
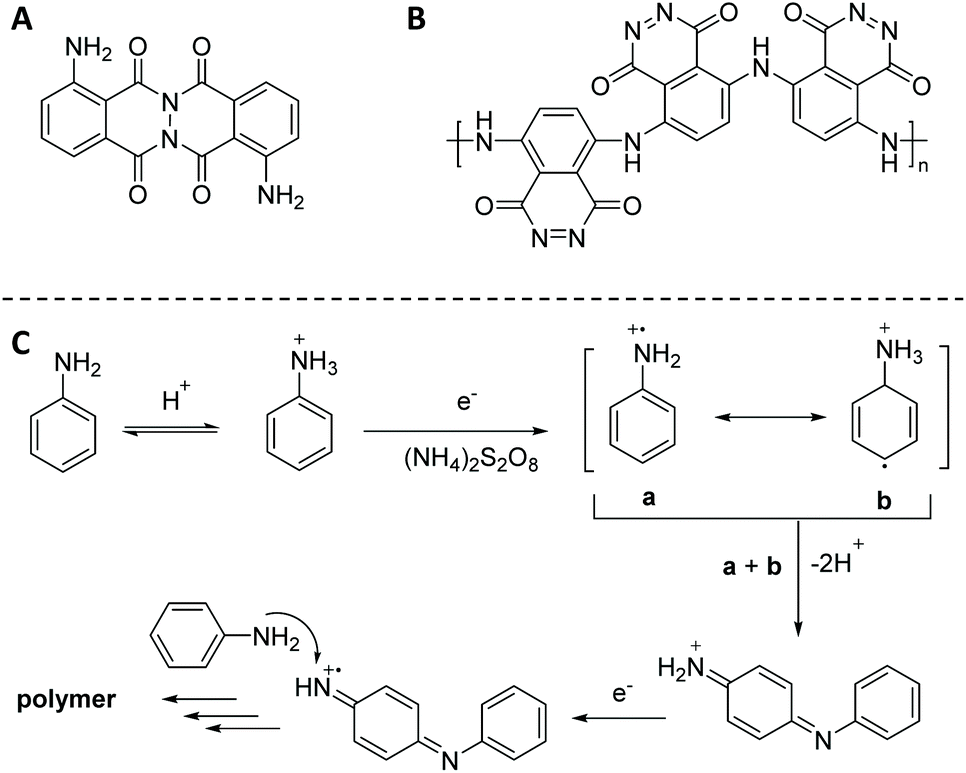
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUntapped toolbox of luminol based polymers
Christina M.
Geiselhart
 ab,
Christopher
Barner-Kowollik
bc and
Hatice
Mutlu
*a
ab,
Christopher
Barner-Kowollik
bc and
Hatice
Mutlu
*a
aSoft Matter Synthesis Laboratory, Institut für Biologische Grenzflächen 3 (IBG-3), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: hatice.mutlu@kit.edu
bInstitute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology (KIT), Engesserstr.18, D-76131 Karlsruhe, Germany
cSchool of Chemistry and Physics, Centre for Materials Science, Queensland University of Technology (QUT), 2 George Street, Brisbane, QLD 4000, Australia
First published on 5th February 2021
Abstract
The objective of the current Perspective article is to highlight the present state of luminol based polymers, with a specific emphasis on how to include luminol derivatives into polymer chains using both electrochemical and chemical techniques with an underline on new synthetic methods. Importantly, the current limitations that limit the expansion of polymeric luminol derivatives will be discussed by drawing attention to the challenges of solubilising monomers, the harsh conditions leading to undesired side reactions during the polymerization process or the necessity of orthogonal post-modification reactions. Importantly, the article discusses the remaining challenges within the field, while suggesting strategies for the advancement of this versatile class of polymers.
 Christina M. Geiselhart | Christina M. Geiselhart received her M.Sc. in Chemistry from the Karlsruhe Institute of Technology (KIT, Germany) in 2017, where she performed her undergraduate studies. Currently, she is a PhD student at the KIT under the supervision of Dr Hatice Mutlu and Prof. Christopher Barner-Kowollik, having spent research time both at the KIT and the Queensland University of Technology (QUT). Her research focuses on the development of macromolecular smart materials with self-reporting properties. |
 Christopher Barner-Kowollik | Christopher Barner-Kowollik, Australian Research Council (ARC) Laureate Fellow, graduated in chemistry from Goettingen University, Germany, Christopher joined the University of New South Wales in early 2000 rising to lead the Centre for Advanced Macromolecular Design in 2006 as one of its directors. He returned to Germany to the Karlsruhe Institute of Technology (KIT) in 2008, where he established and led a Centre of Excellence in soft matter synthesis. He moved to QUT in early 2017 and established QUT's Soft Matter Materials Laboratory. Christopher's research achievements have been recognised by an array of national and international awards including the coveted Erwin Schrodinger Award of the German Helmholtz Association and most recently the United Kingdom Macro Medal. He is a Fellow of the Australian Academy of Science, the Royal Society of Chemistry and the Royal Australian Chemical Institute. |
 Hatice Mutlu | Dr Hatice Mutlu, born in Bulgaria, studied Chemistry at Marmara University and Bogazici University. Subsequently, she obtained her PhD from the Karlsruhe Institute of Technology (KIT) in the group of M. A. R. Meier. After post-doctoral stays in the groups of J.-F. Lutz (ICS-CNRS, Strasbourg) and C. Barner-Kowollik (KIT), respectively, she currently works as a senior researcher at KIT. Her research interest spans from the synthesis of complex macromolecular architectures and functional polymers to the development of new polymer-forming reactions and novel conjugation chemistries with a particular focus on design of novel sulfur-based and self-reporting materials. |
Introduction
Light emission phenomena (i.e. chemiluminescence and fluoresence, CL) have been associated to mythological and cultural events since the beginning of humankind.1,2 Therefore, it is not surprising that chemiluminogens,3 which are known to emit light mostly through oxidation–reduction processes, represent an important class among the wide toolbox of stimuli responsive soft matter,4 particularly as self-reporting materials.5 Indeed, chemiluminogens6 have paved the way towards a variety of advanced technological applications (e.g. sensors,7 bioimaging materials8 and light emitting diodes9 amongst others), due to their high sensitivity and wide dynamic range without the requirement of sophisticated equipment. Nature offers a variety of bioluminogenic systems10–12 (such as fireflies,13 jellyfish14 and squids),15 whereas purely organic and nonbiological chemiluminogens are rare. The frequently-employed synthetic chemiluminogens16 can be categorized mainly into three groups as oxalate esters (Scheme 1A),17 dioxetanes (Scheme 1B)18 and 5-amino-2,3-dihydrophthalazine-1,4-dione (luminol)19,20 derivatives (Scheme 1C). Nevertheless, a literature survey reveals that luminol derivatives that are characterized by blue light emission (λ close to 425 nm) upon oxidative conditions remain as one of the most attractive chemiluminogens, as the covalent conjugation of oxalate esters is generally difficult to achieve. In a similar manner, dioxetanes are labile and prone to undergo decomposition by heat and/or light as a result of intrinsic structural sensitivity.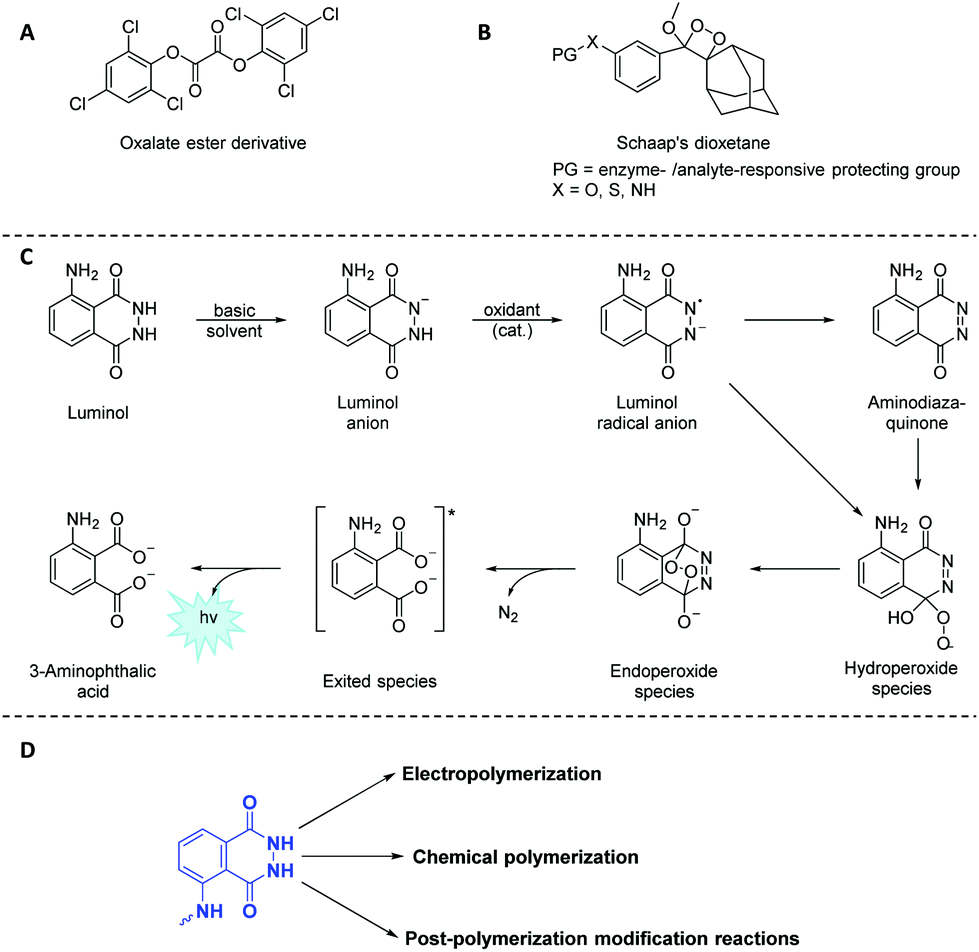 | ||
| Scheme 1 Schematic representation of the frequently-employed synthetic chemiluminogens: (A) oxalate esters, (B) dioxetanes, and (C) 5-amino-2,3-dihydrophthalazine-1,4-dione (luminol). (D) Possible pathways to include luminol derivatives in a polymeric backbone using both electrochemical and chemical techniques in addition to orthogonal post-polymerization modification reactions. | ||
Since its first utilization by Specht to analyze crime scenes,21 luminol and its derivatives face a myriad of applications in the fields of analytical chemistry,22 biotechnology23 and forensic science24 as efficient (electro)chemiluminogen. Inevitably, new designs concerning the modifications of luminol structure have emerged in the recent years in order to tune the chemiluminescence efficiency, intensity, sensitivity, quantum yield or the recognition ability of the resulting chemiluminogens. Recently, a structured literature review25 on the most essential and recent information regarding the chemical features, CL, clinical and nonclinical applications along the future uses of luminol and its small molecule derivatives has been provided.
Considering that polymers play a key role in industry and everyday life due to their tuneable properties, luminol (read-out) polymeric systems have rarely been investigated due to the lack of a comprehensive overview on such polymers. It is also of critical importance to understand the distinctive chemical characteristics of luminol-based polymers, since these characteristics ultimately dictate the performance of the specified polymers in different fields of application ranging from self-reporting sensors to biomedicine. While we make no claim that the list of examples is complete, the selection of examples within the review is selected to show what is synthetically possible. Accordingly, we will first discuss how to include luminol derivatives in a polymeric backbone using both electrochemical and chemical techniques (Scheme 1D) with an emphasis on new synthetic methods. Importantly, we will discuss the present limitations that restrict the full exploitation of polymeric luminol derivatives, specifically (i) the difficulty of solubilising monomers, (ii) the harsh conditions required, leading to undesired side reactions during the polymerization process or (iii) the necessity of orthogonal post-modification reactions (Scheme 1D). Furthermore, we will explore the importance of tailored polymers which fulfil a wide range of applications by adjusting their composition, structure, inherent chemical and physical properties. Finally, we will provide a perspective on future research towards the synthesis of luminol-based polymer derivatives, from both a fundamental and application perspective.
1. Electro-(co)polymerized luminol and its derivatives
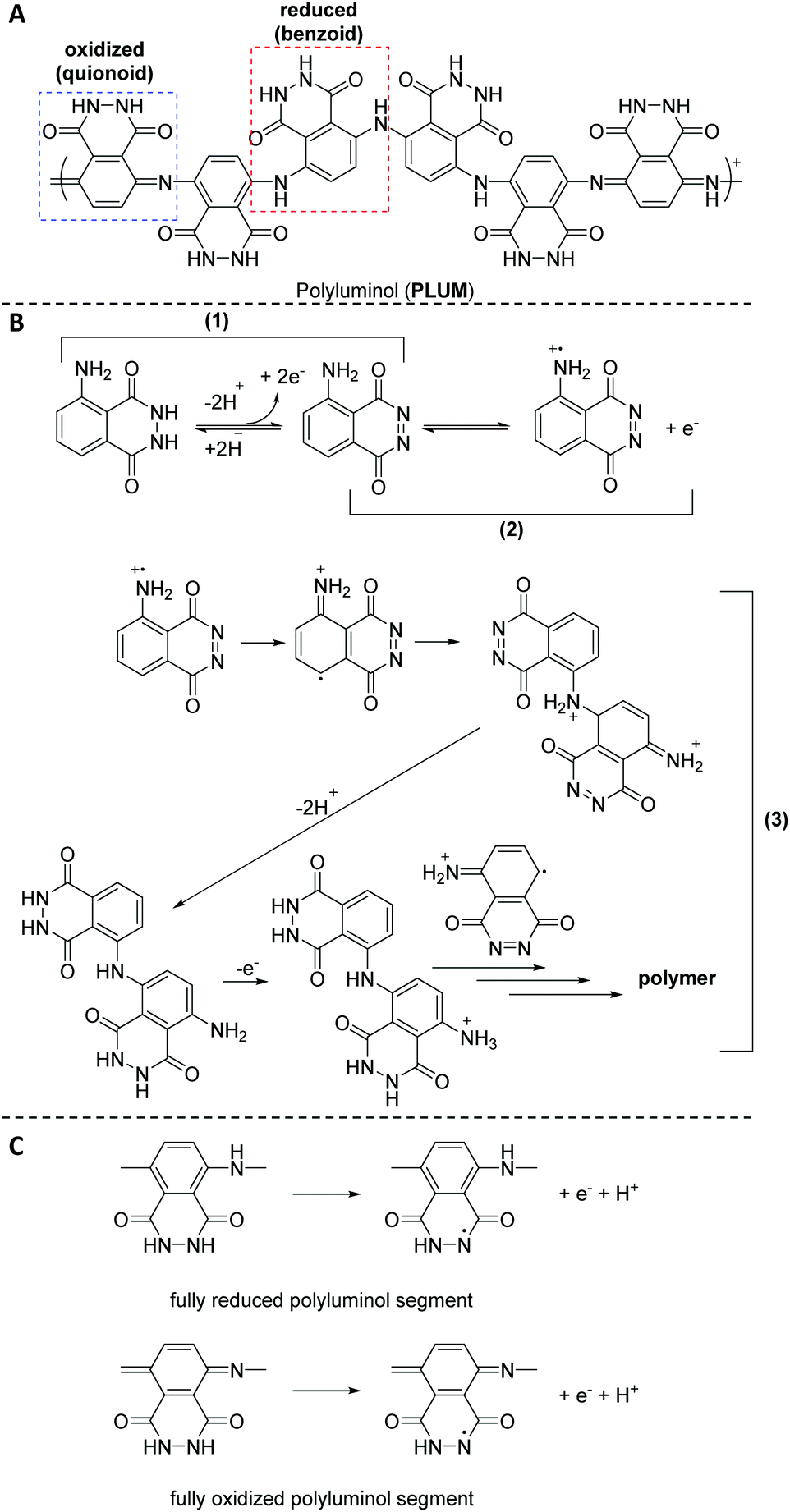 | ||
| Scheme 2 Schematic representation of: (A) polyluminol (PLUM) with reduced (benzoid) and oxidized (quinoid) form, (B) the postulated mechanism of electrochemical polymerization, and (C) benzoid (fully reduced) and quinoid (fully oxidized) polyluminol segments. | ||
Fu and colleagues45 were the first to produce strongly adhering polymeric films by the luminol radical anions during the ECL analysis of luminol in aqueous alkaline solution on the surface of platinum electrodes; however, without any detailed discussion. Subsequently, Chen35et al. explored the electrochemical polymerization behaviour of luminol (0.84 mM) in acidic solution on the surface of platinum electrodes. Among H2SO4, HCl, H3PO4 and acetic acid, H2SO4 was identified as the best acid with the optimal concentration of 0.5 M. The polymerization was observed either under potentiostatic (i.e. in the present of constant potential electrolysis (E = 1.2 V vs. SCE)) or potentiodynamic (cyclic scan from −0.20 to 1.2 V (vs. SCE)) conditions. The obtained polymeric film was directly characterized on the electrode surface by cyclic voltammetry (CV) and surface enhanced Raman scattering (SERS) spectra. The CV analysis revealed that when the second oxidation of luminol oxidation was within the anodic limit potential, two redox processes were observed, respectively at +0.3 and +0.5 V (vs. SCE). Critically, the mechanism of the polymerization has also been discussed. Specifically, SERS analysis indicated that the electropolymerization mechanism includes the following successive stages (Scheme 2B): (1) the electrochemical oxidation of the NH2-group of luminol to form imidogen group; (2) the cation–radical formation upon the initial luminol oxidation (i.e. E > 1.0 V); (3) its isomerization on the radical of quinoid type and recombination of initial and isomerized radical with dimer of “head-to-tail” type formation, which is followed by further dimer oxidation on the NH2-group site with the polycondensate formation. In fact, the aforementioned mechanism is in analogy to the polymerization process of aniline in acidic solution.46 The obtained PLUM was postulated to exist in the form of reduced and oxidized segments (Scheme 2C). Not surprisingly, the PLUM derivative depicted electrochemical activity in acidic solution and inactivity in basic solution in a similar manner to polyaniline. These materials were used as a flavin electroluminescent sensor with good sensibility and selectivity. An important positive aspect in using electropolymerization to obtain PLUM is related to the low solubility of luminol in aqueous solutions, thus by utilizing electropolymerization it was possible to work with low concentrations of luminol in solution. In subsequent work, Lin36 and colleagues employed an electrochemical quartz crystal microbalance (EQCM) in addition to CV analysis to study the growth of the polyluminol film (luminol concentration of 1.0 mM) in situ in acidic solution (0.1 M, i.e. pH of 1.5) on the surface of glassy carbon electrodes, and hence to exploit its possible chemical composition. Interestingly, the cyclic voltammogram under the specified acidic medium was characterized by one well-defined redox couple between +0.05 and +0.9 V (vs. SEC), with formal potential occurring at about +0.48 V (vs. SEC) that has been attributed to the reduced and oxidised forms of PLUM.35,37 The recorded EQCM results indicated a frequency change response with E > 0.75 V (vs. SCE), thus suggesting the oxidation process of luminol, which in turn has triggered the polymerization process. Importantly, the electrode mass recorded for luminol (1.0 mM in 1.0 M H2SO4) increased systematically with the potential cycling, therefore also evidencing that the polymerization took place on the electrode surface. Moreover, it was shown that the PLUM is stable in solutions in a wide range of pH values between 1 and 11, with a formal potential slope of −58 mV pH−1, which is near the Nernst equation slope for the same number of involved protons and electrons.36 Particularly, presenting an ionogenic functional group (with pKa values of the monomer at about 6.35 and 15.21),47 PLUM was considered as a suitable polymer to display self-doping properties with the improved pH dependent characteristics, such as conductivity and electroactivity, in alkaline medium. Most importantly, since the –NH2 group is not involved in the ECL reaction of luminol, the delivered polyluminol derivatives, as their monomer counterpart (i.e. luminol), have acted as an ECL luminophore. The authors also demonstrated that the reported polyluminol film deposited on a glassy electrode was electrocatalytically active for NADH oxidation in acidic and neutral aqueous conditions. Interestingly, the electrocatalytic oxidation current developed from the anodic peak of the redox couple.
Luminol has been polymerized not only at pH 1.5 (acidic), but also at pH 6 (buffered) by maintaining a suitable oxidation potential for the luminol monomer (i.e. potentiostatic conditions) or by using cyclic voltammetry in a suitable positive scan potential region (i.e. potentiodynamic conditions) on a screen-printed electrode (SPE) surface.30 Leca-Bouiver et al. showed that the polyluminol film formed in a pH 6 buffered solution acted as an ECL luminophore, like the luminol monomer when it is used in solution. In other words, H2O2 detection was possible in a wide linear range, extending from 0.79 μM to 1.3 mM. Those results have key potential as the first step towards the preparation of various more elaborate oxidase-based ECL reagentless biosensors that no longer are requiring the addition of luminol in solution. The same authors processed the electropolymerization of luminol also by applying a fixed current at pH 6 under a galvanostatic mode (30 min, 1.25 μA cm−2).31 The effects of current density and polymerization time were evaluated, revealing that when a current density of 5 μA cm−2 was reached, polymerization was not observed as indicated by electrochemical characterization, while longer polymerization times improved the performance until a 30 min optimum value. A biosensor for choline detection based on polyluminol has been developed. To achieve the latter, the synthesized polyluminol has been associated with a choline oxidase (ChOD)-immobilizing silica gel obtained by a sol–gel process. Low micromolar (from 0.4 μM to 0.13 mM) choline concentrations could be detected, thus revealing sensitive disposable and reagentless easy-to-use oxidase-based biosensors that could be extended to the detection of many other oxidase-substrate compounds. Critically, under the experimental conditions, the operational stability of the disposable devices lasted for 7 and 8 assays.
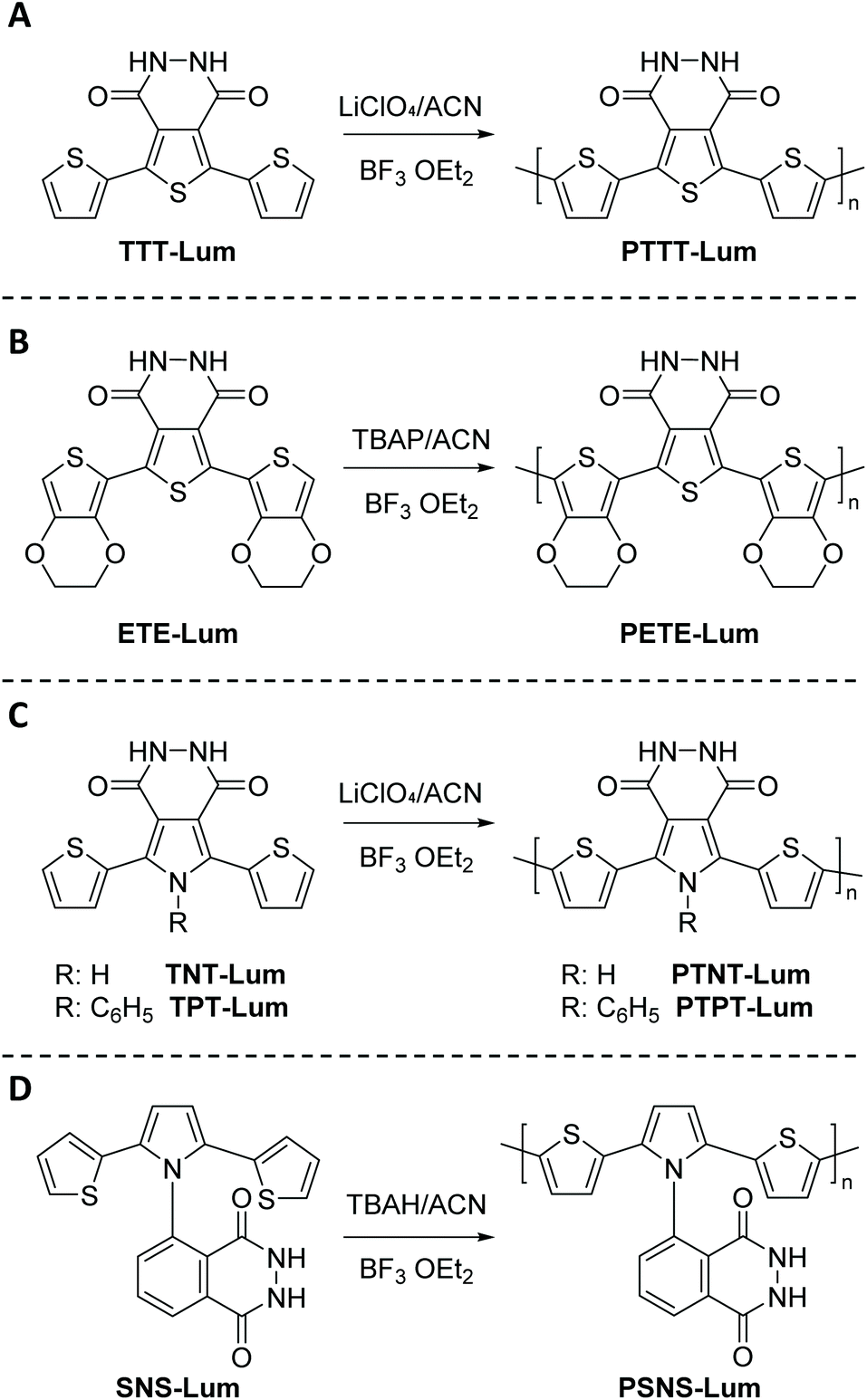 | ||
| Scheme 3 Schematic representation of novel luminol derived CL compounds based on pyridazine, terthienyl, and pyrrole systems, namely (A) 5,7-di-thiophen-2-yl-2,3-dihydro-thieno[3,4-d]pyridazine-1,4-dione (TTT-Lum), (B) 5,7-di-ethylenedioxythiophen-2-yl-2,3-dihydro-thieno [3,4-d] pyridazine-1,4-dione (ETE-Lum), (C) 5,7-di-thiophen-2-yl-2,3-dihydro-1H-pyrrolo[3,4-d]pyridazine-1,4(6H)-dione (TNT-Lum) and 6-phenyl-5,7-di-thiophen-2-yl-2,3-dihydro-1H-pyrrolo [3,4-d] pyridazine-1,4(6H)-dione (TPT-Lum), in addition to (D) (2,3-dihydro-5-(2,5)-di(thiophen-2-yl)-1H-pyrrol-1-yl)phthalazine-1,4-dione (SNS-Lum), with the respective polymers PTTT-Lum, PETE-Lum, PTNT-Lum, PTPT-Lum and PSNS-Lum. | ||
 | ||
| Scheme 4 Schematic representation of: (A) dimeric luminol product obtained via KIO3 mediated oxidation, (B) the postulated polymeric product obtained during the mediated (NH4)2S2O8 oxidative polymerization of luminol, and (C) the chemical oxidative polymerization of aniline, as analogue to mimic the polymerization of luminol. | ||
Subsequently, TTT-Lum was modified with 3,4-ethylenedioxythiophene (EDOT) side groups to deliver 5,7-di-ethylenedioxythiophen-2-yl-2,3-dihydro-thieno [3,4-d] pyridazine-1,4-dione, ETE-Lum, in Scheme 3B, with the aim to lower the oxidation potential of the novel monomer.49 Indeed, the oxidation potential was lowered from 1.10 V for TTT-Lum to 0.88 V for ETE-Lum, since electron rich EDOT units induced the oxidation substantially easier compared to thiophene. Furthermore, it was envisioned that the side group modification ensures the synthesis of linear polymer chains by leaving only one position (C-5) for polymer chain growth. Respectively, the potentiodynamic electro polymerization (cyclic scan from 0.0 V to 1.0 V (vs. Ag/AgCl) with a scan rate of 0.1 V s−1) of 1.0 mM ETE-Lum was performed on an ITO electrode in 0.1 M tetrabutylammonium perchlorate (TBAP)/ACN containing 5% BF3Et2O by volume. Note that during the polymerization process the intensity of light was increased with increasing potential, thus the obtained polymers, PETE-Lum, displayed ECL properties under very low potentials (0.9 V). The well-adhered, thick, and highly stable electro active PETE-Lum, which was soluble in basic aqueous solution, was recognized as a rare example of chemiluminescent polymeric materials bearing a pyridazine unit. PETE-Lum, as polymer film in its neutral state, exhibited a well-defined absorption band at 554 nm (slightly higher than PTT-Lum), which was attributed to π–π* transition. Whereas the band gap, defined as the onset energy for the π–π* transition, was found to be 1.65 eV, which in turn was lower than that of PTTT-Lum (1.74 eV). With PTTT-Lum and PETE-Lum, Onal and Cihaner have confirmed that insertion of pyridazine units into convenient heterocyclic rings (e.g. terthienyl) facilitates not only the synthesis of novel chemiluminogenic systems, but also allow an access to polymeric materials with chemiluminogenic features. Their contribution to this field was extended with the design and synthesis of compounds (i.e. 5,7-di-thiophen-2-yl-2,3-dihydro-1H-pyrrolo[3,4-d]pyridazine-1,4(6H)-dione, TNT-Lum, and 6-phenyl-5,7-di-thiophen-2-yl-2,3-dihydro-1H-pyrrolo [3,4-d] pyridazine-1,4(6H)-dione, TPT-Lum, in Scheme 3C), where the pyridazine unit is fused with a pyrrole ring instead of the thiophene.50 Their approach has also provided the opportunity to systematically compare the properties of the polymers based on thieno- and the pyrrolopyridazine luminol derivatives. Successful electrochemical polymerization of both monomers (e.g. 12.0 mM and 32.0 mM of TNT-Lum and TPT-Lum, respectively) was carried out by repeating potential scanning in the presence of BF3OEt2 in an electrolyte solution of 0.1 M LiClO4 dissolved in ACN. The oxidation peak for PTPT-Lum (1.05 V) was observed at a higher value when compared to PTNT-Lum (0.89 V). These results clearly indicate that in the absence of the hydrogen atom on the N-atom in the pyrrole ring, the polymer PTPT-Lum cannot be planar compared to the polymer PTNT-Lum bearing the hydrogen on the nitrogen in the polypyrrole ring. Nevertheless, it was observed that the intensity of peak currents of both polymer films increased linearly as a function of scan rates, which confirmed that the films well adhered on the electrode surface and the respective redox behaviours were non-diffusional. Furthermore, the respective polymer films of TNT-Lum and TPT-Lum in their neutral states exhibited well-defined π–π* transition absorption bands at 471 nm for PTNT-Lum and 361 nm with a shoulder at 428 nm for PTPT-Lum. The electronic band gap was found to be 2.02 eV for PTNT-Lum eV and 2.16 eV for PTPT-Lum. As expected from a conjugated polymer film, PTNT-Lum and PTPT-Lum also exhibited electrochromic features; e.g. PTNT-Lum was beige in the neutral state and transmissive grey in the oxidized state. In line with these studies, additionally (2,3-dihydro-5-(2,5)-di(thiophen-2-yl)-1H-pyrrol-1-yl)phthalazine-1,4-dione, SNS-Lum, (Scheme 4D) with a pendant luminol tail, was designed and synthesized.51 In a representative polymerization, 0.01 mM of SNS-Lum was electropolymerized, potentiostatically and potentiodynamically, in 0.1 M tetrabutylammonium hexafluorophosphate (TBAH)/ACN electrolyte containing 5% BF3Et2O. Indeed, the oxidation potential of the system was decreased about 0.37 V (from 0.94 V to 0.57 V) by the addition of BF3Et2O, which facilitated the electropolymerization to deliver PSNS-Lum as the first example of conjugated polymers with pendant luminol arms (Fig. 1). Accordingly, a new redox couple appeared after repetitive anodic scans in the CV of ACN-BF3OEt2 mixture, which revealed the formation of the electroactive polymer film. PSNS-Lum exhibited a well-defined reversible redox couple (0.48 V at 0.1 V s−1) (Fig. 1). When a potential of 0.8 V was applied to the coated electrode, the well-adhered electroactive polymer film induced ECL, whereas in the presence of oxygen and an external negative potential (−1.0 V) under neutral conditions, an intense CL emission was observed, thus unveiling the versatility of this novel material for the detection of the reactive oxygen species produced by biological processes.
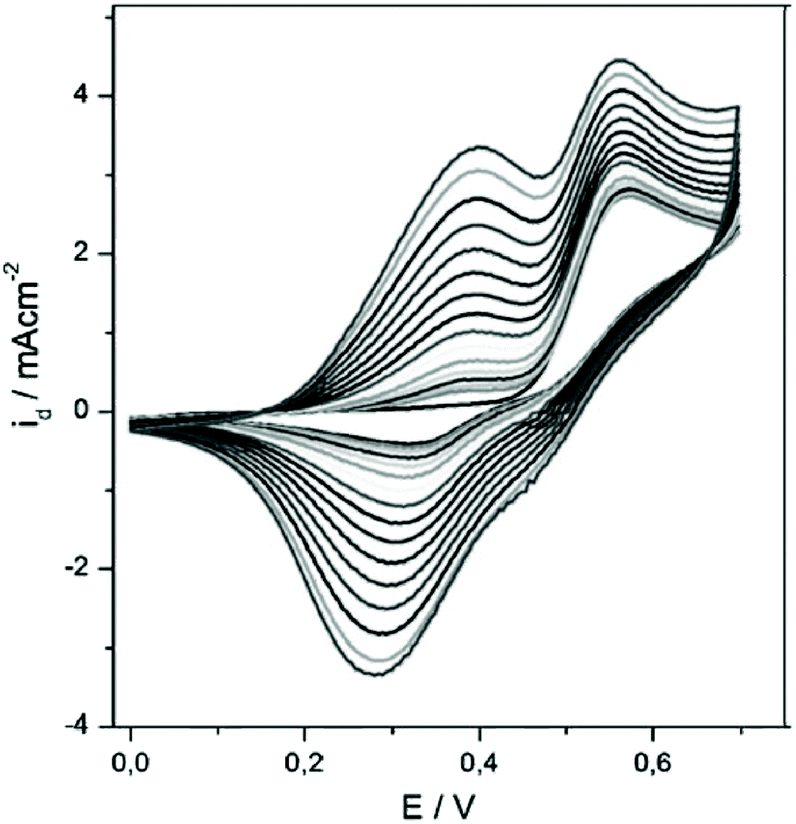 | ||
| Fig. 1 Electropolymerization of 0.01 mM SNS–Lum in 0.1 M TBAH dissolved in a mixture of ACN and BF3Et2O by the volume of 0.5% at 0.1 V s−1 by potential scanning to give PSNS-Lum. Reproduced with permission from ref. 51. Copyright 2010 Wiley–VCH. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :aniline = 1:1, 1:4, 1:20: 1:40 and 1:60, where the luminol concentration was kept constant in all experiments, i.e. 0.5 M). It was observed that luminol, which is oxidized at lower potential than aniline, inhibited the aniline oxidation and consequently affected its incorporation in the film. While the ECL behavior of the poly(luminol–aniline) films was not elaborated, the films showed electroactive and stable behaviour in acidic medium. Herein, the redox process associated with polyluminol was overlaid with polyaniline degradation products, thus presenting a potential shift of about −68 mV pH−1 unit in 1 < pH < 8 and showing pH sensitivity. The presence of proton acceptor/donator groups in the poly(luminol–aniline) structure, as –CO–NH–NH–CO–, introduced additional stability under basic conditions, under which polyaniline films are usually unstable.
:aniline = 1:1, 1:4, 1:20: 1:40 and 1:60, where the luminol concentration was kept constant in all experiments, i.e. 0.5 M). It was observed that luminol, which is oxidized at lower potential than aniline, inhibited the aniline oxidation and consequently affected its incorporation in the film. While the ECL behavior of the poly(luminol–aniline) films was not elaborated, the films showed electroactive and stable behaviour in acidic medium. Herein, the redox process associated with polyluminol was overlaid with polyaniline degradation products, thus presenting a potential shift of about −68 mV pH−1 unit in 1 < pH < 8 and showing pH sensitivity. The presence of proton acceptor/donator groups in the poly(luminol–aniline) structure, as –CO–NH–NH–CO–, introduced additional stability under basic conditions, under which polyaniline films are usually unstable.
For further investigating and improving the ECL properties of the polyluminol films, specific aromatic amine compounds (0.5 M), such as benzidine, were mixed with luminol (1.0 M) in acid solutions (pH 1.5), respectively, and electrocopolymerized under potentiodynamical conditions (−0.20 and 1.0 V) on SPE cells.32 Repetitive cycling of the potential over the oxidation peaks of the composite films produced a progressive increase in the voltammetric peaks at 0.4 and 0.58 V, indicating the build-up of a surface-bound material. Electrodes covered with this copolymer displayed different ECL properties and ECL sensing for the H2O2 with a 6 × 10−11 M detection limit in the presence of 0.1 M borax buffer solution. The poly(luminol-benzidine) composite film showed higher fluorescence quantum yields compared to the PLUM, suggesting that the different ECL performance of the poly(luminol-benzidine) composite films may originate from the modulation of the polymeric benzidine to the photophysical property of the polymeric luminol in the composite films. In another study, 3,3′,5,5′-tetramethylbenzidine was employed as a co-monomer for luminol-polymer formation.53 The polymeric films were grown potentiodynamically with a potential interval between −0.2 and 1.0 V in 0.2 M H2SO4. The optimal luminol-3,3′,5,5′-tetramethylbenzidine ratio was found to be 1:5 with copolymer growth kinetics being 1.6 times faster than luminol. The ECL emission from the copolymer implied the generation of excited polymeric 3-aminophthalate, as suggested by the fluorescent properties observed at 330 and 450 nm, respectively. Most importantly, the copolymer offered a better adherence and ECL intensity than polyluminol.
2. Chemical polymerization of luminol and its derivatives
Chemical polymerization of luminol has been considered as a more practical and economical alternative to electropolymerization, since it provides the possibility to modify and tune the chemical structure of organic redox-active materials for specific properties. In fact, as previously mentioned, the luminol polymerization was only studied during its electrochemical oxidation when substantial amount of polymer (e.g. PLUM) as brittle thin film was formed on the surface of an electrode. Critically, the chemical structure of PLUM was not elucidated in the free state and the respective physio-chemical properties were not elaborated. Therefore, with the purpose to study the possibility of luminol polymerization under different chemical conditions, and further elucidate the chemical structure and properties, the following polymerization methods of luminol will be reviewed: (i) oxidative chemical polycondensation, (ii) microwave (μW) assisted oxidative chemical polymerization, (iii) free-radical polymerization and (iv) miscellaneous polymerization approaches.On the one hand, it was observed that employing an excess of (NH4)2S2O8 has not necessarily influenced the polymerization yield, while the redox potential of KIO3 (e.g. 1.085 V) was insufficient for the oxidation of the luminol amino-group, thus resulting in the lowest yield (i.e. 5.7%). On the other hand, the excess of luminol (0.22 < C(Lum)/M < 0.55) resulted in five times increased isolated product yield (42.5%). Whereas the nature of the solvent did not have any detrimental impact on the yield of luminol polycondensation, the ratio of monomer to solvent was crucial. Accordingly, the maximum polymerization yield (i.e. 42.5%) was reached in the DMSO–water (1:9) solvent in the presence of 0.55 M luminol, which was activated with a 0.22 M (NH4)2S2O8 oxidant system. The authors have utilized elemental analysis, Fourier transform infrared (IR) and Raman spectroscopy in order to elucidate the most probable chemical structure of the luminol oxidative polycondensation products in the presence of the two different oxidants. Particularly, the results of the elemental analysis (e.g. 59.6227% C; 17.3913% N; 3.1005% H and 19.8757% O) revealed that, in fact, the KIO3 mediated oxidation resulted in a dimeric luminol oxidation product (shown in Scheme 4A), as the oxidation occurred on the amide nitrogen atom. Whereas, the replacement of the oxidant with (NH4)2S2O8 resulted in a polymer growing at the site of the NH2-group. Thus, the latter chemical oxidation delivered a PLUM derivative containing azo-groups. While, the chemical structure can be represented as shown in Scheme 4B, the chemical oxidative polymerization of luminol, in analogy to aniline (Scheme 4C),55 is represented by an intricate interplay of consecutive reactions, which are still far from being completely understood. Nevertheless, additional confirmation for the formation of the depicted PLUM derivative were the absent bands of the luminol NH2-group deformation vibrations at 1628 cm−1 and torsional vibrations at 492 cm−1 in the IR spectrum. In a similar manner, the band at 1588 cm−1 could be assigned to the vibration of the azogroup –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– in the spectra of polyluminol. Critically, the absorption bands arising from the aminophthalate derivative—the emitter of the luminol CL – at 1610 and 1400 cm−1, respectively, were absent in the spectra of the PLUM derivatives A and B depicted in Scheme 4. Thus, not only the verification of the lost CL properties, but also the authentication of the postulated mechanism for the polycondensation were elaborated in the context of the CL studies. In fact, the source of luminescence during the luminol oxidation should be the radiative deactivation of 3-aminophthalate, in accordance with the generally accepted scheme of luminescence (Scheme 2C), whereas this phenomenon has not been observed under the mentioned conditions applied for the polyluminol synthesis. Nevertheless, this study disclosed how challenging it is to manipulate the conditions of the chemical oxidative polymerization while maintaining the CL properties of the derived polymers intact. It took more than 15 years until a second attempt was performed to synthesize electrochemically active polyluminol using the oxidative chemical polymerization method reported by Koval'chuk et al.54 Through in-depth analytical characterization via vibrational spectroscopy, UV–Visible (Vis), and X-Ray photoelectron spectroscopy (XPS), which was performed on the same polymer by Lian and colleagues,56 it was established that the polymerization occurred at the primary amine group, and a polymer containing alternating benzoid and quinoid rings connected via amine groups was formed. For instance, the characteristic band of the symmetric and asymmetric stretching mode of the primary amine (NH2) observed at 3454–3418 cm−1 has changed to a single broad peak for the polymer, attributed to the stretching mode of the aromatic secondary amine (NH). In a similar manner, the UV–Vis spectra of the polymeric luminol in a DMSO solution showed a transition at 390 nm, representing the transition of π–π* generated from the benzoid rings57 in addition to a second transition at 619 nm attributed to the excitation state of the quinoid ring structure, similar to the one in polyaniline.58 XPS analysis was conducted to identify changes in the chemical state of the polymer and displayed a distinctive peak at 287.3 eV, which was attributed to the CN groups. The high-resolution spectra of the elemental core level for N 1s, which appeared at binding energy of 400.08 eV, provided more clues on the polymerization reaction and implied clearly that the polymerization occurred at the NH2 group. Particularly, it was observed that the polymer had a broader N 1s peak than luminol with a full width at half of 1.97 versus 1.86. The broadening of the N 1s peak for the polymer indicated an increase in the number of chemical bonds. Importantly, the XPS results provided additional support on the presence of quinoid segments for the polymer, since the deconvolution of N 1s peak showed the presence of a new sub peak at 398.98 eV corresponding to N of the quinoid segment resulting from the polymerization similar to that of polyaniline.59,60 To determine the electrochemical properties with the idea to discover potential applications, the thermal stability and crystallinity of the polymer were investigated using differential scanning calorimetry (DSC) and X-ray diffraction (XRD) spectroscopy. The thermal analysis revealed the semi-crystalline nature of the polyluminol with two thermal events at 99 °C and 152 °C, corresponding to a glass transition and a melting temperature. Furthermore, the XRD patterns of the polymer have confirmed the semi-crystalline structure. The electrochemical redox activity was elaborated on a GC electrode, suggesting a reversible charge transfer process with multiple oxidation peaks corresponding to the different oxidation states of the polymer at 0.43, 0.63 and 0.92 V, respectively, accompanied with reduction peaks found at 0.24, 0.53, and 0.86 V. In fact, the electrochemical behaviour of polyluminol on GC demonstrated its possible use to modify carbon-based electrodes in order to enhance the capability of carbon materials for energy storage.
N– in the spectra of polyluminol. Critically, the absorption bands arising from the aminophthalate derivative—the emitter of the luminol CL – at 1610 and 1400 cm−1, respectively, were absent in the spectra of the PLUM derivatives A and B depicted in Scheme 4. Thus, not only the verification of the lost CL properties, but also the authentication of the postulated mechanism for the polycondensation were elaborated in the context of the CL studies. In fact, the source of luminescence during the luminol oxidation should be the radiative deactivation of 3-aminophthalate, in accordance with the generally accepted scheme of luminescence (Scheme 2C), whereas this phenomenon has not been observed under the mentioned conditions applied for the polyluminol synthesis. Nevertheless, this study disclosed how challenging it is to manipulate the conditions of the chemical oxidative polymerization while maintaining the CL properties of the derived polymers intact. It took more than 15 years until a second attempt was performed to synthesize electrochemically active polyluminol using the oxidative chemical polymerization method reported by Koval'chuk et al.54 Through in-depth analytical characterization via vibrational spectroscopy, UV–Visible (Vis), and X-Ray photoelectron spectroscopy (XPS), which was performed on the same polymer by Lian and colleagues,56 it was established that the polymerization occurred at the primary amine group, and a polymer containing alternating benzoid and quinoid rings connected via amine groups was formed. For instance, the characteristic band of the symmetric and asymmetric stretching mode of the primary amine (NH2) observed at 3454–3418 cm−1 has changed to a single broad peak for the polymer, attributed to the stretching mode of the aromatic secondary amine (NH). In a similar manner, the UV–Vis spectra of the polymeric luminol in a DMSO solution showed a transition at 390 nm, representing the transition of π–π* generated from the benzoid rings57 in addition to a second transition at 619 nm attributed to the excitation state of the quinoid ring structure, similar to the one in polyaniline.58 XPS analysis was conducted to identify changes in the chemical state of the polymer and displayed a distinctive peak at 287.3 eV, which was attributed to the CN groups. The high-resolution spectra of the elemental core level for N 1s, which appeared at binding energy of 400.08 eV, provided more clues on the polymerization reaction and implied clearly that the polymerization occurred at the NH2 group. Particularly, it was observed that the polymer had a broader N 1s peak than luminol with a full width at half of 1.97 versus 1.86. The broadening of the N 1s peak for the polymer indicated an increase in the number of chemical bonds. Importantly, the XPS results provided additional support on the presence of quinoid segments for the polymer, since the deconvolution of N 1s peak showed the presence of a new sub peak at 398.98 eV corresponding to N of the quinoid segment resulting from the polymerization similar to that of polyaniline.59,60 To determine the electrochemical properties with the idea to discover potential applications, the thermal stability and crystallinity of the polymer were investigated using differential scanning calorimetry (DSC) and X-ray diffraction (XRD) spectroscopy. The thermal analysis revealed the semi-crystalline nature of the polyluminol with two thermal events at 99 °C and 152 °C, corresponding to a glass transition and a melting temperature. Furthermore, the XRD patterns of the polymer have confirmed the semi-crystalline structure. The electrochemical redox activity was elaborated on a GC electrode, suggesting a reversible charge transfer process with multiple oxidation peaks corresponding to the different oxidation states of the polymer at 0.43, 0.63 and 0.92 V, respectively, accompanied with reduction peaks found at 0.24, 0.53, and 0.86 V. In fact, the electrochemical behaviour of polyluminol on GC demonstrated its possible use to modify carbon-based electrodes in order to enhance the capability of carbon materials for energy storage.
:PAnis–30:70 showed complete disappearance of the peak associated with the 3-aminophthalate anion as a result of the fluorescence quenching. This was also evident from the lowest quantum yield (Φ) values for PLUM:PAnis–30:70 derivatives synthesized in acidic (Φ = 0.005) and basic media (Φ = 0.002). Nevertheless, bovine serum albumin (BSA) was used as a model protein for performing adsorption studies in order to show the potential of the copolymers as a protein biosensor. UV-Vis analysis displayed a peak broadening from 250 to 300 nm that was attributed to the conformational changes of BSA upon interaction with copolymer particles, which in turn caused the gradual perturbation of the microenvironments around the aromatic amino acid residues of BSA.
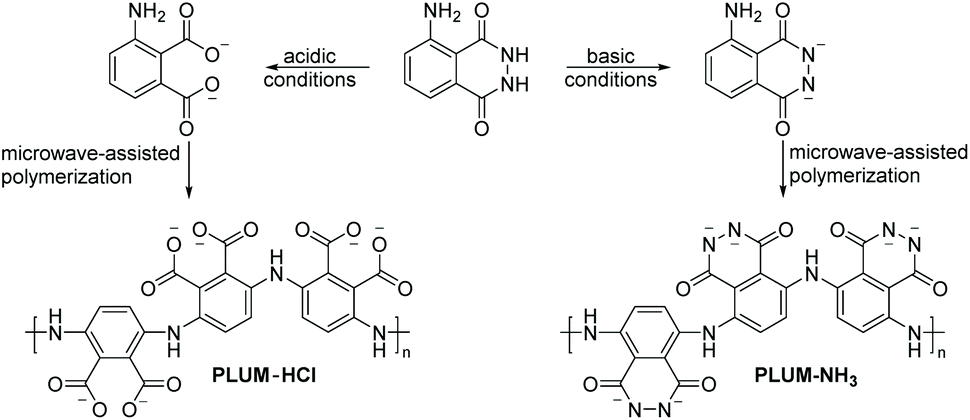 | ||
| Scheme 5 Microwave-assisted oxidative chemical polymerization of luminol (3.0 M) under acidic (0.01 N HCl, left, PLUM-HCl) and basic (0.1 NH3, right, PLUM-NH3) conditions, respectively. In both cases, ferric chloride (1.5 equiv.) was used as the initiator and the reactions were performed for 20 min at 30 °C. | ||
In a subsequent work, the same authors explored the microwave-assisted copolymerization of luminol in the presence of another aniline analogue (i.e. o-phenylenediamine, Opd).62 Thereby, the weight ratios were tuned in order to modulate the CL properties. Importantly, the homo- and copolymerization reactions of luminol (1.8 M) were, for the first time, investigated with benzoyl peroxide (BPO, 25.0 mol%) as an oxidizing agent. Accordingly, the reactions were performed in aqueous media for 15 min at 25 °C in a microwave oven. The homopolymer with a substantially different chemical structure compared to the one obtained via the previously reported μW assisted polymerization (Scheme 6A) was found to have an apparent viscosity average molecular weight (Mv) of 7500 g mol−1 with an intrinsic viscosity (η) of 0.36; since the values of η were observed to be increasing with increasing segments of the aniline derivative, the copolymer with a composition of PLUM:POpd–20:80 was recognized for the highest value of η, i.e. 0.73 and Mv of 12500 g mol−1. Accordingly, in-depth analytical characterization was performed for the homo- and copolymers by employing IR, NMR and UV-Vis techniques. Particularly, the distinguishable NH stretching vibration band for the homopolymer of luminol was detected at 3132 cm−1, while the peak at 1668 cm−1 was correlated to imine stretching vibration. In a similar manner, the peaks associated with quinonoid and benzenoid ring puckering were observed at 1402 cm−1 and 1350 cm−1, respectively. On the one hand, the UV-Vis spectra of the copolymers envisioned the formation of a random copolymer exhibiting composition dependent optical characteristics. On the other hand, the individual electronic states of each monomer were retained as bands at 280 and 425 nm associated with the aniline derivative segment, and the band at 375 nm with the luminol segment (Scheme 6B) were detected. Fluorescence spectroscopy and confocal imaging were also carried out to explore the emission characteristics. The copolymers, which were found to be non-toxic at concentrations as high as 200 μg mL−1, showed composition dependent blue as well as red emission (Scheme 6C). The latter characteristic facilitated that the copolymers are utilized for in vivo imaging of cancer cells. Last but not at least, XRD and TEM analyzes confirmed the morphology of copolymers to be dependent on the ratio of the two monomers in the copolymer, nevertheless indicating a highly organized and crystalline morphology.
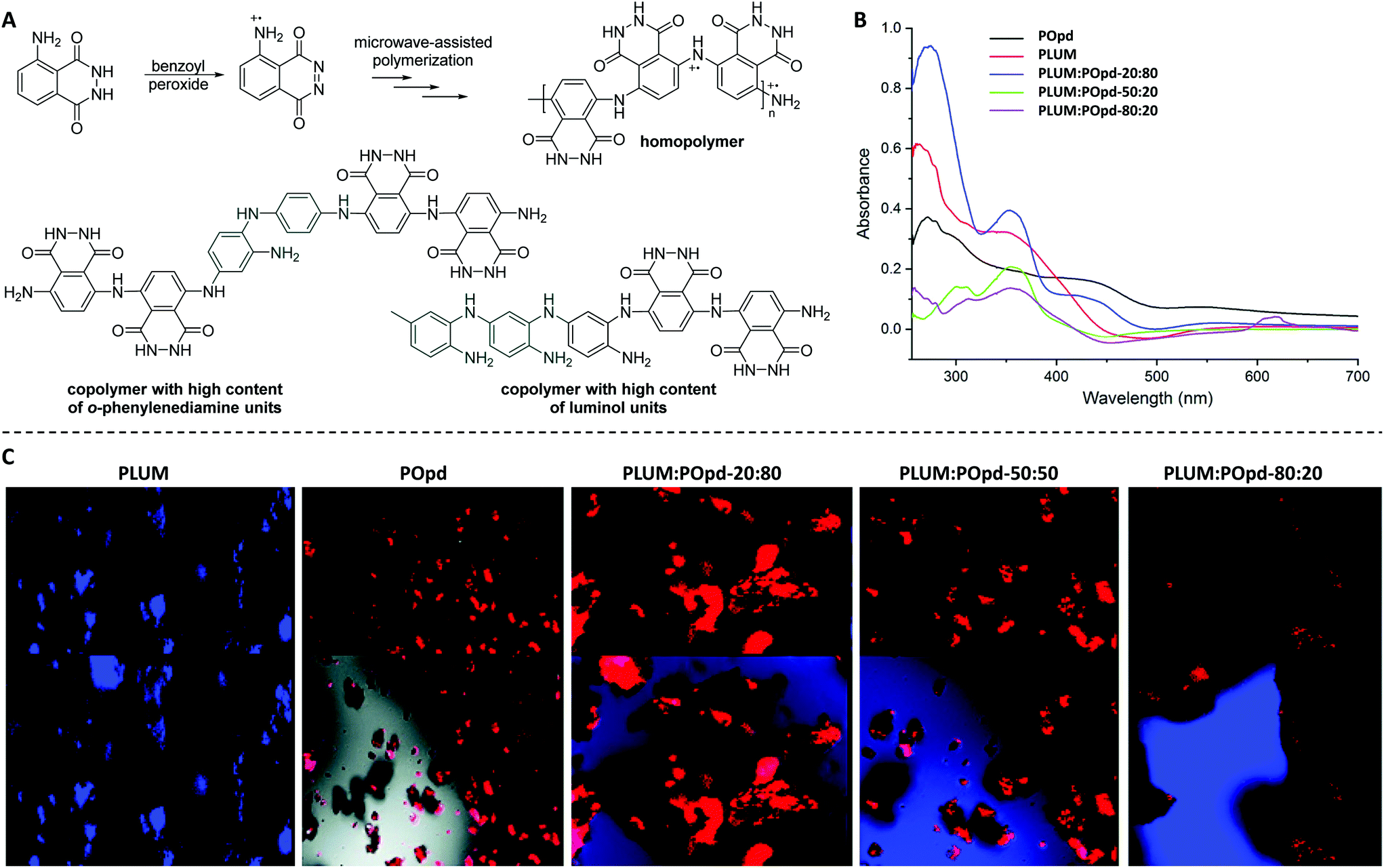 | ||
| Scheme 6 (A) Microwave-assisted oxidative chemical (co)polymerization of luminol (LUM, 1.8 M) in the presence of benzoyl peroxide (25.0 mol%) to deliver homopolymer, and copolymers which are enriched either with o-phenylenediamine (Opd) or luminol. The reactions were performed for 15 min at 25 °C. (B) Comparative UV-visible spectra of homopolymers (POpd and PLUM) and the respective copolymers PLUM:POpd-20/80, PLUM:POpd-50/50, and PLUM:POpd-80/20. (C) Confocal micrographs of PLUM, POpd, PLUM:POpd-20/80, PLUM:POpd-50/50, and PLUM:POpd-80/20, respectively. (B) and (C) are reproduced with permission from ref. 62. Copyright 2018 Royal Society of Chemistry. | ||
Whereas some major disadvantages of microwave-assisted chemistry are still existing (such as the inability to scale up and the limited penetration depth), the results presented above indicate the potential of the method towards the synthesis of luminol based (co)polymers with tuneable morphology and CL properties.
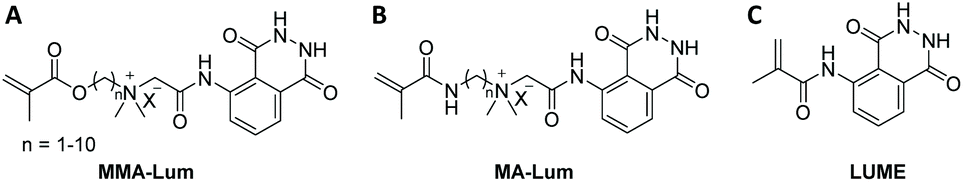 | ||
| Scheme 7 Schematic representation of novel luminol derived CL monomers (A)–(C), which could be copolymerized via conventional free-radical polymerization ((A) and (B)) and single transfer electron living radical polymerization technique (C). | ||
Quite recently (in 2019), the luminol based methacrylamide monomer (LUME) was copolymerized with (hydroxyethyl)methacrylate (HEMA) using single transfer electron living radical polymerization technique (SET-LRP) in the presence of ethyl-α-bromoisobutyrate as initiator to deliver a copolymer with a number average molecular weight of 12800 g mol−1 (Scheme 7C).64 It should be emphasized that the homopolymerization of LUME has failed, and the authors have not provided any reasons for this observation. Nevertheless, luminol and LUME revealed substantially similar UV–Visible spectral patterns with two absorption bands, one at 310 nm and a broad band at 375 nm. However, the copolymer of LUME and HEMA (feed ratio of 1:1) showed a blue shift with a band at 210 nm in addition to a broad band between 310–340 nm. The incorporation of LUME monomeric units into the copolymer was also quantified via UV–VIS spectroscopy according to Beer–Lambert's law, revealing a luminol incorporation of 11.36 g mol−1. Compared to luminol (λ = 435 nm), LUME exhibited a red shifted emission peak at 445 nm due to the change in the electronic environment of LUME, i.e. the acylation of the amino group, which has resulted in the change of the inductive effect. On the contrary, the copolymer demonstrated a blue shift with the CL peaks at 423 and 425 nm, respectively. Nonetheless, under optimal conditions the detection limit of peroxide in solution was found to be 0.057 μM for the copolymer. Accordingly, the copolymer was tested for its ability to detect H2O2 in living cells. Human cervical cancer HeLa cells loaded with 5.0 μM copolymer for 10 min at 37 °C were exposed to fluorescence microscopy. Indeed, the bright field images of the copolymer treated cells indicated that the cells are viable throughout the imaging experiments, thus addressing not only the cell permeability of the copolymer, but also underpinning its effectiveness as hydrogen peroxide fluorescence imaging agents in the cells.
000 g mol−1, whose luminol content has varied from 0.05% to 2.0% (Scheme 8A).67 By virtue of its CL properties, the copolymer has found useful application in the treatment and monitoring of industrial water, as it was reported within the patent invention.
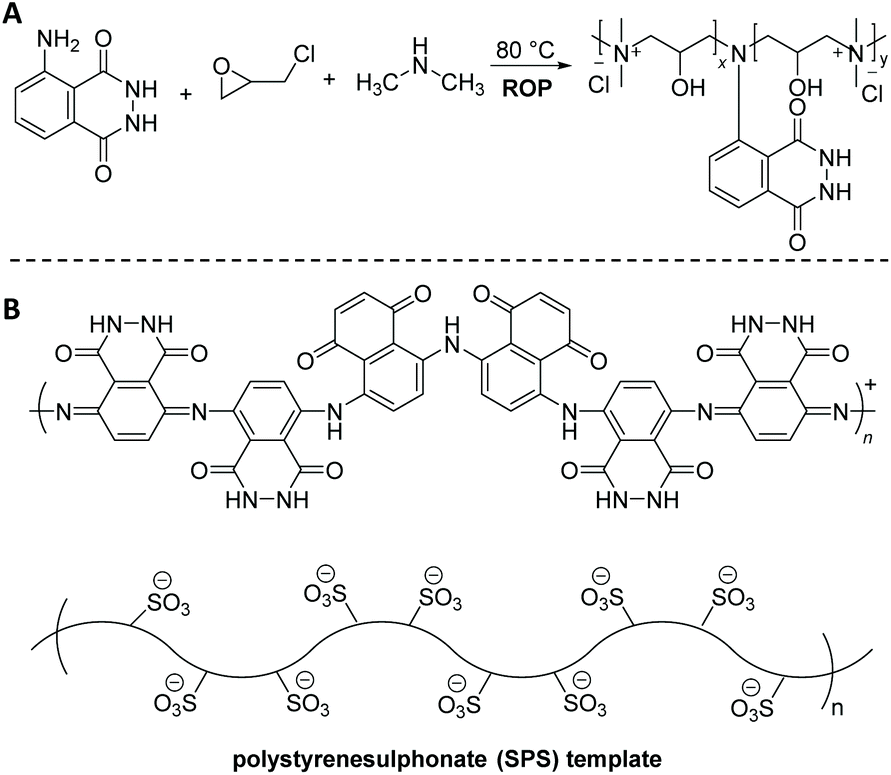 | ||
| Scheme 8 Schematic representation of: (A) the ring-opening copolymerization (ROP) of epichlorohydrin in the presence of luminol and dimethylamine to deliver copolymers with an Mn of 10000 g mol−1 and luminol content that has varied from 0.05% to 2.0%. (B) The enzyme catalyzed polymerization of luminol in the presence of polystyrenesulphonate (SPS) using horseradish peroxidase (HRP) as a biocatalyst in a phosphate buffer at pH 8. | ||
Enzymatic polymerization represents an effective and preferable alternative to conventional chemically-catalyzed processes. It offers significant advantages, summarized in the employed mild reaction conditions mainly in terms of temperature and toxicity, and high selectivity of enzymes, avoiding protection–deprotection strategies and resulting in improved quality/performance of end products. For example, Nabid68 and colleagues developed horseradish peroxidase (HRP) catalyzed polymerization of luminol in the presence of 0.04 mM polystyrenesulphonate (SPS) template to yield a water-soluble PLUM (Scheme 8B). Among the three different pH values (pH = 2, 4, and 8) that have been utilized, pH = 8 showed the most optimum conditions for the adequate synthesis of polyluminol, due to the fact that the activity of the HRP enzyme decreases abruptly in acidic media (e.g. at pH 4, the HRP activity is around 0 after 1 h). The C–H out-of-plane bending located at 833 cm−1 in the IR spectra, which is due to a para-substitution pattern, indicated the presence of a head-to-tail polymerization. Furthermore, the UV-Vis spectra of PLUM displayed the two characteristic absorption peaks around 300 and 348 nm with a hypsochromic shift of 61 nm in accordance to the monomer, i.e. luminol (397 nm). Nevertheless, it is worth to note that the CL reaction was observed from the reaction of PLUM (0.01 mM) dissolved in 0.1 M NaOH aqueous solution with oxidants like H2O2 and Fe3+ (both 1.0 mM), being highly sensitive for Fe3+. Crucially, the solubility of the polymer at all pH conditions makes it a suitable material for the self-assembly into organized structures with biological macromolecules such as enzymes for fabrication of biosensors.
3. Post-polymerization modification reactions towards luminol-based polymers
Post-polymerization modifications are a highly versatile toolbox for the functionalization and modification of macromolecules.69 Thus, the tailor-made manipulation of polymer properties is achieved without changing the polymer backbone. Significant efforts have also been made to develop methods which can be utilized for the synthesis of luminol-modified polymers to meet the demand for the design and construction of efficient CL systems. For example, a literature survey reveals that Kai70,71 and colleagues are the first who have synthesized novel luminol-decorated macromolecular probes based on a natural polymer (polysaccharide), i.e. dextran, via post-polymerization functionalization (Scheme 9). Indeed, dextran as a macromolecule is easily available and biocompatible, and its structure allows simple modification by means of periodateoxidation72 with NaIO4 to produce aldehyde moieties in the macromolecule. The subsequent step was the introduction of a number of luminol molecules into the macromolecule by imine formation, followed by the last step in which the conjugate was reduced (in the presence of sodium borohydride) to obtain a more stable product. In addition to luminol, biotin was also introduced to the system as a key molecule in order to improve the sensitivity of the chemiluminescent macromolecule (i.e. dextran). Elemental analysis confirmed the atomic composition of the synthesized dextran-based luminol–biotin modified (DLB) polymer as: C 43.0, H 5.8, N 4.4, and S 0.17 %, implying that the probe contains 560 luminol units and 34 biotin units in a dextran with 3100 glucose units. The DLB polymer was also analyzed by gel-filtration liquid chromatography with UV detection (λabs = 275 nm) and revealed a molecular weight of 630000 g mol−1. The aqueous solution of the probe gave approximately 8 times higher CL intensity (on a nylon membrane by use of the reagents acetonitrile, tetrapropylammonium hydroxide and H2O2 catalyzed by Fe3+) at the same amount (1.0 fmol) of free luminol, and lasted approximately 80 s. Indeed, this short measurement time was advantageous for saving computer accumulation of enormous signals of CL imaging in the limited capacity of a hard disk. Avidin–biotin systems73 have been used quite often for the fabrication of enzymatic sensors, based on the avidin-modified electrode surface and immobilisation of biotin-labelled enzymes through avidin–biotin complexation. Therefore, a macromolecular assembly was obtained through biotin–avidin interaction in the presence of avidin to deliver an enzyme-free sensor for protein (i.e. Cytochrome P450 3A4 (abbreviated CYP3A4)) detection. Thus, for the efficient sensing of the targeted CYP3A4 protein, macromolecular assembly of the DLB polymer and avidin at an optimal ratio of 1:1 was essential. Indeed, the enzyme-free polymeric chemiluminescent probe gave the highest CL intensity in the presence of at least 190 fmol CYP3A4 protein on a poly(vinylidene difluoride) (PVDF) membrane. To expand the toolbox of natural polymers (e.g. polysaccharides) modified with luminol as CL imaging probe, Kai et al.74 have hypothesized that a highly charged polymer like alginic acid could become a suitable polymer. In fact, in a similar manner to dextran, alginic acid is an easily accessible and biocompatible natural polymer. Therefore, the post-modification of alginic acid with luminol was attained in a similar manner to dextran: (i) oxidation of the polysaccharide with NaIO4 to produce several aldehyde moieties in the macromolecule; (ii) the introduction of a number of luminol molecules into the macromolecule by imine formation; and (iii) the reduction of the conjugate to obtain a more stable product. Since the solubility and hence the CL efficiency of the post-modified natural macromolecule increased with the oxidation degree and luminol amount, one equivalent of NaIO4 (100% oxidation degree) and subsequently two equivalents of luminol per one uronic acid unit in the macromolecule were employed in order to achieve a high substitution yield. Based on gel-filtration liquid chromatography with UV detection, the amount of luminol bound to alginic acid was calculated to be approximately 84 molecules with an average molecular weight of 132000 g mol−1. The CL efficiency of the advanced alginic acid–luminol system was tested in the presence of hemin, tetra-n-propylammonium hydroxide and H2O2. Indeed, the system exhibited improved CL imaging for specific proteins with a detection limit of 15–250 ng in comparison with a commercially available stain kit using either Sypro® ruby fluorescence stain or Coomassie Brilliant Blue colour stain.
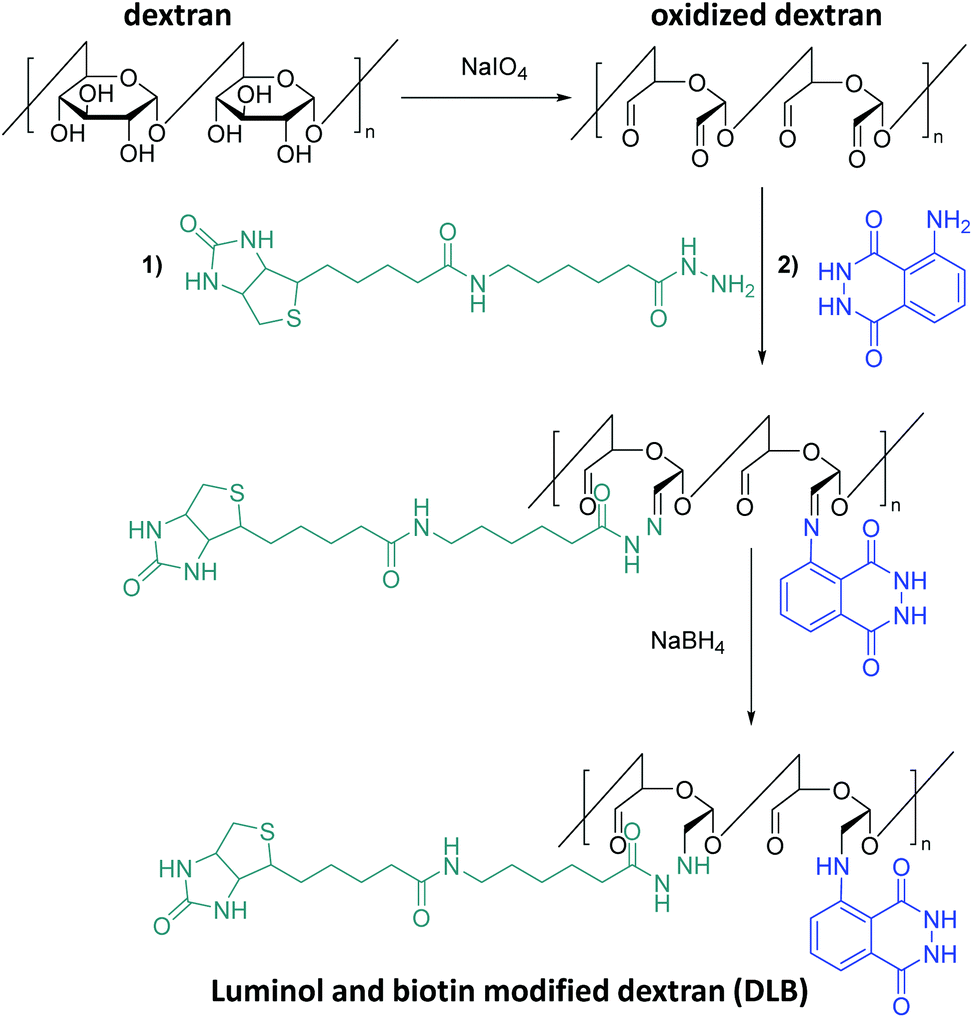 | ||
| Scheme 9 Schematic representation of the post-polymerization modification of dextran: (i) oxidation of the polysaccharide with NaIO4 to produce aldehyde moieties; (ii) the introduction of a number of luminol and biotin molecules into the macromolecule by imine formation; and (iii) the reduction of the conjugate in presence of NaBH4 to obtain a more stable product DLB. | ||
Another naturally occurring polysaccharide, i.e. β-cyclodextrin (β-CD), was also post-modified with luminol75,76 in order to deliver luminescent materials for real-time imaging of acute and chronic inflammatory diseases. The chemical post-modification was achieved by activation of β-CD via 1,1′-carbonyldiimidazole (CDI), followed by conjugation of luminol units on β-CD (Scheme 10A). The obtained materials, i.e. LUM-IMI-CD, displayed amphiphilic properties since the covalently linked luminol units were relatively hydrophilic, whereas the remained imidazole moieties were hydrophobic. Analytical characterization based on NMR spectroscopy and UV–Vis absorbance suggested about 1 luminol and 3 imidazole units covalently linked to each β-CD molecule. Spherical nanoparticles (LUM-IMI-CD-NP) with a mean diameter of 228 ± 19 nm (n = 15), as observed by TEM and differential light scattering (DLS), were obtained via nanoprecipitation/self-assembly of the LUM-IMI-CD derivatives (Scheme 10C). Importantly, these nanoparticles have not only efficiently enabled monitoring the response to reactive oxygen species (ROS, i.e. at 1.0 mM H2O2) with amplified and sustained luminescence (Scheme 10D, with a maximum emission wavelength of ∼440 nm), but also diverse inflammation-associated diseases by its myeloperoxidase (MPO) – responsive luminescence capability, in both cellular environment and in vivo murine models. The response mechanism was simply based on hydrolysis with the respective release of luminol and imidazole derivative (Scheme 10B). It was also hypothesized that those spherical nanoparticles (i.e. LUM-IMI-CD-NP) can also illuminate alcoholic liver injury or acute liver failure (ALF) by targeting the liver injury, thus self-illuminate under diseased conditions.77
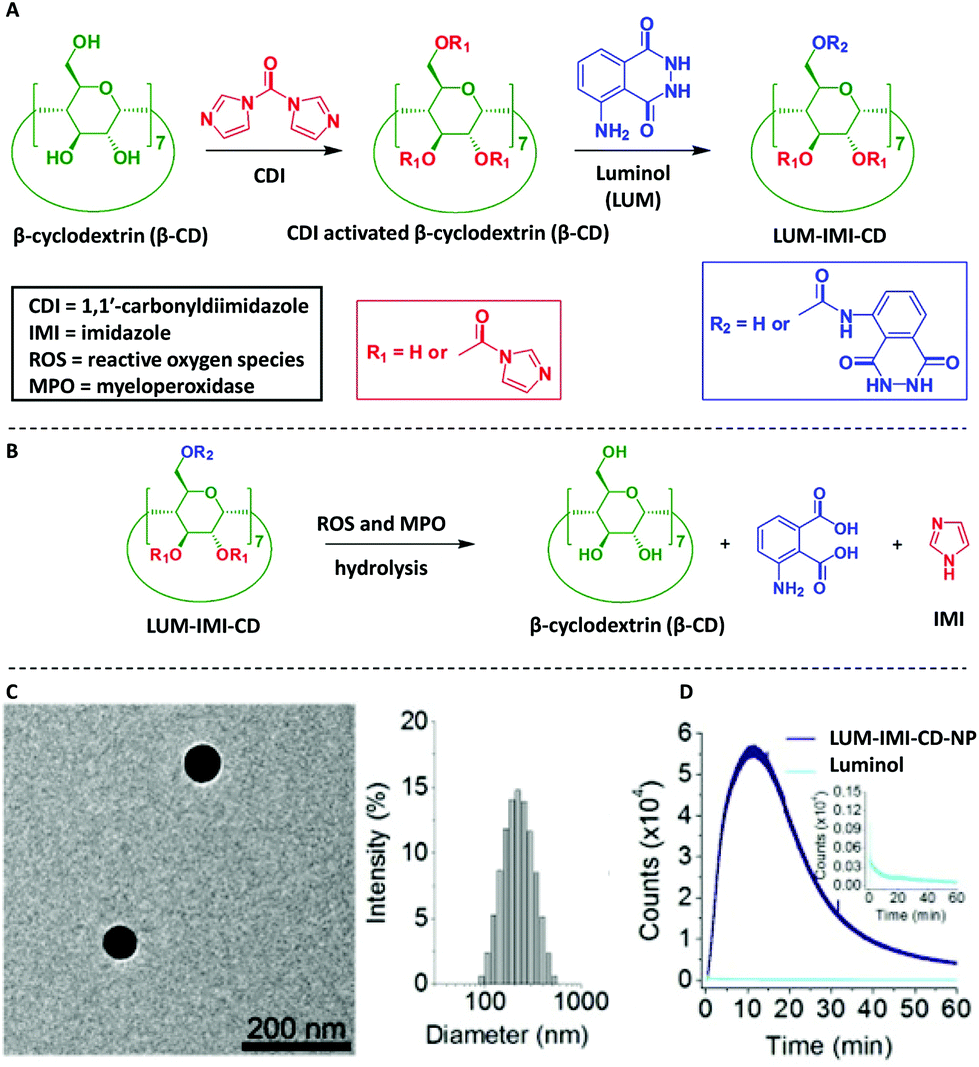 | ||
| Scheme 10 Schematic illustration of synthesis and hydrolysis of a luminescent β-cyclodextrin material, LUM-IMI-CD. (A) The synthetic route of a myeloperoxidase (MPO)-responsive luminescent material by functional modification of β-cyclodextrin. (B) Hydrolysis of the luminescent cyclodextrin material into the parent cyclodextrin compound. (C) Characterization of the MPO-responsive luminescent nanoparticle, LUM-IMI-CD-NP, via TEM and DLS. (D) Time dependent luminescent intensity of LUM-IMI-CD-NP (black line) and luminol (neon blue line) in the presence of H2O2 at 0.5 or 5 mM, respectively. In both cases, LUM-IMI-CD-NP and free luminol with the same content of luminol were used. (C) and (D) are reproduced with permission from ref. 75. Copyright 2017 Elsevier Ltd. | ||
Poly(ethylene glycol) (PEG), as a non-natural biocompatible polymer, is widely used as a gold standard from industrial manufacturing to medicine-oriented research, and therefore it was of crucial importance to deliver a PEG-based system which is modified with a luminol derivative. Accordingly, Zhang and colleagues have synthesized an amphiphilic polymeric conjugate of PEG (i.e. PEG-LUM-C6 in Scheme 11A and B) by sequential coupling reactions with luminol and a fluorescent derivative [chlorin e6 (Ce6)],78 which can further self-assemble into a nanoparticle capable of bioluminescence resonance energy transfer (BRET). While IR analysis displayed the characteristic absorption bands of Ce6, luminol, and PEG in the obtained product, the 1H NMR spectrum revealed that the molar ratio of Ce6, luminol, and PEG in the synthesized conjugate was approximately 2:1:1. Additionally, TEM analysis confirmed the nanoparticle formation with a mean hydrodynamic diameter of 171 nm (polydispersity index 0.29 ± 0.02, Scheme 11C). In the presence of 100 mM H2O2, the luminescence spectrum of the conjugate (0.5 mg mL−1) showed two emission peaks: one at 450 nm, corresponding to the CL of luminol, and the other one at 675 nm, belonging to the fluorescence emission of Ce6. The CL was depended on the concentration of H2O2; in other words, there was no detectable CL when the H2O2 concentration was below 10 mM, and at higher H2O2 concentrations (>100 mM), the luminescence exponentially decreased with time. Nevertheless, compared to free luminol, the nanoparticles formed from the PEG-LUM-C6 conjugate showed a markedly enhanced CL signal (31-fold higher, shown in Scheme 11D), which was attributed to the increased vascular permeability and high accumulation rate at inflammatory sites, thus facilitating real-time, non-invasive self-illuminating imaging system. Furthermore, by using a screened cancer cell line, the team of Zhang could also demonstrate that the PEG-LUM-C6 based nanoparticles are promising CL self-illuminating sensors for selective photodynamic therapy treatment of tumors through in situ triggering via H2O2.79
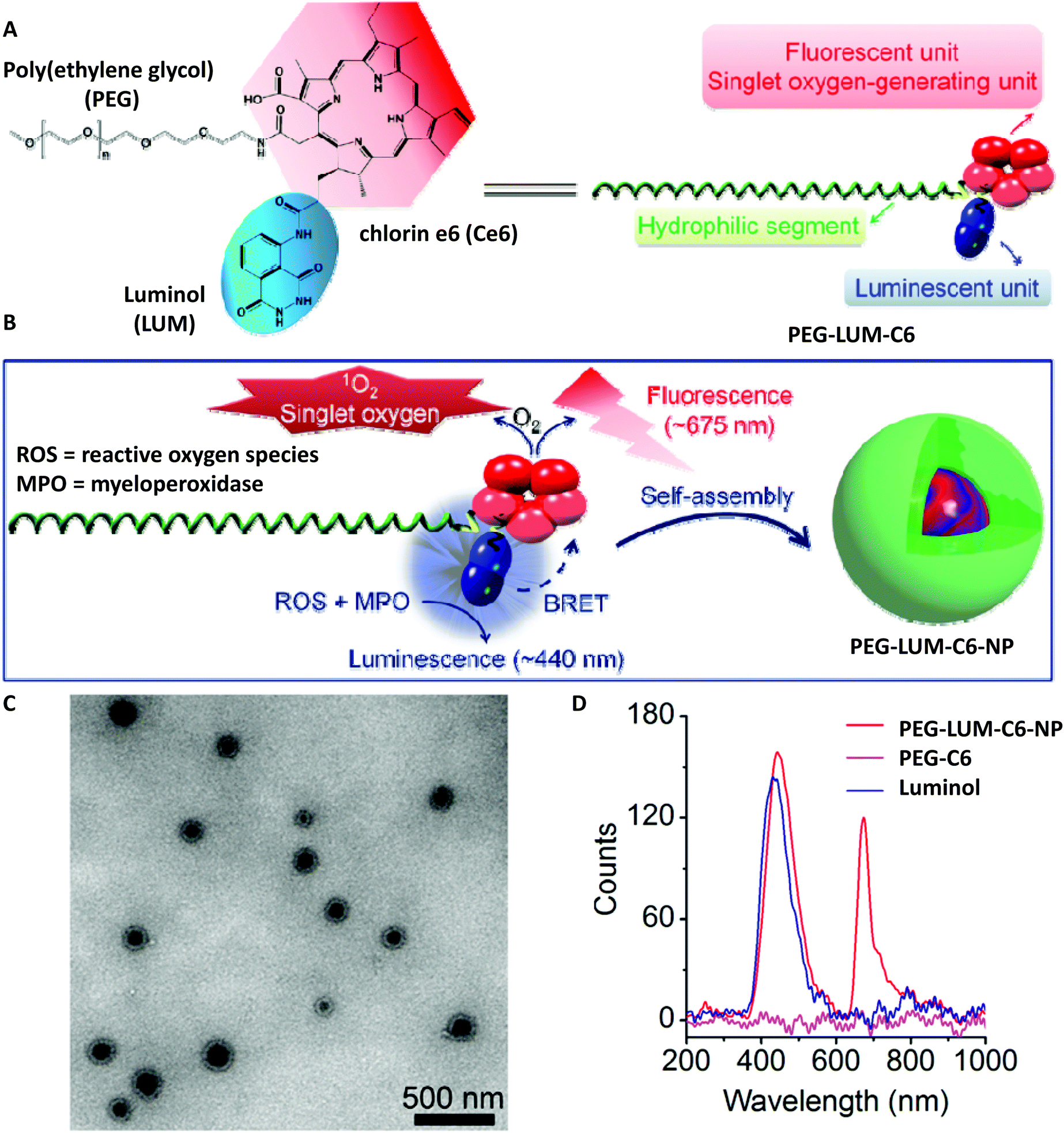 | ||
| Scheme 11 (A) Chemical structure and schematic of a designed amphiphile of PEG-LUM-C6. (B) Sketch of self-assembly of the PEG-LUM-C6 conjugate into a core–shell structured nanoparticle, i.e. PEG-LUM-C6-NP. (C) Transmission electron microscopy (TEM) image of assembled PEG-LUM-C6-NP. (D) Luminescent spectra of a CLP conjugate, a Ce6–PEG conjugate, and luminol in the presence of 100 mM H2O2. Scheme is reproduced with permission from ref. 78. Copyright 2019 American Association for the Advancement of Science. | ||
Alternatively to PEG, poly(methacryloyl chloride) (i.e. PMC) was post-modified with luminol to deliver poly(methacrylamide)-based polymer for ROS sensing in live cell imaging.64 To assess the post-polymerization modification, first PMC with a Mn of 6800 g·mol−1 (Đ = 1.23) was synthesized via conventional free radical polymerization, which was further post-functionalized to deliver the luminol functionalized poly(methacrylamide) homopolymer (PML). Prior to assessing its ability to detect H2O2 in living cells, the detection limit of peroxides in solution was investigated for PML, which was 0.06 μM. Respectively, human cervical cancer HeLa cells were incubated with PHL (5.0 μM) and exposed to fluorescence microscopy at 37 °C, revealing slight blue fluorescence in intracellular regions. In order to generate a prominent increase of bright-blue fluorescence in the intra cellular regions, it was essential that the polymer incubated cells were treated with additional 50.0 μM H2O2 for extra 90 min. Importantly, the reported poly(methacrylamide) was soluble in basic medium as well as polar solvents such as DMSO and DMF, which facilitated to deliver PML-coated electrode surfaces or modified enzymes without the need for laborious electro(co)polymerization of luminol and enzyme immobilization using cyclic voltammetry method.
Quite recently, the synthesis of a self-reporting system with CL output was reported, which could be used to map reactive oxidative stress in the human body without the need for an external trigger.80 Accordingly, a copolymer (LUM-TBD-PMMA) decorated with luminol and a superbase (i.e. 1,5,7-triaza-bicyclo-[4.4.0]dec-5-ene (TBD)), the latter being essential as a coreactant for the CL response of luminol,81 was synthesized via sequential free radical polymerization of pentafluorophenyl acrylate (PFPA) and 4-vinyl benzylchloride (VBC) in the presence of methyl methacrylate (MMA), and subsequent orthogonal post-polymerization modification (Scheme 12A). While the PFPA segment reacted selectively in an orthogonal manner with luminol by virtue of the high reactivity of acrylate PFP-ester derivatives toward aromatic amines,82 the VBC moiety was substituted with TBD. Critically, 19F NMR analysis facilitated to assess that all active PFP-ester moieties were substituted by the luminol. In order to construct a self-reporting CL system, the superbase-driven luminol concept81 was further expanded by constructing supramolecular assemblies (CL-LUM-TB-PMMA-complex-Me-β-CD in Scheme 12B) based on the luminol–TBD–polymer with a tailor-made supramolecular host-molecule, such as the methylated derivative of β-CD (Me-β-CD). It is important to mention that the guanidine-moiety in the essential amino acid L-arginine,83 which is analogue of the guanidine functional unit in TBD, is known to enable selective host–guest inclusion complexes with β-CD, whereas β-CD is also recognized as efficient booster for the CL of luminol.84 Accordingly, insight into the complexation process between copolymer and Me-β-CD was obtained from nuclear Overhauser effect spectroscopy (NOESY shown in Scheme 12C) analysis, which revealed the presence of the following cross-resonances (i.e. NOEs) at: (i) 8.5 ppm assigned to the anticipated dominant dipolar interaction between the Me-β-CD annular protons and the guanidine derivative; (ii) 7.5 ppm that arise from the interactions of the Me-β-CD with the luminol moiety; and (iii) 4.5 ppm, assigned to a complexation of the MMA moieties or further superbase functionalities by the Me-β-CD. UV-Vis analysis of the complex upon the addition of 0.1 mL of 1.0 M H2O2 showed that the adsorption bands associated with the luminol segment appearing at 360 nm and 300 nm, decreased, while a peak at 260 nm arose. Further, DLS and NOESY analysis have clearly demonstrated the impact of the oxidant on the supramolecular complex. In other words, upon the addition of 0.1 mL of 1.0 M H2O2, the hydrodynamic size of the newly formed complex was 47% larger than the one of the parent complex, and close to the size of the parent copolymer (7% difference). The authors postulated that the new complex was formed upon the host–guest interactions between the oxidized luminol and Me-β-CD, in accordance with the results of the NOESY spectrum which revealed a cross-resonance at 10.5 ppm assigned to interactions between the oxidant (H2O2) and Me-β-CD or the polymer backbone. Importantly, the NOESY spectra didn't shown any detectable cross-resonance between Me-β-CD and TBD, respectively (Scheme 12C). Whereas the cross-resonance in the aromatic region (blue box) indicated interactions between the oxidized luminol and Me-β-CD, thus confirming the DLS results. Importantly, the CL emission of the complex was visible by the naked eye, being ∼140 times higher compared to the CL emission of the parent copolymer (Scheme 12D). The authors suggest that their study serves as inspiration for the development of artificial materials for the sensing of critical situations (damages or structural changes) in polymeric materials.
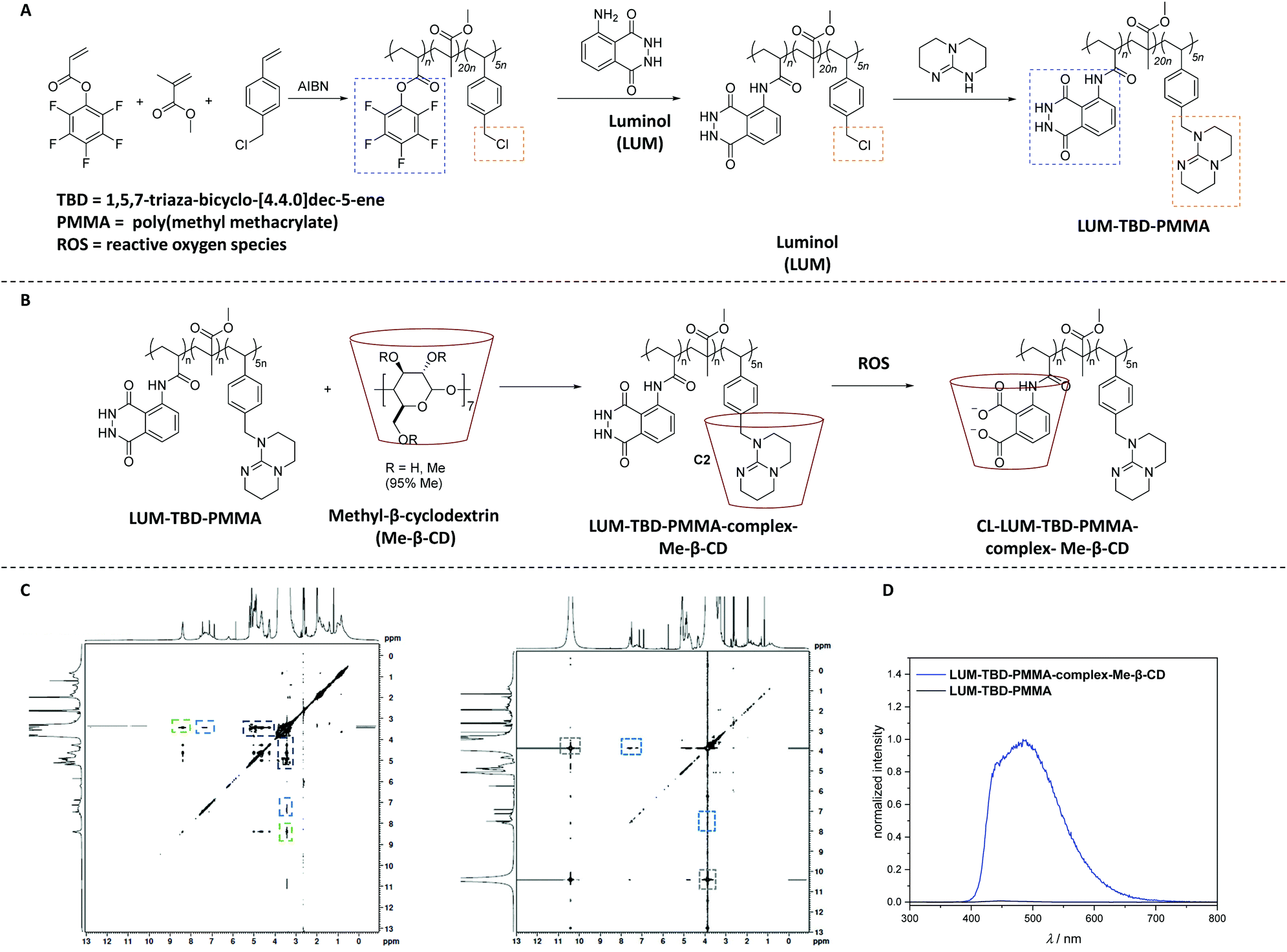 | ||
| Scheme 12 (A) Synthetic route to the luminol–superbase polymers LUM-TBD-PMMA via free radical polymerization and subsequent post-polymerization modification with luminol and the respective superbase. (B) General reaction pathway of the host–guest-complexation reaction to obtain LUM-TB-PMMA-complex-Me-β-CD (the chemical structure of each compound is depicted in solution, i.e. DMSO). Subsequently, the oxidation process is displayed, triggered by the addition of ROS (i.e. H2O2) resulting in the host–guest-complex CL-LUM-TB-PMMA-complex-Me-β-CD (C). NOESY spectrum of LUM-TB-PMMA-complex-Me-β-CD (left) and CL-LUM-TB-PMMA-complex-Me-β-CD + H2O2 (right) in DMSO-d6. The NOESY spectra are recorded at 300 K. (D) CL emission of LUM-TB-PMMA-complex-Me-β-CD and the parent copolymer LUM-TB-PMMA (c = 3.25 × 10−4 mM) in DMSO at ambient temperature, triggered by 0.1 mL of 1.0 M H2O2. The schemes and figures are reproduced with permission from ref. 80. Copyright 2020 Royal Society of Chemistry. | ||
Conclusion and outlook
Underpinned by extensive reports in recent years on luminol utilization for the generation of advanced polymeric materials, it is clear that the future is bright for this versatile class of polymer systems. Nevertheless, the limited number of synthetic methodologies that have been developed for preparing luminol containing polymers is a clear sign that the field has remained in its infancy. Considerable efforts have largely focused on electrochemical and chemical polymerization methods, whereas the toolbox of the numerous opportunities to apply the concepts of classical and modern polymer chemistry to prepare macromolecules with useful properties is still not fully unlocked. The development of new step-growth, addition and condensation polymerization methods that directly utilize luminol remains an important opportunity to prepare polymeric materials with worthwhile CL, ECL and thermomechanical properties. In fact, the investigation of advanced accelerated or catalyzed processes will be of critical importance to afford new materials. For instance, multicomponent polymerization processes are an example of new synthetic methods that can utilize luminol for polymer synthesis that should be further explored. Indeed, it is evident that novel and clever synthetic solutions remain to enhance the scope of methods and materials, which is the key to unlocking the true technological potential of this field.Finally, it should be emphasized, one more time, that the panoramic breadth of the luminol based polymers point to a possible future in which those polymers hold promise for materials science applications. Although concrete application examples remain to be realized, they have the potential to be developed by the provision of advanced luminol containing soft matter materials systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. B.-K. acknowledges continued support from the Karlsruhe Institute of Technology (KIT) in the context of the Helmholtz BioInterfaces in Technology and Medicine (BIFTM) and Science and Technology of Nanosystems (STN) programs. Further, C. B.-K. acknowledges key support from the Australian Research Council (ARC) for funding in the form of a Laureate Fellowship underpinning his photochemical research program as well as the Queensland University of Technology (QUT) for key continued support. H. M. acknowledges Prof. P. Theato (KIT) and Prof. C. Barner-Kowollik (QUT) for their continued mentorship.Notes and references
- R. Boyle, Philos. Trans. R. Soc. London, 1668, 2, 581 Search PubMed.
- E. N. Harvey, The Nature of Animal Light, J. B. Lippincott Company, 1920 Search PubMed.
- M. Vacher, I. F. Galvan, B.-W. Ding, S. Schramm, R. Berraud-Pache, P. Naumov, N. Ferre, Y.-J. Liu, I. Navizet, D. Roca-Sanjuan, W. J. Baader and R. Lindh, Chem. Rev., 2018, 118, 6927 CrossRef CAS.
- M. L. Wei, Y. F. Gao, X. Li and M. J. Serpe, Polym. Chem., 2017, 8, 127 RSC.
- C. M. Geiselhart, H. Mutlu and C. Barner-Kowollik, Angew. Chem., 2021 DOI:10.1002/anie.202012592.
- J.-Y. Koo and G. B. Schuster, J. Am. Chem. Soc., 1977, 99, 6107 CrossRef CAS.
- H.-W. Yeh and H.-W. Ai, Annu. Rev. Anal. Chem., 2019, 12, 129 CrossRef CAS.
- K. Suzuki and T. Nagai, Curr. Opin. Biotechnol., 2017, 48, 135 CrossRef CAS.
- B. Minaev, G. Baryshnikov and H. Agren, Phys. Chem. Chem. Phys., 2014, 16, 1719 RSC.
- T. Goto, Pure Appl. Chem., 1968, 17, 441 Search PubMed.
- Z. Yao, B. S. Zhang and J. A. Prescher, Curr. Opin. Chem. Biol., 2018, 45, 148 CrossRef CAS.
- A. C. Love and J. A. Presche, Cell Chem. Biol., 2020, 27, 904 CrossRef CAS.
- W. W. Coblentz, Phys. Z., 1909, 10, 955 Search PubMed.
- S. H. D. Haddock, M. A. Moline and J. F. Case, Annu. Rev. Mar. Sci., 2010, 2, 443 CrossRef.
- E. A. Widder, Science, 2010, 328, 704 CrossRef CAS.
- M. Yang, J. Huang, J. Fan, J. Du, K. Pu and X. Peng, Chem. Soc. Rev., 2020, 49, 6800 RSC.
- L. Delafresnaye, F. R. Bloesser, K. B. Kockler, C. W. Schmitt, I. M. Irshadeen and C. Barner-Kowollik, Chem. – Eur. J., 2020, 26, 114 CrossRef CAS.
- A. P. Schaap, T.-S. Chen, R. S. Handley, R. DeSilva and B. P. Giri, Tetrahedron Lett., 1987, 28, 1155 CrossRef CAS.
- H. O. Albrecht, Z. Phys. Chem., 1928, 136U Search PubMed.
- E. H. White, O. Zafiriou, H. H. Kagi and J. H. M. Hill, J. Am. Chem. Soc., 1964, 86, 940 CrossRef CAS.
- W. Specht, Angew. Chem., Int. Ed. Engl., 1937, 50, 155 CrossRef CAS.
- C. A. Marquette and L. J. Blum, Anal. Bioanal. Chem., 2006, 385, 546 CrossRef CAS.
- C. Dodeigne, L. Thunus and R. Lejeune, Talanta, 2000, 51, 415 CrossRef CAS.
- E. Baxter, Presumptive Tests and Chemical Enhancements, in Complete Crime Scene Investigation Handbook, CRC Press, 2015, pp. 129–152 Search PubMed.
- P. Khan, D. Idrees, M. A. Moxley, J. A. Corbett, F. Ahmad, G. von Figura, W. S. Sly, A. Waheed and M. I. Hassan, Appl. Biochem. Biotechnol., 2014, 173, 333 CrossRef CAS.
- C. A. Marquette and L. J. Blum, Anal. Bioanal. Chem., 2006, 385, 546 CrossRef CAS.
- C. A. Marquette, B. D. Leca and L. J. Blum, Luminescence, 2001, 16, 159 CrossRef CAS.
- N. Harvey, J. Phys. Chem., 1929, 33, 1456 CrossRef CAS.
- M. M. Richter, Chem. Rev., 2004, 104, 3003 CrossRef CAS.
- A. Sassolas, L. J. Blum and B. D. Leca-Bouvier, Anal. Bioanal. Chem., 2008, 390, 865 CrossRef CAS.
- A. Sassolas, L. J. Blum and B. D. Leca-Bouvier, Sens. Actuators, B, 2009, 139, 214 CrossRef CAS.
- G. Li, X. Zheng and L. Song, Electroanalysis, 2009, 21, 845 CAS.
- Y.-W. Liou and C. M. Wang, J. Electroanal. Chem., 2001, 495, 126 CrossRef CAS.
- K. Chiang Lin, S. Yu Lai and S. Ming Chen, Analyst, 2014, 139, 3991 RSC.
- G. F. Zhang and H. Y. Chen, Anal. Chim. Acta, 2000, 419, 25 CrossRef CAS.
- S. M. Chen and K. C. Lin, J. Electroanal. Chem., 2002, 523, 93 CrossRef CAS.
- Y. T. Chang, K. C. Lin and S. M. Chen, Electrochim. Acta, 2005, 51, 450 CrossRef CAS.
- K. C. Lin and S. M. Chen, J. Electroanal. Chem., 2006, 589, 52 CrossRef CAS.
- V. Ferreira, A. C. Cascalheira and L. M. Abrantes, Electrochim. Acta, 2008, 53, 3803 CrossRef CAS.
- V. Ferreira, A. C. Cascalheira and L. M. Abrantes, Thin Solid Films, 2008, 516, 3996 CrossRef CAS.
- E. De Robertis, R. S. Neves and A. J. Motheo, Mol. Cryst. Liq. Cryst., 2008, 484, 322 CAS.
- T. P. Mendonca, S. R. Moraes and A. J. Motheo, Mol. Cryst. Liq. Cryst., 2006, 447, 383 CAS.
- C. H. Wang, S. M. Chen and C. M. Wang, Analyst, 2002, 127, 1507 RSC.
- J. Ballesta-Claver, J. Ametis-Cabello, J. Morales-Sanfrutos, A. Megía-Fernández, M. C. Valencia-Mirón, F. Santoyo-González and L. F. Capitán-Vallvey, Anal. Chim. Acta, 2012, 754, 91 CrossRef CAS.
- Z. Fu, Z. Lin, S. Huang and M. Liu, Acta Phys. Chim. Sin., 1994, 10, 371 CAS.
- E. M. Genies and C. Tsintavi, J. Electroanal. Chem., 1985, 195, 109 CrossRef CAS.
- J. Schiller, J. Arnhold, J. Schwinn, H. Sprinz, O. Brede and K. Arnold, Free Radic. Res., 1999, 30, 45 CrossRef CAS.
- D. Asil, A. Cihaner and A. M. Önal, Chem. Commun., 2009, 307 RSC.
- N. Atilgan, F. Algi, A. M. Onal and A. Cihaner, Tetrahedron, 2009, 65, 5776 CrossRef CAS.
- M. P. Algi, Z. Oztas, S. Tirkes, A. Cihaner and F. Algi, J. Fluoresc., 2017, 27, 509 CrossRef CAS.
- D. Asil, A. Cihaner, F. Algi and A. M. Onal, Electroanalysis, 2010, 22, 2254 CrossRef CAS.
- W. Chen and G. Xue, Prog. Polym. Sci., 2005, 30, 783 CrossRef CAS.
- J. Ballesta-Claver, M. C. Valencia-Mirón and L. F. Capitán-Vallvey, Anal. Bioanal. Chem., 2011, 400, 3041 CrossRef CAS.
- E. P. Koval'chuk, I. V. Grynchyshyn, O. V. Reshetnyak, R. Y. Gladyshevs'kyj and J. Błażejowski, Eur. Polym. J., 2005, 41, 1315 CrossRef.
- T. Zhang, H. Qi, Z. Liao, Y. D. Horev, L. A. Panes-Ruiz, P. S. Petkov, Z. Zhang, R. Shivhare, P. Zhang, K. Liu, V. Bezugly, S. Liu, Z. Zheng, S. Mannsfeld, T. Heine, G. Cuniberti, H. Haick, E. Zschech, U. Kaiser, R. Dong and X. Feng, Nat. Commun., 2019, 10, 4225 CrossRef.
- J. N'Diaye and K. Lian, J. Electroanal. Chem., 2019, 839, 90 CrossRef.
- T. F. Otero and M. Bengoechea, Langmuir, 1999, 15, 1323 CrossRef CAS.
- R. Pandimurugan and S. Thambidurai, Anal. Methods, 2015, 7, 10422 RSC.
- M. M. Mahat, D. Mawad, G. W. Nelson, S. Fearn, R. G. Palgrave, D. J. Payne and M. M. Stevens, J. Mater. Chem. C, 2015, 3, 7180 RSC.
- S. Golczak, A. Kanciurzewska, M. Fahlman, K. Langer and J. J. Langer, Solid State Ionics, 2008, 179, 2234 CrossRef CAS.
- S. Jadoun, S. M. Ashra and U. Riaz, Polym. Adv. Technol., 2018, 29, 1007 CrossRef CAS.
- U. Riaz, S. Jadoun, P. Kumar, R. Kumar and N. Yadav, RSC Adv., 2018, 8, 37165 RSC.
- P. G. Murray, D. Sproul and A. M. Tseng, WO2000018850A1, 2000.
- S. Deepa and K. Rajendrakumar, ChemistrySelect, 2019, 4, 1158 CrossRef CAS.
- Y. Koyama, M. Umehara, A. Mizuno, M. Itaba, T. Yasukouchi, K. Natsume, A. Suginaka and K. Watanabe, Bioconjugate Chem., 1996, 7, 298 CrossRef CAS.
- R. Klingender, Handbook of Specialty Elastomers, 1st edn, CRC Press, 2008, pp. 245–288 Search PubMed.
- B. Johnson and W. J. Ward, WO1998054569A1, 1998.
- M. R. Nabid, S. S. Taheri, R. Sedghi and S. J. T. Rezaei, Macromol. Res., 2011, 19, 280 CrossRef CAS.
- E. Blasco, M. B. Sims, A. S. Goldmann, B. S. Sumerlin and C. Barner-Kowollik, Macromolecules, 2017, 50, 5215 CrossRef CAS.
- H. Zhang, C. Smanmoo, T. Kabashima, J. Lu and M. Kai, Angew. Chem., Int. Ed., 2007, 46, 8226 CrossRef CAS.
- H. Zhang, T. Shibata, T. Krawczyk, T. Kabashima, J. Lu, M. K. Lee and M. Kai, Talanta, 2009, 79, 700 CrossRef CAS.
- B. Balakrishnan, S. Lesieur, D. Labarre and A. Jayakrishnan, Carbohydr. Res., 2005, 340, 1425 CrossRef CAS.
- A. Jain and K. Cheng, J. Controlled Release, 2017, 245, 27 CrossRef CAS.
- T. Krawczyk, M. Kondo, G. Azam, H. Zhang, T. Shibata and M. Kai, Analyst, 2010, 135, 2894 RSC.
- J. W. Guo, H. Tao, Y. Dou, L. L. Li, X. Q. Xu, Q. X. Zhang, J. Cheng, S. L. Han, J. Huang, X. D. Li, X. H. Li and J. X. A. Zhang, Mater. Today, 2017, 20, 493 CrossRef CAS.
- J. Guo, D. Li, H. Tao, G. Li, R. Liu, Y. Dou, T. Jin, L. Li, J. Huang, H. Hu and J. Zhang, Adv. Mater., 2019, 31, 1904607 CrossRef CAS.
- H. Tao, J. Guo, Y. Ma, Y. Zhao, T. Jin, L. Gu, Y. Dou, J. Liu, H. Hu, X. Xiong and J. Zhang, ACS Nano, 2020, 14, 11083 CrossRef CAS.
- X. Xu, H. An, D. Zhang, H. Tao, Y. Dou, X. Li, J. Huang and J. Zhang, Sci. Adv., 2019, 5, eaat2953 CrossRef.
- H. An, C. Guo, D. Li, R. Liu, X. Xu, J. Guo, J. Ding, J. Li, W. Chen and J. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 17230 CrossRef CAS.
- C. M. Geiselhart, H. Mutlu, P. Tzvetkova and C. Barner-Kowollik, Polym. Chem., 2020, 11, 4213 RSC.
- C. M. Geiselhart, C. W. Schmitt, P. Jöckle, H. Mutlu and C. Barner-Kowollik, Sci. Rep., 2019, 9, 14519 CrossRef.
- M. Eberhardt, R. Mruk, R. Zentel and P. Théato, Eur. Polym. J., 2005, 41, 1569 CrossRef CAS.
- R. Kataky, P. M. Kelly, D. Parker and A. F. Patti, J. Chem. Soc., Perkin Trans. 2, 1994, 2381 RSC.
- R. Maeztu, G. Tardajos and G. González-Gaitano, J. Phys. Chem. B, 2010, 114, 2798 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |