
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
RAFT dispersion polymerization of N,N-dimethylacrylamide in a series of n-alkanes using a thermoresponsive poly(tert-octyl acrylamide) steric stabilizer†
R. R.
Gibson
a,
A.
Fernyhough
b,
O. M.
Musa
c and
S. P.
Armes
 *a
*a
aDainton Building, Department of Chemistry, University of Sheffield, Brook Hill, Sheffield, South Yorkshire S3 7HF, UK. E-mail: s.p.armes@sheffield.ac.uk
bAshland Inc., Listers Mills, Heaton Rd, Bradford, West Yorkshire BD9 4SH, UK
cAshland Inc., 1005 US 202/206, Bridgewater, New Jersey 08807, USA
First published on 19th March 2021
Abstract
Herein we report the reversible addition–fragmentation chain transfer (RAFT) solution polymerization of tert-octyl acrylamide (OAA) in 1,4-dioxane using a trithiocarbonate-based RAFT agent. POAA homopolymers were synthesized with good control (Mw/Mn < 1.22) within 1 h at 70 °C when targeting mean degrees of polymerization (DP) of up to 100. Differential scanning calorimetry studies conducted on a series of five POAA homopolymers indicated a weak molecular weight dependence for the glass transition temperature (Tg), which varied from 67 to 83 °C for POAA DPs ranging from 22 to 99. High blocking efficiencies were observed when chain-extending such homopolymers with OAA, suggesting that most of the RAFT end-groups remain intact. Subsequently, we employed POAA as a steric stabilizer block for the PISA syntheses of spherical nanoparticles in n-heptane via RAFT dispersion polymerization of N,N-dimethylacrylamide (DMAC) at 70 °C. Targeting PDMAC DPs between 50 and 250 resulted in reasonably good control (Mw/Mn ≤ 1.42) and produced well-defined spherical diblock copolymer nanoparticles (z-average diameters ranging from 23 nm to 91 nm, with DLS polydispersities remaining below 0.10) within 5 h. A facile one-pot synthesis route to near-monodisperse 36 nm diameter POAA82-PDMAC100 nanoparticles was developed in n-heptane that provided similar control over the molecular weight distribution (Mw/Mn = 1.19). Unfortunately, POAA85-PDMACx diblock copolymer nanoparticles tended to deform and undergo film formation prior to transmission electron microscopy (TEM) studies. To overcome this problem, ethylene glycol diacrylate (EGDA) was introduced towards the end of the DMAC polymerization. The resulting core-crosslinked POAA85-PDMAC195-PEGDA20 triblock copolymer nano-objects exhibited a relatively well-defined spherical morphology. Interestingly, the colloidal stability of POAA85-PDMACx diblock copolymer dispersions depends on the type of n-alkane. Spherical nanoparticles produced in n-heptane or n-octane remained colloidally stable on cooling to 20 °C. However, the colloidally stable POAA-PDMAC nanoparticles prepared at 70 °C in higher n-alkanes became flocculated on cooling. This is because the POAA steric stabilizer chains exhibit upper critical solution temperature (UCST)-type behavior in such solvents. Nanoparticle aggregation was characterized by variable temperature turbidimetry and dynamic light scattering experiments.
Introduction
Highly hydrophobic polymers are widely used for various applications, including self-cleaning surfaces,1 anti-icing formulations,2 anti-biofouling substrates3–5 and the separation of oil and water.6,7tert-Octyl acrylamide (OAA) is a highly hydrophobic monomer that has been used as a comonomer in commercial formulations (e.g. hair-styling products).8–10 However, there are surprisingly few reports of the homopolymer or its physical properties in the academic literature.11–16Remarkably, there appears to be only a single report of the reversible addition–fragmentation chain transfer (RAFT) solution polymerization of OAA.17 This involved using tin-based RAFT agents, which enabled 119Sn nuclear magnetic resonance (NMR) spectroscopy to be used to monitor chain-end fidelity during RAFT polymerization. Chain extension experiments involving polymerization of OAA using a POAA precursor led to around 80% conversion within 13 h at 60 °C, while gel permeation chromatography (GPC) analysis indicated reasonably narrow molecular weight distributions. However, relatively long induction periods of up to 4.5 h were reported for such tin-based RAFT agents. In principle, using more conventional (i.e. metal-free) RAFT agents should enable the reaction conditions to be further optimized for the synthesis of POAA homopolymers, and perhaps also POAA-based block copolymers. This hypothesis is explored in the current study.
Polymerization-induced self-assembly (PISA) has been examined by many research groups over the past decade or so.18–24 It enables the rational design of diblock copolymer nano-objects such as spheres, worms or vesicles at up to 50% w/w solids in a range of polar and non-polar solvents by systematic variation of the relative volume fraction of each block.25,26 PISA requires the use of a controlled living radical polymerization technique such as RAFT polymerization.27–30 In 2013 Fielding et al. reported the first example of a well-controlled RAFT dispersion polymerization in non-polar media.31 Poly(lauryl methacrylate)-poly(benzyl methacrylate) [PLMA-PBzMA] spheres, worms or vesicles were prepared in n-heptane using a PLMA17 precursor, with BzMA monomer conversions of more than 97% being achieved within 5 h at 90 °C. Subsequently, PLMA-PBzMA diblock copolymer nano-objects were also prepared in n-dodecane, with this higher boiling point solvent enabling the thermoresponsive behavior of PLMA-PBzMA worms to be studied.32
Herein we report the synthesis of a series of well-defined homopolymers via RAFT solution polymerization of OAA in 1,4-dioxane and the subsequent synthesis of POAA-based diblock copolymer nanoparticles via RAFT dispersion polymerization of N,N′-dimethyl acrylamide (DMAC) in various non-polar solvents (n-heptane, n-octane, n-decane, n-dodecane, n-tetradecane or n-hexadecane). Turbidimetry studies indicated interesting differences in the temperature-dependent colloidal stability of such dispersions. An atom-efficient one-pot PISA protocol is demonstrated for the synthesis of POAA82-PDMAC100 nanoparticles prepared in n-heptane.
Experimental
Materials
tert-Octyl acrylamide (OAA; 98% purity) was kindly provided by Ashland Inc. (Delaware, USA) and was used without further purification. N,N-Dimethylacrylamide (DMAC), 2,2′-azobis(2-methylpropionitrile) (AIBN), 1,4-dioxane, n-octane, n-tetradecane, n-hexadecane, CDCl3 and 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (DDMAT) were purchased from Sigma Aldrich UK. n-Heptane, n-decane and n-dodecane were purchased from Alfa Aesar (Heysham, UK). Ethylene glycol diacrylate (EGDA) was purchased from Santa Cruz Biotechnology (Dallas, USA).Synthesis of a POAA85 macro-CTA by RAFT solution polymerization of OAA in 1,4-dioxane
The protocol for the preparation of a POAA85 macro-CTA is described below. OAA (20.11 g, 0.11 mol), DDMAT RAFT agent (0.40 g, 1.10 mmol; target DP = 100), AIBN (18.0 mg, 0.11 mmol; DDMAT/AIBN molar ratio = 10) and 1,4-dioxane (30.79 g, 40% w/w) were weighed into a 100 mL round-bottom flask and degassed under N2 with continuous magnetic stirring for 20 min. The OAA polymerization was allowed to proceed for 60 min in an oil bath set to 70 °C, before quenching by exposing the hot reaction solution to air while cooling to 20 °C. 1H NMR spectroscopy studies indicated a final monomer conversion of 82%. The crude homopolymer was precipitated into excess methanol to remove residual OAA monomer before placing in a vacuum oven at 30 °C for three days to afford a dry yellow powder. The mean DP was calculated to be 85 by end-group analysis using UV spectroscopy (λ = 308 nm). Chloroform GPC analysis indicated an Mn of 9900 g mol−1 and an Mw/Mn of 1.18 using a series of ten near-monodisperse poly(methyl methacrylate) (PMMA) calibration standards.Synthesis of POAA85-PDMACx diblock copolymer nanoparticles via RAFT dispersion polymerization of DMAC in various n-alkanes
A typical protocol for the synthesis of POAA85-PDMAC100 diblock copolymer nanoparticles in n-heptane was conducted as follows: POAA85 macro-CTA (0.30 g, 18.8 μmol), DMAC (0.19 g, 1.88 mmol; target DP = 100) and AIBN (0.30 mg, 1.88 μmol; 0.03 g of a 10 mg g−1 stock solution of AIBN dissolved in DMAC; POAA85/AIBN molar ratio = 10) were dissolved in n-heptane (1.95 g; targeting 20% w/w solids). The glass vial was sealed and degassed via N2 gas for 15 min at 20 °C before being immersed in a pre-heated oil bath for 5 h at 70 °C. The DMAC polymerization was quenched by exposing the hot reaction solution to air while cooling to 20 °C. The resulting diblock copolymer nanoparticles were characterized by 1H NMR spectroscopy in CDCl3, while 0.1% w/w dispersions were prepared by dilution with n-heptane for DLS and TEM studies. Chloroform GPC analysis indicated an Mn of 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 g mol−1 and an Mw/Mn of 1.19 (vs. a series of ten PMMA standards). Other diblock compositions were prepared by adjusting the amount of DMAC monomer to target the desired DP. For these additional syntheses, the volume of the continuous phase was adjusted to maintain an overall copolymer concentration of 20% w/w solids. 1H NMR analysis indicated that at least 98% DMAC conversion was achieved in all cases. POAA85-PDMACx diblock copolymer nanoparticles were also prepared in n-octane, n-decane, n-dodecane, n-tetradecane and n-hexadecane. All synthetic parameters except for the volume of solvent were unchanged. Owing to the differing densities of these n-alkanes, the overall solution volume varied for these formulations.
900 g mol−1 and an Mw/Mn of 1.19 (vs. a series of ten PMMA standards). Other diblock compositions were prepared by adjusting the amount of DMAC monomer to target the desired DP. For these additional syntheses, the volume of the continuous phase was adjusted to maintain an overall copolymer concentration of 20% w/w solids. 1H NMR analysis indicated that at least 98% DMAC conversion was achieved in all cases. POAA85-PDMACx diblock copolymer nanoparticles were also prepared in n-octane, n-decane, n-dodecane, n-tetradecane and n-hexadecane. All synthetic parameters except for the volume of solvent were unchanged. Owing to the differing densities of these n-alkanes, the overall solution volume varied for these formulations.
One-pot synthesis of POAA82-PDMAC100 diblock copolymer nanoparticles via RAFT dispersion polymerization of DMAC in n-heptane
OAA (0.40 g, 2.18 mmol), DDMAT RAFT agent (9.9 mg, 27.3 μmol; target DP = 80) and AIBN (0.40 mg, 2.7 μmol; DDMAT/AIBN molar ratio = 10) were dissolved in n-heptane (0.62 g; targeting 40% w/w solids) in a glass vial. The resulting solution was then degassed for 20 min at 20 °C using a N2 sparge before immersing the reaction vial in a pre-heated oil bath set at 70 °C. After 150 min, 1H NMR studies indicated 98% OAA conversion and a mean DP of 82. Chloroform GPC analysis indicated an Mn of 8100 g mol−1 and an Mw/Mn of 1.16. Next, deoxygenated n-heptane (3.00 mL; targeting 20% w/w solids) was added to dilute the reaction solution containing the POAA82 macro-CTA and then deoxygenated DMAC (0.27 mL, 2.66 mmol; target DP = 100) was also added. The DMAC polymerization was allowed to proceed for 5 h at 70 °C, resulting in a 20% w/w dispersion of POAA82-PDMAC100 diblock copolymer nanoparticles (Mn = 18500 g mol−1 and Mw/Mn = 1.19 by chloroform GPC analysis using PMMA calibration standards).
Synthesis of core-crosslinked POAA85-PDMAC100-PEGDA20 triblock copolymer nanoparticles via sequential RAFT dispersion polymerization of DMAC and EGDA in n-heptane
A typical protocol for the synthesis of core-crosslinked POAA85-PDMAC100-PEGDA20 nanoparticles was conducted as follows: POAA85 macro-CTA (0.40 g, 25.1 μmol), DMAC (0.25 g, 2.51 mmol; target DP = 100) and AIBN (0.40 mg, 2.51 μmol; 0.04 g of a 10 mg g−1 stock solution of AIBN dissolved in DMAC; POAA85/AIBN molar ratio = 10) were dissolved in n-heptane (2.94 g; targeting 20% w/w solids). The glass vial was sealed and degassed under N2 for 15 min at 20 °C before being placed in a pre-heated oil bath set at 70 °C for 195 min. EGDA (0.09 g, 0.50 mmol; target DP = 20; previously degassed with N2 gas at 20 °C) was then added using a deoxygenated syringe/needle. EGDA polymerization was allowed to proceed for 4 h before quenching by exposure of the hot reaction mixture to air while cooling to 20 °C. The resulting core-crosslinked triblock copolymer nanoparticles were diluted with n-heptane to afford a 0.1% w/w dispersion prior to characterization by DLS and TEM.Copolymer characterization
210 ± 170 mol−1 dm3 cm−1.
000 g mol−1) were used for calibration and data were analyzed using Varian Cirrus GPC software supplied by the instrument manufacturer.
Results and discussion
Synthesis of POAA homopolymers by RAFT solution polymerization
A POAA85 homopolymer was prepared by RAFT solution polymerization of OAA in 1,4-dioxane at 70 °C using a DDMAT RAFT agent, see Scheme 1. 1H NMR spectroscopy studies indicated that 82% OAA monomer conversion was achieved within 60 min and the mean DP was estimated to be 85 by end-group analysis using unique proton signals assigned to the RAFT chain-ends (Fig. S1†). This approximate DP was confirmed by UV spectroscopy studies in chloroform. For this calculation (see Fig. S2†), it is assumed that all chains contain a trithiocarbonate end-group and that the molar extinction coefficient, ε, for this chain-end is identical to that of DDMAT, for which ε = 15210 ± 170 mol−1 dm3 cm−1 at 308 nm. In practice, the wavelengths observed for the absorption maxima of these two species differ by just 2 nm (310 nm vs. 308 nm, respectively). This suggests that the corresponding molar extinction coefficients should be very similar.33
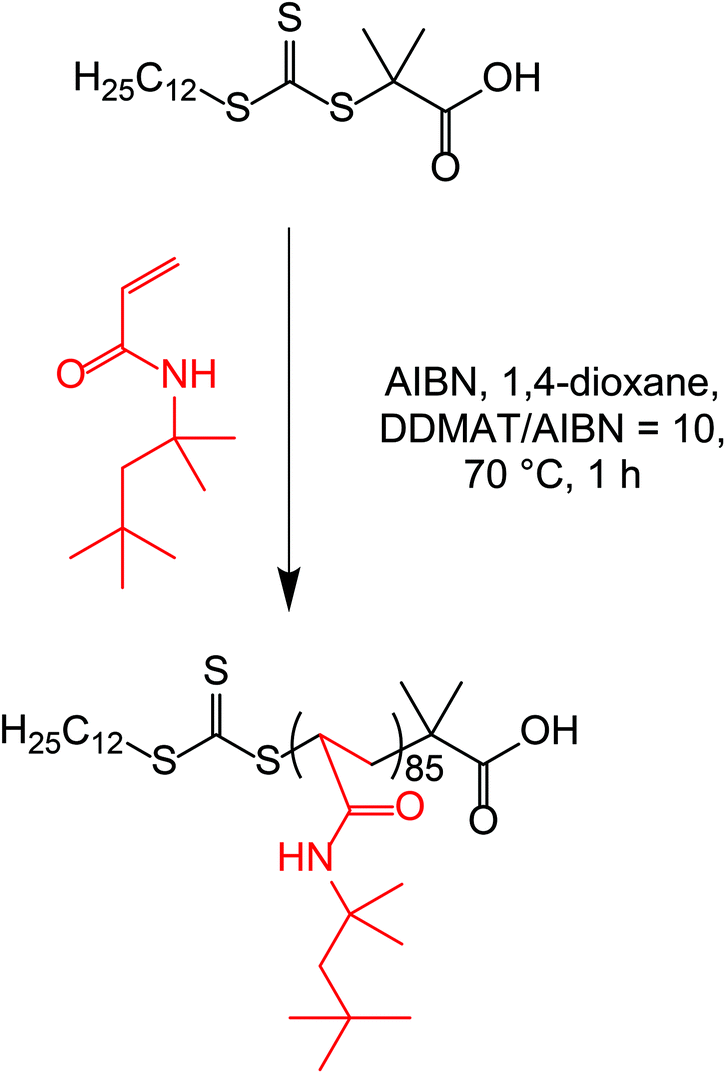 | ||
| Scheme 1 Synthesis of a POAA85 homopolymer by RAFT solution polymerization of tert-octyl acrylamide (OAA) in 1,4-dioxane at 70 °C targeting 40% w/w solids. | ||
Aliquots were periodically extracted during the RAFT homopolymerization of OAA when targeting a DP of 70, with monomer conversions being determined by 1H NMR spectroscopy (Fig. 1a) and molecular weight data being obtained by GPC analysis using chloroform as an eluent. DDMAT/AIBN molar ratios of either 5 or 10 were explored, with a marginally faster rate of polymerization being achieved when using more initiator (Fig. 1). However, the final dispersities and conversions were very similar. Thus, using a DDMAT/AIBN molar ratio of 5.0 afforded 97% conversion, an Mn of 8700 g mol−1 and an Mw/Mn of 1.18 (Fig. S3†), whereas using a DDMAT/AIBN molar ratio of 10 produced 98% conversion, an Mn of 8500 g mol−1 and an Mw/Mn of 1.16, see Fig. 1b. For such homopolymerizations there was either little or no induction period (e.g. just 10 min when using a DDMAT/AIBN molar ratio of 10). In contrast, relatively long induction periods (up to 4.5 h) were reported for the only other literature example of the RAFT homopolymerization of OAA.17 Klumperman and co-workers have attributed similar observations to a so-called initialization process.34 This problem may well be related to the use of an organotin-based RAFT agent by Kulai and co-workers,17 whereas a more conventional trithiocarbonate-based reagent was employed in the present study.
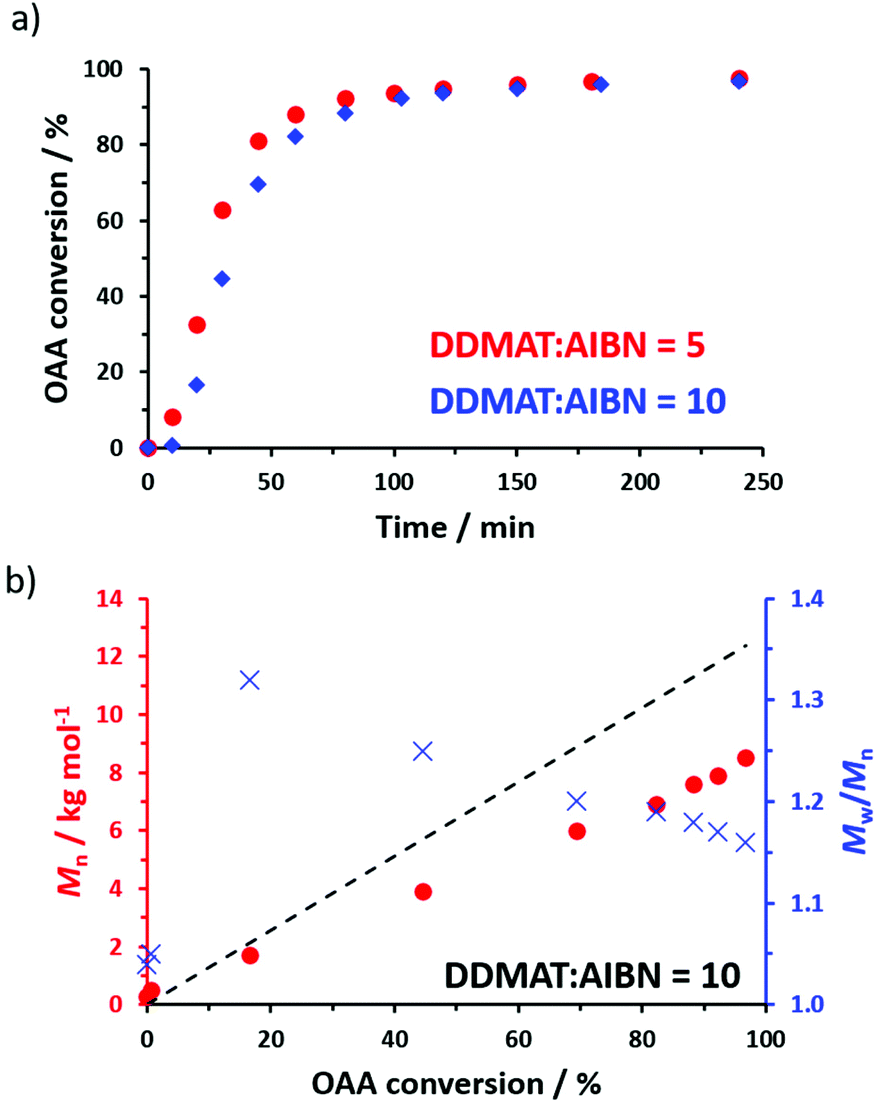 | ||
| Fig. 1 (a) Conversion vs. time curves obtained for the RAFT solution polymerization of OAA at 70 °C in 1,4-dioxane using a DDMAT RAFT agent and AIBN initiator targeting a POAA DP of 70 at 40% w/w solids using a DDMAT/AIBN molar ratio of either 5 or 10. (b) Evolution of Mn and Mw/Mn with conversion observed during the RAFT solution polymerization of OAA at 70 °C in 1,4-dioxane when targeting a POAA DP of 70 at 40% w/w solids using a DDMAT/AIBN molar ratio of 10. The dashed line indicates the theoretical Mn data. The experimental Mn data set falls below this theoretical line owing to a systematic GPC calibration error. | ||
The Tg of each of the five POAAx homopolymers (Table S1†) was determined using differential scanning calorimetry (DSC). The shortest homopolymer (POAA22) had a Tg of 67 °C while the longest (POAA99) had a Tg of 83 °C, indicating the expected weak molecular weight dependence (Fig. 2). OAA monomer is a solid at room temperature, with DSC studies indicating a melting point of around 64 °C (see Fig. S4†). Preliminary attempts to polymerize OAA by RAFT aqueous emulsion polymerization at 70 °C (i.e. above its melting point) using a water-soluble homopolymer precursor, poly(N-(2-acryloyloxy)ethyl pyrrolidone),35 were unsuccessful: there was either no polymerization at all or OAA underwent conventional free radical polymerization, with no RAFT control being achieved. This failure was attributed to the highly hydrophobic nature of OAA, which has five pendent methyl groups. Presumably, this means that its aqueous solubility is simply too low to enable its emulsion homopolymerization. Similar problems are well-documented for other highly hydrophobic monomers such as lauryl methacrylate or stearyl methacrylate.36
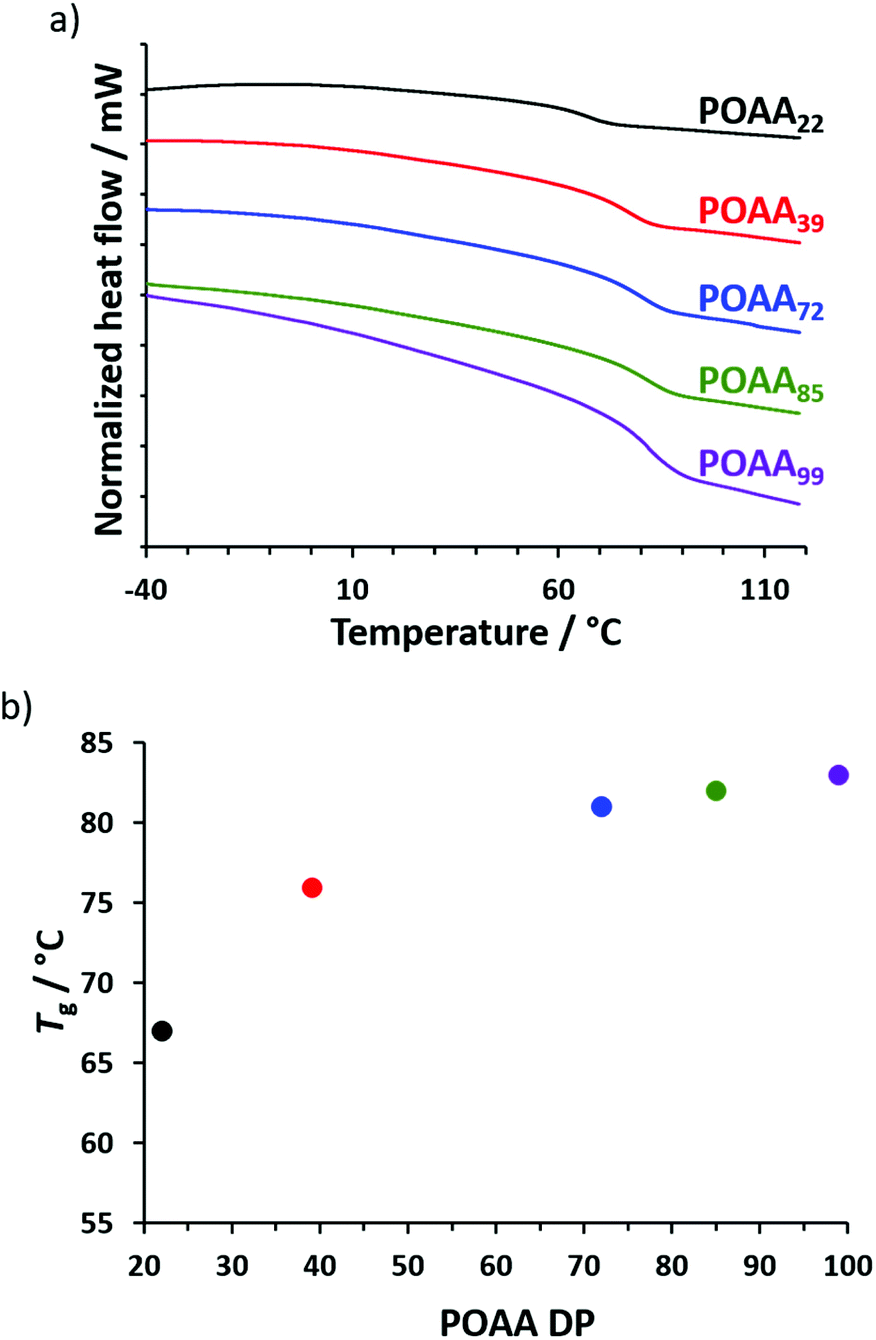 | ||
| Fig. 2 (a) Differential scanning calorimetry (DSC) traces recorded at a heating rate of 10 °C min−1 for a series of five POAAx homopolymers: x = 22 (black), 39 (red), 72 (blue), 85 (green) and 99 (purple). (b) Relationship between Tg and POAA DP plotted for the data shown in (a). | ||
Self-blocking studies were conducted to examine the fidelity of the RAFT end-groups on the POAA85 homopolymer. Accordingly, chain extension experiments targeting POAA DPs of 50, 100 or 150 were performed at 40% w/w solids in 1,4-dioxane. In each case, more than 97% OAA conversion was achieved within 3 h at 70 °C while GPC analysis indicated that the whole molecular weight distribution (MWD) was shifted to higher molecular weight relative to that for the POAA85 precursor (see Fig. 3). Such high RAFT chain-end fidelity augurs well for the synthesis of POAA-based diblock copolymers when using alternative acrylamides for the second-stage polymerization. The observed increase in Mw/Mn after chain extension is comparable to that reported by Kulai and co-workers when performing self-blocking experiments.17
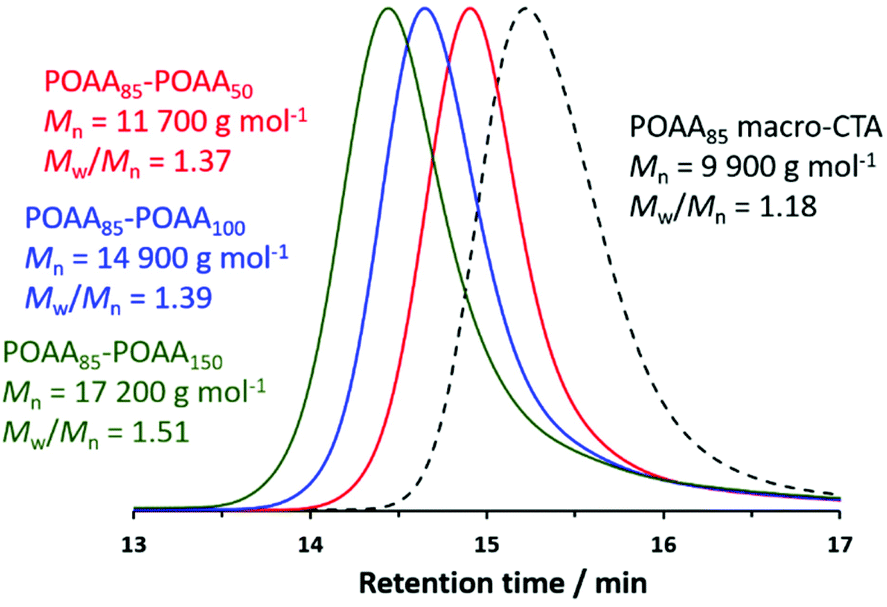 | ||
| Fig. 3 Chloroform GPC curves recorded for a POAA85 precursor and the corresponding chain-extended POAA85-POAAx homopolymers prepared by RAFT solution polymerization of OAA at 70 °C in 1,4-dioxane, where x = 50 (red), 100 (blue) and 150 (green). The unimodal nature of the latter three traces indicates a relatively high blocking efficiency in each case. | ||
RAFT dispersion polymerization of DMAC in n-alkanes using POAA85 as a steric stabilizer block
To prepare sterically-stabilized diblock copolymer nanoparticles, the POAA85 precursor was subsequently chain-extended via RAFT dispersion polymerization of DMAC in n-heptane at 70 °C targeting 20% w/w solids (see Scheme 2). The polymerization kinetics for this chain extension were monitored using 1H NMR spectroscopy (Fig. 4a) while chloroform GPC was used to monitor the evolution in Mn and Mw/Mn (Fig. 4b). Essentially full DMAC conversion was achieved within 5 h and a linear increase in Mn was observed. The final POAA85-PDMAC100 diblock copolymer had an Mn of 21900 g mol−1 and an Mw/Mn of 1.20. These data are consistent with a well-controlled RAFT polymerization.
 | ||
| Scheme 2 Synthesis of a series of POAA85-PDMACx diblock copolymer nanoparticles by RAFT dispersion polymerization of DMAC in n-heptane at 70 °C targeting 20% w/w solids. | ||
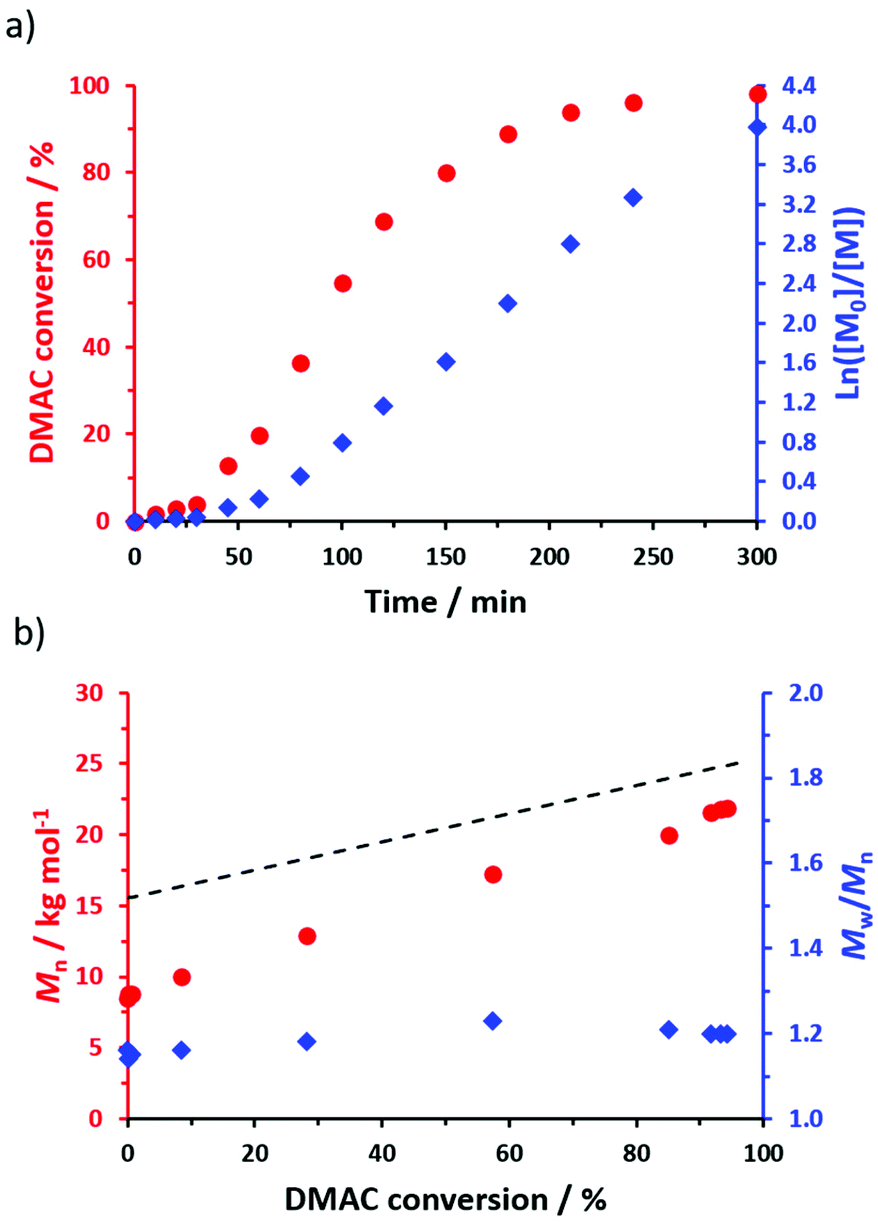 | ||
| Fig. 4 (a) Conversion vs. time curve with the corresponding semi-logarithmic plot obtained for the RAFT dispersion polymerization of DMAC at 70 °C in n-heptane using a POAA85 precursor and targeting a PDMAC DP of 100 at 20% w/w solids (POAA85/AIBN molar ratio = 10). (b) Evolution of Mn and Mw/Mn with DMAC conversion for the same RAFT dispersion polymerization. The dashed line indicates the theoretical Mn data. The experimental Mn data set falls below this theoretical line owing to a systematic GPC calibration error. | ||
A series of POAA85-PDMACx diblock copolymer nanoparticles were prepared in n-heptane (Fig. 5a) with a linear increase in Mn being observed when targeting higher PDMAC DPs. Reasonably good RAFT control was achieved, although a gradual increase in Mw/Mn is discernible when targeting higher PDMAC DPs (see Table S2†). In all cases, high DMAC conversions (≥98%) were achieved as indicated by 1H NMR analysis. A linear relationship was obtained between the z-average nanoparticle diameter determined by dynamic light scattering (DLS) and the target PDMAC DP up to a core DP of 175. Above a target PDMAC DP of 200, somewhat larger nanoparticles were obtained with slightly higher DLS polydispersities (Fig. 5b). For such PISA syntheses, an increase in both nanoparticle diameter and polydispersity can indicate a (partial) change in copolymer morphology, e.g. the presence of some worms rather than just pure spheres.31,37–40 However, close inspection of the corresponding transmission electron microscopy (TEM) images (Fig. S5†) did not provide any evidence for the presence of anisotropic nano-objects.
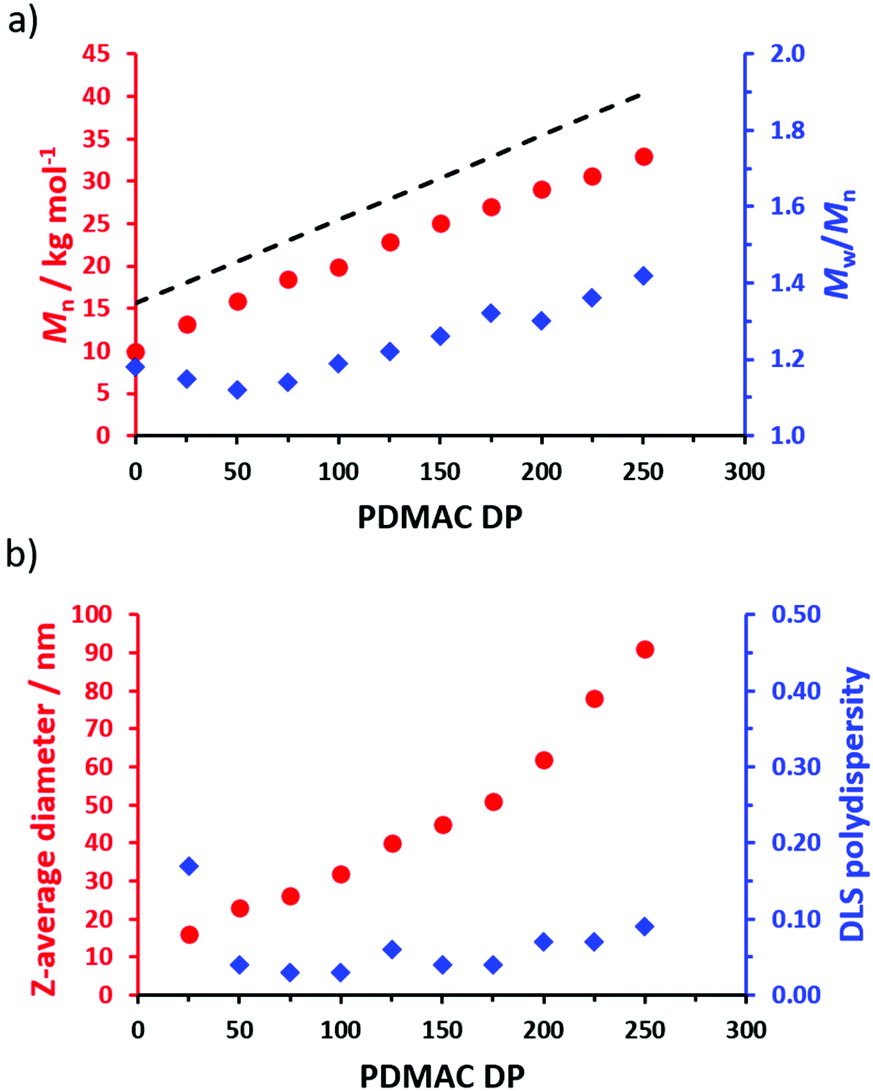 | ||
| Fig. 5 (a) Evolution in Mn and Mw/Mn with target PDMAC DP for a series of POAA85-PDMACx diblock copolymers (refractive index detector with calibration using a series of near-monodisperse PMMA standards). The Mn value for the corresponding POAA85 precursor is also shown as a y-intercept. The dashed line indicated the theoretical Mn data. The experimental Mn data set falls below this theoretical line owing to a systematic GPC calibration error. (b) Evolution in z-average diameter and DLS polydispersity with target PDMAC DP for a series of POAA85-PDMACx nano-objects. Each diblock copolymer dispersion was initially prepared at 20% w/w solids and then diluted to 0.1% w/w solids using n-heptane prior to analysis. | ||
Unfortunately, nanoparticle deformation tended to occur during TEM grid preparation. This problem was not foreseen because the Tg of PDMAC homopolymer has been reported to be 120 °C.41 To address this issue, ethylene glycol diacrylate (EGDA) was added towards the end of the DMAC polymerization when targeting a POAA85-PDMAC195 diblock copolymer. The resulting core-crosslinked POAA85-PDMAC195-PEGDA20 triblock copolymer nano-objects were much more resistant to deformation during TEM grid preparation and exhibited a relatively well-defined spherical morphology (Fig. 6). Moreover, the z-average diameter indicated by DLS studies of these crosslinked nanoparticles was close to that observed for the comparable linear nanoparticles (65 nm vs. 62 nm respectively), see Table S3.† The DLS diameter for the core-crosslinked POAA85-PDMAC195-PEGDA20 spheres was also determined in chloroform. This is a good solvent for both blocks, so nanogel swelling was anticipated under such conditions. Indeed, a significantly larger diameter (90 nm) was observed for such nanoparticles (Table S3†).
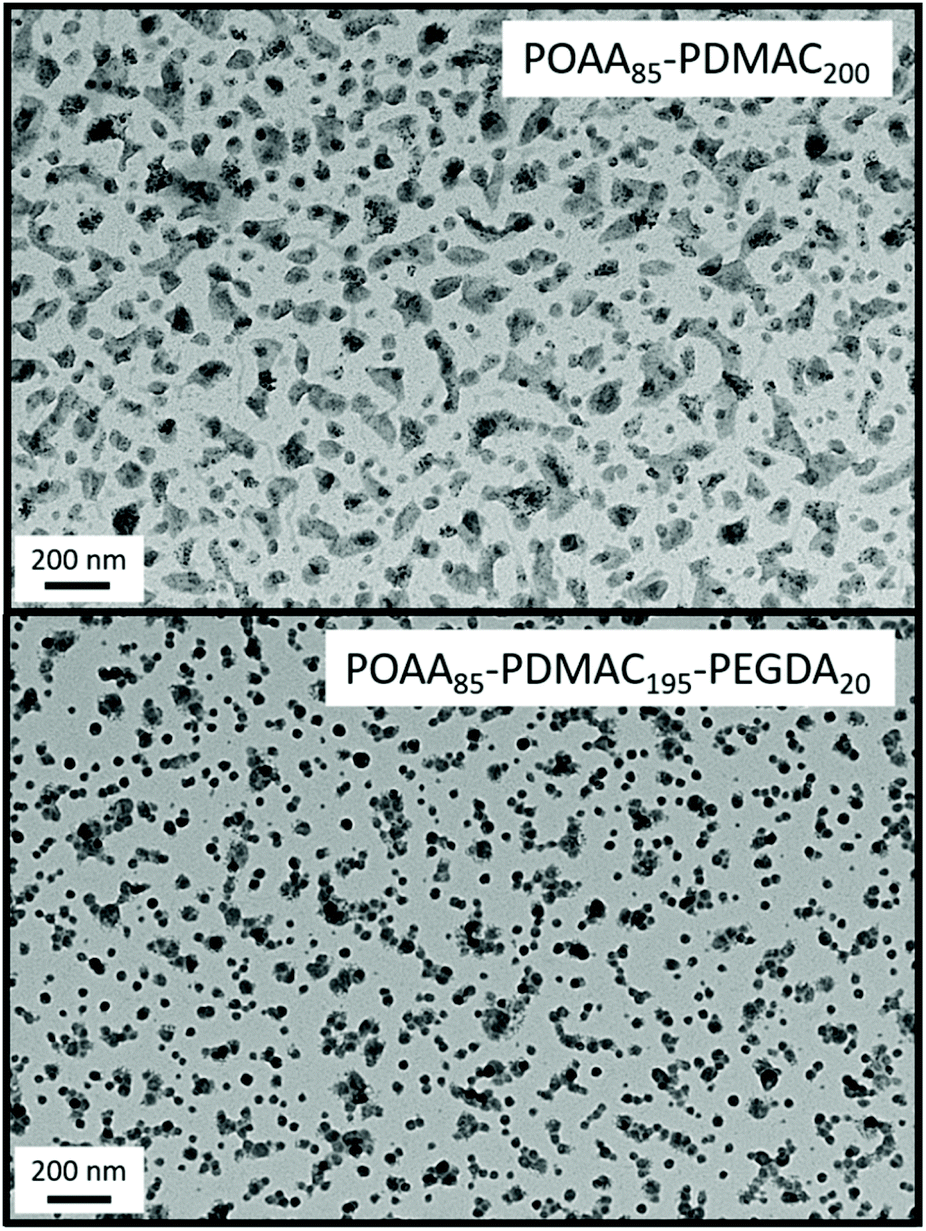 | ||
| Fig. 6 Representative TEM images recorded for linear POAA85-PDMAC200 and core-crosslinked POAA85-PDMAC195-PEGDA20 nano-objects. The latter nanoparticles exhibit a relatively well-defined spherical morphology, whereas the former nanoparticles tend to undergo film formation during TEM grid preparation. | ||
To demonstrate the potential industrial relevance of such PISA formulations, a one-pot synthetic protocol was developed to produce POAA82-PDMAC100 diblock copolymer nano-objects directly in n-heptane (Table S2†). First a POAA82 precursor was prepared by RAFT solution polymerization of OAA in n-heptane when targeting a DP of 80. An OAA conversion of 98% was achieved within 150 min with an Mn of 8100 g mol−1 and an Mw/Mn of 1.16 being indicated by chloroform GPC analysis (Fig. 7). A deoxygenated solution containing DMAC and n-heptane was added to this reaction solution to target POAA82-PDMAC100 nano-objects at 20% w/w solids. The DMAC polymerization was allowed to proceed for 5 h at 70 °C. A final monomer conversion of more than 99% was determined by 1H NMR analysis and chloroform GPC analysis indicated an Mn of 18500 g mol−1 and an Mw/Mn of 1.19 for the final POAA82-PDMAC100 diblock copolymer chains (see Fig. 7). The Mn and Mw/Mn data were comparable to the diblock copolymer nano-objects prepared by a two-step protocol. Similarly, the resulting spherical nanoparticles had a z-average diameter of 36 nm (DLS polydispersity = 0.05) which is consistent with the z-average diameter of 32 nm (DLS polydispersity = 0.03) obtained for the two-pot synthesis, see Fig. 5.
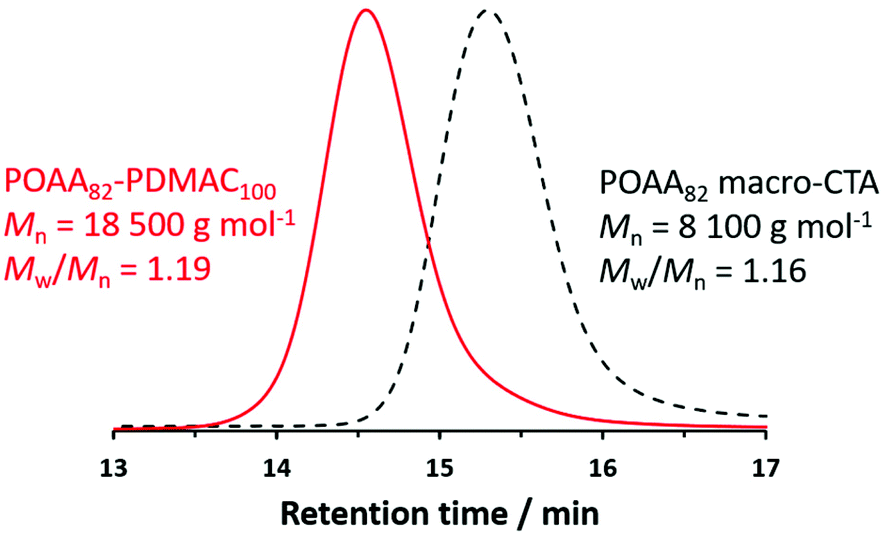 | ||
| Fig. 7 Chloroform GPC curves recorded for the initial POAA82 precursor (98% conversion) and the final POAA82-PDMAC100 diblock copolymer (more than 99% conversion after 5 h at 70 °C) prepared by a one-pot protocol targeting 20% w/w solids via RAFT dispersion polymerization of DMAC in n-heptane. | ||
The PISA synthesis of POAA85-PDMAC150 nanoparticles was also conducted in n-dodecane at 70 °C. As expected, a free-flowing turbid dispersion was observed at this reaction temperature. However, an opaque, free-standing paste was formed on cooling this 20% w/w dispersion to 20 °C (Fig. S6†).
To further examine this unexpected behavior, DLS particle size distributions were determined for POAA85-PDMAC150 nano-objects prepared in either n-heptane or n-dodecane at temperatures ranging from 80 °C to 20 °C (Fig. 8). On cooling a dispersion of POAA85-PDMAC150 nanoparticles prepared in n-heptane, both the z-average diameter (∼46 nm) and DLS polydispersity (∼0.05) remained essentially constant across the whole temperature range. In contrast, for the same nanoparticles prepared in n-dodecane, the apparent particle diameter increased dramatically from 52 nm (40–80 °C) up to 276 nm (20 °C), indicating that aggregation occurs on cooling. Such aggregation was accompanied by a substantial increase in DLS polydispersity (from less than 0.10 to more than 0.57). However, this aggregation proved to be reversible on heating, indicating weak flocculation and minimal hysteresis.
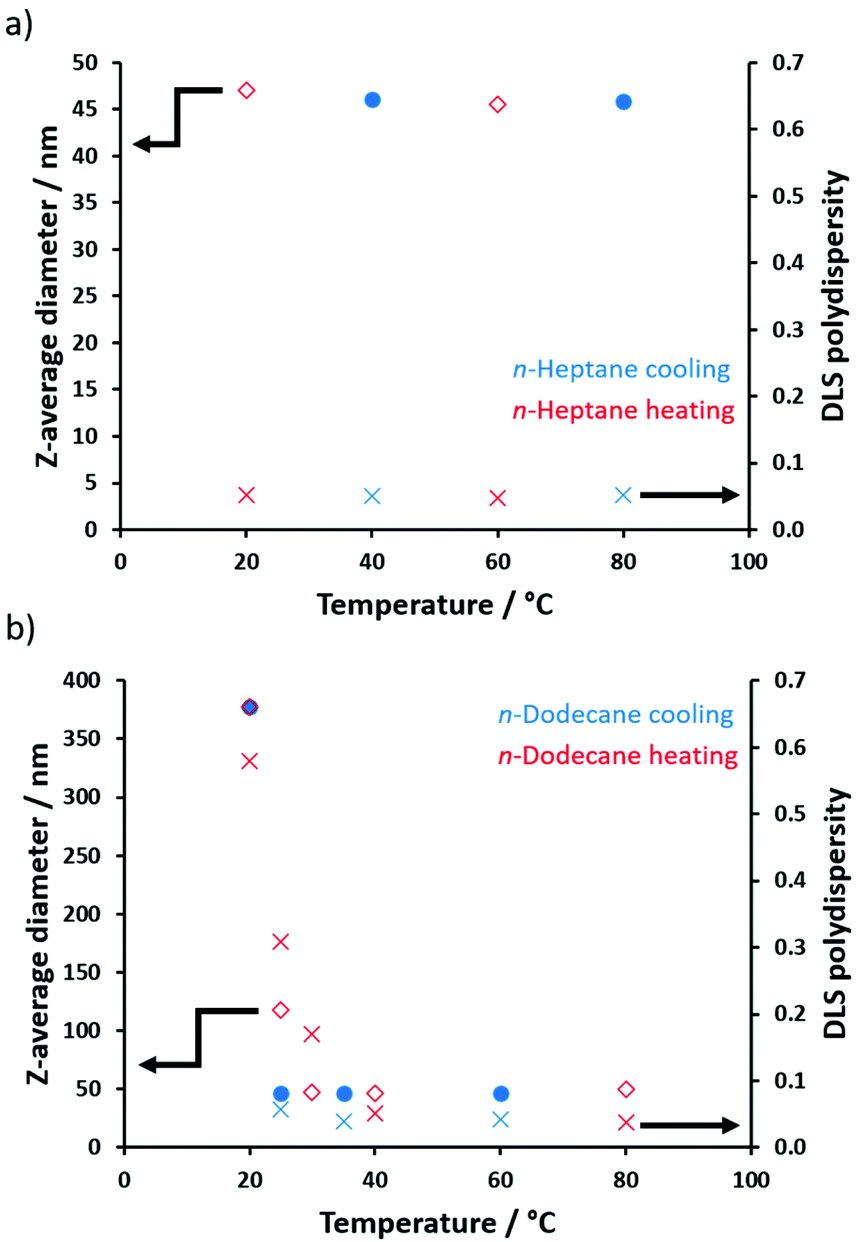 | ||
| Fig. 8 Temperature dependence of the z-average diameter and corresponding DLS polydispersity determined for the following 0.1% w/w copolymer dispersions: (a) POAA85-PDMAC150 nanoparticles prepared in n-heptane cooled from 80 to 20 °C and then heated from 20 to 80 °C; (b) POAA85-PDMAC150 nanoparticles prepared in n-dodecane cooled from 80 °C to 20 °C then heated from 20 °C to 80 °C. These DLS experiments confirm that these nanoparticles exhibit thermoreversible flocculation with minimal hysteresis in n-dodecane but no such aggregation occurs in n-heptane. | ||
In principle, such thermoresponsive behavior might be an example of crystallization-driven aggregation, whereby an initially stable colloidal dispersion becomes aggregated owing to crystallization between neighbouring steric stabilizer chains.42 However, DSC studies indicated no crystallization event when cooling either a 20% w/w dispersion of POAA85-PDMAC150 nanoparticles in n-dodecane or a 20% w/w solution of POAA85 homopolymer in n-dodecane from 120 °C to −50 °C (see Fig. S7†). This suggests that the thermoreversible flocculation observed for the POAA85-PDMAC150 nanoparticles in n-dodecane is simply due to the upper critical solution temperature (UCST)-like behavior of the POAA stabilizer chains, which become less solvated at lower temperature. If this is the case, then POAA85 homopolymer should exhibit UCST behavior, i.e. it should be soluble in n-dodecane at 80 °C but precipitate on cooling to 20 °C.
Perhaps surprisingly, turbidimetry studies conducted on POAA85 homopolymer in various n-alkanes did not provide any evidence for UCST-type behavior. More specifically, this homopolymer remained soluble between 20 and 90 °C when dissolved in n-heptane, n-octane, n-decane and n-dodecane. However, visual inspection confirms that this homopolymer is indeed insoluble in n-tetradecane and n-hexadecane at 20 °C (see Fig. S8†). Unfortunately, we have been unable to determine the critical flocculation temperature for such phase separation via turbidimetry. Moreover, DSC analysis of a 50% w/w solution of POAA85 homopolymer in either n-tetradecane or n-hexadecane indicated no UCST behavior (Fig. S9†) when heating from −60 °C to 100 °C. Instead, only a strong endothermic peak corresponding to the melting point of the solvent is observed at 7 °C for n-tetradecane and 18 °C for n-hexadecane, respectively.
Many polymers exhibit temperature-dependent solubility in various solvents.43–49 For example, UCST behavior is typically observed for polymers in organic solvents, with relatively few examples being reported in aqueous solution.43 A well-documented example of a UCST system is polystyrene/cyclohexane; this polymer is soluble in hot cyclohexane but becomes insoluble on cooling below 35 °C.45–47 According to Imre and co-workers, styrene oligomers exhibit UCST behavior in n-alkanes.48 Similarly, poly(ethylene oxide) exhibits UCST behavior in ethanol.49
POAA85-PDMACx nanoparticles prepared in n-dodecane, n-tetradecane or n-hexadecane invariably formed waxy pastes on cooling, indicating UCST-like thermoreversible flocculation. To further investigate this phenomenon, POAA85-PDMAC150 nanoparticles were prepared directly in turn in each of the six n-alkanes via PISA (Table S2†).
Relatively good RAFT control (high blocking efficiencies, similar Mn values, unimodal MWDs and relatively low Mw/Mn values) was achieved during the RAFT dispersion polymerization of DMAC at 70 °C using a POAA85 macro-CTA in n-heptane, n-octane, n-decane, n-dodecane or n-tetradecane (Fig. 9a). In marked contrast, only poor RAFT control (inefficient chain extension, a bimodal MWD and a relatively high Mw/Mn of 1.58) was observed when the same synthetic protocol was conducted in n-hexadecane. Initially, the DMAC monomer acts as a co-solvent and ensures solubility of the POAA85 precursor in n-hexadecane. However, as the DMAC polymerization proceeds, the monomer concentration falls and the solvency gradually worsens, which leads to nanoparticle flocculation as well as loss of RAFT control. To address this problem, the PISA synthesis of POAA85-PDMAC150 nanoparticles in n-hexadecane was also attempted at 90 °C, which is well above the UCST of 55 °C exhibited by the same (target) nanoparticles in this solvent when cooling at 5 °C min−1 (see Fig. 9b). This latter protocol produced a slightly higher Mn of 20000 g mol−1 but the MWD remained bimodal and relatively broad (Mw/Mn = 1.76).
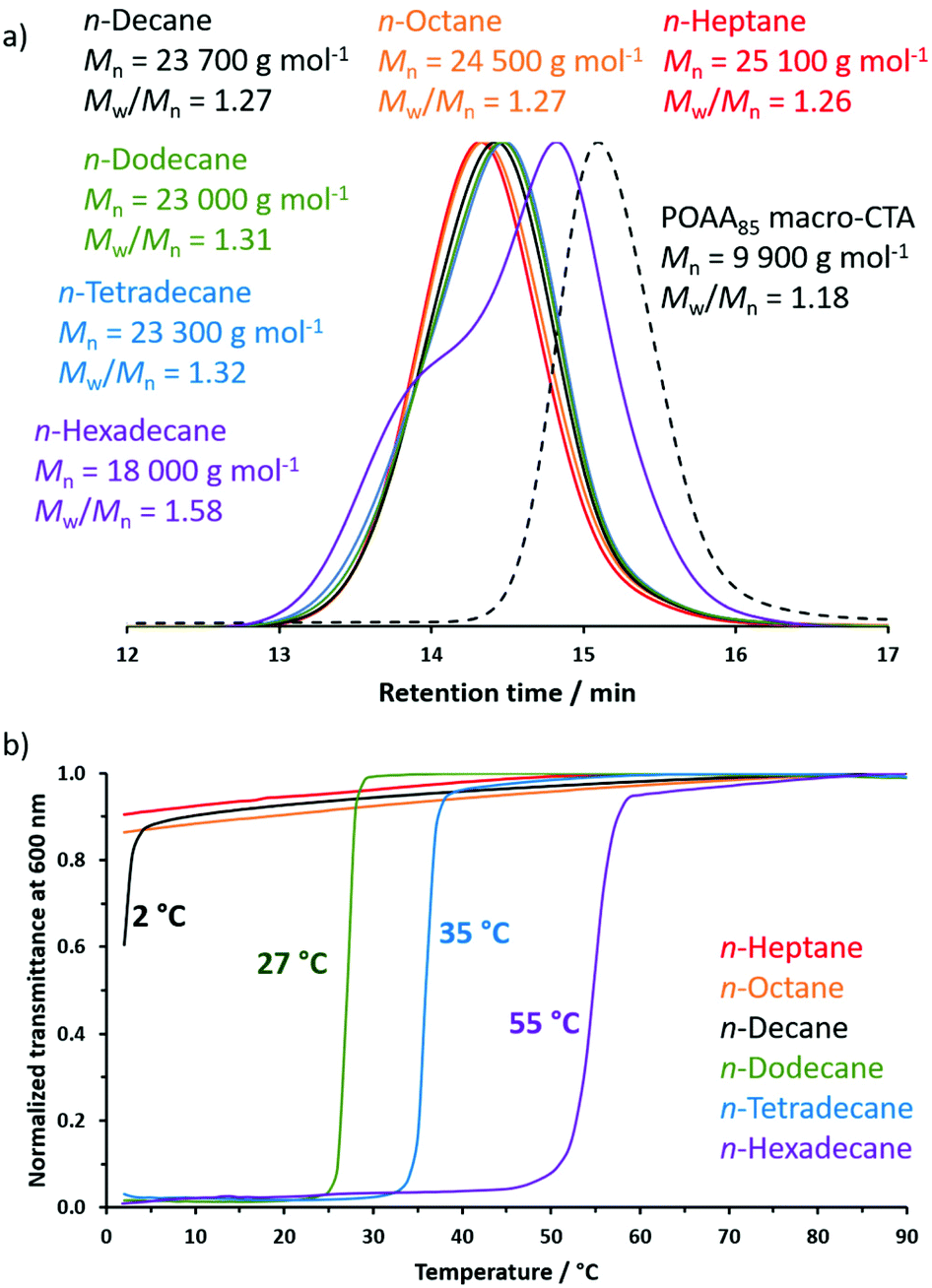 | ||
| Fig. 9 (a) Chloroform GPC curves recorded for a series of POAA85-PDMAC150 diblock copolymers prepared by RAFT dispersion polymerization of DMAC using a POAA85 precursor at 70 °C in n-heptane (red), n-octane (orange), n-decane (black), n-dodecane (green), n-tetradecane (blue) or n-hexadecane (purple). The GPC curve for the POAA85 precursor (dashed black line) is also included as a reference. (b) Normalized transmittance (λ = 600 nm) against temperature curves recorded for 1.0% w/w dispersions of POAA85-PDMAC150 nanoparticles on cooling from 90 °C to 2 °C. | ||
The turbidity of this series of six POAA85-PDMAC150 dispersions was evaluated in turn at an arbitrary wavelength of 600 nm on cooling from 90 to 20 °C (Fig. 9b). Initially, each of these 1.0% w/w dispersions exhibited high transmittance, indicating minimal light scattering and good colloidal stability. In the case of n-hexadecane, when cooling at 1 °C min−1 the dispersion became relatively opaque below 67 °C owing to the onset of aggregation (see Fig. S10† for the effect of varying the cooling rate on the transmittance vs. temperature plot obtained for this copolymer dispersion). On cooling the dispersion further, the nanoparticles sedimented to the bottom of the cuvette, resulting in a final non-zero transmittance (see inset in Fig. S10†). Thus the data shown in Fig. 9b was recorded at a faster cooling rate of 5 °C min−1. In contrast, the critical flocculation temperature observed for such POAA85-PDMAC150 nanoparticles is approximately 35 °C in n-tetradecane and 27 °C in n-dodecane. (N.B. In all cases, nanoparticle light scattering means that such dispersions never become fully transparent even at 90 °C, hence the turbidity data were normalized with respect to the highest transmittance value). Bearing in mind the difference in nanoparticle concentration and cooling rate, the increase in turbidity observed at around 27 °C for n-dodecane is reasonably consistent with the onset of flocculation below 25 °C indicated by DLS studies conducted in the same solvent (see Fig. 8). The onset of nanoparticle aggregation in n-decane was observed below 5 °C, which is close to the minimum temperature for our instrument set-up. Accordingly, an ice bath was used to lower the temperature of this 1.0% w/w nanoparticle dispersion to −1 °C, which resulted in macroscopic precipitation (see Fig. S11†). In contrast, only minimal changes in turbidity were observed for POAA85-PDMAC150 nanoparticles prepared in n-octane and n-heptane, suggesting that colloidal stability is retained in these lower n-alkanes at sub-ambient temperatures.
Conclusions
RAFT solution homopolymerization of a highly hydrophobic acrylamide-based monomer, OAA, has been conducted in 1,4-dioxane. GPC studies confirm that good control over the molecular weight distribution can be achieved when using a suitable trithiocarbonate-based RAFT agent. Five well-defined POAAx homopolymers were prepared by systematically varying the [OAA]/[DDMAT agent] molar ratio and their respective mean DPs were calculated by end-group analysis using UV spectroscopy. DSC studies indicate a modest increase in Tg when targeting higher POAA DPs. A POAA85 precursor was subsequently employed for the RAFT dispersion polymerization of DMAC in n-heptane. A series of sterically-stabilized POAA85-PDMACx diblock copolymer spheres was produced with high DMAC conversions being achieved in all cases (≥98% within 5 h at 70 °C). An increase in both z-average diameter and molecular weight was observed when targeting higher PDMAC DPs, albeit at the expense of reduced RAFT control. TEM studies of the linear diblock nanoparticles proved to be problematic owing to nanoparticle deformation during sample preparation. Thus, EGDA was employed as a bifunctional crosslinker and added towards the end of the DMAC polymerization to produce covalently-stabilized nanoparticles. This strategy enabled a well-defined spherical morphology to be confirmed by TEM while also producing nanogels that swelled when dispersed in chloroform, which is a good solvent for both blocks. Moreover, well-defined POAA82-PDMAC100 nanoparticles could be prepared using an atom-efficient one-pot protocol that augurs well for potential industrial scale-up for such PISA formulations.The temperature-dependent colloidal stability of a series of POAA85-PDMAC150 nanoparticles prepared in n-heptane, n-octane, n-decane, n-dodecane, n-tetradecane or n-hexadecane at 70 °C was investigated using turbidimetry. When prepared in either n-heptane or n-octane, the nanoparticles remained well-dispersed at all temperatures. However, thermoreversible flocculation of the nanoparticles was observed on cooling from 70 °C to 20 °C for the four higher n-alkanes, with progressively higher critical flocculation temperatures being observed when increasing the n-alkyl chain length. This UCST-like behavior is attributed to poor solvation of the POAA85 stabilizer block at lower temperature, which is consistent with the insolubility of this precursor in n-tetradecane and n-hexadecane at 20 °C.
Conflicts of interest
The authors declare a patent application made by Ashland relating to the results described herein.Acknowledgements
EPSRC is thanked for funding a CDT PhD studentship for the first author (EP/L016281). Ashland Specialty Ingredients (Bridgewater, New Jersey, USA) is thanked for partial financial support of this PhD project, for supplying the OAA monomer and for permission to publish this work. SPA also thanks the ERC for a five-year Advanced Investigator grant (PISA 320372) and the EPSRC for an Established Career Particle Technology Fellowship (EP/R003009).References
- D. Y. Kim, J. G. Lee, B. N. Joshi, S. S. Latthe, S. S. Al-Deyab and S. S. Yoon, J. Mater. Chem. A, 2015, 3, 3975–3983 RSC.
- Y. Tang, Q. Zhang, X. Zhan and F. Chen, Soft Matter, 2015, 11, 4540–4550 RSC.
- B. Thallinger, E. N. Prasetyo, G. S. Nyanhongo and G. M. Guebitz, Biotechnol. J., 2013, 8, 97–109 CrossRef CAS PubMed.
- A. Vaterrodt, B. Thallinger, K. Daumann, D. Koch, G. M. Guebitz and M. Ulbricht, Langmuir, 2016, 32, 1347–1359 CrossRef CAS PubMed.
- M. Spasova, N. Manolova, N. Markova and I. Rashkov, Appl. Surf. Sci., 2016, 363, 363–371 CrossRef CAS.
- J. Li, L. Yan, H. Li, J. Li, F. Zha and Z. Lei, RSC Adv., 2015, 5, 53802–53808 RSC.
- K. Jayaramulu, K. K. R. Datta, C. Rösler, M. Petr, M. Otyepka, R. Zboril and R. A. Fischer, Angew. Chem., Int. Ed., 2016, 55, 1178–1182 CrossRef CAS PubMed.
- A. L. Micchelli, G. J. Legato, S. H. Ganslaw, L. D. Schuler and N. Brunswick, US3927199A, 1973.
- R. Krohn and E. Schulze zur Wiesche, WO2020089367, 2020.
- D. Metten, R. Scheffler, J. B. Lange and C. Martinez, WO2018177722, 2018.
- W. Shi and C. P. Palmer, Electrophoresis, 2002, 23, 1285–1295 CrossRef CAS PubMed.
- M. Rumyantsev, O. A. Kazantsev, S. I. Kamorina, D. M. Kamorin and A. P. Sivokhin, J. Mol. Struct., 2016, 1121, 86–92 CrossRef CAS.
- J. F. Bork, D. P. Wyman and L. E. Coleman, J. Appl. Polym. Sci., 1963, 7, 451–459 CrossRef CAS.
- L. M. Gouveia, S. Paillet, A. Khoukh, B. Grassl and A. J. Müller, Colloids Surf., A, 2008, 322, 211–218 CrossRef CAS.
- E. K. Penott-Chang, L. Gouveia, I. J. Fernández, A. J. Müller, A. Díaz-Barrios and A. E. Sáez, Colloids Surf., A, 2007, 295, 99–106 CrossRef CAS.
- A. I. Barabanova, E. V. Bune, A. V. Gromov and V. F. Gromov, Eur. Polym. J., 2000, 36, 479–483 CrossRef CAS.
- I. Kulai, O. Brusylovets, Z. Voitenko, S. Harrisson, S. Mazières and M. Destarac, ACS Macro Lett., 2015, 4, 809–813 CrossRef CAS.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS PubMed.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- J. Zhou, H. Yao and J. Ma, Polym. Chem., 2018, 9, 2532–2561 RSC.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 CrossRef PubMed.
- C. E. Boott, J. Gwyther, R. L. Harniman, D. W. Hayward and I. Manners, Nat. Chem., 2017, 9, 785–792 CrossRef CAS PubMed.
- A. J. Morse, S. P. Armes, K. L. Thompson, D. Dupin, L. A. Fielding, P. Mills and R. Swart, Langmuir, 2013, 29, 5446–5475 Search PubMed.
- A. B. Lowe, Polymer, 2016, 106, 161–181 CrossRef CAS.
- J. Chiefari, Y. K. B. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- L. A. Fielding, J. A. Lane, M. J. Derry, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 5790–5798 CrossRef CAS PubMed.
- K. Skrabania, A. Miasnikova, A. M. Bivigou-Koumba, D. Zehm and A. Laschewsky, Polym. Chem., 2011, 2, 2074–2083 RSC.
- B. Klumperman, E. T. A. van den Dungen, J. P. A. Heuts and M. J. Monteiro, Macromol. Rapid Commun., 2010, 31, 1846–1862 CrossRef CAS PubMed.
- O. J. Deane, O. M. Musa, A. Fernyhough and S. P. Armes, Macromolecules, 2020, 53, 1422–1434 CrossRef CAS.
- S. Rimmer and P. Tattersall, Polymer, 1999, 40, 5729–5731 CrossRef CAS.
- Y. Kang, A. Pitto-Barry, H. Willcock, W. D. Quan, N. Kirby, A. M. Sanchez and R. K. O'Reilly, Polym. Chem., 2015, 6, 106–117 RSC.
- S. L. Canning, V. J. Cunningham, L. P. D. Ratcliffe and S. P. Armes, Polym. Chem., 2017, 8, 4811–4821 RSC.
- V. J. Cunningham, S. P. Armes and O. M. Musa, Polym. Chem., 2016, 7, 1882–1891 RSC.
- S. Qu, R. Liu, W. Duan and W. Zhang, Macromolecules, 2019, 52, 5168–5176 CrossRef CAS.
- X. Xie and T. E. Hogen-Esch, Macromolecules, 1996, 29, 1746–1752 CrossRef CAS.
- M. J. Derry, O. O. Mykhaylyk, A. J. Ryan and S. P. Armes, Chem. Sci., 2018, 9, 4071–4082 RSC.
- J. Seuring and S. Agarwal, Macromol. Rapid Commun., 2012, 33, 1898–1920 CrossRef CAS PubMed.
- D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214–7243 RSC.
- S. Saeki, N. Kuwahara, S. Konno and M. Kaneko, Macromolecules, 1973, 6, 589–593 CrossRef CAS.
- S. A. Vshivkov and A. P. Safronov, Macromol. Chem. Phys., 1997, 198, 3015–3023 CrossRef CAS.
- A. Siporska, J. Szydlowski and L. P. N. Rebelo, Phys. Chem. Chem. Phys., 2003, 5, 2996–3002 RSC.
- A. R. Imre, W. A. Van Hook, B. H. Chang and Y. C. Bae, Monatsh. Chem., 2003, 134, 1529–1539 CrossRef CAS.
- D. L. Ho, B. Hammouda, S. R. Kline and W. R. Chen, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 557–564 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00045d |
| This journal is © The Royal Society of Chemistry 2021 |