
Quinoidal conjugated polymers with open-shell character
Xiaozhou
Ji
 and
Lei
Fang
*
and
Lei
Fang
*
Department of Chemistry, Texas A&M University, College Station, TX 77843-3255, USA. E-mail: fang@chem.tamu.edu
First published on 5th February 2021
Abstract
π-Conjugated polymers composed of quinoidal or proquinoidal building blocks represent an intriguing class of macromolecules in terms of both fundamental research and practical applications. They have received increasing research interest in recent years on account of their distinct structural features and unique properties including open-shell electronic configurations and delocalization of polarons. Investigations of the influences of the quinoidal character on optical, electronic, and magnetic properties demonstrated a great prospect of these polymers in diverse application fields. This review summarizes the recent development of quinoidal conjugated polymers with regard to design, synthesis, fundamental properties, and representative applications, with emphasis on their open-shell character and high-spin materials properties. The challenges and outlook associated with this research field are also discussed.
 Xiaozhou Ji | Xiaozhou Ji received her BS degree from Nanjing University in 2015. In the same year, she joined the group of Dr Lei Fang at Texas A&M University, where she received her PhD degree in chemistry. In 2021, she joined Dr Zhenan Bao's group at Stanford University as a postdoctoral scholar. Her research interests include conjugated molecules and macromolecules, quinoidal π-systems, and doping of organic materials. |
 Lei Fang | Lei Fang is an associate professor in the Department of Chemistry at Texas A&M University. He obtained his BS (2003) and MS (2006) degrees from Wuhan University. He began his PhD study at the University of California Los Angeles, and received the degree from Northwestern University in 2010, mentored by Professor Sir Fraser Stoddart. Subsequently, he spent two and a half years at Stanford University as a postdoctoral scholar working with Professor Zhenan Bao. In 2013, he joined the faculty of Texas A&M University, where he currently leads a multidisciplinary research team focusing on functional organic materials. |
1. Introduction
1.1 Quinoidal character of π-conjugated systems
An organic π-conjugated system is a molecular entity featuring side-to-side interactions of p-orbitals across intervening σ-bonds, typically in the form of alternating single and multiple bonds.1 π-Conjugated polymers have been investigated extensively and employed in a wide range of applications on account of their intriguing electronic and optical properties. Most conjugated polymers contain cyclic π-units in the main chain. These building blocks can be represented by either a Hückle aromatic form or a “quinoidal” form. In the aromatic form, the aromatic (poly)cyclic building blocks are typically connected with one another by single bonds. In contrast, in the quinoidal form, a π-unit exerts no local ring aromaticity and is typically connected with others by double bonds. An aromatic π-conjugated unit can also be described as “proquinoidal” if it possesses a significant resonance contribution from the quinoidal form. Herein, we use the term “quinoidal conjugated polymer” to describe conjugated polymers that exhibit significant quinoidal character due to the presence of quinoidal or proquinoidal building blocks in the backbone. Quinoidal conjugated polymers often exhibit drastically different structural features, such as bond lengths, conformation, open-shell character, etc., compared to their counterparts composed of non-quinoidal aromatic units. Consequently, they can exhibit distinctive electronic, optical, and magnetic properties that are often not accessible in aromatic conjugated polymers, promising for a wide range of intriguing applications.For a certain π-system, the degree or ratio of the quinoidal character, with respect to the aromatic character, can be conveniently quantified using the parameter bond length alternation (BLA), which is defined as the average of the differences in bond lengths between the alternant single and double bonds.2 As an important structural parameter, BLA is related to the electronic properties of a conjugated polymer, such as the bandgap energy [the energy difference between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO)]. To illustrate these concepts, here we take one of the simplest conjugated polymers—polyacetylene (Fig. 1a)—as an example. Ideally, an infinite polyacetylene chain with equal length alternant bonds (zero BLA) possesses the largest π-conjugation, exhibiting zero bandgap energy and metallic properties. Such a metallic state, however, is unstable. Therefore, Peierls distortion takes place to afford two stabilized degenerate forms with |BLA| > 0. As a result, polyacetylene possesses a non-zero energy bandgap and is non-metallic in reality. For an infinite chain of a conjugated polymer constituted by cyclic aromatic π-units, such as poly-para-phenylene (PPP) (Fig. 1b), polythiophene, or polypyrrole, etc., the potential energy curve of the ground state possesses two minima, corresponding to the quinoidal and aromatic forms, respectively. In Fig. 1, BLA is defined as (d2 − d1)/2 as labeled on the structure; therefore, the quinoidal form shows a negative BLA value while the aromatic form exhibits a positive BLA value.3 Peierls stabilization contributes to both minima, while the aromatic form is more favorable than the quinoidal form due to the additional stability imparted by Hückle aromaticity, and thus is typically representative of the polymer ground state. To transform between the quinoidal and aromatic forms, the system would go through a high-energy point at which BLA is close to zero on the potential energy curve. The quinoidal character and hence the BLA are closely related to the bandgap energy of π-conjugated systems.4 In a pioneering study by Brédas, BLA was used as an indicator of the quinoidal character of PPP, polypyrrole, and polythiophene and the calculated bandgaps decrease linearly with increasing quinoidal character.5 In some recent computational study, the correlation between the quinoidal character and bandgaps of conjugated copolymers was explored with new quantum chemical descriptors that possess higher compatibility and universality than the simple BLA parameter.6,7
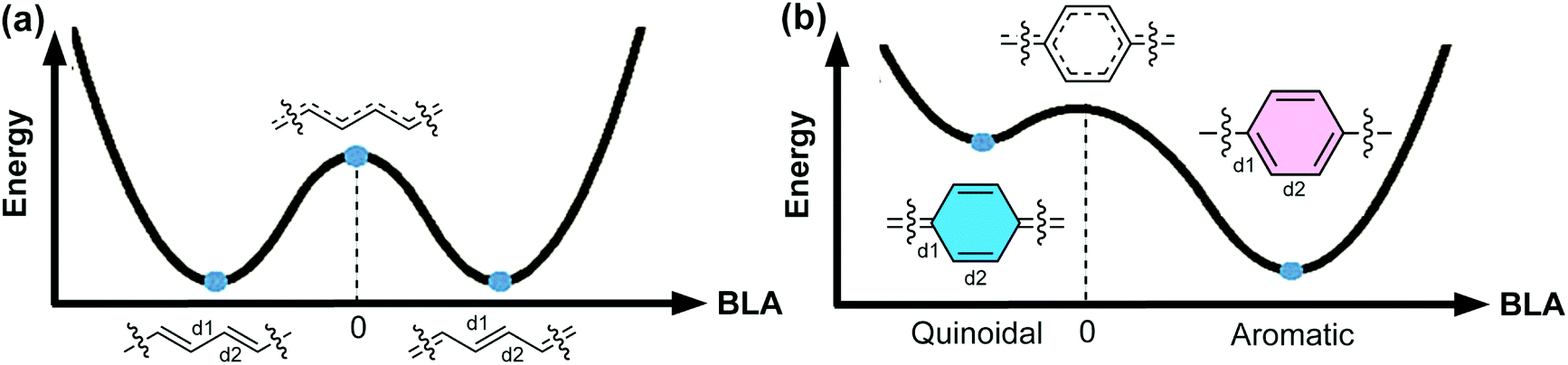 | ||
| Fig. 1 Energy diagrams of the ground states of the infinite chain of (a) polyacetylene and (b) poly-para-phenylene (PPP) as a function of BLA, which is defined as (d2 − d1)/2. Reproduced (adapted) from ref. 8 with permission from John Wiley and Sons. | ||
Apart from BLA, the backbone conformations are often different between a quinoidal conjugated polymer and its aromatic analogue.9 On account of the double-bond connections between cyclic π-units, a quinoidal structure often exhibits higher coplanarity and rigidity, while its aromatic counterpart possesses a higher degree of freedom for torsional rotation about the single bond linkages.10 The coplanarity of the quinoidal structure further facilitates electron delocalization throughout the π-system, contributing to a more extended π-conjugation scale.
1.2 Open-shell character of quinoidal structures
The past four decades have witnessed increasing research interest in quinoidal small molecules and oligomers. The resonance between the closed-shell and open-shell forms of quinoidal molecules has been studied extensively, providing advanced fundamental understanding of the nature of molecular orbitals and spin–spin interactions in these systems.11 Their electronic, optical, and magnetic properties also show promising potential to be applied in organic electronics and spintronics.12–16 Quinoidal molecules with open-shell diradical character are also good candidates for singlet fission processes, which play an important role in the latest development of organic photovoltaic applications.17,18 Related literature reports on small molecules and oligomers have already been summarized in several reviews and accounts.11,15,19–22 Similar to quinoidal small molecules and oligomers, the open-shell diradical character and spin-active states can be derived from quinoidal building blocks in conjugated polymers.11,23 In this article, we focus on quinoidal conjugated polymers with open-shell character.From the perspective of resonance forms, the open-shell diradical character of a quinoidal π-system can be considered as a result of the increased resonance contribution of the diradical form due to the stabilization effect of the original quinoidal ring gaining Hückle aromaticity.24,25 In the energy diagram (Fig. 2), with increasing one-electron overlap integral SAB between two localized orbitals, the singlet molecular orbital is stabilized while the triplet state energy increases, resulting in the progress from open-shell to closed-shell. The energy gap between the singlet and triplet states also changes depending on SAB, leading to varied electronic and magnetic properties. We would like to point out that for quinoidal small molecules and oligomers with defined end groups, the preference of resonance contributions can be critically impacted by the end groups. The impact of end groups in a longer polymeric system, however, is diluted as the molecular weight increases, and the open-shell character is analyzed mainly within the repeating units. Overall, the underlying diradical character often plays an essential role in governing the electronic and magnetic properties of many quinoidal conjugated polymers.
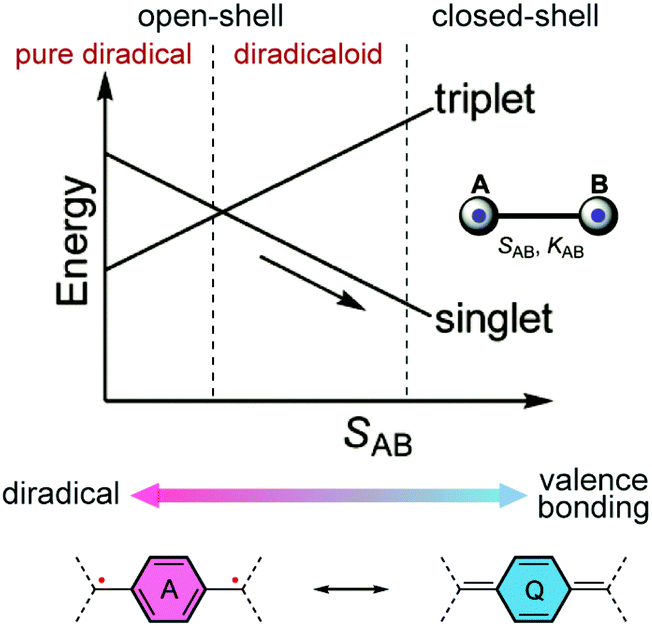 | ||
| Fig. 2 Change of energies of triplet and singlet electron pairs with the increase in the one-electron overlap integral SAB and resonances between open-shell diradical and closed-shell quinoidal structures. Reproduced (adapted) from ref. 11 with permission from the Royal Society of Chemistry. | ||
1.3 Quinoidal polarons/bipolarons in doped conjugated systems
Doped π-conjugated polymers are important organic materials that often exhibit open-shell, high-spin properties. Upon redox doping, electrons are injected into/removed from the neutral polymers, leading to anionic/cationic charged segments. In this process, the aromatic building blocks are partially converted into the quinoidal form, thereby affording drastically changed chemical, electronic, and magnetic properties. For example, without doping, polypyrrole or polythiophene is in a dominant aromatic form (Fig. 3a). The energy gap between the overall π-bonding and π*-antibonding orbitals, corresponding to the valence and conduction bands, respectively, renders semiconducting properties.26 Upon oxidative doping, positive charges are created and local reorganization leads to an ionized quinoidal form. In the solid state, the lattice deformation with a radical cation is termed as a “polaron”, whose energy levels are localized in the forbidden band, leading to a simultaneous shift in the Fermi level and electrical conductivity. At a higher doping level, radical cations in close proximity can combine so that the two neighboring positive charges are coupled in the quinoidal unit.27 Such a doubly-charged quinoidal section can be considered as a “bipolaron” that delocalizes over several rings. In this case, two localized states form in the original bandgap and evolve into two bipolaron bands with increased doping levels. In doped polymers, both high-spin polarons and spinless bipolarons are charge carriers. The low effective mass of polarons and bipolarons and high carrier mobility give rise to the high electrical conductivity of polymers with quinoidal segments,28 while a study on doped poly(3-hexylthiophene) suggested29 that polarons possess higher mobility than bipolarons. Polyaniline is another representative conducting polymer material that can be doped into a quinoidal form. Half-oxidation of the neutral polyaniline affords the quinoidal bipolaron structure, which has been proposed to dissociate into polarons and further delocalize into the polaron lattice, rendering high electrical conductivity (Fig. 3b).30,31 Overall, the quinoidal form is ubiquitous in the doping process of conjugated polymers, and plays an important role in the generation of polarons and bipolarons for the electrical conductivity of conducting polymers.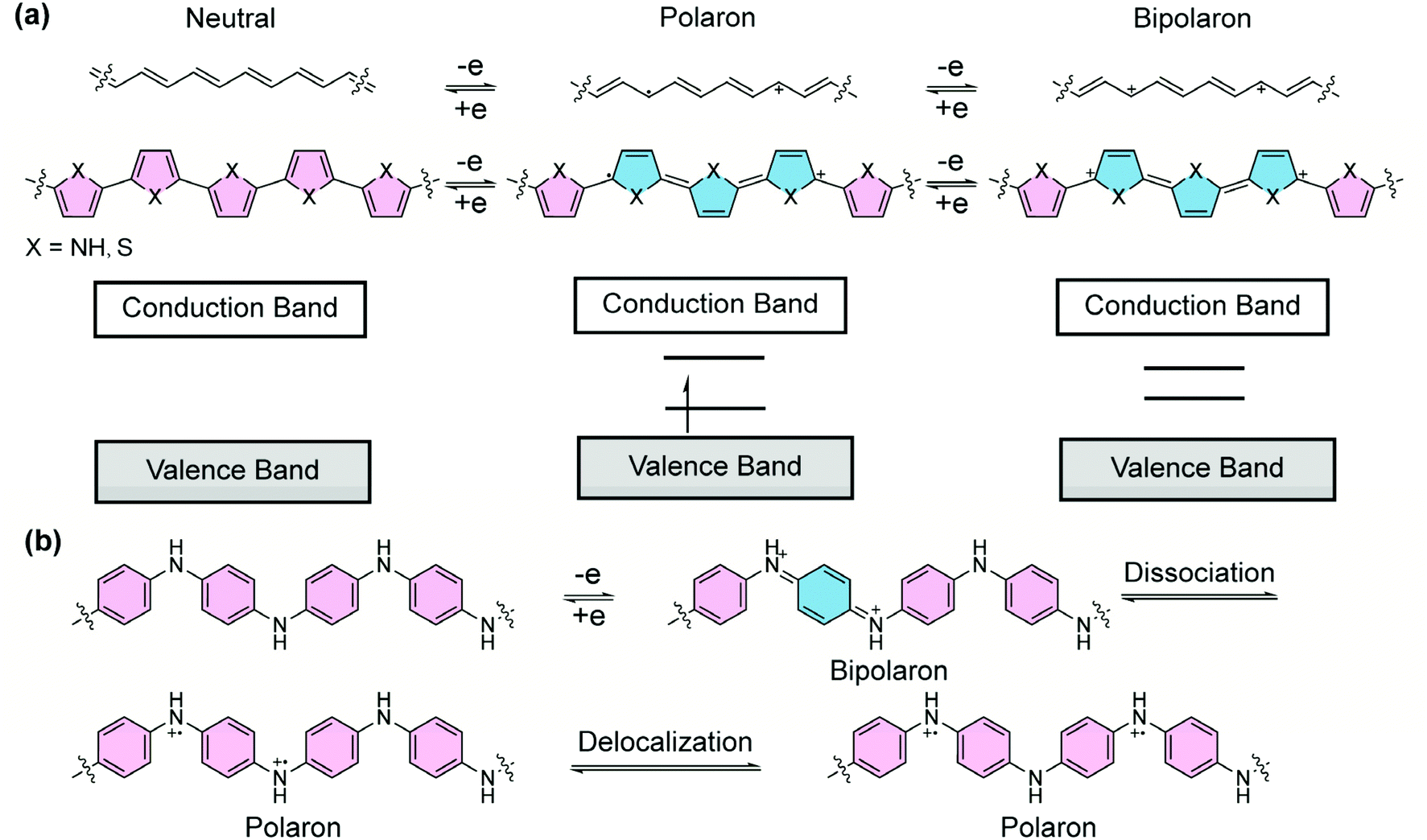 | ||
| Fig. 3 (a) Graphical illustration of the structures of polyacetylene, polypyrrole, and polythiophene in their undoped form and partially quinoidal form after doping and energy diagrams of the neutral, polaron, and bipolaron states. (b) Polyaniline in the undoped form and doped form with polaron–bipolaron equilibrium. | ||
Although the importance of the quinoidal structure has been revealed for a long time, polymeric π-systems composed of quinoidal or proquinoidal building blocks (defined as “quinoidal conjugated polymers” in this article) and the investigations on their open-shell character are less reported due to the challenges associated with their synthesis and stability.17 Despite these challenges, a number of important advances have been made to further our knowledge on the design and synthesis of such polymers and to deepen our understanding of the quinoidal properties in macromolecules for a wide range of applications. In this review article, the progress throughout recent decades in this field is summarized based on quinoidal conjugated polymers with open-shell character. Emphases are placed on the synthesis, key features of quinoidal structures, and representative applications. In contrast to the well-reviewed quinoidal conjugated polymers with narrowed bandgaps commonly applied in organic field-effect transistors and photovoltaic applications,32–35 in this paper, we would like to bring attention to the fundamental properties of quinoidal conjugated structures, including open-shell electronic configurations and state delocalization, and their related applications as high-spin materials.
2. Design and synthesis
In this section, we categorize quinoidal conjugated polymers into two types with (1) proquinoidal and (2) quinoidal building blocks. It is worth noting that the first category can be drawn in fully aromatic formulae in a strict definition. Here they are considered as quinoidal conjugated polymers and are included in this review because of their significant open-shell character and properties associated with their quinoidal resonance forms.2.1 Proquinoidal polymers
A homopolymer consisting of aromatic yet proquinoidal repeating units is expected to exhibit quinoidal characteristics as a result of the significant contribution of its quinoidal resonance form. Such proquinoidal character can be imparted to the main chain by fusing additional conjugated rings in a way that the aromaticity of the additional rings strengthens the quinoidal contribution of the main-chain units. For example, to render a higher proquinoidal nature in polythiophene, Wudl et al. designed and synthesized36 polyisothianaphthene (PITN). A benzene unit is fused to the 3,4-position of each thiophene repeating unit in the polymer, so that the quinoidal resonance form of the thiophene backbone is augmented by the aromatization of the fused benzene units (Fig. 4a).37 Compared to conventional polythiophene, PITN exhibits a drastically decreased bandgap energy (∼1 eV),38,39 which is attributed to the increased quinoidal character with smaller BLA according to computational studies.40 Following this pioneering work, the strategy of imparting the proquinoidal character by fusing an additional aromatic ring has been employed extensively in the synthesis of proquinoidal homopolymers (Fig. 4b). Notable examples include poly(thieno[3,4-b]pyrazine),41–44 poly(thieno[3,4-b]thiophene),45–48 poly(naphtho[2,3-c]thiophene),49,50 poly(dithieno[3,4-b:2′,3′-d]thiophene),51 poly(thieno[3,4-b]quinoxaline),50etc. Many of these polythiophene-derived homopolymers were polymerized by electrochemical or chemical oxidation (Fig. 4c).48 Other methods have also been reported, such as Grignard metathesis polymerization43 and nonoxidative polymerization based on phthalide-derived monomers.52,53 Under oxidative conditions (Fig. 4c), the afforded polymers are in the doped state, containing charged quinoidal structures and featuring narrowed bandgaps and enhanced π-conjugation.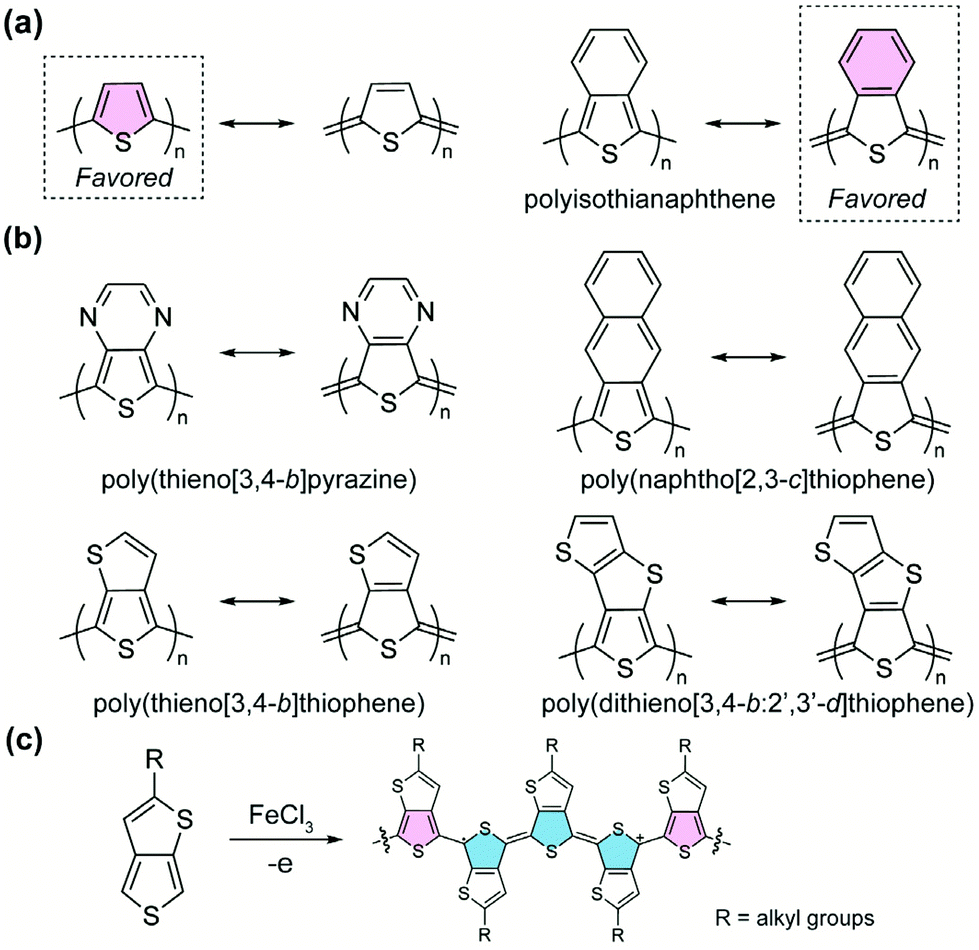 | ||
| Fig. 4 (a) Resonance structures of polythiophene and polyisothianaphthene; (b) representative examples of proquinoidal homopolymers; (c) oxidation polymerization of thieno[3,4-b]thiophene and the doped product with charged quinoidal structures. | ||
Compared to proquinoidal homopolymers, proquinoidal–aromatic alternating copolymers are much more commonly seen in the recent literature. In such a polymer, one can anticipate a significant resonance proportion in which the proquinoidal unit adopts the quinoidal form. In comparison with aromatic homopolymers, the alternating arrangement of proquinoidal and aromatic building blocks usually generates narrower energy bandgaps. Numerous proquinoidal–aromatic copolymers with proquinoidal building blocks, including thieno[3,4-b]pyrazine,44,54,55 thieno[3,4-b]thiophene,14,56–61 dithieno[3,4-b:2′,3′-d]thiophene,62 isothianaphthene,63etc., have been developed as active materials for organic field-effect transistors, photovoltaics, organic light-emitting diodes, and many other applications.
In the syntheses of these polymers, Stille coupling polymerization is the most commonly used method to construct copolymers containing alternating proquinoidal and aromatic repeating units (Fig. 5). A number of comprehensive review articles have been published on this topic.32,33,44,47,64–67 In this context, only several recent representative copolymers with open-shell character are shown here. Benzo[1,2-d:4,5-d′]bistriazole68,69 and benzo[1,2-c:4,5-c′]bis[1,2,5]thiadiazole70–75 are proquinoidal while electron-accepting building blocks. Conjugated polymers including these two moieties, P1–478 and P5 and P6,79 were reported by Tam and Xu. Thiadiazole[3,4-g]quinoxaline is another strong proquinoidal acceptor unit and the derived polymers P7 and P8 were synthesized by Azoulay.76,77 The open-shell diradical character of these proquinoidal building blocks leads to appealing conducting properties in their applications.
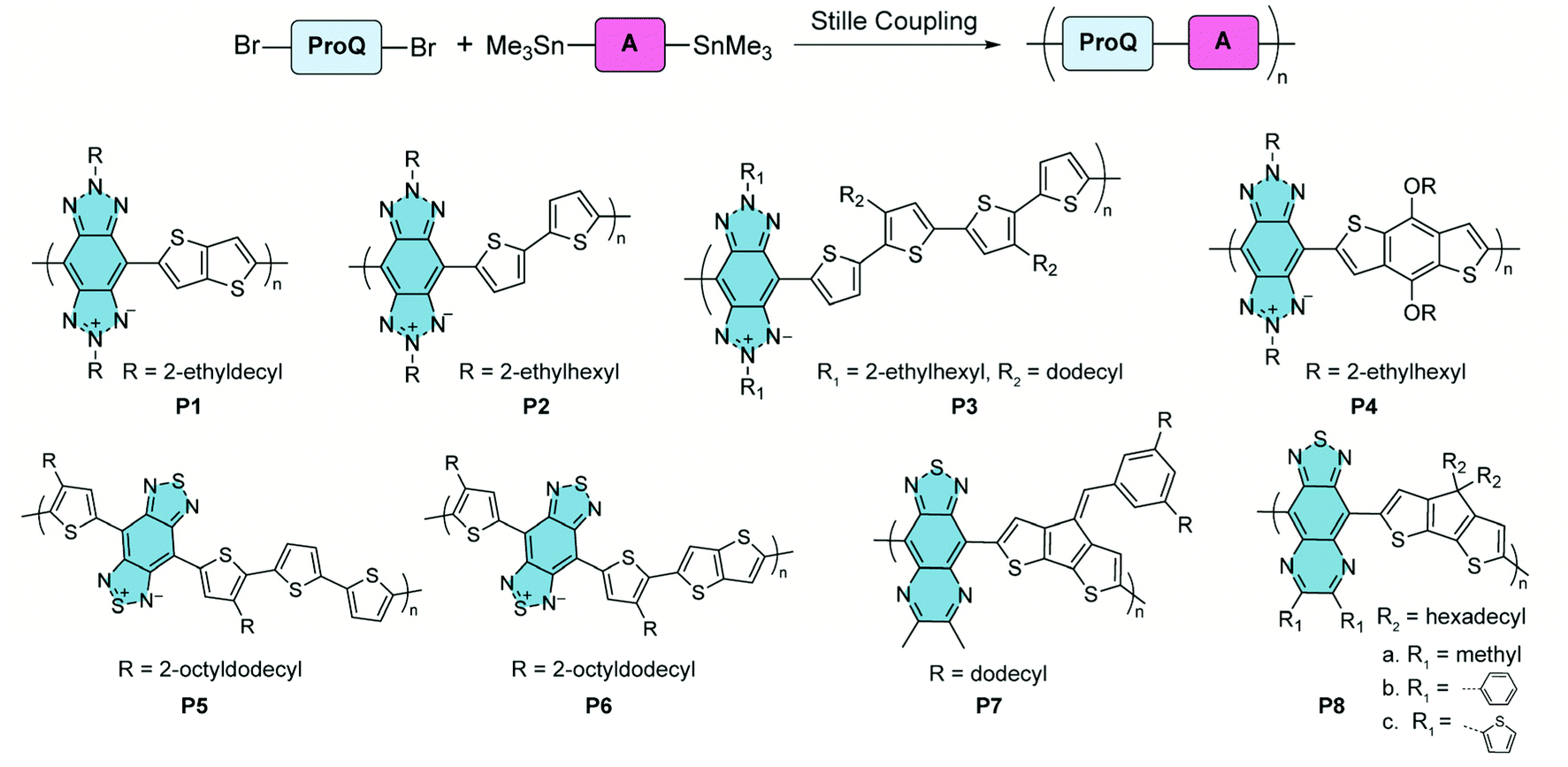 | ||
| Fig. 5 Synthesis and structural formulae of recently reported proquinoidal–aromatic alternating copolymers demonstrating open-shell character. | ||
2.2 Quinoidal–aromatic alternating copolymers
Compared to “proquinoidal” units, “quinoidal” units that adopt a true quinoidal constitution in the ground state are typically less accessible, so that the synthesis of quinoidal–aromatic conjugated polymers requires more careful design to mitigate the stability issue. Two general strategies have been employed for the synthesis: (1) direct polymerization of stabilized quinoidal monomers and (2) post-polymerization modification (e.g. oxidation) of an aromatic polymer precursor into its corresponding quinoidal–aromatic form.Certain quinoidal units derived from thiophenone78,79 and pyrrolidone80–83 can be incorporated into conjugated copolymer backbones by Stille coupling polymerization based on monomers with aromatic terminal groups (Fig. 6). Kim's group synthesized copolymers with isatin-terminated indolone quinoidal moieties. Possible E- and Z-configurational isomers were obtained in thiophene and bithiophene building blocks. When using the 3,4-ethylenedioxythiophene (EDOT)-like repeating units, the noncovalent interactions between sulfur and oxygen atoms lock the conformation of the polymer chain, leading to high coplanarity and stereoregular polymer products. Other quinoidal moieties, such as thieno[2,3-b]thiophene and thiophene dioxide, are also designed and synthesized. The afforded copolymers with low energy bandgaps are usually studied in organic field-effect transistor and organic photovoltaic applications. These works have been reviewed in a recent publication.32 Recently, quinoidal building blocks derived from para-aza-quinodimethane were designed and incorporated into copolymers P9–12 by Liu and coworkers.84–86para-Quinodimethane is a highly reactive quinoidal moiety with large open-shell diradical character. However, good stability is conferred by introducing heteroatoms into the π-system. The effects of different main-chain building blocks and side chain bulkiness on the film morphology and charge transport were investigated systematically by comparing the P9 and P11 series.84,85 By replacing the ether solubilizing groups with cationic triphenylphosphonium groups, P12 was obtained as a conjugated polyelectrolyte with reasonable stability and water-solubility.86
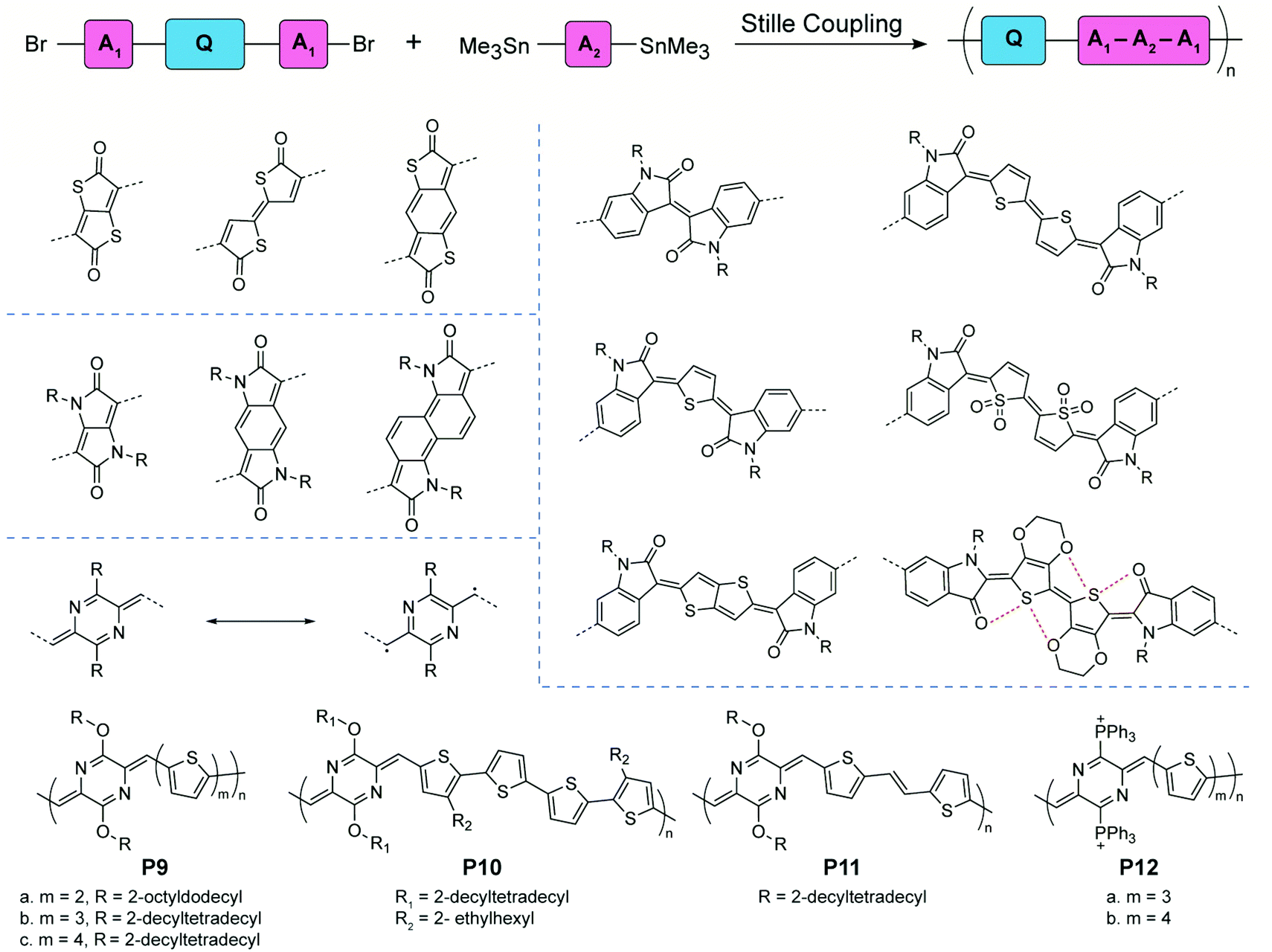 | ||
| Fig. 6 General synthetic scheme, structural formulae of representative quinoidal building blocks, and structures of para-aza-quinodimethane-derived quinoidal–aromatic alternating copolymers. | ||
An alternating quinoidal–aromatic constitution can also be achieved by post-polymerization modification of a fully aromatic precursor. For example, Fang's group designed87 a series of cyclohexadiene-1,4-diimine-derived, quinoidal–aromatic alternating copolymers featuring a ladder-type constitution (Fig. 7). First, a fully aromatic copolymer is synthesized88 by polymerizing para-diaminophenylene- and fluorene-derived monomers. The ladder-type constitution is subsequently constructed by a ring-annulation reaction, affording P13a as a ladder-type analogue of the fully-reduced form of polyaniline (leucoemeraldine). Post-polymerization oxidation converts the para-diaminophenylene units into their quinoidal form, namely, cyclohexa-2,5-diene-1,4-diimine units. The partially oxidized polymer P13b, as an analogue of an emeraldine base, can be obtained under mild oxidation conditions. The fully-oxidized, alternating quinoidal–aromatic copolymer P13c is prepared by peroxide oxidation. The quinoidal structure is confirmed by 13C NMR compared to small molecular models. The redox interconversion between the aromatic form P13a and the quinoidal–aromatic form P13c is highly reversible thanks to the significantly improved robustness of the π-system imparted by the ladder-type constitution.
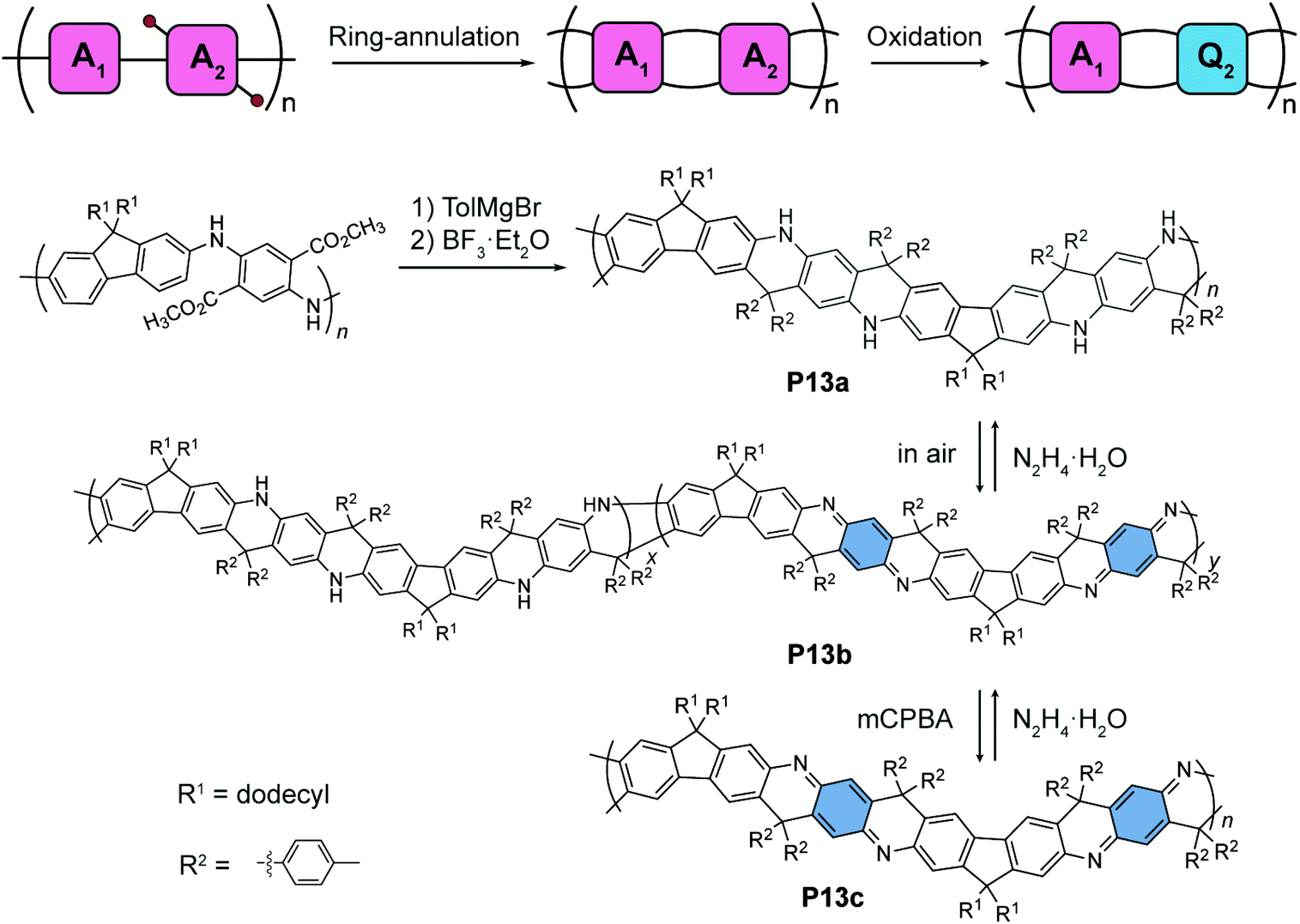 | ||
| Fig. 7 Synthesis of ladder-type polyaniline derivatives P13a–P13c. | ||
Oxidation of methine-bridged polythiophenes represents another interesting strategy for the synthesis of quinoidal–aromatic copolymers (Fig. 8). Chen and Jenekhe synthesized89 such polymers through a two-step route. First, a thiophene-derived monomer is polymerized with benzaldehyde through an acid-catalyzed Friedel–Crafts reaction. Then the resulting polymer is oxidized to introduce the quinoidal thienomethine moieties in P14 and P15.90,91 Each quinoidal moiety contains three diastereoisomers, i.e., E/E, E/Z, and Z/Z, leading to isomeric mixtures in the product. In a recent report, a C–C bulk polycondensation reaction was used on the bisthiophene methine monomer, affording P16 after partial oxidation.92 Other aromatic building blocks, e.g., diketopyrrolopyrrole, could also be connected with thienoquinodimethane units through Stille coupling polymerization, affording the donor–acceptor copolymer P17.93
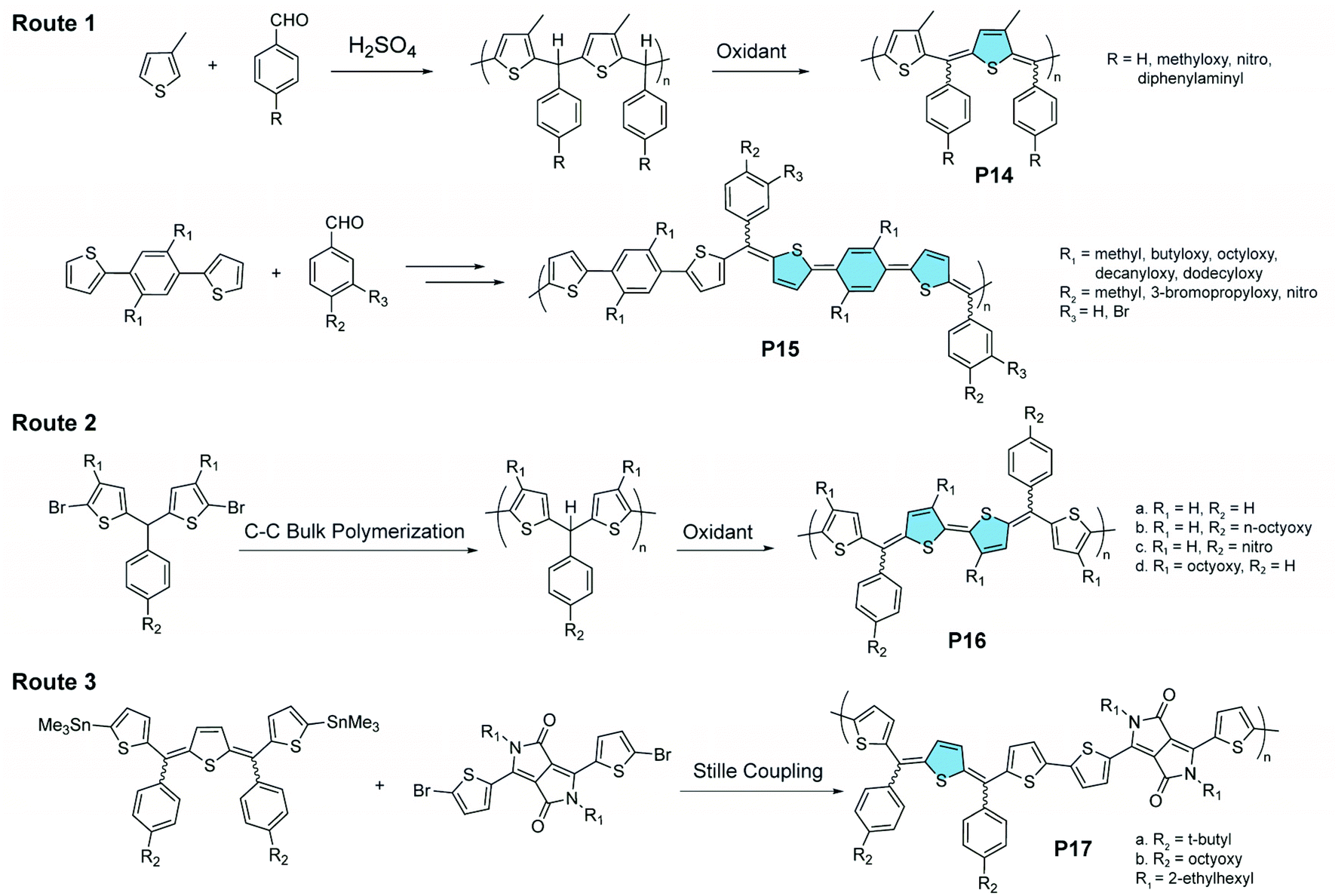 | ||
| Fig. 8 Three synthetic routes to methine-bridged polythiophenes featuring alternating quinoidal and aromatic units on the main chain. | ||
3. Properties and applications
3.1 Open-shell diradical character
The unpaired electrons and their corresponding magnetic moments render intriguing behaviors of quinoidal conjugated polymer that are typically not observable on their closed-shell counterparts. Therefore, the investigation of quinoidal polymers with isolable stability of high-spin states has emerged in diverse research fields, on topics of spin manipulation, organic magnetism, quantum functionalities, and interrelated optoelectronic properties.In proquinoidal–aromatic copolymers such as P5 and P6, the open-shell resonance was characterized, exhibiting increasing electron paramagnetic resonance (EPR) intensities with decreasing temperature, indicating the triplet open-shell ground state (Fig. 9). The smaller temperature dependence of P5 can be seen as evidence of its larger energy gap between the triplet and singlet states. Magnetic susceptibility χ is a sum of three components: antiferromagnetic coupling between polymer chains (χAFM), ferromagnetic coupling from the triplet state (χFM), and temperature-independent paramagnetism arising from coupling between a magnetic ground state and non-thermally populated excited states (χTIP). The coupling constant of antiferromagnetic coupling JAFM (kcal per mole repeating unit) is calculated to be −0.63 for P5 and −0.55 for P6, indicating a more extended spin coupling of P6. According to glazing incidence small- and wide-angle X-ray scattering measurements on thin film samples, the π–π distances of P5 and P6 are 3.45 Å and 3.51 Å, respectively. These data support the hypothesis that smaller π–π distances lead to larger |JAFM| for these proquinoidal polymers.
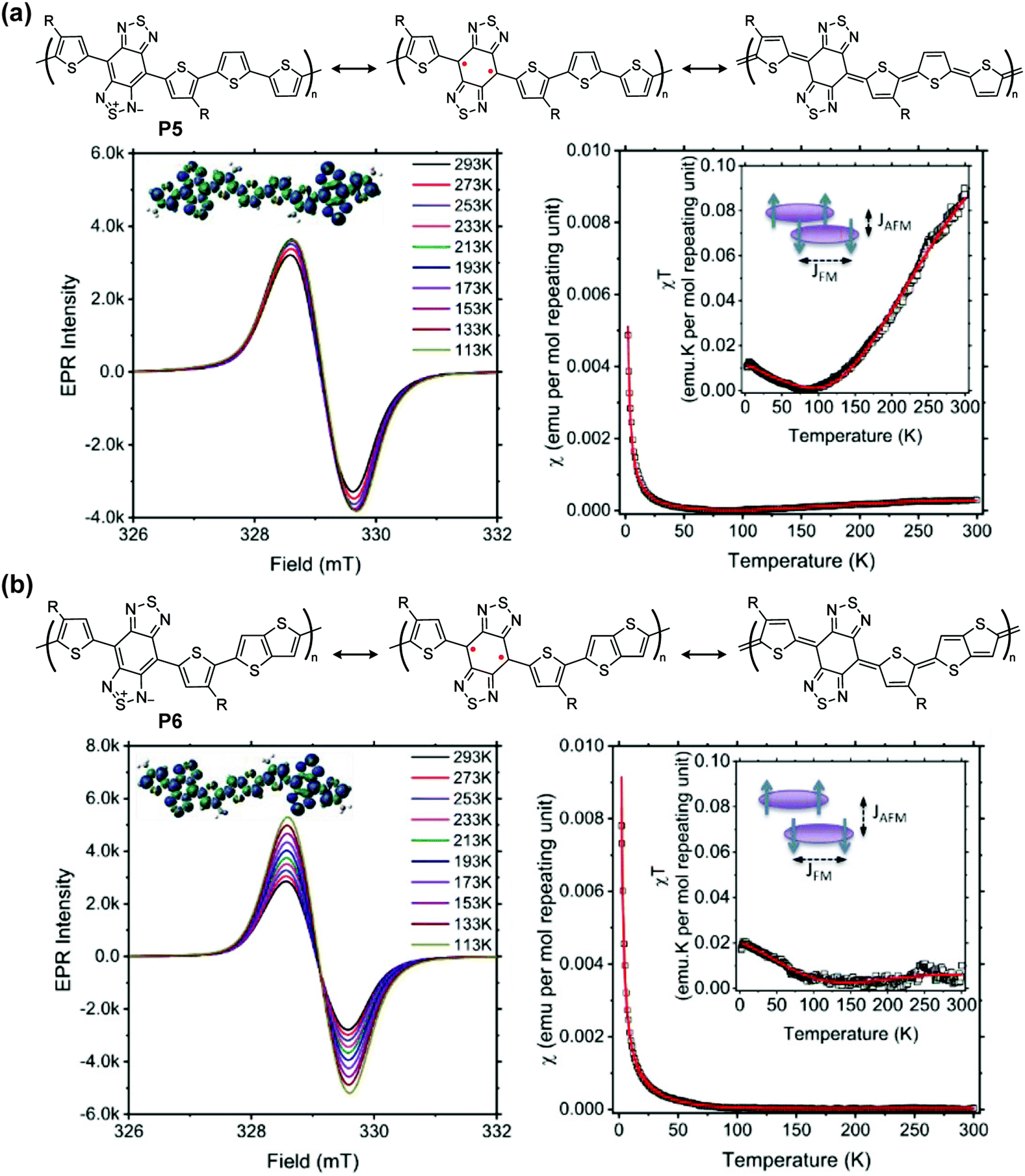 | ||
| Fig. 9 Closed-shell and open-shell structures of P5 (a) and P6 (b), and their variable-temperature-EPR spectra, χ–T and χT–T plots [inset: diagram showing ferromagnetic (FM) and antiferromagnetic (AFM) coupling of spins]. Reproduced from ref. 94 with permission from the American Chemical Society. | ||
For P7 composed of cyclopentadithiophenyl and thiadiazoloquinoxaline units76 (Fig. 10a), a paramagnetic ground state was also observed and the singlet–triplet energy gap (ΔES–T) was calculated to be 9.30 × 10−3 kcal mol−1 (Fig. 10b and c) based on superconducting quantum interference device (SQUID) magnetometry results. The triplet ground state, with weak interchain ferromagnetic interaction, was also found to be independent of the spin concentration. The distribution of spins was characterized by two-pulse spin echo measurements of instantaneous diffusion (Fig. 10d), indicating that the nearest electron spins are approximately distributed in one dimension, as if randomly placed on a linear polymer chain. Similarly, a high spin concentration was also observed for the P8 series with the triplet ground state and weak ferromagnetic coupling.77
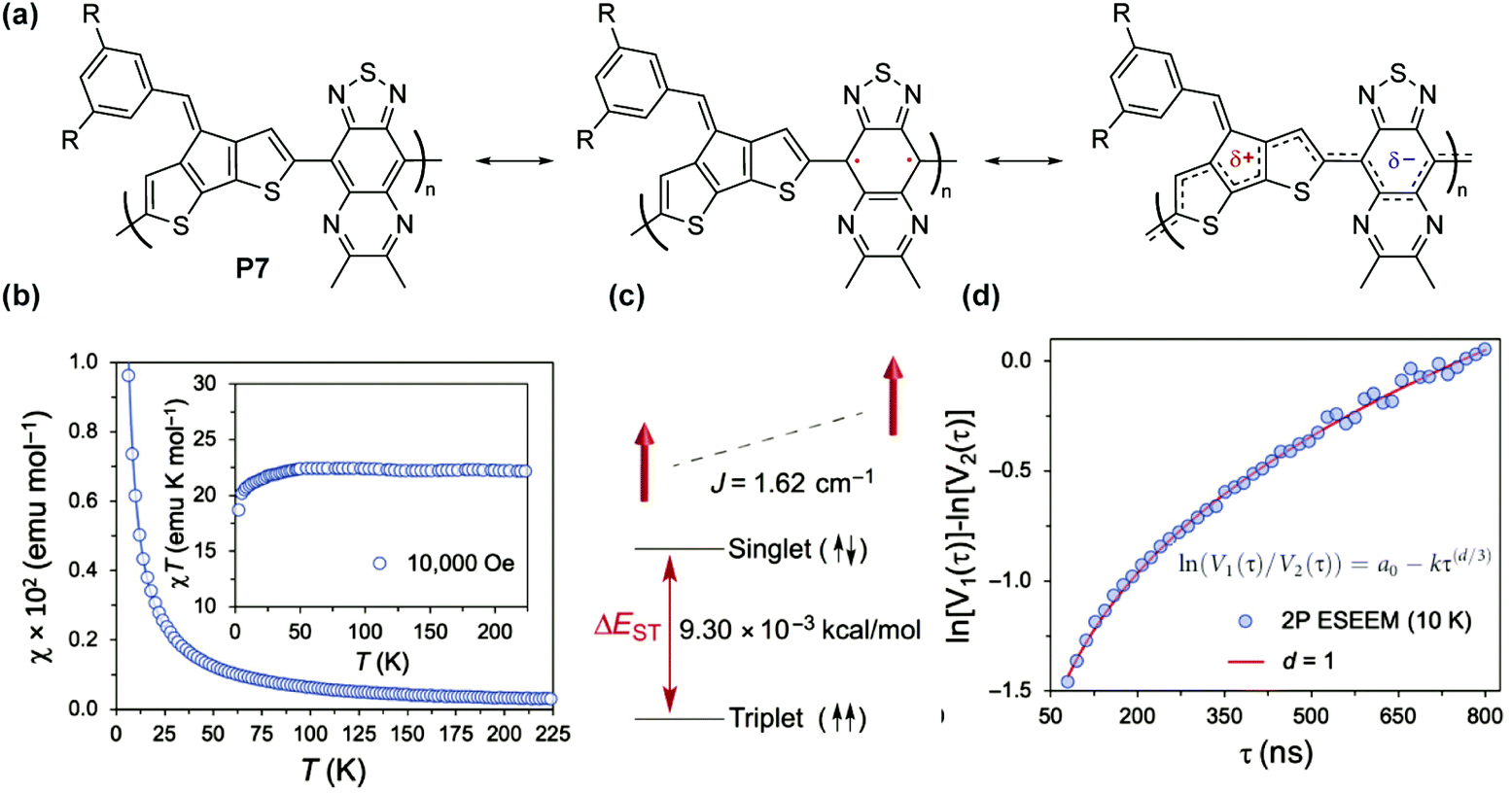 | ||
| Fig. 10 (a) Closed-shell and open-shell resonance forms of P7; (b) χ–T and χT–T plots obtained by SQUID measurement; (c) energy diagram of singlet and triplet states; (d) two-pulse electron spin echo instantaneous diffusion data. Reproduced from ref. 76 with permission from the American Association for the Advancement of Science. | ||
Interestingly, the quinoidal–aromatic copolymer P10 was reported recently as a good singlet fission candidate.95 The fulfillment of energetic requirements for singlet fission is primarily ascribed to the distinctive biradical character of the heterocyclic quinoidal core.96,97P10 was found to produce long-lived triplet pairs efficiently in strongly coupled polymer thin films, showing the promising potential of quinoidal conjugated polymers in developing next-generation singlet-fission-based organic solar cells.
3.2 Delocalization of polarons/bipolarons
Recently, Wang and coworkers reported10 direct observation of quinoidal units in PPP chains as charge carriers. Polymers and oligomers were synthesized on the surface of the Cu (111) substrate. The aromatic (benzenoid) and quinoidal forms can be distinctively visualized by scanning tunneling microscopy (STM) and noncontact atomic force microscopy. Most quinoidal segments are found with a length of around 8 rings. The excitation from the ground state aromatic to the quinoidal forms can be facilitated either by a surface adsorption process, which enforces a planar chain conformation, or by the charge transfer from underlying Cu (111). Using STM with a voltage bias, the intrachain transport of quinoidal segments can be observed (Fig. 11a and b). Interchain transport is also directly observed as the quinoidal segment hopped from one chain to another (Fig. 11c and d) under a bias of 1 mV. These results suggest a high-mobility character of the quinoidal polarons or bipolarons in PPP on the Cu (111) surface.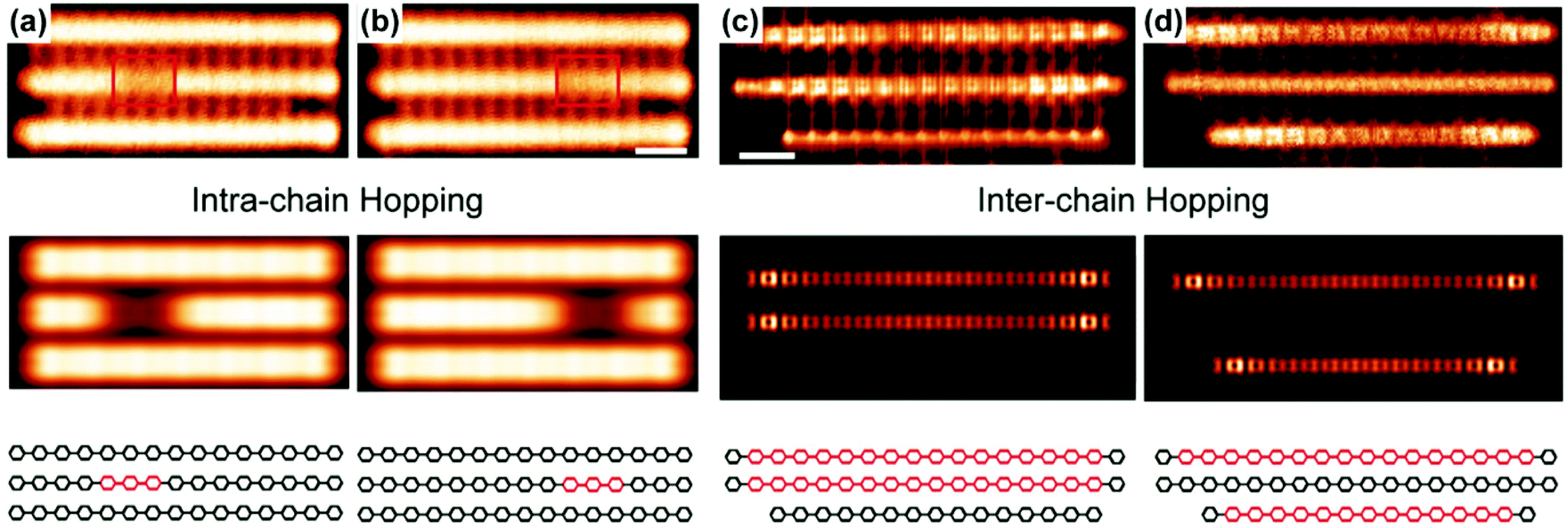 | ||
| Fig. 11 (a and b) (Top) Experimental STM images of PPP chains showing the transport of the quinoidal segment in one of the chains; (middle) simulated STM images; (bottom) structural representation of the three chains shown above. (c and d) (Top) Constant-height STM images of PPP chains showing the hopping of the quinoidal segment from the middle chain to the bottom chain; (middle) simulated current images; (bottom) structural representation of the three chains shown above. Reproduced from ref. 10 with permission from the American Chemical Society. | ||
The high conductivity rendered by the hopping of quinoidal segment has been applied in organic conducting and thermoelectric devices.77 For example, the electrical conductivity of up to 6 S cm−1 can be achieved on proquinoidal poly(thieno[3,2-b]thiophene) (PTT).98 To further enhance the conductivity, Zhu's group developed99 an in situ solution casting polymerization method to prepare doped PTT-Tos with tosylate dopant anions (Fig. 12a).100 The polymer with the shortest alkyl chain (methyl) shows electrical conductivity up to 450 S cm−1 at 300 K and 4444 S cm−1 at 370 K. Different from the metallic conducting poly(3,4-ethylenedioxythiophene) (PEDOT)-tosylate system, the strong temperature dependence of conductivity indicates a thermally activated hopping mechanism of the transport of quinoidal quasiparticles (Fig. 12b). A polaron-dominant carrier transport mode is proposed based on strong EPR signals. In this work, the increased electrical conductivity at higher temperatures also contributes to the enhanced thermoelectric performances (Fig. 12c).
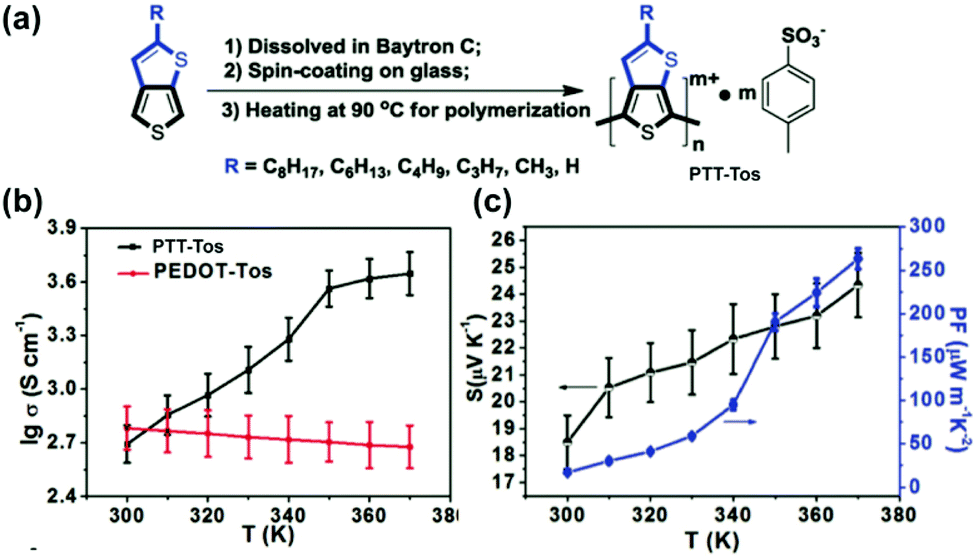 | ||
| Fig. 12 (a) Synthesis and structural formula of PTT-Tos; (b) temperature-dependent conductivity of PTT-Tos compared with PEDOT-Tos; (c) temperature-dependent Seebeck coefficient and power factor of PTT-Tos. Reproduced from ref. 99 with permission from John Wiley and Sons. | ||
Other proquinoidal polymers, such as P5 and P6, have also demonstrated promising properties as electrical conducting and thermoelectric materials after doping. After doping with Fe(III), polaron/bipolaron species are generated in P5 and P6 (Fig. 13a).94 With an increased doping level, a broad absorption band spanning from 1000 nm beyond 3100 nm (transition energy lower than 0.4 eV) emerges and increases, suggesting the presence of highly delocalized polarons/bipolarons (Fig. 13b). The electrical conductivities of P5 and P6 reach 313.9 S cm−1 and 287.0 S cm−1 after doping, respectively. The extended intra- and intermolecular delocalization of polarons or bipolarons also renders high Seebeck coefficients via carrier-induced softening (46.6 μV K−1for P5 and 65.4 μV K−1 for P6). The conductivity and the Seebeck coefficient contribute cooperatively in enhancing the thermoelectric power factor up to 43.5 μW m−1 K−2 for P5 and 65.2 μW m−1 K−2 for P6. Based on DFT calculation results, the formation of stable triplet bipolarons in doped P6 is proposed to be the reason for its large Seebeck coefficient despite the high electrical conductivity.
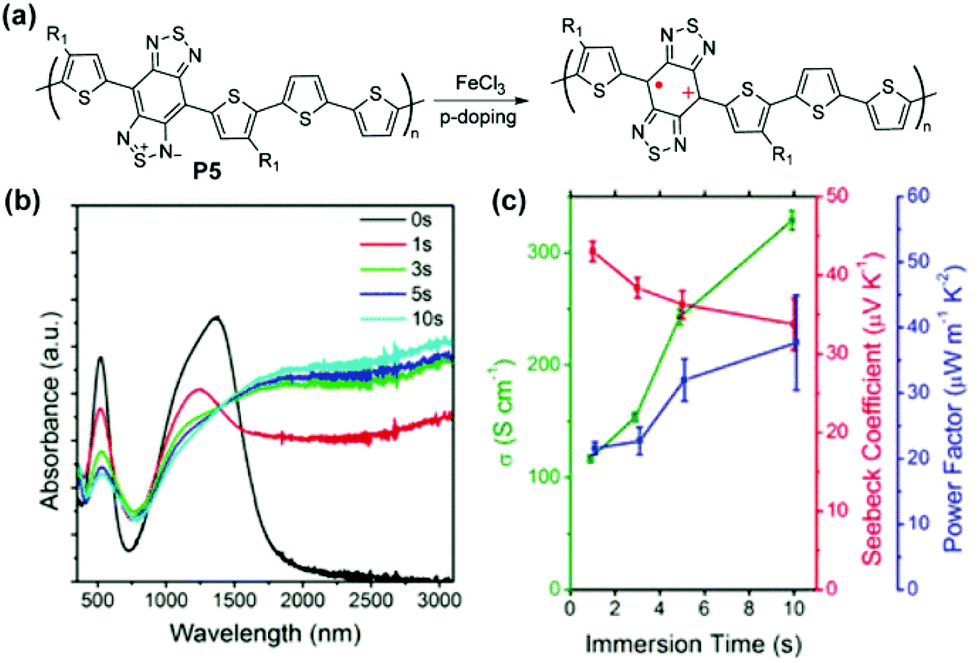 | ||
| Fig. 13 (a) Structural representation of P5 before and after oxidative doping; (b) evolution of thin film UV-vis-NIR absorption spectra of P5 immersed in FeCl3 solution as a function of time; (c) electrical conductivity, Seebeck coefficient, and power factor of P5 as a function of immersion doping time. Reproduced from ref. 94 with permission from the American Chemical Society. | ||
The ladder-type polyaniline analogue P13c with quinoidal building blocks can be doped by acid (Fig. 14a) to generate an analogue of pernigraniline salt P13c-H. For conventional pernigraniline salt, the iminium bonds are labile under ambient conditions so that the polymer is unstable and cannot be extensively investigated for its intrinsic spin properties, while with a ladder-type constitution, the originally labile iminium linkage is stabilized by the extra strand of bonds.101 The good stability of P13c-H enables the comprehensive study of its strong open-shell character by EPR spectroscopy and SQUID magnetometry (Fig. 14b).87 The radical cations are delocalized along the ladder-type backbone with extended conjugation. P13c-H possesses dominant Pauli paramagnetism and negligible Curie spin character in the solid state (Fig. 14c). The high Pauli paramagnetic susceptibility (χPauli) value indicates a significantly enhanced polaron delocalization range, in terms of both intrachain and interchain, and high density of states at the Fermi level.102
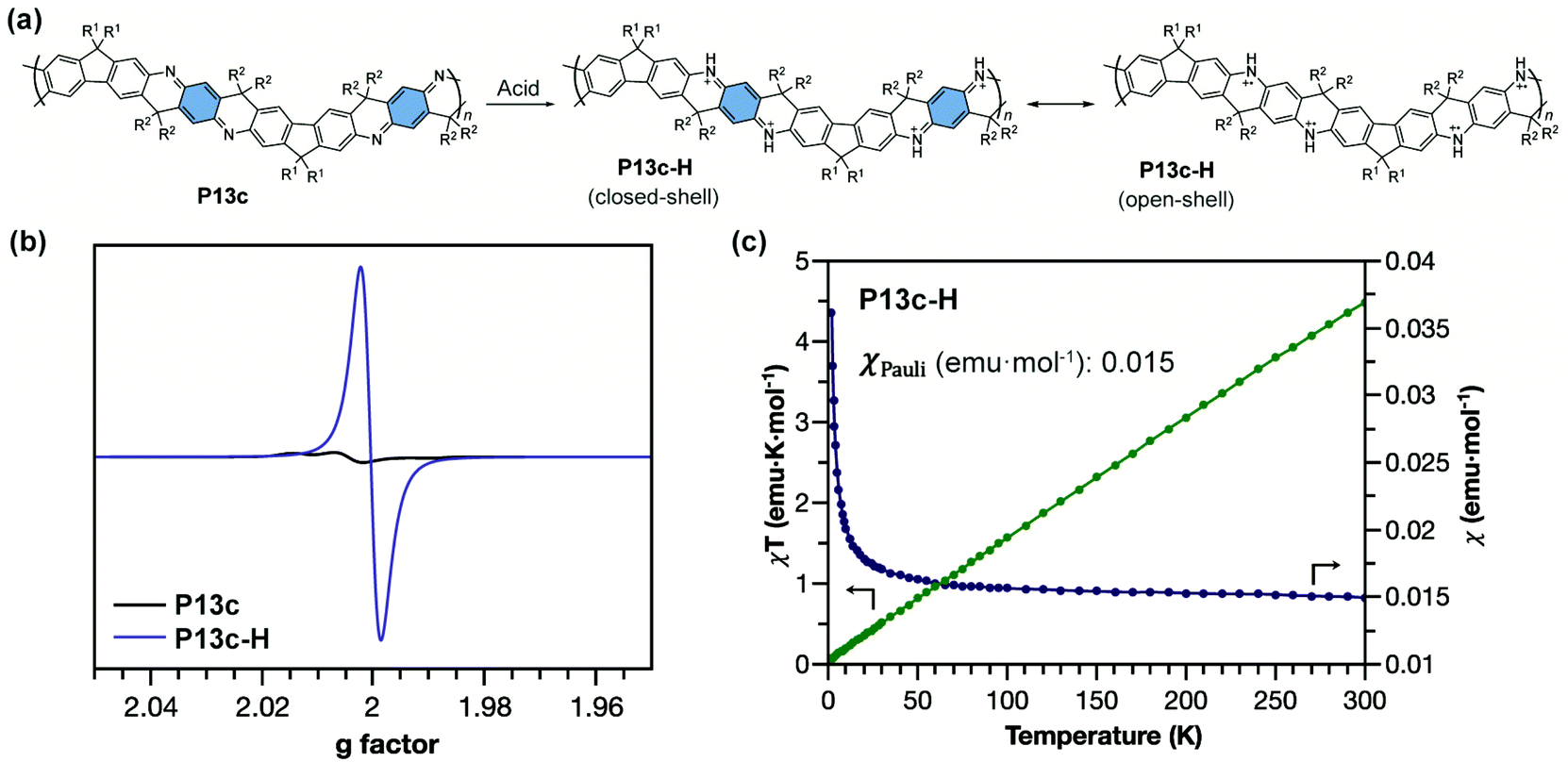 | ||
| Fig. 14 (a) Structural representation of P13c and its acid-doped form P13c-H with closed-shell and open-shell resonance structures; (b) EPR spectra of P13c and P13c-H; (c) χ–T and χT–T plots of P13c-H showing dominant Pauli paramagnetism. Reproduced from ref. 87 with permission from the Royal Society of Chemistry. | ||
3.3 Narrowed bandgaps and tunable energy levels
Energy bandgaps of conjugated polymers are determined by a number of factors, including BLA, aromatic resonance energy, substituents, planarity, and intermolecular interactions.4,103–109 The relationship between energy bandgap and BLA in organic conjugated polymers has been extensively investigated, revealing that quinoidal structures with smaller BLA would efficiently decrease the energy bandgap.4,5,33,103,104,106–110 Besides the small BLA, another important feature of quinoidal structures is their preferred coplanar conformation. The coplanar conformation can further lower the energy bandgap33 by facilitating delocalization of electrons, states, and quasiparticles not only along the backbone but also in between polymer chains through intermolecular electronic coupling. In many cases, quinoidal structures are also the electron-withdrawing units in donor–acceptor systems, leading to intramolecular charge transfer and further reduced energy bandgap. In this context, the quinoidal design has been employed in the development of many conjugated polymer materials for photovoltaic application and field-effect transistors. These examples have been already reviewed in other publications32,33,35,64,110 so that they are not discussed extensively here. For the polymers covered in this review, their parameters and performances associated with these applications are summarized in Table 1.| Ref. | Polymer | E HOMO (eV) | E LUMO (eV) | E g,opt (eV) | μ h (cm2 V−1 s−1) | Devicea | σ intrinsic (S cm−1) |
|---|---|---|---|---|---|---|---|
| a BGBC: Bottom-gate/bottom-contact; BGTC: bottom-gate/top-contact. | |||||||
| 111 | P1 | −4.82 | 1.08 | 0.0011 | BGBC | 10−2 | |
| P2 | −5.01 | −3.60 | 1.22 | 0.092 | 10−3 | ||
| P3 | −5.02 | −3.50 | 1.26 | 0.13 | 10−4 | ||
| P4 | −5.16 | −3.60 | 1.38 | 10−5 | |||
| P5 | −5.06 | −3.79 | 0.70 | 1.8 (ref. 112) | |||
| P6 | −5.09 | −3.84 | 0.52 | 1.0 (ref. 113) | |||
| 76 | P7 | −4.79 | −4.23 | <0.30 | 10−2 | ||
| 77 | P8a | −4.93 | −3.90 | 0.57 | 1.75 × 10−1 | BGBC | 8.18 |
| P8b | −5.06 | −4.19 | 0.59 | 4.50 × 10−4 | 2.89 × 10−3 | ||
| P8c | −4.95 | −4.15 | 0.54 | 1.95 × 10−3 | 3.75 × 10−3 | ||
| 84 | P9a | −4.90 | −3.58 | 1.32 | 0.024 | BGTC | |
| P9b | −5.02 | −3.53 | 1.49 | 0.47 | |||
| P9c | −5.07 | −3.54 | 1.53 | 0.082 | |||
| 95 | P10 | 1.78 | |||||
| 85 | P11 | −5.0 | −3.6 | 1.38 | 0.0039 | BGTC | |
| 86 | P12a | −5.04 | −4.11 | 1.22 | |||
| P12b | −5.07 | −4.09 | 1.33 | ||||
| 87 | P13a | −4.88 | 3.10 | ||||
| P13b | −4.90 | −3.68 | |||||
| P13c | −5.33 | −3.76 | 1.57 | ||||
| 91 | P14 | 1.81–1.95 | |||||
| 90 | P15 | −5.89 to −5.66 | −4.11 to −3.88 | 1.26–2.07 | |||
| 92 | P16a | −5.30 | −3.73 | 1.67 | |||
| P16b | −5.13 | −3.70 | 1.70 | ||||
| P16c | −5.06 | −3.87 | 1.59 | ||||
| P16d | −5.12 | −3.63 | 1.81 | ||||
| 93 | P17a | −5.2 | −4.0 | 1.2 | |||
| P17b | −4.9 | −3.7 | 1.2 | ||||
The low energy bandgap also warrants quinoidal polymers as promising active agents for near-infrared (NIR) photothermal therapy. For example, P12a and P12b possess NIR absorbance edging at 1010 nm and 940 nm, respectively.86 Reasonable water solubility of these polymers can be achieved by installing cationic groups on the polymer backbone. In a diluted aqueous solution of P12b, a significant photothermal effect is observed. The solution temperature increases to 50.6 °C in 5 min under excitation of an 808 nm laser (Fig. 15a), in drastic contrast with the blank solution. The extent and rate of warming can be controlled and employed to cause the death of bacteria through damage to proteins and lipids on the cell membrane. A 95% reduction of the model bacterial colony is observed after 5 min of irradiation when treated with P12 solution (Fig. 15b).
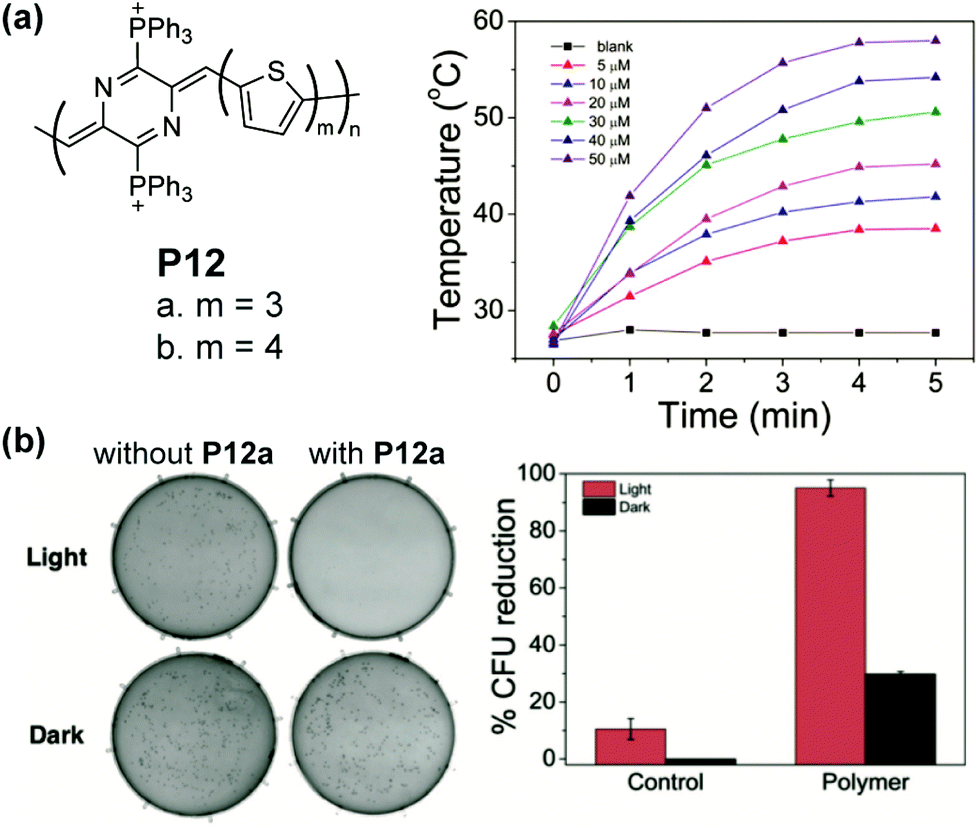 | ||
| Fig. 15 (a) Structure of P12 polymers and the photothermal effect of P12a in diluted solution; (b) photographs of S. aureus colonies with and without P12a both with and without 808 nm laser irradiation for 5 min and quantized colony reduction data. Reproduced from ref. 86 with permission from John Wiley and Sons. | ||
4. Conclusion and perspectives
This review summarizes recent advances on the syntheses, properties, and applications of quinoidal conjugated polymers featuring open-shell character. The quinoidal structure imparts a number of intriguing properties—including but not limited to extended π-conjugation, open-shell character, and polaron/bipolaron delocalization—to these polymers. A wide range of polymers with diverse structural constitutions have been investigated to demonstrate great potential as novel high-spin organic materials. However, various challenges still remain in this research topic. First, the scope of structures and syntheses of (pro)quinoidal monomeric building blocks is relatively narrow compared to traditional aromatic conjugated polymers. Although small energy bandgaps have been extensively demonstrated in quinoidal conjugated polymers, studies on their open-shell character are still scarce. Second, it is still challenging to precisely predict and control energy levels of (pro)quinoidal building blocks due to the complex contribution from BLA, intrachain charge transfer, and Peierls distortion.59 Last but not least, the open-shell character of the quinoidal structure could bring in reactivity and stability issues for fundamental characterization and practical applications.In future research, the discovery and development of novel quinoidal building blocks and synthetic methodologies are desired in order to achieve precision control of the unique properties of quinoidal conjugated polymers. Further fundamental investigations on the open-shell, high-spin character and polaron and/or bipolaron delocalization properties are critical to understand the nature of quinoidal structures and to render high stability in these materials.
In terms of applications, it is anticipated that quinoidal conjugated polymers will play an impactful role in the future development of organic photovoltaics and field-effect transistors, on the basis of the thriving accomplishments made in these fields and recent progress in singlet fission study. Besides, the incorporation of quinoidal units in doped conjugated polymers can be a viable strategy to achieve robust organic materials with metallic ground states.77 Last but not least, advances in the understanding of the open-shell character have also enabled the use of quinoidal polymers in organic thermoelectricity, spintronics, nonlinear optics, etc.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Robert A. Welch Foundation (A-1898) and the President's Excellence Fund of Texas A&M University (X-Grant) for financial support of this work.Notes and references
- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”), Blackwell Scientific Publications, Oxford, 1997 Search PubMed.
- M. Kertesz, C. H. Choi and S. Yang, Chem. Rev., 2005, 105, 3448 CrossRef CAS.
- J. Casado, in Physical Organic Chemistry of Quinodimethanes, Springer, 2017, pp. 209 Search PubMed.
- S. Rasmussen, in Encyclopedia of Polymeric Nanomaterials, ed. S. Kobayashi and K. Müllen, Springer Berlin Heidelberg, Berlin, Heidelberg, 2015, pp. 1155 Search PubMed.
- J. L. Brédas, J. Chem. Phys., 1985, 82, 3808 CrossRef.
- N. Bérubé, J. Gaudreau and M. Côté, Macromolecules, 2013, 46, 6873 CrossRef.
- Y. Hayashi and S. Kawauchi, Polym. Chem., 2019, 10, 5584 RSC.
- P. M. Burrezo, J. L. Zafra, J. T. López Navarrete and J. Casado, Angew. Chem., Int. Ed., 2017, 56, 2250 CrossRef CAS.
- J. Bredas, R. Chance and R. Silbey, Phys. Rev. B: Condens. Matter Mater. Phys., 1982, 26, 5843 CrossRef CAS.
- B. Yuan, C. Li, Y. Zhao, O. Gröening, X. Zhou, P. Zhang, D. Guan, Y. Li, H. Zheng, C. Liu, Y. Mai, P. Liu, W. Ji, J. Jia and S. Wang, J. Am. Chem. Soc., 2020, 142, 10034 CrossRef CAS.
- T. Y. Gopalakrishna, W. Zeng, X. Lu and J. Wu, Chem. Commun., 2018, 54, 2186 RSC.
- R. Lv, S. Geng, S. Li, F. Wu, Y. Li, T. R. Andersen, Y. Li, X. Lu, M. Shi and H. Chen, Solar RRL, 2020, 2000286 CrossRef CAS.
- L. Ren, H. Fan, D. Huang, D. Yuan, C. Di and X. Zhu, Chem. – Eur. J., 2016, 22, 17136 CrossRef CAS.
- C. Zhang and X. Zhu, Acc. Chem. Res., 2017, 50, 1342 CrossRef CAS.
- X. Hu, W. Wang, D. Wang and Y. Zheng, J. Mater. Chem. C, 2018, 6, 11232 RSC.
- D. Huang, H. Yao, Y. Cui, Y. Zou, F. Zhang, C. Wang, H. Shen, W. Jin, J. Zhu, Y. Diao, W. Xu, C. Di and D. Zhu, J. Am. Chem. Soc., 2017, 139, 13013 CrossRef CAS.
- J. Casado, R. P. Ortiz and J. T. L. Navarrete, Chem. Soc. Rev., 2012, 41, 5672 RSC.
- J. Xia, S. N. Sanders, W. Cheng, J. Z. Low, J. Liu, L. M. Campos and T. Sun, Adv. Mater., 2017, 29, 1601652 CrossRef.
- C. K. Frederickson, B. D. Rose and M. M. Haley, Acc. Chem. Res., 2017, 50, 977 CrossRef CAS.
- T. Stuyver, B. Chen, T. Zeng, P. Geerlings, F. De Proft and R. Hoffmann, Chem. Rev., 2019, 119, 11291 CrossRef CAS.
- Y. Li, L. Li, Y. Wu and Y. Li, J. Phys. Chem. C, 2017, 121, 8579 CrossRef CAS.
- T. Kubo, Chem. Lett., 2015, 44, 111 CrossRef.
- J. J. Dressler, M. Teraoka, G. L. Espejo, R. Kishi, S. Takamuku, C. J. Gómez-García, L. N. Zakharov, M. Nakano, J. Casado and M. M. Haley, Nat. Chem., 2018, 10, 1134 CrossRef CAS.
- Z. Zeng, X. Shi, C. Chi, J. T. L. Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578 RSC.
- W. Zeng, H. Phan, T. S. Herng, T. Y. Gopalakrishna, N. Aratani, Z. Zeng, H. Yamada, J. Ding and J. Wu, Chem, 2017, 2, 81 CrossRef CAS.
- A. Moliton and R. C. Hiorns, Polym. Int., 2004, 53, 1397 CrossRef CAS.
- J. L. Brédas and G. B. Street, Acc. Chem. Res., 1985, 18, 309 CrossRef.
- D. Yuan, D. Huang, S. M. Rivero, A. Carreras, C. Zhang, Y. Zou, X. Jiao, C. R. McNeill, X. Zhu, C.-a. Di, D. Zhu, D. Casanova and J. Casado, Chem, 2019, 5, 964 CAS.
- J. Yamamoto and Y. Furukawa, J. Phys. Chem. B, 2015, 119, 4788 CrossRef CAS.
- A. G. MacDiarmid, Angew. Chem., Int. Ed., 2001, 40, 2581 CrossRef CAS.
- J. Scotto, M. I. Florit and D. Posadas, Electrochim. Acta, 2018, 268, 187 CrossRef CAS.
- T. Mikie and I. Osaka, J. Mater. Chem. C, 2020, 8, 14262 RSC.
- C. Liu, K. Wang, X. Gong and A. J. Heeger, Chem. Soc. Rev., 2016, 45, 4825 RSC.
- Y. Liang and L. Yu, Acc. Chem. Res., 2010, 43, 1227 CrossRef CAS.
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607 CrossRef CAS.
- F. Wudl, M. Kobayashi and A. J. Heeger, J. Org. Chem., 1984, 49, 3382 CrossRef CAS.
- I. Hoogmartens, P. Adriaensens, D. Vanderzande, J. Gelan, C. Quattrocchi, R. Lazzaroni and J. L. Brédas, Macromolecules, 1992, 25, 7347 CrossRef CAS.
- F. Wudl, M. Kobayashi, N. Colaneri, M. Boysel and A. J. Heeger, Mol. Cryst. Liq. Cryst., 1985, 118, 199 CrossRef CAS.
- N. Colaneri, M. Kobayashi, A. Heeger and F. Wudl, Synth. Met., 1986, 14, 45 CrossRef CAS.
- J. L. Brédas, A. J. Heeger and F. Wudl, J. Chem. Phys., 1986, 85, 4673 CrossRef.
- M. Pomerantz, B. Chaloner-Gill, L. O. Harding, J. J. Tseng and W. J. Pomerantz, J. Chem. Soc., Chem. Commun., 1992, 1672 RSC.
- D. D. Kenning and S. C. Rasmussen, Macromolecules, 2003, 36, 6298 CrossRef CAS.
- L. Wen, B. C. Duck, P. C. Dastoor and S. C. Rasmussen, Macromolecules, 2008, 41, 4576 CrossRef CAS.
- S. C. Rasmussen, R. L. Schwiderski and M. E. Mulholland, Chem. Commun., 2011, 47, 11394 RSC.
- Y. Deng, B. Sun, Y. He, J. Quinn, C. Guo and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 3459 CrossRef CAS.
- G. A. Sotzing and K. Lee, Macromolecules, 2002, 35, 7281 CrossRef CAS.
- G. A. Sotzing, V. Seshadri and F. J. Waller, in Handbook of Conducting Polymer, ed. T. A. Skotheim and J. R. Reynolds, CRC Press, Boca Raton, 2007, p. 11/1 Search PubMed.
- K. Lee and G. A. Sotzing, Macromolecules, 2001, 34, 5746 CrossRef CAS.
- J. Kürti, P. R. Surján and M. Kertész, J. Am. Chem. Soc., 1991, 113, 9865 CrossRef.
- K. Nayak and D. S. Marynick, Macromolecules, 1990, 23, 2237 CrossRef CAS.
- C. Arbizzani, M. Catellani, M. Mastragostino and M. Cerroni, J. Electroanal. Chem., 1997, 423, 23 CrossRef CAS.
- R. van Asselt, I. Hoogmartens, D. Vanderzande, J. Gelan, P. E. Froehling, M. Aussems, O. Aagaard and R. Schellekens, Synth. Met., 1995, 74, 65 CrossRef CAS.
- I. Polec, A. Henckens, L. Goris, M. Nicolas, M. Loi, P. Adriaensens, L. Lutsen, J. Manca, D. Vanderzande and N. Sariciftci, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1034 CrossRef CAS.
- E. W. Culver, T. E. Anderson, J. T. López Navarrete, M. C. Ruiz Delgado and S. C. Rasmussen, ACS Macro Lett., 2018, 7, 1215 CrossRef CAS.
- Y. Zhu, R. D. Champion and S. A. Jenekhe, Macromolecules, 2006, 39, 8712 CrossRef CAS.
- Y. Liang, D. Feng, Y. Wu, S.-T. Tsai, G. Li, C. Ray and L. Yu, J. Am. Chem. Soc., 2009, 131, 7792 CrossRef CAS.
- Y. Liang, Y. Wu, D. Feng, S.-T. Tsai, H.-J. Son, G. Li and L. Yu, J. Am. Chem. Soc., 2009, 131, 56 CrossRef CAS.
- J. Hou, H.-Y. Chen, S. Zhang, R. I. Chen, Y. Yang, Y. Wu and G. Li, J. Am. Chem. Soc., 2009, 131, 15586 CrossRef CAS.
- N. Kleinhenz, L. Yang, H. Zhou, S. C. Price and W. You, Macromolecules, 2011, 44, 872 CrossRef CAS.
- T. Zheng, L. Lu, N. E. Jackson, S. J. Lou, L. X. Chen and L. Yu, Macromolecules, 2014, 47, 6252 CrossRef CAS.
- W. Zhang, T. Huang, J. Li, P. Sun, Y. Wang, W. Shi, W. Han, W. Wang, Q. Fan and W. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 16311 CrossRef CAS.
- Y. Cai, X. Xue, G. Han, Z. Bi, B. Fan, T. Liu, D. Xie, L. Huo, W. Ma, Y. Yi, C. Yang and Y. Sun, Chem. Mater., 2018, 30, 319 CrossRef CAS.
- J. D. Douglas, G. Griffini, T. W. Holcombe, E. P. Young, O. P. Lee, M. S. Chen and J. M. Fréchet, Macromolecules, 2012, 45, 4069 CrossRef CAS.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666 CrossRef CAS.
- H. J. Son, F. He, B. Carsten and L. Yu, J. Mater. Chem., 2011, 21, 18934 RSC.
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633 CrossRef CAS.
- C. Duan, F. Huang and Y. Cao, J. Mater. Chem., 2012, 22, 10416 RSC.
- T. L. Tam, H. H. R. Tan, W. Ye, S. G. Mhaisalkar and A. C. Grimsdale, Org. Lett., 2012, 14, 532 CrossRef CAS.
- T. L. D. Tam, T. T. Lin and M. H. Chua, Phys. Chem. Chem. Phys., 2017, 19, 15671 RSC.
- T. L. D. Tam and J. Wu, J. Mol. Eng. Mater., 2015, 3, 1540003 CrossRef CAS.
- K. Ono, S. Tanaka and Y. Yamashita, Angew. Chem., Int. Ed. Engl., 1994, 33, 1977 CrossRef.
- A. Thomas, K. Bhanuprakash and K. K. Prasad, J. Phys. Org. Chem., 2011, 24, 821 CrossRef CAS.
- A. Thomas and K. Bhanuprakash, ChemPhysChem, 2012, 13, 597 CrossRef CAS.
- A. Thomas, G. Krishna Chaitanya, K. Bhanuprakash and K. M. M. Krishna Prasad, ChemPhysChem, 2011, 12, 3458 CrossRef CAS.
- J. D. Yuen, R. Kumar, D. Zakhidov, J. Seifter, B. Lim, A. J. Heeger and F. Wudl, Adv. Mater., 2011, 23, 3780 CAS.
- A. E. London, H. Chen, M. A. Sabuj, J. Tropp, M. Saghayezhian, N. Eedugurala, B. Zhang, Y. Liu, X. Gu, B. Wong, N. Rai, M. K. Bowman and J. D. Azoulay, Sci. Adv., 2019, 5, eaav2336 CrossRef CAS.
- L. Huang, N. Eedugurala, A. Benasco, S. Zhang, K. S. Mayer, D. J. Adams, B. Fowler, M. M. Lockart, M. Saghayezhian, H. Tahir, E. R. King, S. Morgan, M. K. Bowman, X. Gu and J. D. Azoulay, Adv. Funct. Mater., 2020, 30, 1909805 CrossRef CAS.
- I. Osaka, T. Abe, H. Mori, M. Saito, N. Takemura, T. Koganezawa and K. Takimiya, J. Mater. Chem. C, 2014, 2, 2307 RSC.
- K. Kawabata, M. Saito, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2016, 138, 7725 CrossRef CAS.
- W. Hong, C. Guo, Y. Li, Y. Zheng, C. Huang, S. Lu and A. Facchetti, J. Mater. Chem., 2012, 22, 22282 RSC.
- P. Deng, L. Liu, S. Ren, H. Li and Q. Zhang, Chem. Commun., 2012, 48, 6960 RSC.
- W. Yue, X. Huang, J. Yuan, W. Ma, F. C. Krebs and D. Yu, J. Mater. Chem. A, 2013, 1, 10116 RSC.
- G. W. van Pruissen, E. A. Pidko, M. M. Wienk and R. A. Janssen, J. Mater. Chem. C, 2014, 2, 731 RSC.
- X. Liu, B. He, C. L. Anderson, J. Kang, T. Chen, J. Chen, S. Feng, L. Zhang, M. A. Kolaczkowski, S. J. Teat, M. A. Brady, C. Zhu, L.-W. Wang and Y. Liu, J. Am. Chem. Soc., 2017, 139, 8355 CrossRef CAS.
- X. Liu, B. He, A. Garzón–Ruiz, A. Navarro, T. L. Chen, M. A. Kolaczkowski, S. Feng, L. Zhang, C. A. Anderson, J. Chen and Y. Liu, Adv. Funct. Mater., 2018, 28, 1801874 CrossRef.
- C. L. Anderson, N. Dai, S. J. Teat, B. He, S. Wang and Y. Liu, Angew. Chem., 2019, 58, 17978 CrossRef CAS.
- X. Ji, M. Leng, H. Xie, C. Wang, K. R. Dunbar, Y. Zou and L. Fang, Chem. Sci., 2020, 11, 12737 RSC.
- Y. Zou, X. Ji, J. Cai, T. Yuan, D. J. Stanton, Y.-H. Lin, M. Naraghi and L. Fang, Chem, 2017, 2, 139 CAS.
- W.-C. Chen and S. A. Jenekhe, Macromolecules, 1995, 28, 454 CrossRef CAS.
- Q. Zhang, J. Feng, K. Liu, D. Zhu, M. Yang, H. Ye and X. Liu, Synth. Met., 2006, 156, 804 CrossRef CAS.
- J. Li, C. Lai, X. Jia, L. Wang, X. Xiang, C.-L. Ho, H. Li and W.-Y. Wong, Composites, Part B, 2015, 77, 248 CrossRef CAS.
- K. Peng, T. Pei, N. Huang, L. Yuan, X. Liu and J. Xia, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1676 CrossRef CAS.
- T. Umeyama, Y. Watanabe, M. Oodoi, D. Evgenia, T. Shishido and H. Imahori, J. Mater. Chem., 2012, 22, 24394 RSC.
- T. L. Tam, G. Wu, S. W. Chien, S. F. V. Lim, S.-W. Yang and J. Xu, ACS Mater. Lett., 2020, 2, 147 CrossRef CAS.
- L. Wang, X. Liu, X. Shi, C. L. Anderson, L. M. Klivansky, Y. Liu, Y. Wu, J. Chen, J. Yao and H. Fu, J. Am. Chem. Soc., 2020, 142, 17892 CrossRef CAS.
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891 CrossRef CAS.
- Z. Sun, Z. Zeng and J. Wu, Acc. Chem. Res., 2014, 47, 2582 CrossRef CAS.
- A. Patra, Y. H. Wijsboom, G. Leitus and M. Bendikov, Chem. Mater., 2011, 23, 896 CrossRef CAS.
- D. Yuan, L. Liu, X. Jiao, Y. Zou, C. R. McNeill, W. Xu, X. Zhu and D. Zhu, Adv. Sci., 2018, 5, 1800947 CrossRef.
- O. Bubnova, Z. U. Khan, A. Malti, S. Braun, M. Fahlman, M. Berggren and X. Crispin, Nat. Mater., 2011, 10, 429 CrossRef CAS.
- X. Ji, H. Xie, C. Zhu, Y. Zou, A. U. Mu, M. Al-Hashimi, K. R. Dunbar and L. Fang, J. Am. Chem. Soc., 2020, 142, 641 CrossRef CAS.
- V. Krinichnyi, A. Konkin and A. Monkman, Synth. Met., 2012, 162, 1147 CrossRef CAS.
- S. Rasmussen, K. Ogawa and S. Rothstein, in Handbook of Organic Electronics and PhotonicsAmerican Scientific Publishers, ed. H. S. Nalwa, American Scientific Publishers, Stevenson Ranch, CA, 2008, vol. 1, p. 1 Search PubMed.
- J. Roncali, Chem. Rev., 1997, 97, 173 CrossRef CAS.
- H. A. M. v. Mullekom, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Mater. Sci. Eng., R, 2001, 32, 1 CrossRef.
- J. Roncali, Macromol. Rapid Commun., 2007, 28, 1761 CrossRef CAS.
- S. Rasmussen and M. Pomerantz, in Handbook of Conducting Polymers, ed. T. A. Skotheim and J. R. Reynolds, CRC Press, Boca Raton, FL, 3rd edn, 2007, p. 12/1 Search PubMed.
- C. L. Chochos and S. A. Choulis, Prog. Polym. Sci., 2011, 36, 1326 CrossRef CAS.
- E. Bundgaard and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2007, 91, 954 CrossRef CAS.
- H. J. Jhuo, P. N. Yeh, S. H. Liao, Y. L. Li, Y. S. Cheng and S. A. Chen, J. Chin. Chem. Soc., 2014, 61, 115 CrossRef CAS.
- T. L. Tam, C. K. Ng, S. L. Lim, E. Yildirim, J. Ko, W. L. Leong, S.-W. Yang and J. Xu, Chem. Mater., 2019, 31, 8543 CrossRef.
- J. Fan, J. D. Yuen, W. Cui, J. Seifter, A. R. Mohebbi, M. Wang, H. Zhou, A. J. Heeger and F. Wudl, Adv. Mater., 2012, 24, 6164 CrossRef CAS.
- J. Fan, J. D. Yuen, M. Wang, J. Seifter, J. H. Seo, A. R. Mohebbi, D. Zakhidov, A. Heeger and F. Wudl, Adv. Mater., 2012, 24, 2186 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |