
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuppressing aggregation induced quenching in anthracene based conjugated polymers†
Daniel G.
Congrave
 a,
Bluebell H.
Drummond
b,
Victor
Gray
bc,
Andrew D.
Bond
a,
Akshay
Rao
b,
Richard H.
Friend
b and
Hugo
Bronstein
*ab
a,
Bluebell H.
Drummond
b,
Victor
Gray
bc,
Andrew D.
Bond
a,
Akshay
Rao
b,
Richard H.
Friend
b and
Hugo
Bronstein
*ab
aDepartment of Chemistry, University of Cambridge, Cambridge, CB2 1EW, UK. E-mail: hab60@cam.ac.uk
bCavendish Laboratory, University of Cambridge, Cambridge, CB3 0HE, UK
cDepartment of Chemistry – Ångström Laboratory, Uppsala University, Box 523, 751 20, Uppsala, Sweden
First published on 3rd March 2021
Abstract
Anthracene is a highly valuable building block for luminescent conjugated polymers, particularly when a large singlet–triplet energy gap (ΔEST) is desired. Unfortunately, the extended π system of anthracene imparts a strong tendency for polymer aggregation, resulting in detrimental effects on its solid state photophysics. A large decrease in photoluminescence quantum yield (PLQY, ΦF) on going from solution to the solid state is especially common, represented in terms of a low ΦR (ΦR = ΦF film/ΦF sol.). Significant and undesirable red-shifting of fluorescence in the solid state is also typical due to processes such as excimer formation. In this work a series of alkylene-encapsulated conjugated anthracene polymers is developed to overcome these challenging problems. We demonstrate a promising material which displays a good solid state PLQY that is effectively unchanged compared to solution measurements (ΦR ∼ 1, ΦF film ∼ 40%), alongside an identical PL 0–0 transition wavelength in solution and thin film. Such a direct transfer of luminescence properties from solution to the solid state is remarkable for a conjugated polymer and completely unprecedented for one based on anthracene.
Introduction
9,10-Disubstituted anthracene derivatives such as 9,10-diphenylanthracene (DPA) and 9,10-bis(phenylethynyl)anthracene (BPEA) are archetypal fluorophores. Diluted into solid hosts or solution they display photoluminescence quantum yields (PLQY, ΦF) close to unity1 alongside narrow Stokes shifts. They also feature large exchange energies (ΔEST). As a result, anthracenes are ubiquitous in many applications such as organic light emitting diodes (OLEDs),2 organic photovoltaics (OPVs),3 chemiluminescence,4 triplet–triplet annihilation upconversion (TTAUC),5,6 singlet fission downconversion (SFDC)1,6 and electrochromics.7 Merging the advantageous photophysical properties of anthracenes with the processability of conjugated polymers should lead to highly desirable materials, and numerous works have sought to achieve this.8–20 Notably, the polymerization of small amounts of anthracene via the 9,10-positions into polyfluorenes has been shown to greatly enhance the color stability of OLEDs due to energy transfer.8,9,13,14 Regioregular copolymers based on 9,10-diarylanthracenes polymerized via the 2,6-positions have also been reported.16,21–23 Additionally, some non-conjugated polymers incorporating anthracene have been investigated.24–26Despite these efforts it has been very challenging to synthesize functional conjugated anthracene polymers.11,12,16–18,20 This is related to the planar extended π system of anthracene, which imparts a strong tendency for aggregation. This causes the photophysical performance of anthracene-based materials to vary greatly between dispersed (e.g. solution) and condensed states. A common phenomenon is aggregation induced quenching (AIQ)15 in the solid state, which typically leads to a very low ratio (ΦR) between thin film and solution PLQYs (ΦR = ΦF film/ΦF sol.). This is a challenging problem for polymeric and small molecule anthracene derivatives alike.23,27–30 For example, the near-unity solution PLQY of DPA is drastically quenched in thin films, affording ΦR ∼ 0.3.30 Even the polymeric examples reported by Chen et al.,23 which consist of highly twisted and bulky DPA-based structures and display some of the highest reported thin film PLQYs for conjugated anthracene polymers, typically lose over half of their photoluminescence (PL) intensity in the solid state (ΦR < 0.4). Anthracene derivatives are also classic examples of molecules that undergo excimer formation in the solid state, which leads to diminished and red-shifted PL.17,30 A further possible PL quenching process is photodimerisation.31 The exact extent of each PL quenching pathway is evidently difficult to predict. Therefore, a synthetic tool to control and/or suppress aggregation in anthracene based conjugated polymers is highly desirable and would allow a previously unattainable class of materials to be accessed.
In this work we develop the synthetic chemistry required to obtain alkylene-encapsulated DPA polymers. As a result, it is possible to realize a blue emitting anthracene based conjugated polymer for which PL quenching in the solid state is essentially absent (ΦR ∼ 1).
Results and discussion
Design and synthesis
Work from our group and others has shown that covalent alkylene encapsulation can increase molecular rigidity and suppress AIQ to afford highly luminescent materials that retain much of their desirable solution photophysics in the solid state.32–36 Kobayashi and coworkers have also proven the viability of such a tactic for preserving the photophysical performance of DPA in powder and pristine films.37 However, covalent encapsulation has never been implemented in anthracene based polymers despite its clear potential to improve their photophysical performance. To achieve this new synthetic chemistry must be developed (Fig. 1). | ||
| Fig. 1 Targeted monomer structural features. | ||
We chose to target polymerization from the phenyl groups of encapsulated DPA as it should afford a twisted backbone, which is expected to help suppress AIQ (Fig. 1).23 The classic synthesis of DPA derivatives involves the treatment of an anthraquinone with two equivalents of an aryl lithium or Grignard reagent to generate a bis-tertiary alcohol, which is then aromatized with SnCl2 or NaH2PO2.38–41 We elected to follow this methodology starting from the anthraquinone derivative 1 (Scheme 1), which was functionalized with four dodecyl chains to ensure the high solubility of the resulting polymers (Scheme S1†). The iodoaryl Ar (a) (Scheme 1a) was selected as a rational synthetic intermediate because the iodide is expected to undergo chemoselective Grignard formation.42 This would allow the installation of aryl groups at the 9,10-positions of anthracene with methoxy protecting groups for encapsulation installed alongside Ar–Br functionality for polymerization. However, upon treatment of 1 at room temperature with an excess of Grignard reagent, formed through transmetallation of Ar (a) with iPr-MgCl,43 the expected bis-tertiary alcohol 2a was not obtained. Instead, the product of single nucleophilic attack (2b) was identified by 1H NMR as the predominant product alongside deiodinated Ar (a) (spectrum S19). A large excess of Grignard reagent (>10 eq.) and prolonged refluxing in THF failed to promote double addition to form 2a. Attempts to improve the nucleophilicity of the organometallic through transmetallation with ‘Turbo Grignard’ (iPr-MgCl·LiCl)44 or n-BuLi rather than iPr-MgCl and addition of TMEDA were also unsuccessful, as was the addition of LaCl3·LiCl45 to improve carbonyl electrophilicity.
 | ||
| Scheme 1 Synthesis of polymer precursors and polymer structures. | ||
The reaction sequence was also attempted with the iodoaryl Ar (b) (Scheme 1a), which incorporates formally electron withdrawing methoxymethyl groups in contrast to the formally electron donating methoxy groups of Ar (a). Unfortunately, the mono addition product was again solely observed by 1H NMR after reaction with anthraquinone. Acidification of the crude reaction mixture and treatment with SnCl2·2H2O also led to no appreciable blue fluorescence, which could indicate the formation of a DPA derivative. It is noted that double addition has been reported upon treatment of anthraquinone with the aryl lithium reagents generated from both 4-bromo-2,6-dimethyliodobenzene46 and 1,3-dimethoxybenzene (Ar (c)).37 We also observed that ortho-lithiated 1,3,5-trimethoxy benzene undergoes successful double addition to anthraquinone in ca. 80% isolated yield (Scheme S5†). Therefore, it appears that the specific combination of the ether and bromo functionalities suppresses the double addition of the organometallics of Ar (a) and Ar (b) to anthraquinones. We tentatively suggest that the additive chelating47 and electron withdrawing48 effects of the ether and Ar–Br functionalities, respectively, could be involved through stabilizing the organometallics and reducing nucleophilicity.
After failing to obtain a pre-functionalised polymerisable monomer, we targeted the substituted DPA 3 (Scheme 1b). It was successfully obtained in 77% overall yield upon treatment of 1 with ortho-lithiated Ar (c), followed by aromatization of the intermediate 2c with SnCl2·2H2O. With DPA 3 in hand, we sought a route to install further functionality for polymerization (Scheme 1c). 3 was firstly converted into the encapsulated derivative 5 after quantitative demethylation to 4 followed by alkylation with 1,7-dibromoheptane. As the phenyl 4-positions of 5 are by far the least sterically encumbered sites on the molecule, we envisaged that they should be prone to functionalization via Ir-catalyzed C–H borylation, the regioselectivity of which is driven predominantly by sterics.49 Indeed, treatment of 5 with [Ir(OMe)(COD)]2, bis(pinacolato)diboron and 3,4,7,8-tetramethyl-1,10-phenanthroline50 regioselectively afforded the bis(pinacol) ester 6, the structure of which was proven unambiguously by single crystal X-ray diffraction (Fig. 2).‡ Although essentially complete conversion of 5 to 6 was observed by 1H NMR, the requirement to purify 6 by flash chromatography to obtain analytically pure material restricted the isolated yield to ca. 50% in our hands. Subsequent treatment of 6 with CuBr2 in mixed solvent conditions fortunately allowed the dibromo monomer 7 to be accessed in 35% yield, despite the fact that 6 is insoluble in highly polar solvents such as MeOH.49,51,52 It is therefore possible to obtain encapsulated DPAs functionalized with either bromide or boronic ester groups for polymerization via the methodology outlined here. The encapsulated DPA 5 can be synthesized in 10 g quantities under typical laboratory conditions, highlighting the promise of obtaining the polymer precursors 6 and 7 on scale. The obligatory high purity of 6 and 7 was confirmed by 1H NMR and elemental analysis.
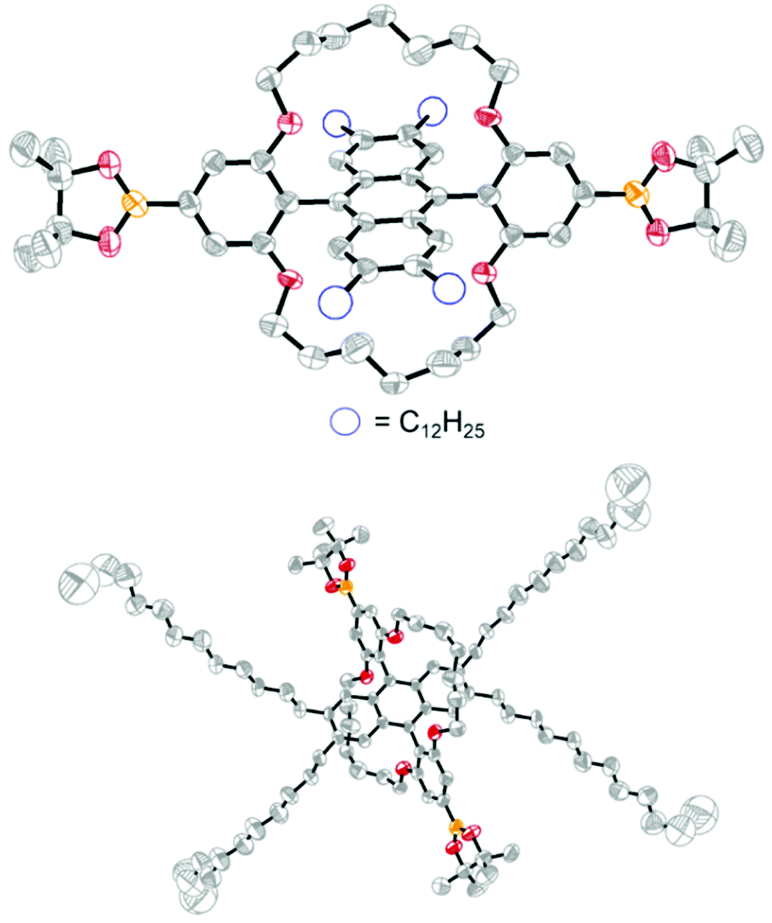 | ||
| Fig. 2 X-ray crystal structure of 6, hydrogen atoms are omitted for clarity. The molecule is situated on a crystallographic inversion centre. | ||
Regioregular copolymers were next synthesized through the Suzuki polycondensation of 6 with either 2,7-dibromo-9,9-(dioctylfluorene) or its arylated analogue, to obtain AF8 and AFP8, respectively (Scheme 1d). They were obtained in respectable number average molecular weights and polydispersity by extraction in a Soxhlet apparatus with chloroform, after washing with acetone and n-hexane to remove oligomeric materials (AF8Mn = 35 kDa, PDI = 1.6, AFP8Mn = 31 kDa, PDI = 1.5). AFP8 was investigated because 9,9-diarylfluorenes are bulkier (to suppress AIQ) and also are more stable under OLED operation than their 9,9-dialkyl analogues,53 increasing the relevance of this system to optoelectronics. The homopolymer AH was next prepared in good molecular weight and polydispersity from 7 under microwave-assisted Yamamoto polymerization conditions followed by the same Soxhlet procedure (AHMn = 66 kDa, PDI = 1.7) (Scheme 1d). AF8 and AFP8 were obtained as light yellow flakes while the higher molecular weight AH was obtained as light yellow fibres. All polymers are well soluble in common aromatic and chlorinated solvents (e.g. toluene, CHCl3 and chlorobenzene).
Photophysical properties
The photophysical properties of the polymers were evaluated in toluene and 1,2-dichlorobenzene solutions and films prepared by spin coating from toluene. Absorption and emission spectra and shown in Fig. 3 and photophysical data are summarized in Table 1. In solution all polymers feature vibronically resolved absorption bands between ca. 370–415 nm, which are assigned to absorption by DPA. An additional band is observed for AF8 and AFP8 at 340 nm, ascribed to absorption by their fluorene comonomers. Absorption for all polymers is clearly red-shifted compared to that of DPA (Abs 0–0 ca. 394 nm),37 indicating that there is electronic communication between monomer units in the ground state. The position of the bands and the steepness of the absorption onsets are well retained in spin coated films for all polymers, indicating that their ground state photophysical properties are effectively unchanged in the solid state. | ||
| Fig. 3 Absorption and PL spectra of the polymers. | ||
| Polymer | State | λ max PL/nm | Φ F /% | Φ R | τ /ns |
|---|---|---|---|---|---|
| a Absolute PLQY measured using an integrating sphere, estimated errors correspond to a relative error of ±5% which is equal or larger to the observed standard error for measurements on three different films. b For thin films this value is the amplitude weighted average of a bi-exponential fit, full data are provided in the ESI (Fig. S1–3, Table S1†). Solution λmax PL data were recorded in toluene. Solution ΦF and τ values were recorded in 1,2-dichlorobenzene (DCB). The PLQY of AF8 was equal within experimental error in toluene and DCB. | |||||
| AF8 | Solution | 432 | 41 ± 2 | ∼1 | 2.94 |
| Film | (432) 457 | 42 ± 2 | 2.28 | ||
| AFP8 | Solution | 432 | 45 ± 2 | 0.73 | 2.90 |
| Film | (435) 459 | 33 ± 2 | 1.62 | ||
| AH | Solution | 429 | 87 ± 4 | 0.25 | 3.80 |
| Film | (431) 458 | 22 ± 1 | 1.52 | ||
In solution AF8 emits in the blue with a respectable PLQY of 41 ± 2% and a fluorescence lifetime (τ) of 2.94 ns. The solution PL of AF8 (PL 0–0 = 432 nm) is red-shifted compared to that of DPA (PL 0–0 ca. 410 nm),37 indicating that there is also electronic communication between monomer units in the excited state. Strikingly, on going from solution to thin film there is no red-shift in the PL 0–0 transition (432 nm) for AF8 and the gradient of the PL onset is practically unchanged. Within experimental error, the solution PLQY of AF8 is also completely maintained in thin film, affording an exceptional ΦR ∼ 1. Such minimal PL quenching is in agreement with a decrease in τ of only 22% in thin film. Clearly, the detrimental effects of aggregation are essentially absent for AF8 in the solid state. Such a direct transfer of luminescence properties from solution to the solid state is remarkable for a conjugated polymer54–57 and, to the best of our knowledge, completely unprecedented for one based on anthracene (Table S3†). We note a slight extension of the PL tail to longer wavelengths in thin film, which we ascribe to interchain energy transfer to lower energy trap sites in the solid state. Such a process does not constitute an additional quenching pathway compared to solution as ΦR ∼ 1 for AF8.
The addition of bulkier aryl rings to the fluorene copolymer AFP8 has a negligible effect on its solution PL properties compared to AF8, as expected.53 However, in thin film the red tail of the PL spectrum is more greatly extended for AFP8 than AF8, and the 0–0 transition of AFP8 is red-shifted by 3 nm compared to solution. The PLQY of AFP8 is also reduced in thin film (33 ± 2% vs. 45 ± 2% in solution), accompanied by a 45% decrease in τ and affording ΦR = 0.73. While the ΦR value of 0.73 for AFP8 is very high and the best reported for a conjugated anthracene polymer before this work,23 increased AIQ in AFP8 compared to AF8 is a surprising result, contrasting with what has been reported for polyfluorenes.53 The extra bulk incorporated into AFP8 compared to AF8 is in the form of phenylene groups, which may provide additional π surfaces to participate in aggregation, while also introducing spiro conjugation.58 We suggest that these factors could contribute to AIQ in the thin films of AFP8.
In solution the fully encapsulated polymer AH exhibits a high PLQY of 87 ± 4%. This is consistent with its longer τ (3.80 ns) compared to the copolymers AF8 and AFP8. However, in thin film the PL broadens and the PLQY is quenched significantly to 22 ± 1%, corroborated by a large decrease of 60% in the τ and affording ΦR = 0.25. Significant AIQ for AH is an unexpected result and indicates that complete alkylene encapsulation of a conjugated polymer is not necessarily sufficient to suppress PL quenching in the solid state. In fact, AF8, for which AIQ is absent in the solid state, clearly incorporates a lower degree of encapsulation than AH. To then add a further level of complexity, the addition of sterically bulky phenylene groups in AFP8 actually promotes AIQ compared to AF8, indicating that two structural features that should benefit solid state emission, encapsulation and steric bulk, are not necessarily additive. These surprising structure–property relationships raise interesting research questions that warrant further investigation in future work.
While the ability of AF8 to retain its solution-state PL properties in thin film is highly attractive and a great advantage of our molecular design, there is room for improvement in PLQY. To determine the cause of the modest solution PLQY of AF8 (41 ± 2%) the rates of radiative (kr) and non-radiative (knr) decay were calculated and compared to that of AH, which displays a significantly higher solution PLQY. While both polymers exhibit similar values of kr (AF8 = 1.4 × 108 s−1, AH = 2.3 × 108 s−1), the rate of non-radiative decay for AF8 is an order of magnitude larger (2.0 × 108 s−1vs. 3.4 × 107 s−1 for AH). Therefore, while copolymerization with 9,9-dioctyl(fluorene) is clearly beneficial for suppressing aggregation in the solid state, it also introduces new pathways for non-radiative decay that lower the intrinsic PLQY of AF8.
Conclusions
Prior to this study aggregation has heavily restricted the development of conjugated anthracene polymers, impeding their solid state photophysics by facilitating aggregation induced quenching (AIQ) and the formation of excimers. Here, new synthetic chemistry was developed to access alkylene-encapsulated DPA polymers. This provided the structural control to develop AF8 as an anthracene polymer that eliminates AIQ and excimer formation in the solid state, displaying ΦR ∼ 1 (ΦR = ΦF film/ΦF sol.) alongside an identical photoluminescence 0–0 transition wavelength in solution and thin film. Such a direct transfer of luminescence properties from solution to the solid state is remarkable for a conjugated polymer and, to the best of our knowledge, completely unprecedented for one based on anthracene.However, our results for the polymers AFP8 and AH reveal that the structure–property-relationships in encapsulated polymers can be surprisingly convoluted – enhancing steric bulk and increasing the extent of encapsulation are by no means tactics guaranteed to achieve improved solid state photophysical performance. Future work should concentrate on improving our understanding of the structure–property relationships between alkylene encapsulation and the PLQYs of conjugated polymers (in both solution and thin film). This could lead to polymers with unity quantum efficiency in the solid state. An anthracene-based example would have particular promise for solid-state triplet–triplet annihilation upconversion5 and direct triplet injection polymer OLEDs.59
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Andrew Danos (Durham University, UK) is acknowledged for help with the solid state PLQY measurements and helpful discussion. Aileen Congreve is acknowledged for allowing use of an integrating sphere at Durham University, UK. VG acknowledges funding from the Swedish Research Council, Vetenskapsrådet 2018-00238. This work was supported by the Engineering and Physical Sciences Research Council (grant no. EP/M005143/1) and the European Research Council (ERC). B. H. D. acknowledges support from the EPSRC Cambridge NanoDTC (grant no. EP/L015978/1).Notes and references
- Y. J. Bae, G. Kang, C. D. Malliakas, J. N. Nelson, J. Zhou, R. M. Young, Y. L. Wu, R. P. Van Duyne, G. C. Schatz and M. R. Wasielewski, J. Am. Chem. Soc., 2018, 140, 15140–15144 CrossRef CAS PubMed.
- H. Park, J. Lee, I. Kang, H. Y. Chu, J. I. Lee, S. K. Kwon and Y. H. Kim, J. Mater. Chem., 2012, 22, 2695–2700 RSC.
- H. Feng, Y. Q. Q. Yi, X. Ke, J. Yan, Y. Zhang, X. Wan, C. Li, N. Zheng, Z. Xie and Y. Chen, Adv. Energy Mater., 2019, 9, 1803541 CrossRef.
- D. J. Vinyard, S. Su and M. M. Richter, J. Phys. Chem. A, 2008, 112, 8529–8533 CrossRef CAS PubMed.
- V. Gray, K. Moth-Poulsen, B. Albinsson and M. Abrahamsson, Coord. Chem. Rev., 2018, 362, 54–71 CrossRef CAS.
- B. Manna, A. Nandi and R. Ghosh, J. Phys. Chem. C, 2018, 122, 21047–21055 CrossRef CAS.
- D. Chandra Santra, S. Mondal and S. Malik, RSC Adv., 2016, 6, 81597–81606 RSC.
- G. Klarner, M. Davey, W.-D. Chen, S. J. Cambell and R. D. Miller, Adv. Mater., 1998, 10, 993–997 CrossRef.
- K. Lee, J.-H. Park, M.-J. Park, J. Lee and H.-K. Shim, J. Nanosci. Nanotechnol., 2011, 11, 4648–4657 CrossRef CAS PubMed.
- Y. Tao, K. Zhang, Z. Zhang, H. Cheng, C. Jiao and Y. Zhao, Chem. Eng. J., 2016, 293, 34–43 CrossRef CAS.
- C. Liu, W. Xu, X. Guan, H. L. Yip, X. Gong, F. Huang and Y. Cao, Macromolecules, 2014, 47, 8585–8593 CrossRef CAS.
- M. S. Taylor and T. M. Swager, Angew. Chem., Int. Ed., 2007, 46, 8480–8483 CrossRef CAS PubMed.
- G. Klärner, J. I. Lee, M. H. Davey and R. D. Miller, Adv. Mater., 1999, 11, 115–119 CrossRef.
- B. D. Marsitzky, J. C. Scott, J. Chen, V. Y. Lee, R. D. Miller, S. Setayesh and K. Müllen, Adv. Mater., 2001, 23, 1096–1099 CrossRef.
- D. A. M. Egbe, S. Türk, S. Rathgeber, F. Kühnlenz, R. Jadhav, A. Wild, E. Birckner, G. Adam, A. Pivrikas, V. Cimrova, G. Knör, N. S. Sariciftci and H. Hoppe, Macromolecules, 2010, 43, 1261–1269 CrossRef CAS.
- M. S. Almeataq, H. Yi, S. Al-Faifi, A. A. B. Alghamdi, A. Iraqi, N. W. Scarratt, T. Wang and D. G. Lidzey, Chem. Commun., 2013, 49, 2252–2254 RSC.
- D. M. E. Freeman, A. Minotto, W. Duffy, K. J. Fallon, I. McCulloch, F. Cacialli and H. Bronstein, Polym. Chem., 2016, 7, 722–730 RSC.
- R. Grisorio, G. P. Suranna, P. Mastrorilli, G. Allegretta, A. Loiudice, A. Rizzo, G. Gigli, K. Manoli, M. Magliulo and L. Torsi, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4860–4872 CrossRef CAS.
- J. W. Jung, F. Liu, T. P. Russell and W. H. Jo, Adv. Energy Mater., 2015, 5, 1500065 CrossRef.
- H. Y. Chen, C. T. Chen and C. T. Chen, Macromolecules, 2010, 43, 3613–3623 CrossRef CAS.
- D. S. Yang, K. H. Kim, M. J. Cho, J.-I. Jin and D. H. Choi, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1457–1467 CrossRef CAS.
- J. W. Jung, F. Liu, T. P. Russell and W. H. Jo, Adv. Energy Mater., 2015, 5, 1500065 CrossRef.
- H. Chen, C. Chen and C. Chen, Macromolecules, 2010, 3613–3623 CrossRef CAS.
- W. Fudickar and T. Linker, Langmuir, 2010, 26, 4421–4428 CrossRef CAS PubMed.
- S. Dumitrescu, M. Grigoraş and C. I. Simionescu, Makromol. Chem., 1983, 184, 2033–2040 CrossRef CAS.
- C. I. Sumionescu, M. Grigoras and V. Barboiu, J. Polym. Sci., Part A: Polym. Chem., 1985, 23, 2089–2098 Search PubMed.
- T. Serevičius, R. Komskis, P. Adomènas, O. Adomènienè, G. Kreiza, V. Jankauskas, K. Kazlauskas, A. Miasojedovas, V. Jankus, A. Monkman and S. Juršènas, J. Phys. Chem. C, 2017, 121, 8515–8524 CrossRef.
- Y. C. Chang, S. C. Yeh, Y. H. Chen, C. T. Chen, R. H. Lee and R. J. Jeng, Dyes Pigm., 2013, 99, 577–587 CrossRef CAS.
- Y. J. Pu, A. Kamiya, K.-i. Nakayama and J. Kido, Org. Electron., 2010, 11, 479–485 CrossRef CAS.
- T. Serevičius, R. Komskis, P. Adomenas, O. Adomeniene, V. Jankauskas, A. Gruodis, K. Kazlauskas and S. Juršenas, Phys. Chem. Chem. Phys., 2014, 16, 7089–7101 RSC.
- L. Zhu, R. O. Al-Kaysi and C. J. Bardeen, J. Am. Chem. Soc., 2011, 133, 12569–12575 CrossRef CAS PubMed.
- A. Leventis, J. Royakkers, A. G. Rapidis, N. Goodeal, M. K. Corpinot, J. M. Frost, D.-K. Bučar, M. O. Blunt, F. Cacialli and H. Bronstein, J. Am. Chem. Soc., 2018, 140, 1622–1626 CrossRef CAS PubMed.
- J. Royakkers, A. Minotto, D. G. Congrave, W. Zeng, A. Patel, A. D. Bond, D. K. Bučar, F. Cacialli and H. Bronstein, J. Org. Chem., 2020, 85, 207–214 CrossRef CAS PubMed.
- K. Sugiyasu, Y. Honsho, R. M. Harrison, A. Sato, T. Yasuda, S. Seki and M. Takeuchi, J. Am. Chem. Soc., 2010, 132, 14754–14756 CrossRef CAS PubMed.
- J. Royakkers, A. Minotto, D. G. Congrave, W. Zeng, A. Hassan, A. Leventis, F. Cacialli and H. Bronstein, Chem. Mater., 2020, 32, 10140–10145 CrossRef CAS.
- J. Royakkers and H. Bronstein, Macromolecules, 2021, 54(3), 1083–1094 CrossRef CAS.
- Y. Fujiwara, R. Ozawa, D. Onuma, K. Suzuki, K. Yoza and K. Kobayashi, J. Org. Chem., 2013, 78, 2206–2212 CrossRef CAS PubMed.
- P. J. Hanhela and D. Brenton Paul, Aust. J. Chem., 1981, 34, 1687–1700 CrossRef CAS.
- P. J. Hanhela and D. Brenton Paul, Aust. J. Chem., 1981, 34, 1669–1685 CrossRef CAS.
- P. J. Hanhela and D. Brenton Paul, Aust. J. Chem., 1981, 34, 1701–1717 CrossRef CAS.
- Z. Zhi, X. Yang, L. Lu and X. Wang, Chem. Educ., 2000, 5, 187–189 CrossRef CAS.
- L. Boymond, M. Rottländer, G. Cahiez and P. Knochel, Angew. Chem., Int. Ed., 1998, 37, 1701–1703 CrossRef CAS.
- P. Knochel, W. Dohle, N. Gommermann, F. F. Kneisel, F. Kopp, T. Korn, I. Sapountzis and V. A. Vu, Angew. Chem., Int. Ed., 2003, 42, 4302–4320 CrossRef CAS PubMed.
- A. Krasovskiy and P. Knochel, Angew. Chem., Int. Ed., 2004, 43, 3333–3336 CrossRef CAS PubMed.
- A. Krasovskiy, F. Kopp and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 497–500 CrossRef CAS PubMed.
- D. Beyer, S. Wang, C. A. Pignedoli, J. Melidonie, B. Yuan, C. Li, J. Wilhelm, P. Ruffieux, R. Berger, K. Müllen, R. Fasel and X. Feng, J. Am. Chem. Soc., 2019, 141, 2843–2846 CrossRef CAS PubMed.
- X. J. Wang, Y. Xu, L. Zhang, D. Krishnamurthy and C. H. Senanayake, Org. Lett., 2006, 8, 3141–3144 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- J. M. Murphy, X. Liao and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 15434–15435 CrossRef CAS.
- S. M. Preshlock, B. Ghaffari, P. E. Maligres, S. W. Krska, R. E. Maleczka and M. R. Smith, J. Am. Chem. Soc., 2013, 135, 7572–7582 CrossRef CAS PubMed.
- A. L. S. Thompson, G. W. Kabalka, M. R. Akula and J. W. Huffman, Synthesis, 2005, 547–550 CAS.
- A. G. Crawford, Z. Liu, I. A. I. Mkhalid, M. H. Thibault, N. Schwarz, G. Alcaraz, A. Steffen, J. C. Collings, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Chem. – Eur. J., 2012, 18, 5022–5035 CrossRef CAS PubMed.
- L. P. Lu, D. Kabra, K. Johnson and R. H. Friend, Adv. Funct. Mater., 2012, 22, 144–150 CrossRef CAS.
- J. Lin, B. Liu, M. Yu, X. Wang, Z. Lin, X. Zhang, C. Sun, J. Cabanillas-Gonzalez, L. Xie, F. Liu, C. Ou, L. Bai, Y. Han, M. Xu, W. Zhu, T. A. Smith, P. N. Stavrinou, D. D. C. Bradley and W. Huang, Adv. Mater., 2019, 31, 1804811 CrossRef PubMed.
- S. Setayesh, A. C. Grimsdale, T. Weil, V. Enkelmann, K. Müllen, F. Meghdadi, E. J. W. List and G. Leising, J. Am. Chem. Soc., 2001, 123, 946–953 CrossRef CAS PubMed.
- D. Vak, C. Chun, C. L. Lee, J. J. Kim and D. Y. Kim, J. Mater. Chem., 2004, 14, 1342–1346 RSC.
- Y. H. Tseng, P. I. Shih, C. H. Chien, A. K. Dixit, C. F. Shu, Y. H. Liu and G. H. Lee, Macromolecules, 2005, 38, 10055–10060 CrossRef CAS.
- S. M. King, S. I. Hintschich, D. Dai, C. Rothe and A. P. Monkman, J. Phys. Chem. C, 2007, 111, 18759–18764 CrossRef CAS.
- A. Salehi, C. Dong, D. H. Shin, L. Zhu, C. Papa, A. T. Bui, F. N. Castellano and F. So, Nat. Commun., 2019, 10, 2305 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, X-ray crystallographic data and additional photophysical data. CCDC 2024118. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1py00118c |
‡ Crystal data. C100H160B2O8, M = 1511.89, monoclinic, a = 19.2620(7), b = 12.5345(5), c = 21.0985(8) Å, β = 110.846(2)°, U = 4760.6(3) Å3, T = 180 K, space group P21/c (no. 14), Z = 2, 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 058 reflections measured, 4996 unique (Rint = 0.089), which were used in all calculations. The final R1 was 0.098 (3116 data with I > 2σ(I)) and wR(F2) was 0.303 (all data). 058 reflections measured, 4996 unique (Rint = 0.089), which were used in all calculations. The final R1 was 0.098 (3116 data with I > 2σ(I)) and wR(F2) was 0.303 (all data). |
| This journal is © The Royal Society of Chemistry 2021 |