
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA study of the application of graphite MALDI to the analysis of short-chain polyethylene glycols†
Ulric
Conway‡
a,
Alexander D.
Warren
ab,
Christopher J.
Arthur
 a and
Paul J.
Gates
*a
a and
Paul J.
Gates
*a
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: paul.gates@bristol.ac.uk
bInterface Analysis Centre, School of Physics, University of Bristol, Tyndall Avenue, Bristol, BS8 1TL, UK
First published on 16th December 2020
Abstract
The main goals in the analysis of synthetic polymers include the determination of the molecular weight, molecular weight distribution and dispersity along with complete characterisation of the chemical structure and end groups. These measurements must be made accurately and efficiently. Matrix-assisted laser desorption/ionisation mass spectrometry is a readily available and frequently used technique for the analysis of synthetic polymers. However, this can be challenging due to issues with matrix interference and analyte suppression as well as the suspected inability of shorter oligomer chains to bind cations with sufficient strength to survive the desorption/ionisation process. This has led to our study where we present the use of carbon-based matrices (specifically colloidal graphite) with polyethylene glycol polymers doped with LiCl as an appropriate and powerful methodology for the successful analysis of low molecular weight polymers.
Introduction
The analysis of synthetic polymers is crucial to several research areas. There are many spectroscopic and analytical techniques which can be applied to this task, including mass spectrometry (MS), however, the diversity of polymers means that no single technique is universally applicable. A combination of techniques is most often applied to gain the desired information. A recent review describes the merits of a wide variety of analytical techniques when applied to synthetic polymer analysis.1“Soft” ionisation methods for MS, such as electrospray ionisation (ESI) or matrix-assisted laser desorption/ionisation (MALDI), where the analyte chains are transferred into the gas-phase without any fragmentation or change in the structure are preferred for polymer analysis. During development, polyethylene glycol (PEG) was chosen as a demonstration analyte to show the potential of ESI to analyse high-mass substances.2 Where ESI falls short though, is in measuring the molecular weight distribution.2,3 ESI shows a clear bias towards longer polymer chains and the observed distribution is also highly sensitive to the potentials and temperatures of the ESI source. This makes it challenging to measure polymer distributions reproducibly with high degree of confidence.3 On the other hand, the simplicity and relative ease of interpretation of MALDI-MS spectra are the key advantages leading to it becoming the method of choice for polymer analysis and the reliable determination of polymer molecular weight distributions.
Due to the diversity and chemical complexity of synthetic polymers, analysing them can be challenging. The goals of the analysis range from the determination of the average molecular weight, molecular weight distribution and measurement of dispersity as well as complete characterisation of the chemical structure and determination of end groups. These tasks become problematic for polymers containing chains that fall below about 500 Da because they are usually suppressed or dominated by the traditional matrices. A further complication is that it is unclear whether these smaller species can be detected at all if they cannot bind to a cation. Both problems result in the distortion of the observed molecular weight distribution towards higher masses.
Two separate studies by Guttman et al. show that MALDI can accurately measure the molecular weight distribution of various polystyrenes by comparing results from over thirteen institutions.4,5 The spectra produced provide an absolute measure of this distribution, which removes the need for mass calibration using polymer standards. This attribute of MALDI makes it an attractive option for any polymer analysis. But MALDI does still have limitations to do with reproducibility and quantification (for example, relative peak areas do not necessarily correlate with the relative amounts of individual components of a mixture – this is especially true for the analysis of polymers).
Most biological polymers are ionised by proton transfer from the more traditional acidic matrices, but synthetic polymers are not easily ionised this way with cationisation by an alkali or transition metal cation being the more common route. This attachment could either occur because of preformed ions in the sample being lifted into the gas-phase by the laser or because of secondary reactions in the desorbed plume. Knochenmuss et al., suggest that gas-phase reactions in the desorbed plume are the most likely route to cationisation.6–9 It is not exactly understood how the choice of the matrix affects the efficiency of the cation attachment but the presence of photoelectrons in the desorbed plume may result in the reduction of metal cations. A separate study by the same group showed that Cu(II) can easily be reduced to Cu(I) by free-electron capture in the gas-phase.10 The ionisation of synthetic polymers via MALDI, therefore, involves a complex interplay between the polymer, the matrix, and any metal cation additives.
Choosing a matrix for the analysis of polymers requires the matching of the polarity of the polymer and the matrix.11 Care is needed here, however, because polymers with a wide distribution of chain lengths may exhibit a change in polarity from the shorter to the longer chains, especially in polymers where the polarity is mainly determined by its end groups. Matrices such as DHB and CHCA, which are popular general matrices, are also commonly used for synthetic polymers. But there is still much investigation into possible novel matrices in this area. Colloidal graphite (CG) applied by airbrush has shown considerable success in the analysis of a wide variety of different low-molecular weight (LMW) analytes.12,13
As mentioned before, synthetic polymers primarily ionise via cation attachment rather than protonation. Therefore, a metal salt is a vital ingredient during sample preparation. Polymers that contain double bonds or aromatic systems, such as polystyrenes and polybutadienes, tend to bind well to Ag+ and Cu+ cations.14 These cations are well known to interact with the π system present in unsaturated hydrocarbons and are generally added in the form of a trifluoroacetate salt.6,10 Saturated polymers which are devoid of heteroatoms, such as polyethylenes and polypropylenes, are challenging to analyse by MALDI, but some studies have shown that silver can also be used in specific circumstances.15,16 Polymers that contain heteroatoms, such as polyethers, tend to ionise well with the addition of alkali metal salts. The cations Na+ and K+ are usually present as impurities from glassware, solvents or even the matrix. In this case, it is not usually necessary to add the salt during sample preparation.
The flexible backbone of PEG allows it to adopt a structure where multiple oxygen atoms can interact with cations. A theoretical study by Ehlers et al. determined the binding energies of various cations to PEGs and claimed it is not possible to detect PEG chains that have fewer than five monomer units.17 The results of their study suggested that there was a dependence on the radius of the cation: longer PEG chains could form a more stable complex when interacting with a larger cation, conversely smaller PEG chains could bind more strongly to smaller cations. This effect has also been observed with similar polymers, such as polymethyl methacrylate18 and polyethylene terephthalate.19 When this is applied to the analysis of PEGs by MALDI, this would mean that the addition of a large alkali metal salt could skew the observed mass distribution towards the higher masses. Shimada et al. studied the cationation affinity of single PEG species – i.e. those that had a uniform length and a specific order of polymerization with no molecular weight distribution.20 This study was able to infer that longer chains prefer larger cations. For example, chain lengths of n = 34–39 were more stable when attached to Cs+, and n = 12–17 were more stable when attached to Na+. Neither of these previous studies considered the behaviour of the LMW PEGs (i.e. with a degree of polymerisation below 8). Ehlers stated that it was not possible to observe PEG units of n < 5, but this work was only conducted using the Na+ cation.17 It might be expected, that according to the trend in cation affinities, that the addition of a Li+ during sample preparation would allow the smaller chains to be seen if they were indeed present. There is already considerable literature precedence for the use of Li+ as a cationising agent for the ESI analyses of PEGs – for example see separate studies by Li and O'Conor.21,22
Experiments on these small chains, regardless of which cation was used, have not been previously reported and so the question of whether it is possible to detect these small chains by MALDI has not been satisfactorily answered. This is most likely due to the matrix used in these studies, DHB, which produces considerable low-mass noise and can entirely suppress the ionisation of LMW analytes – as discussed in our previous paper.12 Graphite-MALDI-MS was developed for the analysis of LMW analytes due to the very low intensity of any matrix ions observed in spectra (often total matrix suppression by the analyte is observed) making it potentially an ideal matrix to study the ionisation of LMW polymers by MALDI-MS.12,23,24
Carbon based matrices have been widely studied in the literature due their highly efficient dispersal of energy to the analyte25 and the tendency to produce spectra that contain very few matrix peaks in the low-mass region.12,23 Applications range from the analysis of small molecules using carbon nanotubes26 and fullerenes27 to the analysis of small peptides using graphite suspensions in glycerol28 or SALDI analysis of thin-layer chromatography plates using powdered graphite suspended in isopropanol.29 There have also been some previous studies on the application to the analysis of LMW polymers.30
Therefore, there are two main aims of this study. The first is to investigate whether small PEG chains, (n < 5) can be detected by MALDI using graphite as a matrix and the second is determining whether this is influenced by the choice of the cation. This study will investigate the general effectiveness of different forms of graphite matrix for analysing LMW PEGs and compare the results obtained to the other commonly used matrices. Single spot imaging will also be performed in an effort to determine the extent matrix:analyte co-crystallisation has on the quality of the results obtained. The overall outcome is to demonstrate the effectiveness of graphite matrices for the analysis of LMW polymers by MALDI-MS and to determine whether graphite gives an accurate reflection of the true distribution of the polymer through the fitting of Gaussian distributions to the results obtained.
This study shows that the application of colloidal graphite as the matrix in conjunction with a suitable metal salt (for example LiCl) avoids the detrimental mass discrimination affects and, as a result, allows for more reliable measurement of the molecular weight distribution for low molecular weight polymers.
Experimental
Instrumentation
MALDI-MS was performed on a 4700 Proteomics Analyzer (Applied Biosystems, Warrington, UK). This is a reflectron Time-of-Flight/Time-of-Flight (TOF/TOF) mass analyser, used in reflectron mode. All spectra were recorded at 1000 shots per spectrum (comprised of 8 sub-spectra, over a 5 s run) at a resolution of approximately 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 FWHM. The laser used was a 200 Hz Nd/YAG at a wavelength of 355 nm with a beam diameter is 50 μm, a pulse energy of 12 μJ and pulse length of less than 500 ps. Calibration (better than 5 ppm) was achieved using the 4700 Proteomics Analyzer Calibration Mixture (Applied Biosystems, Warrington, UK).
000 FWHM. The laser used was a 200 Hz Nd/YAG at a wavelength of 355 nm with a beam diameter is 50 μm, a pulse energy of 12 μJ and pulse length of less than 500 ps. Calibration (better than 5 ppm) was achieved using the 4700 Proteomics Analyzer Calibration Mixture (Applied Biosystems, Warrington, UK).
Chemicals
Sinapinic acid (SA), α-Cyano-4-hydroxycinnamic acid (CHCA) and dithranol (DTH) were all obtained from Sigma Aldrich (Gillingham, UK). 2,5-Dihydroxybenzoic acid (DHB) was obtained from Fisher Scientific (Loughborough, UK). All solvents were HPLC gradient grade purchased from Fisher Scientific (Loughborough, UK). The organic matrices were all prepared as 10 mg mL−1 solutions in methanol:water (2:1, v/v) with 1 μL drops deposited by pipette and dried on the sample plate before the analyte. Lithium chloride, obtained from Sigma Aldrich (Gillingham, UK), was prepared at 1 mg mL−1 in water.
Colloidal graphite (CG) and micronised graphite (MG) were obtained from The Graphite Trading Company (Halesowen, UK). 99.9995% high purity graphite rod (GR) was obtained from Sigma Aldrich (Gillingham, UK) and a Staedtler Mars Lumograph 4B pencil (4B) (Staedtler, Nürnberg, Germany) was purchased from local artists supply shop. CG and MG were each applied by airbrush using the previously published protocol.13 4B and GR were transferred to the sample plate by drawing a line across the required spots ensuring that the spot was fully covered but trying to avoid scratching the plate surface.
PEG200, PEG400 and PEG600 were obtained from Sigma Aldrich (Gillingham, UK). All PEGs were prepared at a concentration of 1 mg mL−1 in methanol:water (2:1, v/v). In the experiments using LiCl, the PEG and salt solutions were mixed (1:1, v/v). In all cases, 1 μL of analyte solution was deposited on top of the previously applied matrices and left to dry.
Methodology
Ten spots were deposited on the target plate for each PEG/matrix combination. For each spot, twenty sub-spectra were measured and averaged to produce a single spectrum per spot. When a polymer chain contributed two peaks to the observed spectrum (e.g. due to sodiation [M + Na]+ and potassiation [M + K]+) the sum of these two peak areas was taken to be the measured abundance of that specific polymer chain length. The peak areas were averaged over the ten spots to obtain the distribution of polymer chains for that PEG sample.Origin was used to fit a Gaussian function to the tabulated data of polymer chains (see eqn (1)).31
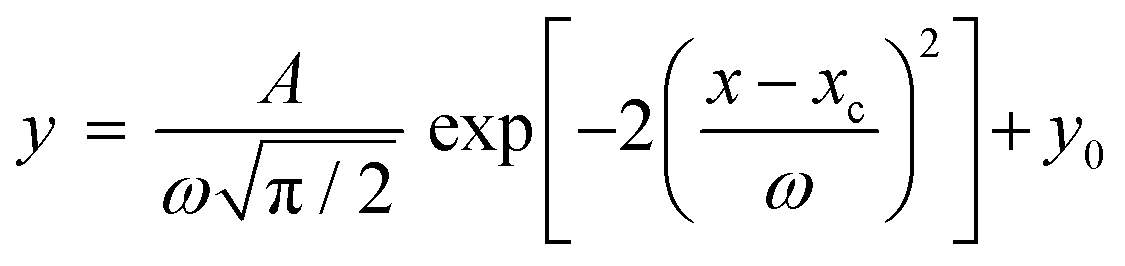 | (1) |
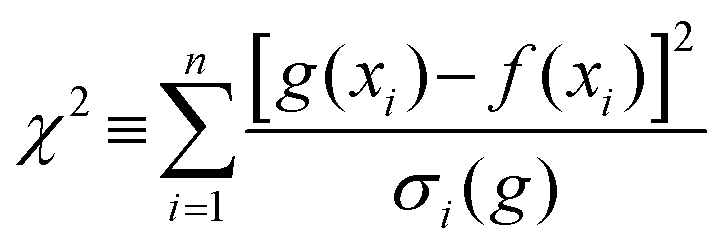 | (2) |
MALDI imaging
MALDI imaging was performed on prepared spots of analyte and matrix. Three PEG polymers (PEG200, 400 and 600) were spotted with five matrices, DHB, DTH, 4B, GR, CG. For imaging, 1 μL of CG suspension in ethanol was deposited by pipette in the same way as the traditional matrices. The imaging parameters where: 255 shots per spectrum at a standard laser Intensity of 3000 and a digitizer bin size of 2 ns. A region encompassing the entire spot was selected and imaged.Results and discussion
ESI and GC-MS analysis
Prior to the MALDI analysis, the PEG samples were analysed by ESI-MS and by GC-MS (see ESI† for spectra and experimental details). For the ESI analysis (see Fig. S1 ESI†) there is clearly a bias towards the longer polymer chains. For PEG200, the distribution runs from n = 4 to n = 15 with the maxima at n = 6 (m/z 305 for [M + Na]+) when it is expected to centre at n = 4. Ions formed by ESI can readily be multiply charged leading to complications in the spectra with overlapping peaks and separate envelopes for each charge state. This is most apparent for PEG600 where doubly and triply charged ions are observed but this is even visible in the PEG200 and PEG400 spectra. The GC-MS study was performed as a sanity check to make sure that the commercial PEG samples had Gaussian distributions. The results are contained in Fig. S2 ESI.† For PEG200, the distribution runs from n = 2 to n = 8 and appears to be highly Gaussian centred around n = 3 and n = 4. For PEG400, the distribution runs from n = 3 to n = 9 and also appears Gaussian centred around n = 6.Methodology
In the first series of MALDI experiments, the performance of several carbon matrices and the organic matrix DHB with PEG400 and PEG600 were considered and compared to the performance of the established polymer matrix DTH.33 The spectra produced were examined to view any matrix associated mass discrimination effects in the ionisation of the polymer solutions, as well as to ascertain the utility of CG for analysis of polymers. MALDI imaging of spots of each solution with each matrix was also carried out (see later).DTH performed well – it produced a wide and even distribution of oligomers for all PEGs and gave high signal-to-noise values. DHB generally performed poorly with the signal intensities being considerably lower. The three carbon-based matrices tested gave more intense spectra than DTH or DHB, however, they showed significant differences to each other in other characteristics, and these are discussed below.
Firstly, there were noticeable variations in the dominant adduct ion present in the spectrum for the 4 samples. This effect was most noticeable with DTH, where the relative abundance of the sodiated and potassiated molecular ions varied greatly with each polymer solution. CG also showed this variation, but peak heights of the sodiated and potassiated species were only marginally different. These variations are a result of various contributions but mainly exist simply due to the variation in cation availability on the surface of each spot. 4B showed potassiation dominating over sodiation for all samples, suggesting that 4B must act as an abundant source of potassium cations to the analyte leading to a preference for cationation with this species. This was also reported in a study by Langley et al. who hypothesised that the source of potassium to be various clays used in the pencil manufacturing process, possibly montmorillonite.24 DHB however, generally showed a preference for sodiation, although some variation was seen. What is notable, is the near absence of protonation for all matrices including DHB. This is indicative of PEG having a higher cation affinity than proton affinity (PA).
Fig. 1 shows representative MALDI-MS of PEG400 with DTH and DHB matrices. Spectrum (a) shows a high level of analyte suppression by the matrix peaks, most notably an intense signal for matrix cation [DTH]+ at m/z = 226. The much greater intensity of matrix peaks compared to those of the analyte is due to preferential ionisation of the matrix. This can occur to such an extent that it may be necessary to rescale the intensity axis to observe the full polymer distribution. This is most problematic for the analysis of polymer mixtures consisting of oligomers in the low-mass range. The additional issue of low-mass overcrowding by the high-intensity matrix peaks is noticeably worse for the analysis employing DHB as the matrix in spectrum (b). Here several very intense matrix signals for [DHB + H − H2O]+ (m/z 137), [DHB + H]+ (m/z 155), [DHB + Na]+ (m/z 177), [DHB + K]+ (m/z 193) and higher mass matrix/salt cluster ions (m/z 383) considerably confuse the low-mass region.
 | ||
| Fig. 1 The MALDI-MS of PEG400 with (a) DTH and (b) DHB as matrices. With DTH [PEGn + K]+ is most abundant, whereas with DHB [PEGn + Na]+ is most abundant. | ||
Analysis of LMW PEGs using colloidal graphite matrix
The next study was a comparison of the performance of graphite matrices to those of some traditional matrices (see Table 1) for the analysis of LMW PEGs. The graphite matrices have all been investigated before for the analysis of a range of LMW analytes4,23,24 but no thorough study has been conducted for LMW PEGs. The traditional matrices are those that are considered the general matrices for MALDI-MS. The most relevance is probably DHB, which is the matrix most often used in the literature for the analysis of LMW PEGs. The specific grades of PEG were chosen so that they would encompass the mass range of most interest (100–1000 Da). This mass region is interesting as it is low enough mass to investigate to what extent graphite matrices have an advantage, if any, over their traditional counterparts.| Graphitic based matrices | Traditional matrices |
|---|---|
| Colloidal graphite (CG) | 2,5-Dihydroxybenzoic acid (DHB) |
| Micronised graphite (MG) | Dithranol (DTH) |
| Graphite rod (GR) | Sinapinic acid (SA) |
| 4B pencil lead (4B) | α-Cyano-4-hydroxycinnamic acid (CHCA) |
Gaussian functions were fitted to the observed distribution for PEG400 using Origin. An example is shown in Fig. 2, which shows the observed polymer distribution and its Gaussian fitting for PEG400. From the function various parameters can be calculated, some of which correspond to physical properties of the polymer. The average value of the Gaussian function, that is, the position of its maximum, can be taken as a measure of the number average molecular weight of the polymer. The goodness of fit was assessed using the reduced chi-square (χ2) value.
 | ||
| Fig. 2 Graph showing the observed distribution of PEG400 analysed with CG matrix. The top horizontal scale gives the corresponding mass for each polymer chain length given on the bottom scale. The graph also shows the fitted Gaussian function (black line). See ESI Tables S1† for further details of the values and fitting parameters. | ||
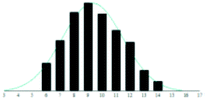
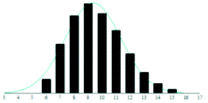
A summary of the data analysis for all the PEG/matrix combinations is presented in Table 2. The dashed horizontal lines separate the graphite-based matrices from the traditional matrices. Also shown in the table are graphical representations of the measured distributions and their corresponding Gaussian fit, to allow examination and comparison of the dataset by eye.
| PEG grade | Matrix | Distribution | Reduced χ2 | Skewness |
|---|---|---|---|---|
| PEG400 | CG |

|
1.3 | 0.11 |
| MG |

|
1.1 | 0.09 | |
| GR |
|
1.4 | 0.15 | |
| 4B |
|
1.4 | 0.18 | |
| DHB |

|
2.6 | 0.29 | |
| SA |

|
2.0 | 0.27 | |
| DTH |
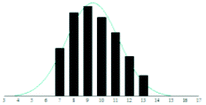
|
2.1 | 0.21 | |
| CHCA |

|
1.9 | 0.19 | |
| PEG600 | CG |

|
1.7 | 0.23 |
| MG |
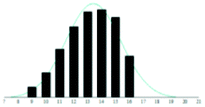
|
2.4 | −0.26 | |
| GR |

|
2.0 | −0.02 | |
| 4B |
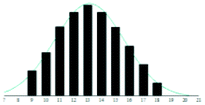
|
1.3 | 0.05 | |
| DHB |
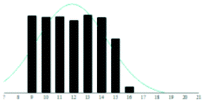
|
5.0 | 0.08 | |
| SA |

|
3.1 | 0.16 | |
| DTH |

|
2.1 | −0.05 | |
| CHCA |

|
3.8 | −0.11 |
The graphite matrices, taken as a group, perform at least as well as the traditional matrices for both the PEG analytes. The values for the reduced χ2 are generally closer to the optimal value χ2 = 1 for the graphite matrices, indicating that a more accurate Gaussian shape was observed for the polymer distribution. Also, some of the larger orders of polymerisation (n ≥ 9), having been unambiguously identified using graphite matrices, were missing from the spectra of the traditional matrices. Notably, DHB, which is well known for its ability to successfully ionise larger PEG analytes, performed poorly at the mass range used in this experiment.
The statistics-based measures described above appear to favour the graphite matrices, but other advantages are not considered by these methods. For example, it is noted in our previous study that the graphite matrices cause little to no interference in the low-mass region, resulting in very clear spectra with a good ion yield (matrix suppression).12 This is also the case in the present study, however, the spectra obtained with traditional matrices are all affected by matrix-related ions in the region of interest; this hampers the reading of the spectra because the unwanted ions dominate the analyte signal (analyte suppression).
Although the graphite matrices perform at least as well as the traditional matrices, the overall performance of the matrices is still questionable. For example, the values for the reduced χ2 are still all above 1 and some are significantly higher. This effect can also be observed by visual inspection of the distributions in Table 2 where the Gaussian function does not smoothly follow the peaks for the abundances of each chain. It is especially apparent for the shorter chain lengths, where the function overestimates the number of observed ions. This is backed up by the values for the skewness of each distribution, shown in the far-right column in Table 2. The values are mostly positive, which means that the distribution is more heavily weighted towards longer chain lengths. This means that there were fewer small polymer chains detected in the spectra than would be expected if the distribution were truly Gaussian.
There could be several explanations for this departure from the expected Gaussian behaviour. It could be that the hypothesis that the distribution of the polymer is Gaussian is incorrect. An ideal polymerisation process would indeed yield a Gaussian distribution, but real reactions are seldom ideal and there may be other factors inherent in the way the sample is prepared for commercial consumption that could affect the distribution. In other words, the smaller chains may simply not be present in the sample and hence will not be seen in the spectra. However, the GC-MS analyses shows that this is not case, and that the PEG200 and PEG400 samples do indeed contain the shorter chain oligomers.
It is also possible that the shorter chains are not being detected because they are unable to bind cations present in the sample tightly enough, thus skewing the distribution towards higher mass polymer chains that can. This observation is consistent with the previous research where it was claimed that the binding energy between the polymer chain and added Na+ or K+ cations is too small to survive the MALDI process.17,20,21 The reason for this appears to be that the cation is too large for the short-chain to be able to form a stable conformation with it. Assuming that this skew in the results is due to undetected low-mass polymer chains, the question must be asked of whether it is possible to detect these small chains using MALDI along with a salt of the smallest alkali metal, for example LiCl.
To test this idea, CG applied by airbrush13 was compared with DHB to analyse PEG200. PEG200 was chosen as the analyte to ensure that there would be an appreciable number of small oligomer chains present. In this sample, the average molecular weight is expected to be around 200 Da, which corresponds to n = 4 being the most abundant chain length. Typical spectra from this experiment are shown in Fig. 3. In spectrum (a), the polymer chains of the type [PEGn + Na]+ can be identified – labelled in green with the value of n, however, the low mass range of spectrum is dominated by ions due to the matrix itself at m/z 137, 155 and 177 (labelled with a red star). These ions are attributed to [DHB + H − H2O]+, [DHB + H]+ and [DHB + Na]+ respectively (along with some higher masses cluster ions). There was also a large variation in the abundances detected for each order of polymerisation, and the overall distribution does not increase smoothly towards the maximum and then decay as would be expected for a Gaussian distribution.
 | ||
| Fig. 3 Positive ion MALDI-MS of PEG200 with a range of matrices. Spectrum (a) is with DHB matrix, (b) is with airbrushed CG matrix and (c) is with airbrushed CG matrix with the PEG being doped with LiCl during the sample preparation. With DHB and CG [PEGn + Na]+ is most abundant, whereas with PEG doped with LiCl, [PEGn + Li]+ is the most abundant. | ||
In spectrum (b), the experiment was repeated with CG as the matrix. Here it is clear that there are no large interferences from matrix ions and the unobstructed distribution of the polymer chain can now be seen. The peaks observed are the cationised polymer chains [PEGn + Na]+ and [PEGn + K]+ from n = 4 to n = 9 where n is the order of polymerisation and labelled in green on the spectrum. However, at the lower end of the spectrum, there appears to be a sudden cut-off in the intensity, where the abundance drops off rapidly when the order of polymerisation decreases below n = 6 and the peak for n = 4 could only be detected in some spectra. This effect was also present in the previous work on larger PEGs (see above) but here it is more pronounced due to the smaller average mass of PEG200.
Spectrum (c) shows PEG200 again with CG matrix but this time with the analyte doped with LiCl. The peaks observed are the cationised polymer chains [PEGn + Li]+ from n = 2 to n = 10 where n is the order of polymerisation labelled in green on the spectrum. There are also peaks resulting from the adduction of LiCl i.e. [PEG[n−1] + LiCl + Li]+ occurring at 2 Da less for each peak. These ‘cluster ions’ are probably due to inhomogeneous drying of the sample spot leading to areas of the spots with higher LiCl concentration and are therefore not considered further. The results show that the PEG chains are readily able to be cationised by lithium down to n = 2. PEG chains of this size were not previously known to be able to be detected by MALDI, but given the right matrix and salt additive, it is possible to get a clear picture of the molecular weight distribution of these polymers, without interference from matrix ions. The base peak in this spectrum corresponds to the n = 4 chain, which has a mass of 194 Daltons, which is as expected for the PEG200 sample and corresponds well with the GC-MS analysis. In the spectra where no lithium has been added (spectrum (b)) the n = 4 peak is barely observed.
MALDI imaging to monitor matrix:analyte co-crystallisation
In addition to the chemical process of ionisation and cationisation, the effect that any variation in matrix:analyte co-crystallisation might have on the ionisation process and resulting spectra is also studied. This is performed through single-spot MALDI imaging of PEG co-spotted with DHB, DTH, and the three carbon matrices (4B, GR and CG) matrices. Fig. 4 shows the imaging results for PEG200 (see ESI† for results with other PEG grades). | ||
| Fig. 4 MALDI imaging of PEG200 with various matrices. The 5 matrices (DHB, DTH, 4B, GR and CG) are all imaged at fixed oligomer masses for the PEG200 (n = 6, 7 and 9) and a monitoring mass for the matrix (m/z 155 for DHB, m/z 226 for DTH and m/z 180 for 4B, GR an CG). A linear colour scale is also provided from red (high intensity) to purple/black (low intensity). The mass spectrum of the same spot is provided on the right-hand side. Matrix peaks for DHB and DTH are indicted by a red star and PEG peaks with the corresponding chain length in red. With DHB [PEGn + Na]+ is most abundant, whereas with DTH [PEGn + K]+ is most abundant. With all graphite-based matrices, a combination of [PEGn + Na]+ and [PEGn + K]+ is observed with the latter being most abundant. | ||
The matrix peaks for the traditional matrices are seen in the resulting single spot MALDI images. The [DHB + H]+ (m/z 155) shows a large variation in intensity over the spot – this is typical behaviour for DHB which has a high tendency to form “sweet spots”.34 Across large areas of the spot, the intensity of the matrix ions is greater than the analyte ions showing considerable analyte suppression. In the spectra, the three identifiable matrix peaks are at least as intense as the analyte, and it is seen that some analyte signal is missing from the spectrum at the low-mass end. This suggests that with DHB, analyte suppression is a dominant process, leading to fewer analyte ions being observed than for any of the other matrices used. Also, the overall intensity of the mass spectrum is about an order of magnitude lower than the others in this study. With DHB (monitor mass m/z 155) the images mirror the poor performance seen in the spectra. A large level of variation over the spot for all three oligomers is shown with both “sweet” and “dead” spots visible, indicating a very inhomogeneous spot. The highest intensity spectra are produced from the edges indicative of the “coffee ring effect” commonly observed with DHB, with the central region producing poor quality low intensity spectra.35 Although there is large amount of variation visible, the areas of highest analyte intensity generally correspond with areas of high matrix intensity, suggesting some degree of matrix:analyte co-crystallisation is taking place. However, there is considerable lateral diffusion of the analyte to the edges of the spot.
With DTH m/z 226 is chosen as the monitor mass as it is the most intense matrix ion observed, generally showing highest intensity at the centre of the spot, with moderate intensity over the rest of the spot. The high-intensity central area is at least twice the intensity of the analyte ions suggesting a degree of analyte suppression is taking place. The molecular weight distribution of the analyte seems more Gaussian than observed with DHB, however fewer lower mass oligomers are observed when compared to DHB and the centre of the distribution is at n = 7 rather than n = 6 with DHB.
DTH produces a uniform signal intensity across the whole of the spot for all three oligomer masses monitored. This suggests an even spread of matrix and polymer over the spot and implies that analyte:matrix co-crystallisation is occurring, producing a highly homogenous spot. In general, the maximum intensity spectra are produced from the central area of the spot as expected. Examination of Fig. 4 shows that the abundance of the different oligomer peaks is also uniform over the spot, which is highly indicative that the oligomers crystallise equally over the spot – i.e. co-crystallisation with the matrix is not dependent on the size of the oligomer, and different oligomers diffuse equally to all areas.
With the carbon-based matrices, m/z 180 (corresponding to C15) was monitored and its images show almost zero abundance. This highlights the excellent level of matrix suppression possible when using carbon matrices. Examination of the spectra leads to the conclusion that there are no matrix ions observed in the mass range studied. However, the three carbon matrices produced significantly different spot characteristics, and this is evident in the contrasting images. Due to their method of application for 4B and GR, analyte:matrix co-crystallisation is not possible, a factor that would suggest spot homogeneity to be poor for these matrices. This is very evident in the case of 4B, which shows the lowest level of homogeneity of the three carbon-based matrices tested. Each oligomer showed a noticeable ring of higher intensity towards the edges of the spot, indicating that lateral diffusion of the analyte is taking place on the plate surface. When compared to DHB, the overall homogeneity and signal intensity compares well, with the area inside the ring producing consistent spectra of medium to low intensity. There are some differences in the images that suggest that a variation in the distribution of differently sized oligomers exists over the spot. GR shows improved performance over that of 4B, with greater homogeneity observed. This is expected since the wax esters and fatty acids that are present in the 4B pencil are not present in GR and as a result the graphite is present at very high purity (>99.9995%). CG produces images with the highest level of homogeneity of the three carbon-based matrices tested (roughly equal to that of dithranol) indicating that analyte:matrix co-crystallisation is occurring which is probably a result of pipette deposition.
With CG, the correlation between the images for different oligomers is also very high, indicating an extremely even distribution of oligomers over the spot surface. This is most probably due to the extremely thin and homogenous layer of carbon deposited, and the absence of any hydrophobic glide agents that are present in 4B. The high correlation between the molecular ion images and the consistently weak image for the matrix (i.e. no diffusion of the matrix is occurring) further indicates that excellent mixing and/or analyte:matrix co-crystallisation is occurring during spot preparation. Finally, CG shows the presence of the shortest chain length PEGs of all the matrices studied here, this is in agreement with the previous results.
Conclusions
The graphite-based matrices are demonstrated to perform similarly to traditional matrices when analysing LMW PEGs by MALDI-MS when comparing their performance with a theoretical function that models the polymer chain distribution, and when using common sample preparation methods. When this performance is coupled with good reproducibility and a lack of interfering matrix-related ions, as is the case with CG, then the matrix offers improved performance compared to those more commonly used, such as DHB.A discrepancy between the theoretical model and the observed results for the smaller chains was interpreted as originating from their low binding energy with the large cations present in the sample, therefore they could not be detected in the mass spectra. PEG200 was therefore analysed with the addition of a LiCl – a smaller cation – to test our hypothesis about cation size and PEG chain length. From the results obtained, it appears that the Li+ can bind strongly enough with the individual polymer chains to survive the desorption/ionisation process and therefore make them visible in the mass spectra. Chains with degree of polymerisation as low as n = 2 were very clearly observed. This is contrary to the theoretical study by Ehlers et al. in which it is claimed that it is not possible to detect PEG chains that have fewer than five monomer units.17 However, in that work, the use of Li+ as the cation was not considered. The fact that we can easily observe n = 2, 3, 4 and 5 and that the distribution of PEG200 is Gaussian suggests that the use of CG doped with LiCl should be the technique of choice to analyse LMW polymers.
Finally, we attempted to demonstrate the effects of analyte:matrix co-crystallisation on the spectral quality through single spot MALDI imaging. The analysis of DHB showed the expected inhomogeneity of the spot with a highly undesirable combination of “sweet” and “dead” spots and considerable analyte suppression. The total intensity was also an order of magnitude lower than the other matrices – despite the same preparation method being employed. DTH perform well, with good spot coverage for both matrix and analyte showing good analyte:matrix co-crystallisation, however, the high intensity of the matrix peak leads to analyte suppression. With the carbon-based matrices, high level of matrix suppression was observed. However, there were considerable difference in the homogeneity of the spots. Although 4B generated the highest intensity spectra, there is an element of skewness to higher massed oligomers leading to an incorrect average mass determination. GR resulted in the highest spot homogeneity, but there was also considerable skewing to higher-massed oligomers. CG showed slightly lower spot homogeneity, with the centre of the spots having the least signal, however, the resulting spectra showed the lowest amount of skewing with even the n = 3 oligomer being visible and the highest intensity for n = 9 and 10.
As a result of this study, the authors proposed that the application of colloidal graphite doped with LiCl as a general methodology for the routine analysis of low molecular weight polymers to avoid skewing the mass distribution to higher masses. With this methodology we were easily able to observe PEG oligomers down to as low as n = 2. This contrasts with the previously published study.17 We believe that this is due to poor cation binding of polymers for Na+ and/or K+. Colloidal graphite also has the added benefit of producing very few detrimental low-mass matrix signals.
Author contributions
UC: Data curation, formal analysis, investigation, methodology, software, visualization, writing – original draft, ADW: Data curation, formal analysis, methodology, CJA: Software, supervision, validation, writing – review & editing, PJG: conceptualisation, methodology, resources, project administration, supervision, validation, visualization, writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the School of Chemistry, University of Bristol for access to the mass spectrometry facility.Notes and references
- K. Jovic, T. Nitsche, C. Lang, J. P. Blinco, K. De Bruycker and C. Barner-Kowollik, Polym. Chem., 2019, 10, 3241 RSC.
- J. Fenn, M. Mann, C. Meng, S. Wong and C. Whitehouse, Science, 1989, 246, 64 CrossRef CAS PubMed.
- L. Latourte, J. Blais, J. Tabet and R. Cole, Anal. Chem., 1997, 69, 2742 CrossRef CAS.
- C. Guttman, S. Wetzel, W. Blair, B. Fanconi, J. Girard, R. Goldschmidt, W. Wallace and D. van der Hart, Anal. Chem., 2001, 73, 1252 CrossRef CAS.
- C. Guttman, S. Wetzel, K. Flynn, B. Fanconi, D. VanderHart and W. Wallace, Anal. Chem., 2005, 77, 4539 CrossRef CAS PubMed.
- R. Knochenmuss, E. Lehmann and R. Zenobi, Eur. J. Mass Spectrom., 1998, 4, 421 CrossRef CAS.
- R. Knochenmuss, Annu. Rev. Anal. Chem., 2016, 9, 365 CrossRef PubMed.
- M. Karas, M. Glückmann and J. Schäfer, J. Mass Spectrom., 2000, 35, 1 CrossRef CAS.
- M. Karas and R. Krüger, Chem. Rev., 2003, 103, 427 CrossRef CAS PubMed.
- J. Zhang, V. Frankevich, R. Knochenmuss, S. D. Friess and R. Zenobi, J. Am. Soc. Mass Spectrom., 2003, 14, 42 CrossRef CAS PubMed.
- A. J. Hoteling, M. L. Piotrowski and K. G. Owens, Rapid Commun. Mass Spectrom., 2020, 34(S2), e8630 CrossRef CAS PubMed.
- A. D. Warren, U. Conway, C. J. Arthur and P. J. Gates, J. Mass Spectrom., 2016, 51, 491 CrossRef CAS PubMed.
- A. D. Warren, D. J. Mitchell and P. J. Gates, Eur. J. Mass Spectrom., 2018, 24, 89 CrossRef CAS PubMed.
- M. Girod, M. Mazarin, T. N. T. Phan, D. Gigmes and L. Charles, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3380 CrossRef CAS.
- R. Chen, T. Yalcin, W. Wallace, C. Guttman and L. Li, J. Am. Soc. Mass Spectrom., 2001, 12, 1186 CrossRef CAS PubMed.
- L. Nyadong, J. P. Quinn, C. S. Hsu, C. L. Hendrickson, R. P. Rodgers and A. G. Marshall, Anal. Chem., 2012, 84, 7131 CrossRef CAS PubMed.
- A. Ehlers, C. de Koster, R. Meier and K. Lammertsma, J. Phys. Chem. A, 2001, 105, 8691 CrossRef CAS.
- A. Hoberg, D. Haddleton, P. Derrick, A. Jackson and J. Scrivens, Eur. J. Mass Spectrom., 1998, 4, 435 CrossRef CAS.
- A. Jackson, H. Yates, W. MacDonald, J. Scrivens, G. Critchley, J. Brown, M. Deery, K. Jennings and C. Brookes, J. Am. Soc. Mass Spectrom., 1997, 8, 132 CrossRef CAS.
- K. Shimada, S. Matsuyama, T. Saito, S. Kinugasa, R. Nagahata and S.-I. Kawabata, Int. J. Mass Spectrom., 2005, 247, 85 CrossRef CAS.
- R. Chen, X. Yu and L. Li, J. Am. Soc. Mass Spectrom., 2002, 13, 888 CrossRef CAS PubMed.
- J. Wei, A. W. T. Bristow and P. B. O’Connor, J. Am. Soc. Mass Spectrom., 2015, 26, 166 CrossRef CAS PubMed.
- C. Black, C. Poile, J. Langley and J. Herniman, Rapid Commun. Mass Spectrom., 2006, 20, 1053 CrossRef CAS PubMed.
- G. J. Langley, J. M. Herniman and M. S. Townell, Rapid Commun. Mass Spectrom., 2007, 21, 180 CrossRef CAS PubMed.
- S. Cha and E. S. Yeung, Anal. Chem., 2007, 79, 2373 CrossRef CAS PubMed.
- S. Y. Xu, Y. F. Li, H. F. Zou, J. S. Qiu, Z. Guo and B. C. Guo, Anal. Chem., 2003, 75, 6191 CrossRef CAS PubMed.
- J. P. Huang, C. H. Yuan, J. Shiea and Y. C. Chen, J. Anal. Toxicol., 1999, 23, 337 CrossRef CAS PubMed.
- J. Sunner, E. Dratz and Y. C. Chen, Anal. Chem., 1995, 67, 4335 CrossRef CAS PubMed.
- S. Peng, M. Elder, N. Ahlmann, N. Hoffman and J. Franzke, Rapid Commun. Mass Spectrom., 2005, 19, 2789 CrossRef CAS PubMed.
- H.-J. Kim, J.-K. Lee, S.-J. Park, H. W. Ro, D. Y. Yoo and D. Y. Yoon, Anal. Chem., 2000, 72, 5673 CrossRef CAS PubMed.
- Origin(Pro) 9.0. OriginLab Corporation, Northampton, MA, USA Search PubMed.
- S. Harrisson, Polym. Chem., 2018, 9, 1366 RSC.
- P. Juhasz and C. E. Costello, Rapid Commun. Mass Spectrom., 1993, 7, 343 CrossRef CAS PubMed.
- Y. Fukuyama, Mass Spectrom., 2015, 4, A0037 CrossRef PubMed.
- R. Deegan, O. Bakajin, T. Dupont, G. Huber, S. R. Nagel and T. A. Witten, Nature, 1997, 389, 827 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01493a |
| ‡ Current address: Kratos Analytical, Trafford Park, Manchester, M17 1GP, United Kingdom. |
| This journal is © The Royal Society of Chemistry 2021 |