
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Proton conducting ABA triblock copolymers with sulfonated poly(phenylene sulfide sulfone) midblock obtained via copper-free thiol-click chemistry†
Marco
Viviani
,
Sebastiaan Pieter
Fluitman
,
Katja
Loos
 and
Giuseppe
Portale
*
and
Giuseppe
Portale
*
Macromolecular Chemistry and New Polymeric Materials, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747AG, Groningen, The Netherlands. E-mail: g.portale@rug.nl
First published on 29th March 2021
Abstract
A series of charged ABA triblock copolymers having sulfonated poly(phenylene sulfide sulfone) (sPSS) as B-block and polystyrene (PS) as A-block have been successfully synthesized using copper-free thiol-click chemistry. One-pot sequential radical addition–fragmentation chain transfer (RAFT) polymerization followed by functionalization with a perfluorinated chain extender (decafluorobiphenyl, DFBP) is used to prepare the PS blocks which are later cliked to the charged sPSS mid-block, synthetized using nucleophilic aromatic substitution polymerization. The proposed synthetic approach ensures good control over the composition of the resulting ABA block copolymers allowing synthesis of block copolymers with well-defined ion exchange capacity (IEC) and nanomorphology. The superstrong segregation regime (χN ≫ 100) of these BCPs generates ordered nanostructures, spanning from spherical to lamellar. All the block copolymers are thermally stable up to 300 °C and are robust against swelling and wetting due to the dimensional stabilization of the ionic domains provided by the PS matrix. The relationship between proton conductivity and nanomorphology is investigated by electrochemical impedance spectroscopy (EIS), revealing the significant impact of self-assembly on the transport properties, reaching a maximum ion conductivity of 50 mS cm−1 at 90 °C and 95% RH in the through-plane direction.
Introduction
Green hydrogen production and fuel cell technology, together with electric storage devices (i.e. batteries), are expected to lead the energy transition toward renewable sources.1–4 Polymer electrolyte (or proton exchange) membrane fuel cells (PEMFC) represent the most efficient devices for energy production combined with a wide range of applications from transportation to portable and stationary power generation.5 The key component of these devices is the polymer electrolyte membrane (PEM) which is responsible for the proton transport from the anode to the cathode.To date, perfluorosulfonic ionomers (PFSI) such as Nafion™, are still the benchmark products and the most used polymers for PEM due to their outstanding proton conductivity at moderate temperatures (0.1 S cm−1 at 30 °C and 80% relative humidity (RH))6 and chemical stability. Despite modest ion exchange capacity (IEC), these polymers show unique ion conductivity thanks to the peculiar phase separation between the super-acidic sulfonic group and the tetrafluoroethylene backbone which promotes the development of a percolated densely sulfonated nanostructure.7,8 However, thermo-mechanical limitations and high hydration requirements limit the optimal operating temperature below 90 °C.9 Moreover, safety and environmental aspects, high fuel crossover and the high cost of production constitute additional problems that prevented the widespread use of PEMFC technology.10,11
To address the aforementioned drawbacks of PFSI membranes, different chemistries mainly based on sulfonated aromatic polymers were investigated as alternative materials for intermediate temperature PEM (70 °C < T < 120 °C).4,12,13 Particular emphasis was given to the investigation of sulfonated aromatic block copolymers due to their stability and possibility to exploit the self-assembly to tune the nanomorphology, improving PEM performance.13–15 Among various factors affecting the stability and the morphologies of the final polymers,16–18 the distribution of highly sulfonated segments along the backbone of multiblock copolymers demonstrated the possibility to obtain percolated structures even at low RH.15,19,20
In this regard, sulfonated poly(phenylene sulfide sulfones) (sPSS) represent an interesting class of polymers suitable for PEM application.21,22 The presence of electron-donating oxygen or sulfur atoms in ortho position to the sulfonic group is known to reduce the hydrolytic stability of sulfonated polymers facilitating their desulfonation.23,24 However, while ether linkages cannot be oxidized without chain scission, the oxidation of the thioether linkage to sulfone (sPSO2) brings consistent advantages in terms of oxidative and dimensional stability under humidified conditions.23,25 Nevertheless, a compromise between IEC and tolerable RH must be considered as excessive swelling or dissolution of the membrane at high IEC are not prevented by simple oxidation of the thioether units.25 The rigidity and high orientation of the –SO2– linkage26,27 provides additional mechanical strength but also exacerbates the brittleness of the polymers.23,28 On the other hand, the sulfide linkage has a shallow rotational barrier that promotes the phase separation of the sPSS and the sulfur atom of the thioether groups can act as an efficient radical scavenger in a fuel cell environment, being readily oxidized to sulfoxide and eventually sulfone in hydrogen peroxide.29,30
Previous works reported the successful implementation of sPSS in multiblock copolymers in combination with different apolar blocks such as poly(arylene ether sulfones)31,32 and poly(arylene sulfide nitrile).33 The obtained results demonstrated superior proton conductivity of the thioether forms compared to the sulfonated analogue.32 However, excessive water uptake affects the performance of the block copolymers at high RH.32 Changing the block copolymer architecture from multiblock to ABA triblock copolymer placing the charged block in the central position has been suggested to prevent excessive swelling and stabilize the ionic domains.13,34 So far, this architecture has been barely explored for proton-conducting polymers34–38 and only recently ABA structure with aromatic charged midblock have been proposed.36 Synthesis of ABA triblock copolymers requires monofunctional external blocks limiting the choice of aromatic A-blocks to Ni-mediated polymerization of poly(phenylene oxide) (PPO) as reported by Guiver et al.34 However, concerning fuel cell applications, metal-catalyzed synthesis poses additional risks as the presence (even in traces) of heavy metals in PEM is known to be detrimental for durability and performance.39,40
To overcome this drawback, we report here a metal-catalyst free approach to synthesize ABA proton conducting triblock copolymers. The copolymers have charged hydrophilic polydisperse sPSS synthesized via nucleophilic aromatic substitution polymerization as mid B-block and hydrophobic narrowly dispersed polystyrene (PS) synthesized via RAFT polymerization as A-blocks.41 RAFT polymerization was used here as an ideal platform for metal-free synthesis and thiol-click chemistries.42–44 We choose PS here to emphasize the phase separation of the resulting block copolymer (via the expected high χ parameter) and, most importantly, because of its good miscibility in polar aprotic solvents such as DMF, DMAc and NMP, that are also good solvents for the sPSS block. Although styrenic polymers are usually not considered ideal materials for PEM, due to their poor oxidative stability,45 several works employed PS as model compound in proton conducting systems37,46–48 and others even reported promising oxidative stability49 and potential applications.50 To avoid metal-catalyzed click-reactions, the blocks were connected using a thiol-fluoro click-chemistry. Decafluorobiphenyl (DFBP) was employed as it readily reacts with thiolate anions under mild conditions51,52 and has been reported to improve nanophase separation when used as a linker for sulfonated block copolymers.53,54 By varying the molecular weight of the constituting blocks, ABA triblock-copolymers with different compositions and different IEC can be obtained, allowing us to explore the nanostructure–property relationship of this new class of charged block copolymers.
Results and discussion
Three PS-b-sPSS-b-PS block copolymers have been prepared and studied here. They are named as BCPx, accordingly to their sPSS content, with x = 1 for the lowest sPSS content and x = 3 for the highest. The length of the charged sPSS midblock is kept constant, while the length of the hydrophobic PS blocks functionalized with DFBP end-group is varied, which was used in a thiol-fluoro click reaction (Scheme 1). For consistency in the numeration, PS blocks were numbered in the same way of the related BCP (i.e. PS1 was used to produce BCP1, etc.). | ||
| Scheme 1 Synthesis of ABA triblock copolymers based on thiol-fluoro click chemistry. Synthesis of PS-DFBP (top panel) and synthesis of BCP (bottom panel). | ||
Synthetic procedures
The PS blocks were synthesized by radical addition–fragmentation (RAFT) polymerization41 using 2-cyano-2-propyl benzodithioate (CPBD) as chain transfer agent (CTA). The dry condition of the reaction limited problems related to the sensitivity towards hydrolysis of this class of CTA.42 The one-pot sequential functionalization is required to limit the presence of active side-products (i.e. alkyl thiols) produced by the aminolysis of trithiocarbonates together with thioureas.55 By varying the [CTA]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Sty] ratio, PS with different molecular weights were synthesized (Table 1). The molecular weight of the resulting polymers was determined by GPC analysis in THF.
:[Sty] ratio, PS with different molecular weights were synthesized (Table 1). The molecular weight of the resulting polymers was determined by GPC analysis in THF.
The aminolysis with hydrazine56 reaction step followed by thiol-halo reaction with decafluorobiphenyl provided the desired PS-DFBP_x (x = 1, 2, 3). The disappearance of the characteristic pink color of the polymer solution gave an indication of the progress of the CTA cleavage, whereas UV-Vis spectroscopy confirmed the disappearance of the characteristic absorption peak of the CTA at 306 nm after 30 min at room temperature in DMF (Fig. 1a).
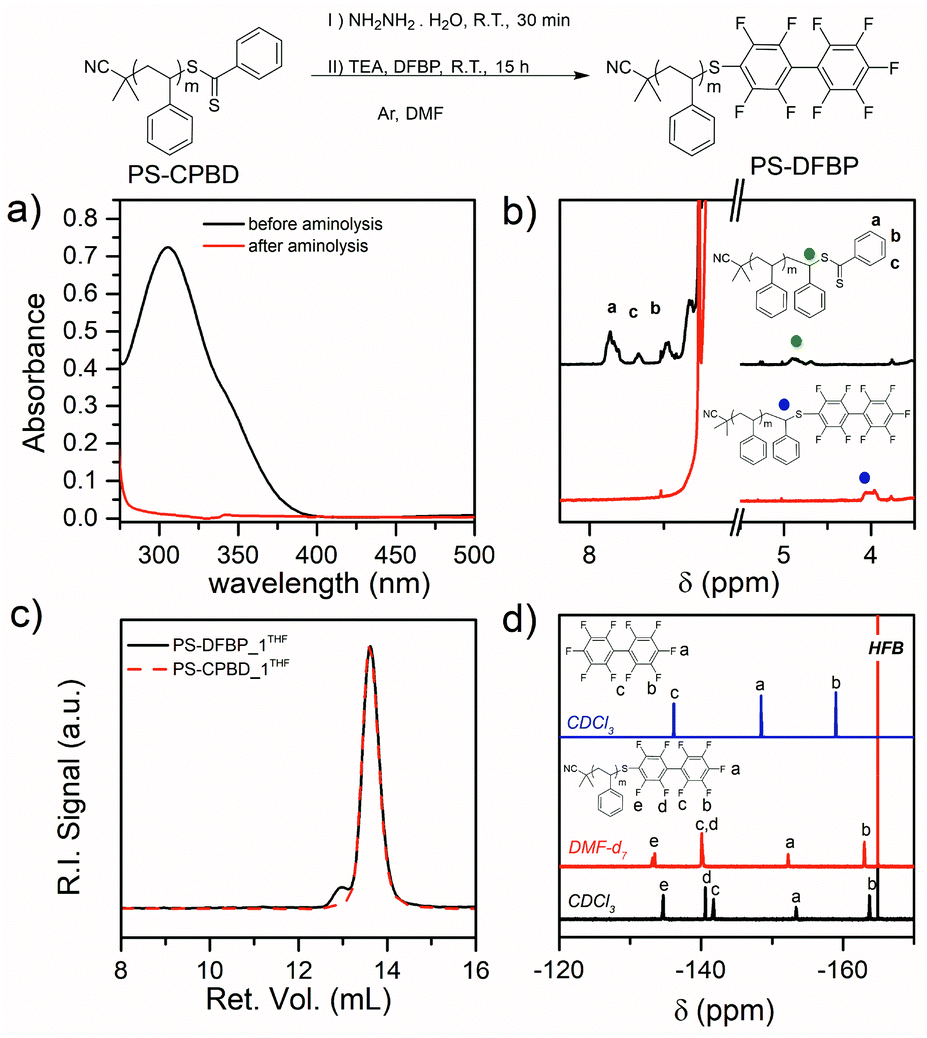 | ||
| Fig. 1 (a) UV-Vis spectra of PS-CPBD before and after aminolysis (4 × 10−5 M in DMF) showing clear disappearance of CPBD absorption peak at 306 nm. (b) 1H NMR spectrum of PS-CPBD before and after aminolysis and derivatization with hydrazine and DFBP respectively (PS-DFBP). (c) Comparison of the GPC eluograms of the PS-DFBP_1 and PS-CPBD_1 in THF. (d) 19F NMR spectra of the DFBP and PS-DFBP the latter in both CDCl3 and DMF-d7 (hexafluorobenzene was used as reference). | ||
Preliminary experiments in our lab (not reported here) have demonstrated the susceptibility of the DFBP towards nucleophilic substitution by primary amines even at room temperatures in DMF. Hence, a large excess of DFBP and significant dilution was adopted to avoid multiple substitutions of the perfluorinated molecule. This condition ensured the formation of the desired PS-DFBP as confirmed by NMR analysis (Fig. 1b and d).
The 1H NMR clearly shows the disappearance of the peaks of the o-, p- and m-protons of the CTA phenyl ring at 7.84, 7.66 and 7.48 ppm, respectively (Fig. 1b). Additionally, the signal of the terminal aliphatic proton at 4.85 ppm completely shifts to 4.00 ppm as a result of the substitution of the DFBP group. The 19F NMR spectrum in CDCl3 exhibit five different peaks at −134.64, −140.59, −141.74, −153.38 and −163.73 ppm corresponding to the terminal nonafluorobiphenyl group (Fig. 1d). The absence of any other peak confirmed the absence of residual DFBP or multiple substitutions on the fluorinated rings. Despite the oxygen-free atmosphere and the use of hydrazine,56 which is known to prevent disulfide formation, a small amount of disulfide coupling occurred as shown by GPC analysis in THF (Fig. 1c) but the dispersity remained low (Đ ≤ 1.2) (Table 1) and the formed disulfide did not interfere with the subsequent click reaction.
Synthesis of BCP1, BCP2 and BCP3 via metal-free thiol-fluoro click reaction
The synthesis of BCPx (with x = 1, 2, and 3) was performed in DMF at 50 °C over 4 days under inert atmosphere in the presence of K2CO3 as base.28,33 Compared to what was reported in literature for the synthesis of multiblock copolymers having DFBP linkers,28,31,33 milder conditions were adopted here in order to limit the extent of side reactions (multiple substitutions). An excess of 3 equivalents of PS-DFBP was employed to ensure full conversion of the sPSS end-groups. Residual salts and unreacted PS-DFBP were removed through extensive washing with water and acetone to obtain pure block copolymers. The NMR analysis confirmed the success of the click reaction (Fig. 2a and b). | ||
| Fig. 2 (a) 1H NMR of BCPx (x = 1, 2, 3) showing the copresence of both the PS and the sPSS peaks highlighted in light blue and light orange, respectively. (b) 19F NMR of the BCP1 in DMF-d7 showing the conversion of the terminal fluorine of the PS-DFBP into the octafluorobiphenyl unit, the presence of peaks a′ and b′ is discussed in the main text. (c) FTIR spectra of the BCPx (x = 1, 2, 3) showing the presence of the characteristic vibrations of the sPSS (dashed lines) and PS-DFBP (dotted lines) blocks. Black arrows indicate the presence of typical DFBP vibration in the PS-DFBP spectrum confirming the functionalization of the PS block. (d) GPC traces in DMF with 0.01 M LiBr of BCP1 with respective PS-DFBP_1 reported for comparison. | ||
1H NMR spectra showed the presence of peaks characteristic of both blocks (Fig. 2a) while 19F NMR analysis proved the success of the click reaction showing the four main peaks of the octafluorobiphenyl moiety at −133.5, −133.8, −139.2 and −139.8 ppm, respectively (Fig. 2b). Additionally, two minor peaks appeared together with the main signals. Considering the excess of PS-DFBP and the unlikely chemical shifts for the di- and tri-substitution of the DFBP with thiolate anion,53 we attributed the additional peaks to the presence of hydroxyl-substitution due to moisture absorption under the alkaline environment.
FTIR spectra of the obtained BCPs show the presence of both the sPSS and the PS characteristic vibrations (Fig. 2c). The broad band visible in the range between 1250 and 1140 cm−1 is due to the sulfonic group stretching vibrations, while the asymmetric stretching of the –SO2– generates the two bands located at 1270 and 1300 cm−1. Additionally, typical signals of poly(p-phenylene sulfides)57 are present at 1093, 1074 and 811 cm−1. The –CH2– aliphatic stretching from the PS block appear at 1485 cm−1 and the monosubstituted benzene out-of-plane deformations at 750 and 730 cm−1. A proportional increase of the sPSS band intensities with its content in the BCP is recognizable (Fig. 2c).
GPC analysis of BCP shows a single peak shifted at lower retention time compared to the PS-DFBP signal, confirming the success of the click reaction (Fig. 2d). Due to the ionic interaction of the BCPs with the column, the molecular weight was calculated based on the wt% composition obtained by 1H NMR (Table 2) using the molecular weight of the PS obtained by GPC analysis in THF.
|
M
NMRna (kg mol−1) |
Đ | f GPCsPSS (wt%) | f NMRsPSS (wt%) | f El.An.sPSS (wt%) |
f
NMRsPSSb (vol%) |
T g (°C) | T d (°C) | |
|---|---|---|---|---|---|---|---|---|
| a Calculated by MGPCn values of PS taking into account the wt% composition of block copolymers obtained by 1H NMR analysis. b Evaluated from the wt% considering the density of the pristine polymers (ρPS = 1.05 g cm−3 (ref. 61) and ρsPSS = 1.53 g cm−3 (ref. 23)) and neglecting the mixing effects.62 c Values in brackets represent Tg values of corresponding pristine PS-DFBP_x (x = 1, 2, 3). d Temperature corresponding to the 5 wt% weight loss. | ||||||||
| BCP1 | 44.1 | 1.27 | 16.5 | 18.4 | 21.1 | 13.4 | 104.3 (101.5)c | 336.1 |
| BCP2 | 32.4 | 1.29 | 22.4 | 24.1 | 27.6 | 17.9 | 103.3 (100.8) | 328.6 |
| BCP3 | 26.2 | 1.36 | 32.1 | 42.8 | 43.9 | 33.9 | 103.4 (96.9) | 316.9 |
The comparison between the compositions of the BCPs calculated based on the Mn obtained by different methods (GPC, 1H NMR and elemental analysis) are also reported in Table 2. Overall, the agreement between the sPSS fraction measured by the different techniques is good, even though slight discrepancies are observed. These differences might be attributed to partial hydroxy-substitution of the DFBP linker and the possible presence of impurities (i.e. alien ions and salts) which are difficult to remove completely in these charged polymers.
Thermal properties
The thermal properties of the BCP in the proton form were analyzed both by DSC and TGA. The results are presented in Fig. 3 and summarized in Table 2. The sPSS homopolymer does not show any Tg up to 250 °C, maximum T explored as the sulfonic group start degrading around 270 °C. Thus, all BCPs show only one glass transition corresponding to the PS blocks and demonstrating strong phase separation between the two blocks. A slight shift to higher temperature is generally observed for the Tg of the PS phase in the BCPs. This is expected due to the rigid nature of the charged block which hinders the free motion of PS chains in the BCP when compared to the homopolymer. | ||
| Fig. 3 (a) DSC thermograms of the block copolymers recorded during the second heating run at 10 °C min−1 under nitrogen atmosphere, the traces of PS-DFBP and sPSS are reported for comparison. (b) TGA thermograms of the block copolymers recorded at 10 °C min−1 under nitrogen atmosphere, both PS-DFBP and sPSS thermograms are reported for comparison. | ||
All BCPs possessed good thermal stability with Td above 300 °C as demonstrated by TGA (Fig. 3b). A decrease in the Td was observed with increasing the sPSS. Being the sulfonic group the weakest point in terms of thermal stability, it is possible to observe that in all cases the BCP structure enhanced the overall thermal stability of the material when compared to the pristine sPSS polymer. From Fig. 3b it is also possible to observe that, since the PS-DFBP has negligible residual mass at 700 °C, the residue of BCPx (x = 1, 2 and 3) is proportional to the sPSS content. In fact, BCP3 displays the highest residual mass among the block copolymers while BCP1 has the least residue.
Nanostructure characterization
The nanomorphology of the charged block copolymers was studied using TEM and SAXS analysis and the results are summarized in Fig. 4 and Table 3. Membranes cast from DMAc and ion-exchanged with H+ or Cs+ were analyzed. Cs+ was used here to enhance the contrast of the ionic domains due to the higher electron density of the Cs+ ions with respect to H+. The effect of the linker between the blocks on the self-assembly of sulfonated aromatic polymers has been reported,28,53,54 with perfluorinated linkers promoting the formation of better phase-separated morphologies. As a result of the rigidity of the sPPS block and of the DFBP linker, a slow casting protocol (40 °C for 3 days) from solutions with dilution below 10 wt% was required to develop well-defined nanostructures.58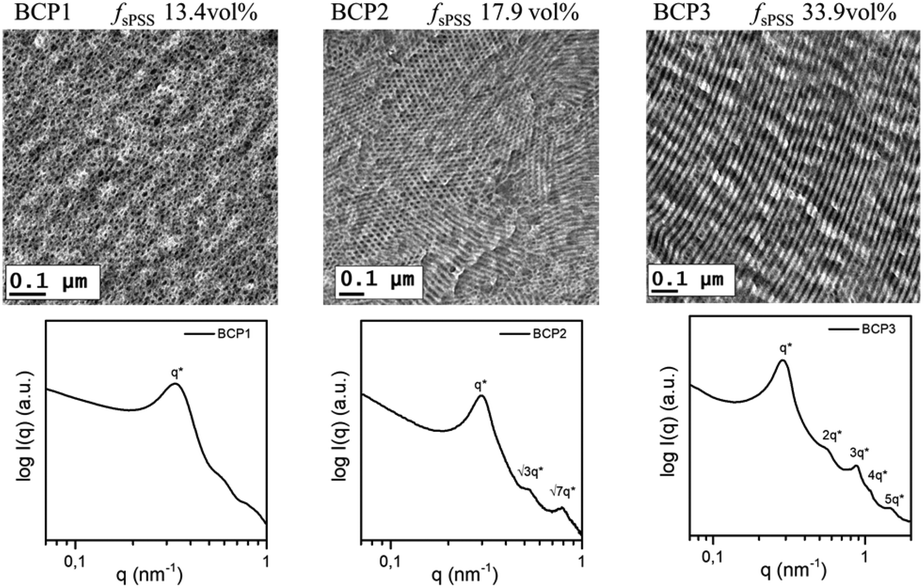 | ||
| Fig. 4 TEM images of the BCP stained with Cs+ ions and the corresponding SAXS profiles obtained from the H-form (BCP1 and BCP3) and Cs-form (BCP2) of the dried membranes at room temperature. | ||
| f sPSS (vol%) | f PS (vol%) | q* (nm−1) | d (nm) | Morph | |
|---|---|---|---|---|---|
| BCP1 | 13.4 | 86.6 | 0.33 | 19.0 | SPH |
| BCP2 | 17.9 | 82.1 | 0.30 | 21.0 | CYL |
| BCP3 | 33.9 | 66.1 | 0.27 | 23.9 | LAM |
All BCPs show enhanced phase separation as a result of the strong incompatibility between the PS and the charged sPSS blocks. For classical block copolymers with strong phase segregation, different morphologies are expected depending on the volume fraction of the B-block.59 For the BCP1 (fspSS = 13.4 vol%), a spherical structure was observed with a periodicity of d = 2π/q* = 19 nm and an average radius of 9.2 nm (estimated using the position of the minima in the SAXS curve and fitting of the SAXS curve, not showed here). SAXS and TEM analysis of BCP2 and BCP3 revealed the existence of two different morphologies. BCP2 (fsPSS = 17.9 vol%) showed formation of hydrophilic cylindrical domains with a SAXS peak sequence typical for the hexagonally packed morphology60 at q*:√3q*:√7q* and with a domain spacing d = 2π/q* = 21 nm (q* = 0.30 nm−1).
Highly oriented lamellar structures already appeared at fsPSS = 33.9 vol% in BCP3. SAXS pattern of BCP3 exhibits scattering maxima at q*, 2q*, 3q*, 4q* and 5q* that correspond to the (100), (200), (300), (400) and (500) scattering reflections of a lamellar structure.
To evaluate the extent of incompatibility between the PS and sPSS blocks, a χ parameter estimation was attempted based on the lamellar spacing of the BCP3 using the formalism developed for monodisperse block-copolymers63 (eqn (1)):
 | (1) |
Similar conclusions can be obtained considering the Hildebrand's solubility parameters estimation (eqn (2)) in combination with the concepts of regular solution theory64 that allows expression of the χ parameter for a couple of homopolymers A and B as:
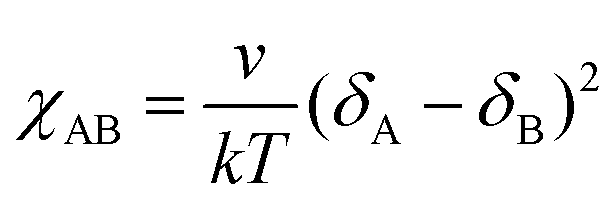 | (2) |
 | (3) |
Dimensional stabilization upon hydration
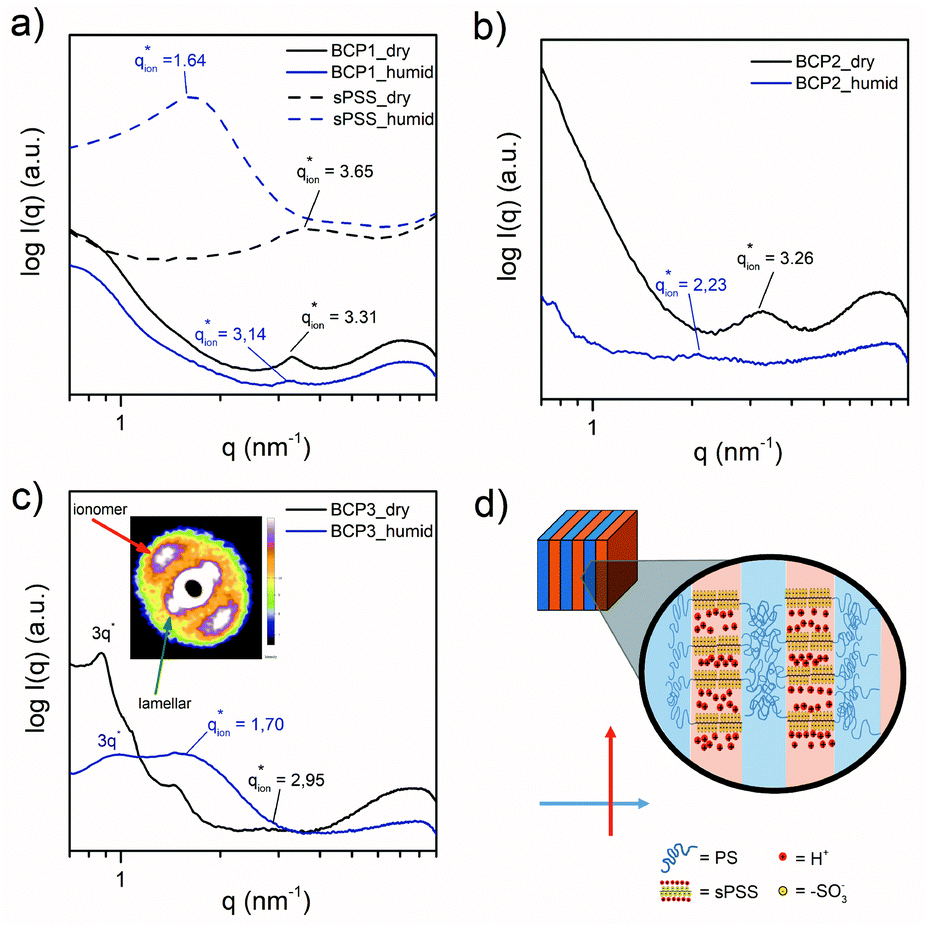
Generally, our PS-b-sPSS-b-PS BCPs were brittle at ambient temperature. However, for BCP1, BCP2 and BCP3, membranes could be obtained and exposed to water vapour or immersed in water, allowing the investigation of the swelling effect on their ionic domain sizes.A medium angle X-ray scattering (MAXS) study of the ionomer peak position in the BCP and the sPSS was performed (Fig. 5). All BCPs show a distinct ionomer peak in the dry state ( ≈ 3.3 nm−1, dion ≈ 1.9 nm) with a peak position similar to the sPSS homopolymer (
≈ 3.3 nm−1, dion ≈ 1.9 nm) with a peak position similar to the sPSS homopolymer (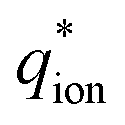 = 3.65 nm−1, dion = 1.7 nm).
= 3.65 nm−1, dion = 1.7 nm).
 | ||
| Fig. 5 MAXS profile at room temperature under dry and hydrated conditions (RH 95%) of (a) BCP1 and sPSS, (b) BCP2 and (c) BCP3 in their protonated form. Inset in (c) quadrant represent the 2D MAXS pattern showing the orthogonal orientation of the ionomer peak with respect to the lamellar direction. (d) Cartoon representing the relative orientation of the lamellae and the ionic domains inside BCP3. | ||
This suggests that the hydrophilic chains preserve their native packing inside the BCP domains. However, under humidified conditions, the three BCPs behave differently. Generally, the ionomer peak position in the BCP shifts to lower q (increase in domain size), but the shift is definitively smaller than the one observed for the sPSS which swells excessively at the limit of dissolution. An ionic domain enlargement below 75% was observed for all the BCPs, sensibly lower if compared to the 123% of the pure sPSS. Thus, the BCP structure greatly reduces the dimensional change in the ionic domain size, with the PS scaffold preventing dissolution but not preventing water molecules to penetrate the sPSS domains and facilitating the hydration of the sulfonic groups.
Further details about the hierarchical structure in these materials can be learned by observation of the 2D X-ray patterns. Interestingly, in the case of BCP3 possessing LAM nanostructure, the MAXS/SAXS pattern clearly shows a strong orthogonal orientation of the ionomer peaks against the lamellar ones (Fig. 5c, inset).
This means that the ionic pathways are orthogonally oriented with respect to the lamellar domains as depicted in Fig. 5d. Our observation further shades light on the structure of sPSS systems that is not well studied so far. Due to the semi-rigid sPSS chain conformation, the ionomer peak directly relates to the interchain distance of adjacent sPSS chain segments. This means that the sPSS chains are highly oriented and self-organized perpendicular to the lamellae directions with ionic groups forming narrow channels perpendicular to the PS “walls”.
This kind of organization might be facilitated here by the stiffness of both the sPSS and the DFBP linker that induced a stretched conformation in the sPSS chains between the PS domains, forcing spontaneous structure alignment during membrane formation. Another interesting observation is that, while for the BCP2 and BCP1 the ionomer peak decreases in intensity upon swelling (suggesting humidity-induced disordering), for BCP3 the ionomer peak intensity is enhanced upon water absorption, as observed for pure sPSS. This means that the peculiar orthogonal arrangement of the sPSS chains with respect to the lamellar planes, favors swelling without compromising the high degree of order of the LAM structure. This is not the case for nanomorphologies with curved interfaces (spherical and cylindrical).
Oxidative stability and proton conductivity
The oxidative stability of the BCP was evaluated using an accelerated test by immersing pieces of membranes for 1 h at 80 °C in Fenton's reagent and the results are reported in Table 4. Interestingly, the highest degradation was observed for the BCP3 that contained the highest weight fraction of sPSS. This might be explained considering the higher hydrophilicity of BCP3 compared to BCP1 and BCP2 that facilitated the reagent penetration inside the membrane. On the contrary, the hydrophobicity induced by the higher content of PS in BCP1 and BCP2 limited the degradation within the considered time interval most probably due to limited mass transfer of the reagent inside the membrane.| Morph | IECtitr.a (meq g−1) |
IECNMRb (meq g−1) |
W.U.c (%) | λ ([H2O]/[SO3H]) | R.W.d (%) | Δd/ddry (%) | |
|---|---|---|---|---|---|---|---|
| a Obtained from titration with NaOH 0.01 N. b Calculated from 1H NMR composition. c Evaluated after immersion in water at 25 °C for 24 h. d Residual weight after 1 h immersion in Fenton's reagent at 80 °C. | |||||||
| sPSS | — | 3.18 | — | — | — | — | 123 |
| BCP1 | SPH | 0.32 | 0.58 | 3.0 | 5 | 97.1 | 5.3 |
| BCP2 | CYL | 0.60 | 0.79 | 10.2 | 9 | 89.9 | 50 |
| BCP3 | LAM | 1.31 | 1.36 | 28.7 | 12 | 54.5 | 74 |
The proton conductivity of these sulfonated polymers is strongly dependent on their ion exchange capacity (IEC). This parameter could be retrieved by back titration of the exchanged proton ions in a salt solution or by evaluation of the 1H NMR composition. The results obtained for BCP1, BCP2 and BCP3 are summarized in Table 4 confirming the increasing trends with increasing sPSS fractions. Generally, an underestimation of the IEC values determined by titration if compared with the value obtained by 1H NMR is observed. This could be ascribed to the limited availability of some –SO3H groups for the H+/Na+ exchange66 in the spherical and cylindrical structures.
The proton conductivity of the selected BCPs was measured in the through-plane direction at 95% RH as a function of temperature between 30 and 90 °C (Fig. 6). As it was not possible to obtain large (cm2) membranes for some of the BCPs, stacks of membrane slices were pelletized and measured in order to investigate in a comparable manner the effect of nanomorphology on the conductivity on all the BCPs.
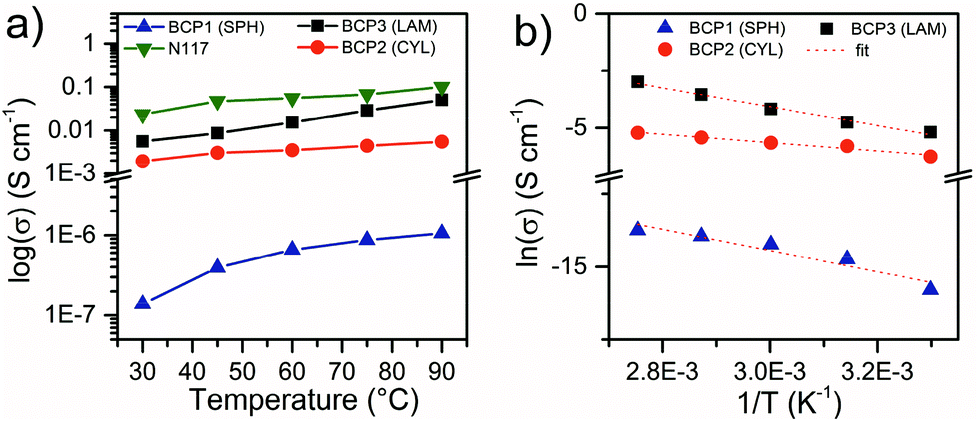 | ||
| Fig. 6 (a) Through-plane proton conductivity of BCP1 BCP2, BCP3 and Nafion™ N117 at 95% RH as a function of temperature. (b) Arrhenius plot of proton conductivity for BCPs. | ||
As expected, the proton conductivity increases with increasing sPSS wt% fraction and temperature. In addition, comparing the results obtained for the three selected BCPs, the dramatic impact of the nanomorphology on the macroscopic proton transport properties is evident. Considering the values at 90 °C, an increase of about 6 wt% of sPSS between BCP2 (CYL) and BCP1 (SPH) generates an increase of more than four orders of magnitude in conductivity. In contrast, a limited increase in conductivity was observed for BCP3 considering the ∼18 wt% increase in fsPSS. These observations can be directly ascribed to the nanostructural features of the block copolymers and also to the limited water uptake of the BCP1 compared to the other two BCPs (Table 4). The spherical morphology of BCP1 has a very low degree of percolation with the ionic domains confined by the PS matrix in closed spheres. Moreover, the limited WU provides minimal hydration level (λ), close to the limit of proton dissociation8 which explains the low conductivity registered. On the other hand, the presence of elongated channels and interconnections between the hydrophilic cylinders in BCP2 (Fig. 4) is primarily responsible for the improved proton conductivity. Additionally, the λ increase corroborates the MAXS observation and the higher accessibility of water to the ionic domains. When it comes to the lamellar structure, considering the difference in sPSS content between BCP2 and BCP3, a large increase in proton conductivity was expected. The relatively low increase in proton conductivity observed here can be rationalized considering that the orientation of a non-negligible portion of the lamellar structures, even if partially randomized in the stack, is orthogonal to the through-plane direction. For the aforementioned reasons, the highest conductivity value measured is about 50 mS cm−1 at RH 95% and 90 °C. This is in the same order of magnitude but lower with respect to the benchmark Nafion™ N117 tested in the same conditions (Fig. 6a). A conductivity test for BCP3 in fully wet conditions at 30 °C provided a conductivity of 20 mS cm−1 which is approximately 4 times higher than the same polymer at 95% RH and comparable to the value obtained at 80 °C (95% RH). This result confirmed the relevance of hydration for proton conductivity in sulfonated block copolymers and the potential of these ABA block copolymers under fully wet conditions.
The proton conductivity values exhibited by our BCP3 (LAM) sample are in the same range, yet slightly lower, than other ABA copolymers with charged mid B-block reported recently in literature by Agudelo et al.36 The difference in conductivity is most probably due by the fact that in ref. 36 the samples have been measured in the fully hydrated state, while our tests were conducted at RH = 95%. Interestingly, a stronger influence of the rigidity of the A-block on the conductivity compared to the influence of the IEC values was reported.36 Similarly, in our case the “glassy” state of the PS block is responsible for the modest conductivity values obtained. Arrhenius plot of proton conductivity (Fig. 6b) shows a linear dependence of the proton conductivity on temperature with activation energies between 16 and 35 eV, comparable to sulfonated poly(phenylene sulfone) copolymers with similar hydration level.67
Conclusions
In this work, we introduced a strategy to synthesize proton conducting ABA triblock copolymers with charged sulfonated midblock flanked by narrowly dispersed polystyrene blocks using copper-free “thiol-click” chemistry. This approach provides unique flexibility and control over the composition and the resulting nanomorphology of the block copolymers and overcomes problems related to the presence of alien metal ions used in common click chemistry without sacrificing control over the block copolymer architecture.A decafluorobiphenyl-functionalized PS was reacted with thiol terminated sPSS to give ABA triblock copolymers. In all cases, insoluble membranes were obtained when fvolsPSS < 50%, thus eliminating problems related to sPSS water solubility. All the BCPs possessed high thermal stability with Td around 300 °C. Nanostructure analysis via X-ray scattering techniques and TEM clearly show strong phase-separation with achievement of ordered nanostructures. Spherical, cylindrical and lamellar nanomorphologies appeared depending on the block copolymer composition. X-ray scattering analysis of the ionomer peak revealed a strong reduction in the swelling of the nanostructure (75% in the case of the lamellar BCP3 compared to 123% of the sPSS) as a result of the dimensional stabilization of the ionic domains provided by the hydrophobic PS matrix. Proton conductivity tested in the through-plane direction revealed the relevance of the nanomorphology on the proton transport, exhibiting Arrhenian behavior and activation energies typically in the range of other sulfonated aromatic polymers. Although further improvements are required in terms of membrane preparation and proton conductivity at reduced humidity, the highest value of 50 mS cm−1@RH95%, 90 °C measured here for the lamellar structure is promising for future applications in polymer membrane fuel cells. Moreover, these strongly phase separated systems could be interesting in the future to produce sensors and other electroactive devices, especially if we consider that some of them show tendency to spontaneous anisotropic alignment of the nanostructure at the macroscopic scale.
Author contributions
The manuscript was written through contributions of all authors. The concept of the project was developed by M. Viviani and G. Portale. Experimental work was conducted by M. Viviani and S.P. Fluitman. Data interpretation and drafting of the manuscript was conducted by M. Viviani and G. Portale. The manuscript was edited by M. Viviani, K. Loos and G. Portale. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Albert J. J. Woortman for GPC analysis, Jur van Dijken for the TGA measurements and Ir. Johans van der Velde for the elemental analysis. G. P. acknowledges the Zernike Institute for Advanced Materials for the startup funding that was used to finance this project.Notes and references
- Y. Ding, Z. P. Cano, A. Yu, J. Lu and Z. Chen, Electrochem. Energy Rev., 2019, 2, 1–28 CrossRef CAS.
- Z. Ma, D. R. MacFarlane and M. Kar, Batteries Supercaps, 2019, 2, 115–127 CrossRef.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS PubMed.
- A. Kraytsberg and Y. Ein-Eli, Energy Fuels, 2014, 28, 7303–7330 CrossRef CAS.
- The US Department of Energy (DOE), Fuel Cell Technol. Off., 2016, 2015, 1–58 Search PubMed.
- C. Ma, L. Zhang, S. Mukerjee, D. Ofer and B. Nair, J. Membr. Sci., 2003, 219, 123–136 CrossRef CAS.
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535–4586 CrossRef CAS PubMed.
- K.-D. Kreuer, S. J. Paddison, E. Spohr and M. Schuster, Chem. Rev., 2004, 104, 4637–4678 CrossRef CAS PubMed.
- K.-D. Kreuer, Chem. Mater., 2014, 26, 361–380 CrossRef CAS.
- M. Rikukawa and K. Sanui, Prog. Polym. Sci., 2000, 25, 1463–1502 CrossRef CAS.
- R. Dams and K. Hintzer, Fluorinated Polymers: Applications, Royal Society of Chemistry, 2017, ch. 1, vol. 2, pp. 1–30 Search PubMed.
- H. Zhang and P. K. Shen, Chem. Rev., 2012, 112, 2780–2832 CrossRef CAS PubMed.
- D. W. Shin, M. D. Guiver and Y. M. Lee, Chem. Rev., 2017, 117, 4759–4805 CrossRef CAS PubMed.
- N. Li and M. D. Guiver, Macromolecules, 2014, 47, 2175–2198 CrossRef CAS.
- Y. A. Elabd and M. A. Hickner, Macromolecules, 2011, 44, 1–11 CrossRef CAS.
- G. He, Z. Li, J. Zhao, S. Wang, H. Wu, M. D. Guiver and Z. Jiang, Adv. Mater., 2015, 27, 5280–5295 CrossRef CAS PubMed.
- T. J. Peckham and S. Holdcroft, Adv. Mater., 2010, 22, 4667–4690 CrossRef CAS PubMed.
- K. Goto, I. Rozhanskii, Y. Yamakawa, T. Otsuki and Y. Naito, Award ACCOUNTS SPSJ Award Accounts, Polym. J., 2009, 41, 95–104 CrossRef CAS.
- K. Nakabayashi, K. Matsumoto and M. Ueda, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3947–3957 CrossRef CAS.
- K. Nakabayashi, K. Matsumoto, T. Higashihara and M. Ueda, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7332–7341 CrossRef CAS.
- F. Wang, J. Mecham, W. Harrison and J. E. McGrath, in American Chemical Society, Polymer Preprints, Division of Polymer Chemistry, 2000, vol. 41, pp. 1401–1402 Search PubMed.
- K. B. Wiles, I. A. Bhanu, F. Wang and J. E. McGrath, in American Chemical Society, Polymer Preprints, Division of Polymer Chemistry, 2002, vol. 43, pp. 993–994 Search PubMed.
- M. Schuster, K.-D. D. Kreuer, H. T. Andersen and J. Maier, Macromolecules, 2007, 40, 598–607 CrossRef CAS.
- S. Takamuku and P. Jannasch, Polym. Chem., 2012, 3, 1202–1214 RSC.
- M. Schuster, C. C. De Araujo, V. Atanasov, H. T. Andersen, K. D. Kreuer and J. Maier, Macromolecules, 2009, 42, 3129–3137 CrossRef CAS.
- I. Baxter, A. Ben-Haida, H. M. Colquhoun, P. Hodge, F. H. Kohnke and D. J. Williams, Chem. – Eur. J., 2000, 6, 4285 CrossRef CAS.
- H. M. Colquhoun, P. L. Aldred, F. H. Kohnke, P. L. Herbertson, I. Baxter and D. J. Williams, Macromolecules, 2002, 35, 1685–1690 CrossRef CAS.
- G. Titvinidze, K.-D. Kreuer, M. Schuster, C. C. de Araujo, J. P. Melchior and W. H. Meyer, Adv. Funct. Mater., 2012, 22, 4456–4470 CrossRef CAS.
- S. Y. Lee, N. R. Kang, D. W. Shin, C. H. Lee, K.-S. S. Lee, M. D. Guiver, N. Li and Y. M. Lee, Energy Environ. Sci., 2012, 5, 9795–9802 RSC.
- D. Zhao, J. Li, M. K. Song, B. Yi, H. Zhang and M. Liu, Adv. Energy Mater., 2011, 1, 203–211 CrossRef CAS.
- F. Schönberger, M. Hein and J. Kerres, Solid State Ionics, 2007, 178, 547–554 CrossRef.
- S. Takamuku and P. Jannasch, Macromolecules, 2012, 45, 6538–6546 CrossRef CAS.
- D. W. Shin, S. Y. Lee, C. H. Lee, K.-S. Lee, C. H. Park, J. E. McGrath, M. Zhang, R. B. Moore, M. D. Lingwood, L. A. Madsen, Y. T. Kim, I. Hwang and Y. M. Lee, Macromolecules, 2013, 46, 7797–7804 CrossRef CAS.
- N. Li, S. Y. Lee, Y.-L. Liu, Y. M. Lee and M. D. Guiver, Energy Environ. Sci., 2012, 5, 5346–5355 RSC.
- M. L. Disabb-Miller, Z. D. Johnson and M. A. Hickner, Macromolecules, 2013, 46, 949–956 CrossRef CAS.
- N. A. Agudelo, J. Palacio and B. L. López, J. Mater. Sci., 2019, 54, 4135–4153 CrossRef CAS.
- T. Saito, H. D. Moore, M. A. Hickner, H. D. Moore and M. A. Hickner, Macromolecules, 2010, 43, 599–601 CrossRef CAS.
- Z. Shao, A. Sannigrahi and P. Jannasch, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4657–4666 CrossRef CAS.
- A. Collier, H. Wang, X. Zi Yuan, J. Zhang and D. P. Wilkinson, Int. J. Hydrogen Energy, 2006, 31, 1838–1854 CrossRef CAS.
- X. Cheng, Z. Shi, N. Glass, L. Zhang, J. Zhang, D. Song, Z.-S. Liu, H. Wang and J. Shen, J. Power Sources, 2007, 165, 739–756 CrossRef CAS.
- J. Chiefari, Y. K. B. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- P. J. Roth, C. Boyer, A. B. Lowe and T. P. Davis, Macromol. Rapid Commun., 2011, 32, 1123–1143 CrossRef CAS PubMed.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2013, 26, 724–744 CrossRef.
- M. A. Hickner, H. Ghassemi, Y. S. Kim, B. R. Einsla and J. E. McGrath, Chem. Rev., 2004, 104, 4587–4612 CrossRef CAS PubMed.
- L. Rubatat, Z. Shi, O. Diat, S. Holdcroft and B. J. Frisken, Macromolecules, 2006, 39, 720–730 CrossRef CAS.
- Z. Shi and S. Holdcroft, Macromolecules, 2005, 38, 4193–4201 CrossRef CAS.
- A. C. C. Yang, R. Narimani, B. J. Frisken and S. Holdcroft, J. Membr. Sci., 2014, 469, 251–261 CrossRef CAS.
- Y. Tamura, L. Sheng, S. Nakazawa, T. Higashihara and M. Ueda, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4334–4340 CrossRef CAS.
- A. Oishi, H. Matsuoka, T. Yasuda and M. Watanabe, J. Mater. Chem., 2009, 19, 514–521 RSC.
- S. Alapour, B. G. De La Torre, D. Ramjugernath, N. A. Koorbanally and F. Albericio, Bioconjugate Chem., 2018, 29, 225–233 CrossRef CAS PubMed.
- C. Langner, J. Meier-Haack, B. Voit and H. Komber, J. Fluor. Chem., 2013, 156, 314–321 CrossRef CAS.
- A. S. Badami, O. Lane, H.-S. Lee, A. Roy and J. E. McGrath, J. Membr. Sci., 2009, 333, 1–11 CrossRef CAS.
- H.-S. Lee, A. Roy, O. Lane, S. Dunn and J. E. McGrath, Polymer, 2008, 49, 715–723 CrossRef CAS.
- X.-P. Qiu and F. M. Winnik, Macromol. Rapid Commun., 2006, 27, 1648–1653 CrossRef CAS.
- W. Shen, Q. Qiu, Y. Wang, M. Miao, B. Li, T. Zhang, A. Cao and Z. An, Macromol. Rapid Commun., 2010, 31, 1444–1448 CrossRef CAS PubMed.
- D. A. Zimmerman, J. L. Koenig and H. Ishida, Spectrochim. Acta, Part A, 1995, 51, 2397–2409 CrossRef.
- M. Lee, J. K. Park, H.-S. Lee, O. Lane, R. B. Moore, J. E. McGrath and D. G. Baird, Polymer, 2009, 50, 6129–6138 CrossRef CAS.
- M. W. Matsen and F. S. Bates, Macromolecules, 1996, 29, 1091–1098 CrossRef CAS.
- W. Takagi, J. Suzuki, Y. Aoyama, T. Mihira, A. Takano and Y. Matsushita, Macromolecules, 2019, 52, 6633–6640 CrossRef CAS.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, Wiley & Sons Inc., 4th edn, 2003 Search PubMed.
- M. J. Park and N. P. Balsara, Macromolecules, 2008, 41, 3678–3687 CrossRef CAS.
- M. W. Matsen and F. S. Bates, J. Chem. Phys., 1997, 106, 2436–2448 CrossRef CAS.
- R. F. Fedors, Polym. Eng. Sci., 1974, 14, 472–472 CrossRef CAS.
- K. P. Mineart, B. Lee and R. J. Spontak, Macromolecules, 2016, 49, 3126–3137 CrossRef CAS.
- K. Krajinovic, T. Kaz, T. Haering, V. Gogel and J. Kerres, Fuel Cells, 2011, 11, 787–800 CrossRef CAS.
- C. C. de Araujo, K. D. Kreuer, M. Schuster, G. Portale, H. Mendil-Jakani, G. Gebel and J. Maier, Phys. Chem. Chem. Phys., 2009, 11, 3305–3312 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full details of the experimental protocols with selected spectra and characterization techniques. See DOI: 10.1039/d1py00094b |
| This journal is © The Royal Society of Chemistry 2021 |