
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterotelechelic poly(propylene oxide) as migration-inhibited toughening agent in hot lithography based additive manufacturing†
Daniel
Grunenberg
a,
Katharina
Ehrmann
 a,
Christian
Gorsche
ab,
Bernhard
Steyrer
c,
Thomas
Koch
c,
Jürgen
Stampfl
c and
Robert
Liska
*a
a,
Christian
Gorsche
ab,
Bernhard
Steyrer
c,
Thomas
Koch
c,
Jürgen
Stampfl
c and
Robert
Liska
*a
aInstitute of Applied Synthetic Chemistry, Technische Universität Wien, Getreidemarkt 9/163, 1060 Vienna, Austria. E-mail: robert.liska@tuwien.ac.at
bCubicure GmbH, Gutheil-Schoder-Gasse 17, Tech Park Vienna, 1230 Vienna, Austria
cInstitute of Materials Science and Technology, Technische Universität Wien, Getreidemarkt 9/308, 1060 Vienna, Austria
First published on 9th February 2021
Abstract
Light-based processing techniques triggering photopolymerization are among the most promising 3D printing technologies due to their benefits regarding resolution, surface quality and build speed. However, the main challenge remains the development of strong and tough materials, since most commercially available photopolymer resins are limited in terms of their thermomechanical performance. We therefore synthesized a heterotelechelic hybrid oligomer based on poly(propylene oxide) (PPO-H), bearing one methacrylic and one addition–fragmentation chain transfer group (AFCT) as end groups. This new compound was successfully implemented as a toughening agent by regulating the network structure via the AFCT mechanism and acted as reactive diluent for highly viscous resins. Formulations containing 10–25 db% (double bond percent) of PPO-H mixed into a commercially available resin (Bomar XR-741MS) were investigated and compared to the corresponding PPO dimethacrylate (PPO-D) mixtures, representing state of the art resins. Full double bond conversion could be reached for PPO-H containing formulations, while shrinkage stress was simultaneously reduced by up to 50% compared to the PPO-D mixtures. Glass transition temperatures decreased with increasing PPO contents. Toughness was enhanced by a factor of 2 (10 db% PPO-H) to 4 (15 db% PPO-H), measured by elongation at break, while decrease in tensile strength remained low (factor of 1.2 and 1.4 for 10 and 15 db% PPO-H, respectively). Impact strength increased by 55 and 92% for 10 and 15 db% PPO-H, respectively. By employing the recently developed Hot Lithography technique, toughened and migration-free 3D printed parts were obtained with PPO-H.
Introduction
Since the pioneering work by Charles W. Hull1 in the 1980s, Additive Manufacturing (AM), commonly also referred to as ‘rapid prototyping’ or ‘3D printing’, has become a rapidly developing and industrially established technique for the production of parts with complex geometries. The basic principle of this method relies on the manufacturing of a 3D object by stacking up individual 2D-layers according to a geometry defined by a computer aided design (CAD) file.2 By now, almost any class of material can be processed by AM, including metals,3 glass,4 ceramics,5–7 and polymers.8–10 In contrast to metals and ceramics, where the technology is mainly limited to sintering techniques, polymers can be processed by a broad variety of AM methods.11 The most important ones among them rely on sintering of individual polymer particles (Selective Laser Sintering, SLS), the extrusion of molten thermoplastic material (Fused Filament Fabrication, FFF or Fused Deposition Modeling, FDM®)12 and inkjet printing.9 Another widely used class of AM methods uses photosensitive resins, which are cured selectively to obtain solid parts. Laser-based stereolithography (SLA),1 digital light processing (DLP),13 two photon polymerization (2PP),14 and continuous liquid interface production (CLIP)15 are relying on this approach.FFF type printers are already available at a few hundred dollars and work with low-cost polymer filaments. In principle, any meltable polymer can be employed, which results in a large diversity of material choice. For printing, the polymer filament has to be extruded through a heated nozzle, which is the reason for the main disadvantages to this technique: building speed is relatively slow and the interlaminar layer adhesion is limited. Furthermore, the printed parts show poor resolution and high surface roughness.16
These limitations can be overcome by SLA or DLP techniques using a laser or light projector to induce photopolymerization of a resin upon excitation with light. In these techniques, a laser in combination with a Galvano-scanner or a digital mirror device is used to induce photopolymerization of a resin upon contact with light. Main benefits of light-based techniques include high spatial resolution, excellent surface quality of the obtained parts, and high building speed.
Despite of all these advantages the main challenge is the limitation in accessible materials with sufficient thermomechanical properties. Typical resins consist of multifunctional acrylates or methacrylates, with methacrylates being preferred due to lower cytotoxicity17 of the monomers and a higher glass transition temperature (Tg) of the final polymer. Especially the toughness of commercially available resins is very limited. This is caused by high crosslinking densities of photopolymer networks combined with inhomogeneous network architectures. Therefore, there is a high demand for resins showing high heat resistance and toughness simultaneously.
Different concepts for the toughening of photopolymers have been reviewed.18 Common approaches include the addition of nanoparticles,19 core–shell particles,20 or rubber-like components.21 The latter method is adapted from non-photopolymers in the plastics industry, producing tough polymers on a megaton scale such as high impact poly(styrene) (HIPS) and acrylonitrile butadiene styrene (ABS). Other promising ways to increase the toughness of photopolymers is the employment of monomers with increased molecular weight (i.e. oligomers) or monomers that exhibit strong intermolecular forces (e.g. hydrogen bonding in urethane methacrylates22). The main barrier for using such materials in lithography-based 3D printing is the fact that these resins are either solid at room temperature or show very high viscosities. Therefore, it is necessary to work at elevated temperatures and to develop a printing setup that is able to deal with highly viscous resins. Such a device has been designed, patented, and introduced as Hot Lithography.23,24 With this setup, which contains a heatable resin vat, photopolymerizable resins can be printed at elevated temperatures (usually ≤120 °C, depending on the temperature stability of the resin) and with viscosities of up to 20 Pa s at the respective temperature. Besides employing higher molecular weight compounds, another widespread concept found in literature is achieving control over the polymerization process and therefore regulating the network structure. The most extensively studied and employed technique in this context is thiol–ene chemistry, which works by inducing a chain transfer during polymerization.25 Despite the high effectiveness of thiol-based additives, storage stability, characteristic odor, and softening of the final polymer are current challenges that need to be overcome in order to make these compounds applicable in a broader sense. Addition–fragmentation chain transfer (AFCT) does not show these limitations of classical thiol–ene chemistry.26 The AFCT process turns the classical chain-growth mechanism into a more step-growth like one, which results in more homogeneous network architectures.27 AFCT therefore gains more and more importance in regulating radical polymerization reactions. We have recently introduced a potent vinyl sulfonate ester-based AFCT agent that shows nearly no delay in reactivity compared to previous generations.28
Based on these findings, we now present the toughening potential of a poly(propylene oxide) (PPO)-based hybrid oligomer. This oligomer is able to regulate the network formation via the AFCT mechanism during Hot Lithography by one end-functionality and is simultaneously hindered from migration by its second end-functionality. While polymerizable difunctional monomers, which contain an allyl sulfide or trithiocarbonate in between these end functionalities, were reported by Bowman and his colleagues,29,30 this is to the best of the authors’ knowledge the first description of a monomer that has both, a polymerizable group and a moiety capable of chain transfer as end groups.
Experimental
Materials and methods
Bisomer PPM 5 LI was provided by GEO Specialty Chemicals and used as received. Ethyl pyruvate (>97%) was purchased from TCI Europe and freshly distilled before use. Methacrylic anhydride, 2,6-di-tert-butyl-4-methylphenol (BHT), 4-methoxyphenol (MEHQ), 4-dimethylaminopyridin (DMAP), hydrochinone, and tosyl chloride were ordered from Sigma Aldrich and used without further purification. Phenothiazine and 1,4-diazabicyclo[2.2.2]octane (DABCO) were obtained from TCI Europe and used as received. For purification after synthesis, aluminium oxide (neutral, Brockmann type I) and silica gel 60 from Carl Roth were used. All solvents were distilled prior to use or were purchased in distilled quality. Concentrated H2SO4 was obtained from Donauchemie AG and used without purification. The photoinitiator ethyl (2,4,6-trimethylbenzoyl) phenyl (TPO-L) was kindly gifted by Lambson Ltd. Bomar XR-741MS (1000 g mol−1) was obtained from Dymax Oligomers & Coatings. The UV absorber UV-1990 was ordered from Eutec Chemical (Taiwan). NMR-spectra were recorded on a Bruker Avance DRX-400 FT-NMR spectrometer at 400 MHz. The signals are reported with their chemical shifts in ppm and fine structure (s = singlet, d = doublet, t = triplet, q = quartet, qn = quintet, sep = septet, m = multiplet). The chemical shifts were referenced by using the respective NMR-solvent [CDCl3 (7.26 ppm)] as internal reference.A self-built metallic heating plate with electric heating and temperature control was used to warm the formulations outside the oven.
Synthesis of poly(propylene oxide) hybrid oligomer (PPO-H)
All syntheses were carried out under inert atmosphere (Ar) using Schlenk technique.1st step: 150.90 g (0.4023 mol, 1.00 eq.) Bisomer PPM 5 LI were weighted into a flame-dried 1 L three-neck round bottom flask equipped with a magnetic stirring bar and vacuum dried under stirring until no bubble formation was visible anymore. 489.28 g (4.214 mol, 10.5 eq.) of freshly distilled ethyl pyruvate were added using a transfer cannula. 4.53 g hydrochinone (3 wt% related to Bisomer PPM 5 LI, inhibitor) were added and dissolved under stirring. After complete dissolution of the hydrochinone, 15 drops of concentrated H2SO4 were added using a Pasteur pipette under vigorous stirring and a counter flow of argon.
A hot distillation bridge was assembled onto the three-neck flask and the solution was stirred at 80 °C and 200 mbar for 47.5 hours in an oil bath. Afterwards, the temperature was raised to 85 °C and the vacuum was lowered to 15 mbar in order to remove excess ethanol and ethyl pyruvate. To remove any remaining traces of ethyl pyruvate, high vacuum (2 × 10–2 mbar) was finally applied until no more distillate was collected.
The remaining reddish-brown reaction mixture was allowed to cool to room temperature and mixed with 1 L of diethyl ether. The mixture was extracted with sat. NaHCO3 solution (2 × 300 mL), deionized water (1 × 300 mL), and brine (2 × 150 mL). The slightly orange organic phase was dried over MgSO4 overnight. Evaporation of the solvent gave an orange-brown oil that was further vacuum dried (2 × 10–2 mbar) at 60 °C for 8 hours. 175.11 g (98%) of the intermediate product 3 were obtained as an orange-brown, transparent, slightly viscous liquid.
2nd step: An apparatus consisting of a 2 L three-neck round bottom flask, a 1 L dropping funnel, and a magnetic stirring bar was flame-dried, evacuated, and refilled with argon three times. 65.34 g (0.5825 mol, 1.50 eq.) DABCO were added to the flask and dissolved in 400 mL of dry methylene chloride. In the dropping funnel, a solution of 88.85 g (0.4661 mol, 1.20 eq.) tosyl chloride in 700 mL was prepared. The intermediate product 3 was then added to the dropping funnel and the whole content was mixed to obtain a homogeneous solution. This solution was added dropwise over two hours at 0 °C and the reaction was continued for 62 hours at room temperature.
The resulting yellow-brown suspension was vacuum filtered over a layer of silica (ca. 3 cm) and concentrated to 1 L. After extraction with 2% aqueous HCl (2 × 300 mL), deionized water (1 × 300 mL), and brine (1 × 300 mL), the organic phase was dried over Na2SO4 overnight. After distilling off the solvent and vacuum drying, the flask was stored at −20 °C to complete the precipitation of a fine solid. The solid side product was removed from the cold suspension by vacuum filtration through a glass frit. The collected brownish oil was filtered over 320 g of aluminium oxide that had been packed with diethyl ether in advance by applying vacuum. A total of 2 L diethyl ether was used to elute the product. After evaporation of the solvent, the filtration process was repeated. 200 ppm of BHT and 200 ppm of MEHQ were added to stabilize the product and the solvent was distilled off. 171.01 g (73%) of PPO-H were obtained as an orange, viscous oil.
1H NMR (CDCl3, 400 MHz, δ, ppm): 0.95–1.37 (m, 16 H; H1), 1.91 (m, 3 H; H2), 2.42 (s, 3 H; H3), 3.15–4.29 (m, 15 H; H4), 5.03 (br s, 1 H; PPO backbone end group H5), 5.47–5.65 (m, 2 H; H6), 6.01–6.18 (m, 2 H, H7) 7.28–7.41 (m, 2 H; H8) 7.71–7.93 (m, 2 H; H9).
Synthesis of poly(propylene oxide)dimethacrylate (PPO-D)
201.68 g (0.538 mol, 1.00 eq.) Bisomer PPM 5 LI were weighted into a flame-dried 500 mL three-neck round bottom flask equipped with a magnetic stirring bar and vacuum dried. 3.28 g (0.027 mol, 0.05 eq.) DMAP and 0.08 g phenothiazine (400 ppm related to Bisomer PPM 5 LI, inhibitor) were added. 160 mL (1.08 mol, 2.00 eq.) of freshly distilled methacrylic anhydride were injected into the flask using a syringe. The reaction was carried out at 70 °C for 48 hours in an oil bath. Afterwards, non-reacted anhydride was distilled off using high vacuum (5 × 10–2 mbar) at 70 °C. The crude mixture was diluted wit 1 L of diethyl ether and extracted with aqueous HCl (2%, 3 × 300 mL), aqueous NaOH (2%, 8 × 300 mL), deionized water (5 × 300 mL), and brine (2 × 300 mL). After drying over Na2SO4 overnight, the organic phase was vacuum filtered over a glass frit filled with a small layer of silica (1 cm) and sand (2 cm) in order to remove any remaining fine particles. Before evaporation of the solvent, 200 ppm of BHT and 200 ppm of MEHQ were added to the filtrate and the product was vacuum dried until no bubble formation was visible any more. 183.73 g (77%) of PPO-D were obtained as a colorless, slightly viscous liquid.1H NMR (CDCl3, 400 MHz, δ, ppm): 1.01–1.35 (m, 16 H; H1), 1.83–2.04 (m, 6 H; H2), 3.11–4.37 (m, 15 H; H3), 4.90–5.31 (br m, 1 H; PPO backbone end group H4), 5.51 (d, 2 H; H5), 6.07 (d, 2 H; H6).
Preparation and moulding of the formulations
1 wt% of TPO-L and the corresponding amounts of Bomar XR-741MS, PPO-H or PPO-D were weighted into a plastic cup and stored at 100 °C for 30 minutes. As the overall amount of double bonds and the amount of AFCT engaging double bonds is slightly obscured by mixing PPO-H with Bomar (two double bonds from Bomar in addition to one regular double bond and one AFCT engaging double bond of PPO-H), all formulation components were calculated in double bond percent (db%), i.e. the relative amount of AFCT double bonds compared to the overall double bonds in the formulation. E.g. 10 db% AFCT content corresponds to a molar ratio Bomar![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PPO-H of 4:1 and to 20 mol% AFCT content of the formulation. As the PPO-D contains no AFCT group, it was added equimolar with respect to the PPO-H mixtures to ensure a fair comparison. The hot cups were mixed with a SpeedMixer™ at 2350 rpm for two minutes and subsequently degassed in a vacuum chamber until no bubble formation was visible any more.
:PPO-H of 4:1 and to 20 mol% AFCT content of the formulation. As the PPO-D contains no AFCT group, it was added equimolar with respect to the PPO-H mixtures to ensure a fair comparison. The hot cups were mixed with a SpeedMixer™ at 2350 rpm for two minutes and subsequently degassed in a vacuum chamber until no bubble formation was visible any more.
The specimens for (thermo)mechanical testing were prepared using a silicone mould. The silicone mould was coated with Teflon spray and stored in an oven at 90–100 °C for ten minutes to remove solvent residues and to preheat the mould. The hot formulations (stored at 90–100 °C) were rapidly poured into the cavities of the preheated mould. Due to the high viscosity of the formulations, sometimes air bubbles were trapped inside the casting. Degassing was realized inside a heated desiccator at 30–100 mbar and was repeated until the cavities were free of air bubbles. After heating the filled moulds in the oven again, excessive formulation was removed using a spatula. The hot mould was then placed on a self-built metallic heating plate (90 °C) and cured for five minutes using a ADJ UV Flood 36 LED with a wavelength of 395–400 nm (6 mW cm−2 at the sample surface) from one side. Post curing was done by irradiation inside an Uvitron IntelliRay 600 broadband UV oven (50 mW cm−2 at the sample surface) for five minutes from both sides.
The cured specimens were polished with sandpaper in order to obtain accurate dimensions for mechanical testing. Prior to the measurements, all samples were stored in a climate chamber at 23 °C and 50% relative humidity for at least four days.
RT-NIR photorheology
Real-time near-infrared (RT-NIR) photorheology was conducted using an Anton Paar MCR302 WESP rheometer with a P-PTD 200/GL Peltier glass plate and a PP08 disposable measuring system. A H-PTD 200 heating mantle was used to conduct the experiments at 90 °C. The rheometer was coupled to a Bruker VERTEX 80 FTIR spectrometer in order to record the RT-NIR spectra.31 Before the measurement, the light intensity at the curing position was determined with an Ocean Optics USB 2000+ spectrometer. For each measurement, the plate gap was fixed to 0.2 mm and 13 μL of the respective formulation were applied. An oscillatory sheer with an angular strain of 1% and a frequency of 1 Hz was used during the procedure. After an equilibration period of 60 s, the LED light source was switched on (300 s, 400 nm, intensity at curing position 4 mW cm−2), irradiating the sample on the glass plate and starting the curing reaction. In order to determine the double bond conversions, an NIR beam was directed through the glass plate onto the sample and analyzed with a mercury cadmium telluride (MCT) detector. NIR spectra were measured at wave numbers from 4000 to 7000 cm−1 with a resolution of 8 cm−1. A set of spectra was recorded 5 s before irradiation and averaged to serve as starting value. To determine the double bond conversion (DBC), the respective double bond signal at ∼6190 cm−1 was integrated and the ratio between start and end value was calculated. Rheological data, i.e. storage and loss modulus as well as the normal force were recorded during polymerization. Furthermore, the gel point (tg) was determined as the crossover between storage (G′) and loss modulus (G′′) in the curve. From these data, also the DBC at the gel point (DBCg) and the time to reach 95% of overall conversion (t95%) were extracted. All measurements were conducted in triplicates and results are given as mean values.Swelling experiments
The swelling studies were carried out with photocured moulded or 3D printed discs in ethanol. All cured samples of varying compositions were studied in triplicates. The samples were initially weighed (mstart) and then kept separately in glass vials at ambient conditions containing 2 mL of absolute ethanol. The solvent was renewed after seven days by decanting the old solvent and replacing it with fresh one. The final swelling weight was taken after 14 days (mswollen). These samples were then dried in a vacuum oven at 60 °C until constant weight (mdry) was obtained. The degree of swelling (S) and the gel fraction (G) were calculated (eqn (1) and (2)) and provided as mean values with standard deviation.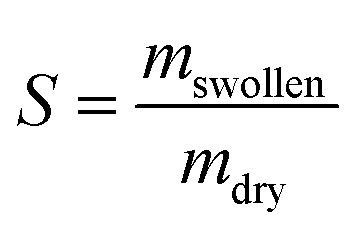 | (1) |
 | (2) |
Dynamic mechanical thermal analysis
An Anton Paar MCR 301 rheometer with a CTD 450 oven and an SRF 12 measuring system was used for dynamic mechanical thermal analysis (DMTA). Test specimens (40 × 5 × 2 mm3) were prepared by moulding or 3D printing and measured in torsion mode with a frequency of 1 Hz and a strain of 0.1%. A normal force of −1 N was applied and the temperature was gradually increased from −100 to 200 °C at a constant heating rate of 2 K min−1. Due to high reproducibility single measurements were performed.Tensile testing
Tensile testing was performed on a Zwick Z050 tensile testing machine at 23 °C. The crosshead speed during the test was set to 5 mm min−1. Five specimens of each formulation were prepared by moulding or 3D printing, sanded, and conditioned at 23 °C and 50% relative humidity for at least four days. The geometry of the samples was in accordance to ISO 527 5B. Means and standard deviations of maximum stress values and elongations at break were calculated. Energies at break were calculated through integration of the area under each stress–strain curve and calculation of mean and standard deviation from these values.Dynstat experiments
Dynstat tests were performed on a Karl Frank Type 573 Dynstat machine. After the unnotched samples (4 × 10 × 15 mm3, prepared by moulding or 3D printing) had been fractured with a hammer, the values were taken from the scale, thereafter converted into kJ and divided by the cross-sectional area of the specimen in m2. At least four specimens were tested for every formulation to obtain means and standard deviations.AM via Hot Lithography
For the 3D printing experiments, a self-built DLP setup as previously introduced by Steyrer et al. was used.32 The printing system is based on a diamond-WXGA light engine with a light intensity of 4 mW cm−2 and a wavelength of 405 nm. A heating unit based on the Hot Lithography technology developed by Cubicure GmbH was mounted onto the DLP setup. All printing and postcuring tests were performed in a yellow-light laboratory. During the printing job the resin vat was heated to 90 °C. The accuracy of the vat temperature was checked with a PT100 contact thermometer in order to secure a temperature of 23 ± 2 °C without heating, and 90 ± 2 °C during the printing job.The mixtures Bomar/PPO-H_10 db%_3D and Bomar/PPO-H_15 db%_3D were prepared by adding 0.1% of the absorber UV-1990 and 1 wt% of the photoinitiator TPO-L to the neat mixtures. Homogenization was achieved by heating to 90 °C whilst mechanical stirring.
The layer thickness was adjusted to 100 μm. For Bomar/PPO-H_10 db%_3D, 25 s of exposure time were necessary to achieve curing. For of Bomar/PPO-H_15 db%_3D, an exposure time of 40 s had to be used. After printing, all specimens were cleaned with a paper towel and postcured in an Uvitron Intelliray 600 UV oven for 2 × 300 s (100% intensity, 175 mW cm−2) from each side.
Results and discussion
In order to overcome the brittleness of most commercially available photoresins, we designed a light-curable heterotelechelic hybrid oligomer (PPO-H) that acts as a non-migrating toughness modifier. By combining an AFCT group and a polymerizable methacrylate group within the same molecule, several advantages are expected. A typical AFCT agent is a low molecular weight component that is added to the formulation and may migrate out of the cured polymer.33 By using our hybrid oligomer instead, a covalent incorporation into the final polymer network and therefore limited migration issues are expected. The high molecular weight further prevents volatility, which can be a challenge when working at elevated temperatures, as it is the case in Hot Lithography.The toughening effect relies on increased homogeneity of the network structure, which is obtained through AFCT regulation.34 By choosing PPO35 as the spacer, which shows a low glass transition of ca. −70 °C, an additional rubber-like toughening effect was anticipated.
For comparison, a corresponding PPO dimethacrylate (PPO-D) was also synthesized, representing the classical difunctional nature of state-of-the art resins, which solely exhibits rubber-like toughening.
Synthesis of PPO oligomers
The starting material for PPO-H and PPO-D, hydroxy-terminated PPO monomethacrylate, bearing five repeating units of propylene oxide on average (Bisomer PPM 5 LI, 1), was commercially available. The PPO hybrid was obtained in a two-step synthesis with 70% yield (Scheme 1).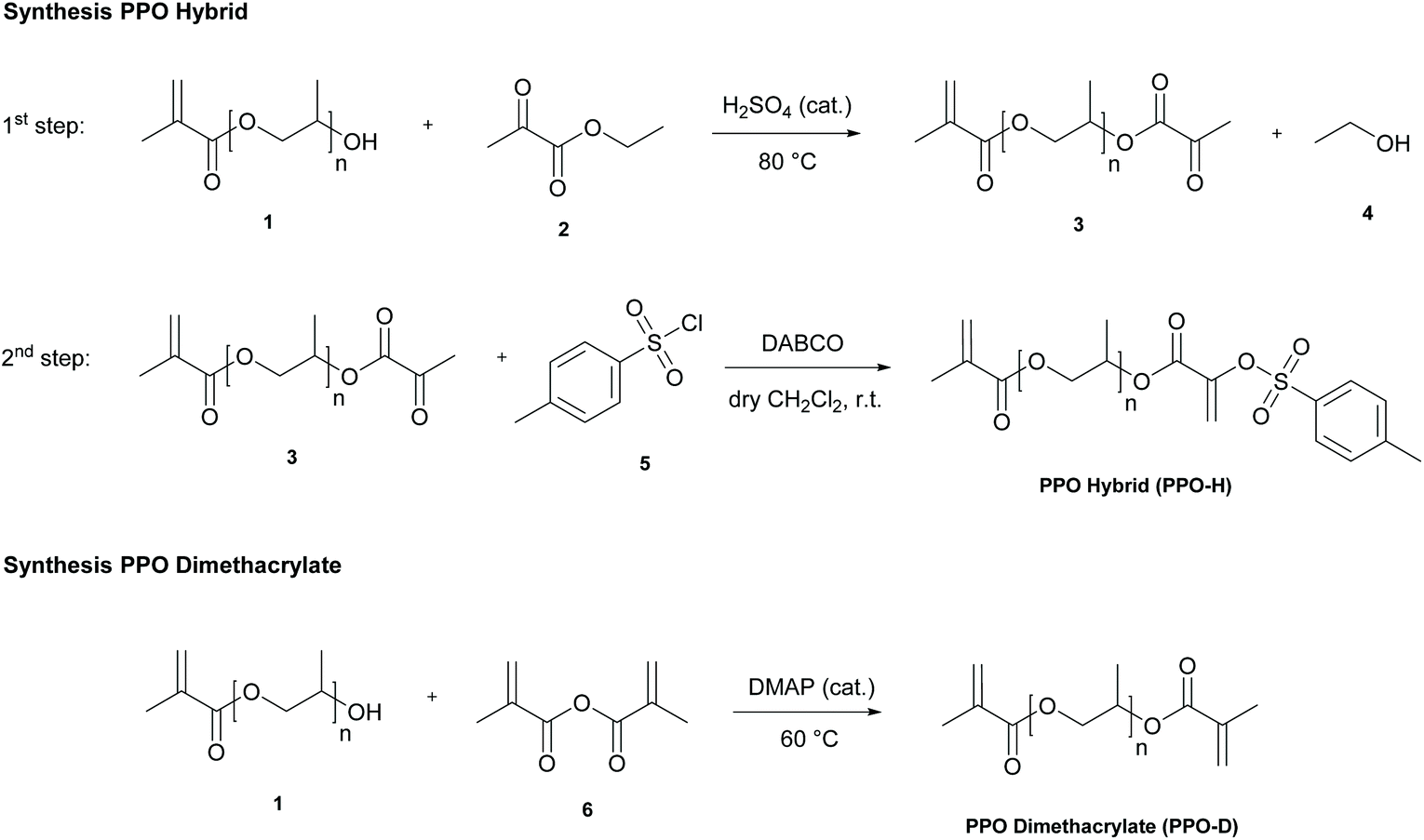 | ||
| Scheme 1 Synthetic routes towards PPO hybrid (PPO-H) and PPO dimethacrylate (PPO-D). DABCO: 1,4-Diazabicyclo[2.2.2]octane. r.t.: Room temperature. DMAP N,N-Dimethylpyridin-4-amine. | ||
The first step involved a transesterification reaction with ethyl pyruvate 2. During this reaction, formed ethanol 4 was continuously distilled off under reduced pressure. The addition of 3 wt% hydrochinone is essential for the reaction, as sulfuric acid can otherwise induce premature polymerization of the (meth)acrylate group.36
1,4-Phenylene bis(4-methylbenzenesulfonate) is therefore formed as a side product by the reaction of tosyl chloride and hydrochinone (see ESI†). Attempts to use smaller amounts of hydrochinone or other stabilizers (phenothiazine) resulted in remarkable increase in viscosity (i.e. polymerization) during the reaction.
During the 2nd step, the intermediate pyruvate 3 is then further converted into a vinyl sulfonate AFCT group.37 Complete removal of excess ethyl pyruvate after the first step is very important, as residues result in the formation of a low molecular weight AFCT compound as a side product.
The PPO dimethacrylate PPO-D was prepared by esterification with methacrylic anhydride 6 in a solvent-free procedure, yielding 87% of product.38 The successful preparation of both compounds on a large scale (>150 g) shows potential for further upscaling.
Preparation of formulations
Due to the low Tg of PPO, photopolymerization of the pure oligomers lead to rubber-like and soft materials. In order to use them for load bearing applications, mixing with a suitable matrix that provides mechanical strength is necessary. Upon a screening of different commercially available photopolymer resins, Bomar XR-741 MS (Bomar), a urethane dimethacrylate, was chosen as the most promising candidate.Formulations containing different amounts of Bomar and PPO-H/PPO-D were prepared in order to investigate the influence of the AFCT content on reactivity and (thermo)mechanical properties (Table 1). Based on previous research, AFCT contents of 10, 15, 20, and 25 double bond percent (db%) were used.28 Db% denotes the relative amount of AFCT double bonds, i.e. AFCT sites, compared to the overall amount of double bonds (AFCT sites and reactive sites) in the formulation. This denotation already accounts for the lower amount of AFCT double bonds per molecule PPO-H compared to Bomar. For example, 25 db% of PPO-H corresponds to a molar ratio Bomar:PPO-H of 1:1 because Bomar contains two mol double bonds (reactive sites) per mol resin while PPO-H contains one mol of double bonds (reactive sites) and one mol of AFCT double bonds (AFCT sites and reactive sites) per mol resin. Therefore, in one mol resin containing equimolar amounts of Bomar and PPO-H, one out of four double bonds is an AFCT site. As PPO-D contains no AFCT group, it was added equimolar with respect to the PPO-H mixtures to ensure a fair comparison.
| Formulation | Composition | Molar ratio [Bomar/PPO] | AFCT content | |
|---|---|---|---|---|
| [mol%] | [db%]a | |||
| a Relative amount of AFCT double bonds to overall double bonds in the formulation. | ||||
| A | Bomar/PPO-H 10 db% | 4/1 | 20 | 10 |
| B | Bomar/PPO-H 15 db% | 2.3/1 | 30 | 15 |
| C | Bomar/PPO-H 20 db% | 1.5/1 | 50 | 20 |
| D | Bomar/PPO-H 25 db% | 1/1 | 50 | 25 |
| E | Bomar/PPO-D “10%” | 4/1 | 20 | — |
| F | Bomar/PPO-D “15%“ | 2.3/1 | 30 | — |
| G | Bomar/PPO-D “20%“ | 1.5/1 | 40 | — |
| H | Bomar/PPO-D “25%“ | 1/1 | 50 | — |
| I | Bomar | — | — | — |
Assessment of photoreactivity via RT-NIR photorheology
Prior to printing on the Hot Lithography machine, it was necessary to evaluate photoreactivity of the formulations. This is crucial because reactivity gives a first indication how the printing parameters should be adjusted. Furthermore, the regulatory ability of the AFCT group needs to be proven. Real-time near infrared photorheology (RT-NIR photorheology) is a powerful tool to monitor the progress of photocuring.31 It combines information from rheology (storage modulus G′, loss modulus G′′) with in situ monitoring of the double bond conversion. Some characteristic values that can be derived from these measurements will now be discussed (Table 2).| Formulation | Composition | t g [s] | DBCg [%] | DBC [%] | t 95% [s] | F Nmin [N] |
|---|---|---|---|---|---|---|
| a Relative amount of AFCT double bonds to overall double bonds in the formulation. b PPO-D content of “x%” is equal to PPO-H content of x db% in wt%, see Table 1. | ||||||
| A | Bomar/PPO-H 10 db%a | 3.8 | 35 | 98 | 58 | 9.1 |
| B | Bomar/PPO-H 15 db%a | 4.7 | 39 | >99 | 44 | 9.3 |
| C | Bomar/PPO-H 20 db%a | 6.4 | 42 | >99 | 47 | 8.7 |
| D | Bomar/PPO-H 25 db%a | 8.4 | 44 | >99 | 41 | 8.0 |
| E | Bomar/PPO-D "10%"b | 3.4 | 36 | 94 | 75 | 12 |
| F | Bomar/PPO-D "15%"b | 3.4 | 35 | 97 | 78 | 13 |
| G | Bomar/PPO-D "20%"b | 3.4 | 27 | 96 | 56 | 15 |
| H | Bomar/PPO-D "25%"b | 3.8 | 31 | 97 | 45 | 16 |
| I | Bomar | 2.8 | 32 | 94 | 100 | 9.6 |
The first characteristic value during curing is the gel point, which is defined as the intersection of G′ and G′′. Two important parameters are derived from this point: the time until gelation (tg, Table 2) and the double bond conversion at the gel point (DBCg, Table 2). Comparison of tg values (Table 2) show that formulations E–H (without AFCT groups) reach the gel point very quickly (<4 s), whereas formulations A–D (with AFCT groups) take longer until they gel (up to 8.4 s). Furthermore, higher amounts of AFCT groups lead to higher gelation times. This is in accordance with prior findings that AFCT groups slightly retard the gelation due to their regulating mechanism.28,39,40 The higher tg of the PPO-H formulations can therefore be seen as a first indication that network regulation takes place. Within the PPO-D series (E–H), there is almost no difference in tg with increasing PPO-D content. Only large amounts of PPO-D (formulation H) retard the gelation by 0.4 s. This can be explained by the viscosities of the monomers: PPO-D is a low viscous liquid (7 mPa s at 90 °C), whereas Bomar is a highly viscous, almost solid resin (1.8 Pa s at 90 °C). The least viscous formulation H therefore stays flexible longer and gels a little later. Regarding 3D printing, a low tg value is desirable as this shortens the time until a layer is solidified and the next layer can be printed. On the other hand, too fast gelation is a sign for an uncontrolled polymerization mechanism resulting in inhomogeneous polymer architecture within the printed parts and leading to incomplete curing and poor toughness. Therefore, a balance between high throughput (low tg) and good mechanical properties (higher tg due to AFCT regulation) needs to be achieved.
DBCg values (Table 2) indicate that the gel point can be shifted to higher conversion by employing PPO-H (up to 44% for Bomar/PPO-H 25 db%). In contrast, both, PPO-D containing formulations and the pure Bomar reference, show lower DBCgs (<36%), which further decrease with increasing PPO-D content due to their higher network densities. Higher DBCgs are associated with reduction in shrinkage stress and postcuring. Shrinkage is a severe challenge in photopolymerization as it is responsible for craze induction and distortion, and can also lead to delamination during 3D printing.41 Low shrinkage resins are therefore highly desirable. The expectation of lower shrinkage of PPO-H compared to PPO-D or Bomar due to a higher DBCg was corroborated through the observation of shrinkage force by following the decreasing normal force (FNmin, Fig. 1E and F, Table 2) the forming network exerts on the stamp during rheology measurements of the curing process. The onset of shrinkage starts when the monomer mixture gels and is transformed from a liquid to a vitrified, glassy state. Shifting the gel point to higher conversion is therefore an effective method of reducing shrinkage stress.42 There seems to be a linear correlation between PPO oligomer concentration and shrinkage stress. The direct comparison of PPO-H and PPO-D reveals that the occurring FN value can be halved by using PPO-H (formulation D vs. H). It has to be noted that the absolute values of FNmin are quite low for all formulations, as the Bomar matrix itself already exhibits low shrinkage due to its dynamic hydrogen bonds between the urethane groups, which can be broken and reformed after rearrangement of the macromolecular chains. Through this rearrangement a lot of shrinkage stress can be compensated.
 | ||
| Fig. 1 Summary of obtained RT-NIR photorheology measurements for the poly(propylene oxide) hybrid monomer (PPO-H, A, C and E) and the poly(propylene oxide) dimethacrylate monomer (PPO-D, B, D and F): storage modulus (G′; A and B), double bond conversion (DBC; C and D), and minimal normal force (FNmin; E and F). | ||
Towards the end of the curing process, another characteristic value can be derived from RT-IR photorheology measurements: the time until which 95% DBC is reached (t95%, Table 2) is used to assess the overall curing speed. Low t95% values are desirable as this results in shorter reaction times enabling a faster printing speed and improved green strength. This is of critical importance since the printed layer not only needs to keep its shape (i.e. to gel) but should also reach a sufficient curing state during printing, ideally close to final conversion. This can then reduce efforts in post-processing of the printed parts, e.g. in a UV oven.
The last important curing parameter is the final double bond conversion (DBC). Achieving full conversion (i.e. >99%) is important to avoid sticky surfaces and migration issues of unreacted monomers, and to achieve good (thermo)mechanical properties. Through addition of the PPO oligomers to the Bomar resin all final conversions can be increased (Fig. 1). In the case of the hybrid PPO-H even full conversion could be achieved. This can again be explained by the reactive diluent effect of the oligomers, leading to a more flexible system.
In summary, RT-NIR photorheology proves the concept of network regulation by the AFCT mechanism successfully. Compared to the state-of-the-art PPO-D, PPO-H containing formulations show increased conversion, while shrinkage stress decreases simultaneously. Additionally, storage modulus G′ values allow a first assessment of the mechanical properties. Fig. 1A shows decreasing storage moduli with increasing PPO-H content. This could be explained by the less dense network structure achieved through PPO-H content, which indicates that the cured polymers become softer. Within the PPO-D series, however, G′ values hardly differ from the Bomar reference (Fig. 1B). This indicates that the rubber-like nature of PPO itself has no significant effect on G′ and the more homogenous network structure through AFCT regulation with PPO-H is responsible for decreased G′.
For further insights into thermomechanical properties of PPO-based monomers with and without AFCT regulation, DMTA and tensile testing of moulded specimens were conducted.
Thermomechanical properties of moulded specimens
For the investigation of the materials themselves, specimens for dynamic mechanical thermal analysis (DMTA) and tensile testing were cured in moulds. DMTA results show that the storage modulus G′ decreases already at lower temperatures and to lower values at the rubber plateau for increasing PPO-H contents (Fig. 2A, Table S1†). | ||
| Fig. 2 Summary of obtained DMTA measurements for the moulded specimens of pure Bomar and Bomar with 10–25 db% poly(propylene oxide) hybrid monomer (PPO-H, A and C) and the poly(propylene oxide) dimethacrylate monomer (PPO-D, B and D): storage modulus (G′; A and B) and loss factor (tanδ; C and D). | ||
In analogy to this observation, the glass transition temperature (Tg) determined by the maximum of the loss factor curve decreases for increasing PPO-H content from 88 to 55 °C (Fig. 2C, Table S1†). Both observations reflect the softening effect of PPO-H as mentioned earlier. Increasing PPO-D contents, however, cause no noteworthy changes in G′ at the rubber plateau and only a minor decrease of Tg (Fig. 2B and D, Table S1†), which confirms the previous interpretation that PPO-H's less dense networks are responsible for softening and not its oligomeric nature, which is a structural feature it shares with PPO-D.
Tensile tests of moulded specimens (Fig. 3, Table S1†) reveal maximum stress values between 63 (10 db%) and 35 MPa (25 db%) for PPO-H/Bomar materials. These values are slightly lower than those for PPO-D/Bomar materials (71–55 MPa for 10–25 db%) and pure Bomar materials (76 MPa), which can be explained by the increasing network density from PPO-H/Bomar to PPO-D/Bomar to Bomar. However, while the elongation at break remains equally low for PPO-D/Bomar and Bomar based polymer networks, a significant increase can be observed for PPO-H/Bomar materials from 15 db% on, up to 112% at 25 db% PPO-H, which clearly is a result of the toughening-effect of the hybrid monomer.
 | ||
| Fig. 3 Ultimate tensile strength (σM); strain at break (εB), and energy at break (EB) for moulded specimens made from pure Bomar and Bomar with 10–25 db% poly(propylene oxide) dimethacrylate (PPO-D) or poly(propylene oxide) hybrid monomer (PPO-H). | ||
Toughness of the materials under continuous strain was calculated from the areas under the stress–strain curves (Fig. 3, Fig. S5 and Table S1†).43,44 While some toughening was anticipated for PPO-D containing materials due to its flexible nature, the obtained energies at break indicate that PPO-D does not increase toughness significantly compared to specimens purely made from Bomar. In fact, if at all, a slight trend of decreasing toughness with increasing PPO-D content is visible, which would suggest that any positive effect of the PPO spacer is overcompensated by the higher crosslink density as a result of PPO addition. On the contrary, PPO-H increases toughness of resulting polymer networks already at a content of 10 db% and a saturation of the effect seems to be reached at a content of 20 db% as a result of increasing strain at break and simultaneous reduction of strength.
To verify these findings, impact strength (a) of unnotched samples was measured by dynstat experiments (Fig. 4, Table S1†). Again, no change in impact strength, i.e. toughness, could be found for PPO-D addition to Bomar, while addition of PPO-H increased toughness already at a content of 10 db%. Interestingly, an increase of PPO-H content from 20 to 25 db% resulted in the highest increase in impact resistance, while stagnation of energy at break increase was observed during tensile testing for these specimens. This is a result of the different loading rates on the material during the two testing methods. Considering the time-temperature superposition principle and that testing at room temperature coincides with the region of the steep drop of the storage modulus, it follows that an increase of loading rate results in a strong increase of modulus and a shift of the dynamic glass transition to higher temperatures. In this case the 25 db% PPO-H containing formulation is still at a more viscous level of the deformation behaviour at the applied impact loading, whereas the formulations with less PPO-H content show a more elastically dominated behaviour under these conditions.
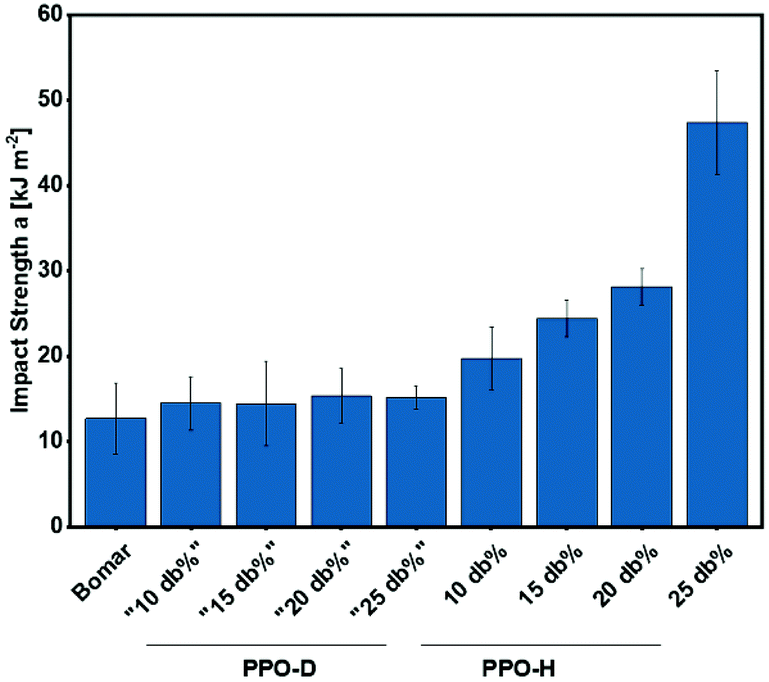 | ||
| Fig. 4 Comparison of impact strength (a) for specimens made from pure Bomar and Bomar with 10–25 db% poly(propylene oxide) dimethacrylate (PPO-D) or poly(propylene oxide) hybrid monomer (PPO-H). | ||
Investigation of monomer migration
To determine migration of PPO-H and PPO-D monomers, swelling tests of 3D printed discs were performed for all materials. Swellability and gel fraction of PPO-H containing polymers were compared to PPO-D containing and pure Bomar-based polymers (Fig. 5, Table S1†). | ||
| Fig. 5 Swellability and gel fraction for moulded specimens made from pure Bomar and Bomar with 10–25 db% poly(propylene oxide) dimethacrylate (PPO-D) or poly(propylene oxide) hybrid monomer (PPO-H), which were incubated in ethanol. | ||
Swellabilities were around 120% for pure Bomar and PPO-D/Bomar based polymers, while swellabilities of PPO-H containing disks increased from 120% to 150% for increasing amounts of PPO-H. This is indicative of the wider network structure of regulated networks obtained with AFCT reagent-containing PPO-H. Gel fractions of PPO-D and Bomar were approximately 100% due to their near-perfect incorporation into the network as difunctional monomers. Polymers containing 10 and 15 db% PPO-H achieved comparable gel fractions close to 100%, and the gel fraction only fell below 90% for the polymer containing the highest amount of PPO-H (25 db%). This proves that migration is severely impaired due to the integration of PPO-H into the network via its methacrylate moiety as intended.
AM via Hot Lithography
The invention of Hot Lithography has raised interest in oligomeric monomers, which exhibit too high viscosities at room temperature for printing (>20 Pa s), but are fluent enough between 80 and 120 °C (<20 Pa s). The new technology uses a heated vat and building platform to decrease viscosities and increase reactivity during the printing process (Fig. 6). The use of oligomers as monomers has therefore become possible to obtain wider and tougher photopolymer networks. | ||
| Fig. 6 Hot Lithography setup for printing (A) and 3D printed specimens from Bomar formulations containing 10 db% poly(propylene oxide) hybrid monomer (PPO-H) (A–C) and corresponding SEM images for detailed layer analysis (D and E). | ||
The new hybrid oligomer PPO-H promises great potential as such a toughening monomer and reactive diluent during 3D printing at elevated temperatures. As shown previously, 10 and 15 db% PPO-H exhibited the best trade-off between mechanical performance and glass transition temperature. Therefore, these formulations were successfully printed with a DLP machine at 90 °C. Complex structures could be printed with excellent resolution (Fig. 6A–D). Scanning electron microscopy (SEM) images of the printed structure confirm this impression of well-defined layers and show that the printed layer thickness is constant (100 μm, Fig. 6E and F).
Thermomechanical and swelling properties of printed specimens
In order to evaluate the printed parts in terms of their thermo-mechanical performance, testing specimens were printed (Fig. S6†), tested via DMTA, tensile testing, and dynstat, and compared to results for the moulded specimens (Fig. 7–10, Table S1†). It should be mentioned that the irradiation time had to be adjusted from 25 s for 10 db%-containing Bomar/PPO formulations to 40 s for 15 db% containing Bomar/PPO formulations due to the longer gelation time at higher PPO-H content (Table 2). DMTA gave equal storage moduli and glass transitions for moulded and printed samples (Fig. 7). | ||
| Fig. 7 Comparison of storage modulus G′ (A) and loss factor tan(δ) (B) of DMTA measurements of the moulded specimens with the 3D printed specimens containing 10 or 15 db% PPO-H. | ||
 | ||
| Fig. 8 Comparison of tensile strength (σM), strain at break (εB), and energy at break (EB) for moulded and 3D printed specimens made from Bomar with 10 or 15 db% poly(propylene oxide) hybrid monomer (PPO-H). | ||
 | ||
| Fig. 9 Comparison of impact strength (a) for moulded and 3D printed specimens made from Bomar with 10 or 15 db% poly(propylene oxide) hybrid monomer (PPO-H). | ||
 | ||
| Fig. 10 Comparison of swellability and gel fraction for moulded and 3D printed specimens made from Bomar with 10 or 15 db% poly(propylene oxide) hybrid monomer (PPO-H). | ||
During mechanical testing, the same trends for material performance were observed with only a slight decrease in stress values for 3D printed samples (<5 MPa: 8% for PPO-H 10% 3D and 15% for PPO-H 15% 3D), which was independent from the formulation composition (Fig. 8). This decrease is within the expected range due to the layered printed structure as opposed to the monolithic moulded structure. Decreases of elongations and energies at break (Fig. 8) as well as of impact strength were higher (Fig. 9).
The results for printed samples as opposed to moulded samples exhibited a slight decrease in swellability (∼5% for both formulations; Fig. 10) and a small increase of the already high gel fraction (∼1% for both formulations; Fig. 10). This confirms that the printed parts are largely free of migrating species in analogy to the moulded samples.
Conclusions
We have shown that the synthesized hybrid oligomer poly(propylene oxide) with one methacrylate and one AFCT end group (PPO-H) is a potent additive for improving the toughness of commercially available photopolymer resins. The toughening mechanism relies on the AFCT end group of PPO-H, which is able to regulate and homogenize the network structure. The other end group of PPO-H is a reactive methacrylate group, which leads to covalent incorporation into the network and therefore migration-free photopolymers. Another benefit of PPO-H over standard low molecular weight reactive diluents (e.g. benzyl methacrylate) is its non-volatility, which is essential for processing at elevated temperatures.Mixtures of PPO-H with a first commercial photopolymer resin (Bomar XR-741MS) showed high reactivity, high double-bond conversions (>99%), and a significant reduction in shrinkage stress (10–50%). While decreases in glass transition temperature were especially observed for PPO-H, a satisfactory trade-off between this decrease and toughening was achieved for 10 and 15 db% PPO-H content (55 and 92% increase in impact strength, Tg of 85 and 76 °C, respectively). In combination with the Hot Lithography technique, PPO-H has proven to be a beneficial additive and reactive diluent in lithography-based 3D printing. These results indicate that PPO-H could also be used with many other available resins in order to improve their (thermo)mechanical properties. Furthermore, it will be also interesting to extend the hybrid concept to other types of oligomers and to investigate the polymer architecture that is formed during photopolymerization more intensively. This will be the focus of our upcoming work.
Author contributions
The manuscript was written through contributions of all authors. The concept was developed by Daniel Grunenberg, Christian Gorsche, and Robert Liska. Experimental work was conducted by Daniel Grunenberg. Mechanical testing was conducted by Thomas Koch. 3D printing experiments were planned by Bernhard Steyrer and Jürgen Stampfl and executed by Bernhard Steyrer. Data interpretation and drafting of the manuscript was conducted by Daniel Grunenberg and Katharina Ehrmann. The manuscript was edited by Katharina Ehrmann, Christian Gorsche, and Robert Liska. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the Austrian Research Agency (FFG) within the Austrian lead project “addmanu” (number 849297) and the project “3DHiPerPolymers” (number 874202) is gratefully acknowledged.Bisomer PPM 5 LI was provided by GEO Specialty Chemicals. The photoinitiator ethyl (2,4,6-trimethylbenzoyl) phenyl (TPO-L) was kindly gifted by Lambson.
Notes and references
- C. W. Hull, UVP Inc., United States Pat, 4575330A, 1986 Search PubMed.
- D. I. Wimpenny, P. M. Pandey and L. J. Kumar, Advances in 3D Printing & Additive Manufacturing Technologies, Springer Publishing Company, 2016 Search PubMed.
- D. Herzog, V. Seyda, E. Wycisk and C. Emmelmann, Acta Mater., 2016, 117, 371–392 CrossRef CAS.
- F. Kotz, K. Arnold, W. Bauer, D. Schild, N. Keller, K. Sachsenheimer, T. M. Nargang, C. Richter, D. Helmer and B. E. Rapp, Nature, 2017, 544, 337–339 CrossRef CAS.
- L. C. Hwa, S. Rajoo, A. M. Noor, N. Ahmad and M. B. Uday, Curr. Opin. Solid State Mater. Sci., 2017, 21(6), 323–347 CrossRef CAS.
- A. de Blas Romero, M. Pfaffinger, G. Mitteramskogler, M. Schwentenwein, C. Jellinek, J. Homa, A. Díaz Lantada and J. Stampfl, Int. J. Adv. Manuf. Technol., 2017, 88, 1547–1555 CrossRef.
- G. Mitteramskogler, R. Gmeiner, R. Felzmann, S. Gruber, C. Hofstetter, J. Stampfl, J. Ebert, W. Wachter and J. Laubersheimer, Addit. Manuf., 2014, 1–4, 110–118 Search PubMed.
- J. W. Stansbury and M. J. Idacavage, Dent. Mater., 2016, 32, 54–64 CrossRef CAS.
- C. Sturgess, C. J. Tuck, I. A. Ashcroft and R. D. Wildman, J. Mater. Chem. C, 2017, 5, 9733–9743 RSC.
- S. Chen, Q. Zhang and J. Feng, J. Mater. Chem. C, 2017, 5, 8361–8365 RSC.
- S. C. Ligon, R. Liska, J. Stampfl, M. Gurr and R. Mulhaupt, Chem. Rev., 2017, 117, 10212–10290 CrossRef CAS.
- S. S. Crump, Stratasys Inc., United States Pat, 5121329A, 1992 Search PubMed.
- P. Tesavibul, R. Felzmann, S. Gruber, R. Liska, I. Thompson, A. R. Boccaccini and J. Stampfl, Mater. Lett., 2012, 74, 81–84 CrossRef CAS.
- M. W. Tibbitt, J. A. Shadish and C. A. DeForest, in Multiphoton Lithography, Wiley-VCH Verlag GmbH & Co. KGaA, 2016, pp. 183–220, DOI:10.1002/9783527682676.ch8.
- J. R. Tumbleston, D. Shirvanyants, N. Ermoshkin, R. Janusziewicz, A. R. Johnson, D. Kelly, K. Chen, R. Pinschmidt, J. P. Rolland, A. Ermoshkin, E. T. Samulski and J. M. DeSimone, Science, 2015, 347, 1349–1352 CrossRef CAS.
- A. Gebhardt and J.-S. Hötter, Additive Manufacturing, Carl Hanser Verlag GmbH & Co. KG, 2016 Search PubMed.
- N. Moszner and U. Salz, Prog. Polym. Sci., 2001, 26, 535–576 CrossRef CAS.
- S. C. Ligon-Auer, M. Schwentenwein, C. Gorsche, J. Stampfl and R. Liska, Polym. Chem., 2016, 7, 257–286 RSC.
- W. C. J. Zuiderduin, C. Westzaan, J. Huétink and R. J. Gaymans, Polymer, 2003, 44, 261–275 CrossRef CAS.
- P. A. Lovell, J. McDonald, D. E. J. Saunders and R. J. Young, Polymer, 1993, 34, 61–69 CrossRef CAS.
- J. L. Amos, J. L. McCurdy and O. R. McIntire, Dow Chemical Co., United States Pat, 2694692, 1954 Search PubMed.
- T. Sterrett, R. Wong and P. Kidd, J. Elastomers Plast., 1986, 18, 187–194 CrossRef CAS.
- J. Ebert and R. Gmeiner, Ivoclar Vivadent AG, TU Wien, WO2016/078838A1, 2016.
- M. Pfaffinger, Opt. Photonik, 2018, 13, 99–101 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- Y. Yagci and I. Reetz, React. Funct. Polym., 1999, 42, 255–264 CrossRef CAS.
- C. Gorsche, K. Seidler, P. Knaack, P. Dorfinger, T. Koch, J. Stampfl, N. Moszner and R. Liska, Polym. Chem., 2016, 7, 2009–2014 RSC.
- L. M. Cox, X. Sun, C. Wang, N. Sowan, J. P. Killgore, R. Long, H.-A. Wu, C. N. Bowman and Y. Ding, ACS Appl. Mater. Interfaces, 2017, 9, 14422–14428 CrossRef CAS.
- C. R. Fenoli and C. N. Bowman, Polym. Chem., 2014, 5, 62–68 RSC.
- C. Gorsche, R. Harikrishna, S. Baudis, P. Knaack, B. Husar, J. Laeuger, H. Hoffmann and R. Liska, Anal. Chem., 2017, 89, 4958–4968 CrossRef CAS.
- B. Steyrer, B. Busetti, G. Harakály, R. Liska and J. Stampfl, Addit. Manuf., 2018, 21, 209–214 CAS.
- C. Gorsche, T. Koch, N. Moszner and R. Liska, Polym. Chem., 2015, 6, 2038–2047 RSC.
- C. Gorsche, M. Griesser, G. Gescheidt, N. Moszner and R. Liska, Macromolecules, 2014, 47, 7327–7336 CrossRef CAS.
- J. E. Mark, Polymer Data Handbook, Oxford University Press, 2009 Search PubMed.
- C. E. Rehberg and C. H. Fisher, J. Am. Chem. Soc., 1944, 66, 1203–1207 CrossRef CAS.
- D. H. R. Barton, C.-Y. Chern and J. C. Jaszberenyi, Tetrahedron, 1995, 51, 1867–1886 CrossRef CAS.
- A. Sakakura, K. Kawajiri, T. Ohkubo, Y. Kosugi and K. Ishihara, J. Am. Chem. Soc., 2007, 129, 14775–14779 CrossRef CAS.
- C. Gorsche, K. Seidler, R. Harikrishna, M. Kury, T. Koch, N. Moszner and R. Liska, Polymer, 2018, 158, 149–157 CrossRef CAS.
- P. Gauss, S. C. Ligon-Auer, M. Griesser, C. Gorsche, H. Svajdlenkova, T. Koch, N. Moszner and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1417–1427 CrossRef CAS.
- C. W. Hull, S. T. Spence, C. W. Lewis, W. Vinson, R. S. Freed and D. R. Smalley, United States Pat, 5772947 A, 1998 Search PubMed.
- P. K. Shah, J. W. Stansbury and C. N. Bowman, Polym. Chem., 2017, 8, 4339–4351 RSC.
- N. Sowan, H. B. Song, L. M. Cox, J. R. Patton, B. D. Fairbanks, Y. Ding and C. N. Bowman, Adv. Mater., 2020, 2007221 Search PubMed.
- W. Brostow, H. E. Hagg Lobland and S. Khoja, Mater. Lett., 2015, 159, 478–480 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Contains 1H NMR data of the synthesized compounds, characterization of the side product during the 2nd synthesis step of PPO-H, tables presenting DMTA, tensile testing, and swelling data of all investigated compounds, and an image of the 3D printed specimen for tensile testing and DMTA (PDF). See DOI: 10.1039/d0py01746a |
| This journal is © The Royal Society of Chemistry 2021 |