundell, J. A. Lawrence III, D. J. Harrigan, S. Lin, T. P. Headrick, J. T. O'Brien, W. F. Penniman and N. Sandler, ACS Macro Lett., 2020, 9, 1363–1368 CrossRef CAS
- V. A. Petrov and N. V. Vasil'ev, Curr. Org. Synth., 2006, 3, 215–259 CrossRef CAS
- J. P. Kennedy and J. A. Hinlicky, Polymer, 1965, 6, 133–140 CrossRef CAS
- N. L. Zutty, J. Polym. Sci., Part A: Gen. Pap., 1963, 1, 2231–2236 CrossRef CAS
- N. G. Gaylord, B. M. Mandal and M. Martan, J. Polym. Sci., Polym. Lett. Ed., 1976, 14, 555–559 CrossRef CAS
- N. Tunoglu and N. Balcioglu, Macromol. Rapid Commun., 1999, 20, 546–548 CrossRef CAS
- N. Balciog`lu and N. Tunog`lu, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 2311–2317 CrossRef
- M. A. Abdulhamid, H. W. H. Lai, Y. Wang, Z. Jin, Y. C. Teo, X. Ma, I. Pinnau and Y. Xia, Chem. Mater., 2019, 31, 1767–1774 CrossRef CAS
- S. Liu, Z. Jin, Y. C. Teo and Y. Xia, J. Am. Chem. Soc., 2014, 136, 17434–17437 CrossRef CAS PubMed
- Y. C. Teo, H. W. H. Lai and Y. Xia, Chem. – Eur. J., 2017, 23, 14101–14112 CrossRef CAS PubMed
- E. V. Bermesheva, A. I. Wozniak, F. A. Andreyanov, G. O. Karpov, M. S. Nechaev, A. F. Asachenko, M. A. Topchiy, E. K. Melnikova, Y. V. Nelyubina, P. S. Gribanov and M. V. Bermeshev, ACS Catal., 2020, 10, 1663–1678 CrossRef CAS
- K. R. Gmernicki, E. Hong, C. R. Maroon, S. M. Mahurin, A. P. Sokolov, T. Saito and B. K. Long, ACS Macro Lett., 2016, 5, 879–883 CrossRef CAS
- J. Lipian, R. A. Mimna, J. C. Fondran, D. Yandulov, R. A. Shick, B. L. Goodall, L. F. Rhodes and J. C. Huffman, Macromolecules, 2002, 35, 8969–8977 CrossRef CAS
- D.-G. Kim, A. Bell and R. A. Register, ACS Macro Lett., 2015, 4, 327–330 CrossRef CAS
- M. Yamashita, I. Takamiya, K. Jin and K. Nozaki, Organometallics, 2006, 25, 4588–4595 CrossRef CAS
- D.-G. Kim, T. Takigawa, T. Kashino, O. Burtovyy, A. Bell and R. A. Register, Chem. Mater., 2015, 27, 6791–6801 CrossRef CAS
- B.-G. Kang, D.-G. Kim and R. A. Register, Macromolecules, 2018, 51, 3702–3710 CrossRef CAS
- S. D. Tsai and R. A. Register, Macromol. Chem. Phys., 2018, 219, 1800059 CrossRef
- H. W. H. Lai, F. M. Benedetti, Z. Jin, Y. C. Teo, A. X. Wu, M. G. D. Angelis, Z. P. Smith and Y. Xia, Macromolecules, 2019, 52, 6294–6302 CrossRef CAS
- X. Ma, H. W. H. Lai, Y. Wang, A. Alhazmi, Y. Xia and I. Pinnau, ACS Macro Lett., 2020, 9, 680–685 CrossRef CAS
- P. Chapala, M. Bermeshev, L. Starannikova, I. Borisov, V. Shantarovich, V. Lakhtin, V. Volkov and E. Finkelshtein, Macromol. Chem. Phys., 2016, 217, 1966–1976 CrossRef CAS
- D. A. Alentiev, S. A. Korchagina, E. S. Finkel'Shtein, M. S. Nechaev, A. F. Asachenko, M. A. Topchiy, P. S. Gribanov and M. V. Bermeshev, Russ. Chem. Bull., 2018, 67, 121–126 CrossRef CAS
- D. A. Alentiev, E. S. Egorova, M. V. Bermeshev, L. E. Starannikova, Y. P. Yampolskii and E. S. Finkelshtein, Polym. Eng. Sci., 2019, 59, 2502–2507 CrossRef CAS
- E. S. Finkelshtein, K. L. Makovetskii, M. L. Gringolts, Y. V. Rogan, T. G. Golenko, L. E. Starannikova, Y. P. Yampolskii, V. P. Shantarovich and T. Suzuki, Macromolecules, 2006, 39, 7022–7029 CrossRef CAS
- J. A. Lawrence III, D. J. Harrigan, C. R. Maroon, S. A. Sharber, B. K. Long and B. J. Sundell, J. Membr. Sci., 2020, 616, 118569 CrossRef
- M. Gringolts, M. Bermeshev, L. Starannikova, Y. V. Rogan, Y. P. Yampol'skii and E. S. Finkel'shtein, Polym. Sci., Ser. A, 2009, 51, 1233 CrossRef
- T. Steinhäusler and W. J. Koros, J. Polym. Sci., Part B: Polym. Phys., 1997, 35, 91–99 CrossRef
- Y. P. Yampolskii, E. S. Finkelshtein, K. L. Makovetskii, V. I. Bondar and V. P. Shantarovich, J. Appl. Polym. Sci., 1996, 62, 349–357 CrossRef CAS
- E. S. Finkelshtein, M. Gringolts, N. Ushakov, V. Lakhtin, S. Soloviev and Y. P. Yampol'skii, Polymer, 2003, 44, 2843–2851 CrossRef CAS
- M. L. Gringol'ts, M. V. Bermeshev, A. V. Syromolotov, L. E. Starannikova, M. F. Filatova, K. L. Makovetskii and E. S. Finkel'shtein, Pet. Chem., 2010, 50, 352–361 CrossRef
- E. S. Finkelshtein, K. Makovetskii, Y. P. Yampol'skii, E. Portnykh, I. Y. Ostrovskaya, N. Kaliuzhnyi, N. Pritula, A. Gol'berg, M. Yatsenko and N. Plate, Makromol. Chem., 1991, 192, 1–9 CrossRef CAS
-
E. Finkelshtein, M. Gringolts, M. Bermeshev, P. Chapala and Y. Rogan, in Membrane Materials for Gas and Vapor Separation, John Wiley & Sons Inc., Chichester, West Sussex, UK, 2017, ch. 6, pp. 143–221 Search PubMed
- M. Bermeshev, B. Bulgakov, L. Starannikova, G. Dibrov, P. Chapala, D. Demchuk, Y. Yampolskii and E. Finkelshtein, J. Appl. Polym. Sci., 2015, 132, 41395 CrossRef
- T. Katsumata, M. Shiotsuki, F. Sanda and T. Masuda, Polymer, 2009, 50, 1389–1394 CrossRef CAS
- M. V. Bermeshev, A. V. Syromolotov, M. L. Gringolts, L. E. Starannikova, Y. P. Yampolskii and E. S. Finkelshtein, Macromolecules, 2011, 44, 6637–6640 CrossRef CAS
- D. A. Alentiev, D. M. Dzhaparidze, P. P. Chapala, M. V. Bermeshev, N. A. Belov, R. Yu. Nikiforov, L. E. Starannikova, Y. P. Yampolskii and E. S. Finkelshtein, Polym. Sci., Ser. B, 2018, 60, 612–620 CrossRef CAS
- M. A. Higgins, C. R. Maroon, J. Townsend, X. Wang, K. D. Vogiatzis and B. K. Long, J. Polym. Sci., 2020, 58, 2644–2653 CrossRef CAS
- H. Tetsuka, K. Isobe and M. Hagiwara, Polym. J., 2009, 41, 643–649 CrossRef CAS
- M. Gringolts, M. Bermeshev, Y. Yampolskii, L. Starannikova, V. Shantarovich and E. Finkelshtein, Macromolecules, 2010, 43, 7165–7172 CrossRef CAS
- T. Corrado and R. Guo, Mol. Syst. Des. Eng., 2020, 5, 22–48 RSC
- V. V. Teplyakov, D. R. Paul, N. B. Bespalova and E. S. Finkel'shtein, Macromolecules, 1992, 25, 4218–4219 CrossRef CAS
- Y. P. Yampol'skii, N. B. Bespalova, E. S. Finkel'shtein, V. I. Bondar and A. V. Popov, Macromolecules, 1994, 27, 2872–2878 CrossRef
- E. S. Finkel'shtein, N. B. Bespalova, E. B. Portnykh, K. L. Makovetskii, I. Y. Ostrovskaya, S. M. Shishatskiy, Y. P. Yampolskii, N. A. Platé and N. E. Kalyuzhnyi, Vysokomol. Soedin., 1993, 35, 489–494 Search PubMed
- J. A. Cruz-Morales, J. Vargas, A. A. Santiago, S. R. Vásquez-García, M. A. Tlenkopatchev, T. de Lys and M. López-González, High Perform. Polym., 2016, 28, 1246–1262 CrossRef CAS
- G. O. Karpov, M. V. Bermeshev, I. L. Borisov, S. R. Sterlin, A. A. Tyutyunov, N. P. Yevlampieva, B. A. Bulgakov, V. V. Volkov and E. S. Finkelshtein, Polymer, 2018, 153, 626–636 CrossRef CAS
- G. O. Karpov, D. S. Bakhtin, M. V. Bermeshev, I. L. Borisov, B. A. Bulgakov, V. V. Volkov and E. S. Finkelstein, Polym. Sci., Ser. B, 2019, 61, 387–394 CrossRef CAS
- I. L. Borisov, T. R. Akmalov, A. O. Ivanov, V. V. Volkov, E. S. Finkelshtein and M. V. Bermeshev, Mendeleev Commun., 2016, 26, 124–126 CrossRef CAS
- J. Vargas, A. A. Santiago, M. A. Tlenkopatchev, M. López-González and E. Riande, J. Membr. Sci., 2010, 361, 78–88 CrossRef CAS
- J. Vargas, A. Martínez, A. A. Santiago, M. A. Tlenkopatchev, R. Gaviño and M. Aguilar-Vega, J. Fluorine Chem., 2009, 130, 162–168 CrossRef CAS
- G. O. Karpov, I. L. Borisov, A. V. Volkov, E. S. Finkelshtein and M. V. Bermeshev, Polymers, 2020, 12, 1282 CrossRef CAS PubMed
- A. I. Wozniak, E. V. Bermesheva, I. L. Borisov, D. I. Petukhov, M. V. Bermeshev, A. V. Volkov and E. S. Finkelshtein, Macromol. Rapid Commun., 2019, 40, 1900206 CrossRef CAS PubMed
- K. D. Dorkenoo, P. H. Pfromm and M. E. Rezac, J. Polym. Sci., Part B: Polym. Phys., 1998, 36, 797–803 CrossRef CAS
- P. P. Chapala, I. L. Borisov, M. V. Bermeshev, V. V. Volkov and E. S. Finkelshtein, Pet. Chem., 2016, 56, 1056–1060 CrossRef CAS
- W. Dujardin, C. Van Goethem, J. A. Steele, M. Roeffaers, I. F. J. Vankelecom and G. Koeckelberghs, Polymers, 2019, 11, 704 CrossRef PubMed
- Y. He, F. M. Benedetti, S. Lin, C. Liu, Y. Zhao, H.-Z. Ye, T. Van Voorhis, M. G. De Angelis, T. M. Swager and Z. P. Smith, Adv. Mater., 2019, 31, 1807871 CrossRef PubMed
- M. Carta, R. Malpass-Evans, M. Croad, Y. Rogan, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Science, 2013, 339, 303–307 CrossRef CAS PubMed
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC
- Y. Zhao, Y. He and T. M. Swager, ACS Macro Lett., 2018, 7, 300–304 CrossRef CAS
- J. Vargas, A. A. Santiago, M. A. Tlenkopatchev, R. Gaviño, M. F. Laguna, M. López-González and E. Riande, Macromolecules, 2007, 40, 563–570 CrossRef CAS
- J. Vargas, A. A. Santiago, J. A. Cruz-Morales, M. A. Tlenkopatchev, T. de Lys, M. López-González and E. Riande, Macromol. Chem. Phys., 2013, 214, 2607–2615 CrossRef CAS
- A. P. Contreras, M. A. Tlenkopatchev, M. del Mar López-González and E. Riande, Macromolecules, 2002, 35, 4677–4684 CrossRef CAS
- M. A. Tlenkopatchev, J. Vargas, M. d. M. López-González and E. Riande, Macromolecules, 2003, 36, 8483–8488 CrossRef CAS
- K. Díaz, J. Vargas, L. F. Del Castillo, M. A. Tlenkopatchev and M. Aguilar-Vega, Macromol. Chem. Phys., 2005, 206, 2316–2322 CrossRef
- J. Vargas, A. Martínez, A. A. Santiago, M. A. Tlenkopatchev and M. Aguilar-Vega, Polymer, 2007, 48, 6546–6553 CrossRef CAS
- J. Pozuelo, M. López-González, M. Tlenkopatchev, E. Saiz and E. Riande, J. Membr. Sci., 2008, 310, 474–483 CrossRef CAS
- I. Rose, M. Carta, R. Malpass-Evans, M.-C. Ferrari, P. Bernardo, G. Clarizia, J. C. Jansen and N. B. McKeown, ACS Macro Lett., 2015, 4, 912–915 CrossRef CAS
- M. Carta, M. Croad, R. Malpass-Evans, J. C. Jansen, P. Bernardo, G. Clarizia, K. Friess, M. Lanč and N. B. McKeown, Adv. Mater., 2014, 26, 3526–3531 CrossRef CAS PubMed
- Y. Zhuang, J. G. Seong, Y. S. Do, H. J. Jo, Z. Cui, J. Lee, Y. M. Lee and M. D. Guiver, Macromolecules, 2014, 47, 3254–3262 CrossRef CAS
- Z.-X. Low, P. M. Budd, N. B. McKeown and D. A. Patterson, Chem. Rev., 2018, 118, 5871–5911 CrossRef CAS PubMed
- S. Kim and Y. M. Lee, J. Nanopart. Res., 2012, 14, 949 CrossRef
- B. Padhi, G. Kang, E. Kim, J. Ha, H. T. Kim, J. Lim and J. M. Joo, ACS Catal., 2020, 10, 1792–1798 CrossRef CAS
- Z. Jin, Y. C. Teo, N. G. Zulaybar, M. D. Smith and Y. Xia, J. Am. Chem. Soc., 2017, 139, 1806–1809 CrossRef CAS PubMed
-
K. A. Günay, P. Theato and H.-A. Klok, in Functional Polymers by Post-Polymerization Modification: Concepts, Guidelines, and Applications, 2013, ch. 1, pp. 1–44 Search PubMed
- M. V. Bermeshev, A. V. Syromolotov, L. E. Starannikova, M. L. Gringolts, V. G. Lakhtin, Y. P. Yampolskii and E. S. Finkelshtein, Macromolecules, 2013, 46, 8973–8979 CrossRef CAS
- B. J. Berron, P. A. Payne and G. K. Jennings, Ind. Eng. Chem. Res., 2008, 47, 7707–7714 CrossRef CAS
- J. P. Planche, A. Revillon and A. Guyot, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 1377–1386 CrossRef CAS
- J. Hyvl, B. Autenrieth and R. R. Schrock, Macromolecules, 2015, 48, 3148–3152 CrossRef CAS
- S. Kovačič, F. Preishuber-Pflügl and C. Slugovc, Macromol. Mater. Eng., 2014, 299, 843–850 CrossRef
- M. Perring and N. B. Bowden, Langmuir, 2008, 24, 10480–10487 CrossRef CAS PubMed
- T. J. Boyd and R. R. Schrock, Macromolecules, 1999, 32, 6608–6618 CrossRef CAS
- M. Perring, T. R. Long and N. B. Bowden, J. Mater. Chem., 2010, 20, 8679–8685 RSC
- S. Meier, F. Stelzer, H. Reisinger, R. Haag, S. Mecking and R. Mülhaupt, Chem. Commun., 2001, 855–856 RSC
- Y. Zhao, J. Chen, W. Zhu and K. Zhang, Polymer, 2015, 74, 16–20 CrossRef CAS
- A. B. Lowe, M. Liu, J. A. Van Hensbergen and R. P. Burford, Macromol. Rapid Commun., 2014, 35, 391–404 CrossRef CAS PubMed
- N. A. Belov, M. L. Gringolts, A. A. Morontsev, L. E. Starannikova, Y. P. Yampolskii and E. S. Finkelstein, Polym. Sci., Ser. B, 2017, 59, 560–569 CrossRef CAS
- A. A. Morontsev, V. A. Zhigarev, R. Y. Nikiforov, N. A. Belov, M. L. Gringolts, E. S. Finkelshtein and Y. P. Yampolskii, Eur. Polym. J., 2018, 99, 340–349 CrossRef CAS
- V. A. Zhigarev, A. A. Morontsev, R. Y. Nikiforov, M. L. Gringolts, N. A. Belov, N. G. Komalenkova, V. G. Lakhtin and E. S. Finkelshtein, Polym. Sci., Ser. C, 2019, 61, 107–119 CrossRef CAS
- J. P. Klein and R. A. Register, J. Polym. Sci., Part B: Polym. Phys., 2019, 57, 1188–1195 CrossRef CAS
- S. D. Drouin, F. Zamanian and D. E. Fogg, Organometallics, 2001, 20, 5495–5497 CrossRef CAS
- A. A. Morontsev, M. L. Gringolts, M. P. Filatova and E. S. Finkelshtein, Polym. Sci., Ser. B, 2016, 58, 695–702 CrossRef CAS
- Y. Yampolskii, N. Belov and A. Alentiev, J. Membr. Sci., 2020, 598, 117779 CrossRef CAS
- R. García-Loma and A. C. Albéniz, Asian J. Org. Chem., 2019, 8, 304–315 CrossRef
- X. He, X. Jiang, Z. Wang, Y. Deng, Z. Han, Y. Yang and D. Chen, Polym. Eng. Sci., 2018, 58, 13–21 CrossRef CAS
- F. Pierre, B. Commarieu, A. C. Tavares and J. Claverie, Polymer, 2016, 86, 91–97 CrossRef CAS
- A. I. Wozniak, E. V. Bermesheva, F. A. Andreyanov, I. L. Borisov, D. P. Zarezin, D. S. Bakhtin, N. N. Gavrilova, I. R. Ilyasov, M. S. Nechaev, A. F. Asachenko, M. A. Topchiy, A. V. Volkov, E. S. Finkelshtein, X.-K. Ren and M. V. Bermeshev, React. Funct. Polym., 2020, 149, 104513 CrossRef CAS
- A. I. Wozniak, I. L. Borisov, E. V. Bermesheva, D. P. Zarezin, A. V. Volkov, E. S. Finkelstein and M. V. Bermeshev, Polym. Sci., Ser. B, 2020, 62, 218–224 CrossRef CAS
- S. Japip, G. R. Lee and T.-S. Chung, Ind. Eng. Chem. Res., 2020, 59, 5315–5323 CrossRef CAS
- R. Swaidan, B. S. Ghanem, E. Litwiller and I. Pinnau, J. Membr. Sci., 2014, 457, 95–102 CrossRef CAS
- Z. Tian, T. Saito and D.-E. Jiang, J. Phys. Chem. A, 2015, 119, 3848–3852 CrossRef CAS PubMed
- M. S. Suleman, K. K. Lau and Y. F. Yeong, Chem. Eng. Technol., 2016, 39, 1604–1616 CrossRef CAS
- K. Vanherck, G. Koeckelberghs and I. F. J. Vankelecom, Prog. Polym. Sci., 2013, 38, 874–896 CrossRef CAS
- J. D. Wind, C. Staudt-Bickel, D. R. Paul and W. J. Koros, Macromolecules, 2003, 36, 1882–1888 CrossRef CAS
- I. Aranda-Suárez, C. Corona-García, A. A. Santiago, S. López Morales, M. Abatal, M. López-González and J. Vargas, Macromol. Chem. Phys., 2019, 220, 1800481 CrossRef
- T. Hong, S. Chatterjee, S. M. Mahurin, F. Fan, Z. Tian, D.-e. Jiang, B. K. Long, J. W. Mays, A. P. Sokolov and T. Saito, J. Membr. Sci., 2017, 530, 213–219 CrossRef CAS
- T. Hong, S. Lai, S. M. Mahurin, P.-F. Cao, D. N. Voylov, H. M. Meyer, C. B. Jacobs, J.-M. Y. Carrillo, A. Kisliuk, I. N. Ivanov, D.-E. Jiang, B. K. Long, J. W. Mays, A. P. Sokolov and T. Saito, Adv. Sustainable Syst., 2018, 2, 1700113 CrossRef
- T. Hong, Z. Niu, X. Hu, K. Gmernicki, S. Cheng, F. Fan, J. C. Johnson, E. Hong, S. Mahurin, D.-E. Jiang, B. Long, J. Mays, A. Sokolov and T. Saito, ChemSusChem, 2015, 8, 3595–3604 CrossRef CAS PubMed
- I. Hossain, D. Kim, A. Z. Al Munsur, J. M. Roh, H. B. Park and T.-H. Kim, ACS Appl. Mater. Interfaces, 2020, 12, 27286–27299 CrossRef CAS PubMed
- D. Kim, I. Hossain, Y. Kim, O. Choi and T.-H. Kim, Polymers, 2020, 12, 1674 CrossRef CAS PubMed
- T. C. Merkel, V. I. Bondar, K. Nagai, B. D. Freeman and I. Pinnau, J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 415–434 CrossRef CAS
- T. Brinkmann, J. Lillepärg, H. Notzke, J. Pohlmann, S. Shishatskiy, J. Wind and T. Wolff, Engineering, 2017, 3, 485–493 CrossRef CAS
- J. Liu, G. Zhang, K. Clark and H. Lin, ACS Appl. Mater. Interfaces, 2019, 11, 10933–10940 CrossRef CAS PubMed
- W. Dujardin, C. Van Goethem, J. A. Steele, M. Roeffaers, I. F. J. Vankelecom and G. Koeckelberghs, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1593–1600 CrossRef CAS
- D. A. Alentiev, D. M. Dzhaparidze, M. V. Bermeshev, L. E. Starannikova, M. P. Filatova, Y. P. Yampolskii and E. S. Finkelshtein, Polym. Sci., Ser. B, 2019, 61, 812–816 CrossRef CAS
- J. T. Vaughn, D. J. Harrigan, B. J. Sundell, J. A. Lawrence III and J. Yang, J. Membr. Sci., 2017, 522, 68–76 CrossRef CAS
- B. A. Bulgakov, M. V. Bermeshev, M. L. Gringol'ts, M. P. Filatova and E. S. Finkel'shtein, Pet. Chem., 2012, 52, 119–122 CrossRef CAS
- Y. P. Yampolskii, N. A. Belov and A. Y. Alentiev, Russ. Chem. Rev., 2019, 88, 387–405 CrossRef CAS
- M. R. A. Hamid and H.-K. Jeong, Korean J. Chem. Eng., 2018, 35, 1577–1600 CrossRef CAS
- Q. Qian, P. A. Asinger, M. J. Lee, G. Han, K. Mizrahi Rodriguez, S. Lin, F. M. Benedetti, A. X. Wu, W. S. Chi and Z. P. Smith, Chem. Rev., 2020, 120, 8161–8266 CrossRef CAS PubMed
- G. Dong, H. Li and V. Chen, J. Mater. Chem. A, 2013, 1, 4610–4630 RSC
- P. P. Chapala, M. V. Bermeshev, L. E. Starannikova, V. P. Shantarovich, N. N. Gavrilova, V. G. Avakyan, M. P. Filatova, Y. P. Yampolskii and E. S. Finkelshtein, J. Membr. Sci., 2015, 474, 83–91 CrossRef CAS
- P. P. Chapala, M. V. Bermeshev, L. E. Starannikova, V. P. Shantarovich, N. N. Gavrilova, Y. P. Yampolskii and E. S. Finkelshtein, Polym. Compos., 2015, 36, 1029–1038 CrossRef CAS
- X. Gao, J. Zhang, K. Huang and J. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 34640–34645 CrossRef CAS PubMed
- I. Hossain, A. Husna, S. Chaemchuen, F. Verpoort and T. H. Kim, ACS Appl. Mater. Interfaces, 2020, 12, 57916–57931 CrossRef CAS PubMed
- Y. Wang, Y. Hasegawa, T. Serikawa, K. Oyaizu and H. Nishide, Chem. Commun., 2020, 56, 964–967 RSC
- Y. Wang, K. Oyaizu and H. Nishide, Pure Appl. Chem., 2020, 92, 871–882 CrossRef CAS
- M. Doğu and N. Ercan, Chem. Eng. Res. Des., 2016, 109, 455–463 CrossRef
- S. Sánchez-Valdes, E. Ramírez-Vargas, L. F. Ramos de Valle, J. G. Martinez-Colunga, J. Romero-Garcia, A. S. Ledezma-Perez, J. Mendez-Nonell, M. E. Castañeda-Flores and A. Morales-Cepeda, Polym. Compos., 2016, 37, 3167–3174 CrossRef
- J. Hutchinson, Prog. Polym. Sci., 1995, 20, 703–760 CrossRef CAS
- M. M. Merrick, R. Sujanani and B. D. Freeman, Polymer, 2020, 211, 123176 CrossRef CAS
- N. Belov, D. Nikolaeva and Y. Yampolskii, Polymer, 2021, 217, 123447 CrossRef CAS
- P. P. Chapala, M. V. Bermeshev, L. E. Starannikova, N. A. Belov, V. E. Ryzhikh, V. P. Shantarovich, V. G. Lakhtin, N. N. Gavrilova, Y. P. Yampolskii and E. S. Finkelshtein, Macromolecules, 2015, 48, 8055–8061 CrossRef CAS
- E. A. Lewis and B. D. Vogt, J. Polym. Sci., Part B: Polym. Phys., 2018, 56, 53–61 CrossRef CAS
- E. A. Lewis, C. M. Stafford and B. D. Vogt, J. Polym. Sci., Part B: Polym. Phys., 2019, 57, 992–1000 CrossRef CAS PubMed
- H. Yin, P. Chapala, M. Bermeshev, A. Schönhals and M. Böhning, ACS Macro Lett., 2017, 6, 813–818 CrossRef CAS
- H. Yin, P. Chapala, M. Bermeshev, B. R. Pauw, A. Schönhals and M. Böhning, ACS Appl. Polym. Mater., 2019, 1, 844–855 CrossRef CAS
- M. A. Kolmangadi, P. Szymoniak, G. J. Smales, D. A. Alentiev, M. Bermeshev, M. Böhning and A. Schönhals, Macromolecules, 2020, 53, 7410–7419 CrossRef CAS
Footnotes |
| † This paper is dedicated to the memory of an outstanding scientist, colleague, and friend, Professor Yuri Yampolskii, who passed away in March 24, 2021. Yuri's transformative work in the field of membranes for gas separation was largely devoted to the in-depth study of membrane properties for a wide range of polymeric materials, and his results have been evaluated by specialists around the world. His personal and scientific interactions with scientists, and non-scientist alike, were both plentiful and productive. |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |