
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The block copolymer shuffle in size exclusion chromatography: the intrinsic problem with using elugrams to determine chain extension success
Kai
Philipps
a,
Tanja
Junkers
 *b and
Jasper J.
Michels
*a
*b and
Jasper J.
Michels
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: michels@mpip-mainz.mpg.de
bPolymer Reaction Design Group, School of Chemistry, Monash University, 19 Rainforest Walk, Building 23, Clayton, Vic 3800, Australia. E-mail: tanja.junkers@monash.edu
First published on 8th April 2021
Abstract
Size exclusion chromatography (SEC) based on direct homopolymer calibration is the preferred method for molecular weight determination in macromolecular synthesis. However, using the same method and calibration in block copolymer (BCP) characterization can lead to an apparent molecular weight reduction when in fact the opposite has taken place. This leads to a situation where researchers often have enough difficulties in interpreting their data, creating false conclusions and deeming successful experiments unsuccessful or vice versa. Here, a selection of block copolymers from the literature is discussed for which such an incoherent change in the retention time has been observed. These examples represent four categories of BCPs for which unexpected SEC results can occur. We discuss the importance of a critical evaluation of SEC and the reasons for the deviating behavior and highlight the danger of the common practice to use elugrams to prove or disprove successful chain extensions in synthesis. Next to a critical evaluation of the various cases, we give recommendations for which other detection and characterization methods may enable accurate block copolymer characterization.
Introduction
Owing to its accessibility and ease of operation, size exclusion chromatography (SEC), also known as gel permeation chromatography (GPC), has long become the preferred method for polymer molecular weight determination. SEC provides the full molecular weight distribution of a sample, from which molecular weight averages and dispersity (Đ) can be derived. Molecular weight determination by SEC makes use of the proportionality between the hydrodynamic volume VH of a (co-)polymer chain in a solvent and the product of the intrinsic viscosity [η] and the molecular weight M:1| VH ∼ [η]·M | (1) |
Here, [η] captures to what extent the polymer chain is swollen by the solvent. For practical reasons, eqn (1) is usually written as:
| HV = [η]·M, | (2) |
Although readily applicable to most homopolymers, molecular weight determination of block copolymers (BCPs) using SEC is generally much less straightforward than typically considered. Besides issues also known to occur for SEC on homopolymers, e.g. associated with aggregation2 or chemical dissimilarity with the calibration standard,3–5 new challenges appear due to the mere fact that the different blocks themselves have different properties and solvation. Consequently, their interaction with both the mobile and stationary phases, as well as their resemblance with the calibration standard, is per definition different. The typical assumption of universal calibration thus does frequently not hold true, despite researchers using this assumption almost without exception in their data interpretation. A step forward is presented by using viscometry, hence allowing for chemically different analytes and standards and by measuring the intrinsic viscosity directly rather than estimating it from calibrations.
The relation between [η] and M is typically expressed via the empiric Mark–Houwink–Sakurada (MHS) equation, which uses the parameters α and K of an unknown analyte for correlation, according to the MHS equation: [η] = KMα. However, even if the MHS parameters are known precisely (which by itself is a challenge and is usually only true for a selected number of homopolymers), an important prerequisite for its use is that the analyte has similar solvation to the calibration standard. Hence, again related to inhomogeneity in both chain length and composition, the outcome of this exercise should be treated with caution when analyzing a BCP. As mentioned, the direct measurement of the intrinsic viscosity somewhat improves the situation in that MHS parameters must not be known. Employing a viscometer, HV can be directly calibrated using standards, and measurement of the intrinsic viscosity of the analyte sample allows for direct determination of its molecular weight. Yet, the inaccuracy associated with the prerequisite of similar solution behavior of chains remains.
A way out of this dilemma is the use of laser light scattering (LS) in a triple detection SEC system, which allows the measurement of the molecular weight directly without relying on knowledge of HV. Regardless, even when combined with an absolute detection technique, such as multi-angle laser light scattering (MALLS), SEC analysis of BCPs exhibits reliability issues since the eluted composition, and hence the scattering contrast (dn/dc), is time-dependent due to the dispersity in the block length. In principle, dn/dc would need to be known for each exact block composition to yield an exact measurement of the molecular weight, which obviously is not feasible. Concentration determination is hence hampered, which also invalidates light scattering results to a certain degree. Furthermore, LS-based methods usually involve not only high cost, but also require more skilled operators and in-depth analysis since scattering data is less straightforward to analyze properly compared to the simpler RI, UV and viscometry detectors.
It is fair to assume that for routine analysis, most labs do not perform light scattering or do not have access to such detectors in the first place. Hence, in the analysis of block copolymers (and other polymer architectures for that matter), researchers typically make basic assumptions and refrain from even determining molecular weights. A typical recommendation is to plot SEC elugrams rather than molecular weight distributions to avoid any ambiguity with calibrations and generally to avoid the above-described dilemmas. A general assumption thereby is that lower elution volumes are correlated to larger molecular weights. Hence, it is widely accepted that a shift to lower elution volumes presents proof for a successful extension in a block copolymerization and that a lack thereof is synonymous with a failure in reaction. This does not necessarily hold true though, as this still assumes a universal-type calibration/correlation to be valid for the investigated polymers. As we discuss here, this can be a dangerous and misleading assumption.
This mini-review is written in part in a tutorial form and gives a concise and accessible discussion on the challenges, approaches and solutions associated with interpreting molecular weight distributions of BCPs from SEC. We intend to provide practical handles and concepts, rather than presenting a lengthy and exhaustive literature survey to allow practitioners to derive meaningful conclusions. We start our discussion by describing examples of BCPs for which SEC actually works quite well, namely, for BCPs comprising flexible blocks that only interact weakly. After that, we discuss a range of examples of BCPs that seem to behave far less ideally due to the fact that the blocks have quite different chemical or physical properties and hence pose significant challenges on the use of SEC for molecular weight determination or even merely proving block extension.
It is particularly worthwhile reviewing such “inhomogeneous” BCPs since their internally cooperative- or multi-functionality is of strong interest for advanced applications, as smart coatings,6–9 drug delivery,10–13 bioimaging,14–16 optoelectronics17–20 and energy harvesting and storage.21–25 We subdivide these non-classical BCPs into four “behavioural categories” and conclude with a brief survey of methods to improve the accuracy of molecular weight estimates. These techniques may either be used in conjunction with SEC or replace it altogether as more viable options. It should be noted that none of the proposed methods represent truly novel concepts, yet SEC is too often used in the literature either as proof or dismissal of success of a reaction, especially in BCP formation without further questioning, and we wish to highlight the problems arising from such conclusions.
Discussion
Classical systems: dual detector approach for BCP molecular weight analysis
The simplest cases are presented by (block-)copolymers of which the monomers form flexible chains and only exhibit weak interactions, i.e. lacking ionic charges, strong dipoles and/or H-bonding capability. In effect, for block-copolymers falling into this category, the covalent link between the different blocks forms the only relevant “hetero-contact”. This enables an interpretation as connected homopolymers26 and therefore allows for molecular weight determination based on a mass fraction-weighted interpolation of the homopolymer SEC calibration curves as typically applied:27log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Mc(V) = ∑wi(V)·logMi(V), Mc(V) = ∑wi(V)·logMi(V), | (3) |
For any BCP obeying this prerequisite, the molecular weight can be established reasonably well with SEC alone.28 However, even for such straightforward systems, one cannot rely on just a single detection method, i.e. typically UV-vis absorption or refractive index (RI) detection. The reason is that for not perfectly alternating copolymers, not only the molecular weight but also the composition is distributed. In such cases, a dual detection method involving for example both the UV-vis and RI detectors can be used.29 The latter records the copolymer concentration, whereas the former records the composition owing to its high chemical specificity.30 Prior to analyzing the copolymer, solutions of the corresponding homopolymers are eluted through both channels in order to predetermine the detector responses, which are comonomer- and instrument-dependent.31,32
The eluted masses mA and mB (e.g. expressed in grams) of comonomers A and B can then be obtained for each “slice” i of the SEC chromatogram by solving the following system of equations for the measured intensities:31
| SUVi = χUVAmA,i + χUVBmB,i | (4) |
| SIRi = χIRAmA,i + χIRBmB,i, | (5) |
 | ||
| Fig. 1 SEC-UV/RI evaluation method on PS-b-PZL: (A) determination of the chemical composition of copolymer fractions from the UV (dashed) and RI (solid) traces; (B) calculation of the concentration and molar mass of copolymer chains, including generation of a copolymer calibration curve (dotted); and (C) calculation of the copolymer mass distribution. Reproduced from ref. 31 with permission from the American Chemical Society, copyright 2003. | ||
Dual detector SEC has shown to give very reasonable estimates for a range of “classical” BCPs, such as polyisobutylene-b-polystyrene (PIB-co-PS),32 polyacenaphthylene-b-isobutylene-b-acenaphthylene (PAc-b-PIB-b-PAc),32 polystyrene-co-poly(butylene terephthalate) (PS-co-PBD),36 polystyrene-b-poly(methyl methacrylate) (PS-b-PMMA),30,31 polystyrene-b-polybutadiene (PS-b-PB)31 and polystyrene-b-polylysine (PS-b-PZL).31 However, as mentioned above, if the hydrodynamic behaviour of the block copolymer differs from the “summed” behaviour of the homopolymers, even dual detector SEC cannot produce reliable results. Early identified examples are cases wherein one or both components carry a bulky side group.32 Besides, a trivial limitation of dual detector SEC is that it does not distinguish between homopolymer contaminants and the copolymer.32
Examples of “non-classical” block copolymers giving deviant SEC analyses
In case SEC is applied to BCPs for which the combination of the blocks gives a behavior that differs strongly from what one would expect based on the linear combination expressed by eqn (3), observations can be counterintuitive. In these situations, care should be taken when using SEC for molecular weight determination, or, as we shall see, even for just collecting qualitative evidence for chain extension or successful synthesis of a BCP. Since SEC analysis is based on the general assumption that the retention time decreases if the hydrodynamic radius of the polymer becomes larger, counterintuitive results are obtained if during coupling or growth of the second block the polymer coil contracts, collapses or exhibits an increased interaction with the stationary phase. Below, we discuss a number of representative examples (overview in Table 1) of “non-classical” functional BCPs that behave deviant in SEC inasmuch an apparent increase in the retention time is observed with increasing molecular weight.| Entry | Polymerc | Polymer type |
![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n,SEC
n,SEC
|
n
|
Đ | Ref. |
|---|---|---|---|---|---|---|
| a Determined from 1H NMR. b Determined by MALDI. c See the main text for abbreviations. | ||||||
| 1 | OHPV5 | Homopolymer | 2.3 | 1.5a,b | 1.02 | 37 |
| 2 | OHPV5-b-PEG12 | Linear BCP | 1.9 | 2.1a | 1.04 | |
| 3 | OHPV5-b-PEG45 | Linear BCP | 3.7 | 3.6a | 1.06 | |
| 4 | OHPV5-b-PEG115 | Linear BCP | 7.7 | 6.6a | 1.05 | |
| 5 | PF–Br | Homopolymer | 6.1 | — | 1.48 | 38 |
| 6 | PF-b-PHFBMA12 | Linear BCP | 9.5 | 9.3a | 1.50 | |
| 7 | PF-b-PHFBMA36 | Linear BCP | 9.6 | 15.7a | 1.60 | |
| 8 | PF-b-PHFBMA39 | Linear BCP | 9.1 | 21.9a | 1.64 | |
| 9 | PF-b-PHFBMA87 | Linear BCP | 8.7 | 29.4a | 1.60 | |
| 10 | PNIPAM | Homopolymer | 23.0 | — | 1.3 | 39 |
| 11 | PNIPAM-b-PPEGA-b-PNIPAM | ABA-type BCP | 14.0 | — | 1.36 | |
| 12 | PS-b-PNIPAM-b-PPEGA | ABCACBA-type BCP | 77.0 | — | 1.45 | |
| 13 | PEI32 | Homopolymer | — | 19.3a | — | 40 |
| 14 | P4VP29-b-PEI32-b-P4VP29 | ABA-type BCP | 18.7 | 25.6a | 1.90 | |
| 15 | P4VP35-b-PEI32-b-P4VP35 | ABA-type BCP | 12.1 | 27.7a | 2.12 | |
| 16 | P4VP85-b-PEI32-b-P4VP85 | ABA-type BCP | 10.5 | 37.8a | 2.17 | |
 | ||
| Fig. 2 SEC traces of OHPVx-b-PEGy polymers, corresponding to entries 1–4 in Table 1; (a) RI signal and (b) UV-vis signal recorded at 490 nm. See the main text for the non-abbreviated nomenclature. Reproduced from ref. 37 with permission from the Royal Society of Chemistry, copyright 2014. | ||
The underlying reason for this discrepancy might in part be related to the fact that the molecular weight of the OHPV homopolymer is likely overestimated (∼50% compared to the value from NMR and MALDI) due to its high rigidity. For a given molecular weight, the associated hydrodynamic volume of poly(p-phenylenevinylene) is hence high in comparison with a more flexible polymer used for SEC calibration (i.e. usually polystyrene).41 This is expressed by the disagreement between the SEC molecular weight average and the estimate obtained by NMR (entry 1). Upon connecting the flexible PEG block, the hydrodynamic properties become more reminiscent of those of the PS calibration standard, leading to an agreement between the SEC and NMR estimates, but perhaps resulting in an apparent reduction in the hydrodynamic volume compared to the OHPV homopolymer. An increase in the retention time due to a higher affinity of the PEG residues with the stationary phase is not likely in view of the apolar column material used in this work37 (crosslinked styrene/divinyl benzene matrix). We note that an increase in the retention time when connecting or growing a flexible block to/from a stiff homopolymer is certainly not always observed42–44 and speculate that it may depend on the length of the latter and/or whether the stiff block is truly in the rod-like limit.
n follows the opposite trend compared to the value obtained from NMR (see Table 1). The authors do not ascribe the anomaly to the stiff–flexible nature of the BCPs, but rather to the poor solubility of the semifluorinated block in the SEC solvent (THF), resulting in an overall collapse of the BCP at an elevated degree of polymerization.
 n = 23000 g mol−1 to 14000 g mol−1. This work39 does not contain comparative values from, for instance, NMR or mass spectrometry (MS). The authors point out that both PNIPAM and PPEGA are known to be strongly susceptible towards hydrogen bonding (although the latter can only accept and not donate H-bonds). They speculate that mutual H-bonding between the different polymer constituents is responsible for contraction of the coil upon block extension, leading to a concomitant reduction in the hydrodynamic volume and an increase in the retention time.
n = 23000 g mol−1 to 14000 g mol−1. This work39 does not contain comparative values from, for instance, NMR or mass spectrometry (MS). The authors point out that both PNIPAM and PPEGA are known to be strongly susceptible towards hydrogen bonding (although the latter can only accept and not donate H-bonds). They speculate that mutual H-bonding between the different polymer constituents is responsible for contraction of the coil upon block extension, leading to a concomitant reduction in the hydrodynamic volume and an increase in the retention time.
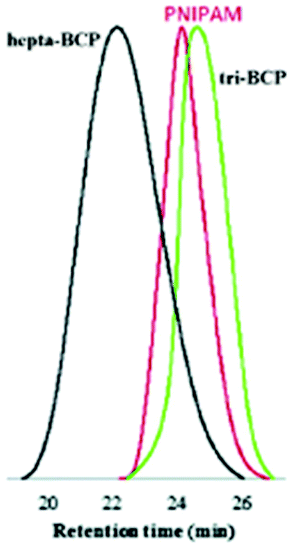 | ||
| Fig. 4 SEC traces corresponding to entries 10–12 in Table 1. The red, green and black traces respectively correspond to the PNIPAM homopolymer, tri-BCP PNIPAM-b-PPEGA-b-PNIPAM and hepta-BCP PS-b-PNIPAM-b-PPEGA-b-PS-b-PPEGA-b-PNIPAM-b-PS (not specifically discussed here). See the main text for the non-abbreviated nomenclature. Reproduced from ref. 39 with permission from the Royal Society of Chemistry, copyright 2014. | ||
 | ||
| Fig. 5 SEC RI elugrams (dotted lines) and the absolute molecular weight (solid curves), determined by GPC–MALLS, of P4VP-b-PEI-b-P4VP tri-BCPs, corresponding to entries 13–16 of Table 1. See the main text for the non-abbreviated nomenclature. Reproduced from ref. 40 with permission from the American Chemical Society, copyright 2020. | ||
Given the fact that PEI and P4VP are flexible polymers, well soluble in the SEC solvent (THF)45,46 and both incapable of donating hydrogen bonds, the reason for the increase in the retention time must be different from the examples discussed above. Indeed, according to the authors, their data carry the signature of (too) strong attractive interaction between the BCP with the stationary phase of the GPC column. Although the authors do not give a suggestion which block is responsible for the effect, the fact that the observed trend relates to the length of the P4VP block seems to identify the culprit. According to the authors, the observation that the molecular weight fraction determined by GPC–MALLS (solid lines in Fig. 5) shows a “U-shaped” time dependence (see Fig. 5) is characteristic for a strong interaction between the polymer analyte and the GPC stationary phase. Unfortunately, the particular column material used in this work is not mentioned. We finally note that, although the interpretation by the authors is certainly plausible, interpreting results generated using only the RI detector or in combination with MALLS requires some care. The reason is that during elution of the BCP, compositional changes associated with the block length distributions are not accounted for. As mentioned in the introduction, if the refractive index of the solution on which both methods rely strongly varies with time, the shape of the elugrams, as well as the MALLS molecular weight estimates, may be strongly impacted.
Alternative methods for molecular weight determination
The cases discussed above represent a small but, in our view, representative selection of examples from the literature reporting anomalous behavior of functional BCPs during SEC analysis. Although the overview is not exhaustive, it probably covers the most important reasons for which single- or even dual-detection SEC methods may yield counterintuitive results: (i) rod–coil BCPs, (ii) blocks exhibiting a (strong) difference in compatibility with the carrier solvent, (iii) strong mutual interactions between blocks and (iv) strong interaction with the stationary phase of one of the blocks.Furthermore, we note that the shape and modality of the SEC elugrams could provide information beyond merely proving a successful BCP synthesis. Deconvolution may allow identifying impurities such as homopolymer contamination55 and side products, e.g. from branching reactions.56 The fact that such impurities can significantly impact the microphase separated morphology of a BCP57 emphasizes the importance of an extended evaluation of the modality in SEC elugrams.
If simple remedies such as a change in temperature or mobile phase composition are ineffective or out of scope, an alternative or additional method is required to determine the molecular weight more accurately. Indeed, most of the examples discussed above present estimates obtained with an alternative approach, such as NMR end-group analysis or MS. One should realize though that where most methods only yield a single value, e.g. for the number- or weight-average molecular weight, GPC has the essential advantage of almost solely being capable of mapping a full distribution. Hence, besides focusing on independent alternative approaches, the remainder of this section also presents methods that can be used in conjunction with SEC to obtain more reliable molecular weight estimates and/or compositional mapping of a BCP sample. Although each method has its own advantages, we will critically discuss disadvantages as well and, where appropriate, assess to what extent the method in question circumvents the anomalies discussed in the previous section.
w). Since the MALLS measurement is independent of the retention time on the GPC column, some of the issues discussed in the previous section are in principle avoided. It should be noted though that for conjugated polymers – as discussed above – MALLS is inherently associated with an error as the incident laser light will cause not only scattering, but potentially also fluorescence. This again can negatively impact the MALLS signals and lead to misinterpretation of data.
Illustratively, Zhao et al. showed that despite exhibiting similar SEC retention times, GPC–MALLS reveals the expected difference in the molecular weight between a poly(oligo(ethylene glycol) monomethyl ether methacrylate) (POEGMA) homopolymer and its equivalent BCP, obtained via a click reaction of the POEGMA block with an azide-functionalized polyfluorene (see Fig. 6).59 Although GPC–MALLS is arguably one of the most powerful tools for molecular weight characterization, we emphasize again that it assumes the sample to be compositionally homogeneous, which imparts a risk when analyzing a BCP that is distributed with respect to the block length (as practically all BCPs are),60 and of which the refractive index of the individual blocks is significantly different.
 | ||
| Fig. 6 SEC traces of alkyne functionalized poly(oligo(ethylene glycol) monomethyl ether methacrylate (POEGMA) and the BCP polyfluorene-b-POEGMA. Reproduced from ref. 59 with permission from the American Chemical Society, copyright 2018. | ||
As an example, Fig. 7 shows the result of a SEC–HPLC analysis of a very inhomogeneous sample based on polystyrene-b-polybutadiene (PS-b-PBD), containing both linear and branched structures, as well as a distribution in the PBD block length. SEC–HPLC identified up to 16 different species in this particular sample.61 Again, as the full analysis is based on an additional parameter or material property (polarity in this case), SEC retention time anomalies can be accounted for, depending on their underlying reason. In view of the challenges discussed above, the example given in Fig. 7 represents a system that is of relatively “low risk”: the “classical” BCP PS-b-PBD comprises two flexible apolar blocks, both incapable of exhibiting strong interactions. Nevertheless, even in case a BCP analyte would exhibit non-straightforward behavior in SEC, the combination with HPLC certainly seems very useful to confirm a successful block extension, coupling of grafting.
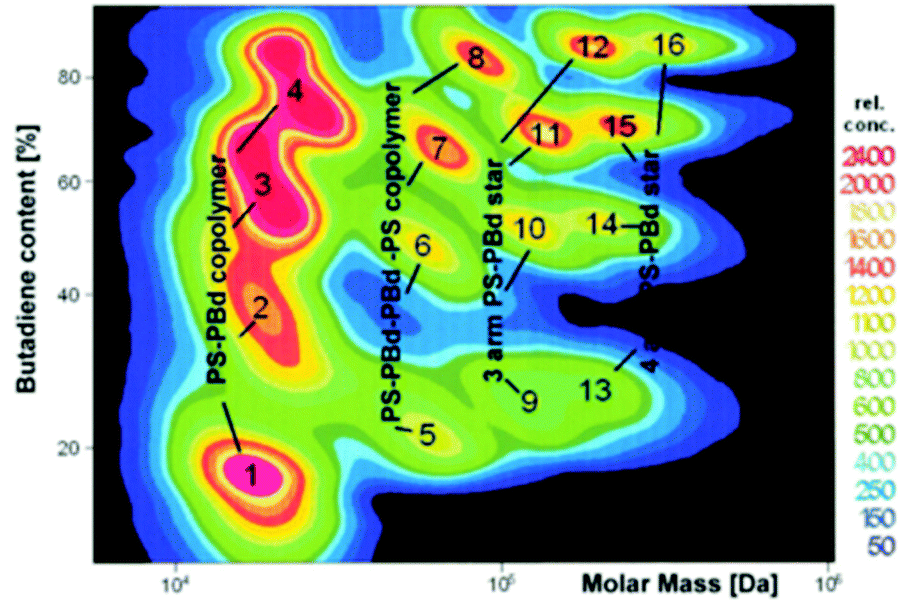 | ||
| Fig. 7 SEC–HPLC 2D chromatography contour map (relative eluted concentration) of a complex mixture of PS-b-PBD, containing both linear and branched structures. The horizontal and vertical axes respectively represent the SEC detected molar mass and the PBD content obtained from HPLC. Reproduced from ref. 26 with permission from the author, copyright 2017. | ||
n). As shown above, in the examples discussed, this direct method for which no calibration is required is also applicable to block-copolymers and in principle applies to all categories mentioned above. A disadvantage compared to SEC, though, is that end group analysis does not yield the full molecular weight distribution. Furthermore, upon block extension, the proton relaxation times increase. As a result, the integration limits become ill-defined due to signal broadening, which increases the uncertainty in the molecular weight estimate. Importantly, end group analysis does per definition not discriminate between a BCP and the equivalent mixture of the separate blocks.
 | ||
| Fig. 8 (a) Calibration curve of P3HT polymers. (b) Comparison of the absolute molecular weight (solid lines) and distributions derived from DOSY (dotted lines). Reproduced from ref. 67 with permission from the American Chemical Society, copyright 2018. | ||
Conclusions
This paper highlights cases from recent literature studies concerning functional, “non-classical” BCPs that give inconsistent results when analyzed with size exclusion chromatography (SEC). Although SEC works relatively well for nonpolar, flexible BCPs, in particular when using dual detector approaches, the method has regularly shown to produce deviant results when the blocks have significantly different physical or chemical properties. This is noteworthy, since such inhomogeneous BCPs are relevant to a range of modern applications, either because of new functions arising from cooperativity between the blocks, or the combination of multiple functionalities within the same polymer. It is easy to misinterpret the BCP size-exclusion data for such systems, and it is important to stay alert for the various effects that can occur in such systems. The common denominator for the examples in this review is an increase in the SEC retention time upon block extension, which produces erroneous estimates when relying on standard calibration. We identify a minimum of four scenarios that lead to this counterintuitive behavior: (i) rod–coil BCPs, (ii) the carrier solvent being a poor solvent for one of the blocks, (iii) strong mutual interactions between different blocks and (iv) strong interaction between the stationary phase and one of the blocks. In such cases, extending or replacing SEC analysis with/by another detection technique is advisable, even for obtaining mere proof for block coupling or extension. We provide a brief but practical overview of alternative methods, but also discuss to what extent they actually circumvent the challenges identified above.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the German Academic Exchange Service (DAAD) and Universities Australia for the financial support in the framework of Project 57446038 “High-Precision Synthesis and Self-Assembly of Semiconducting Block Copolymers”. Open Access funding provided by the Max Planck Society.Notes and references
- Z. Grubisic, P. Rempp and H. Benoit, Rubber Chem. Technol., 1969, 42, 636–640 CrossRef.
- W. Liu, X. Liao, Y. Li, Q. Zhao, M. Xie and R. Sun, Chem. Commun., 2015, 51, 15320–15323 RSC.
- H. C. Hsieh, C. C. Hung, K. Watanabe, J. Y. Chen, Y. C. Chiu, T. Isono, Y. C. Chiang, R. R. Reghu, T. Satoh and W. C. Chen, Polym. Chem., 2018, 9, 3820–3831 RSC.
- L. Y. Shi, Z. Shen and X. H. Fan, Macromolecules, 2011, 44, 2900–2907 CrossRef CAS.
- Z. Yi, Y. Zhang, Y. Chen and F. Xi, Macromol. Rapid Commun., 2008, 29, 757–762 CrossRef CAS.
- C. Ong, Y. Shi, J. Chang, F. Alduraiei, Z. Ahmed and P. Wang, Ind. Eng. Chem. Res., 2019, 58, 4838–4843 CrossRef CAS.
- J. J. Li, Y. N. Zhou and Z. H. Luo, Ind. Eng. Chem. Res., 2014, 53, 18112–18120 CrossRef CAS.
- L. Wu, L. Wang, Z. Guo, J. Luo, H. Xue and J. Gao, ACS Appl. Mater. Interfaces, 2019, 11, 34338–34347 CrossRef CAS PubMed.
- K. Chen, J. Jia, Y. Zhao, K. Lv and C. Wang, Mater. Des., 2017, 135, 69–76 CrossRef CAS.
- Y. L. Lo, M. F. Tsai, Y. Soorni, C. Hsu, Z. X. Liao and L. F. Wang, Biomacromolecules, 2020, 21, 3342–3352 CrossRef CAS PubMed.
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118, 6844–6892 CrossRef CAS PubMed.
- M. Finnegan, G. Mallon, A. Leach and E. Themistou, Polym. Chem., 2019, 10, 5814–5820 RSC.
- K. Ramesh, A. K. Mishra, J. K. Kim, Y. T. Jeong, Y.-S. Gal and K. T. Lim, Materials, 2020, 13, 3713 CrossRef CAS PubMed.
- N. Zaquen, L. Lutsen, D. Vanderzande and T. Junkers, Polym. Chem., 2016, 7, 1355–1367 RSC.
- I. Chandrasiri, D. G. Abebe, M. Loku Yaddehige, J. S. D. Williams, M. F. Zia, A. Dorris, A. Barker, B. L. Simms, A. Parker, B. P. Vinjamuri, N. Le, J. N. Gayton, M. B. Chougule, N. I. Hammer, A. Flynt, J. H. Delcamp and D. L. Watkins, ACS Appl. Bio Mater., 2020, 3, 5664–5677 CrossRef CAS.
- C. Ma, T. Han, M. Kang, E. Liarou, A. M. Wemyss, S. Efstathiou, B. Z. Tang and D. Haddleton, ACS Macro Lett., 2020, 9, 769–775 CrossRef CAS.
- L. C. Hsu, S. Kobayashi, T. Isono, Y. C. Chiang, B. J. Ree, T. Satoh and W. C. Chen, Macromolecules, 2020, 53, 7496–7510 CrossRef CAS.
- F. Ge, Z. Liu, F. Tian, Y. Du, L. Liu, X. Wang, H. Lu, Z. Wu, G. Zhang and L. Qiu, Polym. Chem., 2018, 9, 4517–4522 RSC.
- J. Zhang, K. Kremer, J. J. Michels and K. C. Daoulas, Macromolecules, 2020, 53, 523–538 CrossRef CAS PubMed.
- Z. A. Page, B. Narupai, C. W. Pester, R. Bou Zerdan, A. Sokolov, D. S. Laitar, S. Mukhopadhyay, S. Sprague, A. J. McGrath, J. W. Kramer, P. Trefonas and C. J. Hawker, ACS Cent. Sci., 2017, 3, 654–661 CrossRef CAS PubMed.
- K. M. Meek and Y. A. Elabd, J. Mater. Chem. A, 2015, 3, 24187–24194 RSC.
- A. Pelz, T. S. Dörr, P. Zhang, P. W. De Oliveira, M. Winter, H. D. Wiemhöfer and T. Kraus, Chem. Mater., 2019, 31, 277–285 CrossRef CAS.
- M. Wang, J. Sun, X. Cai, Y. Huang and F. Li, Org. Electron., 2021, 88, 106020 CrossRef CAS.
- H. Idriss, A. Gapin, W. Khelifi, S. Blanc, I. Karamé, S. Chambon, L. Hirsch, A. Bousquet and C. Lartigau-Dagron, Macromolecules, 2020, 53, 9043–9053 CrossRef CAS.
- L. Y. Su, H. H. Huang, Y. C. Lin, G. L. Chen, W. C. Chen, W. Chen, L. Wang and C. C. Chueh, Adv. Funct. Mater., 2020, 2005753, 1–11 Search PubMed.
- P. Kilz, Two-dimensional Chromatography of Copolymers, in Practical Copolymer GPC/SEC and Related Analysis Using Techniques, ed. P. Kilz and D. Held, LCGC, 2017 Search PubMed (accessed at http://www.pss-polymer.com on 2020-05-19).
- P. Kilz and H. Pasch, in Encyclopedia of Analytical Chemistry, John Wiley & Sons, Ltd, Chichester, UK, 2006, pp. 7495–7543 Search PubMed.
- J. M. Sebastian and R. A. Register, J. Appl. Polym. Sci., 2001, 82, 2056–2069 CrossRef CAS.
- A. Revillon, J. Liq. Chromatogr., 1980, 3, 1137–1150 CrossRef CAS.
- V. V. Nesterov, V. D. Krasikov, Y. V. Chubarova and B. G. Belen'kii, Polym. Sci. U.S.S.R., 1982, 24, 1512–1517 CrossRef.
- H. Schlaad and P. Kilz, Anal. Chem., 2003, 75, 1548–1551 CrossRef CAS PubMed.
- Z. Fodor, A. Fodor and J. P. Kennedy, Polym. Bull., 1992, 29, 689–696 CrossRef CAS.
- B. Trathnigg and X. Yan, Chromatographia, 1992, 33, 467–472 CrossRef CAS.
- J. V. Dawkins and M. Hemming, J. Appl. Polym. Sci., 1975, 19, 3107–3118 CrossRef CAS.
- M. I. Malik, G. W. Harding, M. E. Grabowsky and H. Pasch, J. Chromatogr. A, 2012, 1244, 77–87 CrossRef CAS PubMed.
- J. R. Runyon, D. E. Barnes, J. F. Rudd and L. H. Tung, J. Appl. Polym. Sci., 1969, 13, 2359–2369 CrossRef CAS.
- C. Feng, M. Jose Gonzalez-Alvarez, Y. Song, I. Li, G. Zhao, G. Molev, G. Guerin, G. Walker, G. D. Scholes, I. Manners and M. A. Winnik, Soft Matter, 2014, 10, 8875–8887 RSC.
- J. J. Wang, Y. N. Zhou, P. Wang and Z. H. Luo, RSC Adv., 2013, 3, 5045–5055 RSC.
- C. St Thomas, H. Maldonado-Textle, J. N. Cabello-Romero, J. MacOssay, X. Zhang, N. Esturau-Escofet and R. Guerrero-Santos, Polym. Chem., 2014, 5, 3089–3097 RSC.
- Z. Xu, T. Liu, K. Cao, D. Guo, J. M. Serrano and G. Liu, ACS Appl. Polym. Mater., 2020, 2, 1398–1405 CrossRef CAS.
- H. L. Ricks, U. H. Choudry, A. R. Marshall and U. H. F. Bunz, Macromolecules, 2003, 36, 1424–1425 CrossRef CAS.
- S. Barrau, T. Heiser, F. Richard, C. Brochon, C. Ngov, K. Van De Wetering, G. Hadziioannou, D. V. Anokhin and D. A. Ivanov, Macromolecules, 2008, 41, 2701–2710 CrossRef CAS.
- J. T. Wang, S. Takashima, H. C. Wu, Y. C. Chiu, Y. Chen, T. Isono, T. Kakuchi, T. Satoh and W. C. Chen, Adv. Funct. Mater., 2016, 26, 2695–2705 CrossRef CAS.
- Y. Cui, Z. Wang, X. Huang, G. Lu, I. Manners, M. A. Winnik and C. Feng, Macromolecules, 2020, 53, 1831–1841 CrossRef CAS.
- J. V. Facinelli, S. L. Gardner, L. Dong, C. L. Sensenich, R. M. Davis and J. S. Riffle, Macromolecules, 1996, 29, 7342–7350 CrossRef CAS.
- S. C. Tsai, Y. C. Lin, E. L. Lin, Y. W. Chiang and S. W. Kuo, Polym. Chem., 2016, 7, 2395–2409 RSC.
- T. Swift, R. Hoskins, R. Telford, R. Plenderleith, D. Pownall and S. Rimmer, J. Chromatogr. A, 2017, 1508, 16–23 CrossRef CAS PubMed.
- S. Mori, J. Chromatogr. A, 1988, 452, 137–143 CrossRef CAS.
- C. M. Hansen, Hansen Solubility Parameters A User's Handbook, CRC Press, 2nd edn, 2007 Search PubMed.
- J. C. J. F. Tacx, N. L. J. Meijerink and K. Suen, Polymer, 1996, 37, 4307–4310 CrossRef CAS.
- M. I. Malik, T. Mahboob and S. Ahmed, Anal. Bioanal. Chem., 2014, 406, 6311–6317 CrossRef CAS PubMed.
- C. L. Maikawa, A. Sevit, B. Lin, R. J. Wallstrom, J. L. Mann, A. C. Yu, R. M. Waymouth and E. A. Appel, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1322–1332 CrossRef CAS PubMed.
- M. L. Dias, C. Azuma and E. B. Mano, Polym. Bull., 1996, 36, 729–735 CAS.
- Z. Dong, J. Mao, D. Wang, M. Yang, W. Wang, S. Bo and X. Ji, Macromol. Chem. Phys., 2014, 215, 111–120 CrossRef CAS.
- G. Gaucher, M.-H. Dufresne, V. P. Sant, N. Kang, D. Maysinger and J.-C. Leroux, J. Controlled Release, 2005, 109, 169–188 CrossRef CAS PubMed.
- M. Edeleva, Y. W. Marien, P. H. M. Van Steenberge and D. R. D′hooge, Polym. Chem., 2021 10.1039/D1PY00151E , Advance Article.
- A. Vora, R. J. Wojtecki, K. Schmidt, A. Chunder, J. Y. Cheng, A. Nelson and D. P. Sanders, Polym. Chem., 2016, 7, 940–950 RSC.
- B. H. Zimm, J. Chem. Phys., 1948, 16, 1093–1099 CrossRef CAS.
- X. Zhao, K. Deng, F. Liu, X. Zhang, H. Yang, J. Peng, Z. Liu, L. Ma, B. Wang and H. Wei, ACS Biomater. Sci. Eng., 2018, 4, 566–575 CrossRef CAS PubMed.
- G. Gody, P. B. Zetterlund, S. Perrier and S. Harrisson, Nat. Commun., 2016, 7, 10514 CrossRef CAS PubMed.
- P. Kilz, H. Much and G. Schulz, Adv. Chem. Ser., 1995, 247, 223–242 CrossRef CAS.
- P. Groves, Polym. Chem., 2017, 8, 6700–6708 RSC.
- A. L. Buckinx, K. Verstraete, E. Baeten, R. F. Tabor, A. Sokolova, N. Zaquen and T. Junkers, Angew. Chem., Int. Ed., 2019, 58, 13799–13802 CrossRef CAS PubMed.
- M. Badoux, S. Drechsler, S. Pal and A. F. M. Kilbinger, Macromolecules, 2017, 50, 9307–9314 CrossRef CAS.
- S. Huber and S. Mecking, Macromolecules, 2019, 52, 5917–5924 CrossRef CAS.
- J. H. Vrijsen, I. A. Thomlinson, M. E. Levere, C. L. Lyall, M. G. Davidson, U. Hintermair and T. Junkers, Polym. Chem., 2020, 11, 3546–3550 RSC.
- K. Gu, J. Onorato, S. S. Xiao, C. K. Luscombe and Y. L. Loo, Chem. Mater., 2018, 30, 570–576 CrossRef CAS.
- K. Nakabayashi, T. Takahashi, R. Sugawara, C. T. Lo and H. Mori, React. Funct. Polym., 2018, 131, 350–360 CrossRef CAS.
- S. Daripa, K. Khawas, A. Sharma, A. Kumar, B. Pal, S. Das, S. Jit and B. K. Kuila, ACS Appl. Polym. Mater., 2020, 2, 1283–1293 CrossRef CAS.
- D. Marsitzky, M. Klapper and K. Müllen, Macromolecules, 1999, 32, 8685–8688 CrossRef CAS.
- T. Gruendling, S. Weidner, J. Falkenhagen and C. Barner-Kowollik, Polym. Chem., 2010, 1, 599–617 RSC.
- L. Charles, Mass Spectrom. Rev., 2014, 33, 523–543 CrossRef CAS PubMed.
- A. H. Ribeiro, J. Haven, A.-L. Buckinx, M. Beuchel, K. Philipps, T. Junkers and J. J. Michels, Polym. Chem., 2021, 12, 216–225 RSC.
| This journal is © The Royal Society of Chemistry 2021 |