
Understanding the effects of nucleophiles in fast living cationic polymerisation of isobutyl vinyl ether in a microflow system from stability and activity of propagating chains
Dan
Xie
 and
Yangcheng
Lu
*
and
Yangcheng
Lu
*
State Key Laboratory of Chemical Engineering, Department of Chemical Engineering, Tsinghua University, Beijing 100084, China. E-mail: luyc@tsinghua.edu.cn; Fax: +86 10 62773017; Tel: +86 10 62773017
First published on 5th April 2021
Abstract
Previous studies on fast living cationic polymerisation have rarely focused on the effects of nucleophiles, which are crucial in determining a suitable initiation system. In this work, taking the polymerisation of isobutyl vinyl ether (IBVE) as an example, the effects of various nucleophiles, namely ethyl acetate, 1,4-dioxane, 1,3-dioxolane, diethyl ether, and ethyl propionate, on the activity and stability of the propagating chains were systematically examined in a microflow system, in which IBVE-HCl/SnCl4 was selected as the initiation system and 2-hydroxyethyl methacrylate was employed as an effective end-capping agent for 1H NMR spectroscopy measurements. The characteristic time of activity (tact) and the half-life of stability (tdecay) exhibited almost linear proportional relationships, revealing that the nucleophiles did not alter the relative values of activity and stability, acting simply to improve the controllability of the cationic polymerisation process. Furthermore, a density functional theory simulation method was established to calculate the dissociation energy of IBVE/SnCl4/Nus (ΔE). Approximately linear relationships between ln(tact) or ln(tdecay) and ΔE were found, indicating the potential of theoretical calculations to estimate the activities and stabilities of the propagating chains in this process.
1. Introduction
The propagating chains initiated by carbocations in conventional cationic polymerisation processes are usually unstable and tend to undergo irreversible termination and chain transfer reactions via proton elimination.1,2 In the last few decades, researchers have achieved living cationic polymerisation via the introduction of nucleophiles, thereby providing a fundamental base for the preparation of functional living polymers with a desired sequence distribution and well-defined architectures.1,3–12 For example, the living cationic polymerisation process enabled the preparation of various functional poly(vinyl ether)s,13 including thermo-sensitive polymers,14–17 pH/temperature responsive polymers,18,19 amphiphilic block copolymers,20 star-shaped polymers,7,21–23 and stimuli-responsive polymers.24–28 The living initiation system of vinyl ethers usually employed adducts of isobutyl vinyl ether (IBVE) and HCl or acetic acid as initiators, a series of Lewis acids as co-initiators, and ethers or esters as nucleophiles.29,30 The functions of such nucleophiles include stabilisation of the propagating chains, adjusting the acidity of the co-initiator (Lewis acid), and the formation of monomeric Lewis acidic species, which ultimately result in uniform chain initiation and chain propagation.2,31–36 From another point of view, these nucleophiles can affect the formation and tightness of the various active species, such as ion pairs. Typically, loose ion pairs result in lower stabilities and higher activities for the propagating chains, while tight ion pairs result in the opposite effects.Recently, various initiation systems have been developed, including the FeCl3/cyclic ether (1,4-dioxane/1,3-dioxolane/THF),37 SnCl4/cyclic ether (1,4-dioxane) or ethyl acetate,38,39 EtxAlCl3−x/ethyl acetate,21,40 FeBr3/THF, InCl3/1,4-dioxane (DO) or ethyl acetate (EA), ZnCl2/EA or DO, HfCl4/EA, ZrCl4/EA, BiCl3/EA, GaCl3/THF, GeCl4/DO, and TiCl4/EA30 systems. However, the main challenge associated with living cationic polymerisation is that the polymerisation rate is quite slow, which directly restricts its application in industry.32 For example, the TiCl4 and EtAlCl2 system suitable for rapid cationic polymerisation (i.e., 1 s, 100% conversion) can achieve the living cationic polymerisation of IBVE in the presence of ethyl acetate, but it requires 120 and 22 h to reach complete conversion, respectively.21,30,40 Efforts have also been channelled into fast living initiation systems,2,38,39,41 such as the FeCl3/cyclic ether (15 s, 100%) and EtAlCl2/SnCl4/ethyl acetate (2 min, 100%) systems. Furthermore, our previous studies show that accelerated living cationic polymerisation with the reduced addition of nucleophiles can be achieved in a microflow system, since the microflow can ensure the stability of the produced propagating chains over a limited time and space. We also demonstrated that the activity of the propagating chains can be measured using the apparent rate constant (kapp) and characteristic time (t1/2),42,43 and the dissociation energies of the active species combining the nucleophile, calculated by density functional theory (DFT) simulations, showed a relationship with living performance.43,44
In this work, we aim to elucidate the effects of different nucleophiles on the activities and stabilities of the growing propagating chains. For this purpose, five different nucleophiles, namely 1,4-dioxane (DO), 1,3-dioxolane (DOL), ethyl acetate (EA), diethyl ether (Et2O), and ethyl propionate (EP), are used to prepare initiation systems containing SnCl4 and IBVE-HCl, which are suitable for the fast living polymerisation of IBVE in a microflow reaction system. The activities and stabilities of various active species are then measured and the relationship between them determined. In addition, the dissociation energies between SnCl4 and various nucleophiles are determined by DFT simulations to reveal the key role of the nucleophile and how they influence the activity (or the stability) of the propagating chains quantitatively.
2. Materials and methods
2.1 Reagents
IBVE (>99.0%, TCI) was purified by heating under reflux for several hours and distilling over calcium hydride prior to use. Toluene, 1,4-dioxane, 1,3-dioxolane, ethyl acetate, diethyl ether (A.R. grade, Beijing Lanyi Chemical Products) and ethyl propionate (99%, TCI) were subjected to reflux for several hours, distilled over calcium hydride, and stored over 4 Å molecular sieves. 2-Hydroxyethyl methacrylate (97%, Innochem) and SnCl4 (99%, anhydrous, Acros) were used as received without further purification. The adduct of IBVE with HCl (IBVE-HCl) was prepared from the addition reaction of IBVE with HCl.45 All chemicals were stored in a glove box under dry argon.2.2 Characterisation
The number-average molecular weight (Mn), weight-average molecular weight (Mw), and molecular weight distribution (MWD, Mw/Mn) of poly isobutyl vinyl ether (PIBVE) were measured by gel permeation chromatography (GPC). GPC was carried out using a Waters 1515 isocratic HPLC pump connected to three Waters Styragel HT2, HT3, and HT4 columns and a Waters 2414 Refractive Index Detector at 30 °C [Waters; Styragel HT2 (THF), Styragel HT3 (THF), and Styragel HT4 (THF); molecular weight range = 100–10k, 500–30k, 5–600k; particle size = 10 μm; column size = 7.8 mm (internal diameter) × 300 mm; flow rate = 1.0 mL min−1]. 1H NMR spectra were recorded on a Bruker AVANCE III HD 400 spectrometer (400 MHz).2.3 Computational methods
The B3LYP DFT functional was used for all quantum chemical (QC) calculations, which were performed using the Gaussian 16 program. All geometries were optimised using the 6-311G(d,p) basis set, with the exception where the SDD set was used for the Sn atoms. Besides, the addition of a polarisation function (+) for anions (i.e., the SnCl4/Nu complex) was required. The effect of a toluene environment was approximated using the SMD solvation model. The final structure was determined based on analytical frequency calculations carried out on the optimised geometries.2.4 Polymerisation procedure
The polymerisation procedure was carried out in a microflow system. As described in our previous work,42 the flow rates for the monomer, Lewis acid, and MeOH (or 2-hydroxyethyl methacrylate (HEMA)) were 8, 8, and 9.6 mL min−1, respectively. The quenched mixture was washed with dilute hydrochloric acid, an aqueous NaOH solution, and then deionised water to remove the initiator residues. The volatiles were then removed under reduced pressure at room temperature (30 °C) and the residue was vacuum-dried overnight to yield a gummy polymer. The monomer conversions were determined gravimetrically.3. Results and discussion
3.1 Determination of the activities and stabilities of the propagating chains with the addition of various nucleophiles
As mentioned above, the activities of the propagating chains can be represented by the apparent rate constant (kapp) and the characteristic time (t1/2, referred to as tact in this work),42,43 whereby this method requires the monomer concentration data to be obtained at different time intervals prior to reaching 100% monomer conversion. Thus, to obtain the kinetic data for activity determination, the polymerisation reaction of isobutyl vinyl ether (IBVE) with SnCl4 in conjunction with IBVE-HCl in the presence of a low concentration of EA, DO, DOL, Et2O or EP (0.1 M) as the nucleophile was investigated in detail. The results are presented in Table 1, and the obtained GPC profiles are shown in Fig. 1.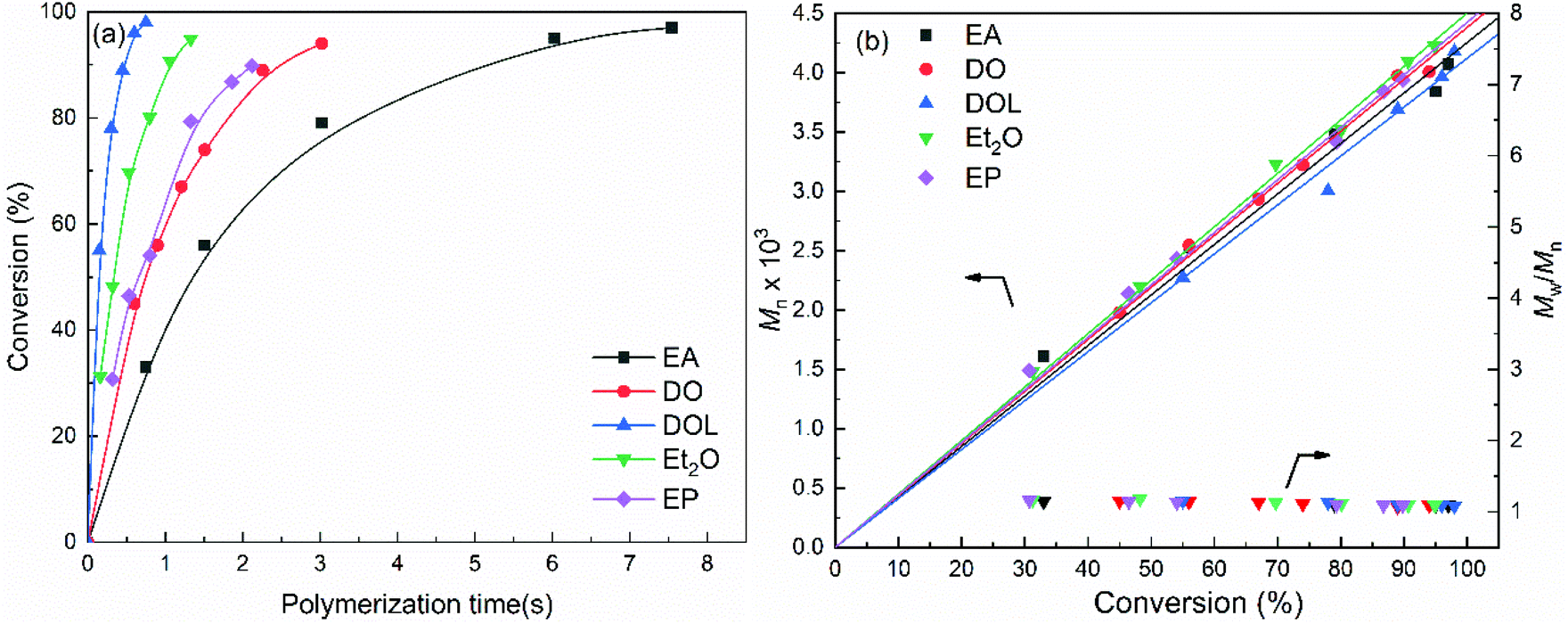 | ||
| Fig. 1 (a) Time–conversion curves for the polymerisation reaction of IBVE with SnCl4, (b) Mn and Mw/Mn for the polymerisation reaction of IBVE (polymerisation conditions: [IBVE]0 = 0.38 M, [IBVE-HCl]0 = 10 mM, [SnCl4]0 = 5.0 mM, [EA/DO/DOL/Et2O/EP] = 0.1 M, in toluene at 0 °C). | ||
| Entry | Nus | t/s | Conv./% | M n,cal/g mol−1 | M n/g mol−1 | M w/Mn | ln([M0]/[M]) |
|---|---|---|---|---|---|---|---|
| All experiments: [IBVE]0 = 0.38 M, [IBVE-HCl]0 = 10 mM, [SnCl4]0 = 5 mM, nucleophile: [EA]0 = 0.1 M for entries 1–5, [DO]0 = 0.1 M for entries 6–11, [DOL]0 = 0.1 M for entries 12–16, [Et2O]0 = 0.1 M for entries 17–22, [EP]0 = 0.1 M for entries 23–28, in toluene at 0 °C. | |||||||
| 1 | EA | 0.75 | 33 | 1260 | 1610 | 1.14 | 0.3988 |
| 2 | 1.51 | 56 | 2170 | 2520 | 1.14 | 0.8321 | |
| 3 | 3.02 | 79 | 3020 | 3480 | 1.10 | 1.5476 | |
| 4 | 6.03 | 95 | 3640 | 3840 | 1.09 | 2.9499 | |
| 5 | 7.54 | 97 | 3740 | 4070 | 1.09 | 3.6088 | |
| 6 | DO | 0.60 | 45 | 1710 | 1980 | 1.15 | 0.5894 |
| 7 | 0.90 | 56 | 2170 | 2550 | 1.14 | 0.8315 | |
| 8 | 1.21 | 67 | 2580 | 2930 | 1.13 | 1.1112 | |
| 9 | 1.51 | 74 | 2860 | 3220 | 1.11 | 1.3606 | |
| 10 | 2.26 | 89 | 3420 | 3970 | 1.07 | 2.2154 | |
| 11 | 3.02 | 94 | 3620 | 4010 | 1.09 | 2.8416 | |
| 12 | DOL | 0.15 | 55 | 2120 | 2270 | 1.15 | 0.8049 |
| 13 | 0.30 | 78 | 2990 | 3000 | 1.13 | 1.5115 | |
| 14 | 0.45 | 89 | 3420 | 3690 | 1.10 | 2.2059 | |
| 15 | 0.60 | 96 | 3690 | 3960 | 1.09 | 3.2560 | |
| 16 | 0.75 | 98 | 3750 | 4180 | 1.08 | 3.7206 | |
| 17 | Et2O | 0.16 | 31 | 1200 | 1483 | 1.16 | 0.3747 |
| 18 | 0.32 | 48 | 1852 | 2200 | 1.18 | 0.6583 | |
| 19 | 0.53 | 70 | 2677 | 3230 | 1.13 | 1.1945 | |
| 20 | 0.80 | 80 | 3076 | 3519 | 1.11 | 1.6147 | |
| 21 | 1.06 | 91 | 3482 | 4100 | 1.09 | 2.3727 | |
| 22 | 1.33 | 95 | 3643 | 4220 | 1.09 | 2.9700 | |
| 23 | EP | 0.32 | 31 | 1180 | 1490 | 1.16 | 0.3671 |
| 24 | 0.53 | 46 | 1784 | 2140 | 1.14 | 0.6247 | |
| 25 | 0.8 | 54 | 2076 | 2430 | 1.13 | 0.7779 | |
| 26 | 1.33 | 79 | 3047 | 3420 | 1.10 | 1.5774 | |
| 27 | 1.86 | 87 | 3332 | 3840 | 1.09 | 2.0227 | |
| 28 | 2.12 | 90 | 3450 | 3940 | 1.09 | 2.2871 | |
As shown in Table 1 entries 1–5, the polymerisation using the SnCl4/EA initiator system gave complete conversion at ∼7.54 s, while the SnCl4/DO, SnCl4/EP, SnCl4/Et2O and SnCl4/DOL initiating systems resulted in complete conversion at 3.02 s, 2.12 s, 1.33 s and 0.75 s, respectively. In addition, it should be emphasised that polymers with a narrow MWD (Mw/Mn = 1.08–1.18) can be produced under all conditions in a microflow system, and as shown in Fig. 1, the Mn value of the polymer is directly proportional to the monomer conversion. This is in good agreement with the calculated value, assuming that the initiator species corresponds to the step-by-step growth of the polymer chain. These results confirmed that under the investigated conditions, the SnCl4/Nu-mediated IBVE polymerisation reactions are invariably living processes. In addition, a nucleophile concentration of 0.1 M was sufficient, and the obtained data were valid for determining the activity of the propagating chains.
In terms of the stability, end-capping analysis is required for the living cationic polymerisation process of IBVE initiated by the SnCl4/X (X = EA, DO, DOL, Et2O or EP) system after reaching 100% monomer conversion. In a pioneering work conducted by Higashimura et al. in 1990, the concentration of living polymers ([P*], the sum of the concentrations of active chains and dormant chains) was determined using sodiomalonic ester as a quencher in the HI/ZnX2 and HI/I2 systems.46,47 Subsequently, they used ln[P*] as a function of time to measure the lifetime of a growing species. However, the synthetic route to sodiomalonic ester is expensive and complex, and no systematic investigations were carried out into the effects of different factors on the lifetime. Since HEMA was reported to be an effective and cheap quencher for living poly(MOVE)s,48 we expected that it could be a suitable and cheap substitute for sodiomalonic ester.
Thus, the reaction was quenched at varying intervals using an excess of HEMA. Fig. 2 shows a typical 1H NMR spectrum for a HEMA-terminated PIBVE, where peaks corresponding to the methacrylate protons at 6.1, 5.6, and 1.9 ppm are clearly visible. The peak intensity ratio of the terminal protons (e) to those of the methylene and methine units of poly(IBVE) (f and g) corresponds to the concentration of living ends. The living polymer chains can be calculated as follows: [living polymer] = [P*] = [M]0 × 103 × conv. × 3e/(f + g), where [M]0 is the initial monomer concentration. In addition, all spectra revealed several chain end structures of side reactions: internal olefin, aldehyde, alkanal, and the structure resulting from β-proton elimination.49 The results of the end-capping analysis and related data are presented in Table 2. It was found that independent of the type of nucleophile, the MWDs of the polymers were always very narrow (Mw/Mn = 1.08–1.13), thereby indicating that the termination process using HEMA was also quantitative and instantaneous.
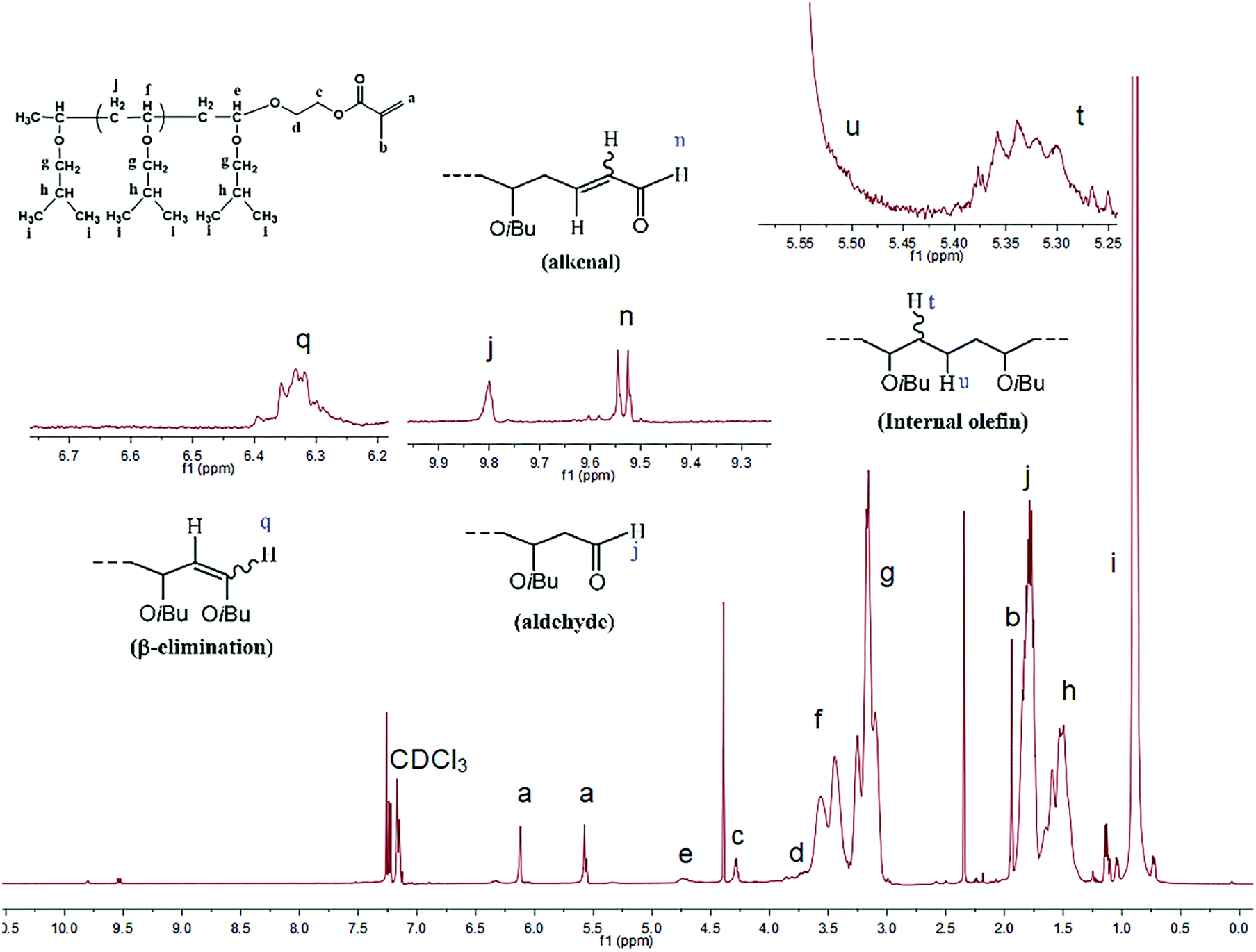 | ||
| Fig. 2 1H NMR spectrum of the HEMA-terminated PIBVE species ([IBVE]0 = 0.38 M, [IBVE-HCl]0 = 10 mM, [SnCl4]0 = 5 mM, nucleophile: [DOL]0 = 0.1 M for entry 18 in Table 2). | ||
| Entry | Nus | t/s | Conv./% | M n,cal/g mol−1 | M n/g mol−1 | M w/Mn | ln[P*] |
|---|---|---|---|---|---|---|---|
| All experiments: [IBVE]0 = 0.38 M, [IBVE-HCl]0 = 10 mM, [SnCl4]0 = 5 mM, nucleophile: [EA]0 = 0.1 M for entries 1–6, [DO]0 = 0.1 M for entries 7–12, [DOL]0 = 0.1 M for entries 13–18, [Et2O]0 = 0.1 M for entries 19–26, [EP]0 = 0.1 M for entries 26–34, in toluene at 0 °C. [P*] = 0.38 × 103 × conv. × 3e/(f + g). | |||||||
| 1 | EA | 60.32 | 100 | 3840 | 4540 | 1.09 | 2.2495 |
| 2 | 75.40 | 100 | 3840 | 4450 | 1.09 | 2.1952 | |
| 3 | 96.51 | 100 | 3840 | 4460 | 1.09 | 2.0766 | |
| 4 | 120.64 | 100 | 3840 | 4220 | 1.10 | 1.9695 | |
| 5 | 156.83 | 100 | 3840 | 3850 | 1.10 | 1.8061 | |
| 6 | 180.96 | 100 | 3840 | 3960 | 1.11 | 1.7147 | |
| 7 | DO | 24.13 | 100 | 3840 | 4590 | 1.07 | 2.2447 |
| 8 | 36.19 | 100 | 3840 | 4550 | 1.08 | 2.1676 | |
| 9 | 48.25 | 100 | 3840 | 4340 | 1.11 | 2.0979 | |
| 10 | 60.32 | 100 | 3840 | 4590 | 1.08 | 1.9357 | |
| 11 | 90.48 | 100 | 3840 | 4410 | 1.09 | 1.7039 | |
| 12 | 120.64 | 100 | 3840 | 4560 | 1.07 | 1.3522 | |
| 13 | DOL | 1.51 | 100 | 3840 | 3830 | 1.09 | 2.1743 |
| 14 | 3.02 | 100 | 3840 | 3680 | 1.10 | 2.1396 | |
| 15 | 6.03 | 100 | 3840 | 3740 | 1.09 | 2.0119 | |
| 16 | 9.05 | 100 | 3840 | 4020 | 1.08 | 1.9099 | |
| 17 | 12.06 | 100 | 3840 | 3810 | 1.09 | 1.8135 | |
| 18 | 15.08 | 100 | 3840 | 3660 | 1.09 | 1.6899 | |
| 19 | Et2O | 6.03 | 100 | 3840 | 4670 | 1.10 | 2.2946 |
| 20 | 9.05 | 100 | 3840 | 4640 | 1.12 | 2.2185 | |
| 21 | 12.06 | 100 | 3840 | 4670 | 1.11 | 2.1790 | |
| 22 | 15.08 | 100 | 3840 | 4640 | 1.11 | 2.0487 | |
| 23 | 24.13 | 100 | 3840 | 4690 | 1.11 | 1.9765 | |
| 24 | 36.19 | 100 | 3840 | 4690 | 1.11 | 1.8265 | |
| 25 | 48.25 | 100 | 3840 | 4630 | 1.13 | 1.9520 | |
| 26 | 60.32 | 100 | 3840 | 4620 | 1.12 | 2.2575 | |
| 27 | EP | 12.06 | 100 | 3840 | 4550 | 1.10 | 1.9577 |
| 28 | 15.08 | 100 | 3840 | 4610 | 1.10 | 2.2822 | |
| 29 | 24.13 | 100 | 3840 | 4590 | 1.09 | 2.1181 | |
| 30 | 36.19 | 100 | 3840 | 4450 | 1.10 | 2.0506 | |
| 31 | 48.25 | 100 | 3840 | 4500 | 1.11 | 1.9316 | |
| 32 | 60.32 | 100 | 3840 | 4530 | 1.10 | 1.8922 | |
| 33 | 90.48 | 100 | 3840 | 4490 | 1.10 | 1.5774 | |
| 34 | 120.64 | 100 | 3840 | 4470 | 1.10 | 1.5489 | |
3.2 Quantitative relationship between the activities and stabilities of the propagating chains
A first-order kinetic model was constructed (Fig. 3) by calculating the apparent polymerisation rate (kapp) from the plot of ln([M]0/[M]) vs. the reaction time. For the SnCl4/EA, SnCl4/DO, SnCl4/EP, SnCl4/Et2O and SnCl4/DOL living initiation systems, kapp was determined to be 0.4871 s−1, 0.9450 s−1, 1.0962 s−1, 2.1973 s−1 and 5.0997 s−1, respectively. Accordingly, tact (i.e., the time required to reach ∼50% conversion at a constant kapp) was found to be 1.42 s, 0.73 s, 0.63 s, 0.32 s and 0.14 s, respectively. In the other words, kapp,DOL ≈ 2.32kapp,Et2O ≈ 4.65kapp,EP ≈ 5.40kapp,DO ≈ 10.47kapp,EA.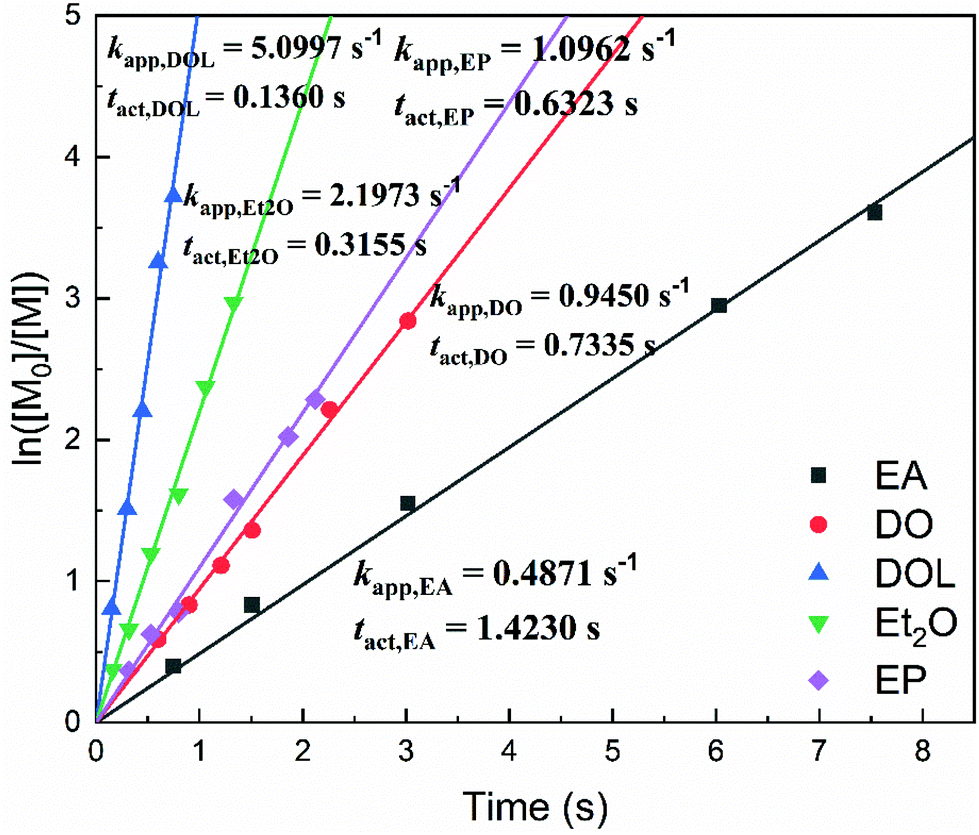 | ||
| Fig. 3 Plot of ln([M]0/[M]) versus time for the SnCl4/X (X = EA, DO, EP, Et2O or DOL) system (polymerisation conditions: [IBVE]0 = 0.38 M, [IBVE-HCl]0 = 10 mM, [SnCl4]0 = 5.0 mM, [EA/DO/DOL/Et2O/EP] = 0.1 M, in toluene at 0 °C). | ||
The ln[P*] values, obtained from these 1H NMR spectra, were then plotted against time, as shown in Fig. 4. The straight ln[P*]–t plots revealed that these decay processes were of the first order in ln[P*], and so can be expressed as follows: −d[P*]/dt = kdecay[P*]. The lifetime (or the half-life, tdecay) of the living polymer ends was then calculated from the slope (kdecay), tdecay = (ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2)/kdecay (tdecay refers to the time at which [P*] is reduced to one-half of the original [IBVE-HCl] in value). In detail, the tdecay is 154.03 s for SnCl4/EA, 74.53 s for SnCl4/DO, 79.67 s for SnCl4/EP, 38.94 s for SnCl4/Et2O, and 19.36 s for SnCl4/DOL, respectively. All of them are significantly shorter than the duration that the stable (living) growing propagating chains present in the HI/I2 system, estimated to range from 40 to 1000 min (17 h).46 It can be noticed that tdecay,EA ≈ 2.07tdecay,DO ≈ 1.93tdecay,EP ≈ 3.96tdecay,Et2O ≈ 7.96tdecay,DOL, which is almost reverse with the quantitative relationship of tact. These results therefore indicate that HEMA reacts quantitatively and specifically with the living chain ends, and is confirmed to be an effective end-capping reagent adaptive to [P*] determination for the first time. It was speculated that the ester oxygen atom of EA stabilised the growing end to a greater extent than cyclic ethers due to stronger coordination or solvation, corresponding to the longest tact being recorded for the SnCl4/EA initiation system.
2)/kdecay (tdecay refers to the time at which [P*] is reduced to one-half of the original [IBVE-HCl] in value). In detail, the tdecay is 154.03 s for SnCl4/EA, 74.53 s for SnCl4/DO, 79.67 s for SnCl4/EP, 38.94 s for SnCl4/Et2O, and 19.36 s for SnCl4/DOL, respectively. All of them are significantly shorter than the duration that the stable (living) growing propagating chains present in the HI/I2 system, estimated to range from 40 to 1000 min (17 h).46 It can be noticed that tdecay,EA ≈ 2.07tdecay,DO ≈ 1.93tdecay,EP ≈ 3.96tdecay,Et2O ≈ 7.96tdecay,DOL, which is almost reverse with the quantitative relationship of tact. These results therefore indicate that HEMA reacts quantitatively and specifically with the living chain ends, and is confirmed to be an effective end-capping reagent adaptive to [P*] determination for the first time. It was speculated that the ester oxygen atom of EA stabilised the growing end to a greater extent than cyclic ethers due to stronger coordination or solvation, corresponding to the longest tact being recorded for the SnCl4/EA initiation system.
 | ||
| Fig. 4 Plot of ln[P*] as a function of time in the IBVE polymerisation reaction initiated by the SnCl4/X (X = EA, DO, EP, Et2O or DOL) system (polymerisation conditions: [IBVE]0 = 0.38 M, [IBVE-HCl]0 = 10 mM, [SnCl4]0 = 5.0 mM, [EA/DO/DOL/Et2O/EP] = 0.1 M, in toluene at 0 °C). | ||
The characteristic time of stability (tdecay) against the characteristic time of activity (tact) can further illustrate the relationship between the activity and stability of the propagating chains intuitively and reveal the effect of the nucleophile essentially, as shown in Fig. 5. The obvious almost linear proportional relationship indicates that for an initiation system the activity and stability values can be calculated from one another, and the effects of nucleophiles on the activities and stabilities may abide by a common rule. This is the first time that the quantitative relationship between these values and the influence of nucleophiles on the activities and stabilities of the propagating chains have been demonstrated experimentally.
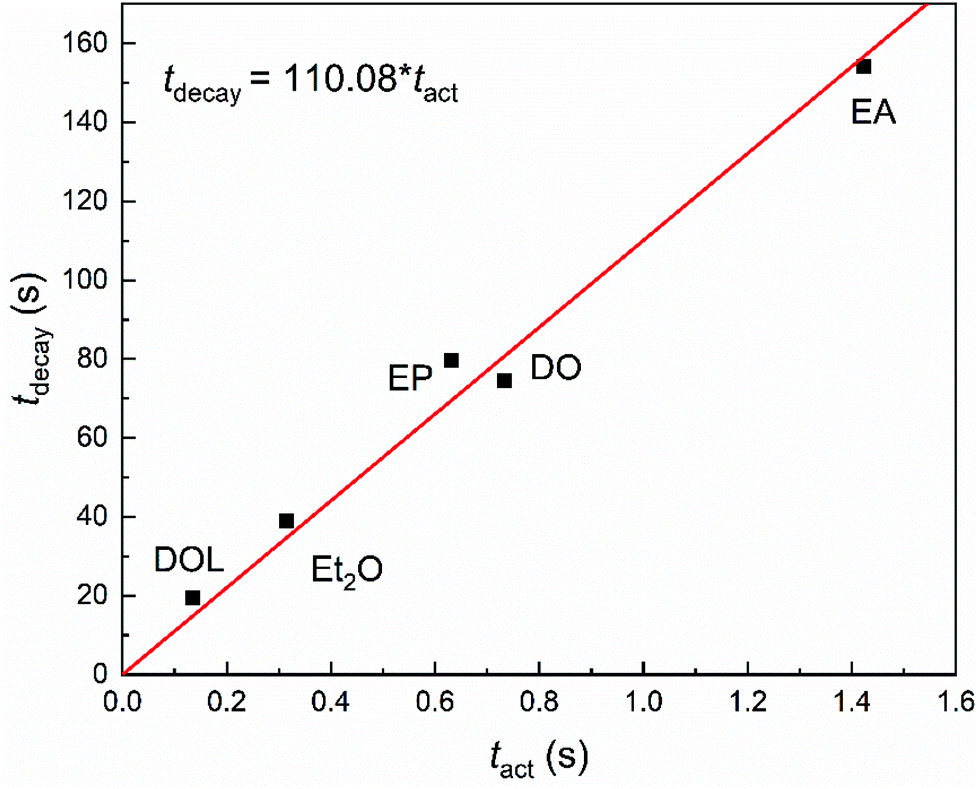 | ||
| Fig. 5 Plot of tdecay as a function of tact in the IBVE polymerisation reactions initiated by the SnCl4/X (EA, DO, DOL, Et2O or EP) system (polymerisation conditions: [IBVE]0 = 0.38 M, [IBVE-HCl]0 = 10 mM, [SnCl4]0 = 5.0 mM, [EA/DO/DOL/Et2O/EP] = 0.1 M, in toluene at 0 °C). | ||
3.3 Correlation between the experimental and simulated data
Our previous report showed that from the perspective of prediction, the dissociation energy of different active species can correspond to the activity (or stability) to some extent.43,44 Herein, we wished to quantify the relationship between the DFT calculation results and experimental values of the activity (or stability), and clarify the nature of different IBVE-HCl·SnCl4·ether ion pairs. Herein, we attempted to establish a more suitable calculation model in DFT simulations with the considerations of more accurate basis sets and the polarization of negative ions, etc. Then the last half of the activation step in the dormant-active equilibrium was calculated, that is, the chain initiation step ([IBVE-HCl·SnCl4·Nu] → [IBVE-HCl·Nu]+ + [SnCl5·Nu]−). In detail, the ionization process of the C–Cl bond (dormant species, IBVE-HCl·SnCl4·Nu complexes) in the growing end produced [IBVE-HCl·Nu]+ and [SnCl5·Nu]− active ions. Then, accompanied by the consumption of monomers, the living chains continue to grow until the terminator is added.As shown in Table 3 and Fig. 6, an ab initio calculation was conducted using Gaussian 16, where it was suggested that the combination of SnCl4 with a nucleophile leads to the formation of a large and stable hexacoordinated anion consisting of five chlorine anions and one additional nucleophile. Due to the large steric hindrance, the introduction of DOL may weaken the interactions with SnCl4 (or IBVE-HCl) to some extent. Accordingly, EA binds more tightly with SnCl4 (or IBVE-HCl), thereby resulting in the most pronounced effects on the activity and stability of the propagating chains in the IBVE-HCl/SnCl4 initiating system, and then a longer lifetime and a lower polymerisation rate. In addition, DFT calculations carried out in a toluene system indicate that the complex of IBVE-HCl·SnCl4·EA has the highest dissociation energy (30.76 kcal mol−1), while the IBVE-HCl·SnCl4·DOL complex exhibits the smallest bond dissociation energy (i.e., 22.65 kcal mol−1). These results indicate that the equilibrium shifts to a greater extent toward the ion pair side in the case of the SnCl4/DOL system, which is in accord with the experimental results. In order to establish a quantitative relationship for prediction from the calculated dissociation energies, an approximately linear kinetic model was obtained by plotting ln(tact) or ln(tdecay) vs. the dissociation energy (Fig. 7). The parallel relationship observed for the ln(t)–ΔE curves shows that the nucleophiles change the absolute values of activity and stability without changing the relative values. It further reveals that nucleophiles mainly affect the frequency of carbocation generation, and the quantitative effects can be calculated well.
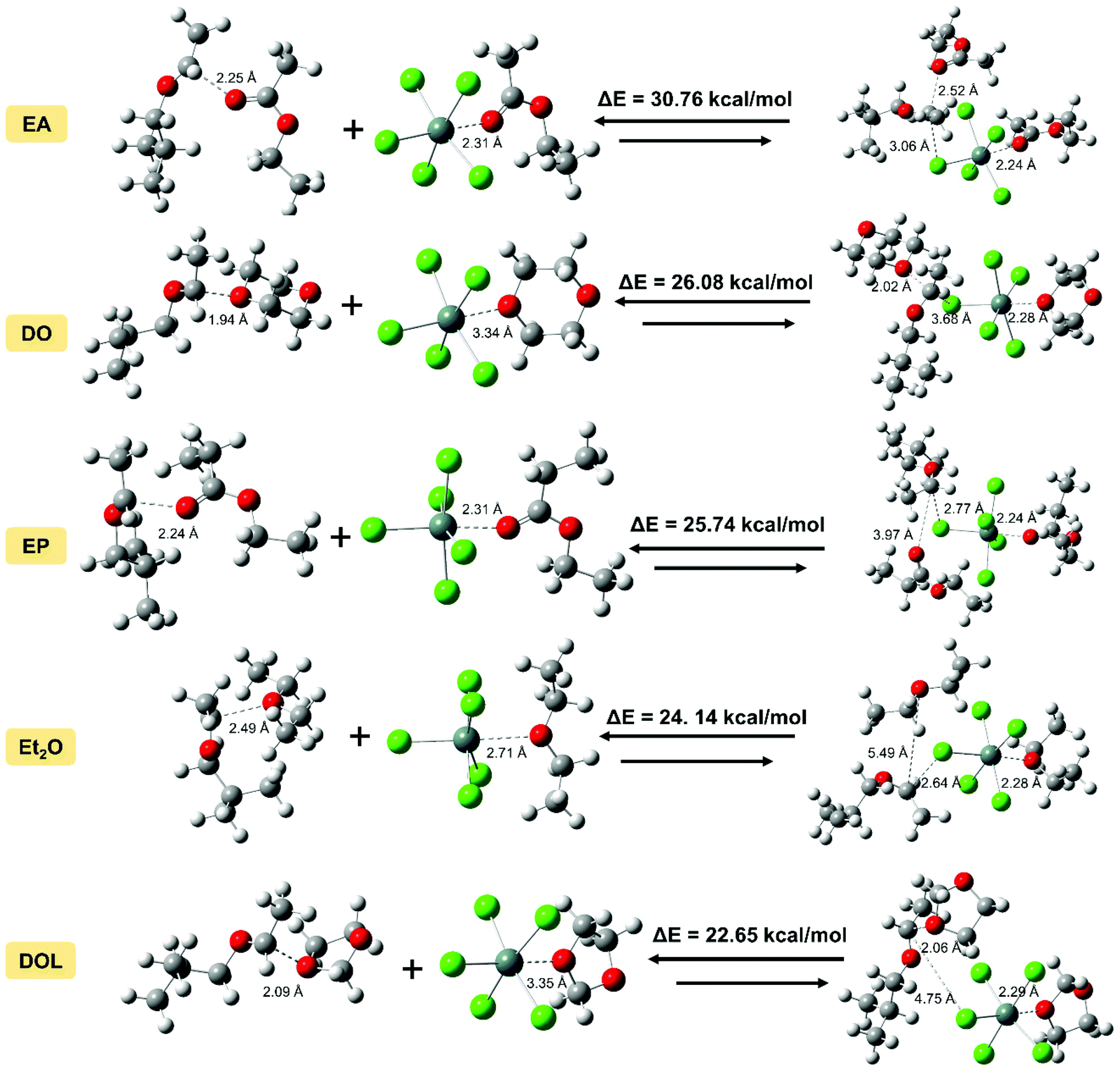 | ||
| Fig. 6 Dissociation energies and optimised structures of IBVE-HCl·SnCl4·Nus calculated using Gaussian 16. (Calculation method: B3LYP/genecp for SnCl5−·Nus, 6-311+G(d,p) for C, H, O, and Cl atoms, SDD for Sn; B3LYP/6-311G(d,p) for IBVE+·Nus; B3LYP/genecp for IBVE-HCl·SnCl4·Nus, 6-311G(d,p) for C, H, O, and Cl, SDD for Sn; em = gd3bj and the SMD solvent model were used for all calculations.) | ||
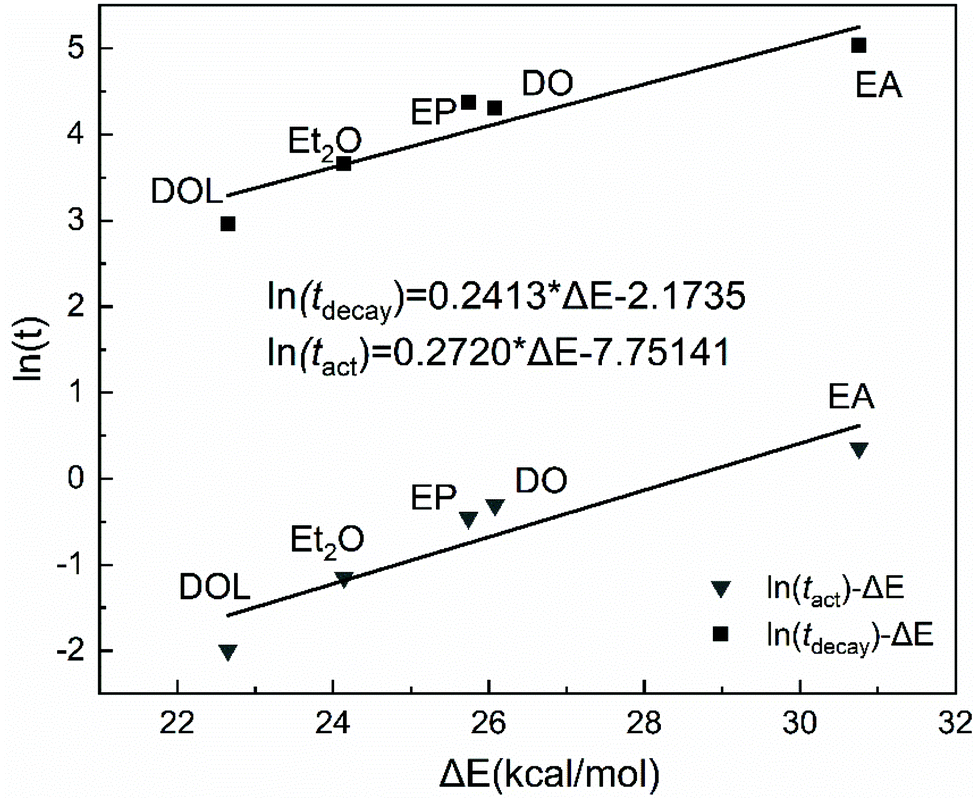 | ||
| Fig. 7 Plots of ln(tact) or ln(tdecay) as a function of the dissociation energy for the SnCl4/X (X = EA, DO, EP, Et2O or DOL) system. | ||
| Energy | E (Hartree) | E (kcal mol−1) |
|---|---|---|
| a B3LYP/genecp for SnCl5−·Nus, 6-311+G(d,p) for C, H, O, and Cl, SDD for Sn; B3LYP/6-311G(d,p) for IBVE+·Nus; B3LYP/genecp for IBVE-HCl·SnCl4·Nus, 6-311G(d,p) for C, H, O, and Cl, SDD for Sn; em = gd3bj and the SMD solvent model were used for all calculations. | ||
| SnCl5−·EA | −2612.7259 | −1639511.6239 |
| IBVE+·EA | −619.4267 | −388696.4441 |
| IBVE-HCl·SnCl4·EA | −3232.2016 | −2028238.8279 |
| Dissociation energy | 0.04902 | 30.7599 |
| SnCl5−·DO | −2612.6838 | −1639485.2164 |
| IBVE+·DO | −619.3790 | −388666.4862 |
| IBVE-HCl·SnCl4·DO | −3232.1043 | −2028177.7837 |
| Dissociation energy | 0.0416 | 26.0812 |
| SnCl5−·DOL | −2573.3490 | −1614802.2184 |
| IBVE+·DOL | −580.0466 | −363985.0131 |
| IBVE-HCl·SnCl4·DOL | −3153.4316 | −1978809.8815 |
| Dissociation energy | 0.0361 | 22.6500 |
| SnCl5−·Et2O | −2538.6458 | −1593025.6153 |
| IBVE+·Et2O | −545.3621 | −342220.1438 |
| IBVE-HCl·SnCl4·Et2O | −3084.0463 | −1935269.8994 |
| Dissociation energy | 0.0385 | 24.1403 |
| SnCl5−·EP | −2652.0534 | −1664190.0033 |
| IBVE+·EP | −658.7570 | −413376.6327 |
| IBVE-HCl·SnCl4·EP | −3310.8514 | −2077592.3758 |
| Dissociation energy | 0.0410 | 25.7398 |
The significance of the above work lies in providing a predictable method for pursuing living/controlled systems. A living system needs not only an inherent controlled reaction, but also a match between mixing characteristic time and reaction characteristic time. The reaction characteristic time can be regulated by the introduction of nucleophiles. In some cases, a conventional system can meet the demand of living polymerisation at the cost of a low production efficiency; however, with stricter requirements, it is necessary to use process enhancing equipment, such as a microflow device with a short mixing time, which results in an improved production efficiency. We also note that nucleophile types do not alter the degree to which the polymerisation reaction deviates from living cationic polymerisation under ideal mixing, and so if such alteration is required, other factors need to be considered. Our ongoing work mainly focuses on the concentration of nucleophiles, solvent type, co-initiator type and monomer type to further reveal the complex effects on the activity and stability of propagating chains.
4. Conclusion
We herein reported the convenient flow determination of the activities and stabilities of propagating chains in fast living cationic polymerization processes in a microflow system, and then investigated the effects of different nucleophiles systematically. More specifically, five nucleophiles, namely ethyl acetate (EA), 1,4-dioxane (DO), diethyl ether (Et2O), ethyl propionate (EP), and 1,3-dioxolane (DOL), were introduced into the IBVE-HCl/SnCl4 initiation system to conduct the accelerated living cationic polymerisation of IBVE in a microflow system. 1H NMR spectroscopy confirmed that 2-hydroxyethyl methacrylate was an effective end-capping agent that can quantitatively terminate and combine with living polymers. Subsequently, the activities and stabilities of the growing propagating chains were presented based on the characteristic time (tact) and half-life (tdecay), respectively. It was found that the IBVE-HCl/SnCl4/EA initiation system resulted in the longest half-life (tdecay,EA = 154.03 s) and the lowest frequency of carbocation generation. In contrast, the IBVE-HCl/SnCl4/DOL initiation systems exhibited shorter half-lives (i.e., tdecay,DOL = 19.36 s). The characteristic time (tact) and the half-life (tdecay) exhibited an approximately linear proportional relationship, revealing that the nucleophiles did not change the relative values of the activity and stability, but simply improved the controllability of the cationic polymerisation process through prolonging tact under limited mixing performance. Furthermore, a density functional theory simulation method was established to calculate the dissociation energy (ΔE) of IBVE-HCl·SnCl4·Nus. An approximately linear relationship between ln(tact) or ln(tdecay) and ΔE was found, indicating the potential of theoretical calculations in estimating the activity and stability of the propagating chains. These findings therefore identify the role of nucleophiles in living cationic polymerisation processes, and may provide a predictive method for establishing an appropriate initiation system.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors are grateful for the financial support of the National Natural Science Foundation of China (21422603, U1662120, 21978152).References
- J. E. Puskas and G. Kaszas, Prog. Polym. Sci., 2000, 25, 403–452 CrossRef CAS.
- S. Aoshima, T. Yoshida, A. Kanazawa and S. Kanaoka, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1801–1813 CrossRef CAS.
- M. Miyamoto, M. Sawamoto and T. Higashimura, Macromolecules, 1984, 17, 265–268 CrossRef CAS.
- Y. Seki, A. Kanazawa, S. Kanaoka, T. Fujiwara and S. Aoshima, Macromolecules, 2018, 51, 825–835 CrossRef CAS.
- M. Kawamura, A. Kanazawa, S. Kanaoka and S. Aoshima, Polym. Chem., 2015, 6, 4102–4108 RSC.
- T. Yoshizaki, A. Kanazawa, S. Kanaoka and S. Aoshima, Macromolecules, 2015, 49, 71–79 CrossRef.
- S. Kanaoka, M. Yamada, J. Ashida, A. Kanazawa and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4594–4598 CrossRef CAS.
- S. Kanaoka, H. Shimomoto, D. Fukami and S. Aoshima, Advances in Fluorine-Containing Polymers, 2012, vol. 1106, pp. 65–79 Search PubMed.
- M. Ciftci, Y. Yoshikawa and Y. Yagci, Angew. Chem., Int. Ed., 2017, 56, 519–523 CrossRef CAS PubMed.
- M. U. Kahveci, F. Oytun and Y. Yagci, Polymer, 2013, 54, 4798–4801 CrossRef CAS.
- M. U. Kahveci, M. Uygun, M. A. Tasdelen, W. Schnabel, W. D. Cook and Y. Yagci, Macromolecules, 2009, 42, 4443–4448 CrossRef CAS.
- M. U. Kahveci and Y. Yagci, Macromolecules, 2011, 44, 5569–5572 CrossRef CAS.
- W. G. S. Reyntjens and E. J. Goethals, Polym. Adv. Technol., 2001, 12, 107–122 CrossRef CAS.
- B. Verdonck, J.-F. Gohy, E. Khousakoun, R. Jérôme and F. Du Prez, Polymer, 2005, 46, 9899–9907 CrossRef CAS.
- S. Sugihara, S. Kanaoka and S. Aoshima, Macromolecules, 2005, 38, 1919–1927 CrossRef CAS.
- K. Kono, T. Ozawa, T. Yoshida, F. Ozaki, Y. Ishizaka, K. Maruyama, C. Kojima, A. Harada and S. Aoshima, Biomaterials, 2010, 31, 7096–7105 CrossRef CAS PubMed.
- H. Shimomoto, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4137–4144 CrossRef CAS.
- Y. Shinke, A. Kanazawa, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 5239–5247 CrossRef CAS.
- Y. Oda, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1207–1213 CrossRef CAS.
- S. Sugihara, S. I. Matsuzono, H. Sakai, M. Abe and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3190–3197 CrossRef CAS.
- T. Shibata, S. Kanaoka and S. Aoshima, J. Am. Chem. Soc., 2006, 128, 7497–7504 CrossRef CAS PubMed.
- H. Shimomoto, D. Fukami, T. Irita, K.-i. Katsukawa, T. Nagai, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1547–1555 CrossRef CAS.
- H. Shimomoto, H. Yoshida, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3703–3709 CrossRef CAS.
- S. Aoshima, Y. Oda, S. Matsumoto, Y. Shinke, A. Kanazawa and S. Kanaoka, ACS Macro Lett., 2013, 3, 80–85 CrossRef.
- J. Motoyanagi, T. Ishikawa and M. Minoda, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3318–3325 CrossRef CAS.
- S. Sugihara, K. Hashimoto, S. Okabe, M. Shibayama, S. Kanaoka and S. Aoshima, Macromolecules, 2004, 37, 336–343 CrossRef CAS.
- T. Yoshida, S. Kanaoka, H. Watanabe and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2712–2722 CrossRef CAS.
- K.-I. Seno, I. Tsujimoto, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6444–6454 CrossRef CAS.
- A. Kanazawa, S. Kanaoka and S. Aoshima, Macromolecules, 2010, 43, 2739–2747 CrossRef CAS.
- A. Kanazawa, S. Kanaoka and S. Aoshima, Macromolecules, 2009, 42, 3965–3972 CrossRef CAS.
- A. Kanazawa, S. Kanaoka and S. Aoshima, Chem. Lett., 2010, 39, 1232–1237 CrossRef CAS.
- S. Aoshima and S. Kanaoka, Chem. Rev., 2009, 109, 5245–5287 CrossRef CAS PubMed.
- S. Aoshima and T. Higashimura, Macromolecules, 1989, 22, 1009–1013 CrossRef CAS.
- Y. Kishimoto, S. Aoshima and T. Higashimura, Macromolecules, 1989, 22, 3877–3882 CrossRef CAS.
- S. Aoshima, K. Shachi and E. Kobayashi, Makromol. Chem., 1991, 192, 1759–1768 CrossRef CAS.
- S. Aoshima, K. Shachi, E. Kobayashi and T. Higashimura, Makromol. Chem., 1991, 192, 1749–1757 CrossRef CAS.
- A. Kanazawa, Y. Hirabaru, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5795–5800 CrossRef CAS.
- T. Yoshida, T. Tsujino, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 468–472 CrossRef CAS.
- T. Yoshida, A. Kanazawa, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4288–4291 CrossRef CAS.
- Y. Shinke, A. Kanazawa, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 5041–5048 CrossRef CAS.
- T. Iwasaki, A. Nagaki and J. Yoshida, Chem. Commun., 2007, 1263–1265, 10.1039/b615159k.
- D. Xie and Y. Lu, Ind. Eng. Chem. Res., 2018, 57, 7441–7449 CrossRef CAS.
- D. Xie and Y. C. Lu, Eur. Polym. J., 2019, 113, 220–228 CrossRef CAS.
- D. Xie, S. Zhu and Y. Lu, RSC Adv., 2020, 10, 5183–5190 RSC.
- M. Kamigaito, Y. Maeda, M. Sawamoto and T. Higashimura, Macromolecules, 1993, 26, 1643–1649 CrossRef CAS.
- W. O. Choi, M. Sawamoto and T. Higashimura, Macromolecules, 1990, 23, 48–53 CrossRef CAS.
- W. O. Choi, M. Sawamoto and T. Higashimura, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 2923–2935 CrossRef CAS.
- W. G. S. Reyntjens and E. J. Goethals, Des. Monomers Polym., 2012, 4, 195–201 CrossRef.
- A. Kanazawa, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3702–3708 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |