
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLinking molecular structure to plant conditions: advanced analysis of a systematic set of mini-plant scale low density polyethylenes†
Kristina Maria
Zentel
 a,
Paul Severin
Eselem Bungu
b,
Harald
Pasch
*b and
Markus
Busch
*c
a,
Paul Severin
Eselem Bungu
b,
Harald
Pasch
*b and
Markus
Busch
*c
aUniversity of Hamburg, Bundesstr. 45, 20146 Hamburg, Germany
bUniversity of Stellenbosch, PO Box X1, Stellenbosch 7602, South Africa. E-mail: hpasch@sun.ac.za
cTechnical University of Darmstadt, Alarich-Weiß-Straße 8, 64287 Darmstadt, Germany. E-mail: markus.busch@pre.tu-darmstadt.de
First published on 27th April 2021
Abstract
Two sample sets of low density polyethylene (LDPE) were investigated and differentiated via comprehensive analysis of their microstructures with specific emphasis on branching. The sample sets were produced under well-defined conditions in a high-pressure mini-plant autoclave reactor and vary with respect to molecular weight and conversion. Focus was laid on analyzing the materials with respect to short- and long-chain branching by NMR, light scattering, viscosimetry and crystallization-based methods. Additionally, selected samples were fractionated preparatively. Temperature rising elution fractionation and solvent gradient fractionation were chosen to fractionate the materials according to crystallizability and molecular weight, respectively. Triple detector SEC provided molecular weight distributions, short chain branching densities, Mark–Houwink and conformation plots of the individual preparative fractions. It was proven that the fractionations were successful with regard to the desired properties and a deeper insight into the heterogeneous, complex microstructure of LDPE and its connection to process conditions was obtained.
Introduction
Polyethylene (PE) exhibits a long-lasting industrial success both for commodity materials as well as niche applications.1–23 Recently, PE has drawn attention in the context of recycling and circular economy.4 Although PE exhibits a very simple molecular structure (just carbon and hydrogen), its properties can be tuned over a very wide range by adjusting its microstructure and molecular topology. The polymeric microstructure of PE is not only governed by molecular weight (Mw) but also by the length and branched density in the material.5 Branches of different lengths and topologies are formed during the polymerization process. While catalytically produced HDPE (high density PE) is mainly linear, LLDPE (linear low density PE) contains short-chain branches (SCB) and LDPE (low density PE) shows a significant SCBs as well as LCBs (long-chain branches) densities. In the case of LDPE, SCBs and LCBs are introduced into the macromolecular structure during the free radical polymerization process via intra- and intermolecular transfer reactions, respectively.6 These branches are formed randomly during the polymerization, so that the resulting material is indeed a mixture of macromolecules with remarkably differing microstructures with respect to molecular weight as well as branching length and density. Consequently, it is of crucial importance to analyze these materials comprehensively in order to establish structure–property relationships, which will eventually be able to link polymer microstructure and branching with product properties such as density, viscosity or crystallinity. While LDPE typically exhibits 10–30 SCBs per 1000 carbon atoms, the LCB density is only 1–3 per 1000 carbon atoms. SCBs affect mainly the density and crystallizability of the material, LCBs lead to a reduced coil radius and influence the flow properties of the material.6,7The heterogeneity of LDPE-type materials is particularly challenging from an analytical point of view. Attempts have been made to comprehensively analyze LDPE and quite a few complementary techniques have been developed and used.5,8,9 Average molecular weights and molecular weight dispersities can be determined by high-temperature size exclusion chromatography (SEC) coupled to a concentration sensitive detector, typically working at 140–150 °C and using 1,2,4-trichlorobenzene as a solvent and mobile phase.10 This experimental setup provides information regarding molecular weight but not branching. Average branching sequences can be measured via quantitative 13C-NMR under high-temperature conditions and different branch types can be identified and grouped into SCBs and LCBs.11–15 For defined ethylene α-olefin copolymers, branches of up to 10 and 16 carbon atoms could be differentiated.13,15,16 In LDPEs the differentiation is more complex due to the simultaneous existence of branches of different chain length. Often branches with carbon number of six or higher is accounted for as LCB, as the chemical shift differences of longer branches are not high enough to be differentiated by NMR.9 However, Zhou et al. recently showed that branches of up to seven carbon atoms could be identified.17
A powerful technique that can correlate branching and molecular weight is triple detector SEC. In addition to a concentration detector, a viscosimetric detector (Visco) and a static light scattering detector (multiangle laser light scattering, MALLS) can be connected to the SEC instrument.18,19 The Visco detector provides the intrinsic viscosity of the sample, which can be plotted as a function of molecular weight to give the so-called Mark–Houwink plot. Intrinsic viscosity increases linearly with molecular weight. Branching leads to a reduced coil radius in solution, which gets more pronounced with increased branching, and leads to reduced viscosities and consequently to a deviation from the linear case. Static light scattering gives the conformation plot (radius of gyration as a function of molecular weight), which can be used to detect LCB due to a reduced coil radius for branched samples.18–22
While these methods are particularly sensitive to LCB, SCB is typically investigated by performing crystallization-based fractionation techniques, such as crystallization analysis fractionation (CRYSTAF)23,24 and thermal fractionation by successive self-nucleation and annealing (SSA).25–29 These techniques are based on the assumption that molecules with a similar degree of branching will co-crystallize, mostly independent of polymer chain length. The resulting polymer crystallinity distribution can then be linked to the branching distribution via correlating the crystallization temperature and the branching content.27–29 However, one has to keep in mind that these methods only apply to the crystalline part of a given material.
Recently, it was shown that a deeper insight into branched PE can be gained by applying a multiple preparative fractionation approach, which consists of preparative fractionation according to crystallinity (preparative temperature rising elution fractionation, pTREF) and preparative fractionation according to molecular weight by either preparative molar mass (pMMF)5,30,31 or preparative solvent gradient fractionation (pSGF).9 The resulting fractions were still heterogeneous with respect to either branching or molecular weight, respectively, and cross-fractionation by combining pTREF/pSGF with triple detector SEC, CRYSTAF and SSA proved to enable a deeper insight into the molecular microstructure.30–32
Unfortunately, most of the studies dealing with the detailed characterization of PE have been conducted on industrial samples. This has the advantage that samples are easily available in large quantities and that they possess industrially relevant microstructures and properties. At the same time, the correlation of microstructure with corresponding reaction and process conditions is not possible, because details of industrial process conditions are usually not disclosed. Moreover, if certain microstructural characteristics (such as branching) are to be investigated systematically or if sensitivities of analytical methods are of interest, suitable samples with a known preparation history are not available.
For this reason, the aim of this publication is the advanced analytical characterization of a systematic set of LDPE samples, which were produced in a mini-plant scale autoclave reactor under well-known and deliberately chosen continuous operation conditions. Polymerization conditions were close to industrial ones with pressures of 2000 bar and a temperature of 250 °C. The produced samples vary with respect to chain length, as they were produced with varying feeds of chain transfer agent, as well as with respect to conversion. These two parameters are interesting because different chain lengths lead to different molecular radii, while different conversions directly translate into different polymer concentrations for the bulk polymerization of LDPE, which are expected to result in diverging transfer reactions and thus different branching.7 The samples were analyzed via comprehensive analytical characterization and the results gave insights into the detection limits and sensitivities of the different techniques. Finally, the findings were directly connected to the polymerization conditions.
Experimental
1,2,4-Trichlorobenzene (TCB), white quartz sand (50–70 MESH), 2-ethoxyethanol, propionic aldehyde and 1,1,2,2-tetrachloro-ethane-d2 used in the study were obtained from Sigma Aldrich. TCB was distilled before use. Chromium(III) acetylacetonate was obtained from Alfa Aesar and xylene from Kimix. Ethene (3.0) was obtained from Airliquide and tert-butyl peroxyactete (50% in aliphates) from Degussa. Irganox 1010 was obtained from Ciba Speciality Chemicals, Switzerland.The investigated PE samples were produced in a high-pressure mini-plant at the TU Darmstadt, Germany. Polymerization experiments were performed in a continuously operated 100 mL stirred autoclave reactor under steady-state conditions. For all experiments the ethylene mass flow was kept constant at 0.556 g s−1, the residence time in the reactor was 90 s and the pressure was held at 2000 bar. More details about the set-up and experimental details can be found in literature.5 Summarized reaction conditions for the experiments performed in the mini-plant can be found in Table 1 and the molecular properties of the samples are given in Table 2. The linear PE reference, PE 105K was purchased from the American Polymer Standard Corporation (Mn = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 g mol−1, Mw = 105000 g mol−1, dispersity 10.1).
400 g mol−1, Mw = 105000 g mol−1, dispersity 10.1).
| Experiment | T (°C) | ṁ CTA (g h−1) | ṁ initiator (10−3 g h−1) | Conversion (%) |
|---|---|---|---|---|
| 03-1 | 245 | 3.64 | 15.01 | 13.9 |
| 03-3 | 245 | 2.45 | 15.01 | 13.2 |
| 03-4 | 240 | 1.22 | 15.01 | 12.5 |
| 04-2 | 250 | 2.92 | 9.01 | 7.9 |
| 05-1 | 248 | 2.92 | 0.00 | 0.7 |
| 06-3 | 245 | 2.92 | 7.10 | 6.2 |
| Sample |
M
na (kg mol−1) |
M
wa (kg mol−1) |
Đ | LCB/1000Cb | SCB/1000Cb | LCB + SCB/1000Cb |
|---|---|---|---|---|---|---|
| a As determined by SEC-IR. b As determined by 13C-NMR. | ||||||
| 03-1 | 17.9 | 142.9 | 7.97 | 2.9 ± 0.2 | 9.1 ± 0.7 | 12 ± 1 |
| 03-3 | 22.1 | 215.7 | 9.76 | 1.8 ± 0.3 | 9.8 ± 1.3 | 12 ± 2 |
| 03-4 | 27.2 | 318.2 | 11.69 | 1.9 ± 0.2 | 9.3 ± 1.0 | 11 ± 2 |
| 04-2 | 18.5 | 113.5 | 6.12 | 1.2 ± 0.2 | 8.8 ± 0.8 | 10 ± 1 |
| 05-1 | 18.9 | 53.8 | 2.84 | 0.08 ± 0.02 | 8.3 ± 0.9 | 8 ± 2 |
| 06-3 | 18.9 | 129.9 | 6.86 | 0.58 ± 0.07 | 9.9 ± 0.8 | 10 ± 1 |
| PE105K | 9.8 | 124.2 | 12.7 | — | — | — |
Triple detector SEC
Size exclusion chromatography (SEC) was performed using a high temperature PolymerChar GPC-IR system (Polymer Char Laboratories Ltd, Valencia, Spain) equipped with an IR5 detector in hyphenation with a Wyatt Dawn Heleos II LS detector (Wyatt Technology Corporation, Santa Barbara, USA) and an online Visco H502 viscometer detector (Polymer Char Laboratories Ltd, Valencia, Spain), an Agilent 1200 autosampler (Agilent Technologies, Santa Clara, USA), three Shodex UT 806M columns, one Shodex UT 807 column and a Shodex UT-G guard column (Showa Denko K.K., Kanagawa, Japan). The reproducibility of the system was checked33 and a broadly distributed reference sample (PE5, Dow, Mw = 115 kg mol−1) was measured to ensure reproducibility after any system changes (calibration, columns, etc.). The columns and detectors were operated at 150 °C. The eluent TCB was distilled prior to its use and stabilized with 2,6-D-tert-butyl-4-methyl phenol (butylhydroxy-toluene, BHT, 0.0125%). A flow rate of 1 mL min−1 was applied. All samples (2 mg) were dissolved in TCB (2 mL) for 1–2 hours at 160 °C and 0.19 mL of the solutions were injected. To ensure dissolution, the solutions were checked to be clear and homogeneous with respect to flow behavior and the prefilter was checked not to be blocked. Linear polystyrene standards (PSS Polymer Standards Service GmbH, Mainz, Germany) with narrow molecular weight distribution (MWD) were used for the IR calibration. Ten standards with a peak maximum ranging from 474 g mol−1 to 4410000 g mol−1 were used. Universal calibration was used to evaluate the Visco detector data. The Mark–Houwink coefficients for linear PE were taken from literature (K = 53 × 10−3 mL g−1 and a = 0.703).34 A mixture of linear PE samples was used as a linear reference for Mark–Houwink plots in the GPC One software (Polymer Char, Valencia, Spain). The mixture consists of five PE samples: 1.02 mg PE1 (PSS, Mw = 181 kg mol−1), 1.05 mg PE2 (University of Konstanz, Mw = 169 kg mol−1), 1.03 mg PE3 (University of Konstanz, Mw = 593 kg mol−1), 0.96 mg PE4 (University of Konstanz, Mw = 1.196 kg mol−1) and 4.48 mg PE5 (Dow, Mw = 115 kg mol−1) were dissolved in 8 mL TCB. The molecular weight dependent radii of gyration of linear PE, which are used as the reference when calculating branching ratios from MALLS data, were taken from Wang et al.35 The molecular weight distributions, as well as the IR and Visco data, were evaluated using Polymer Char's GPC One software. The multiangle laser light scattering (MALLS) data were processed with the Astra software (Wyatt Technology, Santa Barbara, USA, version 6.1.7.17) and a dn/dc value of −0.1040 mL g−1 was used for all samples.35
13C-Nuclear magnetic resonance (NMR)
Prior to the 13C-NMR measurements, the samples (120–150 mg) were dissolved in deuterated 1,1,2,2-tetrachloro-ethane-d2 (TCE-d2) (4.0 g). Based on the study of Zhou et al.36 the solvent contained 0.025 M chromium(III) acetylacetonate as a relaxation agent. Dissolution was carried out at 100 °C for 60 min. To ensure good sample dissolution, the samples was pre-dissolved with uniform flow was observed during sample preparation. The mixture was allow to cool to a clear homogeneous solution. 13C-NMR measurements were carried out in 400 MHz Bruker BioSpin NMR spectrometer (Bruker, Billerica, USA) at a resonance frequency of 100 MHz and a temperature of 100 °C. The recycle delay (D1) was chosen to be 10 s in accordance with prior investigations.33,36 A 30° pulse was applied and an inverse-gated decoupling pulse sequence was chosen. The data were evaluated using the software MestreNova by performing a manual phase correction, a baseline correction via a Bernstein polynomial fit of the third order, using an apodization of 1 Hz and integrating the individual peaks manually via the sum method (summing up the area under the curve). The short chain branched density that includes methyl, ethyl, butyl and amyl branches was calculated according to eqn (1). Here, IBx is the signal intensity of the branch with chain length x and is the sum of peak areas of all carbon atoms.
is the sum of peak areas of all carbon atoms.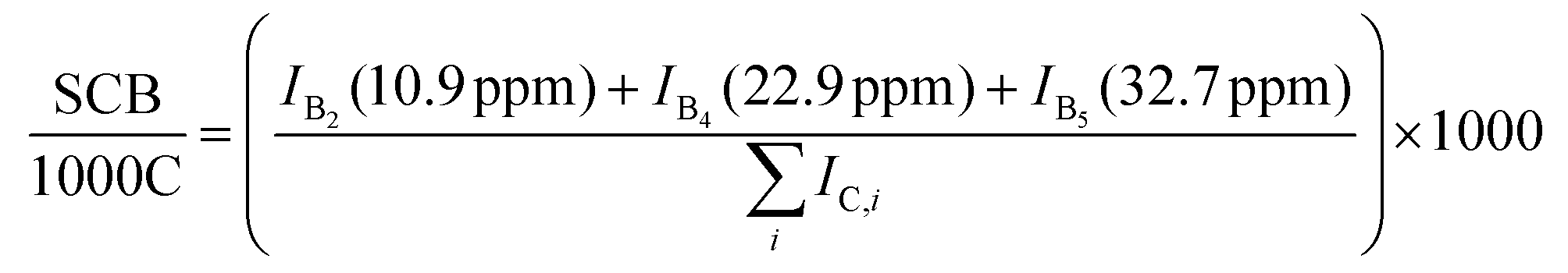 | (1) |
The long chain branched density was calculated using eqn (2), where IB6+ is the signal intensity of all branches with chain length 6 and more and DP is the degree of polymerization as obtained from the number-average molecular weight determined by SEC-visco.
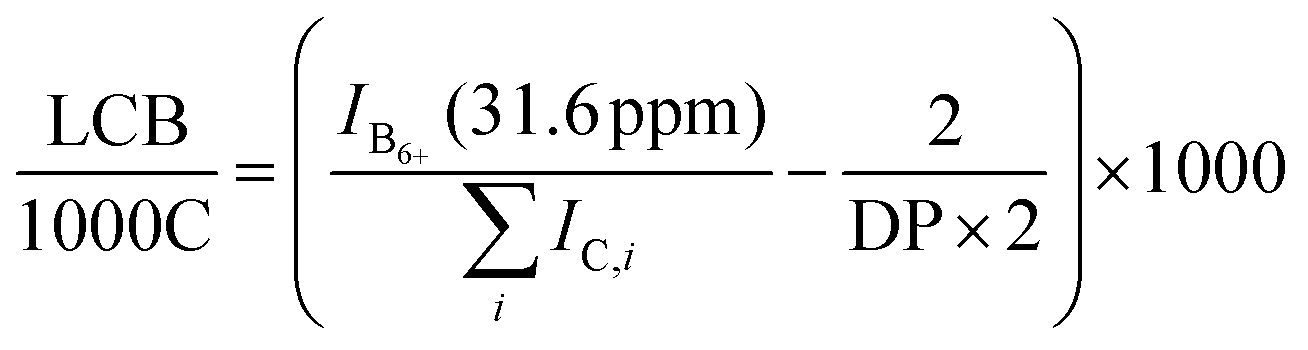 | (2) |
Relative standard derivations (RSD) were calculated according to literature.37 The detailed peak assignment and spectra are given in the ESI.†
Crystallization analysis fractionation (CRYSTAF)
Crystallization from solution analysis was conducted using a 200 Polymer Char (Valencia, Spain) CRYSTAF instrument. Five analytes (17 mg) were simultaneously dissolved in 35 mL TCB in five stainless steel reactors at 160 °C while stirring. Complete dissolution was checked by ensuring 100% sample concentration and regular distribution of the cumulative distribution curves. After complete dissolution of the samples (150 min), the temperature was lowered to 100 °C and stabilized for 60 min. Then, the crystallization step was conducted by slowly reducing the temperature to 30 °C at a steady cooling rate of 0.1 °C min−1 in order to minimize co-crystallization. During the crystallization step, an infrared detector was used to measure the polymer solution concentration as a function of temperature. Differential as well as cumulative chemical composition distributions were recorded.Differential scanning calorimetry (DSC)
The thermal properties of the samples were studied using a TA Instruments Q100 DSC system (TA instruments, New Castle, USA). It was calibrated using an indium metal standard according to standard procedures. Melting as well as crystallization temperatures were determined under the same experimental conditions of a scanning rate of 10 °C min−1 and a temperature range from 10 to 200 °C. All measurements were conducted in a nitrogen atmosphere with a purge flow rate of 50 mL min−1. Polymer samples (5.4 mg) were subjected to a total of three temperature cycles, while the first heating cycle only serves to erase the thermal history of the material. The second cycle (cooling) was used to determine the crystallization temperature and the third cycle (heating) served to investigate the melting temperature. After each cycle, an isothermal step of 2 min was maintained to obtain thermodynamic equilibrium.Successive self-nucleation and annealing (SSA)
The thermal fractionation experiments using successive self-nucleation and annealing (SSA) were conducted in the same instrument and under the same standard conditions as the standard DSC measurements. The following steps were performed to achieve the multiple melting endotherms: In the first step, the thermal history was erased by heating and cooling the sample at a scanning rate of 10 °C min−1 between 0 and 200 °C. Before the cooling cycle started, the temperature was held constant for 5 min. Then, the actual fractionation started: the sample was subjected to a second heating cycle to a selected first self-seeding temperature (Ts,1) based on the region published by Cavallo and others.26,28,29 Then, the temperature was held constant at that temperature for 5 min, before cooling to 1 °C at a rate of 10 °C min−1. Ts,1 was chosen to be 120 °C for all investigated samples. In the third cycle, the sample was heated to a self-seeding temperature (Ts,2), which is lower than Ts,1 by 5 °C and kept isothermally for 5 min before cooling at the same rate. These described heating and cooling cycles are repeated with a stepwise (T = 5 °C) decreasing temperature Ts till 30 °C. After the final step, the sample was heated at a rate of 10 °C min−1 from 0–200 °C to obtain the multiple melting endotherm, which can then be used to determine the methylene sequence length distribution according to Zhang et al.27Polymer fractionation
Some polymer samples were further investigated with respect to their microstructure using fractionation techniques. Preparative temperature rising elution fractionation (pTREF) was used in order to fractionate the samples predominately according to crystallinity and thus branching structure. On the other hand, samples can be fractionated mainly based on molecular weight if a preparative solvent gradient fractionation (pSGF) is applied.Preparative temperature rising elution fractionation (pTREF)
Preparative TREF was carried out using an instrument built in house. A dilute polymer solution (∼1 wt%) was prepared by dissolving 3.0 g LDPE in 300 mL of xylene in a glass reactor at 130 °C under reflux conditions. Irganox 1010 (2 wt%) was added as a stabilizer to prevent thermo-oxidative degradation of the LDPE during the fractionation process. After complete dissolution, the solution was stirred at 130 °C for 2 h to allow the polymer chains to be in an equilibrium state. Then, the reactor was quickly filled with a preheated crystallization support (quartz sand) in order to prevent uncontrolled recrystallization of the polymer. The oil bath was then cooled to ambient temperature (∼25 °C) at a rate of 1 °C h−1, to ensure slow and controlled crystallization of the LDPE under TREF conditions. Subsequently, the polymer-coated sand was transferred into a stainless steel column that was placed in a modified GC oven for temperature-controlled elution. A continuous flow of preheated xylene was used to elute the polymer fractions (∼500 mL per fraction) as the oven temperature was raised at predetermined intervals. A total of four fractions were collected: at 70 °C, 75 °C, 80 °C and 85 °C. The polymer was recovered from the eluted solutions using a rotary evaporator at a vacuum pressure of 30–40 mbar and a bath temperature of 80 °C. Then, the fractions were precipitated and washed with acetone and dried under vacuum to constant weight. The fractionation process was conducted thirteen times to ensure enough material was fractionated.Preparative solvent gradient fractionation (pSGF)
Preparative solvent gradient fractionation was conducted using xylene as the good solvent and 2-ethoxyethanol as the non-solvent. Approximately 9.0 g of the sample stabilized with 2.0 wt% Irganox 1010 were dissolved in 400 mL of xylene at 130 °C and then stirred for 1 h under identical conditions. Then the solution was titrated with an excess of 2-ethoxyethanol (400 mL; yields solvent-non-solvent ratio of the first fraction). The non-solvent was added slowly with the help of a dropping funnel to avoid uncontrolled precipitation. After a complete dosage of the non-solvent, the resulting dispersion was stirred under identical conditions for 1 h. The polymer solution was then introduced into a column packed with glass wool and sea sand, which was tempered at 130 °C. The column was allowed to equilibrate at 130 °C for another hour before being emptied to collect the first fraction. Subsequently, a series of preheated solvent/non-solvent mixtures with increasing xylene contents were added to the column, allowed to equilibrate and then emptied and collected one at a time. A total of 5 fractions were collected with solvent:non-solvent, xylene:2-ethoxyethanol ratios of 50, 54, 58, 60 and 100 vol%. A total time of 90 min was spent with each step in order to minimize the kinetic effects of the dissolution process on the fractionation. Eluted fractions (800 mL) were dried using a rotary evaporator, precipitated in acetone and dried under vacuum to constant weight. The total time required to complete one experiment was approximately 14 hours.
Results and discussion
Bulk samples
Previous studies on the molecular heterogeneity of LDPE often worked with industrial samples.14 This is reasonable because such products are of interest for the market and they are available in large quantities. However, in the present case for a better correlation between the sample preparation and the resulting molecular structure, a set of LDPE samples is investigated, which was produced in a high-pressure mini-plant autoclave reactor. The autoclave reactor was operated in continuous mode and samples were collected under stationary reaction conditions.7The major advantage of this approach is that polymerization conditions can be chosen freely within physically reasonable ranges and are recorded during the experiment. Consequently, differences in the resulting polymeric microstructures and polymer properties can be directly linked to varying polymerization conditions. For this study, a set of six samples was chosen, which was produced under conditions close to industrial relevance. The idea was to produce samples with systematically varying molecular weights. This can be achieved experimentally by adjusting the chain-transfer agent (propionic aldehyde) feed. Samples 03-1, 3-3 and 3-4 represent polymers, which were produced under very similar reaction conditions with respect to temperature, initiator feed and conversion, but decreasing CTA amounts (compare Table 1). Additionally, experiments were performed, which should give polymers with similar average molecular weights (similar CTA feed rates) but in varying conversion ranges. Experimentally this is more challenging, as the initiator feed stream, external heating via the mantle, polymerization rate, reactor temperature and resulting conversion are strongly interconnected. Nonetheless, Table 1 shows that the experiments were conducted successfully and samples with conversions from 0.7% (05-1), 6.2% (06-3), 7.9% (04-2) to 13.9% (03-1) were obtained. From a microstructural point of view, they are interesting because they are expected to show different degrees of long-chain branching. As the high-pressure ethylene polymerization is a bulk polymerization, the conversion is equal to polymer concentration. With increasing polymer concentration, chain transfer to polymer is enhanced and more LCBs should be formed.
The molecular structure and chain heterogeneity of these mini-plant samples were investigated by means of triple detection SEC, 13C NMR, CRYSTAF and SSA. While 13C NMR provides average branches densities, triple detection SEC produces molecular weights and polymer dispersities (Đ) (see Table 2) and subsequently long-chain branching (LCB) distribution characteristics as a function of molecular weight. The MWD curves given in Fig. 1a compare molecular weight characteristics of a set of selected samples produced at 1.2, 2.5 and 3.6 g h−1 CTA mass flow rates and 0.015 g h−1 initiator feed stream (hereafter referred to as CTA samples). As is seen, the peak maxima molecular weight is shifting towards lower molecular weights as the CTA feed stream is increased. This is expected because an increase in CTA concentration ([CTA]) is the typical way of controlling molecular weight in radical ethylene polymerization. The CTA has the ability to donate hydrogen atoms to growing macroradicals, leading to chain termination, while regenerating new radical sites for continuous propagation. Increasing [CTA] increases the rate of chain transfer to the low molecular weight species CTA and subsequently decreases chain length. Added to this, the MWD curves show shoulders in the higher molecular weight region, which become increasingly pronounced with decreasing CTA feed or increasing sample molecular weight (see the marked area in Fig. 1a). This is a characteristic observation for autoclave LDPEs and can be attributed to increased side reactions (transfer to polymer reactions, as well as growth of terminal double bonds) resulting in the formation of high molecular weight highly branched macromolecules.33 As is seen, the concentration of the high molecular weight molecules decreases with an increase in CTA mass flow rate and subsequently the weight-average molecular weights and molecular weight dispersities decrease. On the other hand, Fig. 1b presents MWD curves of a set of selected samples produced at a constant CTA feed stream (2.92 g h−1) and selected four initiator mass flow rates ranging from 0 to 0.015 g h−1 (further referred here as initiator samples). In this case, a very similar peak maximum molecular weight was recorded for all the samples. The number-average molecular weight varies from 17.9 to 18.9 kg mol−1 (see Table 2). However, an increase in the initiator feed stream resulted in an increase in conversion. Such changes are attributed to increased radical concentrations and thus higher polymerization rates.
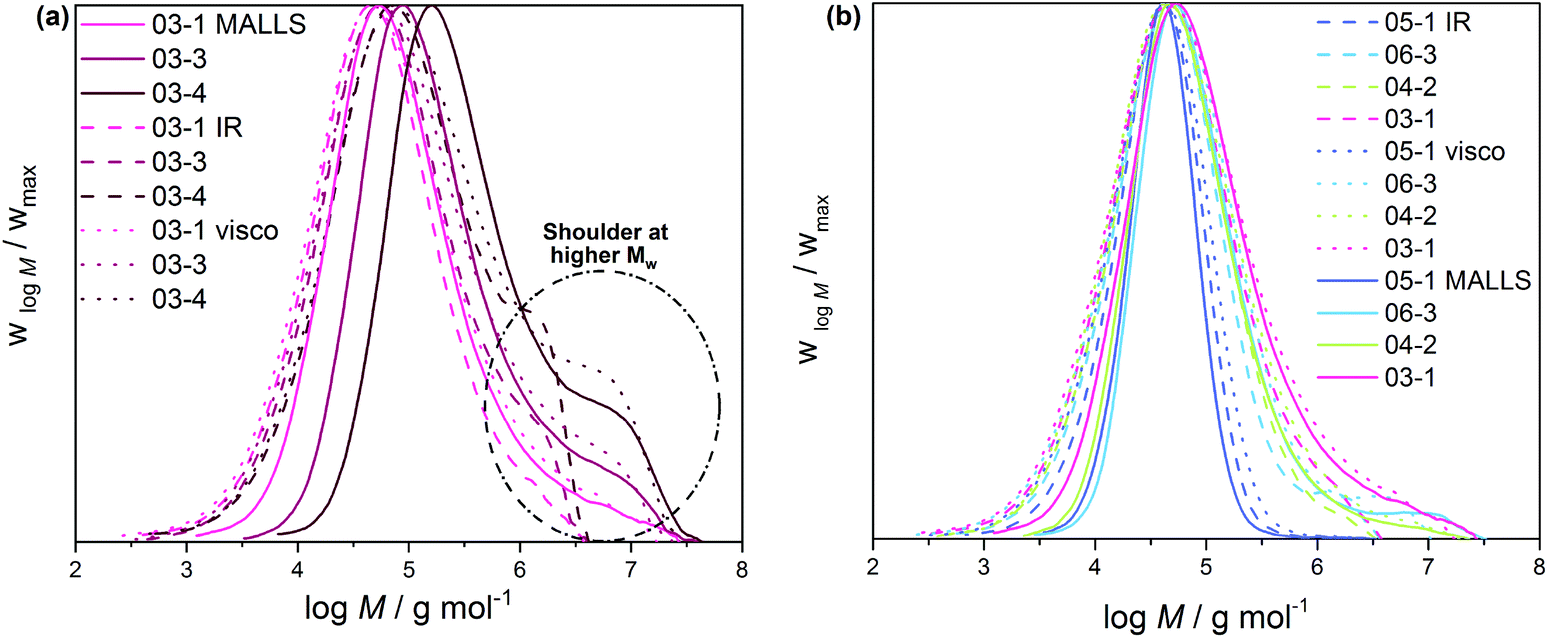 | ||
| Fig. 1 Comparison of molecular weight distributions measured by IR (dashed lines), Visco (dotted lines) and MALLS (solid lines) detectors coupled to SEC. (a) Samples with increasing average molecular weights and (b) samples with conversions from 1% to 13% are compared. Detector delays are corrected. | ||
According to the NMR results in Table 2, both sets of samples display comparable average SCB densities, indicating that CTA and initiator concentrations have no major effect on backbiting. This is in line with the kinetic mechanism of backbiting as a unimolecular reaction and the fact that all polymerizations were conducted at the same temperature (250 °C). In addition to influencing molecular weight and conversion through decreasing CTA and increasing initiator feed streams, respectively, both methods show a marked influence on the molecular weight dispersity as indicated by the broadening of the MWD curves.
To further investigate the influence of [CTA] and [initiator] on the molecular structure, the SEC fractions were analyzed for branching using multiangle laser light scattering (MALLS) and viscometer (Visco) detectors. While MALLS has the ability to measure absolute molecular weights and mean square radii of gyration (Rg), the Visco detector measures molecular weights based on universal calibration and intrinsic viscosities ([η]) of polymers. These quantities (Rg and [η]) always complement each other since their magnitudes are defined by molecular size in solution. It is important to note that the size/radius of macromolecules is influenced by both molecular weight and LCB. As for linear macromolecules, the coil radius is directly correlated to molecular weight. Based on the conformation and Mark–Houwink (M–H) relationships for an ideal linear sample given in eqn (3)35 and eqn (4),34 respectively, a linear correlation between Rg or [η] and molecular weight is confirmed for the linear reference sample PE 105K as shown in Fig. 2. Here, a M–H and conformation intercept (k) of 0.056 and 0.067, respectively, were recorded, while a M–H slope (α) of 0.702 and a conformation slope (a) of 0.503 were obtained.
| Rg = kMa (for an ideal linear PE, k = 0.029, a = 0.57) | (3) |
| [η] = kMα (for an ideal linear PE, k = 0.053, α = 0.703) | (4) |
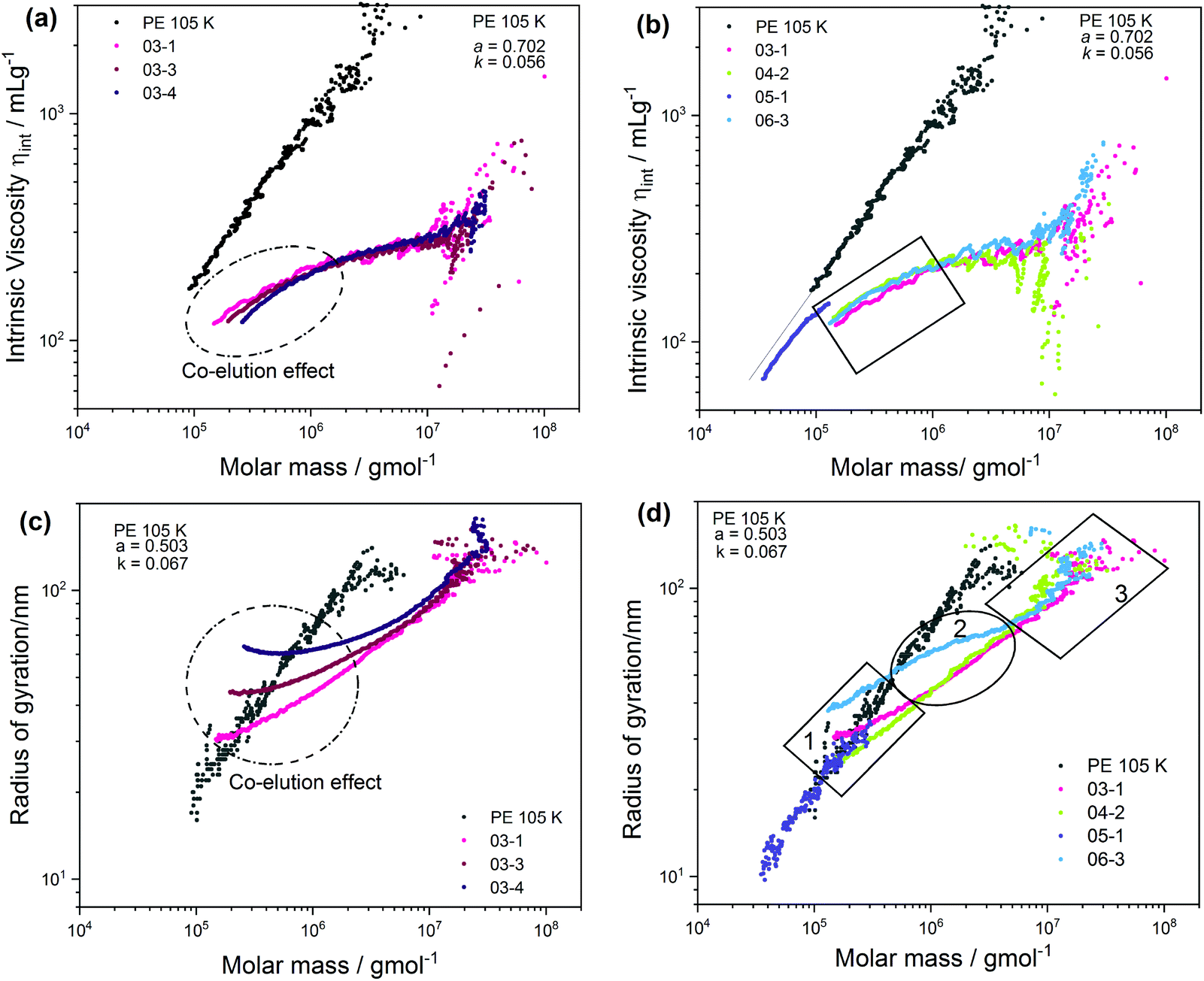 | ||
| Fig. 2 Comparison of Mark–Houwink plots in (a) and (b), measured by SEC-Visco and radii of gyration (c) and (d) measured by SEC-MALLS as a function of molecular weight. Data also compare samples with an increasing average molecular weight in (b) and (d) and increasing conversions from 1% to 13% in (a) and (c). | ||
These values are comparable to those of an ideal linear sample reported in literature.35 In the case of branched macromolecules, chain contraction is enhanced by the presence of LCBs. In such instances, the linear conformation and M–H relationships are not obeyed, especially at the higher molecular weight region, where chain contraction is enhanced due to increasing LCB. These differences have been exploited for many years to (1) distinguish branched macromolecules from their linear counterparts and (2) compare branched polymers of different degrees of LCB. The latter is considered here and the [η] and Rgversus molecular weight plots given in Fig. 2a and c are comparing LCB of the CTA samples. Except for the low molecular weight region where subtle differences are seen, all three samples display superimposing M–H plots, which is more noticeable at the high molecular weight region, indicating similarity in their LCB structure. Hypothetically, the subtle difference seen in the circled area in Fig. 2a is often characterised by the co-elution or anchoring effect in SEC, and is assumed to occur when long side branches of highly branched high molecular weight molecules penetrate the pores of the stationary phase. This cause a delay and force the molecules to elute late at higher elution volumes, which correspond to the low molecular weight region.38 Another or additional source of errors might arise from data treatment.39,40 Due to the high molecular weight sensitivity of the MALS detector, this phenomenon is more pronounced with the conformation plots as indicated in the circled area in Fig. 2.
Interestingly, the co-elution effect is enhanced by molecular weight since anchoring is seen to increase with an increase in the sample molecular weight. This trend is expected because the rate of chain transfer to polymer reactions to generate LCB is significantly increased with an increase in molecular weight. The quantification of LCB by the SEC triple detection concur well with those of 13C NMR shown in Table 2, where, relatively similar average LCBs are reported for the CTA samples.
Also presented in Fig. 2b are M–H plots of the initiator samples. Sample 05-1 is a branched sample that was produced at zero initiator feed stream. Although a slight deviation from the linear plot is seen at the higher molecular weight area, the bulk of the sample displays linear M–H characteristics at a lower intercept to PE 105k, indicating little or no LCB. The shift of the M–H plot to a lower k is indicating the presence of SCB, which is in accordance with 13C NMR results shown in Table 2.
The MH plots of the other initiator samples (03-1, 04-2 and 06-3) deviate more from the linear plots. The deviation becomes increasingly pronounced with an increase in the sample molecular weight and polymer dispersity (see the area in the rectangular box). This observation is contrary to the MH principles of linear polymers and may be associated with the interplay between the effect of molecular weight and branching, and is ascribed to the co-elution effect.
A more complex trend is seen with the plots in Fig. 2d. In this case, the conformation plots are divided into three molecular weight regimes, which are indicated as 1, 2 and 3, corresponding to the low, mid and high molecular weight regions. The strong deviation at the low molecular weight region (1) is due to the co-elution effect. Clear differences in the LCB are seen at the mid molecular weight region (2), where a lower degree of LCB is seen for sample 06-3, which concur well with those of NMR. In the high molecular weight region (3), no conclusion is drawn due to a low signal-to-noise ratio. For information regarding branching distribution heterogeneity, the samples were further investigated by means of CRYSTAF and SSA in solution and in melt, respectively. Typically, CRYSTAF fractionates semicrystalline polymers in TCB based on crystallizability at temperatures between 100 and 30 °C.
For more details on the principle of this technique and its applications to the analysis of LDPE, consult the work of Eselem Bungu et al.5,30,41
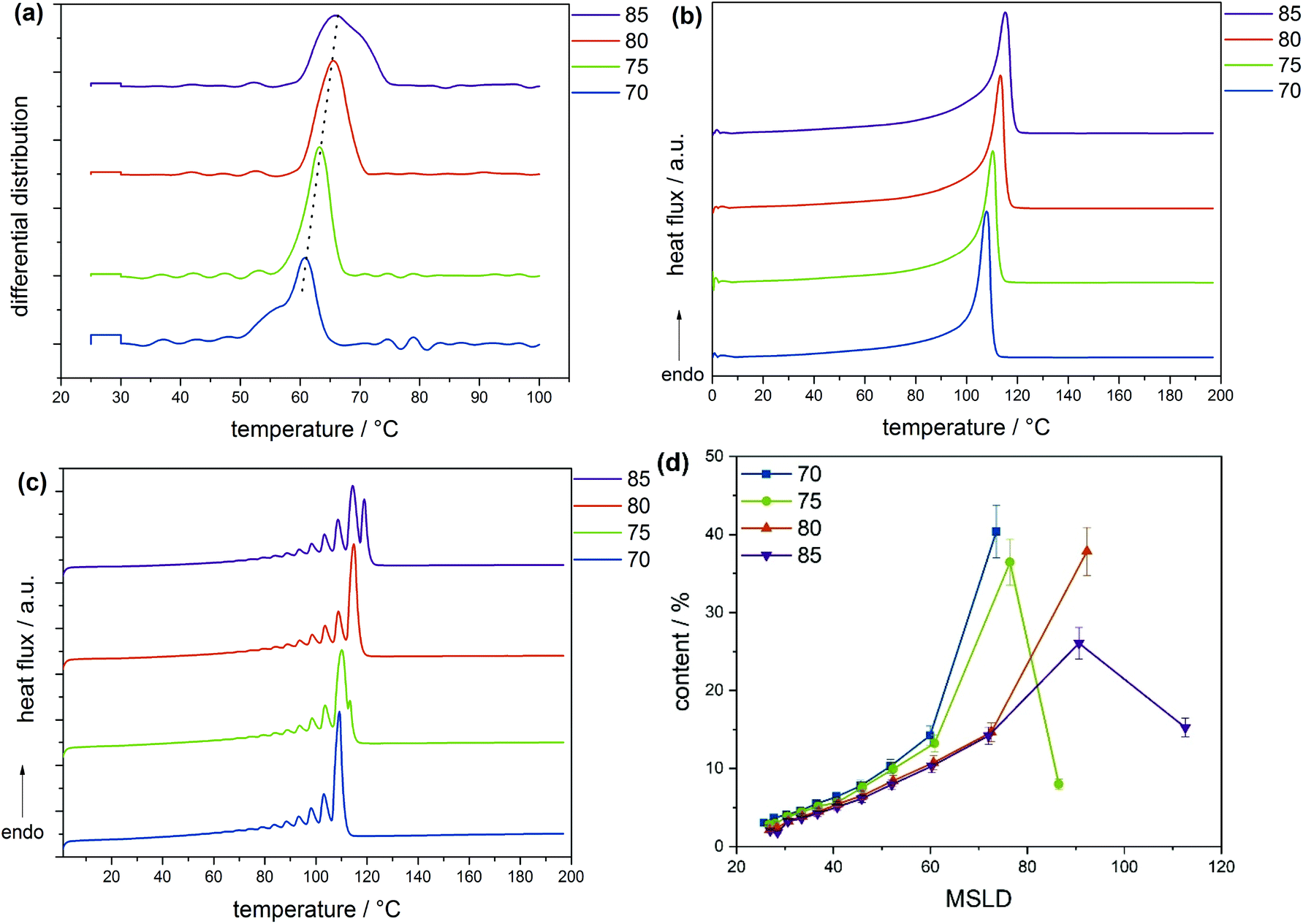
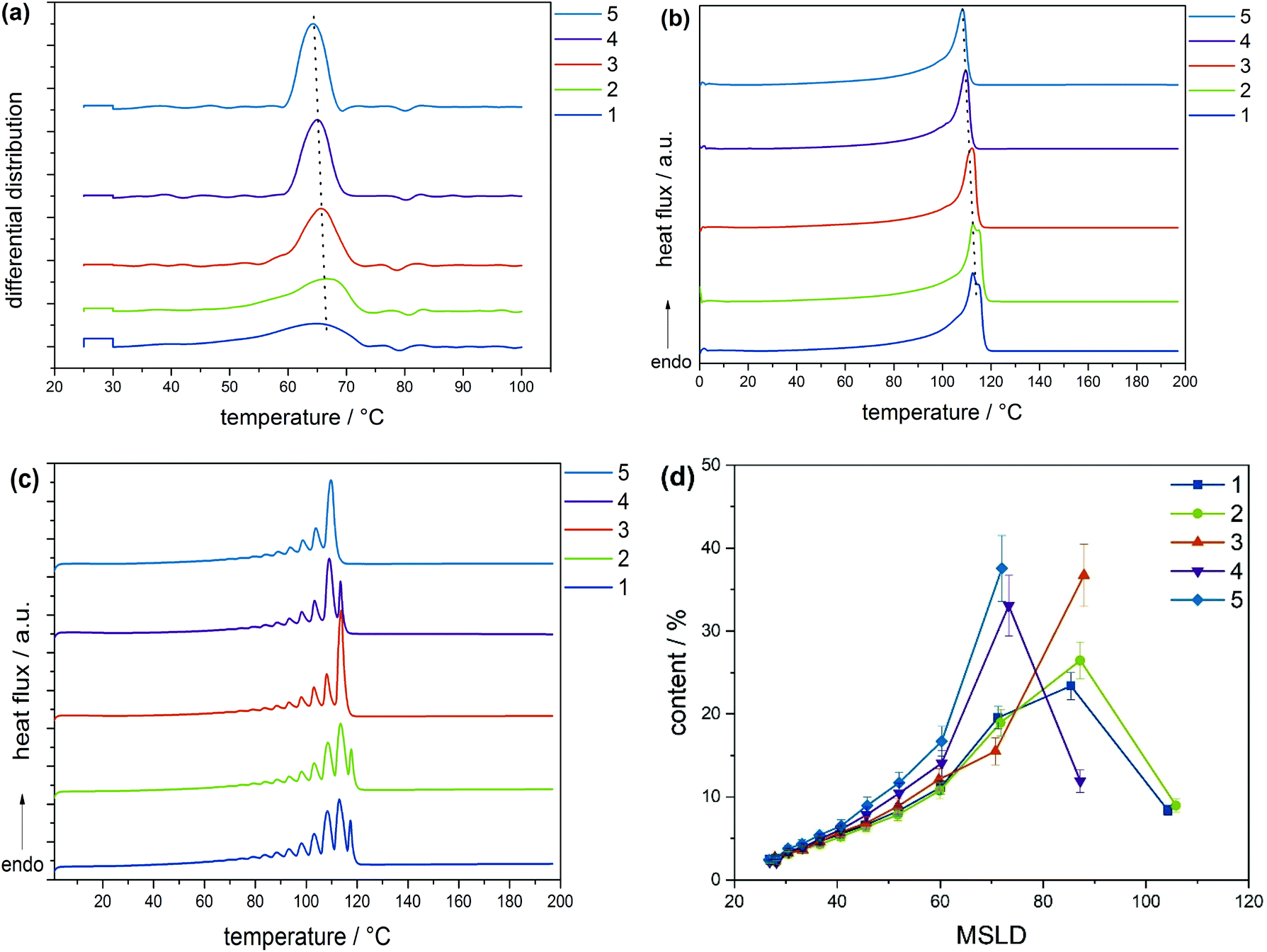
According to the CRYSTAF findings shown in Fig. 3a, the CTA samples demonstrate broad crystallization profiles with peak temperatures around 64 °C, which are characteristic of branched polymers.5 In detailed inspection, sample 03-1 displays a shoulder at lower temperatures. A decrease in intensity of the shoulder is seen with 03-3, which completely disappears for 03-4, indicating an increase in the branching homogeneity. This happens as sample molecular weight and molecular weight dispersity increase due to decreasing CTA feed stream. Considering that, the total average branching is almost identical for the CTA samples; the appearance of a shoulder may be associated with the formation of an increasing amount of low molecular weight molecules of a higher degree of SCB. The remark made here is that, with increasing molecular weight, materials with higher uniformity regarding the number of chain ends are produced, while the lower molecular weight species contain fractions with more chain ends and, therefore, a higher heterogeneity. Also presented in Fig. 3b are CRYSTAF plots of the initiator samples. As is seen, sample 06-3, which exhibits the lowest conversion/LCB, is displaying a unimodal CRYSTAF profile. As the initiator mass flow rate is increased, corresponding to an increase in conversion/LCB, simultaneous growth in a shoulder at a lower crystallization temperature and a slight shift in the peak maximum towards a lower crystallization temperature is seen. Considering this, all three samples display comparable SCB densities, a higher conversion translates to a higher level of LCB and subsequently an increase in the total branching density, and therefore, an increase in the branching heterogeneity. In addition to the CRYSTAF findings, thermal fractionation by SSA was used to investigate and compare chain heterogeneities of the respective sets of samples. This was achieved by observing the crystal size distribution, which is synonymous to branching distribution as a function of melting temperature as was previously shown.29 Presented in Fig. 4 are the SSA melt endotherms and methylene sequence length distribution (MSL) plots of the CTA and initiator samples. As is seen, each endotherm displays 10 distinguishable melting peaks between 70 and 115 °C in agreement with previous studies.29–31 Each peak representing a group of polymer chains with similar thermodynamic stabilities having comparable crystallizable MSL. This can be attributed to similarities in the average branching or chain crystallinity. However, a variation in the number of peaks and/or peak intensities for peaks with similar crystal size characteristics is observed and used to draw a qualitative and quantitative structural comparison between different sample sets. The individual SSA plots shown in Fig. 4a can be used to compare the CTA samples. The three samples display similar numbers of peaks, while peaks with similar MSL characteristics vary in intensity.
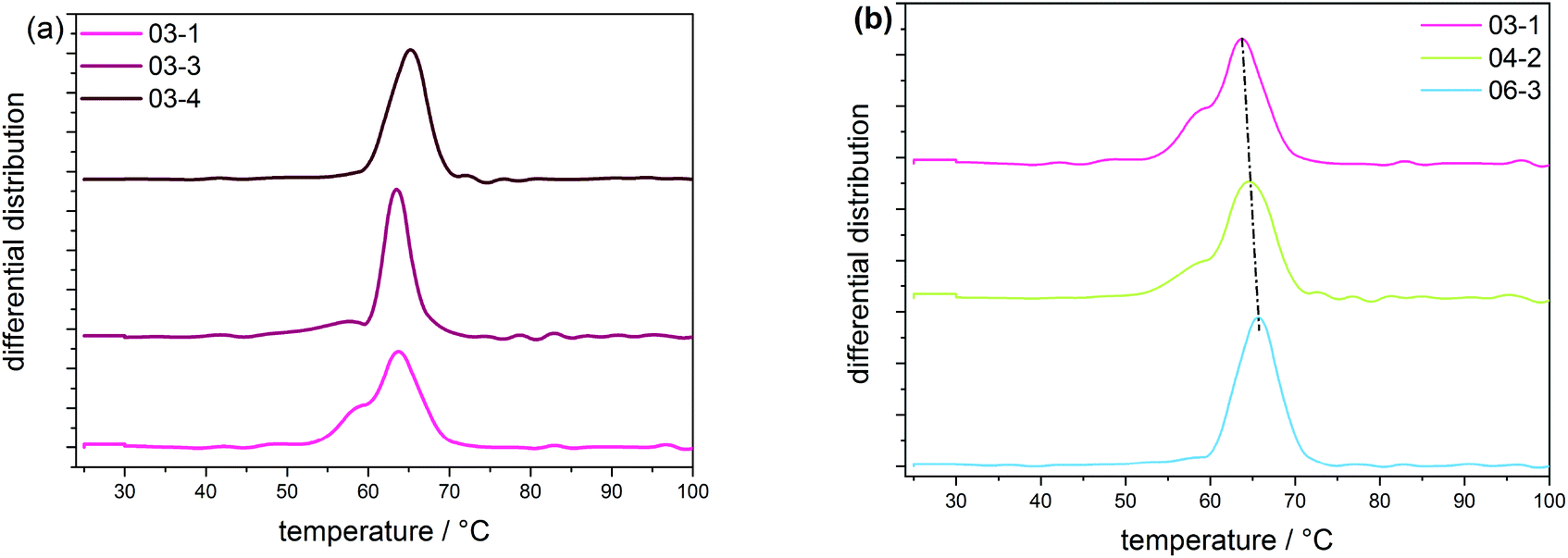 | ||
| Fig. 3 CRYSTAF measurements of samples with (a) increasing average molecular weight and (b) with conversions from 1 to 13%. | ||
 | ||
| Fig. 4 SSA measurements (a) and (b) and methylene sequence length distributions (MSLD) plots (c) and (d) of samples with conversions from 1% to 13% (a) and (c) and increasing average with molecular weights (b) and (d). MSLD plots were obtained from SSA measurements (lines drawn to guide the eye). | ||
These differences are ascribed to differences in the crystal size composition. For more clarity, the MSL and percentage peak content of each peak were measured from the respective peak temperatures and areas, respectively, using the approach previously introduced in literature and references therein.29,31 Plots of MSL versus seed content of the CTA samples are compared in Fig. 4c. They were calculated from the melt endotherms according to literature.27,29 Even though each sample shows a comparable distribution in MSL, the seed content for seeds with similar MSL varies significantly amongst the samples as shown in the area indicated in the rectangular box. As is seen, the seed content for seeds with MSL of approximately 72 carbon atoms (C72) increases from sample 03-1 to 03-4 and concur well with increasing sample molecular weight. Although very similar SCB densities are reported, a decreasing total branching density is seen with increasing seed content. On the other hand, an increase in the seed content is seen with an increase in total branched points and subsequently a decrease in sample molecular weight for the C90 seeds.
This is counter-intuitive because a higher amount of longer MSL molecules should correlate to higher crystallinity and not higher branching. However, taking into account that a higher [CTA] was applied for this sample, the higher C90 content could be ascribed to a higher amount of low molecular weight chains with relatively linear backbone or longer MSL. It is clear that 03-1, which displays the lowest molecular weight, constitutes the highest amount of C90 chains and the lowest of C72 chains, indicating a higher degree of the crystallinity. The decreasing trend for the C90 seeds from sample 03-1 to 03-4 correlates well with the decreasing CTA feed stream, correspondingly, and concurs with the decreasing shouldering in CRYSTAF. Taken into account that all three CTA samples report similar branching, the visible inhomogeneity at the low-temperature low crystalline region in CRYSTAF for samples 03-1 and 03-3 may be attributed to solubility effects induced by the increasing amount of low molecular weight components, that are formed at increasing CTA feed streams. From previous studies, these low molecular weight fractions have shown to exhibit longer MSL and higher melting temperatures even though they display lower CRYSTAF crystallization temperatures.30,31 In a similar way, the MSLD plots of the initiator samples are compared in Fig. 4d. Considering that these samples possess comparable molecular weights but differ significantly in their LCB and subsequently total branching, any variation in the crystal size amount could be affiliated to differences in the degree of branching. As indicated in the rectangular box, the content of the C72 seeds decreases with decreasing branching density and increases with an increase in sample molecular weight. This is an indication that at higher molecular weight, molecules exhibit a higher degree of branching. In the case of the C90 seed, a reverse trend is seen. i.e., an increase in the branching density leads to an increase in the C90 seed content. The C90 seeds contain predominantly the low molecular weight molecules of higher crystallinity and are not necessary influenced by branching. The higher C90 content recorded for sample 03-1 could be influenced by the higher [CTA] flow stream (3.64 g h−1) used, implying a higher amount of the low molecular weight species were formed, in comparison to the case of 06-3 and 04-2 where a low CTA flow stream is applied (2.96 g h−1). For these sample sets, there is a strong interplay between the effects of molecular weight and branching on the crystal size distribution, which makes it challenging to correlate the effect of increasing conversion to the MSL of the polymers. For a clearer interpretation of the polymer microstructure, comprehensive characterization via preparative fractionation techniques will be performed on a selected sample from each sample set. Offline analysis of the different fractions will provide in-depth knowledge on the polymer microstructure.
Preparative fractionation and analysis of mini-plant LDPE
In order to gain deeper insight into the microstructural properties of these mini-plant samples, preparative fractionation was performed on representative samples of each of the different sample sets. Analysis by preparative fractionation is a comprehensive approach of providing in-depth knowledge on polymer microstructure. This approach is used to narrow down the molecular complexity of synthetic polymers by providing fraction library sets of samples with relatively narrow branching and/or molecular weight distributions and of varying crystallinities (branching) and/or molecular weight, respectively. In this case, sample 04-2 of the initiator sample set was selected for pTREF fractionation. This sample displays a molecular weight dispersity of 6.12 and by TREF principle, a range of fractions with varying total branched density and narrow branching distributions are obtained based on crystallizability. 04-2 was preferred for pTREF because of (1) its narrow molecular weight dispersity. In order to minimize the effect of broad MWD on melt rheology, it was imperative to select a sample with relatively narrow molecular weight dispersity.This helps to reduce the possibility of obtaining fractions of very broad molecular weight dispersities and, therefore, minimizes the effect of MWD, while enhancing the impact of branching on the rheological properties. (2) A suitable amount of sample was available for fractionation. For a successful non-linear rheology analysis to be achieved, it requires a substantial amount of material (∼3 g). Based on this, it was paramount to select a sample that could provide suitable amounts of each fraction to enable further rheology measurements. However, measurements regarding the rheology of these fractions is the subject of another study. During the TREF process, four fractions were collected at 70, 75, 80 and 85 °C and were further analyzed by means of triple detection SEC, CRYSTAF, DSC and SSA. While the 70 °C fraction contains macromolecules eluting between 30 and 70 °C, the 85 °C fraction comprises of macromolecules eluting between 80 °C and 140 °C. The percentage composition by mass and the thermo-physical properties of the fractions are summarized in Table 3. In addition, the MWD curves, as well as plots of SCB distribution as a function of molecular weight are compared in Fig. 5a. The SCB distribution shown here was determined by IR coupled to SEC. Although 13C-NMR is the more precise method with respect to absolute numbers, SEC-IR is capable of providing branching information as a function of molecular weight as shown here. It gives semi-quantitative branching information and provides branching density trends over the molecular weight distribution and compare branching between different samples. As is seen, the fractions display unimodal MWD curves with an irregular trend in peak molecular weight as the fractionation temperature increases, indicating the low molecular weight selectivity in the TREF process. With the exception of the 70 °C fraction that displays a molecular weight dispersity of ∼7, the other fractions display relatively narrow molecular weight dispersities when compared to the bulk sample. In addition, the variation in the degree of branching is in accordance with TREF principles i.e. a decrease in the degree of branching with an increase in the fractionation temperature, except for the 85 °C fraction, which shows a higher degree of SCB at the lower molecular weight region. To highlight differences in the LCB characteristics, the MH plots of the fractions are compared in Fig. 5b. On one hand, the plots are overlaid at the low molecular weight region, indicating similarities in the branching structure. At the high molecular weight end, a small shift towards the linear plot is seen for the 85 °C fraction, which is attributed to a decrease in LCB. The mid temperature TREF fractions (75 and 80 °C) overlay on each other but deviate further away from the linear and from the 85 °C fractions, indicating a higher degree of branching. Further information on the LCB characteristics was extracted from the conformation plots presented in Fig. 5c. In this case, plots of the 80 and 85 °C fractions overlay across all molecular weights, indicating fractions with comparable LCB conformation. However, plots of the 75 and 70 °C fractions are seen to deviate further away from the linear plot and towards lower radii of gyration indicating an increase in the degrees of LCB at the low molecular weight region with decreasing fractionation temperatures. Nonetheless, the plots overlay at the high molecular weight region, indicating similarity in the branching characteristics. These results also confirm previous observations that the light scattering technique shows greater sensitivity towards detecting smaller differences in LCB when compared to the viscometer technique. The data from the conformation plots were used to calculate the branching index g using the relationship in eqn (5),35 which states that the ratio of 〈Rg〉2 of a branched and a linear molecule of the same molecular weight is less that 1 (<1) and decreases with an increase in the LCB (1 > g > 0). Based on this relationship, plots of branching ratio g as a function of molecular weight are compared in Fig. 5d. A general trend of decreasing g with an increase in molecular weight is demonstrated.
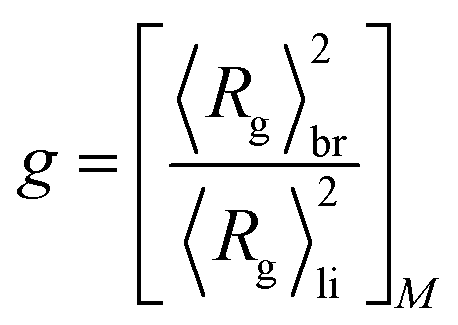 | (5) |
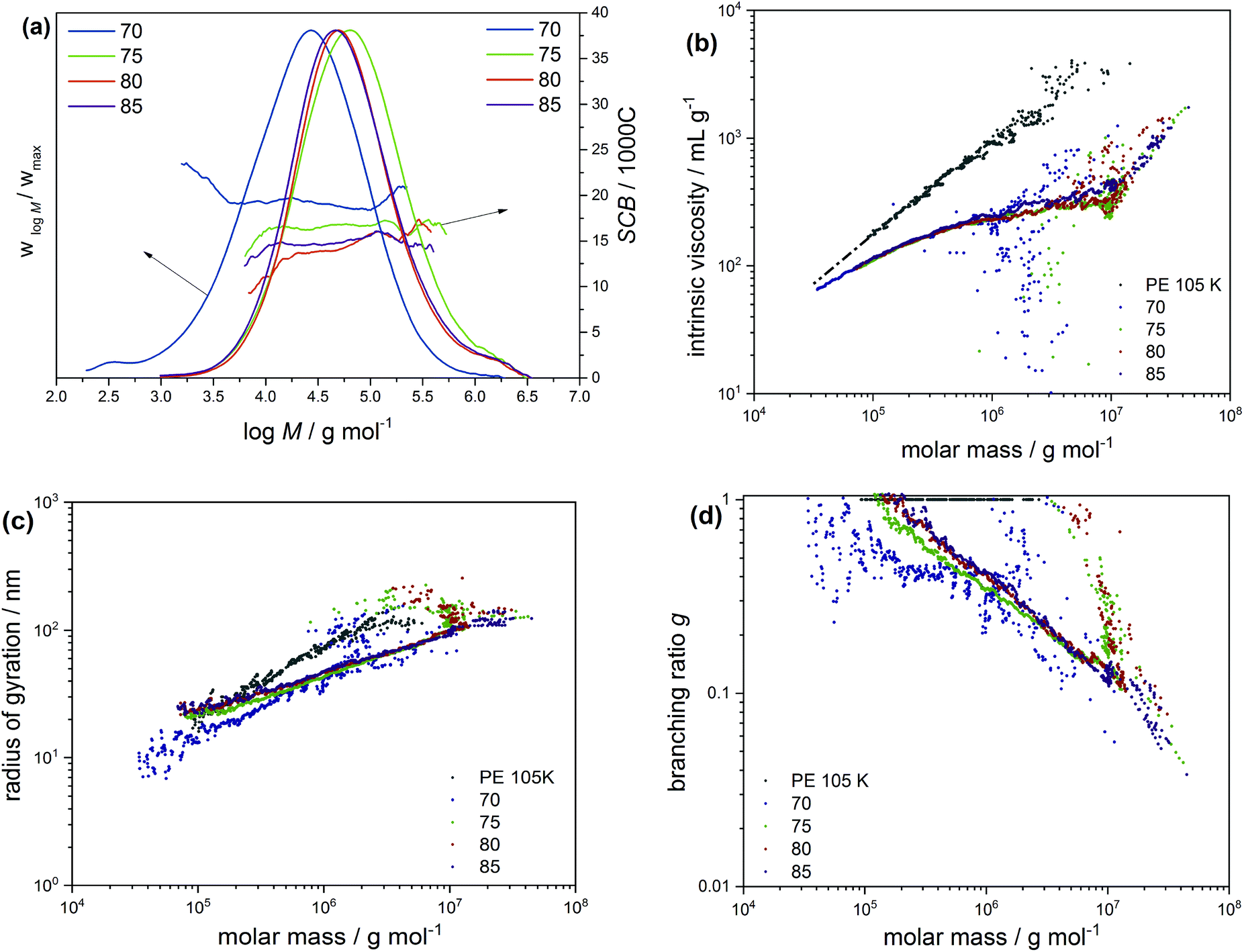 | ||
| Fig. 5 (a) Molecular weight distributions and short-chain branching (SCB/1000C) expressed as a function of molecular weight and determined by SEC-IR. Also illustrated in (b)–(d) are the Mark–Houwink plots, radii of gyration and branching ratios, respectively, demonstrating LCB distribution of the TREF fractions of sample 04-2 as a function of molecular weight. Correspondingly, these data were measured by SEC-Visco and SEC-MALLS. | ||
| Fraction | 70 | 75 | 80 | 85 | 04-2 |
|---|---|---|---|---|---|
| a As determined by SEC-IR. | |||||
| Yield (%) | 24.5 | 33.3 | 32.7 | 8.0 | 100 |
| Yield (g) | 5.95 | 8.06 | 7.93 | 1.93 | — |
| M n (kg mol−1) | 7.2 | 33.6 | 32.1 | 29.9 | 18.5 |
| M w (kg mol−1) | 50.7 | 147.1 | 124.7 | 125.5 | 113.5 |
| Đ | 7.1 | 4.4 | 3.9 | 4.2 | 6.1 |
| T m (DSC) (°C) | 107 | 110 | 113 | 114 | 110 |
| T c (CRYSTAF) (°C) | 60 | 63 | 66 | 66 | 64 |
| SCB/1000Ca | 19.7 ± 1.2 | 16.6 ± 0.7 | 14.4 ± 1.9 | 14.8 ± 0.7 | 19.4 ± 1.3 |
As shown in the low molecular weight region, a decrease in LCB is observed with an increase in the fractionation temperatures from 70 to 80 °C. However, plots of the 80 and 85 °C fractions look identical over the molecular weight range, which is attributed to similarity in the branching structures. Offline analysis of the TREF fractions in solution and melt was conducted by means of CRYSTAF and DSC, respectively. In this section, CRYSTAF crystallization behaviour of the TREF fractions was monitored in TCB and the individual plots are compared in Fig. 6a. As is seen, the 70 °C fraction displays a bimodal crystallization profile as shown by the shoulder at lower temperatures. This is expected since the fraction was collected over a temperature range of 40 °C. Similar bimodal characteristics are seen for the 85 °C fraction, as indicated by the shoulder at higher crystallization temperatures. As was mentioned earlier, this fraction was collected over a broad temperature range of 50 °C. In comparison, the 75 and 80 °C fractions, which were each collected at temperature intervals of 5 °C display unimodal and narrow crystallization profiles. The soluble component, which consists of the highly branched and/or low molecular weight species, is higher in the 70 °C fractions as indicated by the rectangular shape at 30 °C. A general trend of increasing peak crystallization temperatures is seen as the TREF elution temperature increases indicating a decrease in branching. These results concur well with the SEC-IR5 results shown in Fig. 5a, where an increase in the degree section, CRYSTAF crystallization behavior of the TREF fractions was monitored in TCB and the individual plots are compared in Fig. 6a. As is seen, the 70 °C fraction displays a bimodal crystallization profile as shown by the shoulder at lower temperatures. This is expected since the fraction was collected over a temperature range of 40 °C. Similar bimodal characteristics are seen for the 85 °C fraction, as indicated by the shoulder at higher crystallization temperatures. As was mentioned earlier, this fraction was collected over a broad temperature range of 50 °C. In comparison, the 75 and 80 °C fractions, which were each collected at temperature intervals of 5 °C display unimodal and narrow crystallization profiles. The soluble component, which consists of the highly branched and/or low molecular weight species, is higher in the 70 °C fractions as indicated by the rectangular shape at 30 °C. A general trend of increasing peak crystallization temperatures is seen as the TREF elution temperature increases indicating a decrease in branching. These results concur well with the SEC-IR5 results shown in Fig. 5a, where an increase in the degree of SCB is found as a function of deceasing TREF temperatures. The melting characteristics of the fractions were also investigated and the melting endotherms are presented in Fig. 6b. While all the fractions display unimodal melting profiles, the peak melting temperature varies with an increase in the fractionation temperature, indicating an increase in the crystal sizes, which is synonymous to decreasing branched density. For more details regarding the intermolecular crystal size distribution, the fractions were subjected to thermal fractionation by means of SSA. This technique is best known for segregating polymer chains with similar crystallizable methylene sequences. SSA plots of the fractions are compared in Fig. 6c. As is seen, the peak melting temperature for the peaks representing the highest population in each fraction increases with an increase in the fractionation temperature. In a situation where such peaks exhibit similar melting temperatures as in the case of the 80 and 85 °C, a new peak with a lower peak intensity emerges for the higher TREF fraction at a higher melting temperature. This subsequently led to an increase in the total number of peaks indicating an increase in crystal size heterogeneity. For more detailed comparison, plots of MSL versus percentage seed content (peak content) are compared in Fig. 6d. It was found that the MSL for peaks with the highest population increases with an increase in the fractionation temperature corresponding to a decrease in branching. This result correlated well with the TREF principle, which is known to fractionate macromolecules according to their crystallizabilities or degrees of branching. In the case of the CTA sample set, the very broad molecular weight sample 03-4 with a molecular weight dispersity of 11.7 was selected and was fractionated by means of preparative solvent gradient fractionation (pSGF). Presented in Fig. 7a are the MWD curves of the SGF fractions displaying a linear increase in the peak maxima molecular weight as the SGF fraction number increases from 1–5. With the exception of fraction 5 that exhibits molecular weight bimodality with a molecular weight dispersity of approximately 4, the other fractions display narrow unimodal MWD curves with molecular weight dispersities ranging between 1.7 to 2.5 based on the absolute molecular weight. Interestingly, the SCB distribution plots demonstrate comparable branching characteristics as functions of molecular weight below C60 decreasing with increasing TREF temperature indicating a poor selectivity towards branching.
 | ||
| Fig. 6 (a) Chemical composition distribution as determined by CRYSTAF, (b) melting endotherms as measured via DSC, (c) SSA melting endotherms and corresponding MSLD (d) of TREF fractions of sample 04-2 (lines drawn to guide the eye). With increasing fractionation temperatures, MSL increases indicating a higher crystallinity of the material. | ||
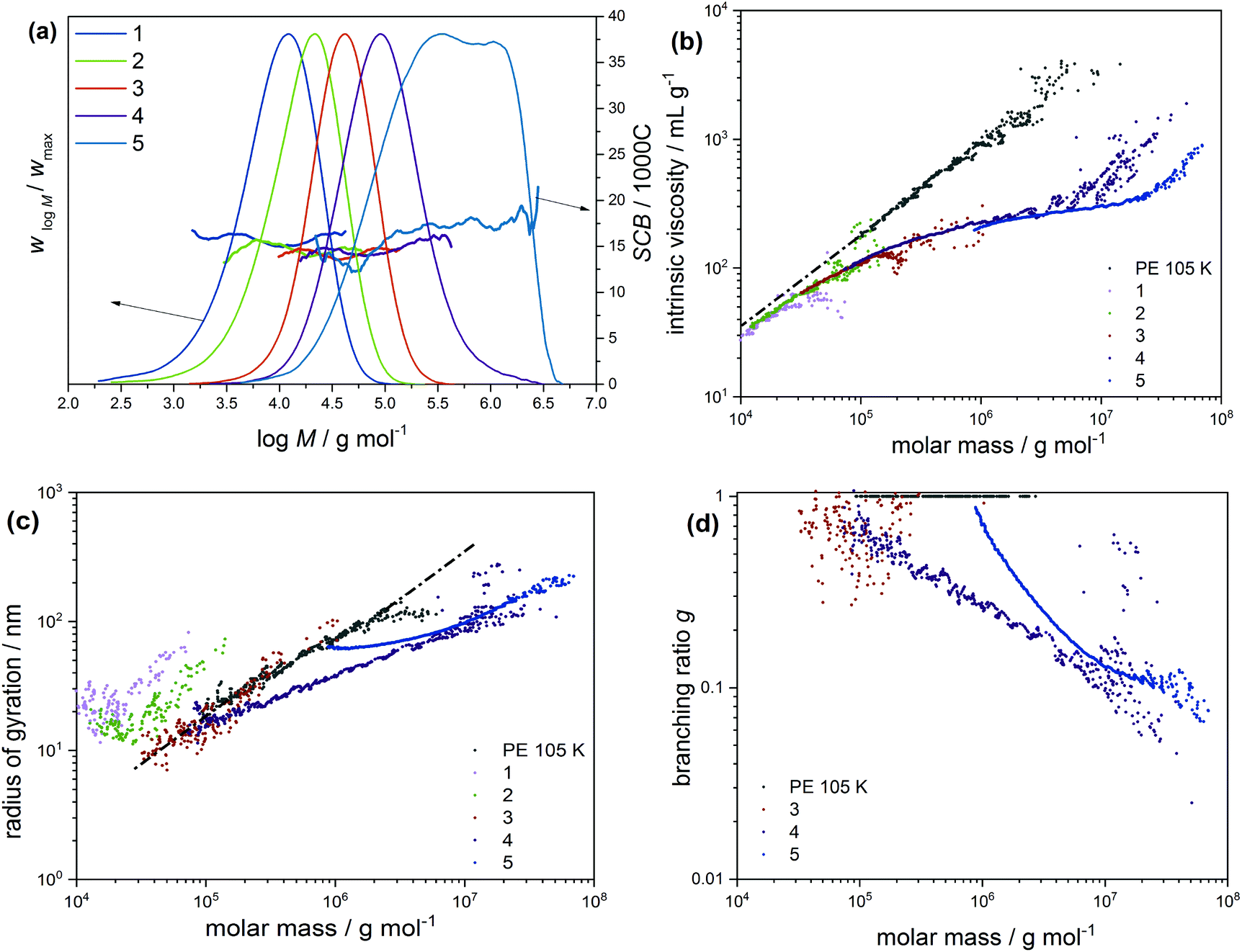 | ||
| Fig. 7 (a) Molecular weight distributions and short-chain branching as a function of molecular weight for the SGF fractions of sample 03-4 as determined by SEC-IR5. Also depicted are the Mark–Houwink (b), the radius of gyration (c) and branching ratio (d) plots illustrating LCB characteristics as a function of molecular weight as determined by SEC-Visco and SEC-MALLS, respectively. | ||
The result of this fractionation clearly satisfies the principles of SGF. The molecular weight characteristics along with the thermo-physical properties are summarized in Table 4. To observe LCB behaviours, the MH plots are compared as shown in Fig. 7b. As is seen in the low molecular weight region, fractions 1 to 3 lie adjacent to the linear plot but at a lower intercept. This behaviour is typical of SCB since the transfer to polymer reaction, which generates LCB, is minimal at the low molecular weight region. Enhanced shifting of the plots away from the linear reference is seen with fractions 4 and 5 and is strictly due to LCB. An increase in the sample molecular weight is indicated by an increase in the deviation from the linear reference. However, subtle differences are found between fractions 4 and 5. To approach these differences with more clarity, the conformation plots were also compared as shown in Fig. 7c. Here, the low molecular weight fractions clearly show linear behaviour while fraction 4 is seen at a lower radius of gyration at the mid molecular weight region. It is important to note that a higher co-elution effect is seen for fraction 5 in this molecular weight region. In the higher molecular weight region, the plots are seen to merge indicating similarity in the LCB. Added to this, plots showing the relationship between molecular weight and branching ratio are compared in Fig. 7d. In this case, the linear polymer has a branching ratio macromolecules at the same molecular weight, fraction 4 exhibits a lower branching ratio as seen at the higher molecular weight region, indicating a higher branching density. The observed differences in the mid molecular weight region are strongly influenced by the co-elution effect and could not be considered for comparison.
| Fraction | 1 | 2 | 3 | 4 | 5 | 03-4 |
|---|---|---|---|---|---|---|
| a As determined by SEC-IR. | ||||||
| Yield (%) | 12.9 | 8.5 | 8.7 | 24.8 | 37.9 | 100 |
| Yield (g) | 3.50 | 2.33 | 2.36 | 6.76 | 10.31 | — |
| M n (kg mol−1) | 5.2 | 9.6 | 29.0 | 61.3 | 153.9 | 27.2 |
| M w (kg mol−1) | 12.8 | 22.2 | 49.1 | 143.8 | 649.0 | 318.2 |
| Đ | 2.5 | 2.3 | 1.7 | 2.4 | 4.2 | 11.7 |
| T m (DSC) (°C) | 112 | 112 | 112 | 109 | 108 | 110 |
| T c (CRYSTAF) (°C) | 65 | 67 | 66 | 65 | 64 | 65 |
| SCB/1000Ca | 15.8 ± 0.5 | 14.6 ± 0.6 | 14.3 ± 0.4 | 14.8 ± 0.7 | 16.8 ± 1.8 | 18.4 ± 2.2 |
In addition, the crystallization and melting distributions of the macromolecules in solution and solid states were investigated by means of CRYSTAF and DSC, respectively. In the solution state, the individual CRYSTAF plots are compared as shown in Fig. 8a. With the exception of sample 1, a drift towards a lower peak crystallization temperatures is seen with an increase in the sample number from 2 to 5, which indicates a small decrease in the degree of branching with increasing sample molecular weight. On the contrary, sample 1, which exhibits a molecular weight of 12.8 kg mol−1, displays a lower peak crystallization temperature and is exempted for the trend. The shift towards a lower crystallization temperature may be ascribed to solubility effects. Analogous to branching, chain ends lead to an increase in solubility. Added to that, the CRYSTAF plots become increasingly broad with a decrease in the sample molecular weight, indicating an increase in the branching/crystal size heterogeneity.
 | ||
| Fig. 8 (a) Chemical composition distribution as determined by CRYSTAF, (b) melting endotherms as measured via DSC, (c) SSA melting endotherms and corresponding MSLD (d) of TREF fractions of sample 03-4. MSL increase slightly with fraction number/molecular weight, which can be attributed to increasing branching in the high molecular weight region (lines drawn to guide the eye). | ||
In the solid state, the melt endotherms of the fractions are compared as shown in Fig. 8b. Just as in the case of CRYSTAF, a decrease in peak melting temperatures is observed with an increase in the sample molecular weight. As is seen, the melting peaks of the low molecular weight fractions 1 and 2 display two peak melting temperatures indicating two distinct polymeric species with higher compositional heterogeneity. These observations concur well with literature. For detailed analysis of the crystal size distributions, the fractions were subjected to thermal fractionation by means of SSA. The DSC–SSA plots are given in Fig. 8c. The number of peaks increases with decreasing fraction molecular weight, indicating increasing crystal size heterogeneity. In the case, in which the same number of peaks is recorded as for fractions 3 and 4, the peak intensity for the seeds at higher temperature increases with a decrease in the fraction molecular weight, indicating an increase in crystallinity. For a more detailed look, plots of MSL versus seed content are compared as shown in Fig. 8d. For seeds with MSL below C60, the seed content is found to increase with an increase in the fraction molecular weight, corresponding to an increase in the degree of branching. Generally, the MSL for seeds with the highest seed content shows a general decrease with increasing sample number, in addition to the decreasing number of seeds, indicating a higher branching structure with increasing branching homogeneity as the sample molecular weight increases. This result correlates well with the CRYSTAF results and also shows that a higher amount of linear components exhibits lower molecular weight as was previously reported.5,9,30
Conclusions
Within this study, a set of LDPE samples was investigated, which was systematically polymerized under well-defined conditions in a high-pressure mini-plant setup. Samples close to industrial relevance were produced at varying CTA feed (CTA samples) and at varying initiator feed (initiator samples) which resulted in systematically varying molecular weights and conversions, respectively. This gave the possibility to characterize LDPE samples with known polymerization history and link the molecular structure to the production conditions. Differences in molecular weights and dispersities were recorded readily via high-temperature triple detector SEC and results followed the expected trends. Branching was investigated via13C-NMR measurements, the Mark–Houwink and conformation plots, which were gained from Visco and MALLS detectors coupled to SEC. The low conversion sample (less than 1% conversion) was found to exhibit little or no LCBs by the 13C-NMR, Visco and MALLS measurements. This can be explained by the enormously reduced transfer to polymer reaction at very low polymer concentration during polymerization. All other samples showed LCB densities between 0.5 and 3 LCB/1000 carbon atoms. Determination of absolute branching densities with 13C NMR measurements was successful but reliable LCB differences between the samples were difficult to observe with Visco or MALLS analysis. While Visco and MALLS are highly sensitive to the presence of long-chain branching, it seems they are less sensitive to differentiate smaller differences in branched density as observed here. However, the Visco detector is powerful in detecting SCBs characteristics for all the investigated samples. Sample heterogeneity and crystallizability very investigated by means of CRYSTAF, DSC and SSA and significant differences could be observed. As a general trend, a lower average molecular weight and a higher conversion lead to samples with a higher microstructural heterogeneity with respect to branching distribution. At the same time, it proved to be very challenging to correlate the influence of conversion to the microstructure of the bulk samples, as they are differing strongly both on MWD as well as branching degrees.To tackle this task, selected samples were fractionates preparatively and the fractions were analyzed in accordance to the bulk samples. The TREF fraction collected at 70 °C showed a higher SCB and a higher microstructural heterogeneity, which was confirmed by triple detector SEC, CRYSTAF, DSC as well as SSA. The fractions collected at higher temperatures were found to be similar with respect to LCB, as was seen by Visco and MALLS measurements, but proved to be more homogeneous with respect to the crystallizabilty (CRYSTAF, DSC, SSA). In contrast, the fractions obtained by SGF where similar with respect to their SCB, but exhibited strongly increasing molecular weights as well as increased LCB with increasing fraction number. The interesting observation drawn from comparing the SSAs and MSLDs of the SGF fractions was a decrease in sample heterogeneity with respect to crystallinity with increasing fraction number, which is synonymous with an increase of molecular weight. For the investigated samples an increase of molecular weight was found to correspond to an increase of LCB, which can be explained by increased transfer to polymer reactions with increasing chain length. This means that low molecular weight materials have a higher crystal size heterogeneity and probably higher crystallinity. This behavior may be attributed to the lower degree of LCBs and consequently longer MS backbones. This finding helps to interpret and understand the results obtained for the bulk samples produced at different conversions: for high conversions but low molecular weights, the effect of molecular weight can exceed the branching effect.
Overall, the possibilities and limitations of various techniques available for the comprehensive analytical characterization of semi-crystalline LDPE were successfully applied and evaluated on a systematic set of mini-plant samples. Another powerful method to investigate long-chain branching in polymers is rheological characterization. The rheological characterization and modelling of the mini-plant samples and especially the preparative fractions will be the focus of a forthcoming publication.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge partial funding of the work by the National Research Foundation (NRF) of South Africa (P. S. E. Bungu) and Fond der chemischen Industrie (FCI) of Germany (K. M. Zentel). We thank Stefan Mecking (University of Konstanz) for providing linear PE samples.Notes and references
- G. Luft, Chem. Unserer Zeit, 2000, 34, 130–199 CrossRef.
- K. S. Whiteley, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011 Search PubMed.
- J. B. P. Soares, T. F. L. Mckenna, Polyolefin Reaction Engineering, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2012 Search PubMed.
- D. S. Achilias, C. Roupakias, P. Megalokonomos, A. A. Lappas and E. V. Antonakou, J. Hazard. Mater., 2007, 149, 536–542 CrossRef CAS PubMed.
- P. S. Eselem Bungu and H. Pasch, Polym. Chem., 2017, 8, 4565–4575 RSC.
- M. Busch, Macromol. Theory Simul., 2001, 10, 408–429 CrossRef CAS.
- K. M. Zentel, J. Degenkolb and M. Busch, Macromol. Theory Simul., 2020, 2000047 Search PubMed.
- P. S. Eselem Bungu and H. Pasch, Polym. Chem., 2018, 9, 1116–1131 RSC.
- Y. Xue, S. Bo and X. Ji, J. Polym. Res., 2016, 23, 131 CrossRef.
- T. Macko, R. Brüll, Y. Zhu and Y. Wang, J. Sep. Sci., 2010, 33, 3446–3454 CrossRef CAS PubMed.
- D. E. Axelson, G. C. Levy and L. Mandelkern, Macromolecules, 1979, 12, 41–52 CrossRef CAS.
- E. F. McCord, W. H. Shaw and R. A. Hutchinson, Macromolecules, 1997, 30, 246–256 CrossRef CAS.
- M. Pollard, K. Klimke, R. Graf, H. W. Spiess, M. Wilhelm, O. Sperber, C. Piel and W. Kaminsky, Macromolecules, 2004, 37, 813–825 CrossRef CAS.
- K. Klimke, M. Parkinson, C. Piel, W. Kaminsky, H. W. Spiess and M. Wilhelm, Macromol. Chem. Phys., 2006, 207, 382–395 CrossRef CAS.
- M. Parkinson, K. Klimke, H. W. Spiess and M. Wilhelm, Macromol. Chem. Phys., 2007, 208, 2128–2133 CrossRef CAS.
- W. Liu, D. G. Ray and P. L. Rinaldi, Macromolecules, 1999, 32, 3817–3819 CrossRef CAS.
- Z. Zhou, S. Pesek, J. Klosin, M. S. Rosen, S. Mukhopadhyay, R. Cong, D. Baugh, B. Winniford, H. Brown and K. Xu, Macromolecules, 2018, 51, 8443–8454 CrossRef CAS.
- I. Suárez and B. Coto, Eur. Polym. J., 2013, 49, 492–498 CrossRef.
- M. I. Malik and H. Pasch, Prog. Polym. Sci., 2014, 39, 87–123 CrossRef CAS.
- P. Debye, J. Appl. Phys., 1944, 15, 338–342 CrossRef CAS.
- B. H. Zimm, J. Chem. Phys., 1948, 16, 1093–1099 CrossRef CAS.
- I. Teraoka, Polymer solutions: an introduction to physical properties, John Wiley & Sons, 2002 Search PubMed.
- B. Monrabal, Macromol. Symp., 1996, 110, 81–86 CrossRef CAS.
- S. Anantawaraskul, J. B. P. Soares and P. M. Wood-Adams, in Polymer Analysis Polymer Theory, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, vol. 182, pp. 1–54 Search PubMed.
- M. Müller, PhD thesis, Niedersächsische Staats-und Universitätsbibliothek Göttingen, 2005.
- A. J. Müller, R. M. Michell, R. A. Pérez and A. T. Lorenzo, Eur. Polym. J., 2015, 65, 132–154 CrossRef.
- M. Zhang and S. E. Wanke, Polym. Eng. Sci., 2003, 43, 1878–1888 CrossRef CAS.
- D. Cavallo, A. T. Lorenzo and A. J. Müller, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 2200–2209 CrossRef CAS.
- P. S. Eselem Bungu, K. Pflug and H. Pasch, Macromol. Chem. Phys., 2020, 2000095 CrossRef CAS.
- P. S. Eselem Bungu and H. Pasch, Polym. Chem., 2019, 10, 2484–2494 RSC.
- P. S. Eselem Bungu, K. Pflug and H. Pasch, Polym. Chem., 2018, 9, 3142–3157 RSC.
- P. S. Eselem Bungu, K. Zentel, S. Hintenlang, M. Busch and H. Pasch, ACS Appl. Polym. Mater., 2020, 2, 5864–5877 CrossRef CAS.
- I. Neuhaus, PhD Thesis, Technische Universität Darmstadt, 2014.
- S. J. O'Donohue and E. Meehan, in Chromatography of Polymers, American Chemical Society, 1999, vol. 731, pp. 52–65 Search PubMed.
- W.-J. Wang, S. Kharchenko, K. Migler and S. Zhu, Polymer, 2004, 45, 6495–6505 CrossRef CAS.
- Z. Zhou, Y. He, X. Qiu, D. Redwine, J. Potter, R. Cong and M. Miller, Macromol. Symp., 2013, 330, 115–122 CrossRef CAS.
- P. Castignolles, R. Graf, M. Parkinson, M. Wilhelm and M. Gaborieau, Polymer, 2009, 50, 2373–2383 CrossRef CAS.
- S. Podzimek, Light scattering, size exclusion chromatography, and asymmetric flow field flow fractionation: powerful tools for the characterization of polymers, proteins, and nanoparticles, Wiley, Hoboken, NJ, 2011 Search PubMed.
- P. Tackx and J. C. J. F. Tacx, Polymer, 1998, 39, 3109–3113 CrossRef CAS.
- P. Tackx and F. Bosscher, Anal. Commun., 1997, 34, 295–297 RSC.
- P. S. Eselem Bungu, K. Pflug, M. Busch and H. Pasch, Polym. Chem., 2018, 9, 5051–5065 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00089f |
| This journal is © The Royal Society of Chemistry 2021 |