
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRAFT miniemulsion polymerisation of benzyl methacrylate using non-ionic surfactant†
Shang-Pin
Wen
 a,
Qi
Yue
a and
Lee A.
Fielding
*ab
a,
Qi
Yue
a and
Lee A.
Fielding
*ab
aDepartment of Materials, School of Natural Sciences, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: lee.fielding@manchester.ac.uk
bHenry Royce Institute, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 15th March 2021
Abstract
Miniemulsion polymerisation has become widely recognised as a versatile and efficient strategy to prepare well-defined polymer latexes. In this article, we report the synthesis of poly(benzyl methacrylate) (PBzMA) latexes via reversible addition–fragmentation chain-transfer (RAFT) mediated miniemulsion polymerisation using non-ionic Lutensol TO 20 surfactant. The influence of hydrophobe, initiator, and RAFT agent on particle diameter, particle number, rate of polymerisation, and molar mass were investigated systematically. In all cases well-controlled RAFT polymerisations were observed, as indciated by kinetic studies and GPC analysis. Higher concentrations of hexadecane lead to an increase in the of number of particles per unit volume and a decrease in PBzMA latex diameter. The rate of polymerisation with respect to initiator concentration follows a power-law relationship Rp α [initiator]1/2 but only negligible differences in molar mass, molar mass dispersity, and particle diameter values were observed for a range of initiator concentrations. Targeting lower PBzMA degrees of polymerisation increases the overall rate of polymerisation and latex diameter.
Introduction
Reversible-deactivation radical polymerisation (RDRP) is a versatile technique for the synthesis of well-defined polymers and has attracted both academic and industrial attention in the past decade.1–3 The three main RDRP techniques are nitroxide-mediated polymerisation (NMP),4–6 atom-transfer radical polymerisation (ATRP),7,8 and reversible addition-fragmentation chain-transfer (RAFT) polymerisation.9–12 These techniques afford the capability to control radical polymerisations to obtain polymers with narrow molecular weight distributions and complex architectures. RDRPs are typically performed in homogeneous media which often require the use of undesirable volatile organic compounds (VOCs) for the control of heat evolution and viscosity of a given polymerisation. However, RDRP performed in heterogeneous conditions, such as emulsion, miniemulsion or dispersion polymerisation, has gained increasing attention in recent years.13–16 Common advantages of heterogeneous polymerisation include the use of water as a continuous phase (as a more environmentally friendly solvent) and low viscosity of the polymerisation medium due to the formation of discrete polymeric particles.17 In particular, RAFT polymerisation has attracted wide attention in the past few decades because it can be applied to an extensive range of functional monomers and be performed under moderate conditions.10 In addition, it has been demonstrated that RAFT polymerisation can be conducted via various heterogeneous routes such as RAFT emulsion polymerisation18,19 and RAFT miniemulsion polymerisation.20–24Miniemulsion polymerisation is a heterogeneous polymerisation technique widely used to prepare well-defined vinyl polymer latexes.25–28 Miniemulsions are normally defined as aqueous dispersions of relatively stable oil droplets (50–500 nm) prepared by applying shear to a system containing water, oil, surfactant and hydrophobe.29 Miniemulsion droplets are considered metastable but their stability during a miniemulsion polymerisation is crucial because it affects the polymerisation kinetics and resulting latex morphology.30,31 Typical miniemulsion formulations are based on anionic surfactants (e.g. sodium dodecyl sulfate, SDS32) in combination with a selectively oil-soluble hydrophobe (e.g. hexadecane,31 dodecyl mercaptan,33 or reactive alkyl methacrylate34). Nevertheless, the influence of cationic and non-ionic surfactants on miniemulsion polymerisation has been reported. For example, Landfester et al.,35 demonstrated polystyrene latexes synthesised using cetyl trimethylammonium bromide (CTAB) cationic surfactant had similar particle sizes when compared to those prepared using SDS. Hecht et al.,36 investigated miniemulsion polymerisation of styrene using a non-ionic surfactant. It was shown that the non-ionic surfactant had slower adsorption and desorption kinetics than SDS, minimising its presence in the continuous phase. This led to newly formed oligomeric species being incorporated within existing droplets before precipitating and thus nucleation of new particles was not observed.
In RAFT miniemulsion polymerisation, a small amount of hydrophobic chain-transfer agent (CTA) is dissolved in the monomer phase prior to emulsification.37,38 Polymerisation control and latex stability in RAFT miniemulsion polymerisation using conventional RAFT agents has been reported utilising relatively high concentrations of surfactant and hydrophobe.18,39,40 However, the use of RAFT agents in miniemulsion polymerisation is sometimes problematic, causing colloidal instability,41,42 broad particle size distributions,20,43 low polymerisation rates,44,45 low monomer conversions32,46,47 and broad molecular weight distributions.48–51 Furthermore, some RAFT miniemulsion polymerisation systems experience inhibition or retardation of the polymerisation.20,44
Acrylic and methacrylic monomers are crucial for manufacturing of a wide range of polymeric materials for a variety of applications.52,53 Among this group of monomers, benzyl methacrylate (BzMA) is a hydrophobic methacrylic monomer which is often used as a substitute for styrene. For example, in RAFT-mediated PISA,54–56 BzMA acts as water-immiscible monomer which exhibits higher polymerisation rates than styrene,9 minimising unreacted monomer in these formulations.57 Furthermore, a wide range of potential applications of PBzMA have been explored, such as: polymeric optical fibres;58 nanoimprinting lithography;59,60 inkjet inks;61 contact lenses;62 coatings and paints;63,64 adhesives;65,66 microtubes;67 monoliths for capillary electrochromatography;68 colour filter photoresist materials;69 stationary phases in liquid chromatography;70,71 and in ionic liquids.72–74 However, to the best of our knowledge, there are relatively few reports which investigate the effects of the RAFT process on the kinetics of miniemulsion polymerisation of BzMA.
Herein we report the preparation of PBzMA via RAFT miniemulsion polymerisation. More specifically, a series of PBzMA latexes have been prepared by systematically adjusting the concentration of hydrophobe, initiator, and RAFT agent (Scheme 1). This has allowed us to extend the knowledge of the rules which govern latex formation during RAFT miniemulsion polymerisation. In this work, we screened chain transfer agents with different hydrophilicity, and surfactants including a conventional anionic surfactant (SDS) and a series of Lutensol TO non-ionic surfactants to ascertain a suitable surfactant for miniemulsion polymerisation of BzMA. The resulting PBzMA latexes were characterised via gravimetry, DLS, GPC, and TEM. Furthermore, we demonstrate that miniemulsion polymerisations with tuneable polymerisation rate, PBzMA molar mass, number of particles per unit volume, and particle diameter can be conducted by altering the concentration of hydrophobe, initiator, or RAFT agent.
 | ||
| Scheme 1 Synthesis of poly(benzyl methacrylate) (PBzMA) latex via RAFT miniemulsion polymerisation at 70 °C using Lutensol TO 20 non-ionic surfactant. Polymerisations were conducted at a dispersed phase content of 20% w/w. | ||
Experimental
Materials
Sodium dodecyl sulfate (SDS, 98.5%), hexadecane (HD, 99%), and azobisisobutyronitrile (AIBN, 98%) were purchased from Sigma-Aldrich (UK) and used as received. The non-ionic Lutensol surfactants, e.g. Lutensol TO 20, which is a saturated iso-C13 alcohol with an ethoxylation block length of 20 units (iso-C13H27O(CH2CH2O)20H, molar mass = 1000 g mol−1), were obtained from BASF (UK) and used as received. Benzyl methacrylate (BzMA, 98%) was purchased from Alfa Aesar (UK) and passed through a column of activated basic alumina to remove inhibitors and impurities before use. Tetrahydrofuran (THF, HPLC grade) was purchased from VWR International (UK) and used as received. Chloroform-d (CDCl3) was purchased from Cambridge Isotope Laboratories (UK). 4-Cyano-4-(2-phenylethanesulfanylthiocarbonyl)sulfanyl pentanoic acid (PETTC) and bis-(2-phenylethanesulfanyl thiocarbonyl) disulphide (PETTCCP) were prepared in-house using previously published methods (ESI†).75,76 Deionised water was used in all experiments.Synthesis of poly(benzyl methacrylate) via RAFT miniemulsion polymerisation
In a typical protocol, the synthesis of PBzMA300via RAFT miniemulsion polymerisation was conducted at a dispersed phase content of 20% w/w, using: BzMA (7.8 g, 44.087 mmol); PETTCCP (41.4 mg, 0.147 mmol); AIBN (4.8 mg, 0.029 mmol, [CTA]/[initiator] = 5); TO 20 (604.0 mg, 7.8% w/w related to BzMA); HD (185.2 mg, 2.4% w/w related to BzMA); and water (31.4 g). The dispersed and the aqueous phases were prepared separately. The dispersed phase was prepared by thoroughly mixing BzMA, HD, AIBN and CTA (PETTC or PETTCCP, if used) by magnetic stirring until homogeneous. The dispersed phase was then added to the aqueous solution of Lutensol TO 20 non-ionic surfactant under vigorous stirring at 800 rpm for 60 min to form a coarse emulsion. This coarse emulsion was ultrasonicated using an ultrasonic processor CPX-750 (Cole Palmer, maximum output power of 750 W) at an amplitude of 70% for 5 min (10 seconds pulse on and 5 seconds pulse off) in an ice-water bath to prevent overheating during miniemulsification. The obtained miniemulsion was then transferred to a 100 mL two-necked round-bottom flask equipped with a condenser and a nitrogen inlet. The reactor contents were deoxygenated by purging with nitrogen for 10 min at room temperature. After deoxygenation, the round-bottom flask was immersed into a preheated oil bath at 70 °C, corresponding to time zero of the polymerisation. The reaction was heated for 240 min and magnetically stirred at 150 rpm. Samples were periodically withdrawn with a degassed needle from the bottom of the flask to monitor the conversion of monomer and the evolution of molar mass. Polymerisations were quenched by cooling to room temperature and exposing to air. Experimental conditions for all the miniemulsion polymerisations performed in this study are given in Tables 1–3.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[PETTCCP]:[AIBN] = 300:1:2.3
:[PETTCCP]:[AIBN] = 300:1:2.3
| HDa/% w/w | Conversionb/% |
M
n
/kg mol−1 |
M
w/Mnc |
D
h
/nm |
N
p
/× 1014 mL−1 |
R
P
/M s−1 |
R
N
/× 10−15 M s−1 |
|---|---|---|---|---|---|---|---|
| a Relative to BzMA monomer. b Determined by gravimetry. c Determined by THF GPC analysis. d Obtained via DLS, PDI values are indicated in brackets. e Calculated using eqn (S1) (ESI†). f Calculated using eqn (S2) (ESI†). g Calculated using eqn (S3) (ESI†). | |||||||
| 1.2 | 98.5 | 36.0 | 1.31 | 338 (0.06) | 1.40 | 0.34 | 2.45 |
| 2.4 | 98.9 | 36.5 | 1.27 | 330 (0.12) | 1.50 | 0.34 | 2.30 |
| 4.8 | 94.6 | 36.0 | 1.29 | 325 (0.10) | 1.57 | 0.33 | 2.10 |
| 7.2 | 95.8 | 34.2 | 1.29 | 299 (0.07) | 2.02 | 0.33 | 1.65 |
| 9.5 | 94.3 | 34.9 | 1.29 | 296 (0.09) | 2.08 | 0.33 | 1.58 |
:[AIBN] ratio at 70 °C for 240 min. The target DP was 300. Polymerisations were conducted at a dispersed phase content of 20% w/w with the concentration of HD and TO 20 fixed at 2.4% w/w and 7.8% w/w relative to BzMA, respectively
| [PETTCCP]:[AIBN] ratio |
Conversiona/% |
M
n
/kg mol−1 |
M
w/Mnb |
D
h
/nm |
N
p
/× 1014 mL−1 |
R
P
/M s−1 |
RNf/× 10−15 M s−1 |
|---|---|---|---|---|---|---|---|
| a Determined via gravimetry. b Determined by THF GPC analysis. c Obtained via DLS, where PDI values are indicated in brackets. d Calculated using eqn (S1) (ESI†). e Calculated using eqn (S2) (ESI†). f Calculated using eqn (S3) (ESI†). | |||||||
| 10 | 60.0 | 24.3 | 1.33 | 268 (0.08) | 2.91 | 0.21 | 0.72 |
| 5 | 91.8 | 36.7 | 1.28 | 306 (0.15) | 1.88 | 0.32 | 1.70 |
| 2 | 96.4 | 37.0 | 1.28 | 311 (0.08) | 1.78 | 0.34 | 1.89 |
| 1 | 98.5 | 38.2 | 1.25 | 303 (0.11) | 1.93 | 0.34 | 1.78 |
| 0.4 | 98.9 | 36.5 | 1.27 | 330 (0.12) | 1.50 | 0.34 | 2.30 |
:[AIBN] ratio was fixed at 5:1. Polymerisations were conducted at a dispersed phase content of 20% w/w with the concentration of HD and TO 20 fixed at 2.4% w/w and 7.8% w/w relative to BzMA, respectively
| Target PBzMA DP | [PETTCCP]a/mmol L−1 | Reaction time/min | Conversionb/% |
M
n
/kg mol−1 |
M
w/Mnc |
D
h
/nm |
N
p
/× 1014 mL−1 |
R
P
/M s−1 |
R
N
/× 10−15 M s−1 |
|---|---|---|---|---|---|---|---|---|---|
| a Concentration relative to the dispersed phase. b Determined by gravimetry. c Determined by THF GPC analysis. d Obtained via DLS, PDI values are indicated in brackets. e Calculated using eqn (S1) (ESI†). f Calculated using eqn (S2) (ESI†). g Calculated using eqn (S3) (ESI†). | |||||||||
| 200 | 28.6 | 240 | 96.3 | 25.4 | 1.24 | 330 (0.12) | 1.49 | 0.34 | 2.25 |
| 300 | 19.1 | 240 | 91.8 | 36.7 | 1.28 | 306 (0.15) | 1.88 | 0.32 | 1.70 |
| 400 | 14.3 | 240 | 84.9 | 43.8 | 1.27 | 291 (0.09) | 2.18 | 0.30 | 1.36 |
| 700 | 8.2 | 360 | 96.5 | 78.4 | 1.29 | 281 (0.07) | 2.67 | 0.22 | 0.84 |
| 800 | 7.1 | 360 | 90.4 | 78.9 | 1.31 | 275 (0.10) | 2.84 | 0.21 | 0.74 |
Characterisation
Monomer conversions during polymerisations were determined by gravimetry. Samples were withdrawn from the reactor at different times and weighed (approximately 1.0 g) in 7 mL vials. After weighing, the samples were immediately quenched with approximately 10 μL of 1% w/w hydroquinone in an ice-water bath. The specimens were placed in an oven and dried at 60 °C to constant weight. Conversions were calculated from the measured dry residue.Dynamic light scattering (DLS) studies were performed using a Malvern Zetasizer Nano ZS instrument equipped with a He–Ne solid-state laser operating at 633 nm using back-scattered light at a scattering angle of 173°. Polymer dispersions without any purification were diluted to approximately 0.1% w/w using deionised water. Samples were analysed using disposable plastic cuvettes at 25 °C. Data were averaged over three consecutive measurements.
Molar mass distributions were assessed using a gel permeation chromatography (GPC) instrument equipped with an Agilent 1260 Infinity pump injection module, an Agilent 1260 Infinity II refractive index detector, and three Phenomenex phenogel columns with a mobile phase of THF at 35 °C. Calibration was achieved using a series of polystyrene standards (ranging from 1 × 103 to 2 × 106 g mol−1).
Transmission electron microscopy (TEM) images were recorded using a FEI Tecnai G2 20 instrument operating at an accelerating voltage of 200 kV and connected to a Gatan 1k CCD camera. Samples for TEM observation were prepared by depositing 3 μL of diluted copolymer dispersion (approximately 0.1% w/w) onto 400 mesh carbon-coated copper grids for 30 min and then carefully blotted using filter paper to remove excess solution. The samples were stained in a vapour space above ruthenium tetroxide (RuO4) solution at room temperature for 7 min.77 Mean nanoparticle diameters were determined using ImageJ software and over 200 randomly selected particles were measured for each sample.
Results and discussion
Surfactant selection for miniemulsion polymerisation of BzMA
In order to ascertain a suitable surfactant for miniemulsion polymerisation of BzMA, several surfactants were screened by monitoring the stability of BzMA miniemulsion droplets obtained after ultrasonication. A conventional ionic surfactant (SDS) and a series of Lutensol TO non-ionic surfactants, including TO 3, TO 8, TO 15, TO 20, and TO 109, were evaluated. All of these surfactants allowed the formation of BzMA miniemulsions, which remained stable over the timescale of a typical miniemulsion polymerisation. However, Lutensol TO 20 formed emulsions that were stable for the longest period of time (over 11 days) and thus was chosen as the optimal non-ionic surfactant from this range to investigate further.Fig. S1 (ESI†) shows critical micelle concentrations (CMC) determined using pendant drop analysis of interfacial tension for SDS and Lutensol TO 20 in deionised water. The CMC for SDS was approximately 9.2 mM, which is in agreement with the literature (∼8 mM).78,79 In contrast, the CMC of TO 20 (∼0.5 mM) was much lower than that of SDS, indicating that TO 20 can act as a more efficient emulsifier.80,81
The difference in diameter between the emulsified droplets and resulting latexes can be used to indicate polymerisation type. For example, if the resulting latex size is significantly smaller than the monomer reservoir droplets, it can be classified as emulsion polymerisation. Whereas if the resulting latex is equal in diameter to the initial droplets, it is classified as miniemulsion polymerisation.82,83 A 1:1 transfer from initial droplets to latexes is possible only when using a suitable surfactant for emulsification in the appropriate concentration range.83 However, the surfactant concentration used in a miniemulsion formulation is generally above the CMC determined in deionised water. This is because the majority of surfactant molecules locate at the monomer/water droplet interface after the emulsification step.36 Therefore, only a small proportion of surfactant molecules dissolve in the continuous phase, meaning the continuous phase surfactant concentration is actually lower than CMC. This prevents micellar nucleation.84,85 For example, Landfester and coworkers reported poly(methyl methacrylate) latexes synthesised via miniemulsion polymerisation using SDS surfactant at concentrations up to 40 mM, which is much higher than the CMC for SDS in pure deionised water (∼8 mM).36,82 The same research group also demonstrated that polystyrene latexes can be synthesised via miniemulsion polymerisation using 0.5–50% w/w SDS surfactant relative to monomer.84
A series of experiments were conducted using a relatively wide range of surfactant concentrations to determine the optimal conditions for miniemulsion polymerisation of BzMA. Fig. S2 and S3 (ESI†) show the evolution of hydrodynamic diameter as a function of time for conventional free-radical miniemulsion polymerisation of BzMA using a range of SDS anionic surfactant and HD hydrophobe concentrations. In most cases, the final PBzMA latex diameter was significantly smaller than the initial miniemulsion droplets. This indicated that regular emulsion polymerisation was taking place as opposed to miniemulsion polymerisation. Fig. S4 (ESI†) shows the mean-diameter of PBzMA latexes synthesised using a wide range of TO 20 non-ionic surfactant concentrations. At surfactant concentrations below 0.5% w/w, a relatively large difference between the monomer droplet and final latex diameters were observed. Above 7.8% w/w surfactant, sub-micron latexes were obtained which had diameters similar to the initial droplet diameter, indicating successful miniemulsion conditions. On increasing the surfactant concentration from 0.1 to 23.3% w/w, the mean latex diameter decreased from approximately 2400 to 164 nm. As expected, higher surfactant concentrations resulted in smaller polymer particles due to the lower surface tension of the system.84 It is noteworthy that the sub-500 nm latexes which had a similar diameter to the initial droplet size were achieved using a TO 20 concentration ≥7.8% w/w (or ≥19 mM). This concentration was approximately 38 times greater than the CMC determined in deionised water (∼0.5 mM). Given the suitability of Lutensol TO 20 for miniemulsion polymerisation of BzMA, this surfactant was utilised in all subsequent miniemulsion polymerisations reported herein.
Comparison of conventional free-radical miniemulsion polymerisation with RAFT-mediated miniemulsion polymerisation
Miniemulsion polymerisation of BzMA was conducted in the presence of two RAFT CTAs (PETTC and PETTCCP, target DP = 300, see Fig. 1 for chemical structures) and without a CTA under otherwise identical conditions. It was observed that PBzMA latexes synthesised via conventional free-radical miniemulsion polymerisation achieved full monomer conversion within 40 min (Fig. S5a, ESI†) whereas the two RAFT-mediated reactions took approximately 60–100 min to reach full conversion. Furthermore, the molar mass of PBzMA was much higher and the molar mass distribution was broader (Mw/Mn > 3.0) in the absence of a CTA than for the polymers synthesised using the RAFT CTAs (Fig. 1, Fig. S5b and Table S2, ESI†). This indicates that RAFT control over BzMA polymerisation was achieved in the presence of both PETTC and PETTCCP RAFT agents.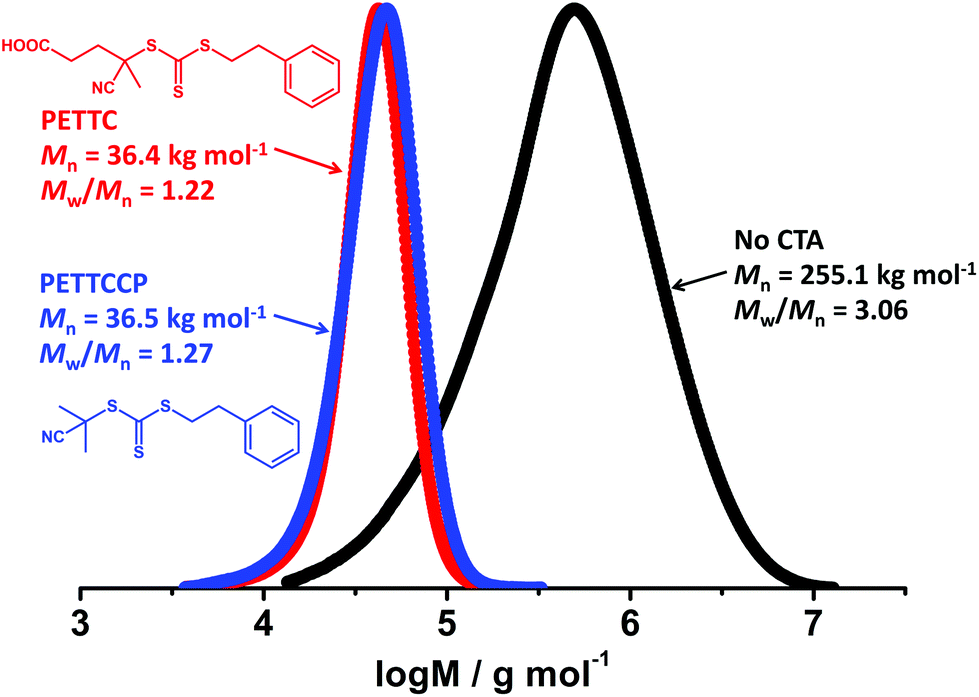 | ||
| Fig. 1 GPC chromatograms for PBzMA synthesised via RAFT miniemulsion polymerisation in the presence of no CTA (black), PETTC (red) and PETTCCP (blue) at 70 °C. The target PBzMA degree of polymerisation (DP) in the presence of RAFT CTAs was 300 and [CTA]/[AIBN] was 0.4. Polymerisations were conducted at a dispersed phase content of 20% w/w with the concentration of HD and TO 20 fixed at 2.4% w/w and 7.8% w/w relative to BzMA, respectively. Monomer conversions using no CTA, PETTC, and PETTCCP CTA were 99.9%, 97.3%, and 99.6%, respectively (Table S2, ESI†). | ||
The mean particle diameter of PBzMA latexes prepared with no CTA and using PETTCCP were 338 nm and 325 nm, respectively. However, the particle diameter was significantly higher (577 nm) when using PETTC. This can be attributed to the PETTC being more hydrophilic than PETTCCP and thus having an increased ability to cross into the aqueous phase. Therefore, PETTC may form a surfactant/cosurfactant structure at the droplet-water interface and lead to an increased particle diameter.33 Nevertheless, the minor difference in particle diameter observed for PBzMA latexes prepared by conventional miniemulsion polymerisation and using PETTCCP, as well as the increased control over polymer molar mass afforded, indicates the suitability of PETTCCP for RAFT-mediated miniemulsion polymerisation of BzMA. Thus, the influence of hydrophobe, initiator and CTA concentration were investigated further.
Influence of hydrophobe concentration
The use of a small quantity of HD as a hydrophobe allows the build-up of osmotic pressure in miniemulsion droplets, providing stability against Ostwald ripening.86 Thus the influence of HD concentration on RAFT miniemulsion polymerisation of BzMA was investigated by varying the HD concentration between 1.2–9.5% w/w, based on BzMA, while the concentration of monomer, initiator, surfactant, and CTA remained unchanged (see Table 1). With increasing HD concentration, the measured PBzMA latex diameter (Dh) decreases from 338 to 296 nm, corresponding to a calculated increase in the number of particles per unit volume (Np). This indicates that with higher HD concentrations, more droplets can be stabilised against Ostwald ripening effects and the average droplet size decreases. However, the influence of HD on polymerisation kinetics was negligible with high monomer conversions being achieved in all cases within 60 min (Fig. 2a). Similarly, no significant differences in PBzMA molar mass evolution were observed (Fig. 2b), with the final latexes having relatively consistent molar masses and molar mass distributions (Table 1). These observations suggest that these miniemulsion polymerisations were under good RAFT control. Furthermore, there was no obvious influence of HD concentration on the overall polymerisation rate (Table 1). However, due to the increased number of particles generated with more HD, the mean polymerisation rate per particle (RN) decreased with increasing HD concentration (Table 1 and Fig. S6, ESI†).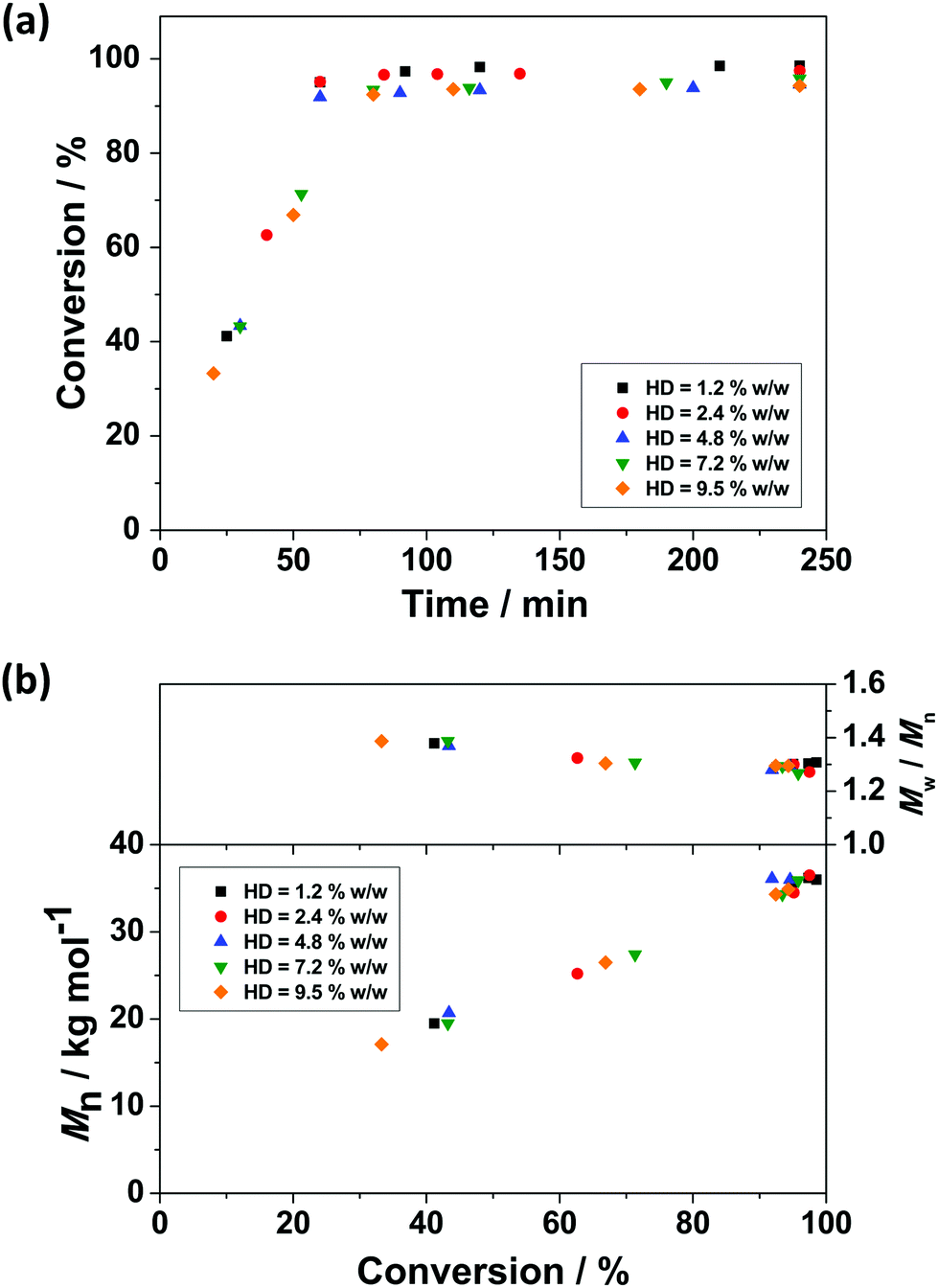 | ||
| Fig. 2 (a) Monomer conversion versus reaction time and (b) Mn and Mw/Mnversus monomer conversion for PBzMA synthesised via RAFT miniemulsion polymerisation with varying HD concentration relative to BzMA at 70 °C. [BzMA]:[PETTCCP]:[AIBN] = 300:1:2.3 and the concentration of TO 20 surfactant was fixed at 7.8% w/w relative to BzMA. | ||
Influence of initiator concentration
The influence of initiator concentration was investigated by varying the [PETTCCP]:[AIBN] ratio whilst keeping the concentration of monomer, hydrophobe, surfactant, and chain transfer agent constant (Table 2). In most cases, >90% monomer conversion was achieved after 240 min (Fig. 3a). Furthermore, increasing the amount of initiator increases the overall rate of polymerisation and follows a power-law relationship Rp α [AIBN]1/2.87–89Fig. 3b shows the approximately linear relationship between ln([M]0/[M]) and reaction time, indicating the polymerisations were first-order with respect to monomer concentration.54Fig. 3c shows the evolution of molar mass and molar mass dispersity (Mw/Mn) versus monomer conversion. It is noteworthy that unlike the polymerisation rate, which was strongly influenced by the initiator concentration, there were negligible differences in the observed molar mass and molar mass dispersity values.
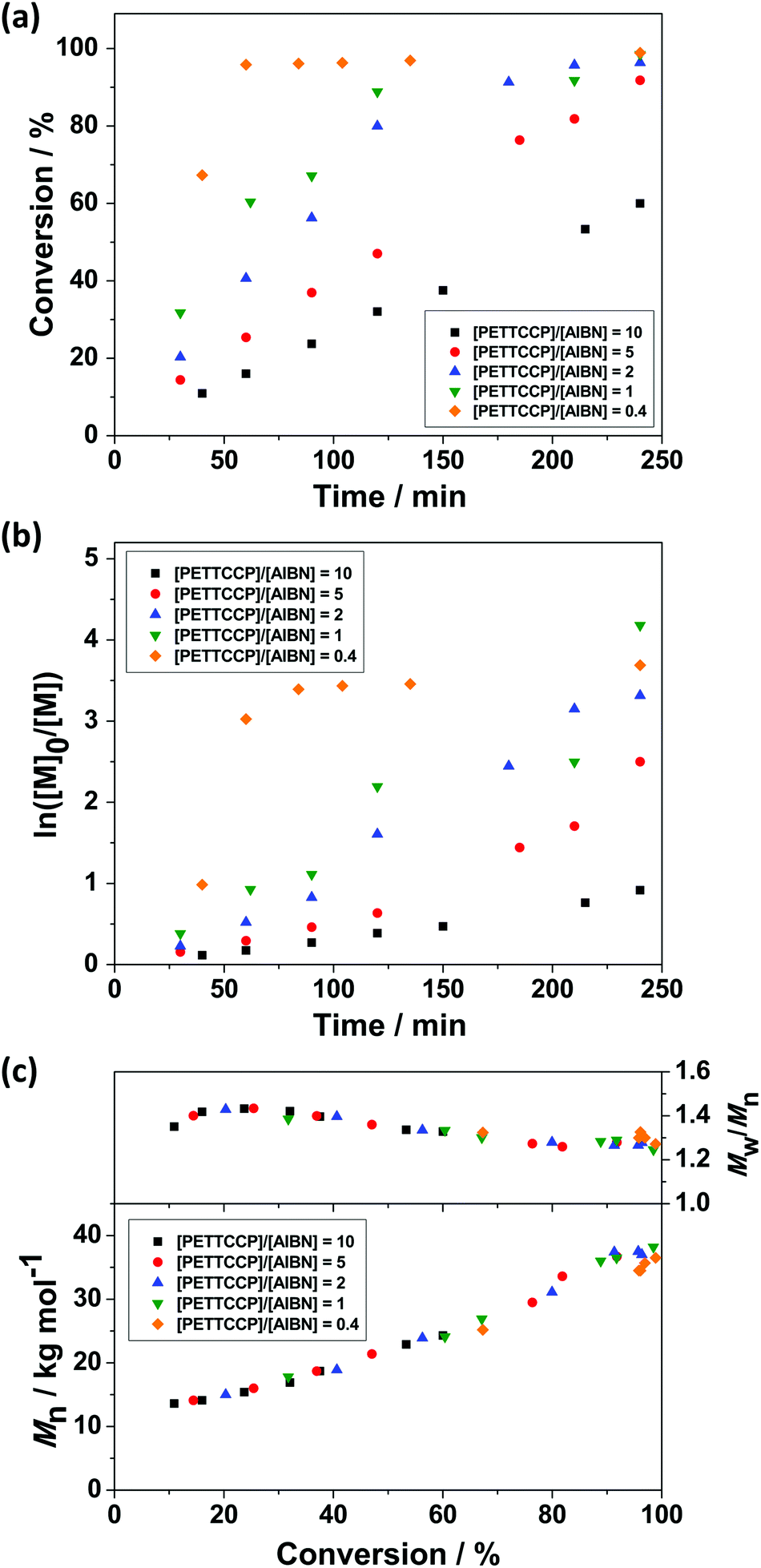 | ||
| Fig. 3 (a) Monomer conversion versus reaction time, (b) semi-logarithmic kinetics, and (c) Mn and Mw/Mnversus monomer conversion for PBzMA synthesised via RAFT miniemulsion polymerisation at 70 °C with varying [PETTCCP]:[AIBN] ratio. The target DP was 300. Polymerisations were conducted at a dispersed phase content of 20% w/w, with the concentration of HD and TO 20 fixed at 2.4% w/w and 7.8% w/w relative to BzMA, respectively. | ||
The relatively linear conversion/time relationships and the pseudo-first-order kinetic plots (Fig. 3) are reasonably consistent with the features expected for a controlled RAFT polymerisation. This indicates that a rapid main equilibrium between the active propagating radicals and thiocarbonylthio capped dormant species was achieved and a constant number of radical propagating chains was formed during polymerisation.88 Additionally, no obvious retardation in these formulations was observed and polymerisations proceeded linearly to high monomer conversions (∼90% after 240 min, Fig. 3a). In all cases, the evolution of molar mass in relation to monomer conversion was close to linear, with relatively narrow molar mass dispersities obtained (Mw/Mn < 1.3). It is noteworthy that positive y-intercepts were observed in all cases after extrapolating the experimental data for Mn to zero conversion. Differences between the GPC calibration standards (polystyrene) and PBzMA may be partially the cause of this positive intercept and deviations from theoretical Mn values.
However, the observed positive y-intercepts may also result from a higher polymerisation rate than that of the addition of growing radicals to the PETTCCP CTA.90,91 Therefore, RAFT control was not established instantaneously, resulting in the rapid formation of moderate molar mass species at low monomer conversions. These observations may be considered as hybrid conventional/living behaviour.92–94 However, this behaviour can be reduced by using a lower ratio of [CTA] to [initiator].91 For example, the y-intercept was approximately 9 kg mol−1 when the [CTA]:[initiator] ratio was 5:1 whereas the y-intercept was approximately 2 kg mol−1 (∼78% less) when the ratio was 0.4. This indicates that higher initiator concentrations allow more rapid CTA activation, resulting in a shorter time in achieving the main RAFT equilibrium. Nevertheless, the molar mass dispersities of the resultant polymers were relatively low, suggesting this hybrid behaviour had a minor effect on the overall RAFT process.
Influence of RAFT agent concentration
The versatility and limitations of RAFT-mediated miniemulsion polymerisation of BzMA were further investigated by varying the PETTCCP concentration. Specifically, the molar concentration of PETTCCP relative to the dispersed phase was varied from 7.1 mmol L−1 (target PBzMA DP = 800) to 28.6 mmol L−1 (target PBzMA DP = 200). A [PETTCCP]:[AIBN] ratio of 5:1 was used whilst keeping the concentration of monomer, hydrophobe, and surfactant constant (Table 3). High monomer conversions (∼90%) were achieved after 240 min (for DP 200, 300, and 400) or 360 min (for DP 700 and 800, Fig. 4a). Interestingly, the polymerisation rate increased with increasing PETTCCP concentration (or decreasing target DP). For example, the polymerisation rate for PETTCCP at 28.6 mmol L−1 relative to the dispersed phase (DP = 200) was approximately 62% faster than for PETTCCP at 7.1 mmol L−1 (DP = 800) (Table 3). In this study, the [CTA]:[initiator] ratio was fixed at 5:1. Thus, increased RAFT CTA concentrations (lower target DP) also meant that the initiator concentration relative to BzMA was also higher. This inevitably led to the faster polymerisation rates observed for lower target DP formulations. It is noteworthy that the rate of polymerisation observed was relatively independent of droplet diameter and rate increases due to compartmentalisation effects were not observed. This can be attributed to the latexes in this work being relatively large (∼300 nm).32,95,96
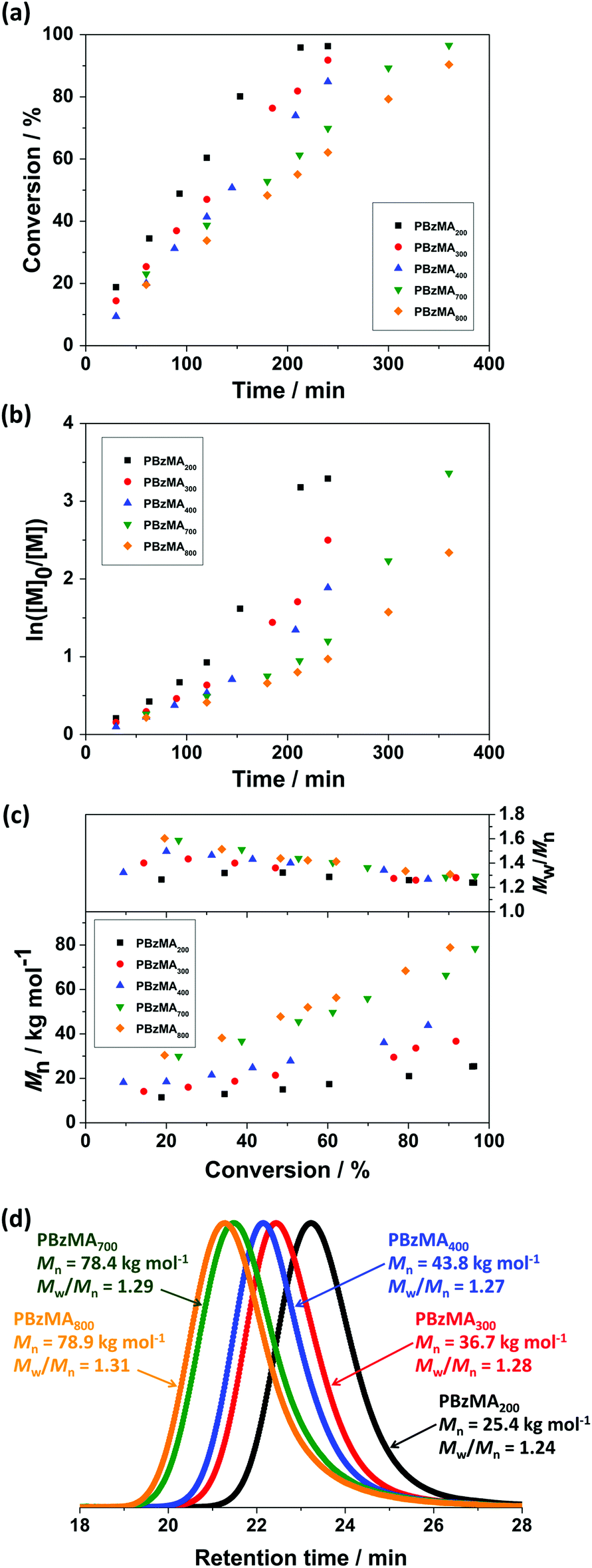 | ||
| Fig. 4 (a) Monomer conversion versus reaction time, (b) semi-logarithmic kinetics, (c) Mn and Mw/Mnversus monomer conversion, and (d) GPC chromatograms for PBzMA synthesised via RAFT miniemulsion polymerisation with varying target DP at 70 °C. The [PETTCCP]:[AIBN] ratio was fixed at 5:1 and polymerisations were conducted at a dispersed phase content of 20% w/w, with the concentration of HD and TO 20 fixed at 2.4% w/w and 7.8% w/w relative to BzMA, respectively. | ||
ln([M]0/[M]) versus reaction time for these RAFT miniemulsion polymerisations is shown in Fig. 4b. The approximately linear relationship in all cases indicates that the reactions were first-order with respect to monomer concentration and thus were well-controlled RAFT polymerisations.54 GPC chromatograms of PBzMA synthesised with various PETTCCP concentrations at full monomer conversion are shown in Fig. 4d. Unimodal and relatively narrow molar mass distributions (Mw/Mn < 1.3) were obtained in all cases, with the Mn clearly increasing with decreasing RAFT agent concentration. The evolution of molar mass and molar mass dispersity (Mw/Mn) versus monomer conversion for PBzMA with various PETTCCP concentrations (target DP 200–800) are shown in Fig. 4c. As the polymerisations progressed, the corresponding dispersity decreased, and the resulting PBzMA had relatively narrow molar mass distributions (Mw/Mn < 1.3) at 90% conversion. It is noteworthy that the molar mass increased approximately linear in all cases, even when targeting high DPs.
The measured PBzMA latex diameter (Dh) decreased from 330 to 275 nm with increasing PETTCCP concentration (Fig. 5). This corresponded to a calculated increase in the number of particles per unit volume (Np) from approximately 1.5 to 2.8 × 1014 mL−1. Furthermore, spherical latexes were obtained in all cases with measured diameters in agreement between TEM and DLS (Fig. 6 and Table 3). It is noteworthy that the number of particles (Np) decreased with increasing RAFT agent concentration (Fig. 5). For example, the Np value for PETTCCP at 28.6 mmol L−1 was approximately 48% less than PETTCCP at 7.1 mmol L−1. This is attributed to the increased viscosity of the oil phase.97 More specifically, formulations with higher PETTCCP concentrations result in droplets which are more viscous. Thus, fewer droplets are produced using the same ultrasonication procedure and results in larger miniemulsion droplets and final PBzMA latexes.
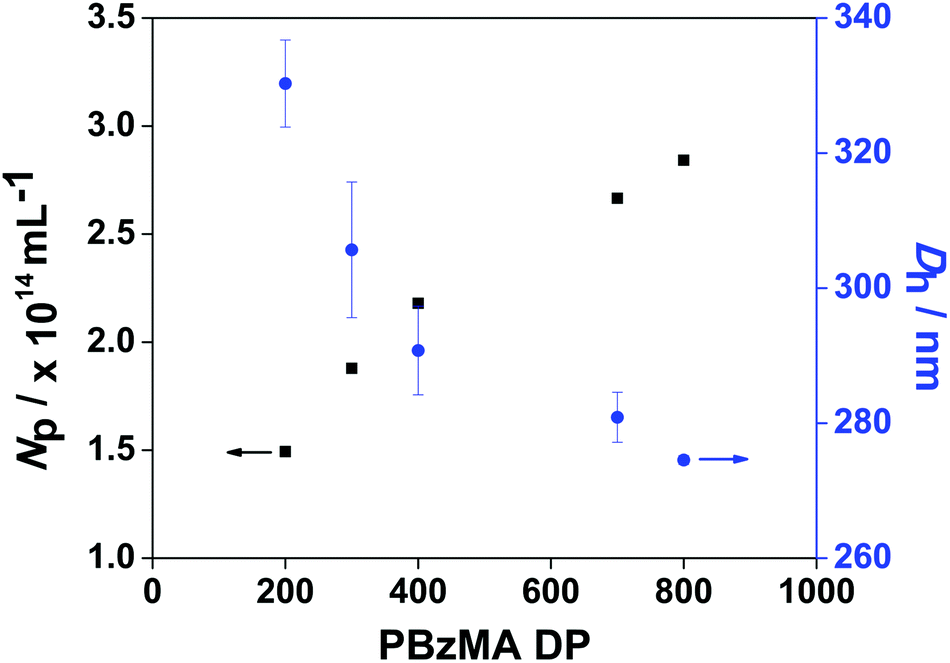 | ||
| Fig. 5 Effect of varying target PBzMA DP on the number of particles (Np) for PBzMA synthesised via RAFT miniemulsion polymerisation at 70 °C. The [PETTCCP]:[AIBN] ratio was fixed at 5:1 and polymerisations were conducted at a dispersed phase content of 20% w/w, with the concentration of HD and TO 20 fixed at 2.4% w/w and 7.8% w/w relative to BzMA, respectively. | ||
 | ||
| Fig. 6 Representative TEM images for PBzMA latexes synthesised via RAFT miniemulsion polymerisation at 70 °C with varying target DP: (a) 200, (b) 300, (c) 700, and (d) 800. The [PETTCCP]:[AIBN] ratio was fixed at 5:1 and polymerisations were conducted at a dispersed phase content of 20% w/w, with the concentration of HD and TO 20 fixed at 2.4% w/w and 7.8% w/w relative to BzMA, respectively. | ||
Finally, PBzMA chain-end stability and fidelity over a 12-month storage period was examined via self-blocking experiments (Fig. S7, ESI†). After being stored at room temperature for over a year, an additional charge of BzMA (target DP of second block = 300) and initiator was added to a dispersion of PBzMA latex (Table 2, entry 3). GPC analysis indicated a relatively good blocking efficiency and the resulting chain-extended PBzMA homopolymer had a unimodal molar mass distribution and relatively low molar mass dispersity (1.39). This indicates that the majority of the trithiocarbonate RAFT chain-ends remained intact and that these PBzMA latexes could undergo efficient chain extension to form second blocks with other monomers to form more complex latex morphologies.98,99
Conclusions
Near-monodisperse PBzMA latexes with controlled particle diameters and polymer molar mass were successfully prepared via RAFT miniemulsion polymerisation. This was achieved using a relatively hydrophobic chain transfer agent (PETTCCP) and non-ionic Lutensol TO 20 surfactant. Stable miniemulsion droplets were obtained and formed PBzMA latexes upon polymerisation. Much narrower molar mass distributions (Mw/Mn < 1.3) were obtained via RAFT miniemulsion polymerisation when compared to conventional miniemulsion polymerisation (Mw/Mn > 3.0). Furthermore, the concentration of hydrophobe, initiator, and chain transfer agent were demonstrated to influence the particle diameter, particle number, rate of polymerisation, and PBzMA molar mass in the final latexes.Increasing the hydrophobe (HD) concentration decreased the PBzMA latex diameter (Dh) and increased the number of particles per unit volume (Np). This indicated that more droplets can be stabilised against Ostwald ripening effects using higher HD concentrations. Furthermore, the mean rate per particle decreased with increasing HD concentration. Increasing initiator (AIBN) concentration increased the overall rate of polymerisation and followed a power-law relationship Rp α [AIBN]1/2. However, only negligible differences in molar mass, molar mass dispersity, and particle diameter values were observed. Increasing RAFT agent (PETTCCP) concentration increased the measured PBzMA latex diameter (Dh) and the overall rate of polymerisation. The changes in latex diameter can be attributed to the increased viscosity of the oil phase, which results in larger miniemulsion droplets. Furthermore, the approximately linear relationship of ln([M]0/[M]) versus reaction time, unimodal GPC chromatograms, relatively narrow molar mass distributions (Mw/Mn < 1.3) and efficient self-blocking indicated that these were well-controlled RAFT miniemulsion polymerisations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
National Chung-Shan Institute of Science and Technology (NCSIST) is thanked for funding a PhD studentship for SPW. EPSRC grant EP/P026095/2 is acknowledged for providing the ultrasonic processor. The University of Manchester Electron Microscopy Centre is acknowledged for access to electron microscopy facilities. This work was supported by the Henry Royce Institute for Advanced Materials, funded through EPSRC grants EP/R00661X/1, EP/S019367/1, EP/P025021/1 and EP/P025498/1.Notes and references
- J. Qiu, B. Charleux and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 2083–2134 CrossRef CAS.
- M. F. Cunningham, Prog. Polym. Sci., 2002, 27, 1039–1067 CrossRef CAS.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 101311 CrossRef CAS.
- G. Moad, E. Rizzardo and D. H. Solomon, Macromolecules, 1982, 15, 909–914 CrossRef CAS.
- M. K. Georges, R. P. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- K. Matyjaszewski, S. Gaynor and J.-S. Wang, Macromolecules, 1995, 28, 2093–2095 CrossRef CAS.
- J. Chiefari, Y. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. Le, R. T. Mayadunne, G. F. Meijs, C. L. Moad and G. Moad, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, J. Chiefari, Y. K. Chong, J. Krstina, R. T. A. Mayadunne, A. Postma, E. Rizzardo and S. H. Thang, Polym. Int., 2000, 49, 993–1001 CrossRef CAS.
- Y. Kwak, A. Goto, Y. Tsujii, Y. Murata, K. Komatsu and T. Fukuda, Macromolecules, 2002, 35, 3026–3029 CrossRef CAS.
- M. J. Monteiro and H. de Brouwer, Macromolecules, 2001, 34, 349–352 CrossRef CAS.
- P. B. Zetterlund, Y. Kagawa and M. Okubo, Chem. Rev., 2008, 108, 3747–3794 CrossRef CAS PubMed.
- M. F. Cunningham, Prog. Polym. Sci., 2008, 33, 365–398 CrossRef CAS.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- P. B. Zetterlund, S. C. Thickett, S. Perrier, E. Bourgeat-Lami and M. Lansalot, Chem. Rev., 2015, 115, 9745–9800 CrossRef CAS PubMed.
- R. G. Gilbert, Emulsion polymerization: a mechanistic approach, Academic Pr, 1995 Search PubMed.
- J. Vosloo, D. De Wet-Roos, M. Tonge and R. Sanderson, Macromolecules, 2002, 35, 4894–4902 CrossRef CAS.
- S. W. Prescott, M. J. Ballard, E. Rizzardo and R. G. Gilbert, Aust. J. Chem., 2002, 55, 415–424 CrossRef CAS.
- M. Lansalot, T. P. Davis and J. P. Heuts, Macromolecules, 2002, 35, 7582–7591 CrossRef CAS.
- K. Matyjaszewski, J. Qiu, N. V. Tsarevsky and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4724–4734 CrossRef CAS.
- M. F. Cunningham, M. Xie, K. B. McAuley, B. Keoshkerian and M. K. Georges, Macromolecules, 2002, 35, 59–66 CrossRef CAS.
- D. Kukulj, T. P. Davis and R. G. Gilbert, Macromolecules, 1997, 30, 7661–7666 CrossRef CAS.
- M. Lansalot, C. Farcet, B. Charleux, J.-P. Vairon and R. Pirri, Macromolecules, 1999, 32, 7354–7360 CrossRef CAS.
- D. Crespy and K. Landfester, Beilstein J. Org. Chem., 2010, 6, 1132–1148 CrossRef CAS PubMed.
- K. Landfester, Angew. Chem., Int. Ed. Engl., 2009, 48, 4488–4507 CrossRef CAS PubMed.
- J. M. Asua, Prog. Polym. Sci., 2014, 39, 1797–1826 CrossRef CAS.
- J. Hu, M. Chen and L. Wu, Polym. Chem., 2011, 2, 760–772 RSC.
- P. A. Lovell and M. S. El-Aasser, Emulsion polymerization and emulsion polymers, John Wiley & Sons, 1997 Search PubMed.
- K. Landfester, Angew. Chem., Int. Ed., 2009, 48, 4488–4507 CrossRef CAS PubMed.
- F. J. Schork, Y. Luo, W. Smulders, J. P. Russum, A. Butté and K. Fontenot, in Polymer particles, Springer, 2005, pp. 129–255 Search PubMed.
- M. Khan, T. R. Guimarães, D. Zhou, G. Moad, S. Perrier and P. B. Zetterlund, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1938–1946 CrossRef CAS.
- D. Mouran, J. Reimers and F. J. Schork, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 1073–1081 CrossRef CAS.
- C. Chern and T. Chen, Colloid Polym. Sci., 1997, 275, 546–554 CrossRef CAS.
- K. Landfester, N. Bechthold, F. Tiarks and M. Antonietti, Macromolecules, 1999, 32, 2679–2683 CrossRef CAS.
- L. L. Hecht, A. Schoth, R. Muñoz–Espí, A. Javadi, K. Köhler, R. Miller, K. Landfester and H. P. Schuchmann, Macromol. Chem. Phys., 2013, 214, 812–823 CrossRef CAS.
- Y. Chong, J. Krstina, T. P. Le, G. Moad, A. Postma, E. Rizzardo and S. H. Thang, Macromolecules, 2003, 36, 2256–2272 CrossRef CAS.
- J. Chiefari, R. T. Mayadunne, C. L. Moad, G. Moad, E. Rizzardo, A. Postma, M. A. Skidmore and S. H. Thang, Macromolecules, 2003, 36, 2273–2283 CrossRef CAS.
- A. Butté, G. Storti and M. Morbidelli, Macromolecules, 2000, 33, 3485–3487 CrossRef.
- J. McLeary, M. Tonge, D. De Wet Roos, R. Sanderson and B. Klumperman, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 960–974 CrossRef CAS.
- G. Qi and F. J. Schork, Langmuir, 2006, 22, 9075–9078 CrossRef CAS PubMed.
- X. Zhou, P. Ni and Z. Yu, Polymer, 2007, 48, 6262–6271 CrossRef CAS.
- L. Yang, Y. Luo and B. Li, Polymer, 2006, 47, 751–762 CrossRef CAS.
- S. W. Prescott, M. J. Ballard, E. Rizzardo and R. G. Gilbert, Macromolecules, 2002, 35, 5417–5425 CrossRef CAS.
- R. W. Simms, T. P. Davis and M. F. Cunningham, Macromol. Rapid Commun., 2005, 26, 592–596 CrossRef CAS.
- J. G. Tsavalas, F. J. Schork, H. de Brouwer and M. J. Monteiro, Macromolecules, 2001, 34, 3938–3946 CrossRef CAS.
- Y. Luo, B. Liu, Z. Wang, J. Gao and B. Li, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2304–2315 CrossRef CAS.
- M. Tonge, J. McLeary, J. Vosloo and R. Sanderson, Macromol. Symp., 2003, 193, 289–304 CrossRef CAS.
- M. Park, K. Kim, A. K. Mohanty, H. Y. Cho, H. Lee, Y. Kang, B. Seo, W. Lee, H. B. Jeon and H. j. Paik, Macromol. Rapid Commun., 2020, 41, 2000399 CrossRef CAS PubMed.
- Z. Huang, P. Pan and Y. Bao, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2092–2101 CrossRef CAS.
- A. V. Fuchs and K. J. Thurecht, Macromol. Chem. Phys., 2015, 216, 1271–1281 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS PubMed.
- Y. Gnanou, L. Leibler and K. Matyjaszewski, Macromolecular Engineering: Precise Synthesis, Materials Properties, Applications, Wiley-VCH, 2007 Search PubMed.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- C. T. Hendley IV, L. A. Fielding, E. R. Jones, A. J. Ryan, S. P. Armes and L. A. Estroff, J. Am. Chem. Soc., 2018, 140, 7936–7945 CrossRef PubMed.
- A. H. Milani, L. A. Fielding, P. Greensmith, B. R. Saunders, D. J. Adlam, A. J. Freemont, J. A. Hoyland, N. W. Hodson, M. A. Elsawy and A. F. Miller, Chem. Mater., 2017, 29, 3100–3110 CrossRef CAS.
- W. Cai, W. Wan, C. Hong, C. Huang and C. Pan, Soft Matter, 2010, 6, 5554–5561 RSC.
- Z. Li, H. Tam, L. Xu and Q. Zhang, Opt. Lett., 2005, 30, 1117–1119 CrossRef CAS PubMed.
- N. Chaix, C. Gourgon, S. Landis, C. Perret, M. Fink, F. Reuther and D. Mecerreyes, Nanotechnology, 2006, 17, 4082 CrossRef CAS PubMed.
- L. J. Guo, J. Phys. D: Appl. Phys., 2004, 37, R123 CrossRef CAS.
- R. P. Held, C. C. Roberts and P. F. McIntyre, Amphoteric dispersants and their use in inkjet inks, US Pat., US7932306B2, 2011 Search PubMed.
- C. González-Chomón, M. Silva, A. Concheiro and C. Alvarez-Lorenzo, Acta Biomater., 2016, 41, 302–311 CrossRef PubMed.
- H. Li, J. Liu, L. Lin, Q. Mu, X. Sun and X. Liu, Polym. Sci., Ser. B, 2014, 56, 855–862 CrossRef CAS.
- E. Takiyama and J. Hasegawa, Stainproofing paint composition and method for producing same, US Pat., US5264466A, 1993 Search PubMed.
- A. C. Chen, D. L. Siegfried and D. S. Mueller, Stain-resistant no-mix orthodontic adhesive, US Pat., US4792577, 1988 Search PubMed.
- M. M. Skoultchi and D. K. Ray-Chaudhuri, Transparent two-part acrylic adhesive composition and the method of use thereof, US Pat., US5310835A, 1994 Search PubMed.
- K. J. Bachus, K. J. Langille, Y. Fu, G. T. Gibson and R. D. Oleschuk, Polymer, 2015, 58, 113–120 CrossRef CAS.
- J. Ou, G. T. Gibson and R. D. Oleschuk, J. Chromatogr., A, 2010, 1217, 3628–3634 CrossRef CAS PubMed.
- C. Yoon, H. S. Kwon, J. S. Yoo, H. Y. Lee, J. H. Bae and J. H. Choi, Color. Technol., 2015, 131, 2–8 CAS.
- A. Aqel, K. Yusuf, Z. A. Al-Othman, A. Y. Badjah-Hadj-Ahmed and A. A. Alwarthan, Analyst, 2012, 137, 4309–4317 RSC.
- A. Aqel, Z. A. ALOthman, K. Yusuf, A. Y. Badjah-Hadj-Ahmed and A. A. Alwarthan, J. Chromatogr. Sci., 2014, 52, 201–210 CAS.
- A. Kharel, C. Hall, P. Černoch, P. Stepanek and T. P. Lodge, Macromolecules, 2020, 53, 885–894 CrossRef CAS.
- K. Hashimoto, Y. Kobayashi, H. Kokubo, T. Ueki, K. Ohara, K. Fujii and M. Watanabe, J. Phys. Chem. B, 2019, 123, 4098–4107 CrossRef CAS.
- T. Ueki, Polym. J., 2014, 46, 646–655 CrossRef CAS.
- S.-P. Wen, J. G. Saunders and L. A. Fielding, Polym. Chem., 2020, 11, 3416–3426 RSC.
- M. Kocik, O. Mykhaylyk and S. Armes, Soft Matter, 2014, 10, 3984–3992 RSC.
- L. A. Fielding, J. A. Lane, M. J. Derry, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 5790–5798 CrossRef CAS PubMed.
- I. Benito, M. Garcia, C. Monge, J. Saz and M. Marina, Colloids Surf., A, 1997, 125, 221–224 CrossRef CAS.
- S. Nilsson, Macromolecules, 1995, 28, 7837–7844 CrossRef CAS.
- L. Zhang, X. Zhang, P. Zhang, Z. Zhang, S. Liu and B. Han, Colloids Surf., A, 2018, 553, 225–229 CrossRef CAS.
- Z. Ujhelyi, F. Fenyvesi, J. Varadi, P. Feher, T. Kiss, S. Veszelka, M. Deli, M. Vecsernyes and I. Bacskay, Eur. J. Pharm. Sci., 2012, 47, 564–573 CrossRef CAS PubMed.
- L. L. Hecht, C. Wagner, K. Landfester and H. P. Schuchmann, Langmuir, 2011, 27, 2279–2285 CrossRef CAS PubMed.
- K. Landfester, N. Bechthold, S. Förster and M. Antonietti, Macromol. Rapid Commun., 1999, 20, 81–84 CrossRef CAS.
- K. Landfester, N. Bechthold, F. Tiarks and M. Antonietti, Macromolecules, 1999, 32, 5222–5228 CrossRef CAS.
- A. J. van Zyl, D. de Wet-Roos, R. D. Sanderson and B. Klumperman, Eur. Polym. J., 2004, 40, 2717–2725 CrossRef CAS.
- H. Huang, H. Zhang, J. Li, S. Cheng, F. Hu and B. Tan, J. Appl. Polym. Sci., 1998, 68, 2029–2039 CrossRef CAS.
- R. J. Young and P. A. Lovell, Introduction to polymers, CRC press, 2011 Search PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- A. Goto, K. Sato, Y. Tsujii, T. Fukuda, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2001, 34, 402–408 CrossRef CAS.
- S. Garnier and A. Laschewsky, Macromolecules, 2005, 38, 7580–7592 CrossRef CAS.
- D. B. Thomas, A. J. Convertine, L. J. Myrick, C. W. Scales, A. E. Smith, A. B. Lowe, Y. A. Vasilieva, N. Ayres and C. L. McCormick, Macromolecules, 2004, 37, 8941–8950 CrossRef CAS.
- E. Rizzardo, J. Chiefari, R. T. Mayadunne, G. Moad and S. H. Thang, Synthesis of Defined Polymers by Reversible Addition—Fragmentation Chain Transfer: The RAFT Process, Controlled/Living Radical Polymerization, ACS Symposium Series, 2000, ch. 20, vol. 768, pp. 278–296, ISBN13: 9780841237070, eISBN: 9780841218215 Search PubMed.
- C. Barner-Kowollik, J. F. Quinn, T. U. Nguyen, J. P. Heuts and T. P. Davis, Macromolecules, 2001, 34, 7849–7857 CrossRef CAS.
- M. C. Davies, J. V. Dawkins and D. J. Hourston, Polymer, 2005, 46, 1739–1753 CrossRef CAS.
- P. B. Zetterlund and D. R. D′hooge, Macromolecules, 2019, 52, 7963–7976 CrossRef CAS.
- H. Tobita, Macromol. Theory Simul., 2009, 18, 108–119 CrossRef CAS.
- Y. Luo, R. Wang, L. Yang, B. Yu, B. Li and S. Zhu, Macromolecules, 2006, 39, 1328–1337 CrossRef CAS.
- V. Cunningham, S. Armes and O. Musa, Polym. Chem., 2016, 7, 1882–1891 RSC.
- Y. Ning, L. Fielding, T. Andrews, D. Growney and S. Armes, Nanoscale, 2015, 7, 6691–6702 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of the synthesis of PETTCCP and PETTC RAFT agents; experimental characterisation details; studies on the synthesis of PBzMA using conventional miniemulsion polymerisation; CMC determination for SDS and Lutensol TO 20; kinetic studies for miniemulsion polymerisation of PBzMA using various RAFT agents; GPC chromatograms from self-blocking experiments. See DOI: 10.1039/d1py00048a |
| This journal is © The Royal Society of Chemistry 2021 |