
Amphiphilic random and random block terpolymers with PEG, octadecyl, and oleyl pendants for controlled crystallization and microphase separation†
Sahori
Imai
a,
Yasuyuki
Ommura
a,
Yuki
Watanabe
b,
Hiroki
Ogawa
bc,
Mikihito
Takenaka
 bc,
Makoto
Ouchi
a and
Takaya
Terashima
*a
bc,
Makoto
Ouchi
a and
Takaya
Terashima
*a
aDepartment of Polymer Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: terashima.takaya.2e@kyoto-u.ac.jp
bInstitute for Chemical Research, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan
cRIKEN Spring-8 Center, Sayo-cho, Sayo-gun, Hyogo 679-5148, Japan
First published on 8th December 2020
Abstract
Controlled crystallization and microphase separation of multi-component copolymers are one possibility to construct precise nanostructures in functional polymer materials. In this paper, we report the crystallization and sub-10 nm microphase separation of amphiphilic random and random block terpolymers in the solid state. By using living radical copolymerization, we designed A/B/C random terpolymers and A/C–B/C random block terpolymers, into which a hydrophobic and crystalline octadecyl group (A), a hydrophobic and amorphous oleyl group (B), and hydrophilic poly(ethylene glycol) (C) were randomly and/or site-selectively introduced as side chains. The crystallization and melting temperatures of A/B/C random terpolymers gradually decreased with increasing content of amorphous oleyl units. The random terpolymers with a relatively small amount of oleyl units induced microphase separation of the side chains to form lamellae with hydrophilic and hydrophobic alternating layers in a domain spacing of about 6 nm, while the lamellar structure was gradually disordered by increasing oleyl groups. In contrast, A/C–B/C random block terpolymers efficiently induced crystallization of the octadecyl groups even in the presence of random copolymer segments with amorphous oleyl units. The random block copolymers opened the possibility of controlling microphase separation from the side chains of the random copolymers and the main chains of the block copolymers.
Introduction
Microphase separation and self-assembly of (co)polymers1–7 are key techniques to construct nanostructures in bulk polymers, thin films, and functional materials for applications including photolithography,8–13 polyelectrolytes and ion conducting materials,14–18 and solar cells.19 Controlling nanostructures to the desired domain size and morphologies is important to design polymer materials with targeted properties and performances. The microphase separation behaviour depends on the design of immiscible segments or associating groups and the primary structures of copolymers such as degree of polymerization (DP), composition, monomer sequence (e.g., block, random and alternating), and branching structures (e.g., star and graft).1–7,20 To create complex nano-scale morphologies, multi-component copolymers such as terpolymers21–31 are more effective than common AB diblock copolymers. By designing incompatible block chains with three different A, B, and C monomers, ABC triblock copolymers21–24 and ABC miktoarm star copolymers25–30 induce selective self-assembly to form finely controlled and complex morphologies in the solid state, e.g., core–shell gyroid,21 sphere-in-lamellae,25 and kaleidoscopic morphologies.27Microphase separation of copolymers is driven by not only self-assembly of polymer chains but also that of side chains. In addition to the use of high χ-low N block copolymers,6,32,33 self-assembly of polymer side chains is an alternative pathway to access sub-10 nm microphase separation structures.34–43 By incorporating highly segregated or selectively associating or crystalline units into side chains, random34–38 and alternating39 copolymers and brush (co)polymers40–43 induce self-assembly of their side chains to give ordered nanostructures in the solid state. Typically, amphiphilic random copolymers bearing hydrophilic poly(ethylene glycol) (PEG) chains and hydrophobic and crystalline octadecyl groups induce microphase separation of their side chains via the crystallization of the octadecyl groups, forming lamellae with a domain spacing of 5–6 nm (Fig. 1a).35 As a unique advantage of such pendant microphase separation, the domain spacing can be controlled just by the copolymer composition (i.e., weight fraction of octadecyl groups); it is independent of the molecular weight and molecular weight distribution of random copolymers. Microphase separation of the amphiphilic random copolymers involves crystallization of the octadecyl groups, while whether such pendant crystallization is essential is still an unsolved matter in designing random copolymers for microphase separation.
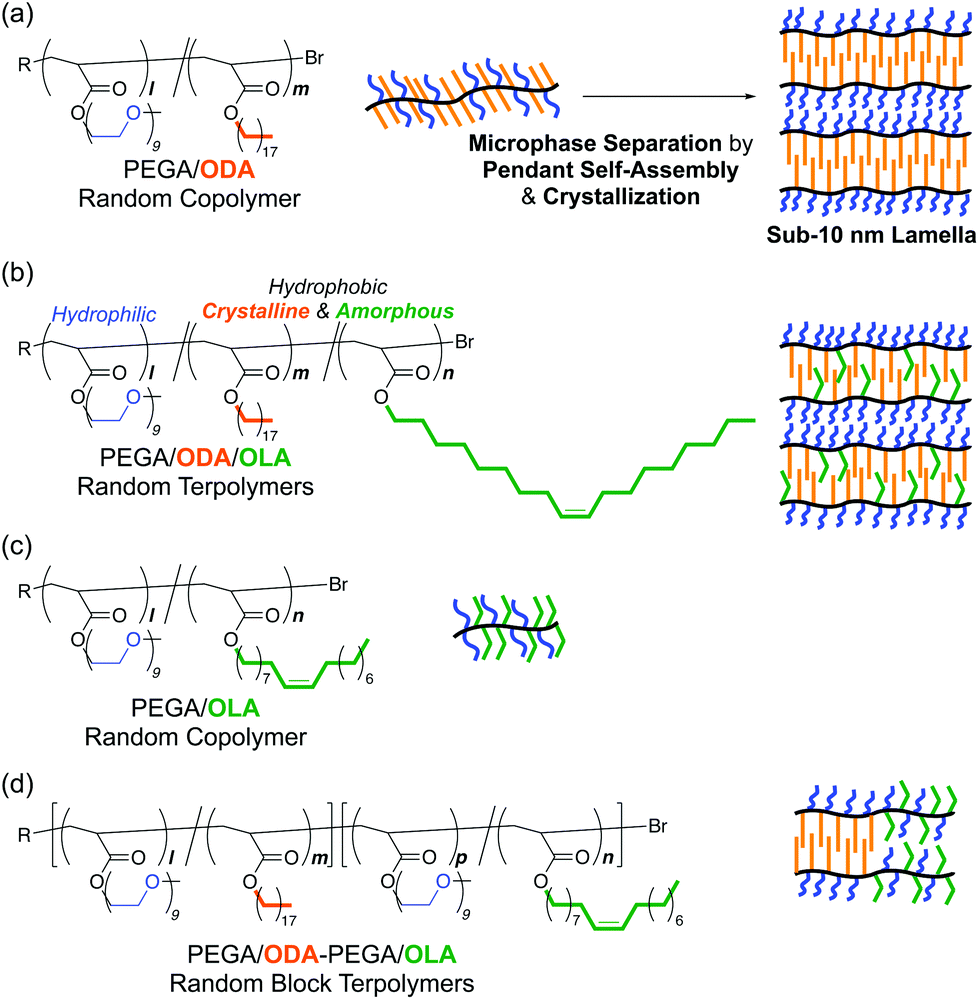 | ||
| Fig. 1 Design of (a) a PEGA/ODA random copolymer (P1), (b) PEGA/ODA/OLA random terpolymers (P2–P6), (c) a PEGA/OLA random copolymer (P7), and (d) PEGA/ODA-PEGA/OLA random block terpolymers (P9 and P10) for controlled crystallization and sub-10 nm microphase separation. | ||
The connection of distinct random copolymers, i.e., marriage of random copolymers and block copolymers, is also one possibility to build well-ordered morphologies in self-assembled materials.44 A/C–B/C random block copolymers potentially induce orthogonal self-assembly of the A/C random copolymer segment and the B/C counterpart. In fact, we found that A/C–B/C amphiphilic random block copolymers carrying distinct two hydrophobic groups (A: a dodecyl group, B: a benzyl group) and hydrophilic PEG side chains (C) formed double core polymers in water via the orthogonal folding of the A/C and B/C segments, respectively.45 These results suggest the possibility that A/C–B/C random block copolymers undergo double microphase separation in the solid state: one is triggered by the side chains of random copolymers and the other is driven by the main chains of block copolymers. The double microphase separation may further allow us to control the domain size of sub-10 nm microphase separation structures in dual directions of side chains and main chains.
Given these backgrounds, we herein report the crystallization and sub-10 nm microphase separation behavior of amphiphilic random or random block terpolymers in the solid state (Fig. 1). This research focused on (1) elucidating the effects of side chain crystallization on microphase separation and (2) controlling the sub-10 nm microphase separation and crystalline domains by tuning the sequence and composition of crystalline/amorphous hydrophobic units. For this, we designed A/B/C random terpolymers and A/C–B/C random block terpolymers, both of which bound a hydrophobic and crystalline octadecyl group (A), a hydrophobic and amorphous oleyl group (B), and hydrophilic PEG (C) as side chains. The amorphous oleyl group (B) consists of 18 carbons identical to the octadecyl group (A) and has the cis-structure of the internal olefin. The crystalline octadecyl (A) and amorphous oleyl (B) units are randomly incorporated into A/B/C random terpolymers, while these units are discretely introduced into the two blocks of A/C–B/C random block terpolymers. These terpolymers were synthesized by living radical copolymerization (Scheme 1).
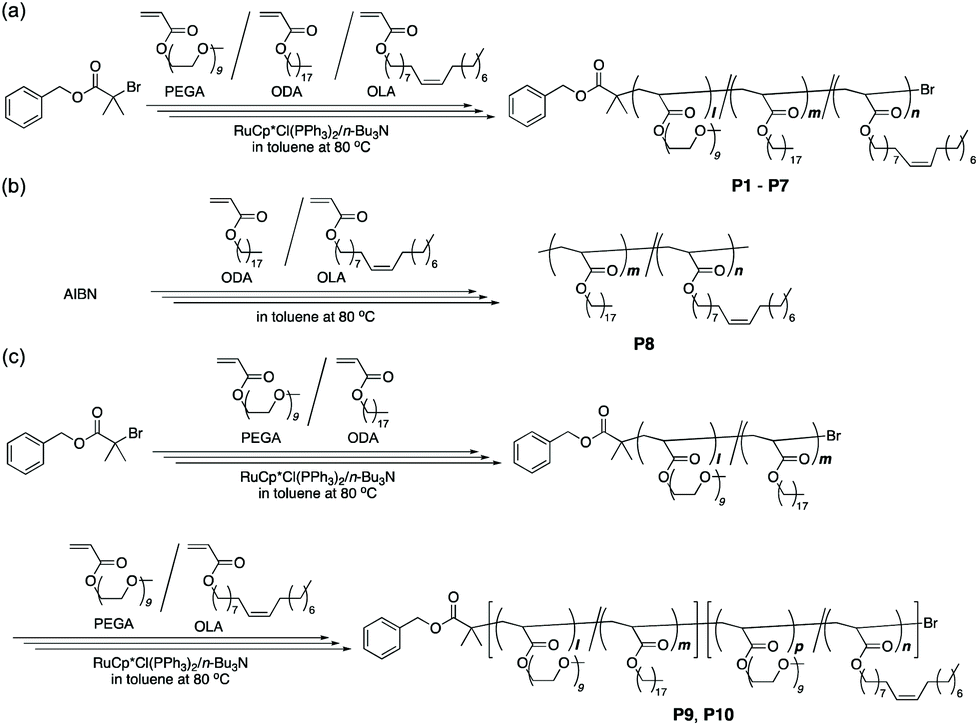 | ||
| Scheme 1 Synthesis of (a) PEGA/ODA/OLA random terpolymers and PEGA/ODA or OLA random copolymers (P1–P7), (b) an ODA/OLA random copolymer (P8), and (c) PEGA/ODA-PEGA/OLA random block terpolymers (P9 and P10) via living or free radical copolymerization. | ||
The crystallization and microphase separation of their terpolymers were examined by differential scanning calorimetry (DSC), X-ray diffractometry (XRD), and small-angle X-ray scattering (SAXS). The crystallinity of A/B/C random terpolymers depended on the content of oleyl groups. The terpolymers showed broad crystallization or melting peaks of the octadecyl groups in their DSC curves, compared with random copolymers bearing PEG and octadecyl groups (no oleyl groups). The crystallization and melting temperatures decreased with increasing content of oleyl groups. The random terpolymers containing a small amount of oleyl groups formed phase-separated lamellar structures with alternating layers of hydrophilic and hydrophobic side chains, while the lamellar structures were disordered by increasing the content of oleyl groups. This indicates that the crystallization of the octadecyl groups is essential for pendant microphase separation into 5–6 nm lamellae. In contrast, A/C–B/C random block terpolymers undergo efficient crystallization of the octadecyl groups even in the presence of random copolymer chains carrying amorphous oleyl groups. Interestingly, the oleyl groups in discrete block segments hardly disturb the crystallization of octadecyl groups. As a result, the random block terpolymers opened the possibility of controlling the microphase separation of the side chains and/or block polymer chains by tuning the length (degree of polymerization) of the block segments.
Results and discussion
Synthesis of amphiphilic random or random block terpolymers
Amphiphilic A/B/C random terpolymers (P2–P6) bearing a hydrophilic poly(ethylene glycol) (PEG) chain (C), a hydrophobic and crystalline octadecyl group (A), and a hydrophobic and amorphous oleyl group (B) were designed to investigate the effects of an amorphous oleyl group on the crystallization and microphase separation of the side chains. As control samples, amphiphilic random copolymers carrying hydrophilic PEG and either a hydrophobic octadecyl group or a hydrophobic oleyl group (P1 and P7) and a hydrophobic random copolymer bearing an octadecyl group and an oleyl group (P8) were prepared. To evaluate the effects of sequence distribution of octadecyl and oleyl groups on crystallization and microphase separation, amphiphilic A/C–B/C random block terpolymers comprising two random copolymers with different hydrophobic groups (P9 and P10) were designed: one block segment consists of a random copolymer with PEG and crystalline octadecyl pendants and the other comprises a random copolymer with PEG and amorphous octadecyl pendants.Amphiphilic random copolymers and terpolymers (P1–P7) were synthesized by ruthenium-catalyzed living radical copolymerization of PEG methyl ether acrylate (PEGA, Mn = 480, 9 average oxyethylene units), octadecyl acrylate (ODA), and oleyl acrylate (OLA) with a ruthenium catalyst [RuCp*Cl(PPh3)2/n-Bu3N] and a bromide initiator (benzyl 2-bromo-2-methyl-propanoate) in toluene at 80 °C (Scheme 1 and Table 1). The mole fraction of PEGA and hydrophobic monomers (ODA + OLA) was set to 40 mol% and 60 mol%, respectively. The ODA content was varied from 60 mol% to 0 mol% by changing the feed ratio of ODA and OLA, while keeping the total fraction of the hydrophobic monomers at 60 mol%.
| Polymer | Monomer | ODAb (mol%) | Time (h) | Conv.c (%) PEGA/ODA + OLA | M n (SEC) | M w/Mnd (SEC) | DPb | l/m/nobsdb |
|---|---|---|---|---|---|---|---|---|
| a P1–P7 were synthesized by ruthenium-catalyzed living radical copolymerization: [PEGA]0/[ODA]0/[OLA]0/[BzMA-Br]0/[RuCp*Cl(PPh3)2]0/[n-Bu3N]0 = 400/600/0/10/1/20 (P1), 400/550/50/10/1/40 (P2), 400/480/120/10/1/40 (P3), 400/420/180/10/1/40 (P4), 400/300/300/10/1/20 (P5), 400/180/420/10/1/40 (P6), and 400/0/600/10/1/20 (P7) mM in toluene at 80 °C. P8 was prepared by free radical copolymerization: [ODA]0/[OLA]0/[AIBN]0 = 300/300/10 mM in toluene at 80 °C. b ODA content (mol%), total degree of polymerization (DP), and l/m/n (DP of PEGA, ODA and OLA) of the polymers determined by 1H NMR. c Monomer conversion determined by 1H NMR with tetralin as an internal standard. d Determined by SEC in THF with PMMA standard calibration. | ||||||||
| P1 | PEGA/ODA | 60 | 47 | 73/74 | 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
1.26 | 100 | 40/60/0 |
| P2 | PEGA/ODA/OLA | 55 | 71 | 66/64 | 19100 |
1.35 | 78 | 31/43/4 |
| P3 | PEGA/ODA/OLA | 48 | 160 | 42/42 | 13900 |
1.19 | 50 | 20/24/6 |
| P4 | PEGA/ODA/OLA | 42 | 160 | 65/64 | 16600 |
1.28 | 79 | 32/33/14 |
| P5 | PEGA/ODA/OLA | 35 | 214 | 68/64 | 18100 |
1.60 | 78 | 31/27/20 |
| P6 | PEGA/ODA/OLA | 25 | 160 | 53/55 | 16900 |
1.27 | 51 | 20/13/18 |
| P7 | PEGA/OLA | 0 | 214 | 39/39 | 12500 |
1.36 | 37 | 14/0/23 |
| P8 | ODA/OLA | 56 | 5 | —/67 | 13600 |
1.48 | — | — |
In all the cases, PEGA and ODA and/or OLA were simultaneously consumed at the same speed, independent of the monomer feed ratio (Fig. S1†). Synchronized consumption of monomers supports the random sequence distribution of both hydrophilic and hydrophobic monomers in terpolymers and copolymers. Upon analysis by size-exclusion chromatography (SEC), the SEC curves of the products shifted to a high molecular weight with increasing monomer conversion (Fig. 2a–c and S1†). Controlled terpolymers and copolymers were obtained (P1–P7: Mn = 12500–21400, Mw/Mn = 1.19–1.60 by SEC in THF calibrated against PMMA standards). A hydrophobic random copolymer without hydrophilic PEG chains (P8) was prepared by free radical copolymerization of ODA and OLA with 2,2-azobis(isobutyronitrile) (AIBN) in toluene at 80 °C (Mn = 13600, Mw/Mn = 1.48).
 | ||
| Fig. 2 SEC curves of (a) P2, (b) P4, (c) P7, their intermediates obtained at earlier conversion (dashed lines, PEGA/ODA + OLA conversion = (a) 16%/16%, (b) 20%/21%, and (c) 12%/12%), and (d) P9 and P9-1st (macroinitiator, dashed line). | ||
P1–P7 were analyzed by 1H nuclear magnetic resonance (NMR) spectroscopy. As shown in Fig. 3a, P4 indicated the proton signals of PEG chains (c: 4.4–4.2 ppm, d: 3.8–3.4 ppm, and e: 3.4–3.3 ppm) and octadecyl and/or oleyl groups (f, j: 4.2–3.8 ppm, g, k: 1.8–1.6 ppm, h, l: 1.5–1.2 ppm, m: 2.1–2.0 ppm, n: 5.5–5.3 ppm, and j, o: 1.0–0.8 ppm), in addition to those of polyacrylate backbones (a, b) and the initiator benzyl ester fragment (p: 5.2–5.0 ppm, Ph group: 7.5–7.3 ppm). In P7, the area ratio of the oleyl olefin protons (n) agreed with that of the other methylene and methyl protons (j, k, o, etc.), meaning that the pendant olefin is intact during copolymerization (Fig. 3b). The direct introduction of the oleyl groups into copolymer side chains without side reactions and gelation was also confirmed by free radical copolymerization of ODA and OLA and 1H NMR analysis of the resulting P8 (Fig. 3c). The total degree of polymerization (DP) and composition (PEGA/ODA/OLA = l/m/n) of P1–P7 were determined by 1H NMR from the area ratio of the respective side chain units to the initiator fragment (Table 1). The l/m/n values were close to those calculated from the feed ratio of the monomers to the initiator and their monomer conversions.
 | ||
| Fig. 3 1H NMR spectra of (a) P4, (b) P7, (c) P8, and (d) P9 in CDCl3 at 25 °C. | ||
Amphiphilic random block terpolymers (P9 and P10) were prepared by ruthenium-catalyzed living radical copolymerization of PEGA and OLA with bromine-capped PEGA/ODA random copolymers and a ruthenium catalyst (Scheme 1c). Here, two PEGA/ODA random copolymers with different DPs were employed as macroinitiators to control the length of the crystalline 1st block segments against that of amorphous 2nd block segments. Well-controlled PEGA/ODA random copolymers were obtained from ruthenium-catalyzed living radical copolymerization of PEGA and ODA with benzyl 2-bromo-2-methyl-propanoate as an initiator (P9-1st: Mn = 13100, Mw/Mn = 1.13, DP = 37, P10-1st: Mn = 19900, Mw/Mn = 1.18, DP = 79, Table 2). Then, the random copolymers were applied to copolymerization of PEGA and OLA with RuCp*Cl(PPh3)2/n-Bu3N in toluene at 80 °C, resulting in well-controlled random block terpolymers (P9: Mn = 24500, Mw/Mn = 1.35, P10: Mn = 29000, Mw/Mn = 1.39, Table 2, Fig. 2d and S1†). The DP and composition (PEGA/ODA-PEGA/OLA = l/m–p/n) of P9 and P10 were determined by 1H NMR (Table 2 and Fig. 3d).
| Polymer | Monomer | ODA/OLAb (mol%) | Time (h) | Conv.c (%) PEGA/ODA or OLA | M n (SEC) | M w/Mnd (SEC) | DPb | l/m–p/nobsdb |
|---|---|---|---|---|---|---|---|---|
| a Random block copolymers were synthesized by ruthenium-catalyzed living radical copolymerization. P9-1st: [PEGA]0/[ODA]0/[BzMA-Br]0/[RuCp*Cl(PPh3)2]0/[n-Bu3N]0 = 400/600/10/1/40 mM in toluene at 80 °C. P9: [PEGA]0/[OLA]0/[P9-1st]0/[RuCp*Cl(PPh3)2]0/[n-Bu3N]0 = 560/840/10/1/40 mM in toluene at 80 °C. P10-1st: [PEGA]0/[ODA]0/[BzMA-Br]0/[RuCp*Cl(PPh3)2]0/[n-Bu3N]0 = 640/960/10/1/20 mM in toluene at 80 °C. P10: [PEGA]0/[OLA]0/[P10-1st]0/[RuCp*Cl(PPh3)2]0/[n-Bu3N]0 = 560/840/10/1/20 mM in toluene at 80 °C. b Hydrophobic monomer content (mol%) in the respective block segments, total degree of polymerization (DP), and l/m–p/n (DP of PEGA/ODA-PEGA/OLA) of the random (block) copolymers determined by 1H NMR. c Monomer conversion determined by 1H NMR with tetralin as an internal standard. d Determined by SEC in THF with PMMA standard calibration. | ||||||||
| P9-1st | PEGA/ODA | 59/0 | 7 | 26/27 | 13100 |
1.13 | 37 | 15/22 |
| P9 | PEGA/OLA | 27/32 | 79 | 23/28 | 24500 |
1.35 | 82 | 15/22–19/26 |
| P10-1st | PEGA/ODA | 59/0 | 7 | 43/43 | 19900 |
1.18 | 79 | 32/47 |
| P10 | PEGA/OLA | 43/17 | 45 | 28/27 | 29000 |
1.39 | 109 | 32/47–12/18 |
Crystallization and microphase separation of random terpolymers
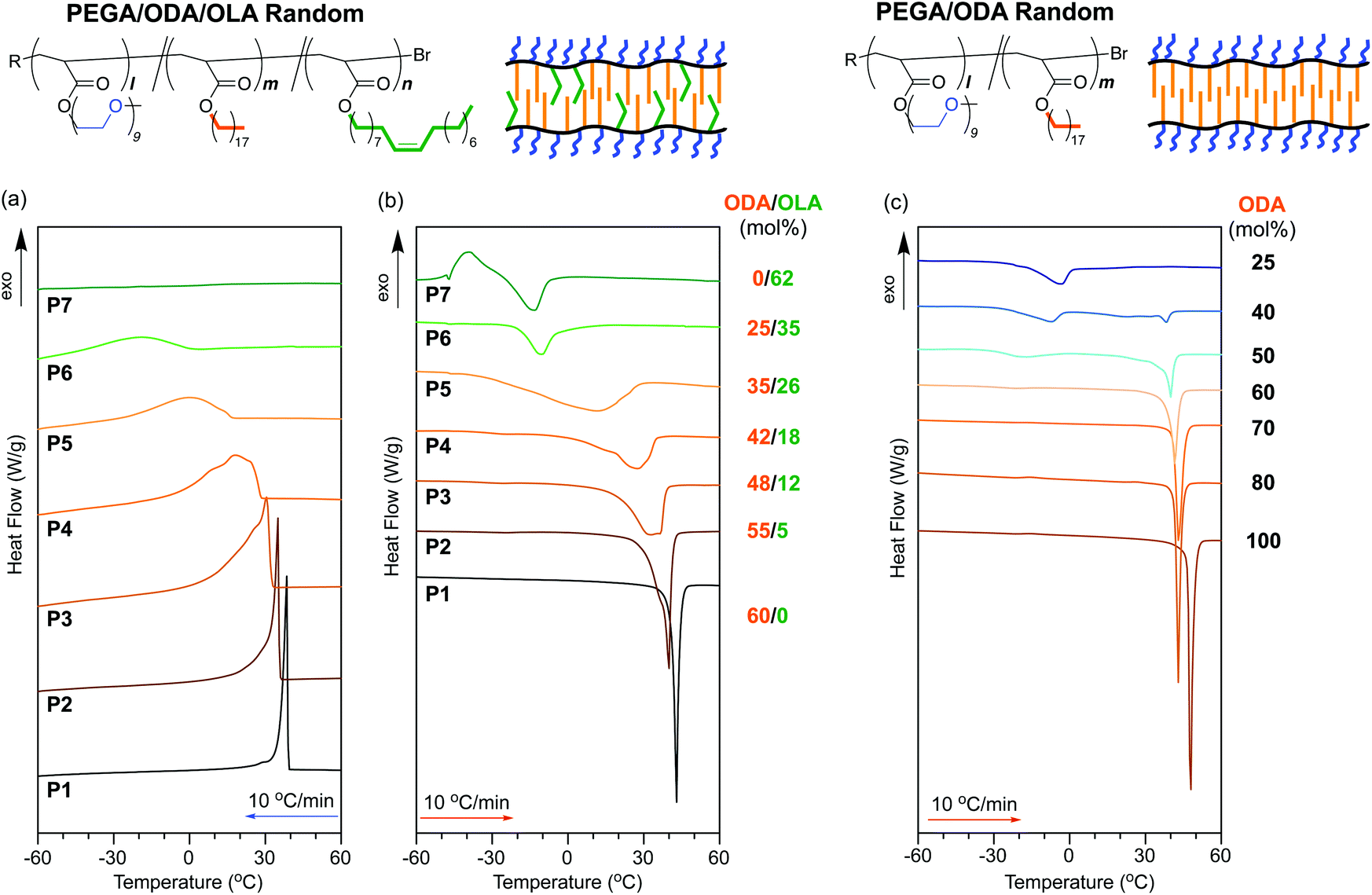 | ||
| Fig. 4 DSC curves recorded for (a) the first cooling scans and (b and c) second heating scans of (a and b) PEGA/ODA/OLA random copolymers (P1–P7: ODA + OLA = 60 mol%) and (c) PEGA/ODA random copolymers (ODA = 25–100%)35 between −80 °C and 150 °C. The cooling and heating rates were −10 °C min−1 and 10 °C min−1, respectively. | ||
A PEGA/ODA random copolymer (P1) sharply showed an exothermic signal originating from the crystallization of the octadecyl groups in the cooling process (crystallization temperature: Tc = 38.5 °C) and an endothermic signal stemming from the melting of the octadecyl groups in the heating process (melting temperature: Tm = 43.0 °C) (Table 3). In contrast, a PEGA/ODA/OLA (40/42/18 mol%) random terpolymer (P4) indicates the broad exothermic and endothermic signals of the octadecyl groups in the cooling and heating processes, respectively (Fig. 4a and b). Both Tc and Tm of P4 were lower than those of P1; the enthalpy of crystallization and melting for P4 was also smaller than the corresponding enthalpy for P1 (Table 3). Similarly, broad exothermic and endothermic signals with lower Tc and Tm were observed in an ODA/OLA random copolymer (P8) (Fig. S2 and S3†). These results importantly suggest that oleyl groups incorporated into P4 affect the crystalline domains of the octadecyl groups in the solid state.
| Entry | Polymer | Monomer | ODA (mol%) | T c (°C) | ΔHca (J g−1) | T m (°C) | ΔHma (J g−1) |
|---|---|---|---|---|---|---|---|
| a The samples of entries 1–11 were analyzed by DSC between the temperature range of −80 °C and 150 °C. The cooling and heating rates were 10 °C min−1 and −10 °C min−1, respectively. The crystallization temperature (Tc) and the enthalpy of crystallization (ΔHc) were obtained from the first cooling scans of the samples from 150 °C. The melting temperature (Tm) and the enthalpy of melting (ΔHm) were obtained from the second heating scans of the samples from −80 °C. b The crystallization temperature (Tc) and the enthalpy of crystallization (ΔHc) were obtained from the second heating scans of the samples from −80 °C. | |||||||
| 1 | P1 | PEGA/ODA | 60 | 38.5 | 52.2 | 43.0 | 52.2 |
| 2 | P2 | PEGA/ODA/OLA | 55 | 35.0 | 50.5 | 39.9 | 50.3 |
| 3 | P3 | PEGA/ODA/OLA | 48 | 30.5 | 44.5 | 32.9 | 43.6 |
| 4 | P4 | PEGA/ODA/OLA | 42 | 18.6 | 34.2 | 27.4 | 34.2 |
| 5 | P5 | PEGA/ODA/OLA | 35 | −0.6 | 27.9 | 11.0 | 27.3 |
| 6 | P6 | PEGA/ODA/OLA | 25 | −21.4 | 16.4 | −10.3 | 16.4 |
| 7 | P7 | PEGA/OLA | 0 | −39.1b | 18.4b | −13.2 | 18.4 |
| 8 | P8 | ODA/OLA | 56 | 13.6 | 35.3 | 23.9 | 35.1 |
| 9 | P9 | PEGA/ODA-PEGA/OLA | 27 | −33.9b | 3.8b | −16.3 | 3.3 |
| 35.6 | 23.0 | 40.7 | 22.8 | ||||
| 10 | P10 | PEGA/ODA-PEGA/OLA | 43 | 38.2 | 37.3 | 42.7 | 37.9 |
| 11 | P1 + P7 | PEGA/ODA + PEGA/OLA | 60 | −37.4b | 9.1b | −12.2 | 9.2 |
| 35.4 | 27.1 | 44.0 | 25.1 | ||||
To investigate the crystallization dependent on oleyl groups in detail, random terpolymers with different ODA/OLA contents (ODA/OLA = 55/5–25/35 mol%, P2–P6) were evaluated. All the samples showed both exothermic and endothermic signals in cooling and heating processes, while the peak broadness, Tc, Tm, and enthalpy of crystallization and melting were dependent on the ODA/OLA ratio (Fig. 4a and b). Both exothermic and endothermic signals gradually broadened with increasing amorphous OLA units. Tc, Tm, and enthalpy of crystallization and melting decreased with increasing OLA units. P5 with 35 mol% ODA exhibited a quite broad endothermic peak from −30 to 30 °C (Tm = 11.0 °C). P6 with 25 mol% ODA, as well as a PEGA/OLA random copolymer (P7), exhibited exothermic and endothermic signals at temperatures lower than 0 °C (Table 3). The crystallization and melting peaks would be derived from PEG side chains and/or trace water included in the samples (e.g., intermediate water).46
In contrast, PEGA/ODA random copolymers with different ODA contents (40–80 mol%) maintained the sharp endothermic signals originating from the melting of the octadecyl groups (Fig. 4c).35Tm of the random copolymers did not so decrease with decreasing ODA mole fraction, different from that of P2–P5 with oleyl groups (Fig. 5a). These results suggest that the broadness of exothermic or endothermic signals and decreasing Tc and Tm in P2–P5 are attributed to the decreasing size of crystalline domains by the sequence distribution of octadecyl units and oleyl units. Since a PEGA/ODA random copolymer with 25 mol% ODA did not show a melting peak of the octadecyl groups, random copolymers and terpolymers containing octadecyl units less than 25 mol% hardly induce crystallization of the octadecyl groups.
 | ||
| Fig. 5 (a) Melting temperatures and (b) enthalpy and (c) entropy of melting of octadecyl groups in PEGA/ODA/OLA random terpolymers (red squares: P2, P3, and P4) and PEGA/ODA random copolymers (black circles,35 black open square: P1) as a function of the ODA content (mol%). | ||
The enthalpy of melting for P2–P4 and PEGA/ODA random copolymers (with over 40 mol% ODA) was linearly proportional to the ODA content (Fig. 5b), namely, the melting enthalpy normalized by the ODA content was constant. This suggests that the octadecyl groups in P2–P4 have crystalline structures similar to those in PEGA/ODA random copolymers. The entropy of melting for P2–P4 was also calculated with the following: ΔSm = ΔHm/Tm. The entropy values for P2–P4 were almost identical, although those for PEGA/ODA random copolymers decreased with decreasing ODA content (Fig. 5c). The entropy of melting is generally related to volume and conformation changes of polymers through phase transition from a crystalline state to a melting state. Thus, this result implies that P2–P4 with 60 mol% hydrophobic content form similar self-assembled nanostructures in the solid state.
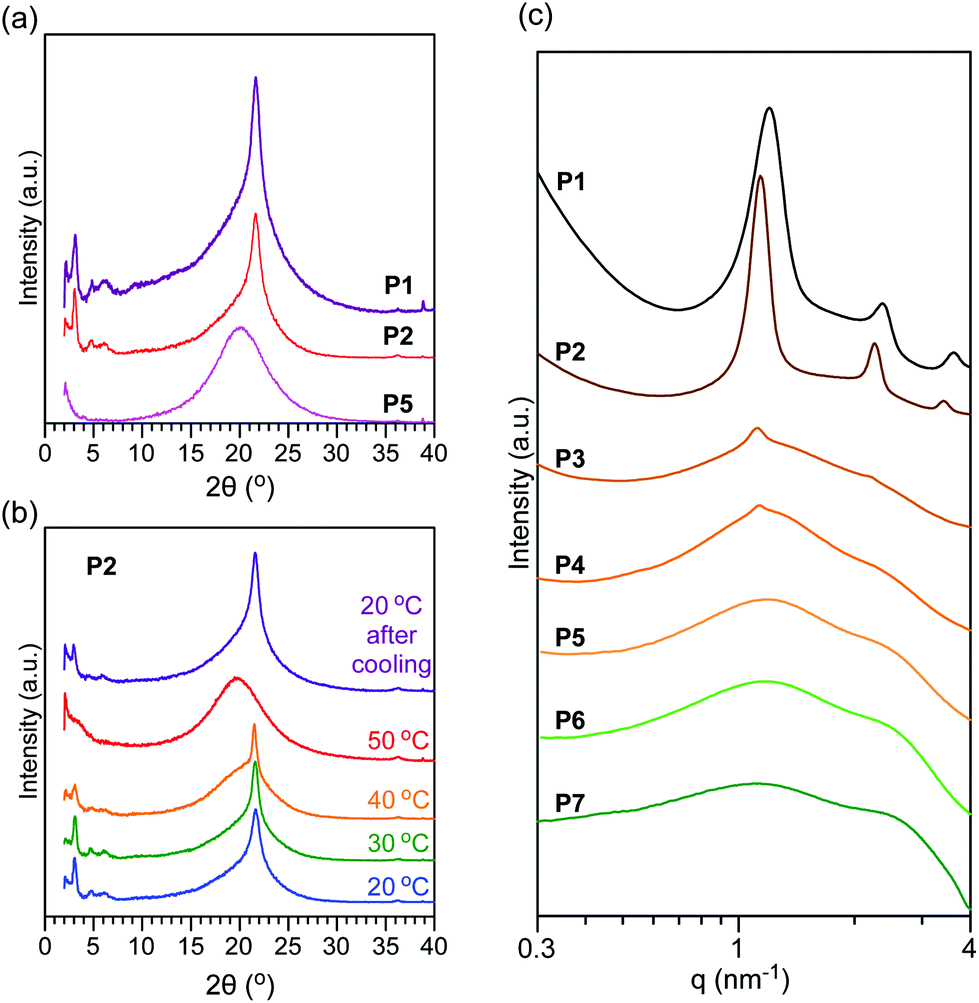 | ||
| Fig. 6 (a) X-ray diffractograms of (a) P1, P2, and P5 at 20 °C and (b) temperature-dependent X-ray diffractograms of P2 at 20, 30, 40 and 50 °C and 20 °C after cooling from 50 °C. (c) SAXS profiles of P1, P2, P3, P4, P5, P6, and P7 at 25 °C. | ||
Temperature-dependent XRD of P2 was conducted between 20 and 50 °C. Upon heating the sample from 20 °C to 40 °C, the height of the sharp peak at 21.6° decreased and an amorphous halo peak gradually overlapped with the sharp peak. At 50 °C, the sharp peak completely disappeared, meaning that the octadecyl groups were disordered. Upon cooling from 50 °C to 20 °C, the octadecyl groups crystallized to show a sharp peak at 21.6° of 2θ. These results indicate that the octadecyl groups of P2 reversibly form a hexagonally packed structure through melting and crystallization processes. It should also be noted that small peaks at around 3° of 2θ were also reversibly observed in this heating and cooling process, suggesting the reversible formation of longitudinal nanostructures.
:2:3) to form lamellar structures. The domain spacing of P1 and P2 was estimated as 5.2 nm and 5.5 nm from the peaks at q = 1.20 nm−1 and 1.14 nm−1, respectively. Considering the size range, the lamellar structure is derived from the microphase separation of PEG side chains and octadecyl and oleyl groups.35 In contrast, P5, P6, and P7 only showed broad peaks at around q = 1 and 2.5 nm−1. In addition to such broad peaks, P3 and P4 showed small integer order peaks (1:2), where the domain spacing was 5.6 nm. The broad peaks in P3, P4, and P5 are attributed to the disordered state via partial or complete melting of octadecyl groups at the SAXS measurement temperature. This is consistent with the melting temperature of the samples. In other words, this result importantly demonstrates that the crystallization of the octadecyl groups is essential for the formation of longitudinal lamellar structures via the microphase separation of random copolymer side chains. In P1, P2, P3, and P4, the domain spacing ranged between 5.2 nm and 5.6 nm. The small difference of the domain spacing implies that oleyl groups with the cis conformation affect the interdigitating and/or end-to-end structures of octadecyl groups in their crystalline domains.42
Crystallization and microphase separation of random block terpolymers
The crystallization and microphase separation of PEGA/ODA-PEGA/OLA random block terpolymers (P9 and P10) were evaluated by DSC and SAXS (Fig. 7). Upon analysis by DSC, P9 and P10 showed sharp endothermic signals derived from the melting of the octadecyl groups in the heating process (Tm = 40.7, 42.7 °C), as well as P1 as a model polymer for the 1st block segments (Fig. 7a). The binary blend of P1 and P7 also showed a similar melting peak at 44.0 °C. The melting peak for octadecyl groups of P9 comprising a short crystalline block exhibited a shoulder at the low temperature region. In P9, P10 and the binary blend of P1 and P7, the enthalpy of melting was proportional to the mole fraction of ODA, although these samples have a relatively low total ODA content below 40 mol% (Fig. 7c). These results indicate that octadecyl groups of random copolymers efficiently induce crystallization even in the presence of random copolymers bearing amorphous oleyl groups. | ||
| Fig. 7 (a) DSC curves recorded for the second heating scans of P1, P7, P9, P10 and the binary blend of P1 and P7 from −80 °C to 150 °C with a heating rate of 10 °C min−1. (b) SAXS profiles of P1, P9, P10 and the binary blend of P1 and P7 at 25 °C. (c) Enthalpy of melting of PEGA/ODA random copolymers (black circles),35 PEGA/ODA/OLA random terpolymers (P2, P3, and P4: gray squares), P9 (red square), P10 (blue square) and the binary blend of P1 and P7 (orange open square) as a function of the ODA content (mol%). | ||
P9, P10 and the binary blend of P1 and P7 were analyzed by SAXS, compared with P1. P10 with a long crystalline 1st block and the binary blend of P1 and P7, as well as P1, showed integer order peaks (1:2:3) stemming from lamellar structures (Fig. 7b). The domain spacing of P10 (d = 5.4 nm) and the binary blend (d = 5.2 nm) was close to that of P1 (d = 5.2 nm). It is interesting that P1 still induces microphase separation into lamellae even in the presence of P7 bearing amorphous oleyl groups. In contrast, P9 with a short crystalline 1st block indicated not only a peak derived from the microphase separation of the side chains at q = 1.14 nm−1 (d = 5.5 nm) but also another peak at q = 0.50 nm−1 whose domain spacing was calculated to be 13 nm (Fig. 7b), although the former microphase separation of the side chains does not give longitudinal structures.
These results suggest that the random block terpolymers competitively induce microphase separation of the side chains and block polymer chains, dependent on the length and/or volume fraction of the 1st and 2nd block segments. Lamellar microphase separation of the crystalline octadecyl groups and PEG chains in the presence of amorphous oleyl-bearing copolymers requires relatively long 1st crystalline random segments. P9 induces double microphase separation in the solid state: one originates from the side chains of hydrophilic PEG and crystalline octadecyl groups in the 1st random segment and the other is probably derived from microphase separation between the crystalline 1st block domains and the amorphous 2nd block domains.47 Since P10 with a long crystalline 1st block did not show such a low q peak, the volume fraction of two blocks would also affect the double microphase separation behaviour. To understand these microphase separation behaviour and morphologies, detailed analyses are now under investigation using various random block copolymers with different chain lengths, compositions, and side chains. These interesting discoveries would open new possibilities to three-dimensional control of nanodomain structures and sizes within solid polymer materials.
Conclusions
In summary, we designed amphiphilic random and random block terpolymers bearing PEG chains, crystalline octadecyl groups, and amorphous oleyl groups to examine the crystallization and microphase separation behaviour. Dual incorporation of octadecyl and oleyl groups as hydrophobic side chains opened the possibility to control not only the crystallization and melting temperatures but also temperature-dependent microphase separation and the crystalline domain sizes and structures in the solid state. In particular, we found the possibility that random block terpolymers comprising a crystalline random segment and an amorphous counterpart induced double microphase separation in different size scales stemming from side chains and polymer chains. The volume fractions of crystalline and amorphous blocks also affected the microphase separation behaviour and longitudinal lamellar structures. Therefore, the controlled crystallization and microphase separation systems using amphiphilic random and random block terpolymers would bring innovation to construct well-defined nanostructures in polymer materials that are potentially applicable to various research fields including nanotechnologies and photolithography.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Society for the Promotion of Science KAKENHI Grants (JP17H03066, JP17K19159, JP19K22218, JP20H02787, and JP20H05219), the Ogasawara Foundation for the Promotion of Science & Engineering, the Noguchi Institute, the Inamori Foundation, and the Tokuyama Science Foundation. We also thank Prof. Ken-ichi Otake (Kyoto University) and Prof. Susumu Kitagawa (Kyoto University) for the XRD measurements. SAXS measurements were performed with proposal no. 2020A1557.Notes and references
- A. J. Meuler, M. A. Hillmyer and F. S. Bates, Macromolecules, 2009, 42, 7221–7250 CrossRef CAS.
- H.-C. Kim, S.-M. Park and W. D. Hinsberg, Chem. Rev., 2010, 110, 146–177 CrossRef CAS PubMed.
- J. K. Kim, S. Y. Yang, Y. Lee and Y. Kim, Prog. Polym. Sci., 2010, 35, 1325–1349 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- A. H. Gröschel and A. H. E. Müller, Nanoscale, 2015, 7, 11841–11876 RSC.
- C. Sinturel, F. S. Bates and M. A. Hillmyer, ACS Macro Lett., 2015, 4, 1044–1050 CrossRef CAS.
- C. M. Bates and F. S. Bates, Macromolecules, 2017, 50, 3–22 CrossRef CAS.
- C. M. Bates, M. J. Maher, D. W. Janes, C. J. Ellison and C. G. Willson, Macromolecules, 2014, 47, 2–12 CrossRef CAS.
- M. Park, C. Harrison, P. M. Chaikin, R. A. Register and D. H. Adamson, Science, 1997, 276, 1401–1404 CrossRef CAS.
- P. Mansky, C. K. Harrison, P. M. Chaikin, R. A. Register and N. Yao, Appl. Phys. Lett., 1996, 68, 2586–2588 CrossRef CAS.
- R. A. Segalman, Mater. Sci. Eng., R, 2005, 48, 191–226 CrossRef.
- C. Park, J. Yoon and E. L. Thomas, Polymer, 2003, 44, 6725–6760 CrossRef CAS.
- W. J. Durand, G. Blachut, M. J. Maher, S. Sirard, S. Tein, M. C. Carlson, Y. Asano, S. X. Zhou, A. P. Lane, C. M. Bates, C. J. Ellison and C. G. Willson, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 344–352 CrossRef CAS.
- W.-S. Young, W.-F. Kuan and T. H. Epps, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 1–16 CrossRef CAS.
- C. E. Sing, J. W. Zwanikken and M. O. de la Cruz, Nat. Mater., 2014, 13, 694–698 CrossRef CAS PubMed.
- R. L. Weber, Y. Ye, A. L. Schmitt, S. M. Banik, Y. A. Elabd and M. K. Mahanthappa, Macromolecules, 2011, 44, 5727–5735 CrossRef CAS.
- M. W. Schulze, L. D. McIntosh, M. A. Hillmyer and T. P. Lodge, Nano Lett., 2014, 14, 122–126 CrossRef CAS PubMed.
- Y. Ye, J.-H. Choi, K. I. Winey and Y. A. Elabd, Macromolecules, 2012, 45, 7027–7035 CrossRef CAS.
- P. D. Topham, A. J. Parnell and R. C. Hiorns, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1131–1156 CrossRef CAS.
- M. W. Matsen, Macromolecules, 2012, 45, 2161–2165 CrossRef CAS.
- T. A. Shefelbine, M. E. Vigild, M. W. Matsen, D. A. Hajduk, M. A. Hillmyer, E. L. Cussler and F. S. Bates, J. Am. Chem. Soc., 1999, 121, 8457–8465 CrossRef CAS.
- T. S. Bailey, H. D. Pham and F. S. Bates, Macromolecules, 2001, 34, 6994–7008 CrossRef CAS.
- J. Chatterjee, S. Jain and F. S. Bates, Macromolecules, 2007, 40, 2882–2896 CrossRef CAS.
- A. Guliyeva, M. Vayer, F. Warmont, A. Takano, Y. Matsushita and C. Sinturel, Macromolecules, 2019, 52, 6641–6648 CrossRef CAS.
- K. Hayashida, N. Saito, S. Arai, A. Takano, N. Tanaka and Y. Matsushita, Macromolecules, 2007, 40, 3695–3699 CrossRef CAS.
- A. T. Lorenzo, A. J. Müller, M.-C. Lin, H.-L. Chen, U.-S. Jeng, D. Priftis, M. Pitsikalis and N. Hadjichristidis, Macromolecules, 2009, 42, 8353–8364 CrossRef CAS.
- Y. Matsushita, K. Hayashida, T. Dotera and A. Takano, J. Phys.: Condens. Matter, 2011, 23, 284111 CrossRef PubMed , 1–12.
- A. Nunns, C. A. Ross and I. Manners, Macromolecules, 2013, 46, 2628–2635 CrossRef CAS.
- Y. Rho, C. Kim, T. Higashihara, S. Jin, J. Jung, T. J. Shin, A. Hirao and M. Ree, ACS Macro Lett., 2013, 2, 849–855 CrossRef CAS.
- T. Higashihara, S. Ito, S. Fukuta, S. Miyane, Y. Ochiai, T. Ishizone, M. Ueda and A. Hirao, ACS Macro Lett., 2016, 5, 631–635 CrossRef CAS.
- J. Masuda, A. Takano, J. Suzuki, Y. Nagata, A. Noro, K. Hayashida and Y. Matsushita, Macromolecules, 2007, 40, 4023–4027 CrossRef CAS.
- J. G. Kennemur, L. Yao, F. S. Bates and M. A. Hillmyer, Macromolecules, 2014, 47, 1411–1418 CrossRef CAS.
- T. Isono, N. Kawakami, K. Watanabe, K. Yoshida, I. Otsuka, H. Mamiya, H. Ito, T. Yamamoto, K. Tajima, R. Borsali and T. Satoh, Polym. Chem., 2019, 10, 1119–1129 RSC.
- D. Neugebauer, M. Theis, T. Pakula, G. Wegner and K. Matyjaszewski, Macromolecules, 2006, 39, 584–593 CrossRef CAS.
- G. Hattori, M. Takenaka, M. Sawamoto and T. Terashima, J. Am. Chem. Soc., 2018, 140, 8376–8379 CrossRef CAS PubMed.
- K. Matsunaga, K. Tanaka and J. Matsui, Chem. Lett., 2018, 47, 500–502 CrossRef CAS.
- E. Barnard, R. Pfukwa, J. Maiz, A. J. Muller and B. Klumperman, Macromolecules, 2020, 53, 1585–1595 CrossRef CAS.
- R. Imanishi, Y. Nagashima, K. Takishima, M. Hara, S. Nagano and T. Seki, Macromolecules, 2020, 53, 1942–1949 CrossRef CAS.
- S. Mete, K. G. Goswami, E. Ksendzov, S. V. Kostjuk and P. De, Polym. Chem., 2019, 10, 6588–6599 RSC.
- M. Matsumoto, M. Takenaka, M. Sawamoto and T. Terashima, Polym. Chem., 2019, 10, 4954–4961 RSC.
- K. Ebata, Y. Hashimoto, K. Ebara, M. Tsukamoto, S. Yamamoto, M. Mitsuishi, S. Nagano and J. Matsui, Polym. Chem., 2019, 10, 835–842 RSC.
- K. Ebata, Y. Hashimoto, S. Yamamoto, M. Mitsuishi, S. Nagano and J. Matsui, Macromolecules, 2019, 52, 9773–9780 CrossRef CAS.
- Y. Kimura, M. Takenaka, M. Ouchi and T. Terashima, Macromolecules, 2020, 53, 4942–4951 CrossRef CAS.
- J. D. Quinn and R. A. Register, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 2106–2113 CrossRef CAS.
- M. Matsumoto, T. Terashima, K. Matsumoto, M. Takenaka and M. Sawamoto, J. Am. Chem. Soc., 2017, 139, 7164–7167 CrossRef CAS PubMed.
- M. Tanaka, T. Motomura, N. Ishii, K. Shimura, M. Onishi, A. Mochizuki and T. Hatakeyama, Polym. Int., 2000, 49, 1709–1713 CrossRef CAS.
- M. Xiang, D. Lyu, Y. Qin, R. Chen, L. Liu and Y. Men, Polymer, 2020, 210, 123034 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, SEC, DSC. See DOI: 10.1039/d0py01505a |
| This journal is © The Royal Society of Chemistry 2021 |