
Synthesis and structural characterization of bio-based bis(cyclic carbonate)s for the preparation of non-isocyanate polyurethanes†
Kamila
Błażek
 a,
Hynek
Beneš
b,
Zuzana
Walterová
b,
Sabina
Abbrent
b,
Arantxa
Eceiza
c,
Tamara
Calvo-Correas
c and
Janusz
Datta
*a
a,
Hynek
Beneš
b,
Zuzana
Walterová
b,
Sabina
Abbrent
b,
Arantxa
Eceiza
c,
Tamara
Calvo-Correas
c and
Janusz
Datta
*a
aGdansk University of Technology, Faculty of Chemistry, Department of Polymers Technology, 11/12 Gabriela Narutowicza Street, 80-233 Gdansk, Poland. E-mail: janusz.datta@pg.edu.pl
bInstitute of Macromolecular Chemistry, CAS, Heyrovského nám. 2, Praque 162 06, Czech Republic
cMaterials+Technologies’ Research Group (GMT), Department of Chemical and Environmental Engineering, Polytechnic School, University of the Basque Country, Pza Europa 1, Donostia-San Sebastian 20018, Spain
First published on 1st February 2021
Abstract
Bio-based cyclic carbonates are of significant research interest as monomers for non-isocyanate polyurethane (NIPU) synthesis. This research describes the synthesis of a series of five-membered bis(cyclic carbonate)s using bio-based polyether polyols (PO3G) with different molecular weights (250, 650 and 1000 g mol−1) and carbon dioxide as green feedstocks. The utilization of CO2 as a source of carbon in the chemical reaction is in agreement with the sustainable chemical industry. Furthermore, in order to support the green and sustainable polymer chemistry approach, the syntheses were attempted under solvent-free conditions. The implemented synthetic methods are focused on the design of processes and final products that minimize negative environmental impact. Detailed chemical structure analysis of synthesized products was performed using a combination of spectroscopy techniques (ATR-FTIR as well as 1D and 2D NMR techniques), mass spectrometry (MALDI-TOF MS) and chromatography analysis (SEC). The formation of the main product with two terminal cyclic carbonates was confirmed and the formed side products were also identified, characterized and quantified. Finally, as a proof of concept, the synthesized bis(cyclic carbonate)s were successfully used for the preparation of NIPU thermosets. Chemical and mechanical properties of the produced materials suggest their high potential for future applications, e.g. as sound absorbing materials.
Introduction
In the field of polyurethane preparation, methods that exclude the use of toxic and hazardous isocyanates have long been sought after.1 One of the most progressive and attractive methods for non-isocyanate polyurethane synthesis (NIPUs) is the ring-opening reaction between five-, six- or seven-membered cyclic carbonates and primary di- or polyamines.2–4 Cyclic carbonates can be used in a wide range of applications as aprotic polar solvents with a high boiling point or as electrolytes in rechargeable lithium-ion batteries, additives, intermediates in the production of polymeric materials (e.g. non-isocyanate polyurethanes and polycarbonates) as well as high-purity chemicals (e.g. glycols, carbamates, pyrimidines, dialkyl carbonates, etc.).5–7Nowadays, the synthesis of cyclic carbonate(s) from an appropriate epoxide and carbon dioxide (CO2) is an active field of research.7 This is mainly due to the necessity of designing new technologies able to mitigate the environmental impact of CO2, which as a greenhouse gas, has a significant impact on an enhanced greenhouse effect. Due to the fact that CO2 is chemically inert, abundant, non-flammable, non-toxic and highly attractive one-carbon (C1) building block, its conversion into valuable chemicals is attracting attention from a wide academic community, industry and society.8 For these reasons, the capture, storage and utilization of CO2 are serious challenges to overcome and move toward sustainable development.9 Another issue is the adoption of rules and practices of green production, ranging from the substitution of hazardous components with more sustainable alternatives for the design and development of completely novel, green polymers.10 Synthesis of new, biomass-derived NIPUs can be accomplished using novel five-membered bis(cyclic carbonate)s prepared by chemical fixation of CO2 from bis-epoxides derived from bio-based feedstocks, such as vegetable oils, bio-based 1,4-butanediol, resorcinol, trimethylolpropane,11 isosorbide,12 sorbitol,13 vanillin,14 glycerol, pentaerythritol,15 furfuryl alcohol,16 gallic acid,17 tannic acid18 and polytrimethylene ether glycols.9,19
While DGEBA (Bisphenol A diglycidyl ether), BDO (1,4-butanediol), PPG (poly(propylene glycol)), PEG (poly(ethylene glycol)) diglycidyl ethers are commercially available products, diglycidyl ethers of other compounds, especially obtained from bio-based resources, require special preparation strategies.20,21 Currently, two main methods for the syntheses of glycidyl ethers with terminal epoxy groups through the reaction with an epihalohydrin are known.22 The first concerns the use of Lewis acids and consists of the halohydrin intermediate process and the dehydrochlorination process. In this type of reaction, halohydrin ether as a side-product may be formed and undesired polymerization reaction can proceed. The second method involves the use of phase-transfer catalysts (PTC), sodium hydroxide solution and organic solvents. Although, by-products formed by this method require special procedures for disposal, which is unfavourable from an industrial point of view.22
In this research study, we have explored the potential of bio-based polyether polyols as precursors for the synthesis of fully bio-sourced NIPUs. First, a series of bis(cyclic carbonate)s with different molecular weights (250, 650 and 1000 g mol−1) was synthesized using a two-step process involving polyol epoxidation and subsequent chemical fixation of CO2. Detailed molecular structure characterization of the prepared intermediates and final products was carried out using ATR-FTIR, 1D- and 2D-NMR, and SEC and MALDI-TOF mass spectrometry. To the best of our knowledge and despite their great potential as monomers for NIPUs, the synthesis of cyclic carbonates has never been reported in such a detailed way. As the last step, the synthesized bis(cyclic carbonate)s were used for the preparation of NIPU thermosets by polyaddition with polyfunctional bio-based amines. The produced crosslinked polyurethane materials were characterized using FTIR, DMTA, TGA and the NIPU network mechanical and swelling properties were determined.
Experimental
Materials
Bio-based polyether polyols, poly(trimethylene oxide) diols – PO3G, Sensatis® with a molecular weight of 250 g mol−1 and PO3G Velvetol® with molecular weights of 650 and 1000 g mol−1 were purchased from Allessa (Germany) and used as received. Epichlorohydrin (99%), sodium hydroxide (NaOH), boron trifluoride-diethyl ether complex (BF3·Et2O) (98%) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 98%) were purchased from Sigma-Aldrich (USA) and used as received. Tetrabutylammonium bromide (TBAB, 99%) was purchased from TCI Chemicals (Belgium) and applied as a catalyst for the cycloaddition of carbon dioxide (CO2). Bio-based dimer diamine, a derivative of dimerized fatty acids, Priamine®1071 was purchased from Croda (Netherlands). All chemicals and solvents used for the analytical methods were of analytical grade and used as received.Synthesis of diglycidyl ethers
The syntheses of diglycidyl ethers (see Fig. 1 – step I) were performed in a 0.5 L glass reactor equipped with a temperature controller, mechanical stirrer and Liebig condenser, according to our previous study.23 In a typical synthesis, the dried polyether polyol PO3G650 (162.5 g, 0.25 mol) and BF3·Et2O (1 wt%) were first mixed and heated to 80 °C. Next, epichlorohydrin (69.39 g, 0.75 mol) was continuously added and the reaction was stirred at 200 rpm at 80 °C. After 8 hours of reaction, the mixture was cooled down to 50 °C. Then, 50% w/w aqueous solution of NaOH (30 g, 1.5 mol) was added dropwise over ca. 5 h. The solid precipitate (formed salts) was removed by filtration under reduced pressure. The product was extracted into ethyl acetate. The mixture was then evaporated to remove solvents and unreacted compounds. The product (ED650) was obtained as a colourless slightly viscous liquid. Similarly, the polyether polyols PO3G250 and PO3G1000 were used for the reactions using the same PO3G/epichlorohydrin molar ratio of 1/3, which led to ED250 and ED1000 products. | ||
| Fig. 1 Scheme of the reaction between polyether polyol and epichlorohydrin (I step) and cycloaddition of CO2 into diglycidyl ether (II step). | ||
Synthesis of bis(cyclic carbonate)s
The synthesized diglycidyl ethers were reacted with carbon dioxide producing five-membered bis(cyclic carbonate)s of PO3G (see Fig. 1 – step II). The reaction was conducted according to our previous study.23 In brief, a 0.5 L three-necked flask equipped with a magnetic stirrer, a temperature controller and an inlet of CO2 was charged with 200 g (0.26 mol) of ED650 (obtained in the previous step) and heated to 110 °C. Next, TBAB (0.5 wt%) as a catalyst was added and the reaction was performed for 30 h under atmospheric pressure with a 100 ml min−1 CO2 gas flow rate. The obtained product was analyzed in detail and used for polyurethane preparation without further purification. At room temperature, the produced bis(cyclic carbonate) from ED650, denoted as DC650 was obtained as a colourless liquid. Similarly, DC250 and DC1000 bis(cyclic carbonate)s were synthesized from ED250 and ED1000, possessing similar organoleptic characteristics.Preparation of non-isocyanate polyurethanes
Non-isocyanate polyurethanes (NIPU)s were prepared via a one-pot polyaddition reaction between bis(cyclic carbonate) and diamine derivatives of dimerized fatty acids (Priamine 1071, amine value of 193 mg KOH per g,23 dimer/trimer content of 3/1 and average amine functionality of 2.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24). In a typical procedure, a mixture of DC650 (51.0 g, 0.06 mol) and Priamine 1071 (41.9 g, 0.07 mol) was mechanically stirred for 3 h at 110 °C in the presence of the DBU catalyst (0.5 wt%). Then, the reaction mixture was placed into a mold and cured at 110 °C for 48 h in a laboratory oven. The prepared NIPU (denoted as NIPU650) was characterized by ATR-FTIR spectroscopy to check the progress of the cyclic carbonate–amine reaction. Similarly, the NIPU250 and NIPU1000 were prepared by the reaction of Priamine 1071 with ED250 and ED1000, respectively. The initial bis(cyclic carbonate)/Priamine 1071 molar ratio of 1:1.2 was always used. The final conversion of the cyclic carbonates was calculated by means of FTIR spectroscopy as the intensity decrease of the C
24). In a typical procedure, a mixture of DC650 (51.0 g, 0.06 mol) and Priamine 1071 (41.9 g, 0.07 mol) was mechanically stirred for 3 h at 110 °C in the presence of the DBU catalyst (0.5 wt%). Then, the reaction mixture was placed into a mold and cured at 110 °C for 48 h in a laboratory oven. The prepared NIPU (denoted as NIPU650) was characterized by ATR-FTIR spectroscopy to check the progress of the cyclic carbonate–amine reaction. Similarly, the NIPU250 and NIPU1000 were prepared by the reaction of Priamine 1071 with ED250 and ED1000, respectively. The initial bis(cyclic carbonate)/Priamine 1071 molar ratio of 1:1.2 was always used. The final conversion of the cyclic carbonates was calculated by means of FTIR spectroscopy as the intensity decrease of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band (at 1790 cm−1) normalized to the C–H band (at about 1370 cm−1) as previously described.25
O band (at 1790 cm−1) normalized to the C–H band (at about 1370 cm−1) as previously described.25
Methods of characterization
All FTIR spectra were recorded on a Nicolet 8700 FTIR spectrophotometer (Thermo Electron Corporation, USA) using the ATR technique (Heated Golden Gate from Specac Ltd). The ATR-FTIR spectra were collected at a resolution of 4 cm−1 with 64 scans per run in the wavenumber range from 4500 to 500 cm−1. High-resolution NMR measurements were recorded on a Bruker 400 MHz Avance Neo spectrometer at the Larmor frequency ν(13C) = 100.624 MHz. The 13C NMR spectra were recorded with a 90° pulse of 10 μs, relaxation delay of 10 s, 4500 scans. 2D gradient heteronuclear single-quantum coherence (gHSQC) measurements were carried out, and the results were recorded on a Varian Unity Inova 500 Plus spectrometer operating at a frequency of 500 MHz using DMSO-d6 as the solvent with pulse field gradients and the spectrum was acquired in the phase-sensitive mode with 1 J(CH) set to 140 Hz. The spectral window for 1H and 13C axes were 4336 Hz and 19482 Hz, respectively. The data were collected in a 1664 × 155 matrix and processed in a 2 K × 1 K matrix. Size exclusion chromatography (SEC) analyses were carried out in THF at 30 °C and a flow rate of 1 mL min−1 using a Thermo Scientific chromatograph equipped with a RefrectoMax 521 refractive index detector and an isocratic Dionex UltiMate 3000 pump. A series of four Phenogel SEC columns with a particle size of 5 μm and pore sizes of 105, 103, 100 and 50 Å was used. Samples were prepared by dissolving in THF (1 wt%) and filtering using nylon filters with 2 μm pore size. The elution times were converted to molecular weight by calibration with polystyrene (PS) standards. MALDI-TOF mass spectra were acquired with an UltrafleXtreme (Bruker Daltonics, Bremen, Germany) in the positive ion reflectron mode, using delayed extraction. The spectra were the sum of 30000 shots with a DPSS, Nd:YAG laser (355 nm, 1000 Hz). External calibration was used. The samples were prepared by the dried droplet method: solutions of the sample (10 mg mL−1) and DHB (2,5-dihydroxybenzoic acid; Sigma-Aldrich, 98%, 20 mg mL−1) as a matrix and of sodium trifluoroacetate (NaCF3COO; Sigma-Aldrich, 10 mg mL−1) as a cationizing agent in DMF (N,N-dimethylformamide, Sigma-Aldrich, biotech. grade, ≥99.9%) were mixed in a volume ratio of 4:20:1. 1 μL of the mixture was deposited on the ground-steel target plate. The drop was dried under an ambient atmosphere. Dynamic mechanical-thermal analysis (DMTA) was performed on a DMA Q800 Analyzer (TA Instruments). The temperature dependence of the complex modulus of rectangular samples (dimensions: 40 × 10 × 2 mm) was measured using a single cantilever mode at a frequency of 10 Hz at temperatures ranging from −100 to 150 °C at a heating rate of 4 °C min−1. Changes in the storage (E′) and loss (E′′) were recorded and the temperature of the alpha transition (Tα) corresponding to glass transition was evaluated as the maximum of the tanδ peak. The number average molar weight between crosslinks (Mc) was calculated as:26,27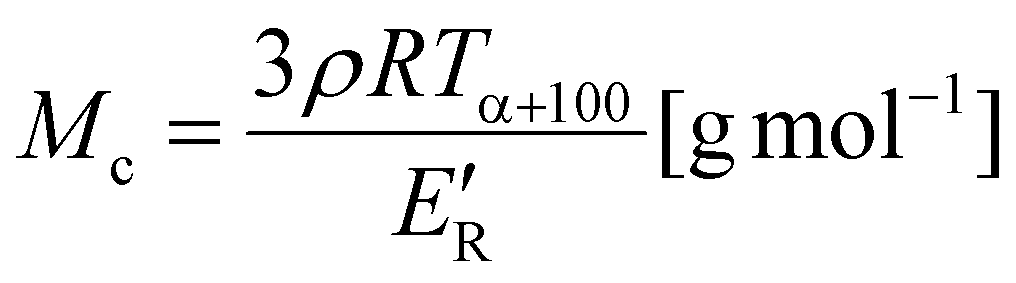 | (1) |
 | (2) |
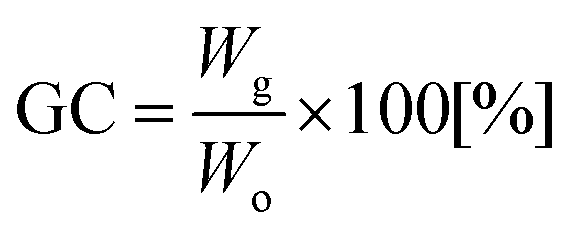 | (3) |
Results and discussion
Synthesis and characterization of diglycidyl ethers (step I)
The reaction between the PO3G polyols and an excess of epichlorohydrin took place in bulk without any solvent addition using the BF3·OEt2 catalyst. It is well known that Lewis acids are efficient catalysts for the addition of epichlorohydrin to polyhydric alcohols.22,28 According to current knowledge, the reaction mechanism involves an initial step of formation of chlorohydrin ether 1 with the simultaneous oxirane ring opening. In a succeeding step, under the influence of the alkaline catalyst (NaOH), hydrogen chloride is eliminated from the chlorohydrin ether during the formation of polyether-based diglycidyl ethers 2.22 The scheme of the reaction between polyether polyols and epichlorohydrin and the subsequent cycloaddition of CO2 into diglycidyl ethers 2 and formation of bis(cyclic carbonate)s 3 is shown in Fig. 1.19,22,23,28–30The chemical structures of the synthesized diglycidyl ethers were first analysed by FTIR (Fig. 2). The FTIR spectra have confirmed the successful epoxidation reaction revealing the formation of oxirane ring moieties (epoxy groups) as two new bands at 908 cm−1 (νC–O of the oxirane group) and 841 cm−1 (νC–O–C of the oxirane group), and the consumption of polyols’ hydroxyl groups – indicated by the almost complete disappearance of the stretching OH band (ν-OH) at 3590–3310 cm−1. The epoxy band intensity was proportional to the epoxy group content and progressively increased (ED1000 < ED650 < ED250) as the molecular weight of the used polyols decreased. All epoxidized samples further contained typical asymmetric and symmetric stretching vibrations of C–H groups (ν-CH2– and ν-CH3) at 2930 and 2850 cm−1, and the stretching vibrations of ether functional groups (ν-C–O–C–) at 1100 cm−1.
 | ||
| Fig. 2 FTIR spectra of the original bio-based polyether polyols (PO3G250, PO3G650 and PO3G1000), the synthesized diglycidyl ethers (ED250, ED650 and ED1000) and the synthesized bis(cyclic carbonate)s (DC250, DC650 and DC1000). | ||
In order to identify the formed products, including side-products MALDI TOF mass spectrometry analysis of the synthesized diglycidyl ethers was conducted. Initially, MALDI TOF mass spectra of the starting materials, i.e., bio-based polyols (PO3G250, PO3G650 and PO3G1000, Fig. S1 in the ESI†), showed a broad distribution of molecular ions (ionized by Na+) with a trimethylene ether repeating unit (Δm/z = 58 Da) and with peak positions revealing a linear structure with OH and H end-groups. After epoxidation, the MALDI-TOF mass spectra of all three synthesized diglycidyl ethers (ED250, ED650, and ED1000, Fig. 3A–C) proved the formation of the structures bearing oxirane end-groups. Owing to the high purity of the original PO3G1000 polyol, the MALDI TOF mass spectrum of ED1000 was smooth and showed three well-distinguished distributions of molecular ions (sodium adducts). The peaks of all three distributions were characterized by a mass increment of 58 Da corresponding to the mass of the repeating unit in poly(trimethylene ether) glycols (PO3G).
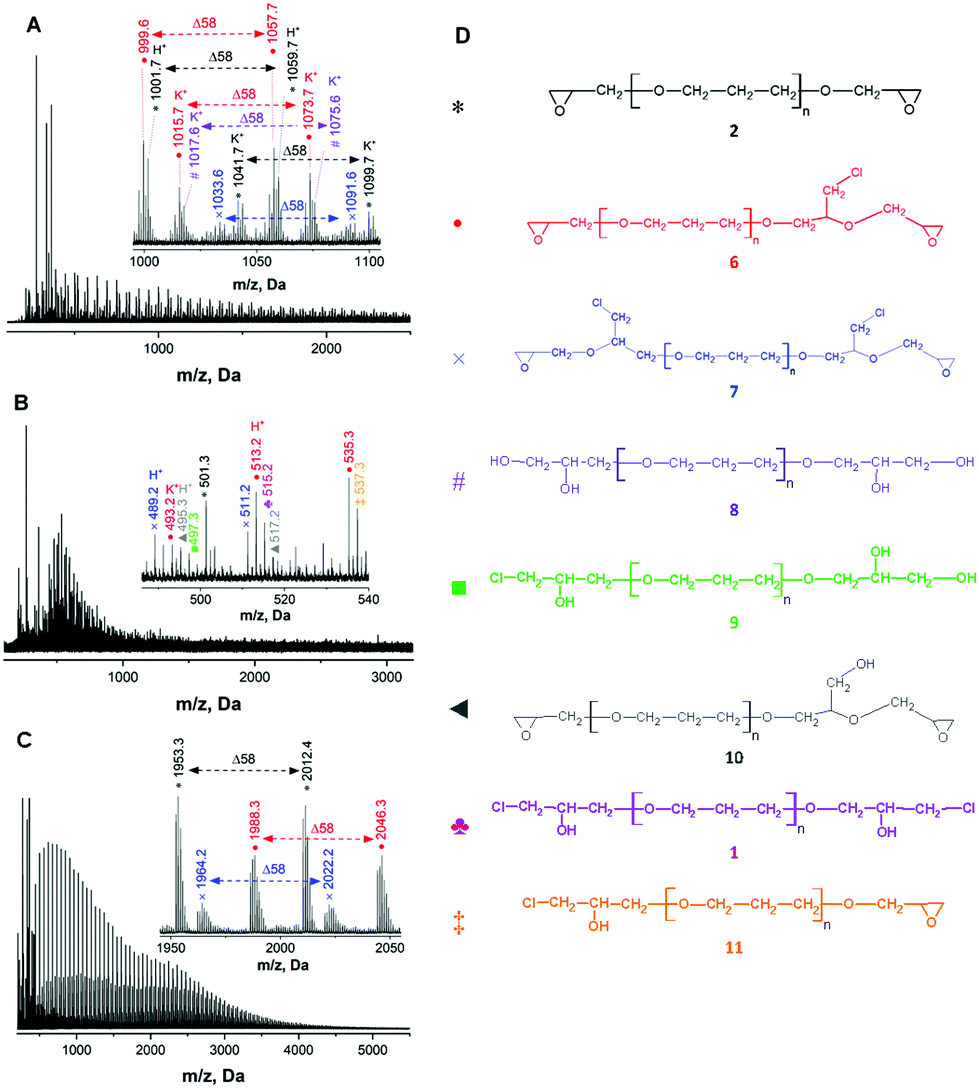 | ||
| Fig. 3 MALDI TOF mass spectra of the synthesized diglycidyl ethers ED 250 (A), ED 650 (B) and ED 1000 (C), and the structures detected by MALDI-TOF in samples (D). The peaks correspond to sodium adducts of molecular ions [M + Na]+ (unless otherwise marked). | ||
The most intensive distribution (marked with black asterisks) corresponded to the targeted diglycidyl ether 2, i.e. the linear PO3G structure with two terminal oxirane rings (Fig. 3D).
Besides this main product, two by-product species were identified to have formed during the epoxidation. These by-products were composed of linear PO3G repeating units (Δm/z of 58 Da) with one (the full red circle marked distribution) or two (the blue cross marked distribution) glycidyl ether terminal groups bearing a chloromethyl side-chain. The possible reaction mechanism of this side reaction involves the initial formation of alkoxy anion 4 and its subsequent reaction with an epoxy group of epichlorohydrin thus producing the chloride-containing alkoxy anion 5 and final elimination of the chloride anion to form glycidyl ether 6 and 7 bearing one or two chloromethyl side-chains (Fig. 4).
 | ||
| Fig. 4 Plausible side reactions that occurred during the diglycidyl ether synthesis. | ||
Moreover, the MALDI TOF mass spectra of ED250 and ED650 also indicated the formation of additional by-products (Fig. 3A and B) assigned to the OH-bearing structures 8, 9 and 10 (Fig. 3D), which were probably formed by the hydrolysis of the oxirane rings (the violet number sign and the green square marked distributions) and the elimination of the chloride anion (the grey triangle marked distribution) as shown in Fig. 4. MALDI TOF mass spectrometry further revealed the presence of chlorohydrin derivatives 1 and 11 in ED650 (Fig. 3B) formed due to incomplete dehydrochlorination.
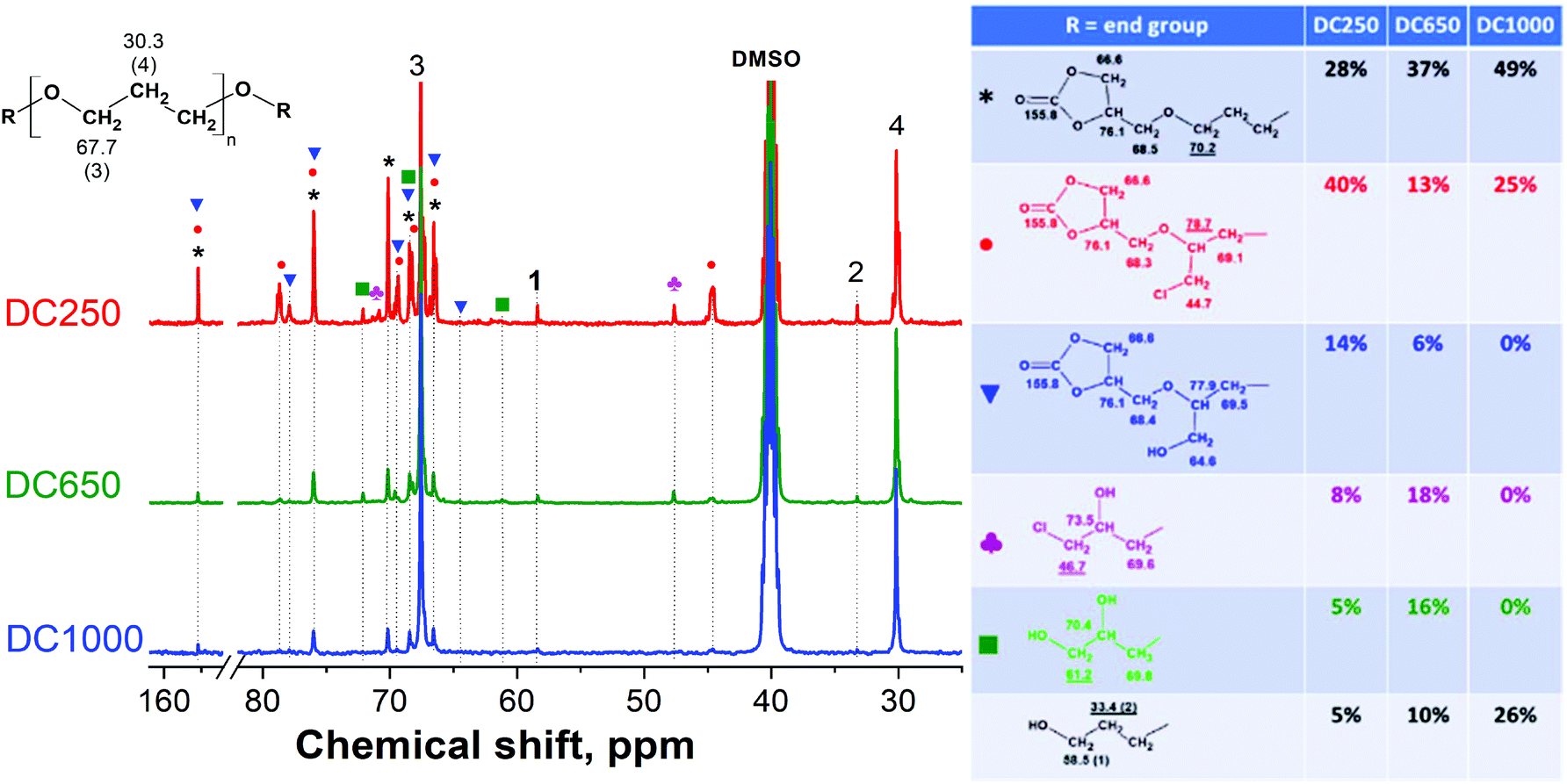
The extent of these side reactions as well as the yield of epoxidation (the epoxy conversion) can be assessed from the 13C NMR results (Fig. 5). Normally, 1H NMR spectra would be used for such analysis.
 | ||
| Fig. 5 13C NMR spectra of the synthesized diglycidyl ethers (ED250, ED650 and ED1000), peak denotation and end group characterization and quantification. | ||
However, in this case, the low abundance and similarity of different end-groups resulted in unresolved, overlapping peaks and 13C NMR had to be used instead. Here, the dominant methylene signals 3 (67.7 ppm) and 4 (30.3 ppm) originate from the trimethylene ether repeating unit of the polymer backbone (also see the 13C NMR spectra of the original PO3Gs in Fig. S2 in the ESI†), while the methylene signals 1 (58.5 ppm) and 2 (33.4 ppm) represent CH2 carbons next to the unreacted OH end-groups. Furthermore, the presence of structures identified by MALDI TOF mass spectrometry (Fig. 3) was verified and quantified by 13C NMR spectroscopy in the form of different existing end-groups (Fig. 5). Firstly, three different glycidyl ether (epoxy) end-groups were identified confirming the formation of the main product 2 (the black asterisk signals) and the by-products with chloromethyl (6, 7; the red circle signals) and hydroxymethyl (10; the blue triangle signals) side-chains.31 Moreover, ED250 and ED650 samples have also been shown to contain the chlorohydrin derivatives 1 and 11 (the magenta trefoil signals), and the OH-bearing structures 8 and 9 (the green square signals).
Since all the 13C NMR signals corresponding to the end-groups were identified, end-group quantification was performed using the sum integral of one non-overlapping signal of each species (the underlined signals in Fig. 5). The sum of three different glycidyl ether end-groups then corresponds to the degree of epoxy conversion, estimated to be 88%, 72% and 29% for ED1000, ED650 and ED250, respectively. This is in very good agreement with the MALDI TOF results confirming the high efficiency and purity of the ED1000 sample.
Synthesis and characterization of bis(cyclic carbonate)s (step II)
The cycloaddition reaction, which led to the formation of bis(cyclic carbonate)s 3 (Fig. 1), was conducted under the optimized conditions as mentioned in detail in our previous study.19 The FTIR spectra of the prepared bis(cyclic carbonate)s DC250, DC650 and DC1000 (Fig. 2) showed the decreasing intensity of the vibration band at 915 cm−1 reflecting the consumption of oxirane rings (epoxy groups). However, the epoxy peak did not completely disappear due to a partial overlapping with the in-plane bending vibration of methyl (–CH3) groups.32 The incorporation of CO2 into the structure of the diglycidyl ether macromolecules was further evidenced by the formation of a distinctive absorption peak at 1800 cm−1, which corresponds to the stretching vibration of carbonyl groups (νCO) of the cyclic carbonates.
For further NIPU preparation via the polyaddition of bis(cyclic carbonate)s with polyamines, it is necessary to convert bisepoxides (prepared in step I) into the bis(cyclocarbonate)s (prepared in step II) in high yield and selectivity by using an environmentally friendly catalyst.33 The TBAB catalyst was previously employed as a green and efficient catalyst for the cycloaddition of CO2 with mono-epoxides34 and aromatic di-epoxides.33 Therefore, it was also adopted for the conversion of 2 into 3 with a complete epoxy group consumption during CO2 cycloaddition, as no residual epoxide peaks remained in the 13C NMR spectra of all synthesized DC samples (the absence of methylene and methine signals of the oxirane ring at ca. 43.9 ppm and 50.8 ppm, respectively, Fig. 6). Moreover, the characteristic carbon signals of cyclic carbonate ring at 155.8 ppm (CO), 76.1 ppm (CH) and 66.6 ppm (CH2)35 confirmed the formation of 3 with the yield of 28% (DC250), 37% (DC650) and 49% (DC1000) respectively. 1H–13C HSQC spectra of the synthesized bis(cyclic carbonate)s (Fig. S3 in the ESI†) helped us to distinguish individual protons and carbons connected to the chlorine atom and OH groups. Fig. 6 shows determination and quantifications of the identified end-groups giving the total content of cyclic carbonate groups (the black asterisk, the red circle and the blue square signals) of 82% (DC250), 56% (DC650) and 74% (DC1000). The methylene signals 1 (58.5 ppm) and 2 (33.4 ppm) representing CH2 carbons next to unreacted OH end-groups indicated the residual amount of unreacted PO3G chains. Additionally, the DC250 and DC650 samples contained certain amounts of chlorohydrin derivatives (1 and 11) and the OH-bearing species (8 and 9), already formed as by-products during the previous step I.
 | ||
| Fig. 6 13C NMR spectra of the synthesized bis(cyclic carbonate)s (DC250, DC650 and DC1000), peak denotation and end group characterization and quantification. | ||
The structure of the main product 3 and the formation of by-products during CO2 cycloaddition were further investigated by MALDI-TOF mass spectrometry (Fig. 7). The MALDI-TOF mass spectra of all synthesized bis(cyclic carbonate)s distinguished five distributions of molecular ions with a mass increment of 58 Da corresponding to the trimethylene ether repeating unit with different end-groups (Fig. 7A–C). The most intensive distribution (the black asterisk marked signals) was assigned to the linear PO3G molecules bearing two terminal cyclic carbonates, which proved the successful formation of the main product – bis(cyclic carbonate)s 3 (Fig. 7D). Two other distributions marked with full red circles and blue crosses were also assigned to bis(cyclic carbonates) species 12 and 13, which contained one and two chloromethyl side chains in terminal groups, respectively. They were formed via CO2 cycloaddition from their epoxidized analogues 6 and 7. It was thus found that the presence of a chlorine atom in the side-chain group did not suppress the reactivity of diglycidyl ethers towards CO2 cycloaddition. The side reactions proceeded to a very low extent in the case of DC1000, while intensive MALDI TOF signals of by-products were observed for DC250 and DC650. In particular, mainly mono(cyclic carbonate) structures bearing only one cyclic carbonate end-group (14–19) were detected. The incomplete CO2 cycloaddition together with the hydrolysis of the oxirane ring leading to the formation of 8, 18 and 19 species were thus confirmed to be the most important side reactions during step II.
 | ||
| Fig. 7 MALDI-TOF mass spectra of the synthesized bis(cyclic carbonate)s DC250 (A), DC650 (B) and DC1000 (C), and the assigned structures (D). The peaks correspond to sodium adducts of molecular ions [M + Na]+ (unless otherwise marked). | ||
Finally, the molecular weight average (Mn) and dispersity (Đ) of the synthesized bis(cyclic carbonate)s were determined using MALDI TOF mass spectrometry and SEC (Table 1 and Fig. S4 in the ESI†). The theoretical Mn of the synthesized bis(cyclic carbonate)s was calculated from the Mn values of the initial bio-based poly(trimethylene glycol)s (PO3G250, PO3G650 and PO3G1000) determined using the 13C NMR (Fig. S2 in the ESI†). The calculation was performed based on the assumption of full conversion and occurrence of no side reactions.
| Sample | M n (theor.) [g mol−1] | M n (SEC) [g mol−1] | Đ (SEC) | M n (MALDI-TOF) [g mol−1] | Đ (MALDI-TOF) |
|---|---|---|---|---|---|
| a PS calibration. | |||||
| DC250 | 460 | 540 | 1.4 | 710 | 1.3 |
| DC650 | 900 | 800 | 2.0 | 790 | 1.5 |
| DC1000 | 1300 | 1200 | 2.2 | 1350 | 1.5 |
Both experimentally determined Mn values correlate well with each other and are in good agreement with the theoretical Mn. The difference in molecular weight confirms the occurrence of side reactions as confirmed by FTIR, NMR and MALDI TOF above. The difference between the theoretical and the determined Mn weights were higher in the case of DC250 than DC650 and DC1000, which agrees well with the above-mentioned findings that the side reactions proceed to a higher extent during DC250 synthesis. Also, the relatively high dispersity of the synthesized bis(cyclic carbonate)s is caused by the high dispersity of the original bio-based polyether polyols (see Table S1 in the ESI†).
Non-isocyanate polyurethanes
As a proof-of-concept application, the successfully synthesized bis(cyclic carbonate)s 3 were subjected to polyaddition with an amine hardener (diamine derivatives of dimerized fatty acids – Priamine 1071) for the preparation of NIPU thermosets 16 (Fig. 8). Generally, for NIPU synthesis, the addition of organic solvent is usually required,33 which makes the whole process technologically pathless and environmentally unfriendly. Herein, in order to make the process as green as possible, NIPU synthesis was realized in bulk with no addition of organic solvents producing mechanically robust, macroscopically homogeneous materials (Fig. S5 in the ESI†).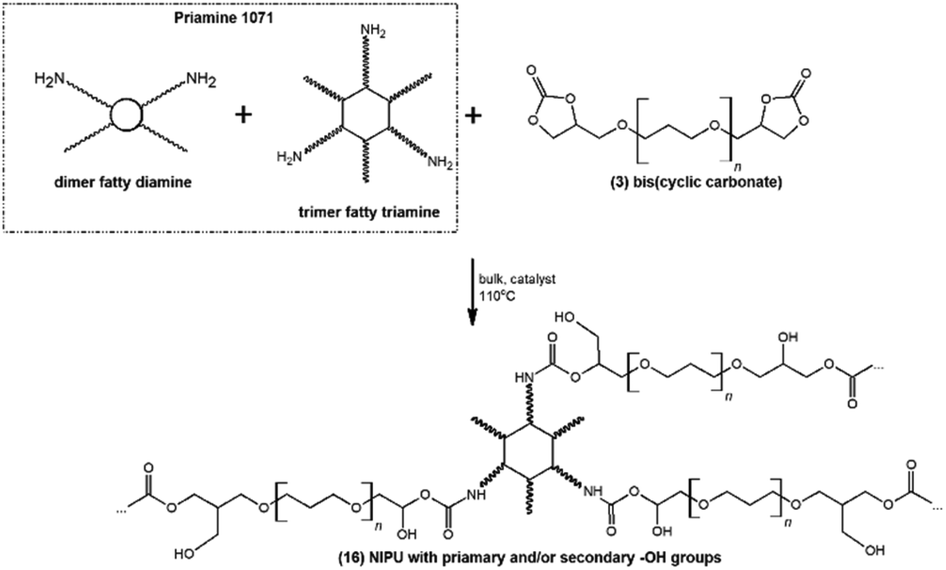 | ||
| Fig. 8 Polyaddition of the synthesized bis(cyclic carbonate)s 3 and bio-based amine hardener Priamine 1071. | ||
FTIR analysis was used to confirm the final structure of crosslinked NIPUs (Fig. 9). It was found that during the polyaddition, the bis(cyclic carbonate)s 3 were quantitatively converted into NIPUs 16 as indicated by the almost complete disappearance of the typical carbonyl group vibration of 3 at about 1790 cm−1. The final conversion of the cyclic carbonate groups was estimated to be 94% (NIPU250), 100% (NIPU650) and 86% (NIPU1000). New peaks appeared at around 3300 cm−1 and 1700 cm−1 corresponding to the O–H (hydroxyl group)/N–H (urethane linkage) vibrations and the CO vibrations of the urethane linkages, respectively.36,37 The formation of urethane linkages was further confirmed by the presence of CO bending (1719 cm−1), N–H bending (1533 cm−1) and C–N stretching (1370 cm−1) bands, and the C–O asymmetric stretching of N–CO–O and C–O–C linkages at about 1245 cm−1.38 The strong absorption band at 1098 cm−1 is related to the ether (C–O–C) vibrations of the PO3G part of NIPUs. The bond assigned to the asymmetric stretching vibrations of N–CO–O and C–O–C of urethane groups were observed at 1245 cm−1. Moreover, the additional peak formation at around 1700 cm−1 for NIPU650 and the carbonyl peak splitting for NIPU1000 can be related to the formation of H-bonded ordered domains,39 as the NIPU structure provides a vast number of H-bonding groups, especially OH groups in the vicinity of urethane linkages.40
 | ||
| Fig. 9 The FTIR spectra of NIPU250, NIPU650 and NIPU1000 non-isocyanate polyurethanes synthesized by the ring-opening polyaddition of the DC250, DC650 and DC1000 bis(cyclic carbonate)s, respectively. | ||
Fig. 10 shows the temperature dependences of the storage (E′) and loss (E′′) moduli and the loss factor (tanδ) of the NIPUs. All NIPUs present a rubbery plateau at a temperature higher than 50 °C confirming their chemically crosslinked structure. The values of storage modulus in the rubbery region (E′R) are indirectly proportional to the average molar mass between crosslinks (Mc) corresponding to the network with the highest density in the case of NIPU250 (Table 2). These results agree well with the swelling degree of NIPUs (SR values in Table 2). While the NIPU1000 network swelled to a high degree due to the presence of long polyether chains between crosslinks, the NIPU250 sample swelled considerably less, confirming the formation of the densest chemically crosslinked network.41 Moreover, the high gel content (GC) of NIPU250 and NIPU650 provide evidence of optimal crosslinking in these materials.42,43 In contrast, NIPU1000 exhibits a low content of gel fraction, which indicates the formation of a large amount of “free species” – structures not chemically bonded to the polyurethane network.44
 | ||
| Fig. 10 The DMTA results of NIPU250, NIPU650 and NIPU1000 non-isocyanate polyurethanes synthesized by the ring-opening polyaddition of the DC250, DC650 and DC1000 bis(cyclic carbonate)s, respectively. | ||
| Sample | Dynamic-mechanical properties | Swelling properties | ||||
|---|---|---|---|---|---|---|
| T α [°C] | tanδmax |
E′R [MPa] | M c [g mol−1] | SR [%] | GC [%] | |
|
T
α is the main (alpha) transition temperature; tanδmax is maximum at tanδ curve; E′R is the storage modulus at the rubbery plateau at Tα + 100 °C; Mc is the molecular weight of the section between the crosslinks; SR is the swelling index; GC is the gel content. |
||||||
| NIPU250 | −4.4/24.2 | 0.73 | 8.26 | 1174 | 334 | 97 |
| NIPU650 | −8.6/13.5 | 0.78 | 3.05 | 3192 | 372 | 87 |
| NIPU1000 | −27.4 | 0.99 | 1.26 | 7737 | 85 | 59 |
During the synthesis of NIPU1000, free (macro)cyclic structures (not covalently incorporated into the polyurethane network) were probably formed as a consequence of the relatively high molecular weight of the DC1000 monomer.45
The tanδ curves of all NIPUs demonstrated a clear maximum (Fig. 10), which corresponds to the main transition temperature (Tα, Table 2). The main transition of the crosslinked NIPUs is connected to the free volume available for network segment relaxation.24,41
The decreasing length of poly(trimethylene oxide) chain in NIPUs shifts the Tα values to higher temperatures due to reduced mobility of the chain segments as a consequence of the increased crosslink density.27 In the case of NIPU250 and NIPU650, the second maximum on tanδ curves appears at 24.2 °C (NIPU250) and 13.5 °C (NIPU650), which suggests a partial (micro)phase separation as a consequence of low miscibility between the long non-polar aliphatic chains of the amine hardener and more polar short chains of the poly(trimethylene oxide) polyol.
Moreover, due to the presence of long pendant chains (fatty acid chains of the amine hardener), all NIPU materials exhibit a broad main transition region and high tanδmax values (Table 2), which demonstrates their high ability to absorb acoustic energy. Generally, tanδ > 0.3 over a broad temperature range is required for efficient vibration damping materials.46–48
As the last step, thermal degradation of NIPUs was investigated using TGA under a nitrogen atmosphere (Table 3 and Fig. S6 in the ESI†). All prepared NIPUs were thermally stable up to ca. 200 °C with the initial decomposition temperature (T5%) of 265, 267 and 286 °C for NIPU250, NIPU650 and NIPU1000, respectively. The thermal degradation of NIPU250 and NIPU650 was shown to proceed in two main steps, typical for these types of polyurethane materials.49 The first weight loss takes place between 250 and 360 °C (Tmax1 at 319 and 323 °C for NIPU250 and NIPU650, respectively) and corresponds to the degradation of the urethane linkages, while the second weight loss is attributed to the decomposition of soft segments – poly(trimethylene oxide) chains (Tmax2 around 460 °C).20,50 In contrast, NIPU1000 decomposes in one broad step with Tmax1 = 393 °C, which reflects the diversity of the decomposed structures24 which is in line with the phase-mixed and homogeneous morphology without segregation into hard/soft domains indicated by DMTA.
| Sample | T 5% [°C] | T 50% [°C] | T 90% [°C] | T max1 [°C] | T max2 [°C] | Solid residues at 650 °C [%] |
|---|---|---|---|---|---|---|
| T 5%, T50%, T90% is a temperature of 5%, 50% and 90% mass loss, respectively; Tmax1 and Tmax2 are the temperatures of the maximum rate of mass loss during the first and the second degradation step. | ||||||
| NIPU250 | 265 | 413 | 458 | 319 | 444 | 0.1 |
| NIPU650 | 267 | 383 | 456 | 323 | 444 | 1.4 |
| NIPU1000 | 286 | 385 | 443 | 393 | — | 2.0 |
The TGA results also clearly reveal that NIPU1000 exhibits the highest initial degradation temperature.51 Taking into account the best thermal stability of DC1000 among the synthesized bis(cyclic carbonate)s (Fig. S7 in the ESI†), the high purity of the synthesized DC1000 is most probably the key factor improving the thermal stability of the final NIPU1000.
Conclusions
In this work, we have explored a green route to non-isocyanate polyurethanes (NIPUs) using only bio-based monomers – tailor-made bis(cyclic carbonate)s crosslinked with the commercially available bio-based amine hardener. Five-membered cyclic carbonates with different molecular weights were first synthesized using a two-step procedure including epoxidation of renewable polyether polyols and subsequent CO2 chemical fixation. Detailed characterization of the synthesized intermediates (diglycidyl ethers) as well as the final bis(cyclic carbonate)s enabled the determination of the extent of side reactions as well as the yield of the main products and their molecular weights. It was found that the first epoxidation step is quite prone to side-reactions, especially to hydrolysis of the oxirane ring and incomplete dehydrochlorination resulting in the formation of chlorohydrine derivatives. On the other hand, the second step of CO2 cycloaddition was found to be highly efficient, leading to a full conversion of epoxy groups into cyclic carbonates. Consequently, the great potential for the use of CO2 was proved in polymer engineering. In order to establish the full application potential of synthesized bis(cyclic carbonate)s, NIPUs were obtained by the reaction between DC and diamine derivatives of dimerized fatty acids. The chemical structure of the synthesized NIPUs was verified by FTIR analysis. Furthermore, the degree of crosslinking (swelling index and gel content), thermal (TGA) and thermo-mechanical (DMTA) properties were determined. The characterization results verify this synthesis route to be satisfactory for obtaining polymer NIPUs with properties suitable for a wide range of applications. In particular, they seem eminently suitable for vibration damping applications.Funding
This work was supported by the National Science Centre, Poland [2018/29/N/ST8/02444] and the Czech Science Foundation [grant number 19-08549S]. Financial support from the University of the Basque Country (UPV/EHU) (GIU18/216 Research Group) is gratefully acknowledged. Tamara Calvo-Correas also thanks the UPV/EHU (ESPDOC19/41).Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
We would like to express our sincere thanks to Katarzyna Szwarc-Karabyka and Paweł Sowiński from the Gdansk University of Technology, Faculty of Chemistry, Nuclear Magnetic Resonance Laboratory, for determining the chemical structures of our products using NMR spectroscopy. The authors would like to express gratitude to Paulina Kosmela PhD from the Gdansk University of Technology, Faculty of Chemistry, Department of Polymers Technology, Poland, for DMTA and TGA analyses. The authors wish to thank Allessa (Germany) for kindly providing bio-based polyols (Sensatis® H250, Velvetol® H650, and Velvetol® H1000) samples used in this study. The authors are also grateful to Croda International (Netherlands) for supplying the diamine derivatives of dimerized fatty acid (Priamine®) samples.Notes and references
- J. Datta and M. Włoch, Polym. Bull., 2016, 73, 1459–1496 CrossRef CAS.
- G. Rokicki, P. G. Parzuchowski and M. Mazurek, Polym. Adv. Technol., 2015, 26, 707–761 CrossRef CAS.
- H. Blattmann, M. Fleischer, M. Bähr and R. Mülhaupt, Macromol. Rapid Commun., 2014, 35, 1238–1254 CrossRef CAS.
- A. Cornille, R. Auvergne, O. Figovsky, B. Boutevin and S. Caillol, Eur. Polym. J., 2017, 87, 535–552 CrossRef CAS.
- C. Calabrese, F. Giacalone and C. Aprile, Catalysts, 2019, 9, 1–30 CrossRef.
- N. Aoyagi, Y. Furusho and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1230–1242 CrossRef CAS.
- G. Liu, G. Wu, J. Chen and Z. Kong, Prog. Org. Coat., 2016, 101, 461–467 CrossRef CAS.
- H. Chang, Q. Li, X. Cui, H. Wang, Z. Bu, C. Qiao and T. Lin, J. CO2 Util., 2018, 24, 174–179 CrossRef CAS.
- K. Błażek and J. Datta, Crit. Rev. Environ. Sci. Technol., 2019, 49, 173–211 CrossRef.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC.
- F. Camara, S. Benyahya, V. Besse, G. Boutevin, R. Auvergne, B. Boutevin and S. Caillol, Eur. Polym. J., 2014, 55, 17–26 CrossRef CAS.
- V. Besse, R. Auvergne, S. Carlotti, G. Boutevin, B. Otazaghine, S. Caillol, J. P. Pascault and B. Boutevin, React. Funct. Polym., 2013, 73, 588–594 CrossRef CAS.
- S. Schmidt, N. E. Göppert, B. Bruchmann and R. Mülhaupt, Eur. Polym. J., 2017, 94, 136–142 CrossRef CAS.
- M. Fache, E. Darroman, V. Besse, R. Auvergne, S. Caillol and B. Boutevin, Green Chem., 2014, 16, 1987–1998 RSC.
- M. Fleischer, H. Blattmann and R. Mülhaupt, Green Chem., 2013, 15, 934–942 RSC.
- Z. Karami, M. J. Zohuriaan-Mehr and A. Rostami, J. CO2 Util., 2017, 18, 294–302 CrossRef CAS.
- G. Liu, G. Wu, J. Chen, S. Huo, C. Jin and Z. Kong, Polym. Degrad. Stab., 2015, 121, 247–252 CrossRef CAS.
- N. Esmaeili, M. J. Zohuriaan-Mehr, A. Salimi and M. Vafayan, Thermochim. Acta, 2018, 664, 64–72 CrossRef CAS.
- K. Błażek, J. Datta and A. Cichoracka, Polym. Int., 2019, 68, 1968–1979 CrossRef.
- J. Nanclares, Z. S. Petrovic, I. Javni, M. Ionescu and F. Jaramillo, J. Appl. Polym. Sci., 2015, 132, 7–14 CrossRef.
- Sigma-Aldrich, http://www.sigmaaldrich.com, (accessed 5 March 2020).
- H.-C. Kang, M. Lee, J. Yoon and M. Yoon, J. Am. Oil Chem. Soc., 2000, 78, 423–429 CrossRef.
- K. Błażek, P. Kasprzyk and J. Datta, Polymer, 2020, 205 DOI:10.1016/j.polymer.2020.122768.
- C. Carré, L. Bonnet and L. Avérous, RSC Adv., 2015, 5, 100390–100400 RSC.
- P. Jutrzenka Trzebiatowska, A. Dzierbicka, N. Kamińska and J. Datta, Polym. Int., 2018, 67, 1368–1377 CrossRef CAS.
- X. He, X. Xu, Q. Wan, G. Bo and Y. Yan, Polymers, 2019, 11, 1026 CrossRef.
- P. Parcheta, E. Głowińska and J. Datta, Eur. Polym. J., 2020, 123 DOI:10.1016/j.eurpolymj.2019.109422.
- J. Hong, D. Radojcic, M. Ionescu, Z. S. Petrovic and E. Eastwood, Polym. Chem., 2014, 5, 5360–5368 RSC.
- O. Kreye, H. Mutlu and M. A. R. Meier, Green Chem., 2013, 15, 1431–1455 RSC.
- S. E. Dechent, A. W. Kleij and G. A. Luinstra, Green Chem., 2020, 22, 969–978 RSC.
- V. Schimpf, J. B. Max, B. Stolz, B. Heck and R. Mülhaupt, Macromolecules, 2019, 52, 320–331 CrossRef CAS.
- J. Ke, X. Li, F. Wang, M. Kang, Y. Feng, Y. Zhao and J. Wang, J. CO2 Util., 2016, 16, 474–485 CrossRef CAS.
- Q. Chen, K. Gao, C. Peng, H. Xie, Z. K. Zhao and M. Bao, Green Chem., 2015, 17, 4546–4551 RSC.
- V. Caló, A. Nacci, A. Monopoli and A. Fanizzi, Org. Lett., 2002, 4, 2561–2563 CrossRef.
- C. J. Whiteoak, A. H. Henseler, C. Ayats, A. W. Kleij and M. A. Pericàs, Green Chem., 2014, 16, 1552–1559 RSC.
- M. Bähr, A. Bitto and R. Mülhaupt, Green Chem., 2012, 14, 1447–1454 RSC.
- M. Janvier, P. H. Ducrot and F. Allais, ACS Sustainable Chem. Eng., 2017, 5, 8648–8656 CrossRef CAS.
- O. Lamarzelle, G. Hibert, S. Lecommandoux, E. Grau and H. Cramail, Polym. Chem., 2017, 8, 3438–3447 RSC.
- A. Sukumaran Nair, S. Cherian, N. Balachandran, U. G. Panicker and S. K. Kalamblayil Sankaranarayanan, ACS Omega, 2019, 4, 13042–13051 CrossRef CAS.
- Y. Ecochard and S. Caillol, Eur. Polym. J., 2020, 137, 109915 CrossRef CAS.
- H. Beneš, A. Paruzel, J. Hodan and O. Trhlíková, J. Renewable Mater., 2018, 6, 697–706 Search PubMed.
- A. Cornille, J. Serres, G. Michaud, F. Simon, S. Fouquay, B. Boutevin and S. Caillol, Eur. Polym. J., 2016, 75, 175–189 CrossRef CAS.
- A. Cornille, S. Dworakowska, D. Bogdal, B. Boutevin and S. Caillol, Eur. Polym. J., 2015, 66, 129–138 CrossRef CAS.
- M. Decostanzi, Y. Ecochard and S. Caillol, Eur. Polym. J., 2018, 109, 1–7 CrossRef CAS.
- K. Dušek and M. Dušková-Smrčková, Macromol. React. Eng., 2012, 6, 426–445 CrossRef.
- K. Mizera, J. Ryszkowska, M. Kurańska and A. Prociak, Polym. Bull., 2020, 77, 823–846 CrossRef CAS.
- T. Wang, S. Chen, Q. Wang and X. Pei, Mater. Des., 2010, 31, 3810–3815 CrossRef CAS.
- S. M. Guillame, H. Khalil and M. Misra, J. Appl. Polym. Sci., 2017, 134, 45 CrossRef.
- P. Kasprzyk, E. Sadowska and J. Datta, J. Polym. Environ., 2019, 27, 2588–2599 CrossRef CAS.
- C. Carré, L. Bonnet and L. Avérous, RSC Adv., 2014, 4, 54018–54025 RSC.
- X. Sheng, G. Ren, Y. Qin, X. Chen, X. Wang and F. Wang, Green Chem., 2015, 17, 373–379 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01576h |
| This journal is © The Royal Society of Chemistry 2021 |