
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual-faced borax mediated synthesis of self-healable hydrogels merging dynamic covalent bonding and micellization†
Xiaohui
Li
ab,
Hatice
Mutlu
 b,
Christian
Fengler
a,
Manfred
Wilhelm
a and
Patrick
Theato
*ab
b,
Christian
Fengler
a,
Manfred
Wilhelm
a and
Patrick
Theato
*ab
aInstitute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology (KIT), Engesserstr.18, D-76131 Karlsruhe, Germany. E-mail: patrick.theato@kit.edu
bSoft Matter Synthesis Laboratory, Institute for Biological Interfaces III (IBG 3), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany
First published on 16th November 2020
Abstract
Intrinsic self-healing hydrogels with excellent mechanical properties are still difficult to achieve. Here, we presented a facile method to integrate dynamic boronate ester bonds and Pluronic F127 (PF127) micelle cross-links into one hydrogel system via a borax catalyzed thiol–ene click reaction and borax–diol chemistry. The hydrogel was capable of elongating up to 340% and self-healing approximately 90% of its initial stress within 24 h. Moreover, the hydrogel could endure a compressive strain of 80% without fracture, and exhibited excellent shape recovery and fatigue resistance ability as determined by the cyclic compressive loading–unloading test at 60% strain, providing a new facile route to design multifunctional hydrogels with spontaneous self-healing ability, good stretchability and excellent compressive properties.
Introduction
Hydrogels are cross-linked soft materials consisting of physically or chemically cross-linked hydrophilic polymers that are swollen but not dissolved in a large amount of water.1 Generally, hydrogels feature excellent biocompatibility, porous structure and tunable stiffness, and their inherent tissue-like nature makes them promising materials for diverse biological applications, such as drug delivery systems,2 tissue engineering,3 superabsorbents,4 biosensors,5 soft biomimetic machines6 and artificial e-skins.7For the preparation of desirable hydrogels, thiol–ene click reactions,8 including radical- and nucleophile-based click reactions and nucleophile/base catalyzed thiol-Michael addition reactions, are commonly utilized due to their distinctive features, such as abundant ene- and thiol-related precursors, mild reaction conditions, tolerance to oxygen/water, and high efficiency with minimal byproducts when optimized.9,10 Initially, Hubbell and co-workers proposed degradable polyethylene glycol (PEG)-based hydrogels using the Michael-type addition reaction, and employed them as well-suitable biomaterials for controlled protein delivery.11 Bowman and co-workers further engineered thiol-maleimide based hydrogels with pendent maleimide moieties as Diels–Alder reaction sites to control the release of peptide sequences.12 Recently, our group proposed a pioneering approach for preparing PEG-based hydrogels via a one-pot borax-catalyzed thiol-acrylate Michael polyaddition reaction. Borax, as a catalyst for the polyaddition reaction between PEG diacrylate and dithiothreitol (DTT), proved to exhibit efficient catalytic ability comparable to phosphate buffer solution (PBS) at room temperature. Meanwhile, boronate ester bonds formed spontaneously (and reversibly), attributed to the complexation between borax and diol groups along DTT, endowing the final hydrogels with self-healing ability and pH/thermoresponsive behaviors.13
Recently, hydrogels featuring autonomous self-healing ability have drawn considerable attention due to their promising potential in mimicking human skin. Commonly, hydrogels can be equipped with self-healing ability by introducing dynamic and reversible bonds, which include metal–ligand coordination,14 imine bonds,15 boronate ester bonds16 and disulfide bonds,17 as well as hydrophobic interactions,18 electrostatic interactions,19 host–guest interactions20 and hydrogen bonding.21 Essentially, these dynamic bonds can undergo a bond scission/regeneration process at the damaged interface, enabling the hydrogels to self-repair under ambient conditions without any external assistance. However, it remains a challenge to prepare self-healing hydrogels with excellent mechanical properties. Recently, this problem has been addressed by integrating two or more physical and chemical dynamic bonds, respectively, into one hydrogel system. For example, Jeon and co-workers designed an extremely stretchable hydrogel with rapid self-healing ability via integrating multiple hydrogen bonds originating from 2-ureido-4-pyrimidone (UPy) dimers, hydrophobic association and encapsulation from sodium dodecyl sulfate (SDS) micelles, respectively. The as-prepared hydrogels were capable of elongating up to 100 times their original length and complete self-healing within 30 s.22 By simply mixing aldehyde-functionalized Pluronic F127 with three-armed acylhydrazine-terminated PEG, Wang and co-authors prepared an ultrastretchable and self-healing hydrogel based on dynamic acylhydrazone bonds in addition to dynamic micelle cross-links. Their hydrogel could be stretched up to 117 times its initial length and displayed a self-healing capacity of up to 85% of its original strength within 24 h.23 While the self-healing hydrogels based on dynamic boronate ester bonds have been intensively developed in recent years,24–26 the combination of dynamic boronate ester bonds with other dynamic physical bonds in one hydrogel system has been rarely investigated.
Therefore, herein, we further explore the possibility of preparing self-healing hydrogels with two distinctive dynamic cross-linking systems through the borax catalyzed thiol-acrylate Michael addition reaction and borax–diol chemistry. To achieve this, a functional copolymer decorated with pendent acrylate and 1,2-diol groups is first synthesized through sequential reversible addition–fragmentation chain transfer (RAFT) polymerization and transesterification modification, followed by acid hydrolysis of the protected 1,2-diols. Subsequently, borax catalyzes not only the thiol–ene reactions between the acrylate moieties and thiol functionalized Pluronic F-127 (PF127-SH) but also facilitates the formation of boronate ester bonds by complexing with 1,2-diols at the same time. Amphiphilic Pluronic F-127 (PF-127) was chosen as a macro-cross-linker due to its distinctive ability to self-assemble into micelles in aqueous solutions,27,28 and the covalent polymerization of PF-127 micelles into a hydrogel system has been proved to be a facile route to enhance the tensile and compressive properties of hydrogels.29–31 In this way, boronate ester bonds and PF127 micelles act as two kinds of dynamic cross-linkers within one hydrogel system. The as-prepared hydrogels are further investigated by evaluating their mechanical properties and self-healing ability.
Experimental section
Materials
2,2′-Azobis(2-methylpropionitrile) (AIBN, 98%, Sigma-Aldrich) was recrystallized from methanol before use; N,N-dimethylacrylamide (DMA, 99%, Sigma-Aldrich) was purified by passing through a basic aluminum column before polymerization. 4-Cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid (CDTPA, 97%, TCI), 2-hydroxyethyl acrylate (HEA, 95%, TCI), isopropylideneglycerol (IPG, 97%, Sigma-Aldrich), 4-(dimethylamino)pyridine (DMAP, 97%, Sigma-Aldrich), Pluronic F-127 (PF127, BioReagent, Sigma-Aldrich), 3-mercaptopropionic acid (99%, Sigma-Aldrich), p-toluenesulfonic acid monohydrate (98.5%, Sigma-Aldrich), dithiothreitol (DTT, BioReagent, Fisher Scientific), borax (97%, Sigma-Aldrich), hydrochloric acid (HCl, 37 wt%, Sigma-Aldrich) and other dry solvents were used as received. Ultrapure water was obtained from GenPure Pro™ (Thermo Scientific). Pentafluorophenyl acrylate was synthesized according to the published literature.32Instruments and characterization studies
All 1H- and 19F-NMR spectra were recorded on a Bruker Avance 400 spectrometer in CDCl3 (400 MHz for 1H with tetramethylsilane as the internal standard and 376 MHz for 19F). 11B-NMR (128 MHz) spectra were recorded on a Bruker Avance 400 spectrometer in D2O. Molecular weights and molecular weight distributions were determined by gel permeation chromatography (GPC) with DMAc as the eluent solvent. FT-IR spectra were recorded on a Bruker Vertex 70 spectrometer with an ATR unit.Rheological measurements
Hydrogel samples were prepared in a cylindrical mold with a diameter of 28 mm to obtain uniform disc-shaped specimens with a height of 4 mm. Rheological measurements were performed on an ARES-G2 system, TA Instruments, with a 28 mm plate–plate geometry. The geometry was lowered until a constant axial force of 0.5 N (812 Pa) was applied to the sample and the temperature was controlled to 25 ± 0.1 °C using a Peltier element (Advanced Peltier System, TA Instruments). Oscillatory amplitude strain sweep over the region from γ0 = 0.1% to 500% was performed prior to determine the linear viscoelastic region under a constant frequency of 1 Hz. Then, oscillatory frequency sweeps were conducted to determine the storage modulus G′ and loss modulus G′′ over the frequency region from 0.2 to 300 rad s−1 at a constant strain of γ0 = 0.1%. A solvent trap was utilized to avoid water evaporation during the tests.Mechanical tests
All mechanical tests were performed on a universal tensile testing machine (TA-XT Plus Texture Analyzer) at room temperature. Compression tests were performed on cylinder-shaped samples with a diameter of 10 mm and a height of 6–7 mm at a crosshead speed of 10% strain per min. Rectangle-shaped samples with a size of 30 × 15 × 2 mm were used for tensile tests at a crosshead speed of 10 mm min−1. The nominal stress (σ) was defined as σ = F/S, where F is the loading force and S is the initial cross-sectional area of the sample. The nominal strain (ε) was defined as ε = Δl/l, where Δl is the length change and l is the initial length. The toughness of the hydrogels was estimated by integrating the area under the stress–strain curves, and the dissipated energy was calculated by the area of hysteresis loops in one cyclic curve.Synthesis of poly(N,N-dimethylacrylamide-co-pentafluorophenyl acrylate) (P(DMA-co-PFPA)) (P1)
The linear copolymer P(DMA-co-PFPA) was synthesized via RAFT polymerization according to the published literature.33 Briefly, N,N-dimethylacrylamide (20 mmol, 2 g), pentafluorophenyl acrylate (2.2 mmol, 0.534 g), RAFT agent CDTPA (0.075 mmol, 30 mg) and AIBN (0.0075 mmol, 1.2 mg) were dissolved in 6 mL of dry 1,4-dioxane in a pre-dried Schlenk tube. The mixture was degassed through three freeze–thaw cycles. The Schlenk tube was then transferred into a pre-heated oil bath at 70 °C and stirred for 16 h. The reaction was terminated by cooling down in liquid nitrogen and then exposure to air. The crude copolymer was obtained through three times precipitation from THF into ice cold n-hexane. The final product was obtained as a yellow solid after being dried in a vacuum oven at 45 °C overnight (2.35 g, yield: 92%).1H-NMR (400 Hz, CDCl3, δ in ppm): 3.18–2.76 (d, 7H, –CH2–CH– from the backbone of PPFPA and –N(CH3)2 from the side chain of PDMA); 2.75–2.54 (s, 1H, –CH2–CH in the backbone of PDMA); 2.0–1.0 (2H, –CH2–CH– in the backbone of PPFPA and PDMA); 0.88 (3H, –CH3 from CTA).
19F-NMR (375 MHz, CDCl3, δ in ppm): −152.54 (ortho), −157.44 (para), −162.05 (meta) from PPFPA.
FT-IR: ν (cm−1): 1780 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ester bond), 1520 (aromatic CC from PFP), 990 (C–F from PFP).
O ester bond), 1520 (aromatic CC from PFP), 990 (C–F from PFP).
GPC: Mn, GPC = 3.09 × 104 g mol−1, Mw/Mn = 1.23.
Synthesis of poly(N,N-dimethylacrylamide-co-ethyl diacrylate-co-isopropylidene ethyl acrylate) (P(DMA-co-EDA-co-IPA)) (P2)
P(DMA-co-EDA-co-IPA) (P2) with the designated molar feeding ratio of EDA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :IPA = 30:70 was synthesized via two-step transesterification modification. First, P(DMA-co-PFPA) (1 g, containing 1 mmol PPFPA) was dissolved in 6 mL of dry DMF in a vial, followed by the addition of DMAP (97.7 mg, 0.8 mmol, 0.8 equiv. of PPFPA). Then, 2-hydroxyethyl acrylate (34.8 mg, 0.3 mmol, 0.3 equiv. of PPFPA) dissolved in 1 mL of dry DMF was added into the vial. The mixture was stirred for 12 h at 80 °C. After that, a slight excess of isopropylideneglycerol (118.9 mg, 0.9 mmol, 0.9 equiv. of PPFPA) dissolved in 1 mL of dry DMF was added and the mixture was stirred for another 15 h at 80 °C. Afterwards, a small amount of the mixture was withdrawn and dialysed against DI water directly for two days for characterization and this P2 sample was obtained as a pale yellow solid after lyophilization. The remaining parts of the mixture were immediately exposed to the deprotection conditions (see below). P(DMA-co-EDA-co-IPA) (P2) with other molar ratios of EDA:IPA was synthesized by the same procedure.
:IPA = 30:70 was synthesized via two-step transesterification modification. First, P(DMA-co-PFPA) (1 g, containing 1 mmol PPFPA) was dissolved in 6 mL of dry DMF in a vial, followed by the addition of DMAP (97.7 mg, 0.8 mmol, 0.8 equiv. of PPFPA). Then, 2-hydroxyethyl acrylate (34.8 mg, 0.3 mmol, 0.3 equiv. of PPFPA) dissolved in 1 mL of dry DMF was added into the vial. The mixture was stirred for 12 h at 80 °C. After that, a slight excess of isopropylideneglycerol (118.9 mg, 0.9 mmol, 0.9 equiv. of PPFPA) dissolved in 1 mL of dry DMF was added and the mixture was stirred for another 15 h at 80 °C. Afterwards, a small amount of the mixture was withdrawn and dialysed against DI water directly for two days for characterization and this P2 sample was obtained as a pale yellow solid after lyophilization. The remaining parts of the mixture were immediately exposed to the deprotection conditions (see below). P(DMA-co-EDA-co-IPA) (P2) with other molar ratios of EDA:IPA was synthesized by the same procedure.
1H NMR (400 MHz, CDCl3, δ in ppm): 6.38 (d, 1H, CH2CH–), 6.17–5.98 (m, 1H, CH2CH–), 5.86 (d, 1H, CH2CH–), 4.74–3.53 (m, 9H, –O–CH2–CH2–O– from PEDA, –C(O)O–CH2–, –CH2–CH(O)–, –CH(O)–CH2– from PIPA), 3.34–2.73 (m, 6H, –N(CH3)2 from PDMA), 2.62 (s, 1H, –CH2–CH– in the backbone of PDMA), 2.40–1.05 (m, 16H, –CH3 from PIPA, –CH2–CH3 from CTA, –CH2–CH– in the backbone of PDMA, PEDA and PIPA), 0.88 (t, 3H, –CH3 from CTA).
FT-IR: ν (cm−1): 1720 (νOC–O– in PEDA and PIPA).
GPC: Mn, GPC = 1.12 × 104 g mol−1, Mw/Mn = 1.89.
Synthesis of poly(N,N-dimethylacrylamide-co-ethyl diacrylate-co-dihydroxypropyl acrylate) (P(DMA-co-EDA-co-DHA)) (P3)
The remaining DMF solution of P2 was transferred into a 50 mL round flask and mixed with 8 mL of 1 M HCl solution. The whole mixture was stirred overnight at room temperature. The final product (P(DMA-co-EDA-co-DHA)) (P3) was purified via dialysis against DI water for 2 days and a pale brown solid was obtained after lyophilization (820 mg, yield: 82%).
1H NMR (400 MHz, CDCl3, δ in ppm): 6.38 (d, 1H, CH2CH–), 6.17–5.98 (m, 1H, CH2CH–), 5.86 (d, 1H, CH2CH–), 4.74–3.53 (m, 11H, –O–CH2–CH2–O– in PEDA, –C(O)O–CH2–, –CH2–CH(O)–, –CH(O)–CH2–, –OH from PDHA), 3.34 to 2.73 (m, 6H, –N(CH3)2 from PDMA), 2.62 (s, 1H, –CH2–CH– in the backbone of PDMA), 2.40–1.05 (m, 10H, –CH2–CH3 from CTA, –CH2–CH– in the backbone of PDMA, PEDA and PDHA), 0.88 (t, 3H, –CH3 from CTA).
FT-IR: ν (cm−1): 3433 (broad, ν–OH– in PDHA), 1720 (νOC–O– in PEDA and PDHA).
GPC: Mn, GPC = 1.98 × 104 g mol−1, Mw/Mn = 1.52.
Synthesis of thiol-terminated Pluronic F127 (PF127-SH)
Thiol-terminated Pluronic F127 (PF127-SH) was synthesized according to a published procedure.34 Briefly, 3-mercaptopropionic acid (2.13 g, 20 mmol), Pluronic F127 (12.5 g, 1 mmol), p-toluenesulfonic acid monohydrate (0.384 g, 2 mmol), and DTT (1.54 g, 10 mmol) were dissolved in 200 mL of dry toluene in a round flask equipped with a Dean–Stark trap and an air condenser. The mixture solution was refluxed for two days at 140 °C until no water was evolved in the Dean–Stark trap. Afterwards, toluene was removed under reduced pressure. The residue was redissolved in 150 mL of DCM, and washed with saturated saline (three times, 50 mL for each). The combined organic solvent was dried with anhydrous MgSO4, condensed and then precipitated into 300 mL of cold ethyl ether. The final product was obtained as a white powder after being dried in a vacuum oven at 45 °C (10 g, yield: 84.6%).1H NMR (400 MHz, CDCl3, δ in ppm): 4.24–4.17 (m, 2H, –CH2–OC(O)–), 3.79–3.70 (m, 2H, –CH2–CH2–OC(O)–), 3.58 (s, 4H, –O–CH2–CH2–O– in the PEO repeating units), 3.53–3.42 (m, 2H, –CH2–CH(CH3)– in PPO repeating units), 3.36–3.28 (m, 2H, –CH2–CH(CH3)– in PPO repeating units), 2.75–2.68 (m, 2H, –CH2–CH2–SH), 2.65–2.58 (m, 2H, –CH2–CH2–SH), 1.62 (t, 1H, –SH), 1.10–1.03 (m, 3H, –O–CH2–CH(CH3)– in PPO repeating units).
FT-IR: ν (cm−1): 1740 (CO ester bond).
Complex hyperbranched sample preparation for 1H-NMR
A dilute solution of P(DMA-co-EDA-co-DHA) (P3, 20 mg, containing 0.006 mmol CH2CH–) and PF127-SH (38 mg, containing 0.0061 mmol SH) was prepared by dissolving the polymers in 1 mL of DI water, and then borax solution (0.1 M, 0.024 mL) was added dropwise into the mixture solution. The whole mixture solution was stirred for 30 minutes at room temperature and then the reaction was quenched immediately in liquid nitrogen. The solution was lyophilized for 24 h; afterwards, 20 mg of the mixture was withdrawn and dissolved in CDCl3 to perform 1H-NMR measurements.
Hydrogel preparation
The hydrophilic copolymer P(DMA-co-EDA-co-DHA) (P3) (100 mg, containing around 0.03 mmol CH2CH– and 0.07 mmol 1,2-diol groups) and PF127-SH (190 mg, containing 0.03 mmol –SH group) were dissolved in 1 mL of ice cold water to form a homogeneous solution. Afterwards, 0.12 mL of 0.1 mol L−1 borax solution was added dropwise into the polymer solution under vigorous stirring at room temperature. The final hydrogels with 20 wt% solid content formed within two minutes. The other hydrogel samples were prepared through the same procedure.
Results and discussion
A functional random copolymer P(DMA-co-EDA-co-DHA) (P3) with pendent acrylate and 1,2-diol groups was synthesized via a combination of reversible addition–fragmentation chain transfer (RAFT) polymerization and sequential trans-esterification modification, followed by acidic deprotection, as illustrated in Scheme 1. First, a reactive parent copolymer P(DMA-co-PFPA) (P1) was prepared by copolymerization of N,N-dimethylacrylamide (DMA) with pentafluorophenyl acrylate through RAFT copolymerization. DMA as a hydrophilic monomer is the major constituent part and hence endows P(DMA-co-PFPA) (P1) with good solubility in aqueous media. The 1H-NMR and 19F-NMR spectra of P(DMA-co-PFPA) (P1) are shown in Fig. 1A and 2A, respectively. The 1H-NMR spectrum displays distinctive signals at 3.18–2.76 ppm originating from PDMA and PPFPA, and broad proton resonance signals at 2.0–1.0 ppm assigned to the backbone of the copolymer P(DMA-co-PFPA) (P1), while the 19F-NMR spectrum shows characteristic peaks at −152.54 ppm, −157.44 ppm, and −162.05 ppm attributed to PPFPA, thus demonstrating the successful synthesis of the desired precursor copolymer P(DMA-co-PFPA) (P1). Besides, the number average molecular weight Mn of P1 was 3.09 × 104 g mol−1 with a narrow molecular weight distribution of Đ = 1.23 as determined by gel permeation chromatography (GPC), which was nearly in accordance with the targeted value (3.39 × 104 g mol−1), suggesting the well-controlled character of RAFT polymerization.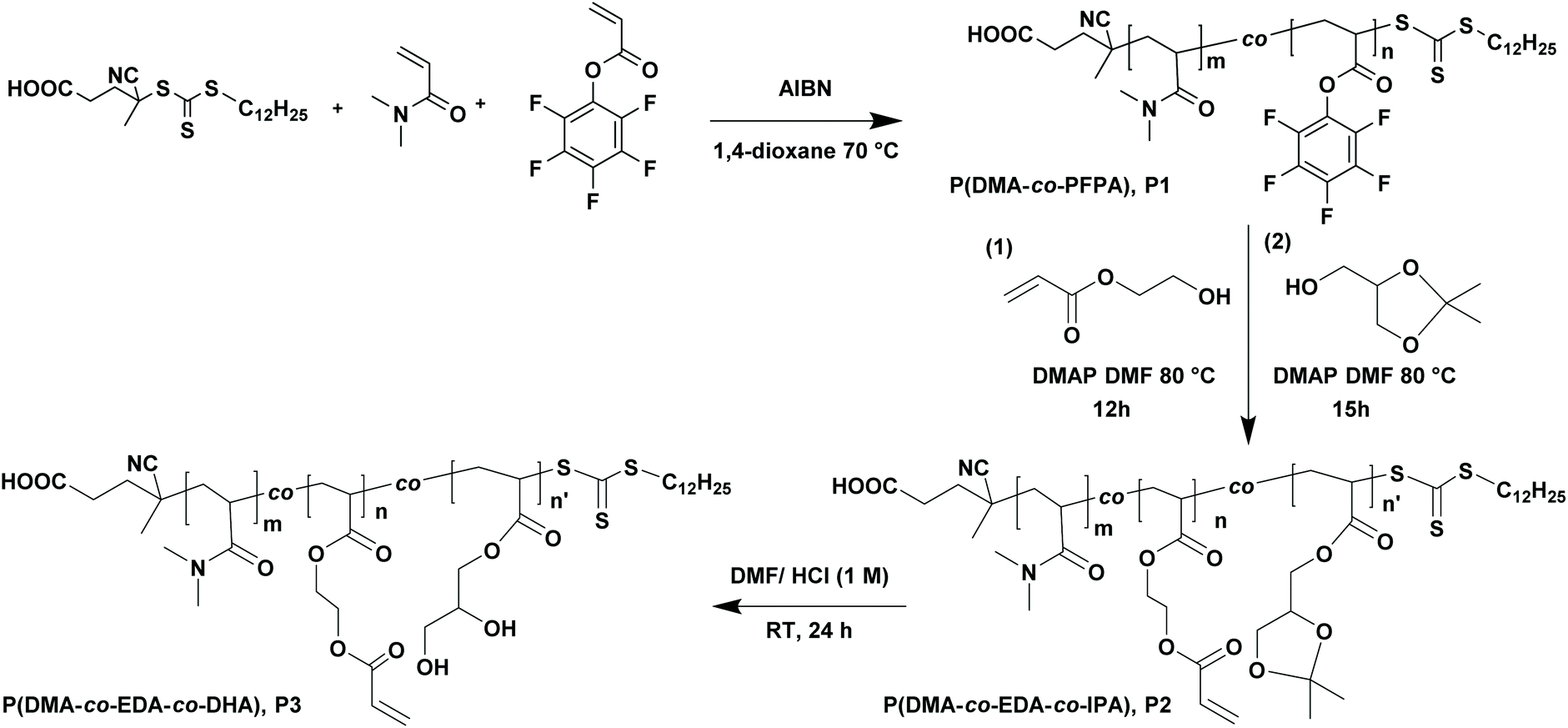 | ||
| Scheme 1 Synthetic routes towards the functional copolymer P(DMA-co-EDA-co-DHA), P3. First, P(DMA-co-PFPA), P1, was synthesized via RAFT polymerization, followed by a sequential trans-esterification modification to yield P(DMA-co-EDA-co-IPA), P2, and acidic deprotection thereof. | ||
 | ||
| Fig. 1 (A) 1H-NMR spectra (400 MHz) of P(DMA-co-PFPA), P1, (B) P(DMA-co-EDA-co-IPA), P2, and (C) P(DMA-co-EDA-co-DHA), P3, in CDCl3. | ||
 | ||
| Fig. 2 (A) 19F-NMR spectra (376 MHz) of P(DMA-co-PFPA), P1, and P(DMA-co-EDA-co-DHA), P3, in CDCl3. (B) FT-IR spectra of P(DMA-co-PFPA), P1, and P(DMA-co-EDA-co-DHA), P3. | ||
According to our previous reports, poly(pentafluorophenyl acrylate) featuring reactive pentafluorophenyl (PFP) ester groups is an effective handle to anchor primary alcohols to polyacrylates via 4-(dimethylamino)pyridine (DMAP) catalyzed trans-esterification modification.35 Thus, we prepared P(DMA-co-EDA-co-IPA) (P2) by sequential substitution of PFP ester groups with 2-hydroxyethyl acrylate and isopropylideneglycerol. As determined by 1H-NMR spectroscopy (Fig. 1B), the proton resonance signals of P(DMA-co-EDA-co-IPA) (P2) at 6.5–5.5 ppm were attributed to unsaturated CH2CH– protons, while new broad peaks at 4.8–3.5 ppm corresponded to the solketal protons of isopropylideneglycerol and –O–CH2–CH2–O– protons of 2-hydroxyethyl acrylate. Besides, the integrals of proton signals at 5.85–5.75 ppm (k) and 4.68–3.50 ppm (l + l′ + m + n + i) were calculated to determine the molar ratio of EDA and IPA of the obtained P(DMA-co-EDA-co-IPA) (P2), which was almost in agreement with the designated molar feeding ratio (Fig. S1, Table S1, ESI†), revealing the successful and well-controlled post-polymerization modification of P1 to yield P2. Furthermore, acetal protecting groups in isopropylideneglycerol could be easily removed via acid hydrolysis, leaving pendent 1,2-diol groups along P(DMA-co-EDA-co-DHA) (P3) polymer chains, which was verified by the complete disappearance of methyl proton signals originating from isopropylideneglycerol at 1.32 ppm and 1.39 ppm in the 1H-NMR spectra of P(DMA-co-EDA-co-DHA) (P3) (Fig. 1C).
The nucleophilic substitution process was also monitored by 19F-NMR and FT-IR spectroscopy. As depicted in the 19F-NMR spectrum of P(DMA-co-EDA-co-DHA) (P3) in Fig. 2A, compared to P(DMA-co-PFPA) (P1), the indicative pentafluorophenyl ester signals at (δ) −152.54 (ortho), −157.44 (para), and −162.05 (meta) ppm completely disappeared after the trans-esterification process, revealing the successful modification. Besides, in the FT-IR spectrum (Fig. 2B), the characteristic pentafluorophenyl ester bands from PPFPA at 1780 cm−1 (CO stretching vibration), 1520 cm−1 (aromatic –C6F5 stretching vibration), and 990 cm−1 (C–F stretching bond) also disappeared after the substitution process. Furthermore, a new CO stretching vibration band at 1720 cm−1 and a broader band at 3433 cm−1 were clearly identified, which were attributed to the new formed ester CO and 1,2-diol groups, further demonstrating the success of ester exchange reaction.
Thiol-terminated Pluronic F127 (PF127-SH) was synthesized through an esterification reaction as previously shown31 and adequately characterized by 1H-NMR and FT-IR spectroscopy (Scheme S1, Fig. S2, Fig. S3, ESI†). By predicting the integral ratio of methyl protons at 1.08 ppm originating from poly(propylene oxide) (PPO) moieties to the characteristic signals of 3-mercaptopropionic acid at 2.62 ppm, the ratio of the thiol functionality was estimated to be around 86%.
Noteworthily, borax is known to be an efficient catalyst for the thiol-acrylate Michael addition reaction13 as well as a reagent in forming dynamic boronate ester bonds with diols.36 First, prior to any hydrogel formation, a reaction of P(DMA-co-EDA-co-DHA) (P3) with PF127-SH in dilute solution was conducted, resulting in presumably highly branched but soluble polymers, which allow subsequent characterization. The catalytic effect of borax on the thiol–ene reaction between P(DMA-co-EDA-co-DHA) (P3) and PF127-SH was confirmed by 1H-NMR spectroscopy. As shown in Fig. 3A, multiple proton resonance signals at 6.5–5.5 ppm assigned to CH2CH– of pendent acrylate groups, as well as the peak at 1.67 ppm attributed to the thiol groups of PF127-SH, totally disappeared in the highly branched polymer after 30 minutes of reaction time, demonstrating that borax was indeed an effective catalyst for thiol-acrylate Michael addition reactions. Meanwhile, the 11B-NMR spectra of the P(DMA-co-EDA-co-DHA) (P3)/borax mixture in deuterated water at different pH values of 3 and 9 were recorded to further indicate the diol/borax complexation involved in the reaction. As illustrated in Fig. 3B, the 11B-NMR spectra at pH 9 exhibited a broad and predominant peak at 16.9 ppm arising from the monoborate B(OH)4− species, as well as a shoulder peak at around 13.2–6.9 ppm which is attributed to the 2:1 complex between the pendent diol group and the tetrahedral boron center. When the pH value was adjusted to 3 by the addition of HCl solution, the abovementioned peaks totally disappeared and a new boron resonance signal at 19.5 ppm appeared with a higher intensity, demonstrating that the complexation structures completely disassembled and borax consequently existed in free B(OH)3 form under acidic conditions.
 | ||
| Fig. 3 (A) 1H-NMR spectra of the borax-catalyzed thiol-acrylate reaction; 1H-NMR (400 MHz) spectra of P(DMA-co-EDA-co-DHA) (P3, bottom blue line), PF127-SH (middle yellow line), and the mixture of P(DMA-co-EDA-co-DHA) (P3), PF127-SH and borax solution in CDCl3 (upper black line). (B) 11B-NMR (128 MHz) spectra of the mixture of P(DMA-co-EDA-co-DHA), P3, with borax solution at pH 3 (dashed dotted line) and pH 9 (solid line) in D2O (polymer concentration: 50 mg mL−1, molar ratio of borax/diol = 1:4). | ||
By simply mixing P(DMA-co-EDA-co-DHA) (P3) and PF127-SH with borax solution, hydrogel formation was observed within several minutes, and the detailed formulations and gelation time are listed in Table S2.† The gelation mechanism involving the two reactions is illustrated in Fig. 4A. Briefly, driven by the hydrophobic PPO block, thiol-terminated amphiphilic triblock copolymers Pluronic F127 (PEO99-PPO65-PEO99, PF127) self-assembled into micelles in water, which serve as macro-cross-linkers to connect the flexible P(DMA-co-EDA-co-DHA) (P3) chains during the gelation process. Once borax is added to a mixed solution of P(DMA-co-EDA-co-DHA) (P3) and Pluronic F127-SH, thiol–ether bonds and tetrahedral boronate ester bonds were formed simultaneously via thiol-acrylate Michael addition reactions and borax–diol complexations, respectively, thus combining dynamic micelle cross-links and dynamic boronate ester bonds within the same hydrogel system. It has to be noted that the as-prepared hydrogel could be compressed, stretched and twisted without any fracture as shown in Fig. 4B. Since a higher concentration of diol groups was critical for designing self-healable hydrogels, we focused on the characterization studies of hydrogel specimens containing P(DMA-co-EDA-co-DHA) (P3) with the designated molar ratio of EDA/DHA = 30:70, which is a suitable ratio for gelation while bearing the maximum content of diol groups (Table S2, ESI†). Unless otherwise stated, the following measurements were perfermed with these specimens.
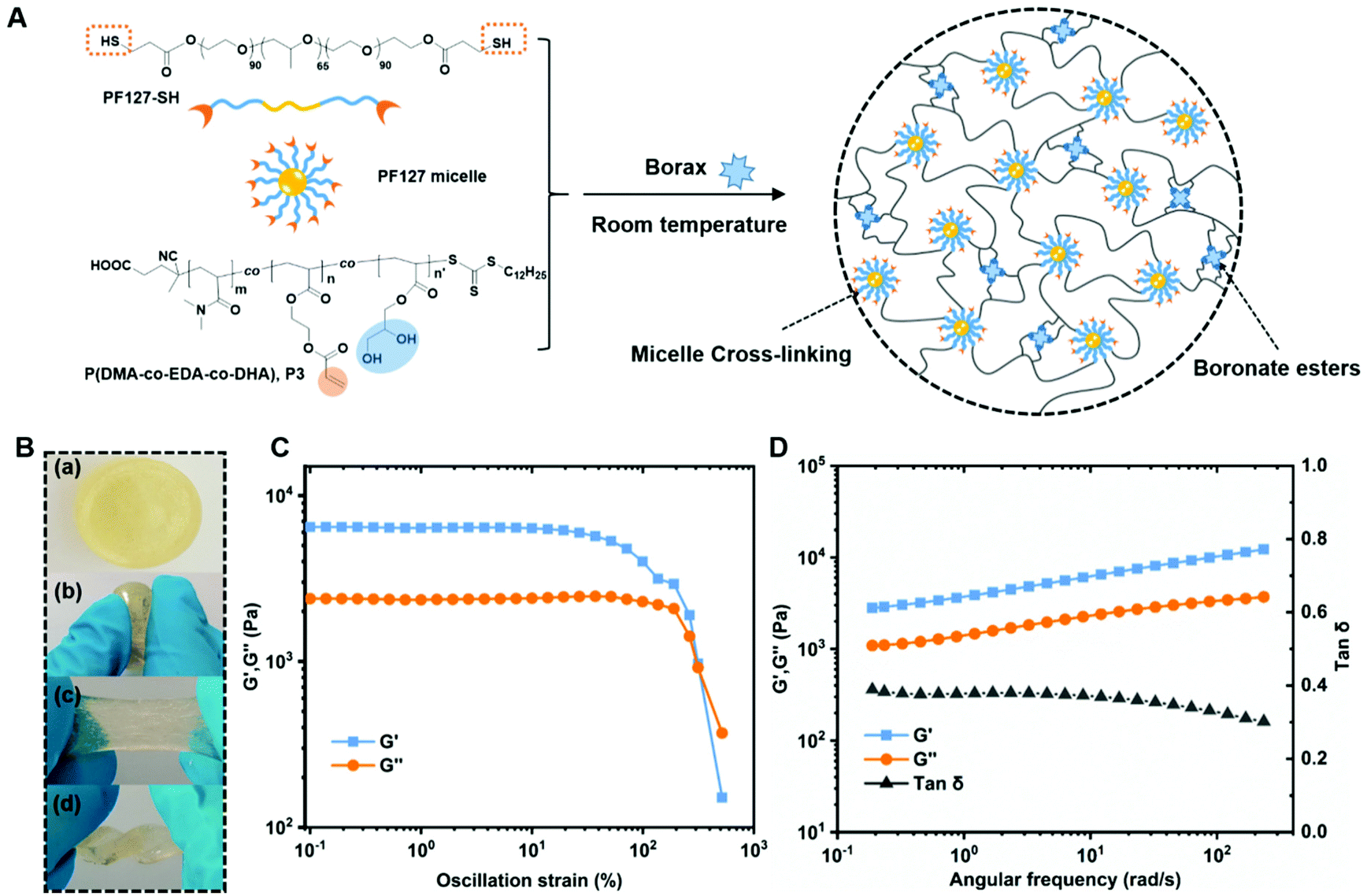 | ||
| Fig. 4 (A) Schematic illustration of the formation of the hydrogels by mixing the as-synthesized P(DMA-co-EDA-co-DHA) (P3), PF127-SH and borax solution at room temperature. (B) Photographs showing the hydrogel under different deformations: original (a), compressed (b), stretched (c) and twisted (d). (C) Oscillation strain-sweep measurement over the strain region from 0.1% to 500%. (D) Frequency sweep measurements of hydrogels over the region from 0.1 to 300 rad s−1. | ||
To further analyze the mechanical properties of the formed hydrogel, a cylindrical sample with a diameter of 28 mm and a thickness of 4 mm was prepared, and tested on a AR-G2 rheometer. First, oscillation amplitude sweeps were performed with a strain range from 0.1% to 500% to determine the linear viscoelastic region of the hydrogel at ambient temperature. As shown in Fig. 4B, the storage modulus (i.e. elastic moduli G′) and loss modulus (i.e. viscous moduli G′′) remained constant as the strain varied from 0.1% to 80%, indicating that the hydrogel could endure the deformation at ambient temperature. While the strain continued to increase, both G′ and G′′ tend to decrease until a crossover point occurred at 320% strain, where the hydrogel network was disrupted upon entering the nonlinear regime. Next, typical frequency sweeps were conducted under a constant strain of 0.1%, as shown in Fig. 4C, and both the storage modulus G′ and the loss modulus G′′ showed a weak frequency-dependent behavior as the characteristic response for viscoelastic materials increased from 3 kPa to 10 kPa and 1 kPa to 3 kPa, respectively. The storage modulus G′ remained larger than the loss modulus G′′ over the entire frequency region, demonstrating its free-standing dominant solid state. Besides, the corresponding loss tangent tan δ = G′′/G′, which was used to quantify the extent of viscous contributions in the material, displayed a value between 0.4 and 0.3, thus further demonstrating the stable gel-like state.
Compressive and tensile tests were consecutively performed to characterize the mechanical properties of the as-prepared hydrogel. As shown in Fig. 5A, the hydrogel could endure a uniaxial compression of up to 80% strain without breaking and recover to its original shape after releasing the load, indicating a good self-recovery property. Besides, cyclic loading–unloading tests under varying compressive strains of 10%, 20%, 30%, 40% and 60%, respectively, were performed without intervals to investigate the hydrogel's energy dissipation behavior. Generally, energy dissipation is illustrated as hysteresis loops in the consecutive loading–unloading cycles, and the corresponding area of a hysteresis loop was used to measure the dissipated energy per unit volume.37 As shown in Fig. 5B, the hysteresis loop areas increased with increasing compressive strain and pronounced hysteresis loops are displayed once the compressive strain exceeded 30%, demonstrating efficient energy dissipation during large strain deformation. Besides, the anti-fatigue character of the hydrogel was further investigated through another five cyclic loading–unloading tests under a constant strain of 60% without any interval, and the corresponding total and dissipated toughness values are listed in Fig. 5D. As shown in Fig. 5C, almost all cyclic curves overlapped with each other and the toughness strength exhibited minimal changes during the last four cycles. The dissipated energy decreased only slightly from the first cycle to the second cycle, and then remained nearly constant for the last four cycles, indicating an outstanding fatigue resistance property. Furthermore, the hydrogel could be stretched up to 340% and the elongated sample could almost recover to its original length after 30 minutes after unloading (Fig. 6B and Fig. S4†). Generally, to dissipate energy upon loading the dynamic character, PF127 micelles could undergo chain slides and disentanglements. The corresponding rearrangements and recombination of the polymer chains under unloading also drive the recovery of deformation. Therefore, the covalent incorporation of PF127 micelles into the hydrogel endowed it with stretchability, and outstanding shape-recovery in addition to fatigue resistance characteristics.
 | ||
| Fig. 5 Compression tests of the prepared hydrogel: (A) compressive stress–strain curves; (B) sequential loading–unloading tests without an interval under varying strains of 10%, 20%, 30%, 40% and 60%, respectively; and (C) consecutive loading–unloading tests without an interval under a constant strain of 60%. (D) The corresponding total and dissipated toughness values calculated from graph C. | ||
 | ||
| Fig. 6 The self-healing characterization of the hydrogels. (A) Photographs showing the cut-and-heal test of the hydrogel: (a) disk-shaped samples with and without a dye were prepared and cut into pieces; (b) two pieces were brought into contact; (c) self-healing for 12 h; (d) self-healing for 24 h. (B) Tensile stress–strain curves of the original hydrogels and hydrogels after self-healing for 24 h. | ||
To illustrate the self-healing behavior of the hydrogel, first qualitative cut-and-heal tests were conducted. As shown in Fig. 6A, disk-shaped hydrogel samples were prepared and one sample was colored with a dye to facilitate an optical contrast in the self-healing experiments. The samples were cut with a razor blade into pieces and then placed back together with the cut surfaces of opposite colored hydrogel samples to allow for a self-recovery process at room temperature. The cut vanished after around 12 h (Fig. 6A (a)–(c)), and nearly became invisible after 24 h, revealing a good self-healing ability. Furthermore, the self-healing efficiency of the hydrogels was quantitatively determined by tensile tests. As shown in Fig. 6B, compared to the original sample, the healed sample exhibited a slightly larger stress at the beginning of deformation after self-healing for 24 h due to inevitable water evaporation, but gradually showed lower stress with the increase of elongation, and finally fractured with approximately 20.8 kPa stress at 320% strain. Compared to the fracture stress of the original sample, which occurred at 23.1 kPa, the self-healing efficiency, defined as the ratio of the fracture stress between the healed and original samples, was estimated to be 90% after a self-healing time of 24 h. In comparison, the self-healing efficiency of the as-prepared hydrogel was comparable to the dynamic acylhydrazone cross-linked poly(ethylene oxide)/PF127 hydrogel (87% within 24 h),23 and was relatively higher than those of the complexation systems between boric acid and a random copolymer bearing a hydroxyl group (∼70% within 24 h).26 Additionally, similar cut-and-heal tests were performed on P2-based hydrogels and the two fragments failed to integrate into one piece after 24 h (Fig. S5, ESI†). Therefore, the self-healing behavior of the hydrogel is mainly attributed to the reversible covalent boronate ester bonding between borax and 1,2-diol groups, which can reform at the damaged interface to repair the separated network. Compared to PF127 micelle-based single networks (such as the P2-based hydrogel in our present work and other hydrogels in the reported literature29,31), the double dynamic network obviously merged the merits of dynamic boronate ester bonds and micellization to obtain multifunctional hydrogels with excellent mechanical properties and good self-healing properties.
Conclusions
To conclude, we proposed an autonomously self-healable hydrogel based on a one-pot borax catalyzed thiol-acrylate Michael addition reaction and borax–diol chemistry. By simply mixing the as-synthesized P(DMA-co-EDA-co-DHA) (P3), thiol-terminated PF127 and borax solution, dynamic micelle cross-links and boronate ester bonds were successfully incorporated into one hydrogel. The fracture and recombination of PF127-SH micelles upon the loading–unloading process endowed the hydrogel with outstanding shape-recovery and fatigue resistance properties as well as moderate stretchability. Besides, the hydrogels were capable of self-healing up to 90% in 24 h due to the spontaneous association of dynamic boronate ester bonds at the damaged interface. Generally, our results illustrated a facile route to prepare self-healable hydrogels with excellent mechanical properties, and we suppose that the present hydrogels might have versatile applications in biological tissue engineering fields, considering the superior biocompatibility of Pluronic F-127 and the low toxicity of the catalyst borax.Conflicts of interest
There are no conflicts to declare.Acknowledgements
X. L. gratefully acknowledges the China Scholarship Council (CSC grant: 201604910618) for financial support for this work. The authors acknowledge financial support from the DFG via project C1 of the SFB1176 “Molekulare Strukturierung weicher Materie”.References
- O. Wichterle and D. Lim, Nature, 1960, 185, 117–118 CrossRef
.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 1–17 Search PubMed
- J. Guo, Y. Kim, V. Xie, B. Smith, E. Watson, J. Lam, H. Pearce, P. Engel and A. Mikos, Sci. Adv., 2019, 5, eaaw7396 CrossRef CAS
- X. Guo, C. Pfeifer, M. Wilhelm, B. Luy and G. Guthausen, Macromol. Chem. Phys., 2019, 220, 1800525 CrossRef
- T. Distler and A. R. Boccaccini, Acta Biomater., 2020, 101, 1–13 CrossRef CAS
- D. Wirthl, R. Pichler, M. Drack, G. Kettlguber, R. Moser, R. Gerstmayr, F. Hartmann, E. Bradt, R. Kaltseis and C. M. Siket, Sci. Adv., 2017, 3, e1700053 CrossRef
- Y.-Z. Zhang, K. H. Lee, D. H. Anjum, R. Sougrat, Q. Jiang, H. Kim and H. N. Alshareef, Sci. Adv., 2018, 4, eaat0098 CrossRef
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC
- D. P. Nair, M. Podgorski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS
- D. L. Elbert, A. B. Pratt, M. P. Lutolf, S. Halstenberg and J. A. Hubbell, J. Controlled Release, 2001, 76, 11–25 CrossRef CAS
- K. C. Koehler, K. S. Anseth and C. N. Bowman, Biomacromolecules, 2013, 14, 538–547 CrossRef CAS
- L. He, D. Szopinski, Y. Wu, G. A. Luinstra and P. Theato, ACS Macro Lett., 2015, 4, 673–678 CrossRef CAS
- L. Shi, P. Ding, Y. Wang, Y. Zhang, D. Ossipov and J. Hilborn, Macromol. Rapid Commun., 2019, 40, 1800837 CrossRef
- Y. Gao, Q. Luo, S. Qiao, L. Wang, Z. Dong, J. Xu and J. Liu, Angew. Chem., Int. Ed., 2014, 53, 9343–9346 CrossRef CAS
- C. C. Deng, W. L. Brooks, K. A. Abboud and B. S. Sumerlin, ACS Macro Lett., 2015, 4, 220–224 CrossRef CAS
- W. J. Yang, X. Tao, T. Zhao, L. Weng, E.-T. Kang and L. Wang, Polym. Chem., 2015, 6, 7027–7035 RSC
- D. C. Tuncaboylu, M. Sari, W. Oppermann and O. Okay, Macromolecules, 2011, 44, 4997–5005 CrossRef CAS
- A. B. Ihsan, T. L. Sun, T. Kurokawa, S. N. Karobi, T. Nakajima, T. Nonoyama, C. K. Roy, F. Luo and J. P. Gong, Macromolecules, 2016, 49, 4245–4252 CrossRef
- G. Sinawang, M. Osaki, Y. Takashima, H. Yamaguchi and A. Harada, Polym. J., 2020, 1–21 Search PubMed
- J. Cao, C. Lu, J. Zhuang, M. Liu, X. Zhang, Y. Yu and Q. Tao, Angew. Chem., 2017, 129, 8921–8926 CrossRef
- I. Jeon, J. Cui, W. R. Illeperuma, J. Aizenberg and J. J. Vlassak, Adv. Mater., 2016, 28, 4678–4683 CrossRef CAS
- P. Wang, G. Deng, L. Zhou, Z. Li and Y. Chen, ACS Macro Lett., 2017, 6, 881–886 CrossRef CAS
- Y. Chen, D. Diaz-Dussan, D. Wu, W. Wang, Y.-Y. Peng, A. B. Asha, D. G. Hall, K. Ishihara and R. Narain, ACS Macro Lett., 2018, 7, 904–908 CrossRef CAS
- M. E. Smithmyer, C. C. Deng, S. E. Cassel, P. J. LeValley, B. S. Sumerlin and A. M. Kloxin, ACS Macro Lett., 2018, 7, 1105–1110 CrossRef CAS
- Y. Chen, W. Qian, R. Chen, H. Zhang, X. Li, D. Shi, W. Dong, M. Chen and Y. Zhao, ACS Macro Lett., 2017, 6, 1129–1133 CrossRef CAS
- L. Arens and M. Wilhelm, Macromol. Chem. Phys., 2019, 220, 1900093 CrossRef CAS
- Z. Liu and P. Yao, Polym. Chem., 2014, 5, 1072–1081 RSC
- Y. n. Sun, G. r. Gao, G. l. Du, Y. j. Cheng and J. Fu, ACS Macro Lett., 2014, 3, 496–500 CrossRef CAS
- J. Qu, X. Zhao, Y. Liang, T. Zhang, P. X. Ma and B. Guo, Biomaterials, 2018, 183, 185–199 CrossRef CAS
- X. Yu, Z. Qin, H. Wu, H. Lv and X. Yang, Macromolecules, 2019, 52, 1249–1256 CrossRef
- M. Eberhardt, R. Mruk, R. Zentel and P. Théato, Eur. Polym. J., 2005, 41, 1569–1575 CrossRef CAS
- S. Lin, A. Das and P. Theato, Polym. Chem., 2017, 8, 1206–1216 RSC
- H. Yu, Y. Wang, H. Yang, K. Peng and X. Zhang, J. Mater. Chem. B, 2017, 5, 4121–4127 RSC
- A. Das and P. Theato, Macromolecules, 2015, 48, 8695–8707 CrossRef CAS
- E. Pezron, A. Ricard, F. Lafuma and R. Audebert, Macromolecules, 1988, 21, 1121–1125 CrossRef CAS
- L. Xu, C. Wang, Y. Cui, A. Li, Y. Qiao and D. Qiu, Sci. Adv., 2019, 5, eaau3442 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01354d |
| This journal is © The Royal Society of Chemistry 2021 |