
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the vesicle-to-worm transition for thermoresponsive block copolymer vesicles prepared via polymerisation-induced self-assembly†
Isabella R.
Dorsman
a,
Matthew J.
Derry‡
 a,
Victoria J.
Cunningham
b,
Steven L.
Brown
b,
Clive N.
Williams
b and
Steven P.
Armes
*a
a,
Victoria J.
Cunningham
b,
Steven L.
Brown
b,
Clive N.
Williams
b and
Steven P.
Armes
*a
aDepartment of Chemistry, The University of Sheffield, Dainton Building, Brook Hill, Sheffield, South Yorkshire, S3 7HF, UK. E-mail: s.p.armes@sheffield.ac.uk
bScott Bader Company Ltd, Wollaston, Wellingborough, NN29 7RL, Northants, UK
First published on 17th February 2021
Abstract
We have previously reported the synthesis of thermoresponsive poly(stearyl methacrylate)-poly(benzyl methacrylate) [PSMA-PBzMA] diblock copolymer vesicles in mineral oil via polymerisation-induced self-assembly (PISA). Such vesicles undergo a vesicle-to-worm transition on heating, which provides an interesting new oil-thickening mechanism (see M. J. Derry, et al., Angew. Chem., 2017, 56, 1746–1750). In the present study, we report an unexpected reduction in dispersion viscosity when heating vesicles of approximately the same composition above a certain critical temperature. Transmission electron microscopy (TEM) studies indicate rich thermoresponsive behavior, with vesicles present at 20 °C, worms being formed at 130 °C and spheres generated at 180 °C, indicating that a worm-to-sphere transition occurs after the initial vesicle-to-worm transition. Moreover, we have also prepared a series of new thermoresponsive diblock copolymer vesicles by RAFT dispersion copolymerization of n-butyl methacrylate (BuMA) with benzyl methacrylate (BzMA) using a poly(stearyl methacrylate) precursor in mineral oil. This model system was developed to examine whether statistical copolymerization of a suitable comonomer (BuMA) could be used to tune the critical onset temperature required for the vesicle-to-worm transition. Indeed, oscillatory rheology studies confirmed that targeting membrane-forming blocks containing up to 50 mol% BuMA lowered the critical onset temperature required to induce the vesicle-to-worm transition to 109 °C, compared to 167 °C for the reference PSMA14-PBzMA125 diblock copolymer. Variable temperature small-angle X-ray scattering (SAXS) experiments confirmed a vesicle-to-worm transition, with the vesicles initially present at 20 °C being converted into worms when heated above 130 °C. Furthermore, a substantial reduction in dispersion viscosity was again observed when heating above the critical onset temperature. TEM and shear-induced polarized light imaging (SIPLI) studies indicate that linear worms are no longer present at 160 °C and 170 °C respectively, suggesting a subsequent worm-to-sphere transition. The thermal transitions studied herein proved to be irreversible on cooling on normal experimental timescales (hours).
Introduction
Block copolymer self-assembly has been widely studied1 and offers a diverse range of both potential and practical applications, including thermoplastic elastomers,2 toughening agents for epoxy resins,3 the dispersion of diesel soot4 or pigment particles,5 drug delivery,6 and cell culture matrices.7,8 Traditionally, such self-assembly is achieved in dilute solution (<![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1% w/w) using various post-polymerisation processes, such as a solvent switch,9,10 a pH switch11,12 or thin film rehydration.13,14 Over the past ten years, there has been considerable interest in the growing field of polymerisation-induced self-assembly (PISA).15–19 This technique involves chain extension of a soluble precursor block in a suitable solvent using a second monomer that polymerizes to form an insoluble second block, thus producing sterically-stabilized diblock copolymer nanoparticles in situ. The final copolymer morphology obtained via PISA is primarily dictated by the relative volume fractions of the stabilizer and core-forming blocks, as indicated by the geometric packing parameter (P) introduced by Israelachvili and co-workers.20 The most common copolymer morphologies, in order of increasing P, are spheres,21,22 worms,23,24 vesicles25,26 and lamellae.27,28
1% w/w) using various post-polymerisation processes, such as a solvent switch,9,10 a pH switch11,12 or thin film rehydration.13,14 Over the past ten years, there has been considerable interest in the growing field of polymerisation-induced self-assembly (PISA).15–19 This technique involves chain extension of a soluble precursor block in a suitable solvent using a second monomer that polymerizes to form an insoluble second block, thus producing sterically-stabilized diblock copolymer nanoparticles in situ. The final copolymer morphology obtained via PISA is primarily dictated by the relative volume fractions of the stabilizer and core-forming blocks, as indicated by the geometric packing parameter (P) introduced by Israelachvili and co-workers.20 The most common copolymer morphologies, in order of increasing P, are spheres,21,22 worms,23,24 vesicles25,26 and lamellae.27,28
PISA offers three decisive advantages over conventional self-assembly techniques. First, PISA produces nanoparticles directly during synthesis, which means that post-polymerization processing is not required.16,18 Second, the rate of polymerization usually increases significantly after micellar nucleation, which enables very high monomer conversions to be achieved within relatively short reaction times compared to the equivalent solution polymerizations.29–31 Third, PISA syntheses can be conducted in a wide range of solvents (aqueous,32 polar33,34 or non-polar35–38 solvents, silicone oils,39,40 ionic liquids,41etc.) at copolymer concentrations of up to 50% w/w.30,42 Thus, PISA is in principle an attractive technique for industrial scale-up.43
PISA has been successfully conducted using reversible-deactivation radical polymerization (RDRP) techniques such as ATRP44–46 and NMP.47–49 However, reversible addition–fragmentation chain transfer (RAFT) polymerization has been preferred for most PISA syntheses.15,19,50 RAFT-mediated PISA allows the convenient synthesis of a wide-range of well-defined functional block copolymer nanoparticles in water, lower alcohols or n-alkanes.16,51,52 In this context, RAFT dispersion polymerization is of particular interest since it can provide convenient access to diblock copolymer nano-objects that exhibit thermoresponsive behavior. For example, block copolymer worms prepared via such formulations often exhibit a reversible worm-to-sphere transition when subjected to either heating (if prepared in ethanol53 or n-alkanes36,37,54) or cooling (if prepared in aqueous solution).24,55–57 At sufficiently high copolymer concentration, the initial worms form a soft, free-standing physical gel as a result of multiple inter-worm contacts, which produces a percolating 3D network.58 The worm-to-sphere transition leads to in situ degelation, because the non-interacting spheres form a free-flowing dispersion.54
Although well-known in the surfactant literature,59,60 vesicle-to-worm transitions have only recently been demonstrated for block copolymers.61–67 For example, we reported two examples of a single thermoresponsive diblock copolymer prepared by RAFT aqueous dispersion polymerization that can form spheres, worms or vesicles simply by varying the solution temperature. However, if the structure-directing block is poly(2-hydroxypropyl methacrylate), such thermal transitions can require relatively long time scales.66 In striking contrast, minimal hysteresis is observed when using a hydrophobic block comprising mainly poly(4-hydroxybutyl acrylate), which was attributed to the significantly greater mobility of the more flexible acrylic backbone.67
Of particular relevance to the present study, Derry et al. reported that poly(stearyl methacrylate)-poly(benzyl methacrylate) [PSMA-PBzMA] vesicles prepared by RAFT dispersion polymerization of BzMA in mineral oil exhibited thermoresponsive behaviour on heating. More specifically, a 10% w/w dispersion of PSMA13-PBzMA96 vesicles underwent a vesicle-to-worm transition above 135 °C. Variable temperature 1H NMR spectroscopy studies indicated that this change in copolymer morphology was the result of surface plasticization of the membrane-forming PBzMA block by hot solvent, thereby effectively increasing the effective volume fraction of the stabilizer block and hence reducing the packing parameter for the copolymer chains.64 In principle, this morphological transition might provide an interesting high temperature oil-thickening mechanism for automotive engine oils or cosmetics formulations.
In the present study, we revisit this PSMA-PBzMA system to examine what happens on heating well above the critical onset temperature required for the vesicle-to-worm transition. Moreover, we investigate whether statistical incorporation of an appropriate comonomer into the membrane-forming block (see Scheme 1) would enable tuning of the critical onset temperature required for a vesicle-to-worm transition. n-Butyl methacrylate (BuMA) was selected as a suitable comonomer. Given the relatively low glass transition temperature (Tg) of poly(n-butyl methacrylate),68 this should enable the critical onset temperature required for the vesicle-to-worm transition to be tuned by statistical copolymerization of BuMA with BzMA. In contrast, copolymerization of BzMA with either methyl methacrylate or ethyl methacrylate would raise, rather than lower, the copolymer Tg. Moreover, statistical copolymerization of lauryl methacrylate with BzMA is also likely to be problematic because this could result in a soluble second block and hence no self-assembly behavior.
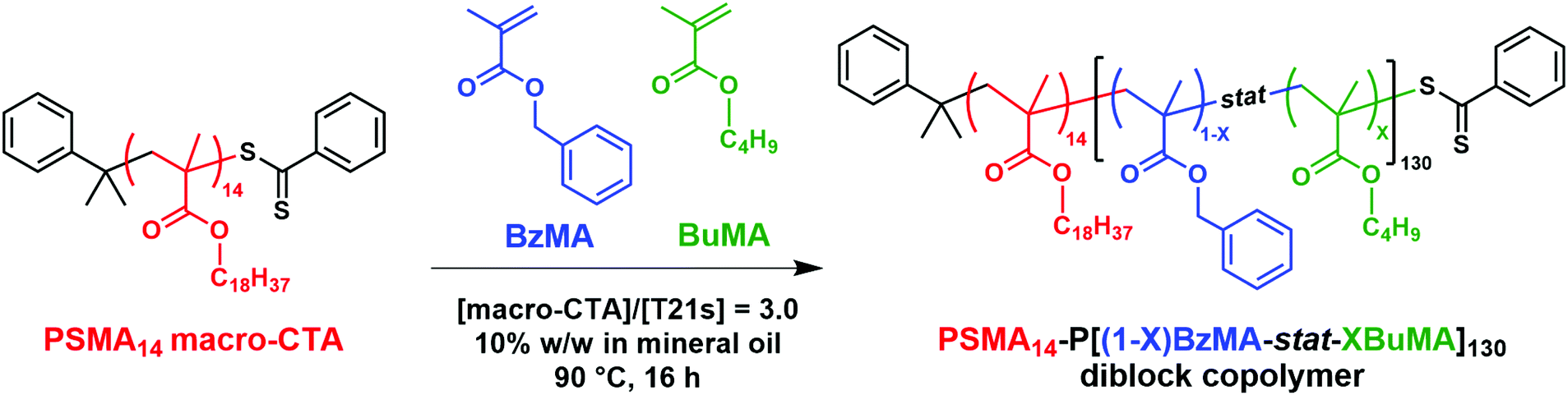 | ||
| Scheme 1 Synthesis of a poly(stearyl methacrylate)-poly(benzyl methacrylate-stat-n-butyl methacrylate) [PSMA-P(BzMA-stat-BuMA)] diblock copolymer via RAFT dispersion copolymerization of benzyl methacrylate (BzMA) and n-butyl methacrylate (BuMA) using a PSMA14 precursor targeting 10% w/w solids in mineral oil at 90 °C. (X and 1 − X denote the mole fractions of the BuMA and BzMA comonomers respectively within the membrane-forming block.) | ||
Experimental
Materials
All reagents were purchased from Sigma-Aldrich (UK) at the highest possible purity and were used as received unless stated otherwise. tert-Butyl peroxy-2-ethylhexanoate (T21s) initiator was purchased from AzkoNobel (The Netherlands). Tetrahydrofuran (THF), n-dodecane and toluene were purchased from Fisher Scientific (UK). Deuterated methylene chloride (CD2Cl2) was purchased from Goss Scientific (UK). API Group III mineral oil (viscosity = 3.1 cSt at 100 °C) was kindly provided by The Lubrizol Corporation Ltd (Hazelwood, Derbyshire, UK).Synthesis of poly(stearyl methacrylate)-poly(benzyl methacrylate) (PSMA-PBzMA) diblock copolymer vesicles
The poly(stearyl methacrylate) macromolecular chain transfer agent (PSMA macro-CTA) used in the initial studies reported herein was the same sample as that reported by Derry and co-workers in 2017, hence its synthetic protocol has been previously reported.64 End-group analysis by 1H NMR spectroscopy indicated a mean degree of polymerization (DP) of 13. This PSMA13 macro-CTA was then used for the synthesis of PSMA13-PBzMA97 vesicles at 10% w/w solids in mineral oil via RAFT dispersion polymerization of BzMA as described below. Benzyl methacrylate (BzMA; 0.226 g; 1.28 mmol), T21s initiator (0.555 mg; 2.57 μmol; dissolved at 10.0% v/v in mineral oil) and PSMA13 macro-CTA (0.060 g; 12.8 μmol; macro-CTA/initiator molar ratio = 5.0; target PBzMA DP = 100) were dissolved in mineral oil (2.58 g). The reaction mixture was sealed in a 14 mL glass reaction vial and purged with nitrogen gas for 30 min. The deoxygenated solution was then placed in a pre-heated oil bath at 90 °C for 16 h (final BzMA conversion = 97%; Mn = 19900 g mol−1, Mw/Mn = 1.10).
Synthesis of poly(stearyl methacrylate) macromolecular chain transfer agent (PSMA macro-CTA)
A second PSMA macro-CTA was synthesised by the RAFT solution polymerization of stearyl methacrylate using the following protocol. Stearyl methacrylate (SMA; 30.7 g; 90.5 mmol), cumyl dithiobenzoate (CDB; 4.93 g; 18.1 mmol), 2,2′-azobisisobutyronitrile (AIBN; 594 mg; 3.6 mmol; CDB/AIBN molar ratio = 5.0) and toluene (54.2 g) were added to a 250 mL round-bottomed flask. The sealed vessel was purged with nitrogen for 30 min and placed in a pre-heated oil bath at 70 °C for 3.5 h. The resulting PSMA macro-CTA (SMA conversion = 85%; Mn = 6200 g mol−1; Mw = 6900 g mol−1; Mw/Mn = 1.12) was purified by precipitation into excess ethanol (twice). The mean DP of this macro-CTA was calculated to be 14 using 1H NMR spectroscopy by comparing the integrated signals corresponding to the aromatic protons of the dithiobenzoate end-groups with those assigned to the two oxymethylene ester protons of the SMA repeat units. This is close to the mean DP of 13 reported by Derry et al.64Synthesis of poly(stearyl methacrylate)-poly(benzyl methacrylate-stat-n-butyl methacrylate) diblock copolymer nanoparticles
A typical PISA synthesis of PSMA14-P(0.5BzMA-stat-0.5BuMA)130 nanoparticles via RAFT dispersion polymerization at 10% w/w solids was conducted as follows. A 14 mL glass reaction vial was charged with PSMA14 macro-CTA (0.060 g; 12.0 μmol) and mineral oil (2.78 g). The reaction mixture was placed in a 70 °C laboratory oven for 2 min to aid dissolution. On cooling to 20 °C, the vial was charged with benzyl methacrylate (BzMA; 0.137 g; 0.778 mmol; 50 mol%), n-butyl methacrylate (BuMA; 0.111 g, 0.778 mmol, 50 mol%) and T21s initiator (0.900 mg; 3.99 μmol; dissolved at 10% v/v in mineral oil; macro-CTA/initiator molar ratio = 3.0; target DP = 130). The reaction mixture was sealed and purged with nitrogen gas for 30 min with magnetic stirring. The deoxygenated solution was then placed in a pre-heated oil bath at 90 °C for 16 h (final BzMA conversion = 98%; final BuMA conversion = 92%; Mn = 21600 g mol−1; Mw/Mn = 1.10).
1H NMR spectroscopy
1H NMR spectra were recorded in either CD2Cl2 (to determine the mean DP for the PSMA precursor) or CDCl3 (for all other spectra) using a Bruker AV1-400 MHz spectrometer. Typically 64 scans were averaged per spectrum.Gel permeation chromatography (GPC)
GPC was used to assess (co)polymer molecular weight distributions (MWDs). The GPC set-up comprised two 5 μm (30 cm) mixed C columns and a WellChrom K-2301 refractive index detector operating at 950 ± 30 nm. The THF mobile phase contained 2.0% v/v triethylamine and 0.05% w/v butylhydroxytoluene (BHT) with a toluene flow-rate marker at a flow rate of 1.0 mL min−1. A series of eleven near-monodisperse poly(methyl methacrylate) (PMMA) standards (Mp values ranging from 800 to 988000 g mol−1) were used for calibration.
Dynamic light scattering (DLS)
DLS studies were performed using a Zetasizer Nano ZS instrument (Malvern Instruments, UK) at a fixed scattering angle of 173°. Copolymer dispersions were diluted to 0.10% w/w using n-dodecane prior to light scattering studies at 25 °C. The intensity-average diameter and polydispersity were calculated by cumulants analysis of the experimental correlation function using Dispersion Technology Software version 6.20. Data were averaged over ten runs each of thirty seconds duration.Transmission electron microscopy (TEM)
TEM studies were conducted using a FEI Tecnai G2 spirit instrument operating at 80 kV and equipped with a Gatan 1k CCD camera. The relatively low glass transition temperature conferred by the n-butyl methacrylate comonomer (Tg of PBuMA homopolymer = 20 °C) made imaging more difficult compared to PSMA-PBzMA nano-objects.64 Prolonged exposure to the high-energy electron beam during TEM studies can cause deformation and/or degradation of the copolymer nano-objects. To aid retention of the original nanoparticle morphology, 0.10% w/w copolymer dispersions were cooled to 3 °C before placing a single droplet onto pre-cooled carbon-coated copper grids and allowing to dry at 3 °C overnight within a laboratory refrigerator.39 The following day, the prepared grids were exposed to ruthenium(VIII) oxide vapor for 7 min at 20 °C prior to analysis. This heavy metal compound acts as a positive stain for the core-forming block to improve electron contrast. Ruthenium(VIII) oxide was prepared as follows: ruthenium(IV) oxide (0.30 g) was added to water (50 g) to form a black slurry; addition of sodium periodate (2.0 g) with continuous stirring produced a yellow solution of ruthenium(VIII) oxide within 1 min at 20 °C. The 0.10% w/w dispersions of PSMA-PBzMA nano-objects (prepared by dilution using n-dodecane) were placed on grids at 20 °C and allowed to dry for 30 min before following the above staining protocol.In order to study the thermally-induced morphological transitions, a sample vial containing two drops (approximately 30 mg) of a 10% w/w dispersion in mineral oil was placed in a pre-heated oil bath at the desired temperature (e.g. from 130 °C to 180 °C), allowed to equilibrate for 10 min, diluted with n-dodecane (preheated to the same temperature), and then allowed to dry on a grid following the protocol described above.
Oscillatory rheology
An Anton Paar MCR502 rheometer equipped with a variable temperature Peltier plate and hood and a 50 mm 2° aluminium cone was used for all experiments. The distance between the cone and plate was 207 μm. The storage (G′) and loss (G′′) moduli were measured as a function of temperature at a fixed strain of 1.0% and an angular frequency of 10 rad s−1. A 20–180–20 °C thermal cycle was conducted at a heating/cooling rate of 2 °C min−1.Shear-induced polarized light imaging (SIPLI)
The instrument design and general experimental set-up has been previously reported by Mykhaylyk and co-workers.69 SIPLI experiments were conducted on a 10% w/w dispersion of PSMA14-P(0.5BzMA-stat-0.5BuMA)130 nano-objects in mineral oil at an applied shear rate of 200 s−1 during a temperature ramp experiment conducted at a heating rate of 5.0 °C min−1.Small angle X-ray scattering (SAXS)
SAXS patterns were recorded at a synchrotron source (Diamond Light Source Ltd, Beamline I22, Didcot, Oxfordshire, UK) using monochromatic X-ray radiation (λ = 0.0995 nm, with q ranging from 0.002 to 2.0 nm−1, where q = 4πsinθ/λ is the length of the scattering vector and θ is one-half of the scattering angle) and a Dectris (Dectris AG, Switzerland) Pilatus P3 Hybrid silicon pixel detector. Glass capillaries of 2.0 mm diameter were used as a sample holder and the sample temperature was controlled using a HFSX350-CAP heating/cooling capillary holding stage (Linkam Scientific Instruments Ltd, Tadworth, UK), with 2 min being allowed for thermal equilibration prior to data collection. Scattering data were reduced using standard routines from the beamline70 and were further analyzed using Irena SAS macros for Igor Pro.71 Water was used for the absolute intensity calibration. Measurements were conducted on a 1.0% w/w dispersion of either PSMA13-PBzMA97 or PSMA14-P(0.5BzMA-stat-0.5BuMA)130 nano-objects in mineral oil.
Results and discussion
Synthesis and characterisation of PSMA13-PBzMA97 diblock copolymer vesicles
Using the same PSMA13 precursor as synthesized by Derry et al. and targeting the same diblock copolymer composition as previously reported,64 PSMA13-PBzMA100 vesicles were targeted in mineral oil at 10% w/w solids. A BzMA conversion of 97% was achieved within 16 h at 90 °C, as judged by 1H NMR spectroscopy, which agrees well with the 96% conversion achieved after 5 h at 90 °C in the original study. THF GPC analysis showed that the resulting diblock copolymer chains (Mn = 19900 g mol−1) exhibited a narrow molecular weight distribution (Mw/Mn = 1.10) and the whole molecular weight distribution curve was well-shifted from that of the PSMA13 macro-CTA (see Fig. S1†). These data suggest that good RAFT control and a high blocking efficiency was achieved. As expected, TEM studies indicated a predominantly vesicular morphology, while a z-average diameter of 133 nm and a polydispersity of 0.09 was obtained by DLS studies (see Fig. 1b and c, respectively).
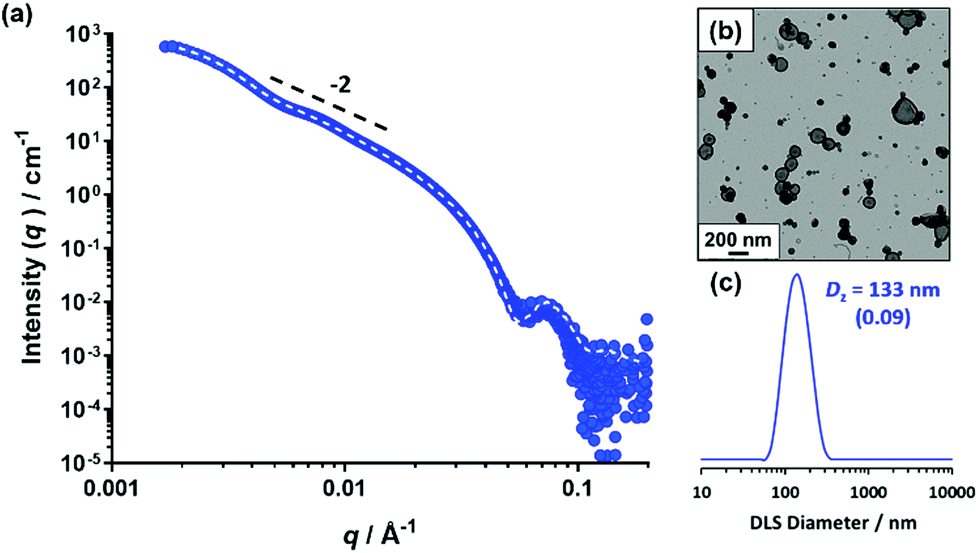 | ||
| Fig. 1 (a) Small-angle X-ray scattering (SAXS) pattern recorded for a 1.0% w/w dispersion of PSMA13-PBzMA97 vesicles in mineral oil at 20 °C. Dashed line indicates the data fit obtained using a well-known vesicle scattering model. A low q gradient of −2 is shown as a guide to the eye. (b) Representative TEM image and (c) DLS particle size distribution obtained for a 0.10% w/w dispersion of PSMA13-PBzMA97 vesicles at 20 °C. | ||
SAXS studies on a 1.0% w/w dispersion at 20 °C produced the characteristic pattern expected for vesicles (see Fig. 1a): the low q gradient is approximately −2 and there are local minima corresponding to the outer vesicle dimensions at q ≈ 0.005 Å−1 and the vesicle membrane thickness, Tmembrane, at q ≈ 0.05 Å−1. Fitting this SAXS pattern to a well-known vesicle model72 gave an overall vesicle diameter, Dvesicle, of 103 ± 43 nm and a vesicle membrane thickness, Tmembrane, of 9.8 ± 0.4 nm.
These data are in good agreement with TEM and DLS observations and are also close to values reported by Derry and co-workers for PSMA13-PBzMA96 vesicles characterized by SAXS at 20 °C.64
Variable temperature studies of PSMA13-PBzMA97 diblock copolymer vesicles
The PSMA13-PBzMA96 vesicles reported by Derry and co-workers64 exhibited thickening behaviour on heating owing to a vesicle-to-worm transition. However, Derry et al. did not explore the effect of further heating above the critical temperature that is required to induce this morphological transition.The PSMA13-PBzMA97 vesicle dispersion prepared in this current study was investigated by oscillatory rheology. A temperature sweep from 20 °C to 190 °C was performed within the linear viscoelastic region (strain amplitude = 1.0%, angular frequency = 10 rad s−1) at a heating rate of 2 °C min−1. The storage modulus (G′) increases by more than five orders of magnitude on heating above 153 °C (see Fig. 2). The cross-over of the storage and loss moduli, also known as the critical gelation temperature (CGT), is observed at 155 °C, above which the dispersion acts as a viscoelastic gel (since G′ > G′′).
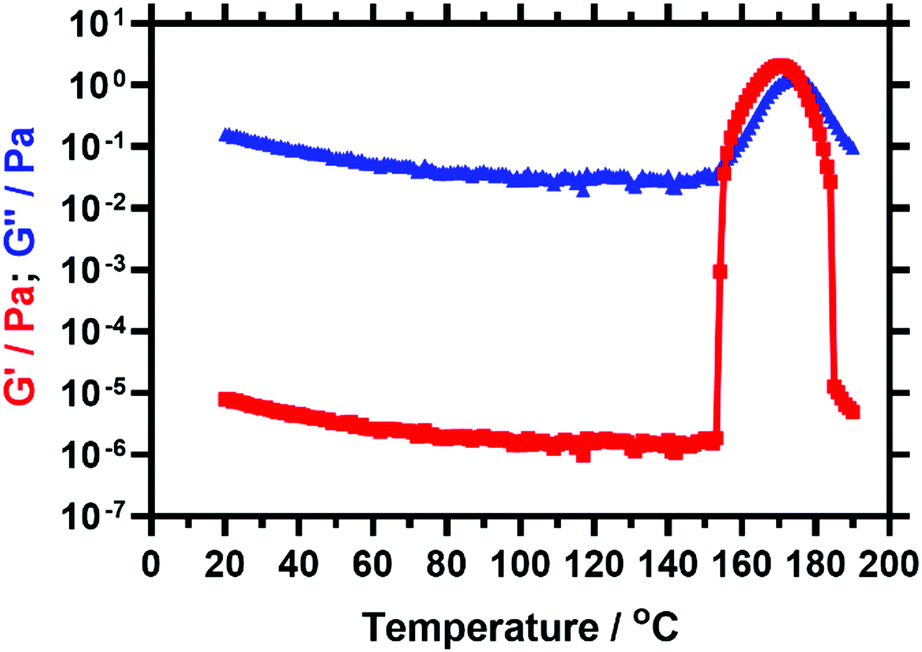 | ||
| Fig. 2 Temperature dependence of the storage modulus (G′, red squares) and loss modulus (G′′, blue triangles) observed for a 10% w/w dispersion of PSMA13-PBzMA97 nano-objects in mineral oil when heating from 20 to 190 °C at 2 °C min−1. This experiment was conducted at 1.0% strain and an angular frequency of 10 rad s−1. | ||
For TEM analysis, 10% w/w PSMA13-PBzMA97 dispersions were heated to the desired temperature prior to dilution to 0.1% w/w using n-dodecane that had been preheated to the same temperature. This sample preparation protocol was adopted to ensure kinetic trapping of the copolymer morphology produced at the designated elevated temperature.64 The image taken of the sample prepared by this method at 150 °C indicated the presence of worms, thus confirming that the enhanced viscosity measured is the result of a thermally-induced vesicle-to-worm transition (see Fig. 3a and b). Since the original study by Derry et al. in 2017, we have demonstrated that the free-standing gels formed by block copolymer worms at ambient temperature most likely arise from multiple contacts between neighbouring worms, rather than from worm entanglements.73 The same inter-worm interactions account for the enhanced dispersion viscosity observed in the present study.
 | ||
| Fig. 3 Transmission electron micrographs obtained for a 0.1% w/w dispersion of PSMA13-PBzMA97 nanoparticles showing (a) vesicles at 20 °C, (b) worms at 150 °C and (c) spheres at 180 °C. | ||
Interestingly, on further heating to 190 °C, a maximum value in G′ is observed, followed by a substantial reduction in viscosity. TEM images recorded for a dilute PSMA13-PBzMA97 dispersion after drying at 180 °C confirm a predominantly spherical morphology under such conditions (see Fig. 3c). This indicates a worm-to-sphere transition that is driven by further surface plasticization of the structure-directing block.37,54
This is not the first time that a single diblock copolymer has been shown to exhibit all three common copolymer morphologies (i.e. spheres, worms and vesicles) simply by varying the solution temperature. We have recently published two examples of amphiphilic diblock copolymers that display this behaviour in aqueous solution.66,67 Moreover, Lodge et al., reported that a dilute solution of polystyrene-polydimethylsiloxane diblock copolymer nano-objects in diethyl phthalate underwent morphology transitions from vesicles to cylinders to spheres on heating.74
Synthesis of PSMA14-P[(1 − X)BzMA-stat-XBuMA]130 diblock copolymer nanoparticles
A PSMA14 precursor was chain-extended via RAFT dispersion copolymerization of benzyl methacrylate (BzMA) and n-butyl methacrylate (BuMA) to generate a series of PSMA14-P[(1 − X)BzMA-stat-XBuMA]130 diblock copolymer nano-objects in mineral oil at 10% w/w solids (see Scheme 1).It is perhaps worth mentioning that the overall target DP of the second insoluble structure-directing block was increased from 100 (as previously targeted for the PSMA13-PBzMA97 vesicles) up to 130. This was to account for the incorporation of the BuMA monomer (142 g mol−1), which has a lower molecular weight than BzMA (176 g mol−1). Otherwise, the reduction in the relative volume fraction of the structure-directing block would result in a lower packing parameter, P, and hence favor a worm morphology.75
A kinetic study of the synthesis of PSMA14-P(0.5BzMA-stat-0.5BuMA)130 vesicles via RAFT dispersion copolymerization of BzMA with BuMA was conducted at 90 °C (see Fig. S2†). Fig. S2a† shows the individual conversion vs. time curves determined for the BzMA and BuMA comonomers respectively during their statistical copolymerization, as determined by 1H NMR spectroscopy. Although the BzMA reacts slightly faster than the BuMA, these two comonomers exhibit comparable reactivities, suggesting a near-statistical copolymerization. As shown in Fig. S2b,† an overall comonomer conversion of 94% was achieved within 6 h and the corresponding semi-logarithmic plot indicated first-order kinetics with respect to monomer. Gel permeation chromatography (GPC) analysis indicated a linear evolution of molecular weight with conversion (Fig. S2c†). As a result, all RAFT dispersion copolymerizations reported herein were conducted at 90 °C for 16 h to maximize the comonomer conversion. An assigned 1H NMR spectrum recorded for the final reaction mixture in CDCl3 when targeting PSMA14-P(0.5BzMA-stat-0.5BuMA)130 at 10% w/w solids in mineral oil under such conditions is shown in Fig. S3.†
The mole fraction of BuMA, X, was systematically varied, see Table 1. When targeting BuMA mole fractions of up to 0.70, ≥97% BzMA conversion and ≥89% BuMA conversion was achieved within 16 h at 90 °C, as judged by 1H NMR spectroscopy. However, somewhat lower comonomer conversions were obtained when targeting a BuMA mole fraction of 0.80 (91% BzMA and 82% BuMA, respectively). The 1H NMR spectra shown in Fig. S4† confirm that the structure-directing block contains a higher proportion of BuMA relative to BzMA when increasing the target BuMA mole fraction from 0.30 to 0.50. THF GPC analysis confirmed that a relatively narrow molecular weight distribution was achieved in all cases (Mw/Mn ≤ 1.16). Efficient chain extension was confirmed by the unimodal nature of the molecular weight distribution curves observed for such diblock copolymers, which were systematically shifted to higher molecular weight compared to that of the PSMA14 precursor, see Fig. 4a.
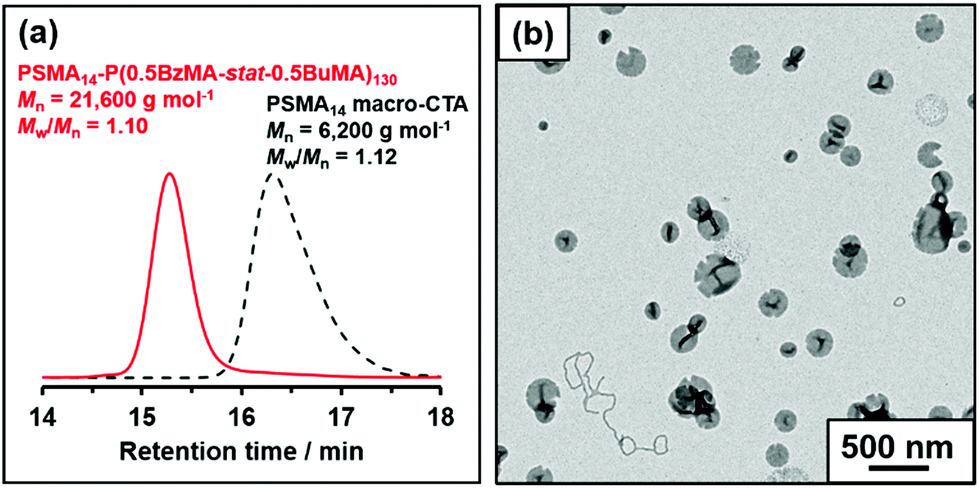 | ||
| Fig. 4 (a) THF GPC traces recorded for PSMA14-P(0.5BzMA-stat-0.5BuMA)130 and the corresponding PSMA14 macro-CTA precursor. (b) Representative TEM image recorded for a dried 0.1% w/w dispersion of PSMA14-P(0.5BzMA-stat-0.5BuMA)130 nano-objects at 20 °C showing predominantly vesicles and a few worms. | ||
| Target mole fraction BuMA (X) | 1H NMR | THF GPC | DLS | TEM | |||
|---|---|---|---|---|---|---|---|
| BzMA conv. (%) | BuMA conv. (%) | M n (g mol−1) | M w/Mn | D z (nm) | PDI | Predominant morphology | |
| 0.00 | 97 | n/a | 23500 |
1.09 | 94 | 0.07 | Vesicles |
| 0.10 | 97 | 92 | 22000 |
1.11 | 112 | 0.06 | Vesicles |
| 0.20 | 97 | 90 | 21800 |
1.11 | 141 | 0.06 | Vesicles |
| 0.30 | 98 | 92 | 21500 |
1.12 | 198 | 0.24 | Vesicles |
| 0.35 | 98 | 92 | 21300 |
1.13 | 208 | 0.22 | Vesicles |
| 0.40 | 97 | 91 | 21300 |
1.12 | 305 | 0.23 | Vesicles |
| 0.45 | 97 | 90 | 20800 |
1.11 | 236 | 0.21 | Vesicles |
| 0.50 | 98 | 92 | 21600 |
1.10 | 318 | 0.21 | Vesicles |
| 0.60 | 97 | 89 | 19900 |
1.14 | 251 | 0.31 | Worms |
| 0.70 | 97 | 89 | 19800 |
1.16 | 113 | 0.19 | Worms |
| 0.80 | 91 | 82 | 16800 |
1.14 | 28 | 0.09 | Spheres |
The predominant morphology for these diblock copolymer nano-objects was determined by TEM analysis (see Fig. S5†), with DLS providing additional information regarding the particle size distribution. As expected based on the prior study by Derry and co-workers,64 the PSMA14-PBzMA130 diblock copolymer formed well-defined vesicles with a number-average diameter of approximately 75 nm as judged by TEM, with DLS reporting a z-average diameter of 94 nm and a polydispersity index of 0.07.
Introducing up to 45 mol% BuMA comonomer into the core-forming block produced progressively larger, more polydisperse vesicles, as judged by DLS (see Table 1). At 50 mol% BuMA, TEM (and DLS) studies revealed a mixed phase comprising predominantly vesicles with some worms (see Fig. 4b). Moreover, further increasing the BuMA content up to 60, 70 or 80 mol% produced mixed vesicle and worm, pure worm or spherical morphologies respectively, as judged by TEM. Again, this is because partial replacement of BzMA with the less massive BuMA comonomer reduces the relative volume fraction of the structure-directing block compared to that of the PSMA stabilizer. This lowers the packing parameter, P, for the copolymer chains, which in turn favors the worm or sphere morphology.75
Variable temperature rheology studies of a series of PSMA14-P[(1 − X)BzMA-stat-XBuMA]130 diblock copolymer vesicles
A series of five dispersions of PSMA14-P[(1 − X)BzMA-stat-XBuMA]130 vesicles (X = 0.30 to 0.50) were targeted at 10% w/w solids in mineral oil while systematically increasing the BuMA content within the membrane-forming block from 30 to 50 mol%. Each dispersion was characterized by oscillatory rheology and a 10% w/w dispersion of PSMA14-PBzMA125 vesicles was also evaluated as a reference sample. This target composition was preferred to PSMA14-PBzMA130 because its temperature-dependent behavior was partially observable within the 20 °C to 180 °C range that could be accessed in rheology studies (see Fig. S6e†).A temperature sweep from 20 °C to 180 °C was performed within the linear viscoelastic region (strain amplitude = 1.0%, angular frequency = 10 rad s−1) at a heating rate of 2 °C min−1. For all the vesicular dispersions studied, a sharp increase in complex viscosity (η*) was observed on heating above 100 °C (see Fig. 5). TEM analysis confirmed that, as first reported by Derry et al. and further investigated in the present study for PSMA13-PBzMA97 vesicles, this enhanced viscosity is the result of a thermally-induced vesicle-to-worm transition.64
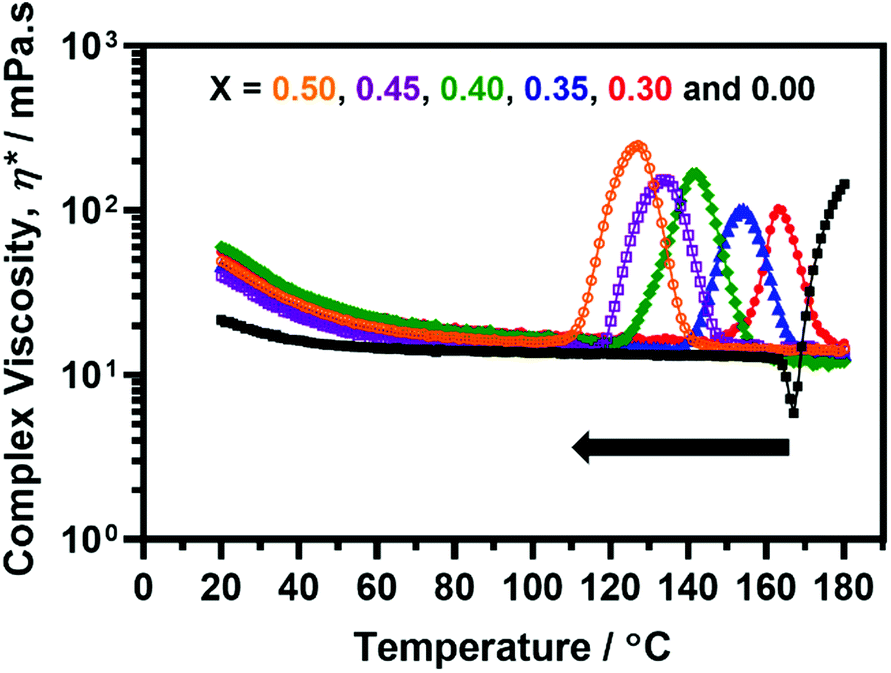 | ||
| Fig. 5 Temperature dependence of the complex viscosity (η*) observed for a series of 10% w/w dispersions of PSMA14-P[(1 − X)BzMA-stat-XBuMA]130 nano-objects containing varying proportions of BuMA within the membrane-forming block on heating from 20 °C to 180 °C at 2 °C min−1. Data were obtained at 1.0% strain using an angular frequency of 10 rad s−1. Data obtained for a reference sample of PSMA14-PBzMA125 vesicles are also shown (black filled squares). | ||
The data shown in Fig. 5 indicate that a series of complex viscosity maxima are observed on further heating. Moreover, the critical temperature required to reach these maximum values can be systematically lowered simply by increasing the relative proportion of BuMA comonomer. Unfortunately, the complex viscosity maximum for the PSMA13-PBzMA125 reference is not fully observable within the 20 °C to 180 °C range. However, if the onset temperature for the upturn in complex viscosity at 167 °C is used for comparative purposes, then targeting a membrane-forming block comprising 50 mol% BuMA lowers this critical temperature by almost 60 °C compared to the PSMA13-PBzMA125 reference vesicles. An even greater reduction in temperature is observed compared to PSMA13-PBzMA130 vesicles, for which an onset temperature of approximately 180 °C is predicted (see Fig. S6†).
Indeed, Fig. 6 confirms that there is a linear relationship between the critical temperature required to achieve maximum viscosity and the proportion of BuMA comonomer within the membrane-forming block. Strikingly, targeting a copolymer composition comprising 50 mol% BuMA lowered this critical temperature by 30 °C compared to when targeting a copolymer containing 30 mol% BuMA. This suggests that incorporating BuMA comonomer into the vesicle membrane facilitates its enhanced plasticization by hot solvent (in this case, mineral oil), which in turn enables the vesicle-to-worm transition to occur at lower temperature.
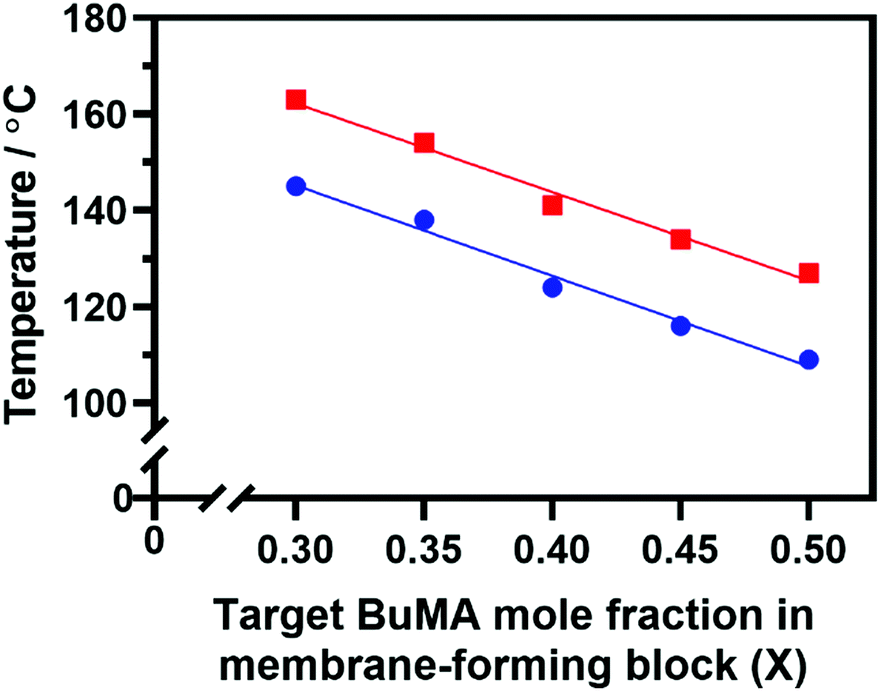 | ||
| Fig. 6 Effect of systematically varying the BuMA mole fraction of the membrane-forming block on (i) the critical onset temperature for vesicle-to-worm transition (blue circles) and (ii) the critical temperature required to achieve maximum complex viscosity (red squares) as indicated by oscillatory rheology studies of a series of PSMA14-P[(1 − X)BzMA-stat-XBuMA]130 nano-objects. | ||
As already noted above for the PSMA13-PBzMA97 diblock copolymer studied, a substantial reduction in complex viscosity is observed for each of the five diblock copolymers on heating well above the critical onset temperature required to induce the vesicle-to-worm transition. This finding is illustrated in Fig. 7, which shows the temperature dependence of the storage modulus (G′) and loss modulus (G′′) for PSMA14-P(0.5BzMA-stat-0.5BuMA)130. At 20 °C, G′′ (6.6 × 10−1 Pa) comfortably exceeds G′ (3.3 × 10−5 Pa), which is typical for a free-flowing dispersion. There is an abrupt increase in G′ at 109 °C, with a maximum G′ of 2.5 Pa being observed at 128 °C. This latter value is five orders of magnitude greater than that measured at 20 °C and is comparable to the G′ of ∼1 Pa previously reported by Derry et al. for a 10% w/w dispersion of PSMA13-PBzMA96 nano-objects.64 The cross-over between the G′ and G′′ curves occurs at 114 °C, which corresponds to the critical gelation temperature (CGT) and is comparable to the critical onset temperature indicated in Fig. 5. G′ exceeds G′′ between 115 °C and 126 °C, which is characteristic of elastic, solid-like behavior. On further heating, G′ is substantially reduced to around 8 × 10−6 Pa, which is comparable to that recorded at around 100 °C. This suggests that a second morphological transition occurs. Bearing mind the results obtained for the PSMA13-PBzMA97 system above, the obvious explanation is a worm-to-sphere transition driven by further surface plasticization of the structure-directing block.37,54
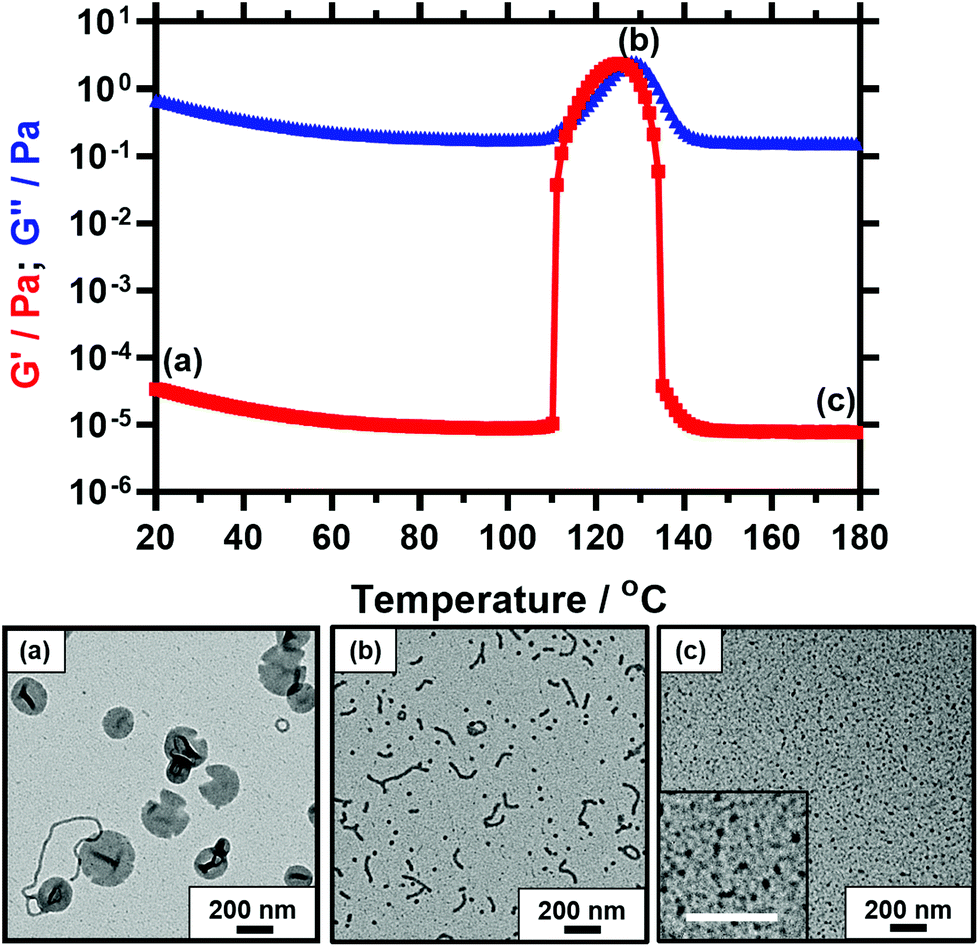 | ||
| Fig. 7 Temperature dependence of the storage modulus (G′, red squares) and loss modulus (G′′, blue triangles) observed for a 10% w/w dispersion of PSMA14-P(0.5BzMA-stat-0.5BuMA)130 nano-objects in mineral oil when heating from 20 to 180 °C at 2 °C min−1. This experiment was conducted at 1.0% strain and an angular frequency of 10 rad s−1. Similar rheological data were obtained at a heating rate of 5 °C min−1 (data not shown). Representative TEM images recorded after drying 0.10% w/w dispersions of PSMA14-P(0.5BzMA-stat-0.5BuMA)130 nano-objects: (a) vesicles (plus worms) at 20 °C, (b) worms (plus spheres) at 130 °C, (c) spheres at 180 °C. All scale bars represent 200 nm. | ||
Fig. 7 shows three representative TEM images recorded for copolymer dispersions diluted to 0.1% w/w at 20 °C, 130 °C and 180 °C, as indicated by labels (a), (b) and (c) shown on the corresponding viscosity-temperature plot. At 20 °C, the predominant copolymer morphology is PSMA14-P(0.5BzMA-stat-0.5BuMA)130 vesicles, along with a few worms. Characteristic folds can be observed by TEM, which indicate vesicle collapse under the ultrahigh vacuum conditions required for this imaging technique. Interestingly, the edges of these vesicles have an unusual physical appearance that suggests an embrittlement effect. This may be the result of TEM grid preparation at 3 °C, which is well below the expected Tg for the membrane-forming block (PBuMA Tg = 20 °C; PBzMA Tg = 54 °C).68 A mixed phase comprising relatively short worms and spheres is observed at 130 °C, while relatively small spheres are visible at 180 °C. These TEM observations are consistent with the rheological data. Further TEM analysis confirmed that these short worms remained stable for at least 1 h at 130 °C. It is also noteworthy that, for the image recorded at 130 °C, the mean worm width is comparable to the sphere diameter. This suggests that the spheres are generated via a budding mechanism from worm ends, as postulated by Fielding and co-workers.54 Moreover, the spheres formed at 180 °C appear to be smaller than those obtained at 130 °C, which suggests a lower aggregation number. This is consistent with studies of thermally-annealed spherical nanoparticles prepared via PISA in non-polar media recently reported by Cornel et al.76 This suggests that these spherical nanoparticles are likely to be in equilibrium with the corresponding molecularly-dissolved copolymer chains at elevated temperature.4,76,77
To assess the thermal stability of the copolymer chains above 150 °C, a 10% w/w dispersion in mineral oil was subjected to a 20–180–20 °C thermal cycle and subsequently analyzed by GPC using a refractive index detector. The chromatogram recorded for the thermally-annealed copolymer was very similar to that of the original copolymer prior to heating (see Fig. S7†). This indicates that minimal copolymer degradation occurs on heating to 180 °C. Interestingly, GPC analysis of the same copolymer using a UV detector (λ = 302 nm) confirmed that 73% of its RAFT end-groups were removed during this thermal cycle. This is consistent with prior work by Moad and co-workers, who demonstrated that thermolysis is a viable method for the removal of trithiocarbonate and dithiobenzoate end-groups from methacrylic polymers.78,79
One reviewer of this manuscript noted the relatively low comonomer conversion (92%) achieved for BuMA when targeting PSMA14-P(0.5BzMA-stat-0.5BuMA)130 nano-objects. However, an oscillatory rheology experiment conducted in the presence of an additional 8% unreacted BuMA confirmed that this comonomer had minimal effect on the thermal transitions reported herein (see Fig. S8†).
The (ir)reversibility of these thermally-induced morphological transitions was investigated by cooling a 10% w/w dispersion of PSMA14-(0.5PBzMA-stat-0.5BuMA)130 nano-objects to 20 °C immediately after an initial 20–180 °C heating ramp during the temperature-dependent oscillatory rheology studies. There was a significant increase in both the storage and loss moduli on cooling: G′ increased by nine orders of magnitude, resulting in a turbid free-standing gel (see Fig. S9a†). TEM images recorded for this dispersion after this 20–180–20 °C thermal cycle indicated a mixture of vesicles and worms, with a significantly higher proportion of the latter nano-objects (see Fig. S9b and S9c†). Similar observations were made after subjecting PSMA13-PBzMA97 vesicles to a thermal cycle.
Variable temperature SIPLI Study
These morphological transitions were also examined using Shear-Induced Polarized Light Imaging (SIPLI). This technique combines rotational rheology with a polarized light source and has been recently used to demonstrate the in situ alignment of highly anisotropic nano-objects such as block copolymer worms at a certain critical rate of applied shear.69Fig. 8 shows how the viscosity of a 10% w/w dispersion of PSMA14-P(0.5BzMA-stat-0.5BuMA)130 nano-objects varies with temperature, along with representative digital images recorded at a constant shear rate at specific temperatures.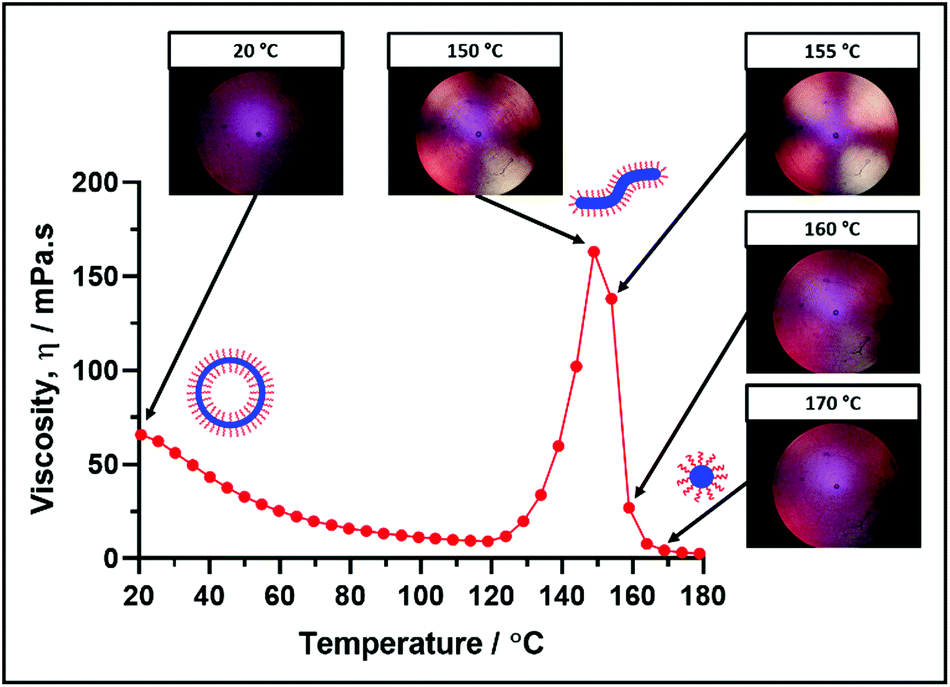 | ||
| Fig. 8 Temperature dependence of the dispersion viscosity and corresponding shear-induced polarized light images (SIPLI) recorded for a 10% w/w dispersion of PSMA14-P(0.5BzMA-stat-0.5BuMA)130 nano-objects on heating from 20 to 180 °C at a rate of 5 °C min−1. Experimental conditions: Shear rate = 200 s−1, 0.50 mm sample gap. | ||
The polarized light image recorded under constant shear at 20 °C appears dark and featureless because PSMA14-P(0.5BzMA-stat-0.5BuMA)130 forms isotropic vesicles at this temperature. In contrast, a distinctive Maltese cross is observed at the same shear rate at 150 °C, which indicates the shear-induced alignment of anisotropic worms. This temperature approximately corresponds to that required for the maximum dispersion viscosity. This Maltese cross is also present at 155 °C, but becomes much fainter at 160 °C and has almost completely disappeared at 170 °C. This suggests that the anisotropic worms have been transformed into isotropic spheres at 170 °C.
It is noteworthy that the critical temperatures required to induce vesicle-to-worm and worm-to-sphere transitions suggested in Fig. 8 do not match those indicated by the oscillatory rheology data shown in Fig. 7. In fact, these thermally-induced transitions occur at significantly lower temperatures (∼20 °C) in the oscillatory rheology experiments. As recently postulated by Byard et al. for a similar doubly thermoresponsive diblock copolymer system, it seems likely that the continuous applied shear employed in the SIPLI studies facilitates both thermal transitions.67 Moreover, a similar explanation has been proposed by Mendes and Menon for the vesicle-to-worm transition exhibited by small molecule surfactants.59
Variable temperature SAXS studies
SAXS patterns were recorded as a function of temperature for a 1.0% w/w dispersion of PSMA14-P(0.5BzMA-stat-0.5BuMA)130 vesicles originally prepared at 10% w/w solids (see Fig. 9).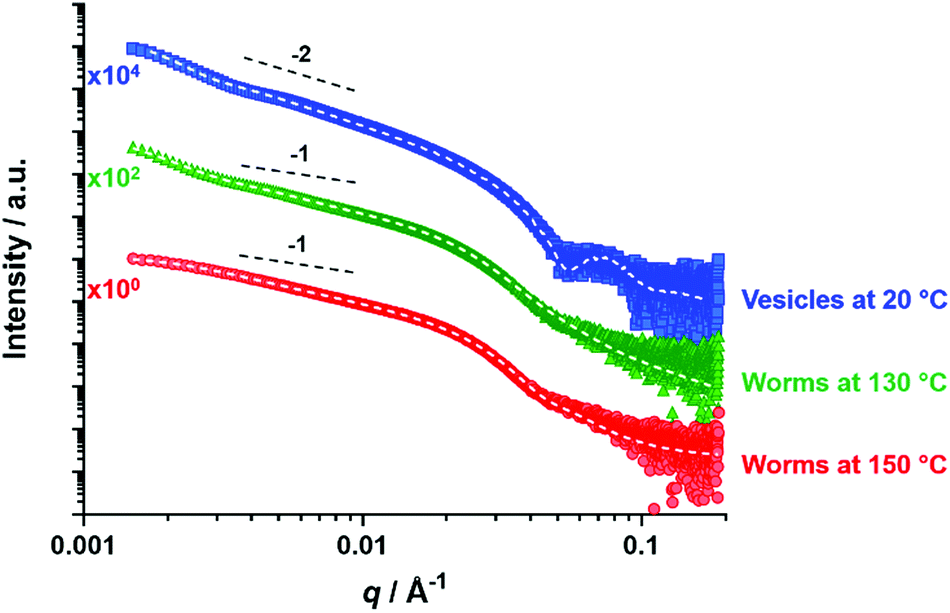 | ||
| Fig. 9 Representative SAXS patterns and data fits obtained for PSMA14-P(0.5BzMA-stat-0.5BuMA)130 vesicles at 20 °C (upper curve, blue squares), PSMA14-P(0.5BzMA-stat-0.5BuMA)130 worms at 130 °C (middle curve, green triangles) and at 150 °C (lower curve, red circles). Dashed lines indicate the data fits obtained using the relevant scattering model. Gradients of −2 and −1 shown as a guide to the eye. | ||
The low q gradient in such I(q) vs. q plots (where I(q) is the scattering intensity and q is the scattering vector) is characteristic of the predominant copolymer morphology.80 Thus the pattern recorded at 20 °C has a low q gradient of approximately −2 and can be satisfactorily fitted using an appropriate vesicle model.72 In contrast, patterns recorded at 130 °C and 150 °C exhibit low q gradients of approximately −1 and can be satisfactorily fitted using a worm-like micelle model,81 (the former pattern requires incorporation of a unified fit to account for the slight upturn at low q,82–84 which is an indication of worm branching). Thus, SAXS studies confirm the vesicle-to-worm transition for this statistical block copolymer system, which is consistent with TEM and SIPLI observations. However, SAXS patterns recorded at 180 °C and 200 °C could not be fitted using either spherical micelle or worm-like micelle models (see Fig. S10†). In this case, it seems likely that the worm-to-sphere transition was not observed because the time allowed for thermal equilibrium was too short. Further experiments would be required to confirm this hypothesis but this is beyond the scope of the current study.
Conclusions
The thermoresponsive behavior of PSMA13-PBzMA97 vesicles prepared by RAFT-mediated PISA at 10% w/w in mineral oil was revisited. Temperature-dependent rheology studies indicated a sharp reduction in the dispersion viscosity on heating above the critical temperature required for the previously reported vesicle-to-worm transition.64 This is attributed to a subsequent worm-to-sphere transition owing to further surface plasticization of the structure-directing PBzMA block, which is supported by the TEM observation of spherical nanoparticles for this copolymer dispersion after heating to 180 °C.Furthermore, the PISA synthesis of PSMA14-P(BzMA-stat-BuMA)130 vesicles was achieved via RAFT dispersion copolymerization of BuMA with BzMA when targeting 10% w/w solids in mineral oil. Introducing BuMA comonomer into the membrane-forming block significantly lowered the critical temperature required to induce a vesicle-to-worm transition from 167 °C to 109 °C, as determined by oscillatory rheology. This morphological transition was confirmed by TEM, SIPLI and SAXS studies. A five-fold increase in G′ was observed above the critical temperature, which is comparable to that previously reported for PSMA13-PBzMA96 vesicles.64 In principle, lowering this critical temperature should enable a wider range of oil-thickening applications to be explored. In practice, higher final comonomer conversions are certainly desirable prior to commercial exploitation. However, we note that residual comonomer appears to have minimal effect on the behavior of these thermoresponsive block copolymer nano-objects.
The thermal transitions reported herein proved to be irreversible on cooling within normal experimental timescales (hours). Moreover, the 10% w/w PSMA14-P(0.5BzMA-stat-0.5BuMA)130 dispersion exhibited a significantly greater viscosity after a 20–180–20 °C thermal cycle compared to the original vesicle dispersion. TEM analysis of the annealed dispersion revealed a mixture of worms and vesicles at 20 °C. It is perhaps noteworthy that such irreversibility may be advantageous for certain applications if a permanent thickening effect is desired.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Samuel A. Harrison is thanked for synthesizing the PSMA14 macro-CTA used in this work. EPSRC is acknowledged for a Centre for Doctoral Training (CDT) grant that funded a PhD studentship for I. R. D. Scott Bader Ltd is thanked for partial support of this CDT PhD studentship and for permission to publish this work. S. P. A. acknowledges an EPSRC Established Career Particle Technology Fellowship grant (EP/R003009). We gratefully acknowledge Diamond Light Source for granting synchrotron beamtime (SM19852, SM21776) and thank the personnel of I22 for their kind assistance.References
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- M. Matsuo, T. Ueno, H. Horino, S. Chujyo and H. Asai, Polymer, 1968, 9, 425–436 CrossRef CAS.
- J. Liu, Z. J. Thompson, H. J. Sue, F. S. Bates, M. A. Hillmyer, M. Dettloff, G. Jacob, N. Verghese and H. Pham, Macromolecules, 2010, 43, 7238–7243 CrossRef CAS.
- D. J. Growney, O. O. Mykhaylyk and S. P. Armes, Langmuir, 2014, 30, 6047–6056 CrossRef CAS.
- S. Creutz, R. Jérôme, G. M. P. Kaptijn, A. W. Van Der Werf and J. M. Akkerman, J. Coat. Technol. Res., 1998, 70, 41–46 CrossRef CAS.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS.
- K. A. Simon, N. J. Warren, B. Mosadegh, M. R. Mohammady, G. M. Whitesides and S. P. Armes, Biomacromolecules, 2015, 16, 3952–3958 CrossRef CAS.
- I. Canton, N. J. Warren, A. Chahal, K. Amps, A. Wood, R. Weightman, E. Wang, H. Moore and S. P. Armes, ACS Cent. Sci., 2016, 2, 65–74 CrossRef CAS.
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CrossRef CAS.
- P. L. Soo and A. Eisenberg, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 923–938 CrossRef CAS.
- V. Bütün, S. P. Armes and N. C. Billingham, Polymer, 2001, 42, 5993–6008 CrossRef.
- F. Liu and A. Eisenberg, J. Am. Chem. Soc., 2003, 125, 15059–15064 CrossRef CAS.
- D. R. Arifin and A. F. Palmer, Biomacromolecules, 2005, 6, 2172–2181 CrossRef CAS.
- J. R. Howse, R. A. L. Jones, G. Battaglia, R. E. Ducker, G. J. Leggett and A. J. Ryan, Nat. Mater., 2009, 8, 507–511 CrossRef CAS.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS.
- D. Le, D. Keller and G. Delaittre, Macromol. Rapid Commun., 2019, 40, 1800551 CrossRef.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS.
- C. Liu, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2020, 11, 3673–3689 RSC.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- E. J. Cornel, S. van Meurs, T. Smith, P. S. O'Hora and S. P. Armes, J. Am. Chem. Soc., 2018, 140, 12980–12988 CrossRef CAS.
- O. J. Deane, O. M. Musa, A. Fernyhough and S. P. Armes, Macromolecules, 2020, 53, 1422–1434 CrossRef CAS.
- S. J. Byard, M. Williams, B. E. McKenzie, A. Blanazs and S. P. Armes, Macromolecules, 2017, 50, 1482–1493 CrossRef CAS.
- N. J. W. Penfold, J. R. Whatley and S. P. Armes, Macromolecules, 2019, 52, 1653–1662 CrossRef CAS.
- C. Gonzato, M. Semsarilar, E. R. Jones, F. Li, G. J. P. Krooshof, P. Wyman, O. O. Mykhaylyk, R. Tuinier and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 11100–11106 CrossRef CAS.
- R. R. Gibson, E. J. Cornel, O. M. Musa, A. Fernyhough and S. P. Armes, Polym. Chem., 2020, 11, 1785–1796 RSC.
- P. Yang, L. P. D. Ratcliffe and S. P. Armes, Macromolecules, 2013, 46, 8545–8556 CrossRef CAS.
- X. Wang, J. Zhou, X. Lv, B. Zhang and Z. An, Macromolecules, 2017, 50, 7222–7232 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS.
- M. J. Derry, L. A. Fielding and S. P. Armes, Polym. Chem., 2015, 6, 3054–3062 RSC.
- E. R. Jones, M. Semsarilar, P. Wyman, M. Boerakker and S. P. Armes, Polym. Chem., 2016, 7, 851–859 RSC.
- C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such and B. S. Hawkett, Macromolecules, 2005, 38, 2191–2204 CrossRef CAS.
- W. M. Wan, C. Y. Hong and C. Y. Pan, Chem. Commun., 2009, 5883–5885 RSC.
- W. M. Wan, X. L. Sun and C. Y. Pan, Macromolecules, 2009, 42, 4950–4952 CrossRef CAS.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- Y. Pei, O. R. Sugita, L. Thurairajah and A. B. Lowe, RSC Adv., 2015, 5, 17636–17646 RSC.
- Y. Pei, L. Thurairajah, O. R. Sugita and A. B. Lowe, Macromolecules, 2015, 48, 236–244 CrossRef CAS.
- E. Guégain, C. Zhu, E. Giovanardi and J. Nicolas, Macromolecules, 2019, 52, 3612–3624 CrossRef.
- M. J. Rymaruk, S. J. Hunter, C. T. O'Brien, S. L. Brown, C. N. Williams and S. P. Armes, Macromolecules, 2019, 52, 2822–2832 CrossRef CAS.
- M. J. Rymaruk, C. T. O'Brien, S. L. Brown, C. N. Williams and S. P. Armes, Macromolecules, 2020, 53, 1785–1794 CrossRef CAS.
- Q. Zhang and S. Zhu, ACS Macro Lett., 2015, 4, 755–758 CrossRef CAS.
- V. J. Cunningham, A. M. Alswieleh, K. L. Thompson, M. Williams, G. J. Leggett, S. P. Armes and O. M. Musa, Macromolecules, 2014, 47, 5613–5623 CrossRef CAS.
- M. Destarac, Polym. Chem., 2018, 9, 4947–4967 RSC.
- G. Wang, M. Schmitt, Z. Wang, B. Lee, X. Pan, L. Fu, J. Yan, S. Li, G. Xie, M. R. Bockstaller and K. Matyjaszewski, Macromolecules, 2016, 49, 8605–8615 CrossRef CAS.
- K. Wang, Y. Wang and W. Zhang, Polym. Chem., 2017, 8, 6407–6415 RSC.
- M. Obeng, A. H. Milani, M. S. Musa, Z. Cui, L. A. Fielding, L. Farrand, M. Goulding and B. R. Saunders, Soft Matter, 2017, 13, 2228–2238 RSC.
- C. Dire, S. Magnet, L. Couvreur and B. Charleux, Macromolecules, 2009, 42, 95–103 CrossRef CAS.
- A. Darabi, A. R. Shirin-Abadi, J. Pinaud, P. G. Jessop and M. F. Cunningham, Polym. Chem., 2014, 5, 6163–6170 RSC.
- A. Darabi, P. G. Jessop and M. F. Cunningham, Macromolecules, 2015, 48, 1952–1958 CrossRef CAS.
- M. Lansalot and J. Rieger, Macromol. Rapid Commun., 2019, 40, 1800885 CrossRef.
- X. Wang and Z. An, Macromol. Rapid Commun., 2019, 40, 1800325 CrossRef.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., 2020, 59, 8368–8392 CrossRef.
- Y. Pei, N. C. Dharsana, J. A. Van Hensbergen, R. P. Burford, P. J. Roth and A. B. Lowe, Soft Matter, 2014, 10, 5787–5796 RSC.
- L. A. Fielding, J. A. Lane, M. J. Derry, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 5790–5798 CrossRef CAS.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS.
- R. Verber, A. Blanazs and S. P. Armes, Soft Matter, 2012, 8, 9915–9922 RSC.
- J. Tan, Y. Bai, X. Zhang and L. Zhang, Polym. Chem., 2016, 7, 2372–2380 RSC.
- J. R. Lovett, M. J. Derry, P. Yang, F. L. Hatton, N. J. Warren, P. W. Fowler and S. P. Armes, Chem. Sci., 2018, 9, 7138–7144 RSC.
- E. Mendes and S. V. G. Menon, Chem. Phys. Lett., 1997, 275, 477–484 CrossRef CAS.
- H. M. L. Davies and J. R. Manning, J. Am. Chem. Soc., 2006, 128, 1060–1061 CrossRef CAS.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS.
- J. R. Lovett, N. J. Warren, S. P. Armes, M. J. Smallridge and R. B. Cracknell, Macromolecules, 2016, 49, 1016–1025 CrossRef CAS.
- R. Deng, Y. Ning, E. R. Jones, V. J. Cunningham, N. J. W. Penfold and S. P. Armes, Polym. Chem., 2017, 8, 5374–5380 RSC.
- M. J. Derry, O. O. Mykhaylyk and S. P. Armes, Angew. Chem., Int. Ed., 2017, 56, 1746–1750 CrossRef CAS.
- R. Deng, M. J. Derry, C. J. Mable, Y. Ning and S. P. Armes, J. Am. Chem. Soc., 2017, 139, 7616–7623 CrossRef CAS.
- L. P. D. Ratcliffe, M. J. Derry, A. Ianiro, R. Tuinier and S. P. Armes, Angew. Chem., Int. Ed., 2019, 58, 18964–18970 CrossRef CAS.
- S. J. Byard, C. T. O'Brien, M. J. Derry, M. Williams, O. O. Mykhaylyk, A. Blanazs and S. P. Armes, Chem. Sci., 2020, 11, 396–402 RSC.
- W. A. Lee and R. A. Rutherford, in Polymer Handbook, ed. J. Brandrup and E. H. Immergut, Wiley, 2nd edn, 1975, pp. III–147 Search PubMed.
- O. O. Mykhaylyk, N. J. Warren, A. J. Parnell, G. Pfeifer and J. Laeuger, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 2151–2170 CrossRef CAS.
- B. R. Pauw, A. J. Smith, T. Snow, N. J. Terrill and A. F. Thünemann, J. Appl. Crystallogr., 2017, 50, 1800–1811 CrossRef CAS.
- J. Ilavsky and P. R. Jemian, J. Appl. Crystallogr., 2009, 42, 347–353 CrossRef CAS.
- J. Bang, S. Jain, Z. Li, T. P. Lodge, J. S. Pedersen, E. Kesselman and Y. Talmon, Macromolecules, 2006, 39, 5583 CrossRef CAS.
- J. R. Lovett, M. J. Derry, P. Yang, F. L. Hatton, N. J. Warren, P. W. Fowler and S. P. Armes, Chem. Sci., 2018, 9, 7138–7144 RSC.
- S. Abbas, Z. Li, H. Hassan and T. P. Lodge, Macromolecules, 2007, 40, 4048–4052 CrossRef CAS.
- K. E. B. Doncom, L. D. Blackman, D. B. Wright, M. I. Gibson and R. K. O'Reilly, Chem. Soc. Rev., 2017, 46, 4119–4134 RSC.
- E. J. Cornel, G. N. Smith, S. E. Rogers, J. E. Hallett, D. J. Growney, T. Smith, P. S. O'Hora, S. Van Meurs, O. O. Mykhaylyk and S. P. Armes, Soft Matter, 2020, 16, 3657–3668 RSC.
- E. J. Cornel, P. S. O'Hora, T. Smith, D. J. Growney, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2020, 11, 4312–4321 RSC.
- A. Postma, T. P. Davis, G. Moad and M. S. O'Shea, Macromolecules, 2005, 38, 5371–5374 CrossRef CAS.
- B. Chong, G. Moad, E. Rizzardo, M. Skidmore and S. H. Thang, Aust. J. Chem., 2006, 59, 755–762 CrossRef CAS.
- O. Glatter and O. Kratky, Small Angle X-ray Scattering, Academic Press, London, 1982, vol. 130 Search PubMed.
- J. S. Pedersen, J. Appl. Crystallogr., 2000, 33, 637–640 CrossRef CAS.
- G. Beaucage and D. W. Schaefer, J. Non-Cryst. Solids, 1994, 172–174, 797–805 CrossRef CAS.
- G. Beaucage, J. Appl. Crystallogr., 1995, 28, 717–728 CrossRef CAS.
- G. Beaucage, J. Appl. Crystallogr., 1996, 29, 134–146 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: THF GPC analysis of the PSMA13-PBzMA97 and PSMA13 macro-CTA; kinetic data obtained during the synthesis of PSMA14-P(0.5BzMA-stat-0.5BuMA)130; assigned 1H NMR spectra; TEM images for PSMA14-P[(1 − X)BzMA-stat-XBuMA]130 recorded at 20 °C (where X = 0.5 to 0.8); complex viscosity vs. temperature plot for PSMA14-PBzMA125 and PSMA14-PBzMA130 vesicles; additional TEM images and DLS data; THF GPC analysis of PSMA14-P(0.5BzMA-stat-0.5BuMA)130 before and after heating to 180 °C; complex viscosity vs. temperature plot for PSMA14-P(0.5BzMA-stat-0.5BuMA)130 nano-objects in the presence or absence of additional BuMA comonomer; temperature dependence of the storage and loss moduli on cooling PSMA14-P(0.5BzMA-stat-0.5BuMA)130 nano-objects from 180 °C to 20 °C, with corresponding TEM images recorded before and after this thermal cycle; further SAXS patterns recorded for PSMA14-P(0.5BzMA-stat-0.5BuMA)130 nano-objects at various temperatures. See DOI: 10.1039/d0py01713b |
| ‡ Present address: Aston Institute of Materials Research, Aston University, Birmingham, B4 7ET, UK. |
| This journal is © The Royal Society of Chemistry 2021 |