
Clarification of the effects of topological isomers on the mechanical strength of comb polyurethane†
Daisuke
Aoki
 and
Hiroharu
Ajiro
*
and
Hiroharu
Ajiro
*
Graduate School of Materials Science, Nara Institute of Science and Technology, 8916-5 Takayama-cho, Nara 630-0192, Japan. E-mail: ajiro@ms.naist.jp
First published on 21st December 2020
Abstract
We designed and synthesized a topological isomer with comb and linear polyurethanes (PUs), bearing one, two, and three units of oligo(ethylene glycol) (OEG) at the side chain. Surprisingly, comb PUs with OEG side chain lengths of two and three units showed about twice as much stress at the break as those of the corresponding linear isomers. The analysis of rheological and structural parameters revealed that the comb PU in this study was more suitable for maintaining entanglement concerning the side chain length than that of the conventional comb polymer. Since the comb PU showed high zero shear viscosity like the conventional comb polymer, it was found that there is molecular chain friction due to the side chain. Therefore, it is assumed that both the entanglement compatibility and the molecular chain friction resulted in an enhancement of the mechanical strength, which was achieved by the topological isomer.
Introduction
It is essential to develop the technology of tailoring properties of a single macromolecular material for industrial applications. The topologies of polymers, such as comb,1 branched,2,3 star,4 and cyclic,5,6 provide a range of material properties based on the arrangement of polymer chains. It is known that there are topological effects in biomacromolecules, such as glycoproteins7 and polysaccharides,8 on the performance of functions controlling their lubrication, shock absorption, cytoprotection, and viscoelasticity. As for artificial materials, it is also known that the topology of polymer chains can control the mechanical properties, such as in commercialized vibration damping materials9 and polyethylene plastics.10In general, it is believed that the branched structure makes the material flexible, when the mechanical properties of branched and linear polymers are compared. For example, it is well known that the branched structure of polyethylene decreases the stiffness and increases the ductility. The control of these mechanical properties allows for industrial application in a wide range of fields.10,11 Incidentally, amorphous polymers without a strong intermolecular interaction do not usually exhibit a significant change in the mechanical properties on stress–strain curves due to the branched topology. The long-chain branch structure of the polycarbonate, which is a typical amorphous polymer, shows almost the same stress–strain curve as the linear structure.12 It has been reported that long-chain branched polycarbonates show almost the same stress–strain curves as linear structures except for their impact enhancement and environmental stress cracking properties. These changes in the mechanical properties are attributed to the polymer topology which could influence complex factors such as entanglement, crystallinity, and morphology.13–15
Among branched polymers, comb polymers have recently attracted attention for helping the understanding of the mechanical and rheological properties of the branched topology by the quantification of the molecular structure defined by the backbone length, side chain length, and grafting density.1,15,16 The unified scaling model beyond the chemical structure can describe the rheological properties, such as the entanglement plateau modulus,15,17 zero shear viscosity,18 strain hardening factor,18 and relaxation time, allowing the prediction of mechanical properties19 in soft materials.
On the other hand, it has been reported that the comb topology in block copolymers with significantly different chemical properties of the backbone and side chains achieves both high toughness and strength. For example, in the polyisoprene-g-polystyrene system, a typical thermoplastic elastomer, the comb topology tends to delay the rise of the stress–strain curve and make materials flexible and tough.20–22 In particular, the series of multi-graft copolymers improves the tensile stress by increasing the number of branch points because the elastic backbone effectively links multiple glassy domains.22,23 Besides, in thermoplastics materials, the grafted polylactide (PLA) series composed of a flexible backbone and crystalline PLA as the side chains have an attractive molecular structure that increases strain at break, and improves toughness while maintaining the strength of PLA.24–26 Furthermore, the topological effect of the comb block copolymer is remarkable in the nanocomposite composed of the silica nanoparticle modified with the comb block copolymer brush.27 The phase-separated structure as a driving force mainly explains such toughening mechanisms in comb block copolymers. In other words, the comb topology modifies the microstructure on the comb block copolymers and enables the addition of ductility for polymeric materials.20–27 However, no topological mechanical enhancement has been reported for a homogeneous amorphous material without phase separation.
We have reported that the comb PU consisting of the rigid hydrogen bonding 4,4′-methylenebis-(cyclohexyl isocyanate) (HMDI) backbone and the soft oligo ethylene glycol (OEG) side chains surprisingly exhibits an enhancement of mechanical strength over the linear PU.28
We are interested in this comb polyurethane because the topological difference in the soft segment position between the backbone and the side chain causes mechanical enhancement, which is a homogeneous amorphous polymer that shows only one glass transition temperature. Thus, this novel structure has the potential for topological mechanical enhancement via different mechanisms involving conventional comb block polymers because of the conflict between the backbone cohesiveness and the side chain plasticity. However, it was difficult to closely investigate the topological mechanical enhancement and its mechanism in the previous study, because there is undoubted influence on the presence of methyl groups on the backbone and a different molecular weight of the monomer. Therefore, it was essential to proceed with a rational approach to understanding the topological mechanical enhancement in comb PU using the conception of the isomeric system between the comb and linear PU.
In this study, we redesigned and synthesized the reported comb PU at an atomic scale and established the topological isomeric PU system between PU-g-OEGm and PU-l-OEGm+2 consisting of HMDI and OEG (Fig. 1), in order to accurately reveal the topological effect. Since the ratio of long OEG units against the HMDI component lowers the glass transition temperature, the unit number m of the side chain for PU-l-OEGm+2 was set to m = 1, 2, 3 units, which allows tensile tests at room temperature. Therefore, the PU derivatives in this work consist of three isomer pairs–PU-g-OEG1 and PU-l-OEG3 (m = 1), PU-g-OEG2 and PU-l-OEG4 (m = 2), and PU-g-OEG3 and PU-l-OEG5 (m = 3). Furthermore, we investigated the rheological properties to clarify the mechanism of topological mechanical enhancement that does not appear in the morphology.
 | ||
| Fig. 1 Schematic image of topological isomers. | ||
Experimental section
Materials
HMDI, triethylene glycol monomethyl ether, diethylene glycol monomethyl ether, ethylene glycol monomethyl ether, pentaethylene glycol, tetraethylene glycol, triethylene glycol, p-toluenesulfonic acid monohydrate, p-toluenesulfonyl chloride, and dibutyltin dilaurate (DBTDL) were purchased from Tokyo Chemical Industry Co. Ltd, Japan. Glycerol, sodium hydroxide solution (5N), hydrochloric acid solution (5N), potassium hydroxide, and sodium hydride in paraffin liquid (60 wt%) were obtained from Nacalai Tesque, Inc. Benzaldehyde was supplied from Wako Pure Chemical Industries. Anhydrous tetrahydrofuran (THF) as a polymerization solvent was purchased from Kanto Chemical Co., Inc.Characterization
1H NMR spectra were recorded in CDCl3 by a JEOL JNM-ECX400 spectrometer. DSC profiles were measured using a Hitachi DSC6200 model with a second heating cycle from −100 to 200 °C at a rate of 10 °C min−1. The Fourier transform infrared spectrometry (FT-IR) spectra were recorded by an IRAffinity-1S spectrometer (Shimadzu). A size exclusion chromatograph (SEC) was equipped with an RI-2031 Plus Intelligent RI detector (JASCO), an AS-2055 Plus Intelligent Sampler, a PU-2080 Plus Intelligent HPLC pump, a CO-2065 Plus Intelligent Column oven, and a commercial column (TSKgel SuperH3000 and GMHXL, Tosoh Corporation) with a narrow dispersed polystyrene standard (Shodex) in THF as an eluent at 40 °C.Tensile test
Tensile testing was performed using a Shimadzu EZ-SX test instrument for three dog bone test pieces cut from films using a punching blade JIS K6251 with a crosshead speed of 10 mm min−1. The films were obtained using a heat press machine MNP – 001 (AS ONE) (Fig. S10a†).Rheological test
Rheological testing was performed using a rheometer (Kinexus, Malvern) for a round shaped disk (Φ = 7 mm, d = 1 mm) (Fig. S10c†). Samples were loaded onto parallel plates with diameter = 10 mm. The dynamic frequency sweep analysis was conducted with a strain of 0.1% in a frequency range of 0.01–100 rad s−1 at temperatures from 20 °C to 160 °C. The polymer density (ρ) was calculated from the compression molded cylinder plastics (Fig. S10c†).Results and discussion
Synthesis and characterization of polyurethane
To construct the topological isomer system between the comb and linear PUs, we needed to design novel diol monomers. We focused on the glycerol skeleton for grafting OEG as the side chains because the addition of glycerol itself and the methyl end group on the composition formula matches the two units of OEG. As to which alcohol on glycerol to use to introduce OEG, we chose the secondary one because it is wise to leave the highly reactive primary alcohols for polymerization, and they were named diol-g-OEGm. We then synthesized PU derivatives by polyaddition between the HMDI determining polymer backbone rigidity and the diol-g-OEGm or commercial OEGm+2 determining polymer topology. The polystyrene (PS) equivalent number average molecular weight (Mn) of a series of synthesized PUs was estimated by SEC. They yielded various values from 5000 Da to 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da due to the delicate difference of monomer ratio (Fig. S7†).
000 Da due to the delicate difference of monomer ratio (Fig. S7†).
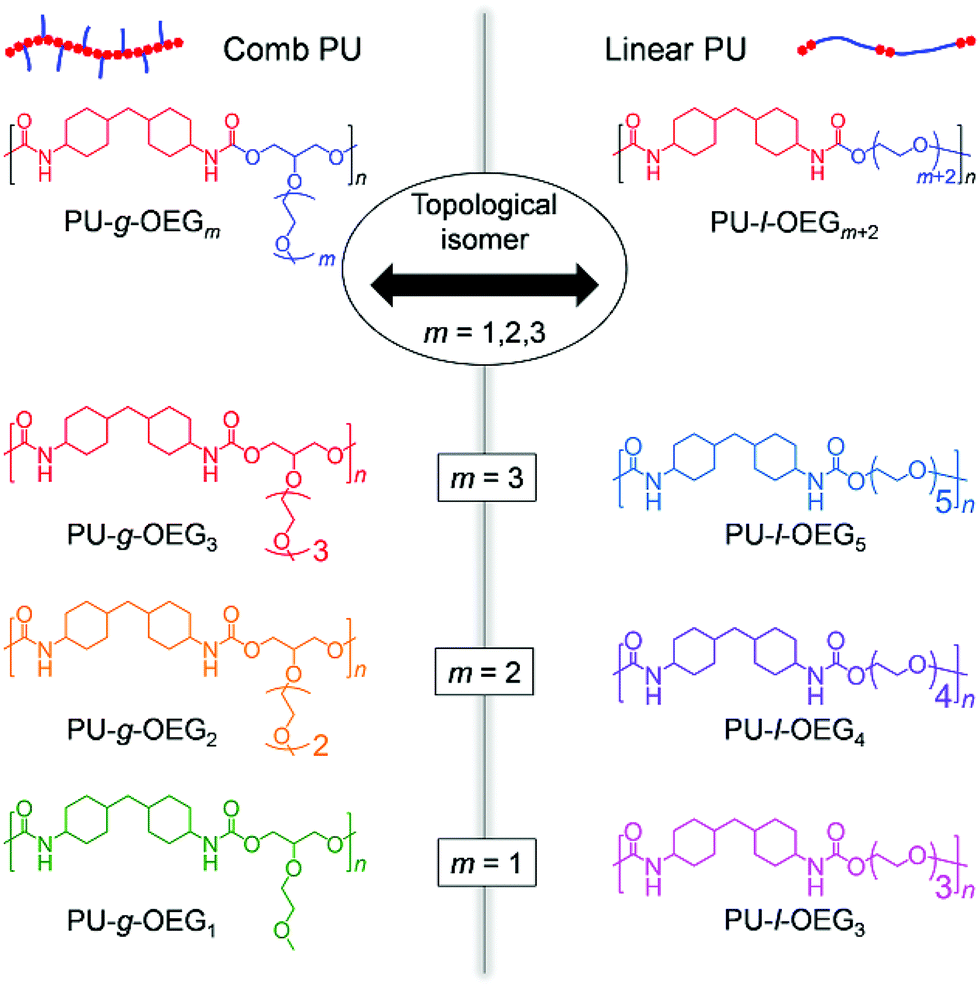
The characteristics of the comb and linear PUs were compared using the samples with Mn of about 20000 Da (PS equivalent). PU-g-OEG2 and PU-l-OEG4 in MALDI-TOF MS exhibited the same peak width as each other, meaning that these two polymers are structural isomers (Fig. 2a). 1H NMR spectra of these PUs identified the difference between PU-g-OEG2 and PU-l-OEG4 by the presence of the methyl group in the OEG side chain at 3.4 ppm although the peak width of MALDI-TOF MS was the same (Fig. 2b).
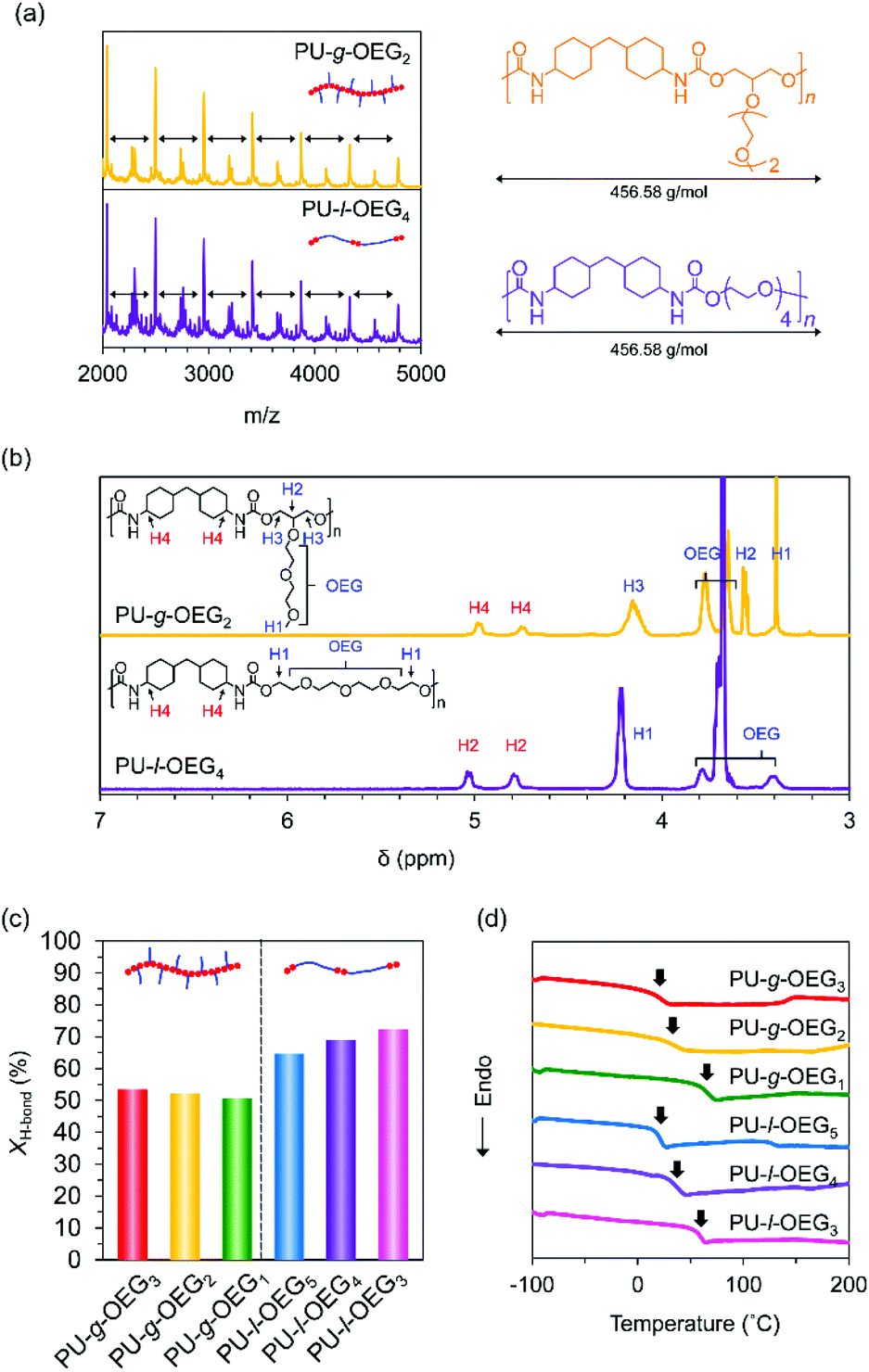 | ||
Fig. 2 (a) MALDI-TOF MS spectra for PU-g-OEG2 and PU-l-OEG4. (b) 1H NMR spectra for PU-g-OEG2 and PU-l-OEG4. (c) v(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O free) proportion for FT-IR spectra of PU derivatives. (d) DSC curves of polyurethane derivatives. O free) proportion for FT-IR spectra of PU derivatives. (d) DSC curves of polyurethane derivatives. | ||
In addition, the hydrogen bonds of urethane were observed at around 1700 cm−1 peak area ratio of CO stretching vibration in the FT-IR spectra (Fig. S9†). This CO stretching vibration at 1700 cm−1 belonged to the ordered hydrogen-bonded CO stretching vibration at 1682 cm−1, the disordered hydrogen-bonded CO stretching vibration at 1697 cm−1, and the free CO stretching vibration at 1715 cm−1 by the hydrogen bond mode between urethane bonds.29 The hydrogen bonding proportion between urethane bonds (XH-bond) was calculated by the following formula:
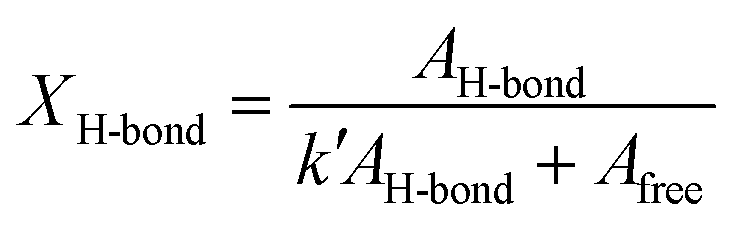
where AH–bond is the sum of peak areas of the hydrogen-bonded CO stretching vibration at 1682 cm−1 and 1697 cm−1, Afree is the peak areas of the free CO stretching vibration at 1715 cm−1, k′ is the ratio between absorption coefficients of the hydrogen-boned and free CO stretching vibration in the urethane bond. The peak areas were obtained from the resolved Gaussian curves and k′ = 1.71 was employed, based on the literature.30 The XH-bond was 53% for PU-g-OEG3, 52% for PU-g-OEG2, 51% for PU-g-OEG1, 65% for PU-l-OEG5, and 69% for PU-l-OEG4, and 72% for PU-l-OEG3 (Fig. 2c). The amount of hydrogen bonding between urethane bonds was topologically dominant and did not change significantly with the number of m. Therefore, although the OEG side chain inhibited hydrogen bonds between the urethane bonds on the comb PU, it was possible to maintain 50% or more of the hydrogen bonds up to an OEG length of at least three units, suggesting that the backbones were extremely close to each other despite the presence of side chains.
Interestingly, the glass transition temperature (Tg) of the series of PUs tended to depend on the ratio of diol and diisocyanate rather than on topology. The Tg value of each isomer pair was 58 °C for PU-g-OEG1 and 56 °C for PU-l-OEG3 (m = 1), 27 °C for PU-g-OEG2 and 27 °C for PU-l-OEG4 (m = 2), 9 °C for PU-g-OEG3 and 17 °C for PU-l-OEG5 (m = 3) (Fig. 2d). The gap of Tg values between comb and linear PUs was 8 °C at maximum with m = 3, suggesting that long OEG side chains enhance plasticization. This non-significant difference of polymer topology in thermal properties within the range of the number of 3 units in the OEG side chain enables the essential comparison of mechanical and rheological properties.
Mechanical strength properties
Tensile tests were conducted on PUs with hot-pressed films for the Mn of about 20000 Da (PS equivalent) at m = 1, 2, 3. The isomeric systems with the comb and linear PU showed topological mechanical enhancement except for m = 1 (Fig. 3a–c). The stress at the break (σb) of the comb and linear PUs was about twice the difference from 21 MPa to 34 MPa (PU-l-OEG5 and PU-g-OEG3, m = 3) and from 13 MPa to 26 MPa (PU-l-OEG4 and PU-g-OEG2, m = 2) (Fig. 3a and b). In the longest OEG length m = 3 system, the mechanical enhancement behaviour of comb PU against linear PU was observed, although the difference between the Tg and ambient temperature of 20 °C is 9 °C for PU-g-OEG3, which is lower than 3 °C for PU-l-OEG5 (Fig. 3a). PU-g-OEG2 also exhibited the mechanical enhancement behaviour against PU-l-OEG4, where σb and σy of PU-g-OEG2 were almost two-fold, and εb is half (Fig. 3b). The high intermolecular friction between side chains of PU-g-OEG3 and PU-g-OEG2 described later caused higher strain hardening behaviour after yielding than the corresponding linear PU. In contrast, PU-g-OEG1 was a typical brittle polymer with low σb and εb that were not comparable to PU-l-OEG3, and a similar mechanical behaviour to the no side chain PU-l-PDO consisting of 1,3-propanediol and HMDI was observed (Fig. 3c). This transition of mechanical properties, like disappearing extremely short side chains, between PU-g-OEG1 and PU-g-OEG2 implied that there is a threshold of the length of the side chain to provide the toughness for the comb PU by OEG side chains.
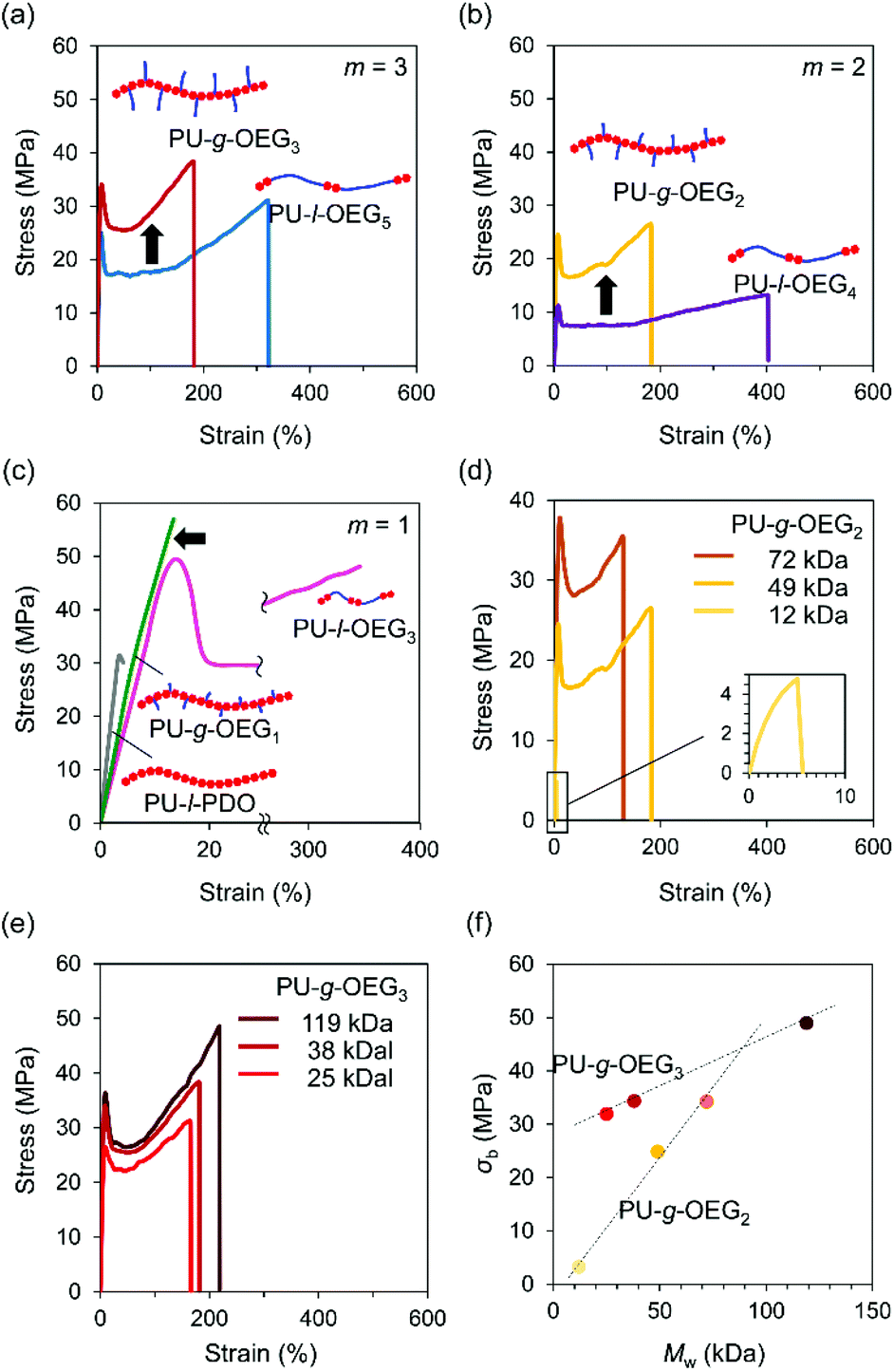 | ||
| Fig. 3 Stress-strain curves for m = 3 system (PU-g-OEG3 and PU-l-OEG5) (a), m = 2 system (PU-g-OEG2 and PU-l-OEG4) (b), m = 1 system (PU-g-OEG1 and PU-l-OEG3) and PU-l-PDO (c) with various molecular weights for PU-g-OEG3 (d), PU-g-OEG2 (e), and plots for stress at break (σb) vs. the weight-average of molecular weight (Mw) (f) with a crosshead speed of 10 mm min−1 (n = 3). | ||
Furthermore, we investigated the relationship between the weight average molecular weight (Mw) and σb. The σb values of PU-g-OEG3 and PU-g-OEG2 increased at different rates as the Mw increased (Fig. 3d and e). Interestingly, the breaking strength of PU-g-OEG3 and PU-g-OEG2 showed a linear correlation with Mw without a decay function convergence at least up till Mw of 100000 Da (PS equivalent) (Fig. 3f). Since the breaking strength of the thermoplastic polymers is inherently related to the damping function with respect to the average molecular weight, it is speculated that the convergence region of breaking strength of the comb PUs exists at a higher value than the breaking strength obtained in this study.
Rheological properties
Next, we move to our investigation of the rheological properties, in order to elucidate the physical factors related to the mechanical strength enhancement. First, to clarify the cause of brittleness due to the length of the OEG side chain on comb PU, the apparent activation energy (Ea) for the slippage of polymer chains was estimated by the Arrhenius plot. The intersection frequency for G′ and G′′ curves of large-scale polymer chain movement with each temperature condition gives an Arrhenius plot (Fig. S12–S18†). Fig. 4 shows the Ea value of each PU derivative. The Ea values of the m = 3 system (PU-g-OEG3 and PU-l-OEG5) and m = 2 system (PU-g-OEG2 and PU-l-OEG4) were 147, 135, 164, and 160 kcal mol−1, respectively. The close Ea values for the pair of PU-g-OEG3 and PU-l-OEG5, PU-g-OEG2, and PU-l-OEG4 suggested a correlation in the number of OEG units rather than the topology, as well as the glass transition temperature. However, the Ea of PU-g-OEG1 was significantly higher than that of PU-l-OEG3, indicating that PU-g-OEG1 is highly resistant to large-scale motion. In other words, the highest Ea of PU-g-OEG1 means the stiff slippage of polymer chains, suggesting that the polymer chains of PU-g-OEG1 were not sufficiently resistant to elongation in the tensile test timescale and lead to rapid destruction. In our presumption, this relationship between Ea and brittleness may be related to the hydrogen bond exchange rate during slippage.31,32 Besides, because the Ea of PU-g-OEG1 was comparable to the Ea of PU-l-PDO, the brittle backbone model polymer, consisting of HDMI and 1,3-propanediol, one unit of the side chain of PU-g-OEG1 may no longer contribute to the smooth motion of the backbone. Therefore, it is speculated that at least two units of side chains for comb PU are required for the smooth slip to acquire the ductile fracture property.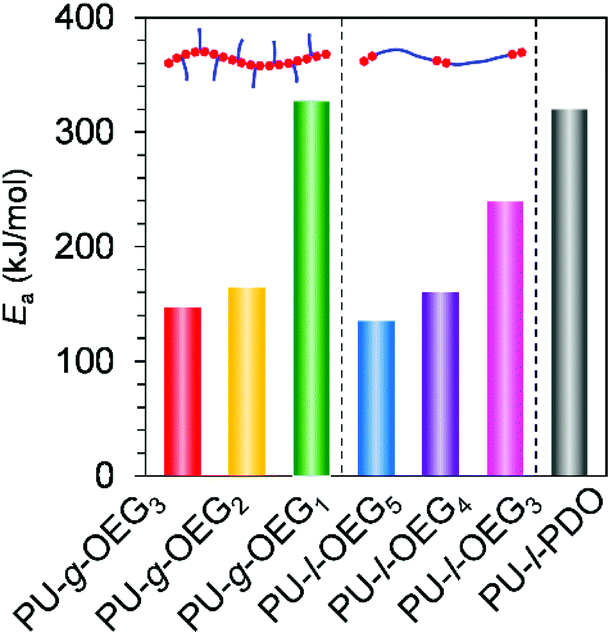 | ||
| Fig. 4 Apparent activation energies (Ea) of polyurethane derivatives for the slippage of polymer chains. | ||
Although the comparison of Ea was not involved in the topological mechanical enhancement of the comb PUs, the entanglement plateau modulus (G0N) of the comb PUs estimated by Van Gurp-Palmen curves33 indicated an inherent nature (Fig. S19†). The entanglement of polymer chains is interrelated with the mechanical properties of materials and is quantified by the average molecular weight between entanglement points (Me) given by the following formula:
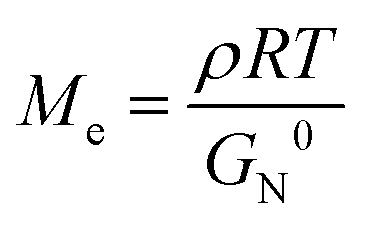
Table 1 summarizes the entanglement properties of PU derivatives. Because the side chains of comb polymers dilute the entanglement of polymer chains and exhibit a low G0N, the comb PUs also showed lower G0N than the corresponding linear PUs.
| Entry | Sample |
G
0Na (MPa) |
M
ea (kg mol−1) |
ρb (kg m−3) |
|---|---|---|---|---|
| a Determined by Van Gurp-Palmen curves. b Calculated by cylinder plastics. | ||||
| 1 | PU-g-OEG3 | 0.88 | 3.3 | 1.17 × 103 |
| 2 | PU-g-OEG2 | 0.98 | 2.8 | 1.12 × 103 |
| 3 | PU-g-OEG1 | 0.95 | 2.6 | 0.99 × 103 |
| 4 | PU-l-OEG5 | 1.4 | 2.1 | 1.22 × 103 |
| 5 | PU-l-OEG4 | 1.2 | 2.1 | 1.03 × 103 |
| 6 | PU-l-OEG3 | 0.95 | 2.7 | 1.03 × 103 |
| 7 | PU-l-PDO | 1.7 | 1.5 | 1.01 × 103 |
The Me values of the m = 3 system (Table 1, entries 1 and 4) and m = 2 system (Table 1, entries 2 and 5) showed higher values in the comb PU thane in the linear PU, suggesting that the side chains diluted the entanglement of the comb PU. There was no significant difference in the Me of m = 1 system (Table 1, entries 3 and 6) because the contribution of the OEG component to the overall polymer was too small. The Me of the comb PU takes a larger value as the side chain length, as with conventional comb polymers (Table 1, entries 1–3). Moreover, the Me of the linear PU decreased with increasing m value because the entanglement was formed owing to the flexible OEG backbone chain and approached the PEG Me34 as OEG units increased (Table 1, entries 4–6). On the other hand, PU-l-PDO without OEG showed the lowest Me, suggesting a high potential entanglement of hydrogen-bonded backbones (Table 1, entry 7).
In order to have a more quantitative discussion of the entanglement properties of comb PU, we conducted an analysis using structural parameters. Since well-studied typical comb polymers polymerized by vinyl monomers have the same molecular chain length, the structural parameters (side chain length nsc, side chain density ng) are described by the degree of polymerization.1,16,17 To compare with the rheological properties established for comb polymers, the structural parameters of comb PU were set by converting the number of atoms into the degree of polymerization of the vinyl polymer (Table S1 and Fig. S20†).
Fig. 5 shows a plot of structural parameters (1 + nsc/ng), which means Me and side chain density. While Me increases as three-halves power, a square, or a cube with the increase of the structural parameter (1 + nsc/ng) in the conventional comb polymer,15,17 the comb PU showed a different slope of the first power. This difference means that the suppression of entanglement due to side chains is less likely to occur as compared to that in conventional comb polymers because the rigid backbone consisting of cyclohexane has hydrogen bonding properties and exhibits strong cohesiveness that is common in PUs. That is, it is presumed that the comb PU is a molecular structure capable of specifically maintaining the entanglement. Finally, we investigated the zero-shear viscosity (η0), which means the friction between polymer chains.1,15 Since the zero-shear viscosity is a numerical value that depends on the molecular weight, the friction properties derived from the molecular structure were estimated by plotting η0 at several different molecular weights (Fig. 6). The straight line connecting the plots of the comb PU is located at a significantly higher η0 compared to the straight line of the corresponding linear PU as shown in Fig. 6a, whereas it is only slightly higher in Fig. 6b. That is, the comb PU has a higher frictional force than the linear PU, and the longer the side chain, the more significant the increase in the frictional force, which shows the same tendency as the conventional comb polymers.1,18 Therefore, the comb PU showed the same rheological properties as the conventional comb polymers in that it has high friction, but differs in conventional comb polymers in that the entanglement is maintained. Since it has a unique rheological property that has high entanglement and high friction that could not coexist up to now in conventional comb polymers, it is presumed that comb PU exhibits topological mechanical enhancement.
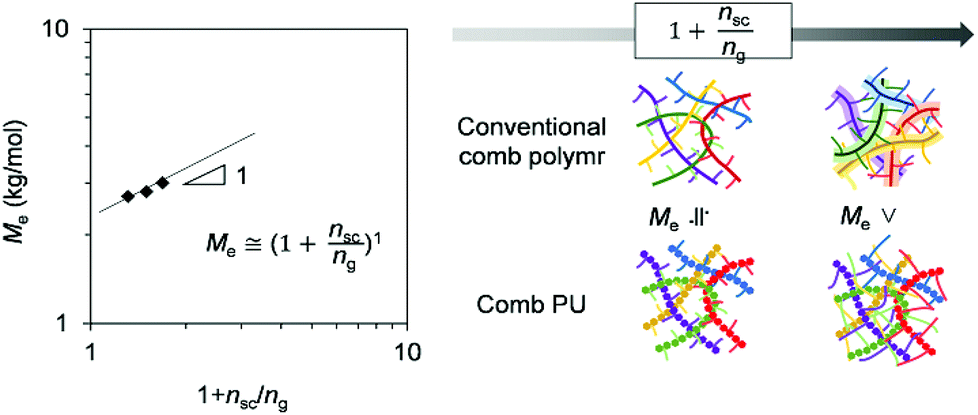 | ||
| Fig. 5 Normalized entanglement modulus as a function of the structural parameter. | ||
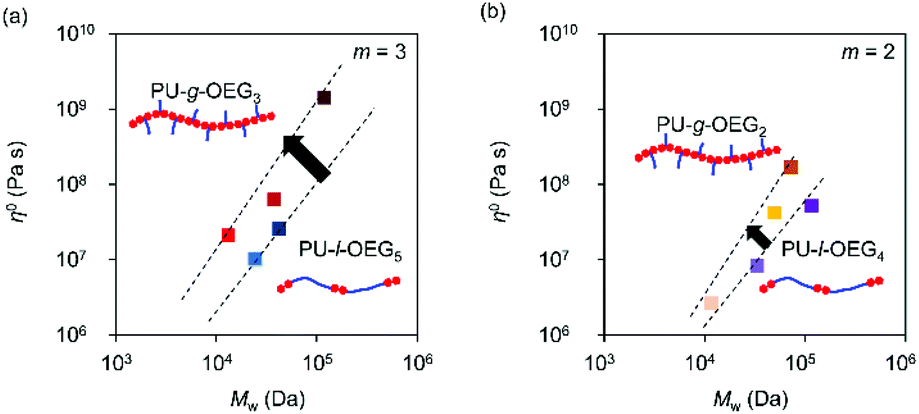 | ||
| Fig. 6 Zero shear viscosity (η0) vs. Mw for the m = 3 system (a) and m = 2 system (b) (Tref = Tg + 50 °C). | ||
Conclusions
In conclusion, we designed and synthesized systems in which comb PU and linear PU are topological isomers. The synthesized comb PU showed topological mechanical enhancement in two or three units of OEG side chains. This enhanced mechanical phenomenon was explained by the high entanglement maintaining ability and frictional force of the comb PU. This topologically mechanical enhanced technology in PU, which is one of the multi-block copolymers, has the potential to improve the mechanical properties of the existing high-strength materials such as the spider fibre.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DA is grateful for the Grant-in-Aid for JSPS Fellows, JP19J15174. This work is partly supported by Grant-in-Aid for Scientific Research (B), JP20H02799.References
- M. Abbasi, L. Faust and M. Wilhelm, Adv. Mater., 2019, 31, 1806484 CrossRef.
- B. I. Voit and A. Lederer, Chem. Rev., 2009, 109, 5924–5973 CrossRef CAS.
- B. Mu, T. Liu and W. Tian, Macromol. Rapid Commun., 2019, 40, 1800471 CrossRef.
- J. M. Ren, T. G. McKenzie, Q. Fu, E. H. Wong, J. Xu, Z. An and G. G. Qiao, Chem. Rev., 2016, 116, 6743–6836 CrossRef CAS.
- M. Romio, L. Trachsel, G. Morgese, S. N. Ramakrishna, N. D. Spencer and E. M. Benetti, ACS Macro Lett., 2020, 9, 1024–1033 CrossRef CAS.
- F. M. Haque and S. M. Grayson, Nat. Chem., 2020, 1–12 Search PubMed.
- A. G. Bajpayee and A. J. Grodzinsky, Nat. Rev. Rheumatol., 2017, 13, 183 CrossRef CAS.
- V. Kontogiorgos, Adv. Colloid Interface Sci., 2019, 270, 28–37 CrossRef CAS.
- K. Urayama, Polym. J., 2008, 40, 669–678 CrossRef CAS.
- M. Stürzel, S. Mihan and R. Mülhaupt, Chem. Rev., 2016, 116, 1398–1433 CrossRef.
- Y. Zhao, Y. Zhu, G. Sui, F. Chen, Q. Zhang and Q. Fu, RSC Adv., 2015, 5, 82535–82543 RSC.
- X. Han, Y. Hu, M. Tang, H. Fang, Q. Wu and Z. Wang, Polym. Chem., 2016, 7, 3551–3561 RSC.
- T. Kida, Y. Hiejima and K. H. Nitta, Polym. Int., 2018, 67, 1335–1340 CrossRef CAS.
- T. Kida, Y. Hiejima and K. H. Nitta, Macromolecules, 2019, 52, 4590–4600 CrossRef CAS.
- C. R. López-Barrón, A. H. Tsou, J. R. Hagadorn and J. A. Throckmorton, Macromolecules, 2018, 51, 6958–6966 CrossRef.
- S. S. Sheiko and A. V. Dobrynin, Macromolecules, 2019, 52, 7531–7546 CrossRef CAS.
- H. Liang, B. J. Morgan, G. Xie, M. R. Martinez, E. B. Zhulina, K. Matyjaszewski and A. V. Dobrynin, Macromolecules, 2018, 51, 10028–10039 CrossRef CAS.
- M. Abbasi, L. Faust, K. Riazi and M. Wilhelm, Macromolecules, 2017, 50, 5964–5977 CrossRef CAS.
- M. Vatankhah-Varnosfaderani, W. F. Daniel, M. H. Everhart, A. A. Pandya, H. Liang, K. Matyjaszewski and S. S. Sheiko, Nature, 2017, 549, 497–501 CrossRef.
- W. Wang, W. Lu, A. Goodwin, H. Wang, P. Yin, N. G. Kang and J. W. Mays, Prog. Polym. Sci., 2019, 95, 1–31 CrossRef CAS.
- H. Wang, W. Lu, W. Wang, P. N. Shah, K. Misichronis, N. G. Kang and J. W. Mays, Macromol. Chem. Phys., 2018, 219, 1700254 CrossRef.
- Y. Zhu, E. Burgaz, S. P. Gido, U. Staudinger, R. Weidisch, D. Uhrig and J. W. Mays, Macromolecules, 2006, 39, 4428–4436 CrossRef CAS.
- Y. Duan, M. Thunga, R. Schlegel, K. Schneider, E. Rettler, R. Weidisch and N. Hadjichristidis, Macromolecules, 2009, 42, 4155–4164 CrossRef CAS.
- J. Zhang, D. K. Schneiderman, T. Li, M. A. Hillmyer and F. S. Bates, Macromolecules, 2016, 49, 9108–9118 CrossRef CAS.
- M. J. Maher, S. D. Jones, A. Zografos, J. Xu, H. J. Schibur and F. S. Bates, Macromolecules, 2018, 51, 232–241 CrossRef CAS.
- I. N. Haugan, B. Lee, M. J. Maher, A. Zografos, H. J. Schibur, S. D. Jones and F. S. Bates, Macromolecules, 2019, 52, 8878–8894 CrossRef CAS.
- H. Shimamoto, C. H. Cheng, K. Kamitani, K. Kojio, Y. Higaki and A. Takahara, Macromolecules, 2019, 52, 5963–5970 CrossRef CAS.
- D. Aoki and H. Ajiro, Macromolecules, 2017, 50, 6529–6538 CrossRef CAS.
- S. Nozaki, S. Masuda, K. Kamitani, K. Kojio, A. Takahara, G. Kuwamura and S. Yamasaki, Macromolecules, 2017, 50, 1008–1015 CrossRef CAS.
- M. M. Coleman, K. H. Lee, D. J. Skrovanek and P. C. Painter, Macromolecules, 1986, 19, 2149–2157 CrossRef CAS.
- S. Seiffert and J. Sprakel, Chem. Soc. Rev., 2012, 41, 909–930 RSC.
- Y. Yanagisawa, Y. Nan, K. Okuro and T. Aida, Science, 2018, 359, 72–76 CrossRef CAS.
- M. Van Gurp and J. Palmen, Rheol. Bull., 1998, 67, 5–8 Search PubMed.
- L. J. Fetters, D. J. Lohse, D. Richter, T. A. Witten and A. Zirkel, Macromolecules, 1994, 27, 4639–4647 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01391a |
| This journal is © The Royal Society of Chemistry 2021 |