
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling the crystallinity and solubility of functional PCL with efficient post-polymerisation modification†
Cinzia
Clamor
a,
Beatrice N.
Cattoz
b,
Peter M.
Wright
b,
Rachel K.
O'Reilly
 *a and
Andrew P.
Dove
*a
*a and
Andrew P.
Dove
*a
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK. E-mail: r.oreilly@bham.ac.uk; a.dove@bham.ac.uk
bInfineum UK Ltd, Milton Hill, Abingdon OX13 6BB, UK
First published on 2nd March 2021
Abstract
Poly(ε-caprolactone) is a semi-crystalline biocompatible polymer with good mechanical properties. Its crystallinity also uniquely enables poly(ε-caprolactone) to be used in different applications, from the development of 3D scaffolds for tissue engineering to advanced nanoparticle design. However, the lack of side-chain functionality in the polymer backbone prevents further functionalisation, thereby limiting the potential to alter physico-chemical properties. Herein, we report the well-controlled ring-opening polymerisation (ROP) of ε-allyl caprolactone using Mg(BHT)2(THF)2 to furnish an allyl-functionalised PCL suitable for post-polymerisation modification. The isolated polymer could then be quantitatively post-functionalised via the efficient thiol–ene addition using a variety of commercially available alkyl thiols. The effect of alkyl chain length on bulk polymer properties was investigated, and demonstrates the potential to tune solubility and crystallinity of poly(ε-caprolactone) in a simple and efficient fashion. Lipophilic, functional polyesters that possess crystallinity are rare but this feature is crucial to exploit further applications of biocompatible polyesters such as for drug delivery or as a robust scaffold for tissue engineering.
Introduction
The use of poly(ε-caprolactone) (PCL) for biomedical applications has attracted increasing interest, owing to its excellent biocompatibility, controlled biodegradation and robust mechanical properties.1–5 In particular, its semi-crystalline nature enables PCL to be used in various applications ranging from the development of 3D scaffolds for tissue engineering to the design of nanoparticles of controlled shape and precisely defined dimensions.5–8 The preparation of PCL via ring-opening polymerisation (ROP) has been widely investigated using various catalytic systems such as organic,9–12 metal-based,13–17 and enzymatic catalysts,18–20 achieving polymers with controlled molecular weights and narrow dispersities.21 However, in order for PCL to increase applicability in material and pharmaceutical sciences, it is essential to achieve precise control over its physical–chemical properties in an accessible manner. While copolymerisation with other lactones or physical blending with other polymers can be used to tailor properties of PCL formulations,22,23 the lack of functional pendant groups within the polymer backbone precludes further functionalisation, limiting the potential to engineer novel materials in a specific and precise fashion.Post-polymerisation modifications represent an attractive strategy for the preparation of polymers bearing a diverse range of functionalities from a single monomer, enabling the introduction of functional moieties that are incompatible with ring-opening polymerisation conditions.24–26 In particular, the use of simple and robust thiol-based click chemistries has attracted considerable attention owing to their rapid reactivity, orthogonality and versatility of conjugation, allowing the incorporation of a wide range of substituents on the polymer backbone through reaction with terminal alkenes (thiol–ene)27–29 and terminal alkynes (thiol–yne).30,31 Moreover, the thiol–ene addition has been explored in materials chemistry for the preparation of elastomers,32 nanoparticles,33 and tissue engineering scaffolds34 exploiting the versatility of this chemistry to obtain different materials with tuneable properties. Notably, several studies have investigated the introduction of alkene functionalities on polyester backbones, however access to polyesters in which the semi-crystallinity of the native polyesters is retained is limited. For example, Hawker and co-workers reported the preparation of a series of alkene-functionalised polymers using thermal and photo-initiated thiol–ene chemistry, comparing the efficiency and orthogonality of thiol–ene additions on PCL using a wide range of thiols, but the thermal properties of the resultant structures were not thoroughly investigated.35 Heise and co-workers employed thiol–ene chemistry for the preparation of side-chain functional poly(macro)lactones, however functionalisation with primary alcohol and amide substituents disrupted the semi-crystalline structure of the native poly(macro)lactone, leading to amorphous materials.36
Herein, we report a facile method for the preparation of post-functionalised PCL to provide lipophilic polyesters, which are semi-crystalline for some formulations containing sufficiently long alkyl side chains. The synthesis of the lipophilic polyesters was obtained via ring-opening polymerisation of an allyl-functional caprolactone monomer which was subsequently functionalised via thiol–ene addition using a range of commercially available alkyl thiols. Polymer solubility and thermal properties were evaluated and the effect of the alkyl chain length on these properties was investigated. This study presents a straightforward method to tune lipophilicity and crystallinity of PCL by post-polymerisation functionalisation in a simple and efficient manner.
Experimental section
Materials
All chemicals and solvents, unless otherwise stated, were purchased from Sigma-Aldrich, Fisher Scientific and Alfa Aesar and used without further purification. Silica gel (pore size 40 Å) was obtained from Fisher Scientific and used as received. Deuterated benzene was ordered from Apollo Scientific, degassed by repeated freeze-pump-thawing and then dried under reflux over sodium and benzophenone before storing under an inert atmosphere of N2. Pentane was dried over CaH2 for two days, distilled under N2, degassed by freeze-pump-thawing (3 cycles) and stored in a N2-filled glovebox. Dry toluene and dichloromethane were deoxygenated with N2 and passed over an alumina column in a solvent tower delivery system. ε-Allyl-ε-caprolactone was synthesised as reported,37 dried over CaH2 for 24 h (and this was repeated 1× with fresh CaH2), distilled under reduced pressure and stored under an inert atmosphere. 4-Methoxybenzyl alcohol was dissolved in dry dichloromethane and dried over CaH2 for 24 h. Excess CaH2 was removed by cannula filtration and the dichloromethane was distilled off under reduced pressure before storing the dry alcohol under an inert atmosphere of N2. Diphenyl phosphate was dried under reduced pressure at 50 °C for 3 days. Irgacure 819 photoinitiator was obtained from BASF and stored in a light-free environment prior to use. Amberlyst A21 free base was washed repeatedly with methanol and dried under reduced pressure prior to use.General considerations
All polymerisations were performed under an inert nitrogen atmosphere in a glovebox unless otherwise stated. All other chemical manipulations were performed using standard Schlenk-line techniques. 1H NMR and 13C NMR spectra were recorded on a Bruker DPX-300, DPX-400 or HD500 spectrometer. Chemical shifts are reported in ppm. Solvent residual signals were used as reference (CDCl3, 1H: δ = 7.26 ppm for CHCl3, 13C: δ = 77.16 ppm for CDCl3; C6D6, 1H: δ = 7.16 ppm for C6D5H and 13C: δ = 128.06 ppm for C6D6). The signals are characterised as s = singlet, d = doublet, dd = doublet in doublet, ddd = doublet in doublet in doublet, t = triplet, q = quartet and m = multiplet. The coupling constant J is recorded in Hertz (Hz). Size exclusion chromatography (SEC) measurements were taken on an Agilent 1260 Infinity II Multi-Detector GPC/SEC System fitted with RI, ultraviolet (UV, λ = 309 nm), and viscometer detectors. The polymers were eluted through an Agilent guard column (PLGel 5 μM, 50 × 7.5 mm) and two Agilent mixed-C columns (PLGel 5 μM, 300 × 7.5 mm) using CHCl3 (buffered with 0.5% v/v NEt3) as the mobile phase (flow rate = 1 mL min−1, 40 °C). Number average molecular weights (Mn), weight average molecular weights (Mw) and dispersities (ĐM = Mw/Mn) were determined using Agilent GPC/SEC software (vA.02.01) against a 15-point calibration curve (Mp = 162–31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 87000 g mol−1) based on poly(styrene) standards (Easivial PS-M/H, Agilent). Molecular weights (Mn), weight average molecular weights (Mw) are given in g mol−1. Matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry (MALDI-ToF MS) analysis was performed on a Bruker Ultraflex Extreme mass spectrometer using a nitrogen laser delivering 2 ns pulses at 337 nm with positive ion ToF detection performed using an accelerating voltage of 25 kV. trans-2-[3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene] malononitrile (DCTB) was used as a matrix (40 g L−1 solution in THF), with sodium iodide (NaI) used as a cationic agent (1 g L−1 solution in THF). Analyte (1 g L−1 solution in THF) was mixed with DCTB and NaI solutions (20 μL of each) and 1 μL was applied to form a thin matrix-analyte film. All samples were calibrated against a 3000 g mol−1 poly(ethylene glycol) standard. The thermal characteristics of the polymers were determined using differential scanning calorimetry (DSC) on a STARe DSC3 system from Mettler Toledo and analysed in 40 μL aluminium pans from −100–200 °C at a heating rate of 10 °C min−1 for two heating/cooling cycles or 1 °C min−1 for one heating/cooling cycle. UV-Vis spectroscopy data was collected on an Evolution 350 UV-Vis spectrophotometer equipped with a Xenon Flash Lamp light source and Dual Matched Silicon Photodiodes detector. Quartz cells (170–2000 nm) from Hellma with two polished sides were used for examining the absorption spectral data by using Thermo INSIGHT-2 v.10.0.30319.1 software. A thermostat and 8-cell Peltier system with precise temperature control between 0 °C and 90 °C was coupled with an Evolution 350 UV-Vis spectrophotometer to record the temperature-dependent transmittance spectra of the sample. For all measurements, n-dodecane was used as the solvent.
87000 g mol−1) based on poly(styrene) standards (Easivial PS-M/H, Agilent). Molecular weights (Mn), weight average molecular weights (Mw) are given in g mol−1. Matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry (MALDI-ToF MS) analysis was performed on a Bruker Ultraflex Extreme mass spectrometer using a nitrogen laser delivering 2 ns pulses at 337 nm with positive ion ToF detection performed using an accelerating voltage of 25 kV. trans-2-[3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene] malononitrile (DCTB) was used as a matrix (40 g L−1 solution in THF), with sodium iodide (NaI) used as a cationic agent (1 g L−1 solution in THF). Analyte (1 g L−1 solution in THF) was mixed with DCTB and NaI solutions (20 μL of each) and 1 μL was applied to form a thin matrix-analyte film. All samples were calibrated against a 3000 g mol−1 poly(ethylene glycol) standard. The thermal characteristics of the polymers were determined using differential scanning calorimetry (DSC) on a STARe DSC3 system from Mettler Toledo and analysed in 40 μL aluminium pans from −100–200 °C at a heating rate of 10 °C min−1 for two heating/cooling cycles or 1 °C min−1 for one heating/cooling cycle. UV-Vis spectroscopy data was collected on an Evolution 350 UV-Vis spectrophotometer equipped with a Xenon Flash Lamp light source and Dual Matched Silicon Photodiodes detector. Quartz cells (170–2000 nm) from Hellma with two polished sides were used for examining the absorption spectral data by using Thermo INSIGHT-2 v.10.0.30319.1 software. A thermostat and 8-cell Peltier system with precise temperature control between 0 °C and 90 °C was coupled with an Evolution 350 UV-Vis spectrophotometer to record the temperature-dependent transmittance spectra of the sample. For all measurements, n-dodecane was used as the solvent.
General procedure for synthesis of ε-allyl-ε-caprolactone (εCL)
To a solution of 2-allylcyclohexan-1-one (5.00 g, 36.1 mmol) in dry CH2Cl2 (180 mL) was added NaHCO3 (4.50 g, 54.3 mmol) followed by m-CPBA (77%, 9.77 g, 43.6 mmol) at 0 °C. The resulting mixture was allowed to warm to room temperature and stirred for 48 h. The reaction was cooled to 0 °C, filtered over Celite and washed with 10% solution of sodium thiosulfate (3 × 100 mL), sodium hydrogen carbonate (3 × 100 mL), brine (3 × 100 mL) and dried over MgSO4. The mixture was concentrated in vacuo and the crude product was purified by column chromatography using hexane/ethyl acetate (gradient elution, 4:1 to 1:1) to yield the desired product (3.88 g, 25.2 mmol, 70%) as a light-yellow oil. Analytical data is in agreement with literature:371H NMR (400 MHz, CDCl3, 299 K, ppm): δ = 5.82 (ddt, J = 17.2, 10.3, 7.1 Hz, 1H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.24–5.00 (m, 2H, CHCH2), 4.27 (dt, J = 8.6, 6.2 Hz, 1H, (CO)OCH), 2.75–2.51 (m, 2H, CH2(CO)O), 2.46 (ddd, J = 14.1, 7.3, 5.8 Hz, 1H, CHCH2CHCH2), 2.31 (dt, J = 14.0, 6.8 Hz, 1H, CHCH′2CHCH2), 2.01–1.80 (m, 3H, CH2CH2CH2), 1.64–1.35 (m, 3H, CH′2CH′2CH′2); 13C NMR (101 MHz, CDCl3, 299 K, ppm) δ = 175.9 ((CO)O), 133.4 (CH2CHCH2), 118.2 (CH2CHCH2), 79.9 ((CO)OCH), 40.6 (CH2CHCH2), 34.9 (CH2(CO)OCH), 33.8 ((CO)OCHCH2), 28.3 (CHCH2CH2CH2), 23.0 (CHCH2CH2CH2).
CH2), 5.24–5.00 (m, 2H, CHCH2), 4.27 (dt, J = 8.6, 6.2 Hz, 1H, (CO)OCH), 2.75–2.51 (m, 2H, CH2(CO)O), 2.46 (ddd, J = 14.1, 7.3, 5.8 Hz, 1H, CHCH2CHCH2), 2.31 (dt, J = 14.0, 6.8 Hz, 1H, CHCH′2CHCH2), 2.01–1.80 (m, 3H, CH2CH2CH2), 1.64–1.35 (m, 3H, CH′2CH′2CH′2); 13C NMR (101 MHz, CDCl3, 299 K, ppm) δ = 175.9 ((CO)O), 133.4 (CH2CHCH2), 118.2 (CH2CHCH2), 79.9 ((CO)OCH), 40.6 (CH2CHCH2), 34.9 (CH2(CO)OCH), 33.8 ((CO)OCHCH2), 28.3 (CHCH2CH2CH2), 23.0 (CHCH2CH2CH2).
General procedure for synthesis of poly(ε-allyl-ε-caprolactone) (AεCL)
Using standard glovebox techniques, a solution of Mg(BHT)2(THF)2 (293 mg, 0.48 mmol, 0.5 mol%) and 4-methoxy benzyl alcohol (26.0 mg, 0.19 mmol, 0.02 eq.) in dry toluene (1 mL) was added to a solution of ε-allyl-ε-caprolactone (1.50 g, 9.73 mmol) in dry toluene (8.70 mL). After stirring for 4 h at 50 °C, the ampoule was removed from the glove box and the reaction was quenched by the addition of two drops trifluoroacetic acid (TFA). The solution was precipitated into cold n-hexane (3×) and dried under vacuum to afford the polymer as a light-yellow oil. 1H NMR and 13C NMR are in agreement with literature:381H NMR (300 MHz, CDCl3, 299 K, ppm): δ = 6.93–6.84 (m, 2H, CHaromatic), 5.72 (ddt, J = 16.4, 10.5, 7.1 Hz, 49H, CHCH2), 5.18–5.01 (m, 109H, CHCH2), 4.91 (p, J = 6.2 Hz, 49H, (CO)OCH), 3.81 (s, 3H, OCH3), 2.27 (m, 206H, CHCH2CHCH2, (CO)OCH2), 1.73–1.46 (m, 202H, CH2CH2CH2), 1.44–1.14 (m, 98H, CH2CH2CH2); 13C NMR (101 MHz, CDCl3, 299 K, ppm): δ = 172.1 (O(CO)),137.9 (CHCH2), 122.0 (CHCH2), 77.2 (OCHCH2), 42.9 (CH2CHCH2), 38.7 (O(CO)CH2), 37.5 (OCHCH2), 29.2 (O(CO)CH2CH2, O(CO)CH2CH2CH2); SEC (CHCl3): Mn = 12800, Mw = 15900, ĐM = 1.24.
General procedure for radical thiol–ene click reaction
Poly(ε-allyl-ε-caprolactone) (500 mg, 59.0 μmol) was dissolved in chloroform (4 mL) and thiol (9.55 mmol, 3 equiv. per olefin) was added, followed by Irgacure 819 (134 mg, 0.32 mmol, 0.1 equiv. per olefin). The solution was irradiated with a UV lamp (315–400 nm) at room temperature for 2–50 min, precipitated into cold MeOH (3×) and dried under vacuum to obtain the thiol-functionalised poly(ε-allyl-ε-caprolactone):Poly(ε-allyl-ε-caprolactone) functionalised with 2-ethyl hexanethiol: 1H NMR (300 MHz, CDCl3, 299 K, ppm): δ = 6.90–6.84 (m, 2H, CHaromatic), 5.02 (s, 2H, CHaromatic–CH2), 4.85 (p, J = 6.0 Hz, 60H, (CO)OCH), 3.79 (s, 3H, OCH3), 2.46 (dd, J = 7.7, 6.2 Hz, 263H, –CH2SCH2), 2.25 (t, J = 7.6 Hz, 133H, (CO)OCH2), 1.88–1.15 (m, 1596H, CH2), 0.87 (dt, J = 9.8, 6.7 Hz, 497H, CH3); 13C NMR (101 MHz, CDCl3, 299 K, ppm): δ = 173.0 (O(CO)), 73.1 (CHO), 39.4 (SCH2CH), 36.5 (CH2SCH2CH), 34.2 ((CO)CH2), 33.6 (CH2SCH2CH), 32.9 (CH2), 32.4 (CH2), 32.2 (CH2), 28.6 (CH2), 25.3 (CH2), 25.2 (CH2), 24.7 (CH2), 22.8 (CH2), 13.9 (CH3), 10.6 (CH3); SEC (CHCl3): Mn = 16400, Mw = 21300, ĐM = 1.30.
Poly(ε-allyl-ε-caprolactone) functionalised with 1-hexanethiol: 1H NMR (300 MHz, CDCl3, 299 K, ppm): δ = 6.82 (d, J = 8.7 Hz, 2H, CHaromatic), 4.97 (s, 2H, CHaromaticCH2), 4.87–4.70 (m, 55H, (CO)OCH), 3.74 (s, 3H, OCH3), 2.42 (td, J = 7.3, 4.9 Hz, 242H, CH2SCH2), 2.21 (t, J = 7.6 Hz, 125H, COOCH2), 1.67–1.37 (m, 550H, CH2), 1.40–1.11 (m, 361H, CH2), 0.88–0.72 (m, 144H, CH3); 13C NMR (101 MHz, CDCl3, 299 K, ppm): δ = 173.3 (O(CO)), 73.5 (CHO), 34.6 (CH2SCH2), 34.0 (CH2SCH2), 33.3 (CH2), 32.3 (CH2), 32.1 (CH2), 31.6 (CH2), 29.8 (CH2), 28.8 (CH2), 25.51 (CH2), 25.1 (CH2), 22.7 (CH2), 14.2 (CH3); SEC (CHCl3): Mn = 14700, Mw = 19500 ĐM = 1.33.
Poly(ε-allyl-ε-caprolactone) functionalised with 1-octanethiol: 1H NMR (300 MHz, CDCl3, 299 K, ppm): δ = 6.88 (d, J = 8.7 Hz, 2H, CHaromatic), 5.03 (s, 2H, CHaromatic–CH2), 4.87 (p, J = 6.7 Hz, 57H, (CO)OCH), 3.80 (s, 3H, OCH3), 2.57–2.42 (m, 244H, CH2SCH2), 2.27 (t, J = 7.6 Hz, 132H, (CO)OCH2), 1.77–1.45 (m, 638H, CH2), 1.44–1.16 (m, 795H, CH2), 0.94–0.80 (m, 200H, CH3); 13C NMR (101 MHz, CDCl3, 299 K, ppm): δ = 173.6 (O(CO)), 73.8 (CHO), 34.8 (CH2SCH2), 34.3 (CH2SCH2), 33.6 (CH2), 32.6 (CH2), 32.3 (CH2), 32.2 (CH2), 30.1 (CH2), 29.6 (CH2), 29.6 (CH2), 29.4 (CH2), 25.8 (CH2), 25.3 (CH2), 23.1 (CH2), 14.7 (CH3); SEC (CHCl3): Mn = 13800, Mw = 18500, ĐM = 1.34.
Poly(ε-allyl-ε-caprolactone) functionalised with 1-decanethiol: 1H NMR (300 MHz, CDCl3, 299 K, ppm): δ = 4.87 (p, J = 6.1 Hz, 59H, (CO)OCH), 3.81 (s, 3H, OCH3), 2.49 (td, J = 7.2, 5.2 Hz, 253H, CH2SCH2), 2.27 (t, J = 7.6 Hz, 131H, COOCH2), 1.74–1.45 (m, 718H, CH2), 1.27 (d, J = 4.9 Hz, 1207H, CH2), 0.96–0.78 (m, 210H, CH3); 13C NMR (101 MHz, CDCl3, 299 K, ppm): δ = 173.6 (O(CO)), 73.9 (CHO), 34.5 (CH2SCH2), 34.0 (CH2SCH2), 32.3 (CH2), 32.1 (CH2), 29.9 (CH2), 29.7 (CH2), 29.5 (CH2), 29.1 (CH2), 25.5 (CH2), 25.1 (CH2), 22.8 (CH3); SEC (CHCl3): Mn = 17 500, Mw = 123500, ĐM = 1.34.
Poly(ε-allyl-ε-caprolactone) functionalised with 1-dodecanethiol: 1H NMR (300 MHz, CDCl3, 299 K, ppm): δ = 5.03 (s, 2H, CHaromatic–CH2), 4.86 (p, J = 6.1 Hz, 53H, (CO)OCH), 3.80 (s, 3H, OCH3), 2.59–2.38 (m, 234H, CH2SCH2), 2.27 (t, J = 7.6 Hz, 125H, (CO)OCH2), 1.84–1.11 (m, 1970H, CH2), 1.00–0.76 (m, 266H, CH3); 13C NMR (101 MHz, CDCl3, 299 K, ppm): δ = 173.6 (O(CO)), 73.71 (CHO), 34.8 (CH2SCH2), 34.3 (CH2SCH2), 33.6 (CH2), 32.6 (CH2), 32.3 (CH2), 30.1 (CH2), 30.0 (CH2), 29.8 (CH2), 29.7 (CH2), 29.6 (CH2), 29.4 (CH2), 28.9 (CH2), 25.7 (CH2), 25.3 (CH2), 25.2 (CH2), 23.1 (CH2), 14.3 (CH3); SEC (CHCl3): Mn = 19 300, Mw = 24800, ĐM = 1.29.
Poly(ε-allyl-ε-caprolactone) functionalised with 1-hexadecanethiol: 1H NMR (300 MHz, CDCl3, 299 K, ppm): δ = 6.88 (d, J = 8.6 Hz, 2H, CHaromatic), 5.04 (s, 2H, CHaromatic–CH2), 4.88 (m, 56H, (CO)OCH), 3.81 (s, 3H, OCH3), 2.58–2.41 (m, 229H, CH2SCH2), 2.27 (t, J = 7.6 Hz, 143H, (CO)OCH2), 1.25 (s, 2900H, CH2), 0.98–0.75 (m, 205H, CH3); 13C NMR (101 MHz, CDCl3, 299 K, ppm): δ = 173.2 (O(CO)), 73.4 (CHO), 34.5 (CH2SCH2), 34.0 (CH2SCH2), 33.3 (CH2), 32.3 (CH2), 32.0 (CH2), 29.8 (CH2), 29.7 (CH2), 29.5 (CH2), 29.4 (CH2), 29.3 (CH2), 29.1 (CH2), 28.6 (CH2), 25.5 (CH2), 25.1 (CH2), 24.9 (CH2), 22.8 (CH2), 14.2 (CH3); SEC (CHCl3): Mn = 22500, Mw = 30150, ĐM = 1.34.
Results and discussion
Monomer synthesis and polymerisation
The allyl-functionalised lactone, ε-allyl-ε-caprolactone (AεPCL), was synthesised from the corresponding 2-allylcyclohexanone via Bayer-Villiger oxidation using meta-chloroperoxybenzoic acid (m-CPBA) as an oxidant (ESI, Fig. S1†). We initially screened the ROP of AεPCL using 4-methoxybenzyl alcohol as initiator ([M0]/[I0] = 50) and DPP as a catalyst (10 mol%), at 25 °C. DPP was chosen as a catalyst since it was also found to efficiently catalyse the ROP of CL and afford polymers with high molecular weight and narrow dispersities.12 The polymerisation was monitored by 1H NMR spectroscopy using the disappearance of the diagnostic OCOCH monomer resonance (δ = 4.2 ppm) and the appearance of OCOCH polymer resonance (δ = 4.9 ppm). Under these reaction conditions, the polymerisation was sluggish, reaching a monomer conversion of 70% after 330 h (Table 1, entry 1 & ESI, Fig. S2†). The kinetics of the polymerisation could be slightly improved by increasing the reaction temperature to 50 °C, affording a monomer conversion of 88% after 140 h (Table 2, entry 2). To further decrease the polymerisation time, we employed Mg(BHT)2(THF)2 as the catalyst, which has been previously reported to be highly active for the ROP of ε-caprolactone and other lactones.16,39,40 Similarly, Mg(BHT)2(THF)2 was found to be comparatively more active for the ROP of AεPCL, leading to 95% monomer conversion in just 8 h, however the dispersity of the final polymer was slightly higher than the DPP-catalysed reactions (Table 1, entry 3). Nevertheless, the polymerisation kinetics displayed a linear relationship between Mn and conversion and low dispersity, indicative of a well-controlled polymerisation (Fig. 1).
 | ||
| Fig. 1 Number-average molecular weight (Mn) and dispersity (ĐM = Mw/Mn) plotted against monomer conversion for the ROP of ε-allyl-ε-caprolactone using Mg(BHT)2(THF)2 and 4-methoxybenzyl alcohol as initiator ([M0]/[I0] = 50). Dispersity determined by SEC analysis in CHCl3 (0.5% NEt3), calibrated against polystyrene standards. | ||
| Entry | Catalyst (mol%) | Reaction temperature (°C) | Reaction time (h) | Monomer conversionb (%) | M n (kg mol−1) | Đ M |
|---|---|---|---|---|---|---|
| a Polymerisations were performed in benzene-d6 with [M0]/[I0] = 50 and [M]0 = 1 M, using 4-methoxybenzyl alcohol as initiator. b Determined by 1H NMR spectroscopy. c Determined by SEC analysis in CHCl3 (0.5% NEt3), calibrated against polystyrene standards. | ||||||
| 1 | DPP (10) | 25 | 330 | 70 | 11.9 | 1.07 |
| 2 | DPP (10) | 50 | 141 | 88 | 11.8 | 1.10 |
| 3 | Mg(BHT)2(THF)2 (5) | 50 | 8 | 90 | 12.8 | 1.24 |
| Entry | Alkyl thiol | Irradiation time (min) | M n theoretical (kg mol−1) | M n NMR (kg mol−1) | M n SEC (kg mol−1) | Đ M | T m (°C) | T c (°C) | ΔHme (J g−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Thiol–ene addition of poly(ε-allyl-ε-caprolactone). (i) Irgacure 819 (0.1 eq. per olefin), alkyl-thiol (3 equiv. per olefin), chloroform, UV light (λ = 315–400 nm), 22 °C. b Determined by integrating end-groups using 1H NMR spectroscopy (CDCl3, 298 K). c Determined by SEC analysis in CHCl3 (0.5% NEt3) as eluent and calibrated against polystyrene standards. d T m and Tc were determined by differential scanning calorimetry and was obtained from the 2nd heating cycle and cooling cycle, respectively. e Total enthalpy of melting (ΔHm) was calculated by integration of the first-order endothermic transition. | |||||||||
| 1 | 1-Hexanethiol | 50 | 15.1 | 15.1 | 14.7 | 1.33 | — | — | — |
| 2 | 1-Octanethiol | 15 | 16.6 | 17.2 | 13.8 | 1.34 | — | — | — |
| 3 | 2-Ethyl hexanethiol | 30 | 16.6 | 18.1 | 16.4 | 1.30 | — | — | — |
| 4 | 1-Decanethiol | 15 | 18.1 | 19.4 | 17.5 | 1.34 | −28 | −43 | −13.43 |
| 5 | 1-Dodecanethiol | 5 | 19.3 | 19.0 | 19.3 | 1.29 | 4 | −8 | −31.76 |
| 6 | 1-Hexadecanethiol | 2 | 22.8 | 23.2 | 22.5 | 1.34 | 35 | 29 | −47.11 |
Analysis of the polymer using 1H NMR spectroscopy confirmed the PCL backbone with pendant allyl moieties (δ = 5.72 and 5.18 ppm) and well-defined end-group signals (δ = 7.31, 6.93 and 3.81 ppm) attributed to the 4-methoxybenzyl alcohol initiator. These were used to calculate molecular weight, Mn NMR = 8.1 kg mol−1, which is close to the theoretical molecular weight, Mn theor = 7.9 kg mol−1 (ESI Fig. S3†). Size exclusion chromatography (SEC) analysis of the polymer showed a higher dispersity (ĐM = 1.24), compared to the DPP-catalysed reaction, and a larger than predicted molecular weight (Mn SEC = 12.8 kg mol−1), which can be explained by poor compositional correlation between AεPCL and the poly(styrene) standards (Fig. 2a).
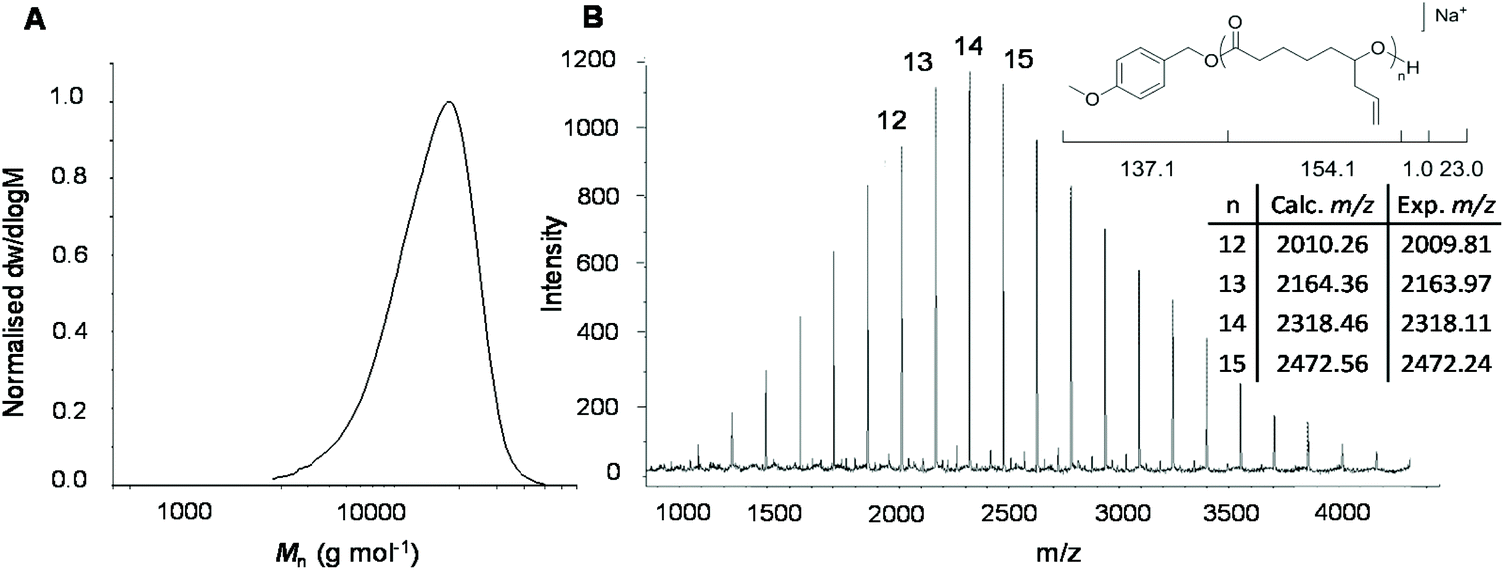 | ||
| Fig. 2 (a) Size exclusion chromatogram of the molecular weight distribution of AεPCL using 4-methoxybenzyl alcohol as initiator ([M0]/[I0] = 50). Molecular weight was determined against poly(styrene) standards using CHCl3 (0.5% NEt3) as eluent. (b) MALDI ToF-MS of AεPCL initiated by 4-methoxybenzyl alcohol. | ||
To corroborate the end-group fidelity that was observed in 1H NMR spectroscopic analysis, matrix-assisted laser ionisation time-of-flight mass spectrometry (MALDI-ToF MS) was performed on a short-chain AεPCL sample (Degree of Polymerisation (DP) = 14). The distribution plot shows a primary distribution with a spacing of m/z 154.16 that is in excellent agreement with the repeating monomer unit. The signal at m/z 2318 represents a DP 14 polymer chain carrying a sodium ion and the 4-methoxybenzyl alcohol end-group (Fig. 2b).
Post-polymerisation functionalisation of poly(ε-allyl-ε-caprolactone)
Post-polymerisation functionalisation of AεPCL was investigated via photoinitiated thiol–ene addition on the pendant allyl-functionality using a selection of commercially available alkyl thiols (Scheme 1, Table 2). To study the overall lipophilicity of the functionalised polyesters, thiols with a linear alkyl chain (6–16 carbon length) were primarily assessed. However, a branched thiol was also reacted with the polymer to compare the solubility and crystallinity to its linear analogue. After irradiation with UV light (λ = 315–400 nm), initially for 15 min, the polymer was precipitated into methanol to afford a functionalised PCL (Scheme 1, Table 2).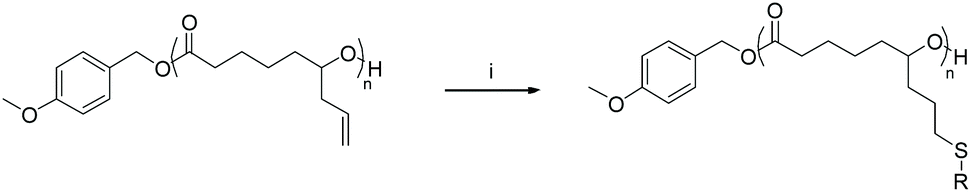 | ||
| Scheme 1 Synthesis of thiol-functionalised poly(ε-allyl-ε-caprolactone). Conditions: (i) IRGACURE 819, alkyl-thiol, chloroform, UV light (315–400 nm), RT. | ||
Analysis of the thiol–ene reactions by 1H NMR spectroscopy indicated if a quantitative reaction had occurred by monitoring the disappearance of the allyl proton signals (ESI Fig. S4–S9†). While the branched 2-ethyl hexanethiol required an additional 15 min irradiation (30 min in total), the hexanethiol required a total of 50 min irradiation for complete conversion. Interestingly, analysis of the AεPCL samples functionalised with 1-dodecane- and 1-hexadecanethiol observed an impurity around δ = 2.6 ppm, which was likely formed as a consequence of reactions between the allyl groups on the polymer backbone. We therefore sought to optimise reaction conditions for the thiol–ene addition of 1-hexadecanethiol. Changing the solvent from CHCl3 to THF or varying the thiol to alkene group ratio had little effect on the formation of reaction side products (ESI, Fig. S6–S9†). However, reducing the irradiation time (t = 2 min) significantly improved the purity of the functionalised polymer and still afforded complete conversion of AεPCL (Table 2, entry 6). The functionalisation was also confirmed by SEC analysis where we observed a noticeable shift to higher molecular weight for the post-functionalised AεPCLs compared to native polymer and without significant change in dispersity (Fig. 3a). Not surprisingly, the molecular weight of the functional polymers was positively correlated to the alkyl chain length of the thiol (for example, Mn SEC = 22.5 kg mol−1 for C16 and Mn SEC = 14.7 kg mol−1 for C6) (Table 2). 1-Dodecane thiol-functionalisation was also complete at a lower irradiation time of only 5 min. These activities are in line with the more electron-rich thiols being more active for radical addition.41
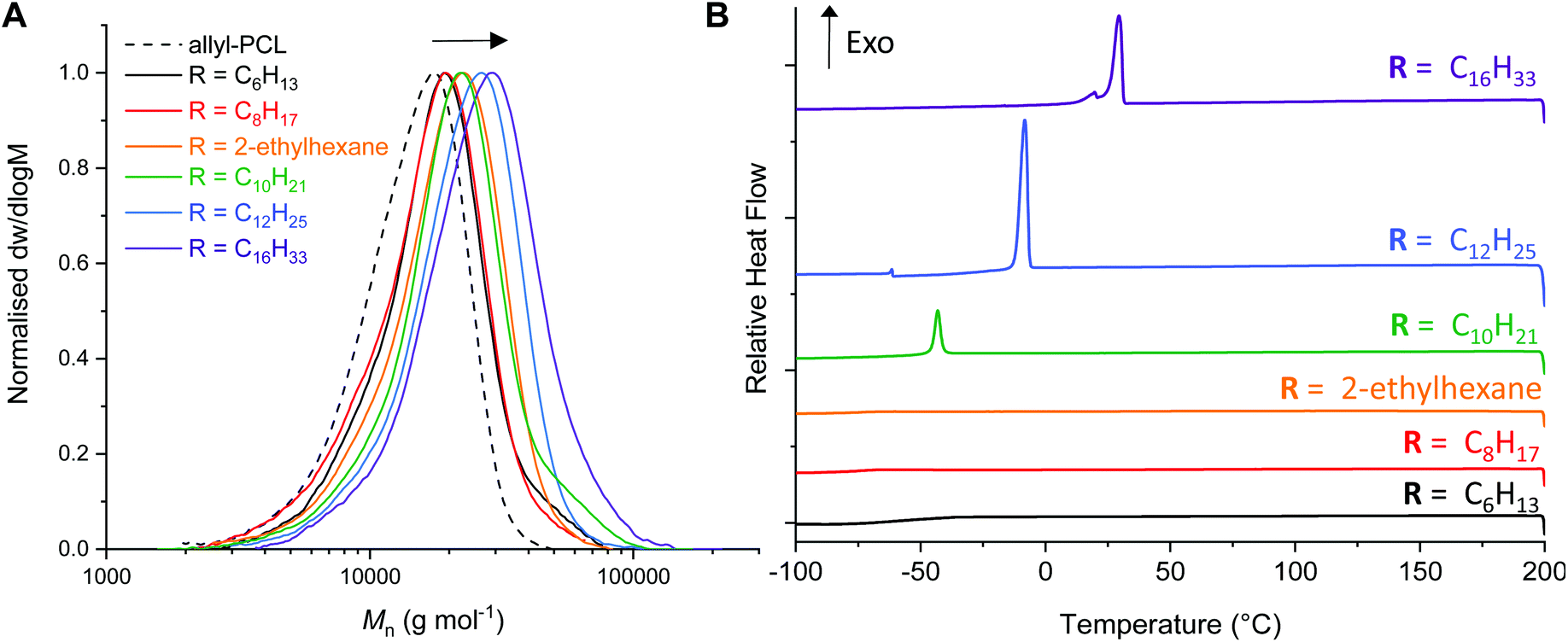 | ||
| Fig. 3 (a) Poly(ε-allyl-ε-caprolactone) functionalised with various thiols: 1-hexanethiol, 1-octanethiol, 2-ethyl hexanethiol, 1-decanethiol, 1-dodecanethiol and 1-hexadecanethiol, compared to poly(ε-allyl-ε-caprolactone). Size exclusion chromatogram with molecular weights determined against poly(styrene) standards using CHCl3 (0.5% NEt3) as eluent. (b) Differential scanning calorimetry (DSC) for 2nd cooling cycle (200 to −100 °C, 10 °C min−1) showing crystallisation of the polymer. | ||
Since the pendant-functionalised polyesters possess a crystallisable alkyl side chain, we investigated the thermal properties of the functional polyesters by differential scanning calorimetry (DSC) (Fig. 3b, ESI Fig. S10†). Even though the crystallinity is likely associated to the alkyl side chain, some of the bulk polymers were nonetheless semi-crystalline which is an atypical behaviour for any backbone functionalised PCL. While C6-, C8- and ethylhexanethiol functionalised PCL are completely amorphous, C10-, C12- and C16-PCL exhibited a sharp first-order melting/crystallisation transition. The melting temperature increased with alkyl chain length, from Tm of −28 °C to 35 °C for C10PCL and C16PCL, respectively (Table 2, entries 4–6 & ESI Fig. S10†). The same trend was observed for the crystallisation temperatures (Tc), increasing from −43 °C to 29 °C for C10PCL and C16PCL, respectively (Fig. 3b). Unsurprisingly, the crystallinity increased with alkyl chain length, albeit a minimum length of C10 carbon atoms was required to induce crystallinity. These findings are in agreement with previous reports, where cooperative organisation of alkyl pendant chains increase effective chain packing and overall order in the polymers to induce crystallinity.42,43 Being able to effectively tune polymer crystallisation by only varying the alkyl chain functionality demonstrates the possibility of customising the physical and mechanical properties of PCL, which is highly sought-after for a wide range of industrial applications.44
Polymer solubility is an important aspect in allowing control over self-assembly behaviour, polymer microstructure and material performance.45 While enhancing water solubility has been widely studied, increasing solubility in hydrophobic solvents is less well studied. Hence, we expected that the ability to quantitatively graft various alkyl thiols onto the PCL backbone would enable increased solubility in non-polar solvents compared to PCL. In order to study the effect of the alkyl chain length on the solubility behaviour, polymer solutions prepared in n-dodecane (1–122 mg mL−1) were analysed by UV-Vis spectroscopy using variable temperature sweeps (20–95 °C). A detection wavelength of 500 nm was chosen since the dissolved polymer does not show any absorption at this wavelength and hence a low transmittance would only indicate poor solubility (Fig. 4). The polyester functionalised with the shortest alkyl chain (C6) showed poor solubility under 50 °C at all concentrations (Fig. 4a). Increasing the alkyl chain length by two carbon atoms to C8 improved the solubility at low concentration, but solubility was still poor for 40 mg mL−1 and 100 mg mL−1, only becoming soluble near 50 °C (Fig. 4b). The branched version (ethylhexanethiol) showed similar behaviour to the linear C8 analogue with only a slightly improvement at 100 mg mL−1 which suggests that branching does not have a significant effect on solubility, which is surprising considering the greater disorder and poorer chain packing behaviour that would be expected as a consequence of the branching (Fig. 4c). The polyester functionalised with 1-decanethiol displayed full solubility at 1 mg mL−1 concentration, however it was only sparingly insoluble over the entire temperature range at higher concentrations (Fig. 4d). Such behaviour was unexpected, especially considering the good solubility of the C6 and C8 analogues at higher temperatures and as such we sought to probe this result further using dynamic light scattering (DLS). Initially, study of the 2-ethylhexane, C8 and C10 functionalised polymers in n-dodecane (at 40 mg mL−1) at both 20 and 90 °C revealed that while 2-ethylhexane- and C8-functionalised PCL solutions contain aggregates at 20 °C, these are greatly diminished at 90 °C (ESI, Fig. S11 and S12†). In contrast, the C10-functionalised PCL solution contained aggregates at both 20 and 90 °C (ESI, Fig. S13†) and furthermore displayed a strong Tyndall effect (ESI, Fig. S14†) – observations that were repeated on n-octane and n-decane (ESI, Fig. S15 & 16†). These observations are consistent with the UV-Vis solubility analyses and suggest that there is a delicate balance between solubility and aggregation behaviour (possibly due to favourable side chain/solvent interactions) that contribute to scattering and diminished transmittance.
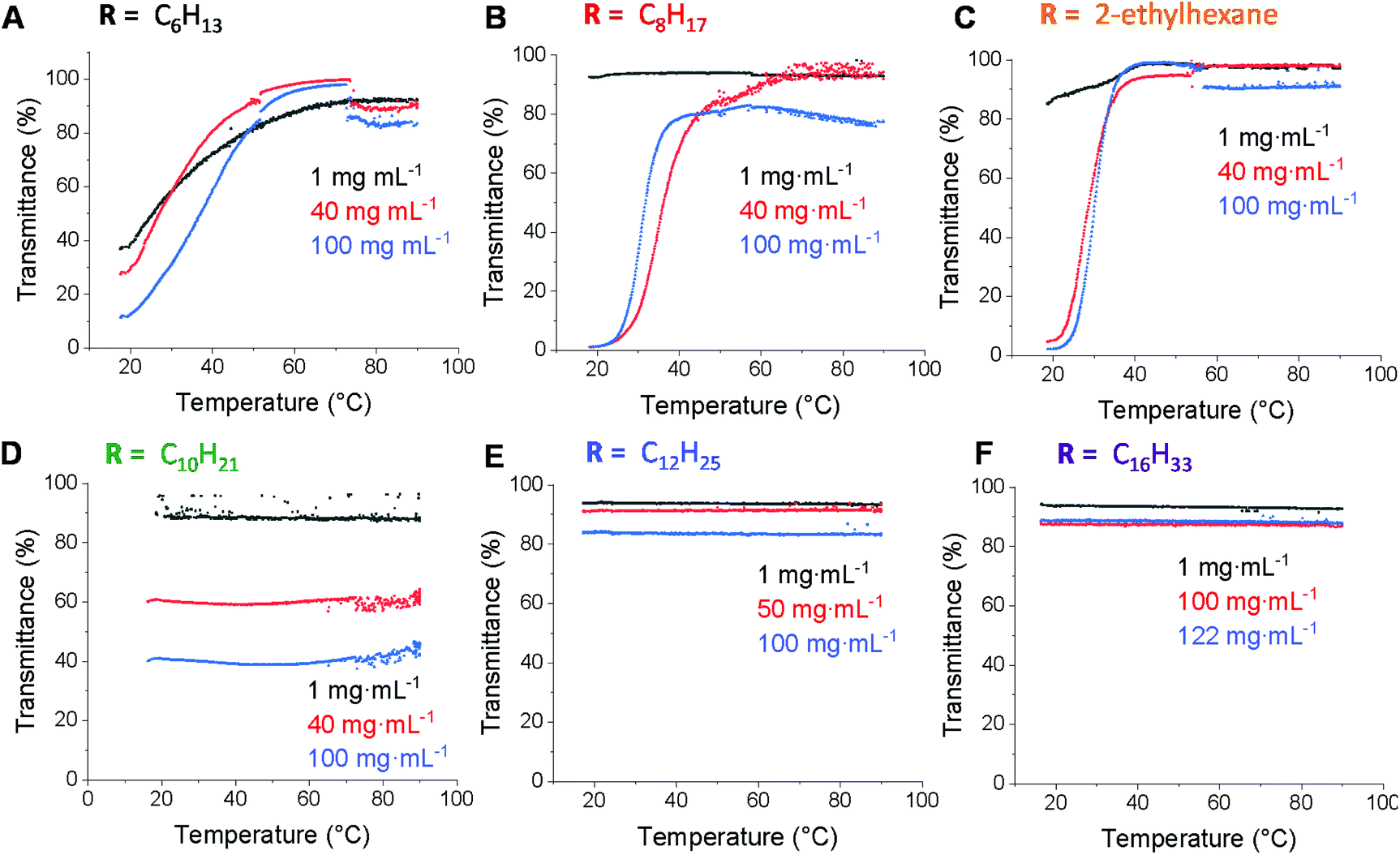 | ||
| Fig. 4 UV-Vis transmittance spectra as function of temperature and concentration in n-dodecane for poly(ε-allyl-ε-caprolactone) functionalised with (a) 1-hexanethiol, (b) 1-octanetiol, (c) 2-ethyl hexanethiol, (d) 1-decanethiol, (e) 1-dodecanethiol and (f) 1-hexadecanethiol (heating rate = 1 °C min−1 from 20–95 °C). | ||
For polymers with longer alkyl pendant chains (C12 and C16) good solubility in n-dodecane at all concentrations and temperatures was observed, as indicated by the high transmittance (Fig. 4e and f), most likely due to the enhanced solubility from increased alkyl chain length. This shows that functionalisation with alkyl substituents of different chain lengths can effectively tune the lipophilicity of PCL, with solubility in n-dodecane improving with alkyl chain length.
Conclusions
The ring-opening polymerisation of an allyl-functionalised ε-caprolactone to afford PCL with a pendant allyl group was explored using DPP and Mg(BHT)2(THF)2 as catalysts. The latter catalyst was more active and provided good control over molecular weight as indicated by the linear growth of the polymer chains and low dispersity. Subsequent post-polymerisation functionalisation of the pendant allyl motif by photo-initiated thiol–ene addition showed this to be an effective and facile method for the preparation of a range of alkyl functionalised polyesters with divergent physicochemical properties. As the alkyl chain length increased on the PCL backbone, we observed increasing solubility in n-dodecane and the polymers became semi-crystalline when the alkyl length reached 10 carbon atoms in length which is likely a consequence of effective chain interactions among the alkyl pendant groups. These findings demonstrate the potential to influence and tune the physical and thermal properties of PCL by simply varying the alkyl functionality, opening a new route for the development of novel materials in a simple and precise fashion. In particular, this affords a method to tailor polymer microstructure and solubility43 which should be important for exploring PCL in new nanostructures and (bio)materials applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C.C. acknowledges funding from Infineum UK Ltd.Notes and references
- R. A. Gross and B. Kalra, Science, 2002, 297, 803–807 CrossRef CAS.
- V. R. Sinha, K. Bansal, R. Kaushik, R. Kumria and A. Trehan, Int. J. Pharmacol., 2004, 278, 1–23 CrossRef CAS PubMed.
- D. R. Chen, J. Z. Bei and S. G. Wang, Polym. Degrad. Stab., 2000, 67, 455–459 CrossRef CAS.
- P. Joshi and G. Madras, Polym. Degrad. Stab., 2008, 93, 1901–1908 CrossRef CAS.
- D. W. Hutmacher, T. Schantz, I. Zein, K. W. Ng, S. H. Teoh and K. C. Tan, J. Biomed. Mater. Res., 2001, 55, 203–216 CrossRef CAS PubMed.
- M. C. Arno, M. Inam, Z. Coe, G. Cambridge, L. J. Macdougall, R. Keogh, A. P. Dove and R. K. O'Reilly, J. Am. Chem. Soc., 2017, 139, 16980–16985 CrossRef CAS.
- M. J. Jenkins, K. L. Harrison, M. M. C. G. Silva, M. J. Whitaker, K. M. Shakesheff and S. M. Howdle, Eur. Polym. J., 2006, 42, 3145–3151 CrossRef CAS.
- Y. Qian, Z. Yao, X. Wang, Y. Cheng, Z. Fang, W.-E. Yuan, C. Fan and Y. Ouyang, Cell Proliferation, 2020, 53, e12730 CrossRef.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS PubMed.
- P. V. Persson, J. Schröder, K. Wickholm, E. Hedenström and T. Iversen, Macromolecules, 2004, 37, 5889–5893 CrossRef CAS.
- K. Makiguchi, T. Satoh and T. Kakuchi, Macromolecules, 2011, 44, 1999–2005 CrossRef CAS.
- L. F. Sánchez-Barba, A. Garcés, M. Fajardo, C. Alonso-Moreno, J. Fernández-Baeza, A. Otero, A. Antiñolo, J. Tejeda, A. Lara-Sánchez and M. I. López-Solera, Organometallics, 2007, 26, 6403–6411 CrossRef.
- E. Martin, P. Dubois and R. Jérôme, Macromolecules, 2003, 36, 7094–7099 CrossRef CAS.
- A. Kowalski, J. Libiszowski, T. Biela, M. Cypryk, A. Duda and S. Penczek, Macromolecules, 2005, 38, 8170–8176 CrossRef CAS.
- H.-J. Fang, P.-S. Lai, J.-Y. Chen, S. C. N. Hsu, W.-D. Peng, S.-W. Ou, Y.-C. Lai, Y.-J. Chen, H. Chung, Y. Chen, T.-C. Huang, B.-S. Wu and H.-Y. Chen, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2697–2704 CrossRef CAS.
- X. Wang, A. Thevenon, J. L. Brosmer, I. Yu, S. I. Khan, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2014, 136, 11264–11267 CrossRef CAS.
- C. Bao, X. Xu, J. Chen and Q. Zhang, Polym. Chem., 2020, 11, 682–686 RSC.
- S. Kobayashi, H. Uyama and M. Ohmae, Bull. Chem. Soc. Jpn., 2001, 74, 613–635 CrossRef CAS.
- J. Engel, A. Cordellier, L. Huang and S. Kara, ChemCatChem, 2019, 11, 4983–4997 CrossRef CAS.
- O. Nuyken and D. S. Pask, Polymers, 2013, 5, 361–403 CrossRef.
- Q. Liu, S. Yuan, Y. Guo, A. Narayanan, C. Peng, S. Wang, T. Miyoshi and A. Joy, Polym. Chem., 2019, 10, 2579–2588 RSC.
- A. Sarasam and S. V. Madihally, Biomaterials, 2005, 26, 5500–5508 CrossRef CAS PubMed.
- R. J. Williams, I. A. Barker, R. K. O'Reilly and A. P. Dove, ACS Macro Lett., 2012, 1, 1285–1290 CrossRef CAS.
- A. W. Thomas and A. P. Dove, Macromol. Biosci., 2016, 16, 1762–1775 CrossRef CAS.
- S. Tempelaar, L. Mespouille, O. Coulembier, P. Dubois and A. P. Dove, Chem. Soc. Rev., 2013, 42, 1312–1336 RSC.
- A. W. Thomas, P. K. Kuroishi, M. M. Pérez-Madrigal, A. K. Whittaker and A. P. Dove, Polym. Chem., 2017, 8, 5082–5090 RSC.
- J. W. Chan, C. E. Hoyle, A. B. Lowe and M. Bowman, Macromolecules, 2010, 43, 6381–6388 CrossRef CAS.
- B. D. Fairbanks, D. M. Love and C. N. Bowman, Macromol. Chem. Phys., 2017, 218, 1700073 CrossRef.
- J. W. Chan, J. Shin, C. E. Hoyle, C. N. Bowman and A. B. Lowe, Macromolecules, 2010, 43, 4937–4942 CrossRef CAS.
- D. Konkolewicz, A. Gray-Weale and S. Perrier, J. Am. Chem. Soc., 2009, 131, 18075–18077 CrossRef CAS PubMed.
- B. Nottelet, G. Tambutet, Y. Bakkour and J. Coudane, Polym. Chem., 2012, 3, 2956–2963 RSC.
- J. Zou, C. C. Hew, E. Themistou, Y. Li, C.-K. Chen, P. Alexandridis and C. Cheng, Adv. Mater., 2011, 23, 4274–4277 CrossRef CAS PubMed.
- Y. Zou, L. Zhang, L. Yang, F. Zhu, M. Ding, F. Lin, Z. Wang and Y. Li, J. Controlled Release, 2018, 273, 160–179 CrossRef CAS PubMed.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063–7070 CrossRef CAS.
- Z. Ates, P. D. Thornton and A. Heise, Polym. Chem., 2011, 2, 309–312 RSC.
- A. Chen, J. Xu, W. Chiang and C. L. L. Chai, Tetrahedron, 2010, 66, 1489–1495 CrossRef CAS.
- D. Mecerreyes, R. D. Miller, J. L. Hedrick, C. Detrembleur and R. Jérôme, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 8700–8875 CrossRef.
- J. A. Wilson, S. A. Hopkins, P. M. Wright and A. P. Dove, Polym. Chem., 2014, 5, 2691–2694 RSC.
- J. A. Wilson, S. A. Hopkins, P. M. Wright and A. P. Dove, Macromolecules, 2015, 48, 950–958 CrossRef CAS.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- S. Zhou, Y. Zhao, Y. Cai, Y. Zhou, D. Wang, C. C. Han and D. Xu, Polymer, 2004, 45, 6261–6268 CrossRef CAS.
- S. Mete, K. G. Goswami, E. Ksendzov, S. V. Kostjuk and P. De, Polym. Chem., 2019, 10, 6588–6599 RSC.
- U. Kumar, T. Kato and J. M. J. Frechet, J. Am. Chem. Soc., 1992, 114, 6630–6639 CrossRef CAS.
- D. S. Boucher, J. Appl. Polym. Sci., 2020, 137, 48908 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional spectral data of the polymers prepared. See DOI: 10.1039/d0py01535k |
| This journal is © The Royal Society of Chemistry 2021 |