
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural design of pyrene-functionalized TEMPO-containing polymers for enhanced electrochemical storage performance†
Wenwen
Xue
ab,
Hatice
Mutlu
 b,
Hongjiao
Li
c,
Wolfgang
Wenzel
c and
Patrick
Theato
*ab
b,
Hongjiao
Li
c,
Wolfgang
Wenzel
c and
Patrick
Theato
*ab
aInstitute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology (KIT), Engesserstr. 18, D-76131, Karlsruhe, Germany. E-mail: patrick.theato@kit.edu
bSoft Matter Synthesis Laboratory – Institute for Biological Interfaces 3 (IBG 3), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, D-76344, Eggenstein-Leopoldshafen, Germany
cInstitute of Nanotechnology, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 6th April 2021
Abstract
We demonstrate the importance of rational structural design of pyrene-functionalized radical (i.e. 2,2,6,6-tetramethyl-1-piperidinyloxy, TEMPO) copolymers for enhanced electrochemical performance by providing insightful guides for designing high-performance polymer-based electrodes for energy storage applications.
1. Introduction
Organic electrode materials (e.g. carbonyl compounds,1 organosulfur compounds,2 radical polymers,3 imine compounds,4etc.) as alternatives to transition-metal-based materials have attracted great attention over the years owing to their structural variations, along environmentally benign and cost-effective characteristics.5 Due to the excellent rate capability and extraordinary cycle life,6 radical containing polymers (RCP), e.g. poly(2,2,6,6-tetramethylpiperidinyloxy-4-yl methacrylate) (PTMA), have received tremendous attention; however, two major drawbacks are hindering their further application. One is the poor electrical conductivity of RCPs (e.g. 10−11 S cm−1 for PTMA),7 which impedes electron transport throughout the electrode layer, resulting in low discharging capacity; and another being the solubility of RCPs in conventional organic electrolytes, e.g. carbonate-based electrolytes, leading to a rapid self-discharge and capacity loss.8 To tackle these problems, various strategies have been attempted. For instance, the former challenge could be resolved by in situ carbon incorporation in the monomer phase prior to polymerization to obtain a nanoscale homogeneous polymer composite electrode with improved electrical conductivity.9 The latter challenge could be resolved by either crosslinking the radical polymers10 or immobilizing the RCPs onto the current collector,11 porous substrates or nanoparticles.12 Alternatively, there are methods that implement distinctive strategies to combat the aforementioned challenges in a synergetic manner. For example, PTMA was covalently anchored onto the surface of highly conductive carbon agents (HCAs), e.g. carbon nanotubes (CNTs) and graphene,13 to yield composites with improved electrical conductivity and rate capability. Analogously, molecular-level anchoring of multiple-pyrene functionalized RCPs on a HCA has resulted in enhanced electrochemical performance through π–π interactions between pyrene functional groups and HCAs.14 Since the covalent bonding method is synthetically tedious and may result in electrodes with impaired conductivity, the “molecular glue” strategy based on non-covalent π–π interactions is more appealing.14bAccordingly, the present study was motivated by the interest in developing a practical synthetic method for rational design of pyrene-functionalized radical polymers featuring 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) sites applicable in energy-related applications. Particularly, we aimed to reveal the impact of the nature of the spacer group (i.e. amide vs. ester) through which the pyrene moiety is tethered to the polymer backbone. In fact, to meet the demands of efficient energy storage, recently two types of hydrophobic polymeric structures with covalently bonded pyrene and TEMPO units have been reported.14 Both synthetic methods rely on the conventional post-oxidation method of piperidine derived precursor polymers obtained via controlled radical polymerization. In both cases, TEMPO conversions were in the typical range of 60–80%,8b and the pyrene moiety was separated from the polymer backbone by a methylene-ester linkage, i.e. –(CH2)–(CO)–O–. Compared to methylene-ester linkages, amide linkers appear to be more appealing with respect to stability and electronic properties (C–N bond order greater than 1), which result from the restricted rotation, low chemical reactivity and extended π-conjugation with planar conformations.15 Moreover, synthetically, amide bonds are significant building blocks for functional polymers, which can be efficiently achieved via amidation or amide transformation.16 Nevertheless, the understanding of the effects of amide bonds as tethering spacers on the characteristics of TEMPO–pyrene copolymers has remained unrevealed. Thus, in order to gain insight, polyacrylamide based radical polymer derivatives, i.e. poly[(1-pyrene acrylamide)-ran-(4-TEMPO acrylamide)] (P(PyAm-ran-TAm), e.g.P1 and P2 in Scheme 1) decorated with varying amide-bonded pyrene, were synthesized and the respective electrochemical properties were investigated by comparing with poly[(1-pyrenemethyl acrylate)-ran-(4-TEMPO acrylamide)] (P(PyMA-ran-TAm), e.g.P3 and P4 in Scheme 1), in which the pyrene moiety was inserted through a methylene-ester linker, in order to permit the evaluation of the spacer effect in the absence of strong electronic coupling with the aromatic unit. The copolymers (P1–P4) were further fabricated into electrodes through noncovalent π–π stacking between pyrene groups and the planar surface of CNTs in order to reveal their potential as energy storage materials.
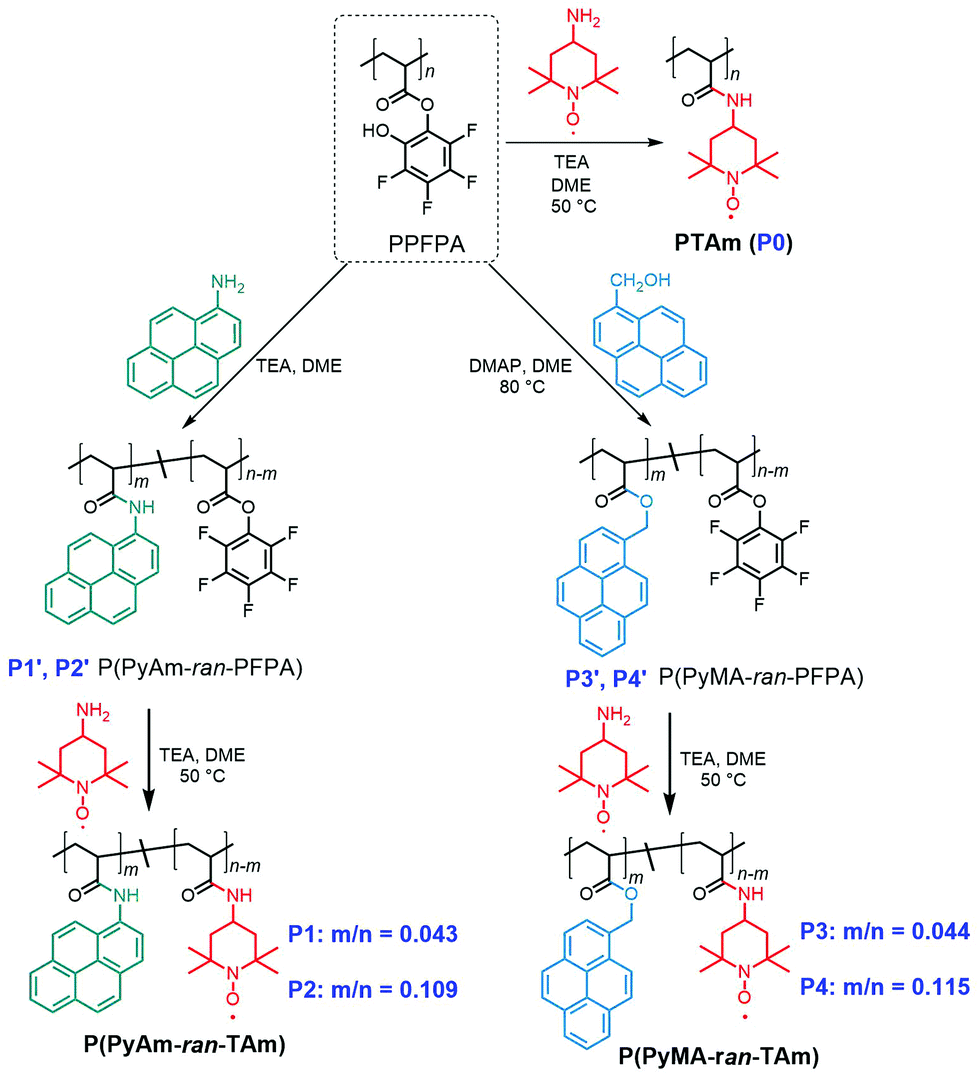 | ||
| Scheme 1 Synthetic routes towards (A) PTAm (P0), (B) P(PyAm-ran-TAm) (P1 and P2) and (C) P(PyMA-ran-TAm) (P3 and P4), respectively. | ||
2. Results and discussion
To efficiently incorporate TEMPO and different pyrene moieties (e.g. 1-pyrene acrylamide, PyAm, and 1-pyrenemethyl acrylate, PyMA) into the polymeric structure, while keeping the degree of polymerization and dispersity in principle the same, post-polymerization modification via an activated ester (namely pentafluorophenyl, PFP ester) was employed (Scheme 1). It is worth noting that the versatile features of post-polymerization modification can not only provide polymers with a similar degree of polymerization and dispersity, but also equip polymers with various functionalities in a well-controlled manner. As molecular weight plays an important role in the electrochemical properties of polymers,17 with post-polymerization modification, such impact can be neglected and only the influence from functional groups, e.g. methylene-ester linked pyrene and the amide-bonded pyrene, was taken into account, thus resulting in a precise study.In the first step, poly(pentafluorophenyl acrylate) (PPFPA), with a number average molecular weight (Mn) of 23.3 kDa and a dispersity (Đ) of 2.2, was synthesized via free-radical polymerization. In order to emphasize the influence of the amide-bonded pyrene moiety on the final material properties, initially a polyacrylamide backboned homopolymer, PTAm (Scheme 1, P0),18 with high radical content (96.2%) was prepared by reacting a PPFPA homopolymer with 4-amino-TEMPO. Furthermore, multi-pyrene PTAm samples (P1 and P2) were prepared via sequential one-pot post-modification of PPFPA with 1-aminopyrene and afterwards 4-amino-TEMPO. Random copolymer structures were targeted to favour the π–π interactions with the CNTs. By varying the reaction temperature during the aminolysis step (50 °C and 70 °C, respectively) of PPFPA with 1-aminopyrene, the pyrene content within the resulting P(PyAm-ran-TAm) could be altered. As shown in Fig. 1A, the UV/vis absorbance maxima for P2 solution (0.05 mg mL−1) in dimethylacetamide (DMAc) at 276 nm (S0 → S2), 326 nm (S0ν=0 → S1ν=1) and 342 nm (S0ν=0 → S1ν=0), corresponding to the two electronic transition moments of the pyrene group, was higher than that of P1. Based on the calibration plot of UV/vis absorption at 289 nm for 1-aminopyrene (Fig. S1†) via Beer–Lambert law,19 the amide-bonded pyrene content within P1 and P2 was determined to be 4.3% and 10.9%, respectively (Table 1). Whereas for P3 and P4, the pyrene content was regulated via the molar feed ratio between the 1-methanolpyrene nucleophile and PPFPA, and was found to be 4.4% and 11.5%, respectively (Table 1), based on the calibration plot of 1-pyrenemethanol at 344 nm (Fig. S1†). Fluorescence quenching of pyrene was observed when TEMPO moieties were introduced to P2, as shown in Fig. S2.† Nevertheless, for all samples (e.g.P1–P4), the content of the radical (i.e. TEMPO), was also calculated according to the spins recorded on an electron paramagnetic resonance (EPR) spectrometer (see Table 1) in DMAc solutions (0.99 mg mL−1), which was in agreement with the results of UV/vis analysis. P0, P1 and P3 exhibited a classic Lorentzian shape (Fig. 1B) due to the strong intermolecular spin-exchange interaction arising from the close spatial proximity between the paramagnetic units.20 Compared to P0, the intensity of P1 and P3 is relatively low, indicating a lower TEMPO content, whereas for P2 and P4, apart from the dominated broad Lorentzian-like component, a triplet splitting was also observed. The latter clearly implies a decrease in average radical–radical spacing induced by an increase in pyrene units along the polymer chain.20
 | ||
| Fig. 1 (A) UV/Vis spectra of pyrene-functionalized TEMPO containing polymers (P1–P4, c = 0.05 mg mL−1); (B) EPR spectra of P0–P4 and PPFPA-based precursor copolymers (P2′ and P4′) (c = 0.99 mg mL−1). Both spectra were recorded in DMAc at ambient temperature. (C) FTIR spectra of P2′, P4′ and P0–P4. | ||
| Samples | Pyrene contenta (%) | TEMPO contentb (%) |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (kDa) c (kDa) |
Đ |
R
Hd (nm) |
|---|---|---|---|---|---|
| a By UV/vis spectroscopy. b By EPR. c By size exclusion chromatography against the PMMA standard. d Based on number-size distribution via DLS with an error of 0.97 nm. | |||||
| PPFPA | — | — | 23.3 | 2.2 | 8.4 ± 1.8 |
| P0 | 0 | 96.2 | 48.5 | 3.0 | 4.2 ± 0 |
| P1 | 4.3 | 92.5 | 37.8 | 3.1 | 4.5 ± 1.0 |
| P2 | 10.9 | 87.3 | 30.9 | 2.8 | 3.6 ± 0.4 |
| P3 | 4.4 | 91.3 | 22.0 | 4.4 | 3.9 ± 0.4 |
| P4 | 11.5 | 85.1 | 20.8 | 4.4 | 4.9 ± 0.4 |
Importantly, it should be noted that all polymers analyzed in this work were derived from the same PPFPA parent polymer, thus possessing the same chemical and analytical information with regard to the polymer backbone structure with the same degree of polymerization and dispersity (Table 1). Indeed, as exhibited in FTIR spectra (Fig. 1C), the characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band at 1782 cm−1 and the arene CC band at 1515 cm−1 for the PFP ester disappeared completely in P0–P4. Meanwhile, new peaks at 1460 cm−1, 1530 cm−1, 1650 cm−1, and 1735 cm−1 corresponding to the vibrations of N–O stretching, N–H bending, CO (amide) stretching (P0–P2), and CO (new ester) stretching (P3 and P4), respectively, were observed as an indication of successful polymer modification. In comparison, P(PyAm-ran-PFPA) (P2′) and P(PyMA-ran-PFPA) (P4′), which are the direct precursor polymers of P2 and P4, respectively, exhibited decreased infrared absorption at 1782 cm−1 (CO, activated ester) aside from the appearance of amide CO (1650 cm−1) and newly formed ester CO (1735 cm−1) characteristic bands, respectively.
O band at 1782 cm−1 and the arene CC band at 1515 cm−1 for the PFP ester disappeared completely in P0–P4. Meanwhile, new peaks at 1460 cm−1, 1530 cm−1, 1650 cm−1, and 1735 cm−1 corresponding to the vibrations of N–O stretching, N–H bending, CO (amide) stretching (P0–P2), and CO (new ester) stretching (P3 and P4), respectively, were observed as an indication of successful polymer modification. In comparison, P(PyAm-ran-PFPA) (P2′) and P(PyMA-ran-PFPA) (P4′), which are the direct precursor polymers of P2 and P4, respectively, exhibited decreased infrared absorption at 1782 cm−1 (CO, activated ester) aside from the appearance of amide CO (1650 cm−1) and newly formed ester CO (1735 cm−1) characteristic bands, respectively.
The evolution of the size distribution of the relevant polymer solutions in DMAc was determined via dynamic light scattering (DLS, Fig. 2A). PPFPA showed a number-weighted size distribution (RH) of 8.4 nm (Table 1), while P0 exhibited a smaller size by 4.2 nm, which might have resulted from the hydrogen bonding between the adjacent amide bond among repeating units. With 4.3% of amide-bonded pyrene content, assessed via UV/vis analysis, P1 displayed an extended chain conformation as a result of the randomly incorporated amide-bonded pyrene moieties, and hence showed a number-weighted size distribution of 4.5 nm. Intriguingly, as the pyrene content increased from 4.3% to 10.9% in P(PyAm-ran-TAm), the size of the polymer coil became smaller, which was 3.6 nm for P2 (with amide-bonded pyrene). The latter indicated that the entanglements of polymer chains were locked by multiple π–π interactions of the pendant pyrene groups, which induced and maintained a coiled structure, whereas, for P(PyMA-ran-TAm), an increase in RH by 1 nm was observed with the increase of pyrene content from 4.4% to 11.5%. This may arise from the decreased rigidity of the polymer structure associated with the methylene-ester spacer.
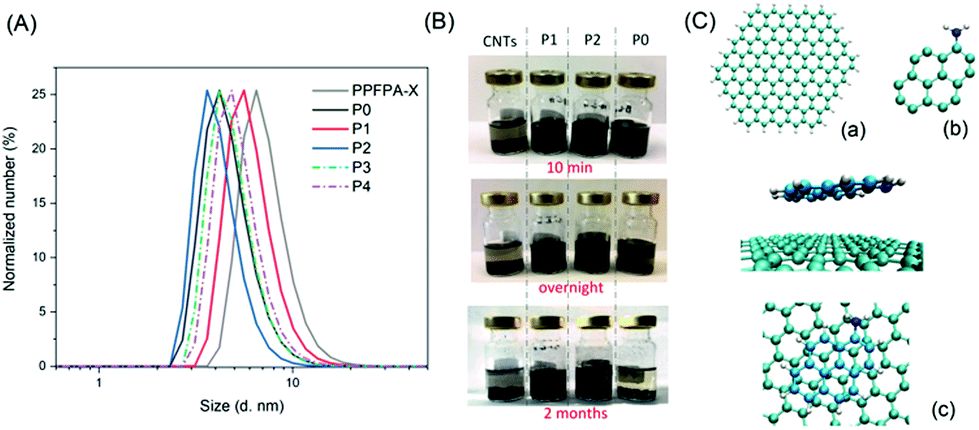 | ||
| Fig. 2 (A) DLS traces of PPFPA and P0–P4. All traces were recorded in DMAc (c = 0.67 mg mL−1) at ambient temperature. (B) Composite suspensions of P0–P2 (3.0 mg) and CNTs (3.0 mg) in THF (4.5 mL) prepared with a homogenizer. Suspensions at the extreme left are blank samples with CNTs (3.0 mg) in THF (4.5 mL). (C) Molecular models of (a) the graphene layer, (b) 1-amino-pyrene and (c) adsorption geometry of 1-amino-pyrene/graphene. | ||
As the next step is the fabrication of the TEMPO-pyrene copolymers into electrodes through noncovalent π–π stacking between pyrene groups and CNT, the existence of such π–π interactions was examined. As shown in Fig. 2B, with the aid of a homogenizer, CNTs were well dispersed in the two amide-bonded pyrene copolymer (P1 and P2) solutions, without apparent sedimentation even after 2 months. Conversely, a rapid settling of CNTs was observed within minutes after homogenizing. For P0 solution, the suspension state could only be maintained for a bit more than one hour. Taken together, these results suggest that the introduction of pyrene moieties into the PTAm-based polymer structure indeed assists in intramolecular π–π interactions with CNTs, which may facilitate the effective immobilization of polymer chains on the surface of CNTs. To compare the interaction between each type of functional group on the polymer and CNTs, quantum mechanical calculations based on Møller–Plesset perturbation theory were performed (see ESI 1.3†).14b,21 Indeed, both 1-amino-pyrene and 1-pyrenemethanol prefer to lie almost flat on the graphene flake, which differs from 4-amino-TEMPO (Fig. 2C and S3†), and show much higher interaction with CNTs compared to 4-amino-TEMPO (Table S1†). Besides, 1-amino-pyrene displayed more affinity towards the graphene layer than 1-pyrenemethanol with interaction energy, ΔE, values of −0.022 eV and 0.006 eV, respectively. The latter implies that, theoretically, 1-aminopyrene/CNTs possess a stronger interaction than 1-pyrenemethanol/CNTs.
To gain a better understanding on the structure–electronic property relationship of the two pyrene containing reactive precursor polymers i.e.P2′ and P4′, cyclic voltammetry (CV) was conducted. To avoid the influence of CNTs on CV,22 no CNTs were added in cathodes for CV measurements. Regarding P2′, redox peaks at 3.55 V were detected with slightly enhanced intensity as the scan number increased from 1 to 5 (Fig. 3A). Similarly, redox peaks were observed for P3′, while a migrating oxidation peak was detected towards the 5th scan, accompanied by a decrease in both oxidation and reduction peaks (Fig. 3B). Compared to the methylene-ester linked pyrene polymer, i.e.P4′, the polymer with amide pyrene moieties, i.e.P2′, is relatively more stable, which may be due to the cross conjugation of the amide bond and pyrene units.23 These results emphasize the significant influence of the structural linkage on the electrochemical properties of pyrene moieties.
 | ||
| Fig. 3 CV curves of (A) P2′ and (B) P4′ at a scanning rate of 0.1 mV s−1 within the potential range of 3.2–4.0 V vs. Li/Li+. The cathodes were composed of the active polymer/CB/PVdF = 10/80/10 (wt%). | ||
However, the CV curves of P2 and P4 showed one pair of redox peaks at around 3.6 V, corresponding to the redox reaction between TEMPO and oxoammonium, and no apparent redox peaks were detected for the amide-bonded pyrene derivatives (Fig. 4A). The latter could result from either the overlap of redox peaks for amide-bonded pyrene and TEMPO derivatives, as both appeared at around 3.6 V, or the low current intensity arising from the amide-bonded pyrene. Nevertheless, a narrow anodic and cathodic peak separation was observed for both P2 and P4, which was 29.3 mV and 34.2 mV, respectively.
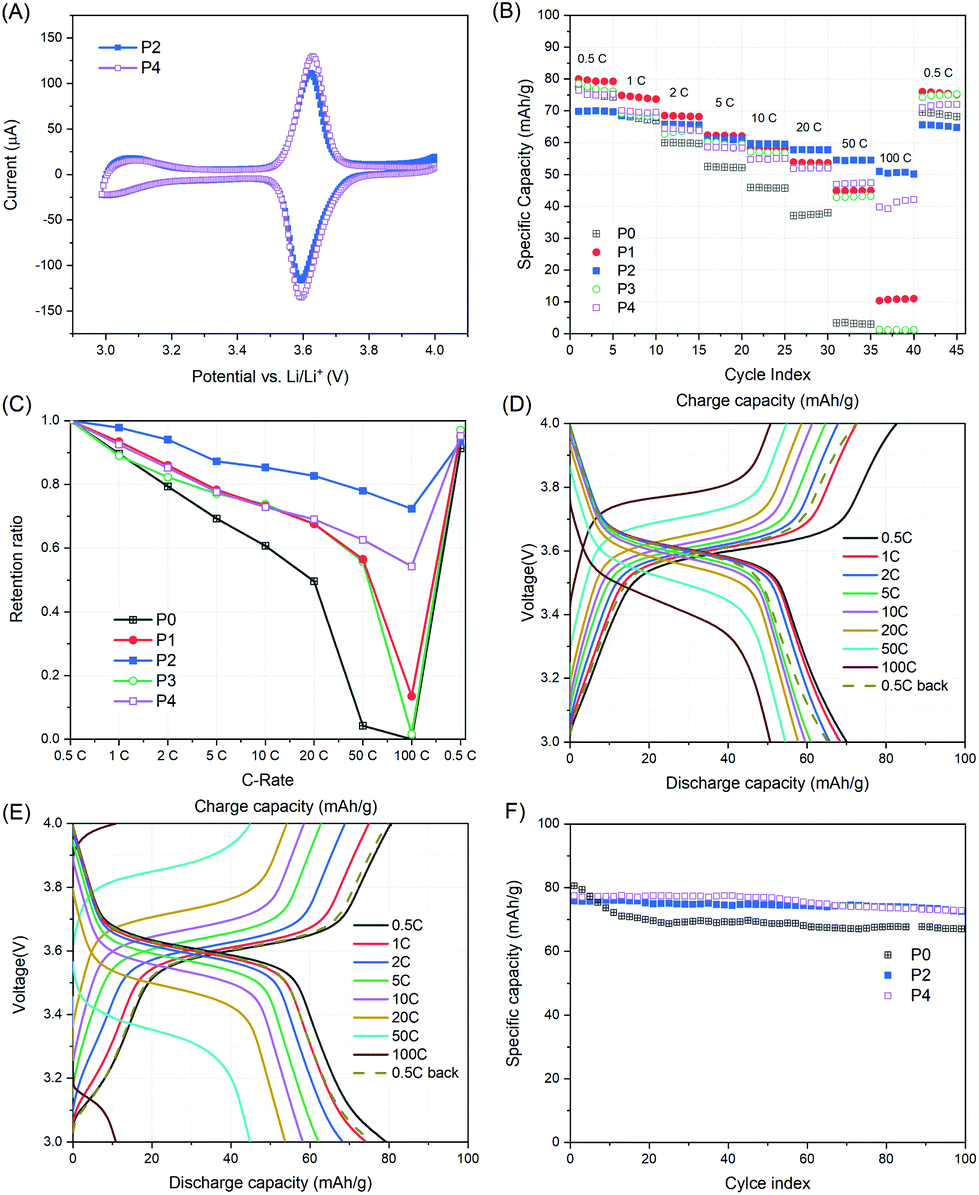 | ||
| Fig. 4 (A) Cyclic voltammetry curves of P2 and P4 at a scan rate of 0.1 mV s−1 within the potential range of 3.2–4.0 V vs. Li/Li+. (B, C) Rate performance of P0–P4 at various C-rates; (D, E) charge–discharge profiles of P1 and P2, respectively. (F) Cycling performances of P0, P2 and P4 at 1C. The cathodes were composed of the active polymer/CNTs/CB/PVdF = 10/10/70/10 (wt%). | ||
The rate performances for P0–P4 were investigated from 0.5C to 100C, as shown in Fig. 4B and C. At 0.5C, P1 delivered a capacity of 80.0 mA h g−1, which was slightly higher (3–4%) than those of P0 and P3. As the C-rate increased stepwise from 0.5C to 20C, the discharge capacity of P0, P1 and P3 steadily decreased (∼10%, 6% and 6% per step, respectively), while from 50C to 100C, a dramatic decline (∼7 and 9 times higher for P1 and P3, respectively) was observed. However, for P2 and P4, a steady but relatively low decline (4% and 6% per step, respectively) was observed in the whole course of 0.5C to 100C. Due to the slower decay rate, although the TEMPO content of P2 and P4 was lower than that of P1 and P3, their discharge capacities exceeded those of P1 and P3 at 10C and 20C, respectively. When cycled back at 0.5C, all samples exhibited high capacity retention (over 91.3%). Therefore, the rate performance in terms of capacity retention is in the order of P2 > P4 > P1 > P3 > P0. Besides, distinct voltage plateaus at around 3.6 V were observed for all samples at various C-rates (Fig. 4E, F and S4†). These results indicate that the incorporation of pyrene units into PTAm indeed results in enhanced rate performance by π–π stacking between pyrene groups and the planar surface of the CNT arising from improved conductivity. Moreover, due to the stable redox properties of the amide-bonded pyrene unit and its higher π–π stacking interaction with CNTs, P(PyAm-ran-Tam) showed better rate performance than P(PyMA-ran-Tam). Furthermore, the higher the pyrene content within the active polymer, the higher the retention ratio that could be retained for the resulting cathode.
Apart from rate performances, the cycle performances of P2 and P4 at 1C were also investigated (Fig. 4F). P0 exhibited the highest initial capacity of 80.6 mA h g−1. However, a fast capacity decline of 13.3% was observed in the following 20 cycles, which might have resulted from the partial dissolution of P0 in the electrolyte. P2 and P4 showed improved cycle lives with only 4.4% and 5.9% capacity loss after 100 cycles, implying the good anchor effect of pyrene moieties on CNTs.
It is worth noting that the ideal charge–discharge profiles for P2–P4 in the voltage range of 3–4 V could hardly be achieved at a low C-rate (e.g. 0.1C) due to the occurrence of side reactions at high voltages (e.g. 3.9–4 V). This can be evidenced by the evolution of coulombic efficiency (CE) at various C-rates and the voltage profiles at 0.1C (Table 2 and Fig. 5). Table 2 reveals that the CE for all samples at 0.5C was on the lower side compared to other C-rates, especially for copolymers with methylene-ester linkages and high pyrene content. For each sample, the CE grew gradually till 99.5% or even higher as the C-rate increased. Below 20C, the CE follows the order of P0 ≈ P1 > P2 > P3 > P4. These results indicate that side reactions accompanied at a low C-rate (e.g. ≤0.5C) for pyrene–TEMPO copolymers, especially the ones with methylene-ester linkages and high pyrene content, i.e.P3 and P4. Moreover, the voltage profiles of P2–P4 at 0.1C showed a plateau at high voltages (3.9–4.0 V) (Fig. 5A and S4†), while P0 and P1 did not (Fig. 5B and S4†). CV measurements of P4 in potentiostatic mode at a low scan rate of 0.025 mV s−1, corresponding to similar charging–discharging time at 0.1C, revealed that oxidation occurred at a voltage higher than 3.9 V (Fig. 5B); while it was not the case at a higher scan rate, e.g. 0.1 mV s−1, as shown in Fig. 4A. Altogether, we postulate that pyrene–TEMPO copolymers with amide–pyrene linkages do have an advantage over methylene-ester linked copolymers as the latter are more prone to side reactions at low C-rates. Furthermore, there exists a balance point on the pyrene content of P(PyMA-ran-Tam) between 4.3% and 10.9%, at which not only battery performance can be improved, but also side reactions can be minimized.
 | ||
| Fig. 5 (A) Voltage profiles of P1 and P4 at 0.1C. (D) CV curve of P4 at a scan rate of 0.025 mV s−1. The cathodes were composed of the active polymer/CNTs/CB/PVdF = 10/10/70/10 (wt%). | ||
| Samples | 0.5C (%) | 1C (%) | 2C (%) | 5C (%) | 10C (%) | 20C (%) | 50C (%) | 100C (%) |
|---|---|---|---|---|---|---|---|---|
| P0 | 98.2 | 99.2 | 99.7 | 100 | 99.9 | 100 | 99.8 | 100 |
| P1 | 98.1 | 99.0 | 99.2 | 99.1 | 99.5 | 99.5 | 99.8 | 99.5 |
| P2 | 84.6 | 94.6 | 97.4 | 94.5 | 97.2 | 98.4 | 99.5 | 99.6 |
| P3 | 79.4 | 84.8 | 90.4 | 97.2 | 98.3 | 98.8 | 99.5 | 87.9 |
| P4 | 72.6 | 87.0 | 92.6 | 96.1 | 97.3 | 98.5 | 99.3 | 99.5 |
3. Conclusions
In summary, theory and experiment have been used to study the effect of the structural design of multiple pyrene functionalized TEMPO containing polymers to improve battery performance. Through post-polymerization modification via an activated ester, amide-bonded pyrene and TEMPO moieties were efficiently incorporated into the same polymer chain. We have revealed that, compared to methylene-ester linked pyrene, the amide-bonded pyrene containing polymers showed relatively stable redox peaks at around 3.55 V vs. Li/Li+. Moreover, due to the higher π–π interaction with CNTs, pyrene–TEMPO copolymers with amide linkages cause better improvement of the battery performance, especially rate performance, than methylene-ester linked pyrene–TEMPO copolymers. Whereas high pyrene content may induce side reactions at low C-rates, e.g. <0.5C, which is even worse on copolymers with methylene-ester linkages. Nevertheless, we have highlighted the often-overlooked role of through-space electrostatic substituent effects via amide-bonded TEMPO–pyrene copolymers, which could assist in the design of nitroxide radical structures for different applications, such as catalysis and batteries.4. Experimental section
4.1 Materials and methods
All solvents and reagents used in synthetic procedures were purchased from Sigma-Aldrich, Fisher Scientific, ABCR, TCI, and VWR and used as received unless otherwise stated. Deuterated solvents were purchased from Eurisotop. Poly(vinylidene fluoride) (PVdF, Solef 6020/100), carbon black super P (CB), carbon nanotubes (CNTs, Elicarb), N-methyl-2-pyrrolidone (NMP), electrolyte LP30 (1.0 M LiPF6 in ethylene carbonate/dimethyl carbonate = 50/50 (v/v)) and lithium foil (0.75 mm thick) were purchased from Solvay, Timcal, Thomas Swan, ACROS Organics, Sigma-Aldrich and Alfa Aesar, respectively. All materials and substrates were used as received unless otherwise noted. A separator (Celgard H2013) was received from Celgard LLC. Yields refer to isolated and purified products. NMR spectra were recorded on a Bruker Ascend 400 MHz NMR spectrometer in deuterated solvents. Chemical shifts were reported in ppm with TMS as an internal standard. EPR spectra were collected on a Bruker EMAXnano. The measurement was performed in dimethylacetamide (DMAc) using a capillary with an inner diameter of 0.9 mm. FTIR spectroscopy was conducted on a Bruker Vertex70 spectrometer in an attenuated total reflectance (ATR) setup. Size exclusion chromatography (SEC) data were obtained by utilizing a PL-SEC 50 Plus with a sample concentration of about 2 mg mL−1 in DMAc. For calculating the molecular weight and Đ, PMMA was used for calibration. Ultraviolet-visible (UV/vis) absorptions were recorded on a Varian UV-Visible Spectrophotometer CARY 300 BIO with both monomers (0.001–0.02 mg mL−1) and polymers (0.05 mg mL−1) dissolved in DMAc and analysed in a quartz cuvette within a scanning range from 200 to 800 nm. Fluorescence spectrometry measurements were performed on a Varian Fluorescence Spectrophotometer CARY Eclipse with an excitation wavelength of 342 nm. Fluorescence spectra were measured in a range of 350 to 650 nm in steps of 1 nm. Dynamic light scattering (DLS) analysis was carried out on a Zetasizer Nano ZS light scattering apparatus (Malvern Instruments, UK) with a polymer solution of 1 mg mL−1 in DMAc. Thermogravimetric analysis (TGA) was performed on a TGA55 analyser (TA instruments) from room temperature to 700 °C at a scan rate of 10 °C min−1 under a nitrogen atmosphere. All electrochemical tests were conducted on 2032-type coin cells assembled in an argon-atmosphere glove box. Cyclic voltammetry (CV) measurements were performed on a potentiostat system (PGSTAT128N Metrohm Autolab) at a scan rate of 0.1 mV s−1 or 0.025 mV s−1. Both galvanostatic charge/discharge tests were conducted on an Arbin BT2143 battery cycler at room temperature in a range of 3.0–4.0 V vs. Li/Li+. The applied C-rate currents were calculated based on the theoretical specific capacity of PTAm, which is 118 mA h g−1.Quantification of the pyrene groups incorporated into the polymeric structure was based on UV/vis absorption data.19 The extinction coefficients for 1-aminopyrene at 289 nm and for 1-pyrenemethanol at 344 nm were determined using a calibration curve in DMAc to ensure an adequate solvation of the pyrene derivatives, which is a linear line by fitting the absorbance maximum as a function of the solution concentration (see Fig. S1†). The slope of the line corresponds to the extinction coefficient, which was determined to be 22876 cm−1 M−1 and 41167 cm−1 M−1, respectively, according to Beer–Lambert law (see eqn (1)).
 | (1) |
4.2 Synthetic procedures
PPFPA was synthesized according to ref. 25. A solution of PFPA (5.00 g, 21.00 mmol) and azobisisobutyronitrile (AIBN, 34.50 mg, 0.21 mmol) in dry 1,4-dioxane (5 mL) was firstly added into a Schlenk tube. Three freeze–pump–thaw cycles were performed to degas the solution. The flask was transferred to a preheated oil bath at 70 °C and stirred overnight. Afterwards, the reaction mixture was exposed to air to quench the polymerization. The solution was diluted with chloroform and precipitated from methanol to afford a white solid. The white solid was re-dissolved in chloroform and re-precipitated from methanol for another two cycles. After drying under vacuum at 40 °C for 72 h, the polymer was obtained in 90% yield. 1H NMR (400 MHz, CDCl3) δ 3.27–2.70 (m, 1H), 2.55–1.69 (m, 2H). 19F NMR (377 MHz, CDCl3) δ −153.19 (s), −157.14 (d, J = 279.3 Hz), −162.20 (s). IR (ATR mode): 1782 cm−1 (CO stretch of PFP-ester), 1516 cm−1 (aromatic ring stretch).
4.3 DFT calculations
The graphene flake was modeled as a large aromatic hydrocarbon with 130 carbon atoms and the edges terminated with hydrogen atoms. TEMPO, 1-amino-pyrene, and 1-pyrenemethanol molecules were located in the center of the flake to avoid the electronic influence from the edge carbons. Geometry optimization and ground state search were carried out within the TurboMole 7.3 program package [TURBOMOLE V7.3 2018. A Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007. http://www.turbomole.com (TURBOMOLE GmbH, 2007)] at the MP2 level of theory with cc-pVDZ basis sets. The interaction energy (ΔE) between the molecular and graphene flakes was defined as the energy difference between the graphene/molecular complex and infinitely separated fragments (graphene/1-amino-pyrene, 1-pyrenemethanol and 4-amino-TEMPO). As confirmed by the literature,26 MP2 methods could give a reliable description of the adsorption of small organic molecules on graphene. The interaction energy for each system is listed in Table S1.†4.4 Preparation of electrodes and coin cell assembly
To eliminate the influence of CNTs on CV measurements (3.2–4.0 V) of coin cells fabricated from P2′ and P3′, cathodes were prepared without CNTs. The active polymer (10 mg, 10 wt%), conductive carbon (80 mg, 80 wt%) and PVdF (10 mg, 10 wt%) were mixed in NMP overnight to afford a homogeneous slurry, followed by blade coating of the slurry onto aluminium foil (thickness 30 μm) and drying in an oven at 80 °C overnight.The active polymer material (P0–P4, 10 mg) and CNTs (10 mg) were dispersed in a mixture of NMP (0.4 mL) and tetrahydrofuran (THF, 2 mL) with the aid of a homogenizer (IKA, T 10 basic ULTRA-TURRAX). A suspension of carbon black (70 mg) in NMP (1.3 mL) was prepared as well. Afterwards, the two aforementioned colloidal dispersions were vigorously mixed together and put on a rotary evaporator to degas and remove the THF and a part of NMP at 40 °C. In the end, PVdF (10 mg) solution in NMP (10 wt%) was added and homogenized gently to obtain a bubble-free and uniform slurry.
Composite cathodes were formed by blade coating of the above-mentioned slurries onto aluminium foil and drying at room temperature and 80 °C under vacuum sequentially. The typical loading of the active polymer/CNT hybrid cathode was 1.75 ± 0.15 mg cm−2. The thickness of the hybrid electrodes was about 50 μm. The coin cell (CR2032) was fabricated by stacking the polymer-based cathode, lithium metal anode and a separator film with the LP30 electrolyte in an argon filled glove box (MBraun) where both O2 and H2O contents were less than 0.1 ppm.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support has been provided in part by the China Scholarship Council (CSC grant No.: 201504910677), STIBET DAAD, DFG (grant No. EXC-2082/1-390761711) and European Union's Horizon 2020 research and innovation programme (grant No.: 957189). The authors thank Prof. P. Levkin (KIT) for access to DLS equipment.References
- L. Zhu, G. Ding, L. Xie, X. Cao, J. Liu, X. Lei and J. Ma, Chem. Mater., 2019, 31, 8582 CrossRef CAS.
- Y. NuLi, Z. Guo, H. Liu and J. Yang, Electrochem. Commun., 2007, 9, 1913 CrossRef CAS.
- K. Nakahara, S. Iwasa, M. Satoh, Y. Morioka, J. Iriyama, M. Suguro and E. Hasegawa, Chem. Phys. Lett., 2002, 359, 351 CrossRef CAS.
- T. Matsunaga, T. Kubota, T. Sugimoto and M. Satoh, Chem. Lett., 2011, 40, 750 CrossRef CAS.
- Y. Lu and J. Chen, Nat. Rev. Chem., 2020, 1 Search PubMed.
- T. Janoschka, M. D. Hager and U. S. Schubert, Adv. Mater., 2012, 24, 6397 CrossRef CAS PubMed.
- Y. Zhang, A. Park, A. Cintora, S. R. McMillan, N. J. Harmon, A. Moehle, M. E. Flatté, G. D. Fuchs and C. K. Ober, J. Mater. Chem. C, 2017, 6, 111 RSC.
- (a) K. Nakahara, J. Iriyama, S. Iwasa, M. Suguro, M. Satoh and E. J. Cairns, J. Power Sources, 2007, 165, 398 CrossRef CAS; (b) K. Zhang, Y. Hu, L. Wang, J. Fan, M. J. Monteiro and Z. Jia, Polym. Chem., 2017, 8, 1815 RSC.
- A. Vlad, J. Rolland, G. Hauffman, B. Ernould and J. F. Gohy, ChemSusChem, 2015, 8, 1692 CrossRef CAS PubMed.
- (a) T. Suga, H. Konishi and H. Nishide, Chem. Commun., 2007, 1730 RSC; (b) S. Wang, A. M. G. Park, P. Flouda, A. D. Easley, F. Li, T. Ma, G. D. Fuchs and J. L. Lutkenhaus, ChemSusChem, 2020, 13, 2371 CrossRef CAS PubMed.
- Y.-H. Wang, M.-K. Hung, C.-H. Lin, H.-C. Lin and J.-T. Lee, Chem. Commun., 2011, 47, 1249 RSC.
- (a) Y. Kim, C. Jo, J. Lee, C. W. Lee and S. Yoon, J. Mater. Chem., 2012, 22, 1453 RSC; (b) H.-C. Lin, C.-C. Li and J.-T. Lee, J. Power Sources, 2011, 196, 8098 CrossRef CAS.
- (a) B. Ernould, M. Devos, J.-P. Bourgeois, J. Rolland, A. Vlad and J.-F. Gohy, J. Mater. Chem. A, 2015, 3, 8832 RSC; (b) Y. Li, Z. Jian, M. Lang, C. Zhang and X. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 17352 CrossRef CAS PubMed.
- (a) K. Zhang, Y. Hu, L. Wang, M. J. Monteiro and Z. Jia, ACS Appl. Mater. Interfaces, 2017, 9, 34900 CrossRef CAS PubMed; (b) N. Hergué, B. Ernould, A. Minoia, R. Lazzaroni, J. F. Gohy, P. Dubois and O. Coulembier, Batteries Supercaps, 2018, 1, 102 CrossRef.
- (a) G. Fischer, Chem. Soc. Rev., 2000, 29, 119 RSC; (b) B. M. Bulheller, A. Rodger and J. D. Hirst, Phys. Chem. Chem. Phys., 2007, 9, 2020 RSC.
- J. Ren, H. Liu, X. Zhang, Y. Hu, G. Zhou and T. Masuda, Polym. Chem., 2020, 11, 3427 RSC.
- K. Zhang, Y. Hu, L. Wang, J. Fan, M. J. Monteiro and Z. Jia, Polym. Chem., 2017, 8, 1815 RSC.
- W. Xue, H. Mutlu and P. Theato, Eur. Polym. J., 2020, 130, 109660 CrossRef CAS.
- R. M. Arnold, G. R. Sheppard and J. Locklin, Macromolecules, 2012, 45, 5444 CrossRef CAS.
- D. C. Bobela, B. K. Hughes, W. A. Braunecker, T. W. Kemper, R. E. Larsen and T. Gennett, J. Phys. Chem. Lett., 2015, 6, 1414 CrossRef CAS PubMed.
- P. Lazar, F. Karlicky, P. Jurečka, M. S. Kocman, E. Otyepková, K. R. Šafářová and M. Otyepka, J. Am. Chem. Soc., 2013, 135, 6372 CrossRef CAS PubMed.
- Z. Song, T. Xu, M. L. Gordin, Y.-B. Jiang, I.-T. Bae, Q. Xiao, H. Zhan, J. Liu and D. Wang, Nano Lett., 2012, 12, 2205 CrossRef CAS PubMed.
- (a) L. Ji, I. Krummenacher, A. Friedrich, A. Lorbach, M. Haehnel, K. Edkins, H. Braunschweig and T. B. Marder, J. Org. Chem., 2018, 83, 3599 CrossRef CAS PubMed; (b) E. M. Espinoza, J. A. Clark, J. B. Derr, D. Bao, B. Georgieva, F. H. Quina and V. I. Vullev, ACS Omega, 2018, 3, 12857 CrossRef CAS PubMed.
- R. De Coen, N. Vanparijs, M. D. Risseeuw, L. Lybaert, B. Louage, S. De Koker, V. Kumar, J. Grooten, L. Taylor and N. Ayres, Biomacromolecules, 2016, 17, 2479 CrossRef CAS PubMed.
- A. Das and P. Theato, Macromolecules, 2015, 48, 8695 CrossRef CAS.
- P. Lazar, F. Karlicky, P. Jurečka, M. S. Kocman, E. Otyepková, K. R. Šafářová and M. Otyepka, J. Am. Chem. Soc., 2013, 135, 6372 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01421d |
| This journal is © The Royal Society of Chemistry 2021 |