
Hierarchical self-assembly of miktoarm star copolymers with pathway complexity†
Jie
Xiao
a,
Qun
He
*a,
Minjun
Yang
a,
Haoquan
Li
a,
Xiandeng
Qiu
a,
Binghua
Wang
b,
Bin
Zhang
 b and
Weifeng
Bu
*ac
b and
Weifeng
Bu
*ac
aKey Laboratory of Nonferrous Metals Chemistry and Resources Utilization of Gansu Province, State Key Laboratory of Applied Organic Chemistry, and College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou, 730000, China. E-mail: hequn@lzu.edu.cn; buwf@lzu.edu.cn
bSchool of Materials Science and Engineering, Zhengzhou University, Zhengzhou, 450002, China
cState Key Laboratory of Solid Lubrication, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, China
First published on 9th December 2020
Abstract
The hierarchical self-assembly of molecular building blocks provides promising opportunities toward the exploration of functional soft materials with structural programmability. Here, we report the hierarchical self-assembly behaviors of amphiphilic miktoarm star copolymers with pathway complexity. In our experiments, supramolecular miktoarm star copolymers with a cluster core of [α-SiW12O40]4− and four polystyrene-block-poly(ethylene glycol) cations (PSn-b+-PEGm, n = 17, 26, 39, 57, 81; m = 45, SEW-1–5) are used as a model of amphiphilic miktoarm stars. When dispersed in the selective THF/methanol and toluene/methanol mixture solvents, the miktoarm stars of SEW-2–5 self-assemble to form bundled fibers, sheet-like assemblies, and hollow spheres. These complex structures are packed by reverse cylindrical or spherical micelles having [α-SiW12O40]4−/PEG45 cores and PSn coronas, wherein the micelle building blocks are originally formed by SEW-2–5 in the nonselective solvents THF and toluene. These hierarchically self-assembled structures do not resemble micelle-like nano-objects formed by amphiphilic copolymers in selective solvents. The mechanism behind such unconventional aggregates is presumably due to intra- and inter-micelle van der Waals attractions occurring under poor solvent conditions for the PSn coronas. The difference is that SEW-3–5 self-assemble into normal vesicles with a PSn core and a [α-SiW12O40]4−/PEG45 corona in the chloroform/methanol mixture solvent. The reverse cylinders, rices, and spheres originally fabricated from SEW-3–5 in chloroform experience molecular reorganization for such normal vesicles in the present selective solvents. Moreover, SEW-1 forms normal lamellae in all the methanol based mixture solvents. The results presented herein not only enable us to reconsider the self-assembly behaviors of amphiphilic miktoarm stars in solution, but also provide opportunities for constructing advanced functional materials with high-level structural hierarchy.
Introduction
The hierarchical self-assembly of molecular building units provides intriguing opportunities to create functional soft materials with increasing levels of structural sophistication.1–5 The design inspiration originates from natural or biological self-assembled systems across multiple length scales with exceptional precision.6–8 Currently, a major challenge in this realm is to develop rational design principles, under which multiple hierarchical levels can be precisely bridged and modulated by noncovalent interaction patterns. According to previous research, block copolymer self-assembly is a well-established supramolecular paradigm to create micellelike aggregates in selective solvents.9–22 Among these aggregates, insoluble blocks gather together to produce a micellar core that is colloidally stabilized by a micellar corona comprising soluble blocks. Increasing the relative volume fractions of insoluble blocks will lead to a sequential morphological variation from spheres to cylinders or rods and then to bilayered vesicles or lamellae.10,12,16–22 The resulting block copolymer micelles can be further used as secondary building blocks for higher-level self-assembly.1,4,5 For example, cross-linked diblock copolymer nanoparticles, including spheres, worms, or vesicles, can form stable Pickering emulsions or colloidosomes with sizes up to hundreds of micrometers.23–26 By manipulating the solvent quality for the blocks, diblock copolymer micelles can be reshaped into anisotropic nanoparticles, leading to the fabrication of nano-sheets,27 supracolloidal chains,28–32 and block supracolloidal chains.28 In the case of ABC triblock copolymers, dynamic patchy micelles can be readily prepared by sequentially controlling the solvent quality for the blocks and the supramolecular cross-linking between the blocks.33–39 The resulting patchy units can further self-assemble or co-assemble into micrometric colloidal polymer chains33,34,36–39 and bilayered38 and network structures.37 It should be noted that all of these complex assemblies above are colloidally stable in solution and thus belong to the conventional classification of micellar structures.Similarly, amphiphilic star copolymers, including block copolymer stars40 and miktoarm stars,41 can also self-assemble in selective solvents to generate micellelike aggregates.42–51 Notably, the resulting micellar structures are much more complicated than those formed by linear block copolymers. For example, star block copolymers can self-assemble into nanoparticle clusters42 and lacunal43 and bicontinuous nanospheres.42 Moreover, stepwise self-assembly of μ-ABC miktoarm star terpolymers results in multiple levels of structural hierarchy. Typical hierarchical morphologies include segmented worm-like micelles,44,45 polygonal bilayered sheets,45 patchy nanofibrils,46 and woodlouse-like particles.47 Such increasing complexities in morphology originate from the topological constraints of the polymer arms converging at a common point by one end in the star copolymers.
Very recently, we reported a series of cluster-based supramolecular star block copolymers52 and miktoarm star copolymers53 which could self-assemble into micelle-like aggregates in nonselective good solvents. These unexpected self-assembly behaviors are attributed to the topological restrictions leading to appropriately increasing incompatibility between different arms.
Herein, we report the hierarchical self-assembly behaviors of miktoarm star copolymers with high pathway complexity. The miktoarm stars are composed of a polyoxometalate core (POM, [α-SiW12O40]4−, d = 1.04 nm) that is encapsulated by four polystyrene-block-poly(ethylene glycol) cations through central 1,2,3-triazolium groups (PSn-b+-PEGm, n = 17, 26, 39, 57, 81; m = 45, SEW-1–5, Fig. 1 and Table 1).53 As previously reported, SEW-1 self-assembles into bilayered nano-sheets with a PS17 core and a [α-SiW12O40]4−/PEG45 corona in the nonselective solvents toluene, tetrahydrofuran (THF), or chloroform.53 However, for SEW-2–5, reverse micellar structures with a [α-SiW12O40]4−/PEG45 core and a PSn (n = 26, 39, 57, 81) corona are observed. With increasing molecular weight of the PSn arm, the micelle morphologies evolve successively from cylinders to spheres.
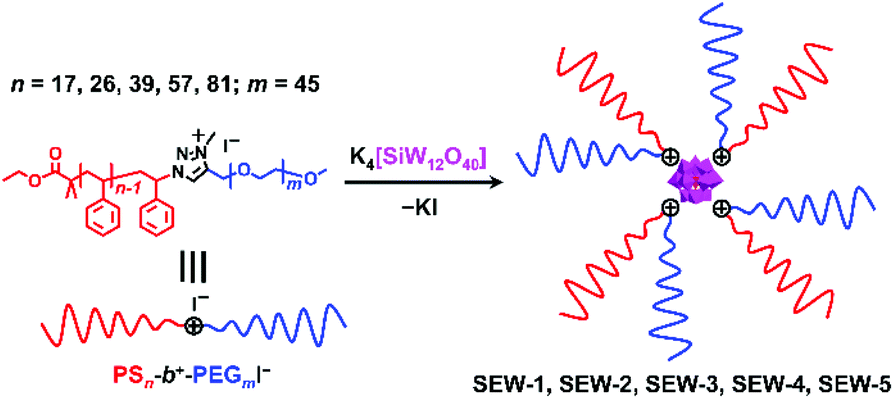 | ||
| Fig. 1 The preparation of POM-based amphiphilic miktoarm star copolymers and their molecular structures. The miktoarm star polymers of SEW-1–5 featured a POM core that was further enwrapped by four PSn-b+-PEGm cations through the central 1,2,3-triazolium groups (n = 17, 26, 39, 57, 81; m = 45). | ||
| Sample code |
M
n,PS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (n)b g mol−1
(n)b g mol−1 |
M n,PEG (m)c g mol−1 | PDId | POM core |
|---|---|---|---|---|
| a The number-average molecular weights of the PS arms (Mn,PS) were obtained by gel permeation chromatography (GPC). b The degrees of polymerization of the PS arms (n) were obtained by 1H NMR. c The PEG arm was commercially available, which was further confirmed by GPC (Mn,PEG) and 1H NMR (m). d The polydispersity indexes (PDI) of PSn-b+-PEGm were obtained by GPC. | ||||
| SEW-1 | 2000 (17) | 2000 (45) | 1.12 | [α-SiW12O40]4− |
| SEW-2 | 2900 (26) | 2000 (45) | 1.12 | [α-SiW12O40]4− |
| SEW-3 | 3900 (39) | 2000 (45) | 1.17 | [α-SiW12O40]4− |
| SEW-4 | 5600 (57) | 2000 (45) | 1.06 | [α-SiW12O40]4− |
| SEW-5 | 8500 (81) | 2000 (45) | 1.10 | [α-SiW12O40]4− |
Upon adding methanol into the micelle solutions above, the solvent quality for PSn and PEGm arms can be weakened and improved, respectively (Tables S1–S3†).54 When dispersed in the selective THF/methanol and toluene/methanol mixture solvents, SEW-2–5 self-assemble to create a series of hierarchical architectures, such as bundled fibers, sheetlike assemblies, and hollow spheres. These complex architectures are packed by the above reverse cylindrical or spherical micelles still retaining [α-SiW12O40]4−/PEG45 cores and PSn coronas. These high-level structural hierarchies do not resemble those conventional micelle-like aggregates fabricated from amphiphilic macromolecules in selective solvents. In contrast, for SEW-3–5, normal vesicles with a PSn core and a [α-SiW12O40]4−/PEG45 corona are obtained via molecular reorganizations in chloroform/methanol mixture solvents. As expected, SEW-1 forms normal bilayered nano-sheets in all methanol based mixture solvents.
Results and discussion
SEW-2 self-assembled in THF/methanol mixture solvents into bundle-like fibers
SEW-2 was first dissolved in a vial with THF, and then methanol was added with vigorous stirring (v/v of THF/methanol = 1:1). The final concentration of SEW-2 was kept at 0.5 mg mL−1. At that moment, the mixture solvent was typically good and poor for the PEGm and PSn arms, respectively (Table S3†).54 The resulting dispersion was turbid at the initial stage. Upon standing quiescently for 2 h, a white precipitate formed at the bottom of the vial (Fig. S1†). This colloidal instability inspired us to further probe the hierarchical morphology of SEW-2via both bright-field transmission electron microscopy (BF-TEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) observations. In the HAADF-STEM images, heavy metal-containing nanodomains appeared much brighter than dark carbon matrix backgrounds. Typical BF-TEM images are shown in Fig. 2a–c, in which SEW-2 formed fiberlike aggregates with a micrometric length. The width ranged from 60 to 120 nm, which was much larger than the total width of the cylindrical micelle formed in THF (15.4 nm).53 In the meantime, the energy dispersive X-ray (EDX) spectrum indicated the existence of tungsten in the nanofibers (Fig. 2d). This suggested that these fibrous nanostructures were bundled aggregates of the cylindrical micelles, which were further confirmed usinghigher-resolution TEM images (Fig. 2b and c). The dark (in the BF-TEM image of Fig. 2b) or white nanostrands (in the HAADF-STEM image of Fig. 2c) were alternately parallel-arranged with the gray domains. The dark or white nanostrands occupied a width of 5.0 ± 0.3 nm and were consequently assigned to the micellar core of the cylinders, while the gray domains were accordingly assigned to the PS26 corona. Moreover, the [α-SiW12O40]4− clusters were restrained at the interface between the PEG45 core and the PS26 corona (Fig. 2). The outer and in-between corona thicknesses were determined to be 6.7 ± 0.4 and 5.6 ± 0.2 nm, respectively (Table 2).
 | ||
| Fig. 2 BF-TEM (a and b) and HAADF-STEM images (c) show that SEW-2 self-assembled to form bundled fibers in the THF/methanol mixture solvent with a methanol percentage of 50 vol%. The inset (a) shows a water contact angle of 125°. The EDX spectrum of the bundle-like fiber was indicative of the presence of tungsten (d). The fibrous aggregates were stable even after annealing the dispersion for 24 h (e and f). | ||
| Volume ratio of methanol |
D
CDa (nm) |
D
CTb (nm) |
σ (chains per nm2) |
D
DMd (nm) |
|---|---|---|---|---|
| a Core diameter of cylindrical micelles. b Corona thickness. c Grafting density of the PS26 chain. d Distance between the micellar cores in the bundles. e Data from the previous work.53 The contour length of the PS26 chain was (0.25 × 26 = 6.5 nm). | ||||
| 0e | 6.3 | 4.6 | 0.51 | — |
| 50% | 5.0 | 6.7 | 0.64 | 5.6 |
| 90% | 4.8 | 6.5 | 0.67 | 5.4 |
It is well known that the surfaces grafted with PS are typically hydrophobic,55 whereas PEG-coated surfaces are relatively hydrophilic.56,57 To further confirm the surface composition of the bundled nanofibers, the casting film of SEW-2 obtained from the THF/methanol mixture was further characterized by water wetting. The resulting static contact angle of water was determined to be 125°, indicative of a highly hydrophobic surface (Fig. 2a). This was consistent with the above assignment of the outer corona as the PS26 chains. To verify this unusual aggregation scenario, the mixture suspension was further annealed at 65 °C for 24 h. The resulting BF-TEM images showed the formation of similar bundlelike nanofibers (Fig. 2e and f). Comparable nanofibers of SEW-2 also formed in the THF/methanol mixture solvent with a methanol composition of 90 vol% (Fig. S2† and Table 2). However, the miktoarm star of SEW-2 did not self-assemble into normal micellar structures with a PS26 core and a [α-SiW12O40]4−/PEG45 corona in the selective solvents of the THF/methanol mixture, but unexpectedly formed bundled nanofibers with a PS26 outer corona. Such aggregation behaviors were compatible with the aforementioned colloidal instability in solution. This was presumably due to the formation of stable cylinderical micelles by SEW-2 in THF.53
Compared with the cylinderical micelles formed in THF,53 the bundles showed three apparent changes under the present solvent conditions. (i) The width of the micellar core was reduced from 6.3 to 5.0 and finally to 4.8 nm (Table 2). Correspondingly, the grafting density of the PS26 chain (σ) increased from 0.51 to 0.65 and finally to 0.67 chains per nm2. (ii) In both BF-TEM and HAADF-STEM images, the PS26 coronas were observed clearly under the THF/methanol solvent conditions, while they were totally invisible in the cylinders obtained from the good solvent THF.53 (iii) The thickness of the exterior corona was consistent with the contour length of the PS26 chain (0.25 × 26 = 6.5 nm),58 while the interior part was only slightly thinner than this contour length. It was, therefore, inferred that the outer PS26 chains were in a fully extended state and the interior PS26 chains were fully interdigitated. Packing parameters such as the thicknesses of the cylindrical cores and coronas and the grafting densities of the PS26 chains are outlined in Table 2.
As already documented by theoretical considerations,59,60 when the solvent quality changes from good to poor, homopolymeric chains grafted on solid surfaces exhibit conformational transitions from stretched coils to collapsed globules. In the former conformation, entropic repulsions between the segments of the chains dominate, while in the latter case, intra-chain van der Waals attractions play a major role. This would lead to considerable shrinkage of the chains in poor solvents as compared to those in good solvents. Such chain constrictions have been experimentally observed by TEM when polymer micelles or vesicles were dispersed in solvents that are relatively poor for PS coronal chains.61–64 The corresponding ionic cores consisting of [α-PW12O40]3−/poly(4-vinylpyridinium methyl)61,62 or [Pt(D2bzimpy)Cl]+/poly(acrylate)63,64 (D2bzimpy = 2,6-bis(N-dodecylbenzimidazol-2′-yl)pyridine) were found to synchronously show serious shrinkage. The PS chains between the ionic cores were fully interpenetrated as a result of inter-micelle van der Waals attractions.61–64 In the present study, the full interdigitation of the PS26 chains and the recognizable compactness of the cylindrical cores were presumably attributed to the inter-cylinder van der Waals attractions between the PS26 chains in the THF/methanol mixture solvents, where the solvent quality was weakened for the PS26 arm (Fig. 2). However, the fully extended PS26 chains were completely opposite to the aforementioned contractive trends of the grafted chains onto solid surfaces in solvents with worsened quality.59–64
Of note is that the cross-sectional areas of the PS26 chains (A = 1/σ = 1.96, 1.54, and 1.49 nm2) were rather comparable to the molecular areas obtained from the surface pressure−area isotherms of PS-based amphiphiles at the air–water interface.65,66 On the other hand, similar stretched PS chains were also observed in the micellar cores of the micellelike aggregates fabricated from PS-(carboxylic acid-functionalized polyhedral oligomeric silsesquioxane, APOSS) amphiphiles.67 This elongated chain behavior was attributed to the rigid APOSS heads and their strong electrostatic repulsions. These features were similar to the highly stretched hydrophobic tails commonly found in the micellelike assemblies of small-molecule surfactants and lipids.68 Actually, these PS tails were comparable to those small amphiphiles in length. Therefore, we tentatively considered the fully stretched configuration as a result of the reduced interfacial area of the PS26 chain under the present solvent conditions (Table 2).
This unusual self-assembly behavior raised an interesting question of whether the present approach was universal to fabricate hierarchically ordered structures of the miktoarm star copolymers. To clarify this question, we further investigated the solution self-assembly processes of the miktoarm stars in other selective solvents such as toluene/methanol and chloroform/methanol.
Self-assembly of SEW-2 in toluene/methanol mixture solvents resulted in the fabrication of sheetlike assemblies
The turbid dispersion of SEW-2 in the toluene/methanol mixture solvent (v/v = 1/1, 0.5 mg mL−1) precipitated over time, analogous to that in the THF/methanol mixture solvent. As mentioned above, this colloidal instability was unusual because of the amphiphilic feature of SEW-2. Typical BF-TEM images showed that SEW-2 formed free-standing sheets with planar sizes of several micrometers (Fig. 3a and b). The water contact angle of the resulting sheet was 102°, indicating that the surface of the sheet was hydrophobic (Fig. 3a). Moreover, the magnified TEM images indicated that the sheetlike assemblies consist of the cylindrical cores of [α-SiW12O40]4−/PEG45 alternately with the PS26 coronas (Fig. 3c). The widths of the core and corona were 6.0 and 8.3 nm, respectively (Table S4†).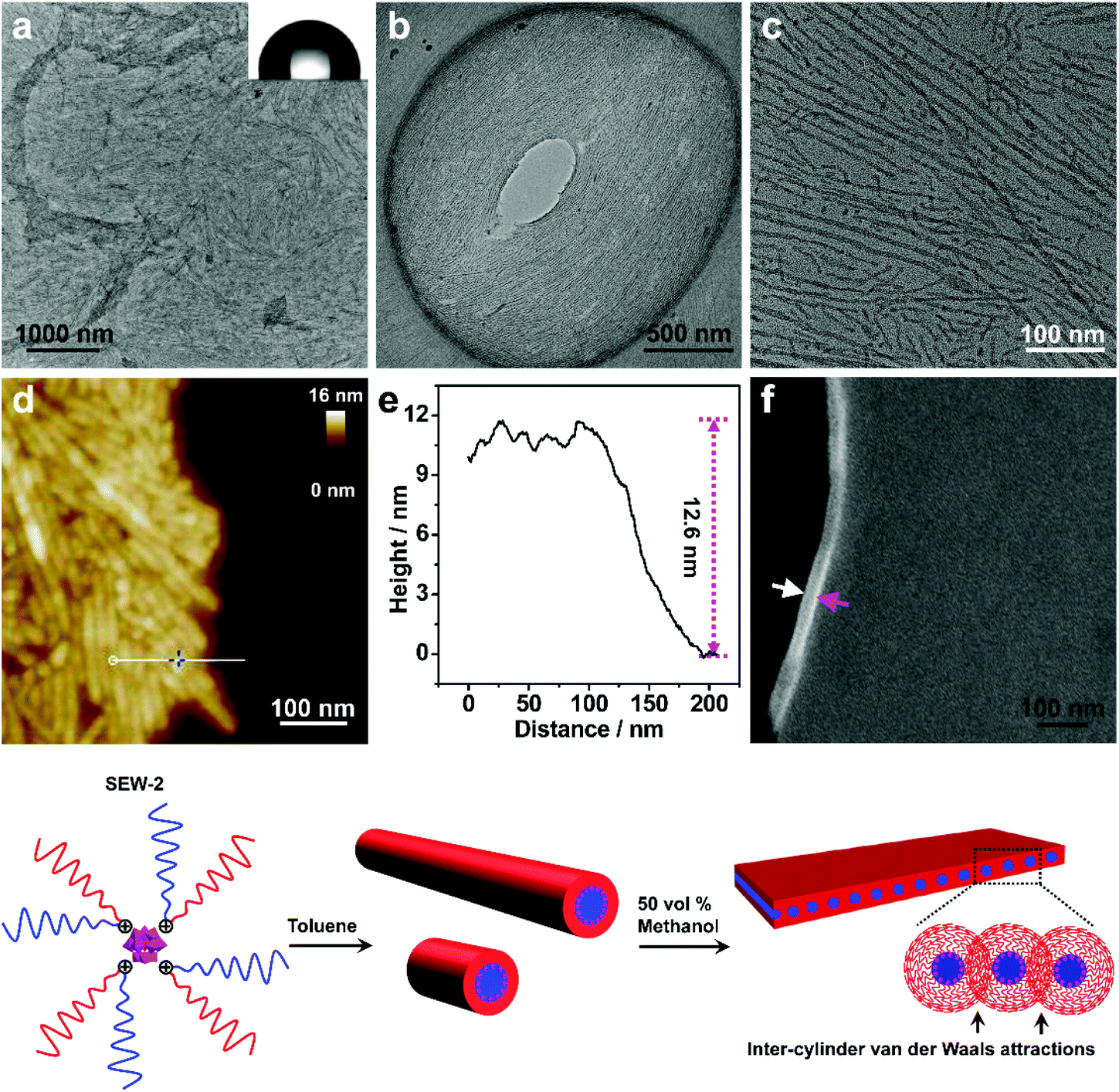 | ||
| Fig. 3 (a, b, and c) BF-TEM images show that SEW-2 self-assembled to generate sheet-like assemblies in the toluene/methanol mixture solvent with a methanol percentage of 50 vol%. The inset (a) shows a water contact angle of 102°. A topographic AFM image (d) and its corresponding height profile (e) are shown for the sheet-like assemblies of SEW-2. (f) As indicated by the arrows in the SEM image, the cross-sectional area of the sheet was clearly captured. | ||
The sheetlike assemblies and cylindrical micelles were further examined by atomic force microscopy (AFM) imaging (Fig. 3d). The thickness of the sheet and the total width of the cylinder were estimated to be 12.6 and 15.7 nm, respectively (Fig. 3e). These two values were similar to the total width of the cylindrical micelle obtained from the TEM images (6.0 + 8.3 = 14.3 nm). Moreover, the cross-sectional area of the sheet was captured by scanning electron microscopy (SEM), where the observed thickness was 26 nm (Fig. 3f). To avoid electric charging during the SEM imaging measurement, both the sides of the sheetlike assemblies were coated with 5 nm thick gold films using a mild ion-sputter. The subtraction of the gold layers (5 × 2 = 10 nm) yielded a 16 nm thickness for the naked sheet, which was consistent with the aforementioned TEM and AFM imaging results. These collective data showed that the sheetlike assemblies were unilamellar in nature (Fig. 3). Meanwhile, the surface of the sheet was uniform (Fig. 3d). Of note is that the thickness of the PS26 stripe (8.3 nm) was smaller than the two-fold contour length of the PS26 chain (6.5 × 2 = 13 nm). This suggested that the PS26 chains were almost fully interpenetrated as a result of the characteristic inter-cylinder van der Waals attractions, leading to the formation of a free-standing sheet in the toluene/methanol mixture solvent (v/v = 1/1). Accordingly, this class of sheetlike assemblies was typically aligned through side-to-side stacking of the cylindrical micelles in a two dimensional form.
This free-standing sheet was distinctly different from the aforementioned bundle-like nanofiber formed in the THF/methanol mixture solvents. This means that the solvent molecules (toluene versus THF) showed a significant impact on these two hierarchically self-assembled processes. Presumably, the toluene molecules could lead to the generation of more anisotropic van der Waals attractions than the THF molecules in solution. As previously reported,53 cylindrical micelles coexisted with rice-like micelles in the toluene solution of SEW-2, and the micelle number ratio was ca. 1:1. However, the rice-like micelles almost disappeared in the present sheetlike assemblies (Fig. 3a–c). This was attributed to better solvation of the PEG45 arm in the toluene/methanol mixture solvent than just in toluene. Therefore, the relative volume of the inner PEG45 block presumably showed an appreciable increase, leading to a morphological change from rice-like to cylindrical micelles. In contrast, upon increasing the methanol content to 90 vol%, SEW-2 self-assembled into bundled nanofibers (Fig. S3 and Table S5†), similar to those found in the THF/methanol mixture solvent (Fig. 2, S2,† and Table 2). Therefore, it was reasonable to find a coexistence of free-standing sheets and bundled nanofibers when SEW-2 was dispersed in the mixture solvent with an intermediate methanol content of 75 vol% (Fig. S4†). This sheet-to-bundle transition suggested that the anisotropic degree of the inter-cylinder van der Waals attractions decreased with the increasing methanol content in the toluene/methanol mixture solvent.
Analogous to SEW-2, the miktoarm stars of SEW-3–5 also self-assembled to generate sheetlike assemblies in the toluene/methanol mixture solvent (v/v = 1/1, Fig. S5–S7, and Table S4†), wherein the cylindrical micelles were aligned via both side-to-side and end-to-end stacking forms through inter-cylinder van der Waals attractions. The rice-like or spherical micelles were no longer visible in the sheetlike assemblies under the present solvent conditions. These scenarios were consistent with the morphological change from rice-like to cylindrical micelles occurring in the case of SEW-2 (Fig. 3). These solvent-tunable morphological transitions encouraged us to further investigate the self-assembly behaviors of SEW-3–5 in toluene/methanol mixture solvents with worse solvent quality.
SEW-5 self-assembled into unilamellar hollow nanostructures in the toluene/methanol mixture solvent
When the methanol content was increased to 75 vol%, SEW-5 self-assembled to produce spherical nanostructures with an average diameter of 160 nm. The outside gray coronas were accordingly assigned to the PS81 arms with a thickness of 9.6 ± 2.4 nm (Fig. 4a and b, and Table S6†). The inset of Fig. 4a shows a water contact angle of 89°, consistent with the above assignment. Moreover, several broken hollow aggregates were captured evidently. These situations were further confirmed by HAADF-STEM imaging (Fig. 4c and d). White nanospheres with a diameter of 6.2 ± 1.8 nm were identified and isolated clearly in the central parts of the spherical aggregates. Consequently, the resulting nanospheres and their intervals (9.0 ± 3.2 nm) were attributed to the micellar cores of [α-SiW12O40]4−/PEG45 and the micellar coronas of PS81, respectively. The diameter of the white core was smaller than that of the micellar core (8.8 nm) of SEW-5 in toluene.53 The PS81 corona thickness at the intervals was consistent with that of the outside PS81 corona. This was due to the full interpenetration of the PS81 chains as a result of the inter-micelle van der Waals attractions. It should be noted that the corona thickness was much smaller than the fully extended length of the PS81 chain (20.25 nm). This situation was in contrast to the highly stretched PS26 chains found in the bundle-like fibers formed by SEW-2 (Fig. 2, S2–S4, S8,†Tables 2 and S5†). Therefore, the shrinkage of the PS81 chain was likely due to its polymeric character and consistent with intra-micelle van der Waals attractions occurring among the segments of the PS81 chains under this worsened solvent quality condition.59–64 | ||
| Fig. 4 BF-TEM (a and b) and HAADF-STEM images (c and d) show that SEW-5 self-assembled to yield unilamellar hollow aggregates in the toluene/methanol mixture solvent with a methanol content of 75 vol%. The inset (a) shows a water contact angle of 89°. The original spherical micelles were still retained onto the hollow nanostructures. The lumen architectures were further confirmed by SEM imaging (e and f). Similar hollow nanostructures were also captured in the case of SEW-3, as revealed by both BF-TEM (g and h) and SEM images (i). | ||
Altogether, the spherical micelles of SEW-5 formed in toluene were further used as second building blocks to further construct hollow nanostructures, in which the spherical micelles retained their core–corona structures and were distributed uniformly (Fig. 4). The total wall thickness of the unilamellar hollow aggregates was calculated to be (9.6 × 2 + 6.2 = 25.4 nm, Table S6†). Such hollow nanostructures were further confirmed by SEM imaging, where holes were clearly observed (Fig. 4e and f). Moreover, in a tilted hole, the wall of the hollow nanostructure was observed, and the total wall thickness was 34.3 nm (Fig. 4f). After subtracting the gold layers on both the sides of the wall (10 nm), the net wall thickness was calculated to be 24.3 nm. This value is in good agreement with the wall thickness of the hollow nanostructures obtained from the aforementioned TEM observation (25.4 nm). Comparable hollow aggregates were also observed when SEW-5 was dispersed in the toluene/methanol mixture solvent with a methanol percentage of 90 vol% (Fig. S9†). Analogous to SEW-5, SEW-3 can also form unilamellar hollow nanostructures in the toluene/methanol mixture solvent (v/v = 1/3), as indicated in both the BF-TEM (Fig. 4g and h) and SEM images (Fig. 4i).
These hollow nanostructures looked like bilayered vesicles in appearance. But actually, the hollow nanostructures were constructed by packing a monolayer of the spherical micelles onto the surface of a hollow sphere under the weakened solvent quality conditions for the PSn arm (n = 39, 81). Conventionally, when spherical micelles were transformed to vesicular structures, amphiphilic macromolecules must be realigned into bilayered forms.10,12,16–22 However, in the present hollow aggregates, the spherical micelles still maintained their core–corona structures. The formation of such an unusual pattern was presumably due to the intra- and inter-micelle van der Waals attractions in the toluene/methanol mixture solvents with high methanol concentrations (75 vol% and 90 vol%). Under these worse solvent conditions; however, sheetlike assemblies obtained in the toluene/methanol mixture solvent (v/v = 1/1) were not observed. This was presumably due to the more unfavorable contact between the micellar precursors and the solvent mixtures, leading to the decreasing anisotropic degree of the intra- and inter-micelle van der Waals attractions.
Self-assembly of SEW-3–5 yielded normal vesicles in chloroform/methanol mixture solvents
The dispersion of SEW-4 (chloroform/methanol = 1/1, 0.5 mg mL−1) was stable at least for 15 days, which was different from those colloidally unstable dispersions obtained from both the THF/methanol and toluene/methanol mixture solvents. Typical BF-TEM images showed that SEW-4 self-assembled into regular vesicular aggregates (Fig. 5a and b). The wall thickness was 14.8 ± 3.6 nm, while the total diameter of the vesicle ranged broadly from 60 to 280 nm. Moreover, dark fringes (ca. 1 nm) were observed on both the outside and inside edges of the vesicle walls (Fig. 5c). The vesicular feature was further supported by the corresponding HAADF-STEM images, where the vesicular walls were covered by white fringes (ca. 1 nm, Fig. 5d). Therefore, the dark or white fringes were assigned to [α-SiW12O40]4− clusters. The contact angle of water for the casting film was 70° (Fig. 5a), which was much smaller than the water contact angles obtained from the surfaces of the nonconventional bundle- and sheetlike assemblies and hollow spheres (Fig. 2–4 and S6†). By contrast, this smaller value (70°) was in the range of water contact angles on the thin films that were grafted with PEG chains (35–75°).56,57 These collective data indicated that the regular vesicles were comprised of PS57 cores and [α-SiW12O40]4−/PEG45 coronas (Fig. 5). The vesicular nature was further proved by SEM imaging (Fig. 5e and f). The observed wall thickness was 40.4 ± 5.6 nm. After removing the thickness of the [α-SiW12O40]4−/PS57 wall (14.8 nm, Table S7†) and gold layers on both the sides of the wall (10 nm), the thickness of the PEG45 corona was calculated to be 7.8 nm. Similar regular vesicles of SEW-4 were also observed in the chloroform/methanol mixture solvent with methanol contents of 75 vol% (Fig. S10a†) and 90 vol% (Fig. S10b and c†).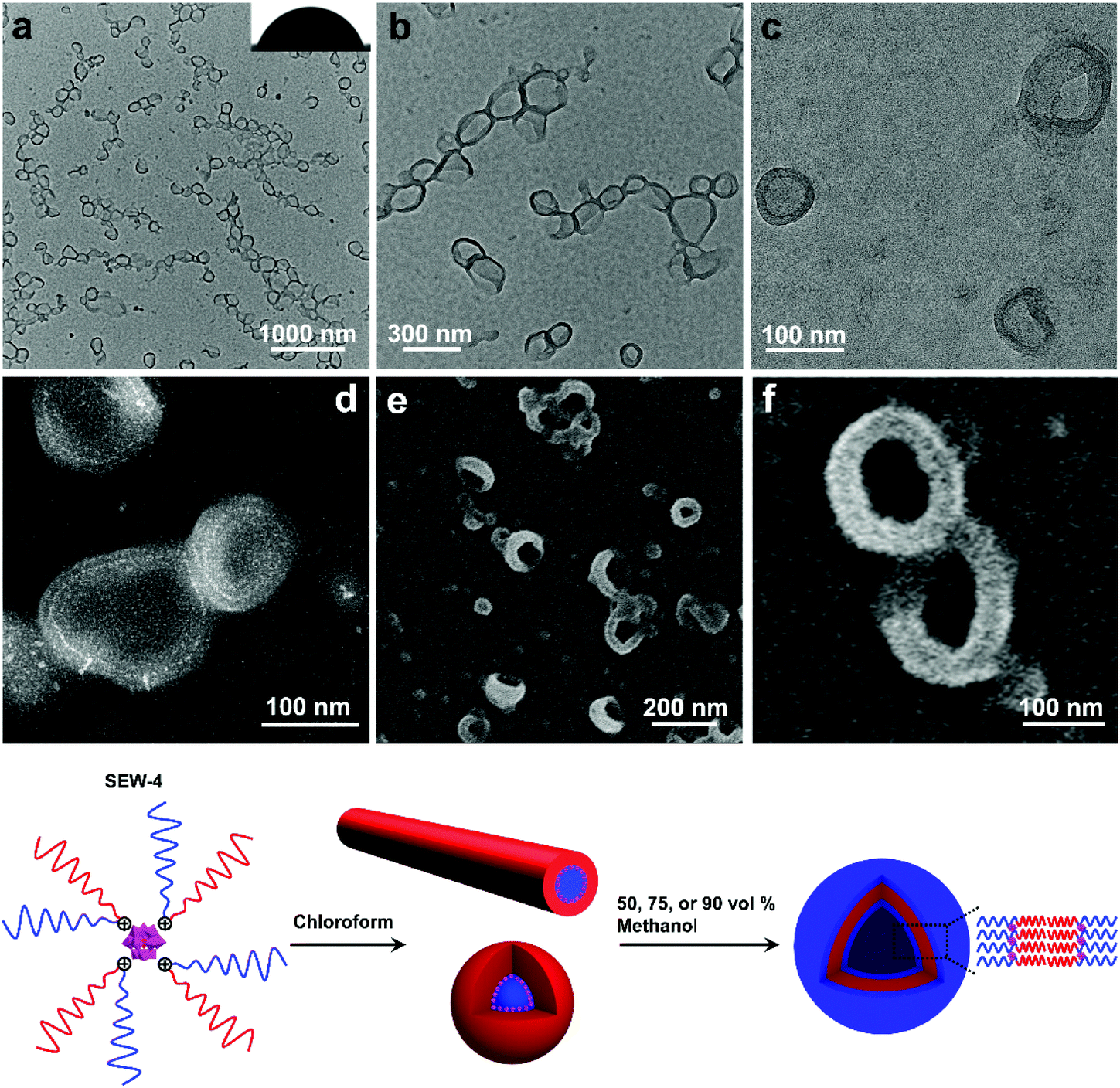 | ||
| Fig. 5 BF-TEM (a–c), HAADF-STEM (d), and SEM images (e and f) show that SEW-4 self-assembled into regular bilayered vesicles with a PS57 core and a [α-SiW12O40]4−/PEG45 corona in the chloroform/methanol mixture solvent with a methanol content of 50 vol%. The inset (a) shows a water contact angle of 70°. | ||
According to the previous data, the dispersion of SEW-4 in the nonselective solvent chloroform showed a coexistence of cylindrical and spherical micelles with a [α-SiW12O40]4−/PEG45 core and a PS57 corona.53 However, in the present selective solvents, SEW-4 self-assembled to yield regular vesicles with a PS57 core and a [α-SiW12O40]4−/PEG45 corona. The core–corona structure was completely reversed. To clarify this self-assembly evolution and mechanism, SEW-4 was further dispersed in the chloroform/methanol mixture solvents with methanol compositions of 10 vol% and 25 vol%. In the former BF-TEM images, vesicular aggregates coexisted with multi-micelle aggregates (Fig. 6a–d). Moreover, several vesicles were substantially attached with solid spheres. And also, isolated vesicles were observed clearly. Therefore, the miktoarm star of SEW-4 was in the process of transforming the original reverse micelles53 to the present regular vesicles. Further increasing the methanol percentage to 25 vol% caused a complete transformation of reverse micelles into regular vesicles with diameters up to ca. 500 nm (Fig. 6e and f).
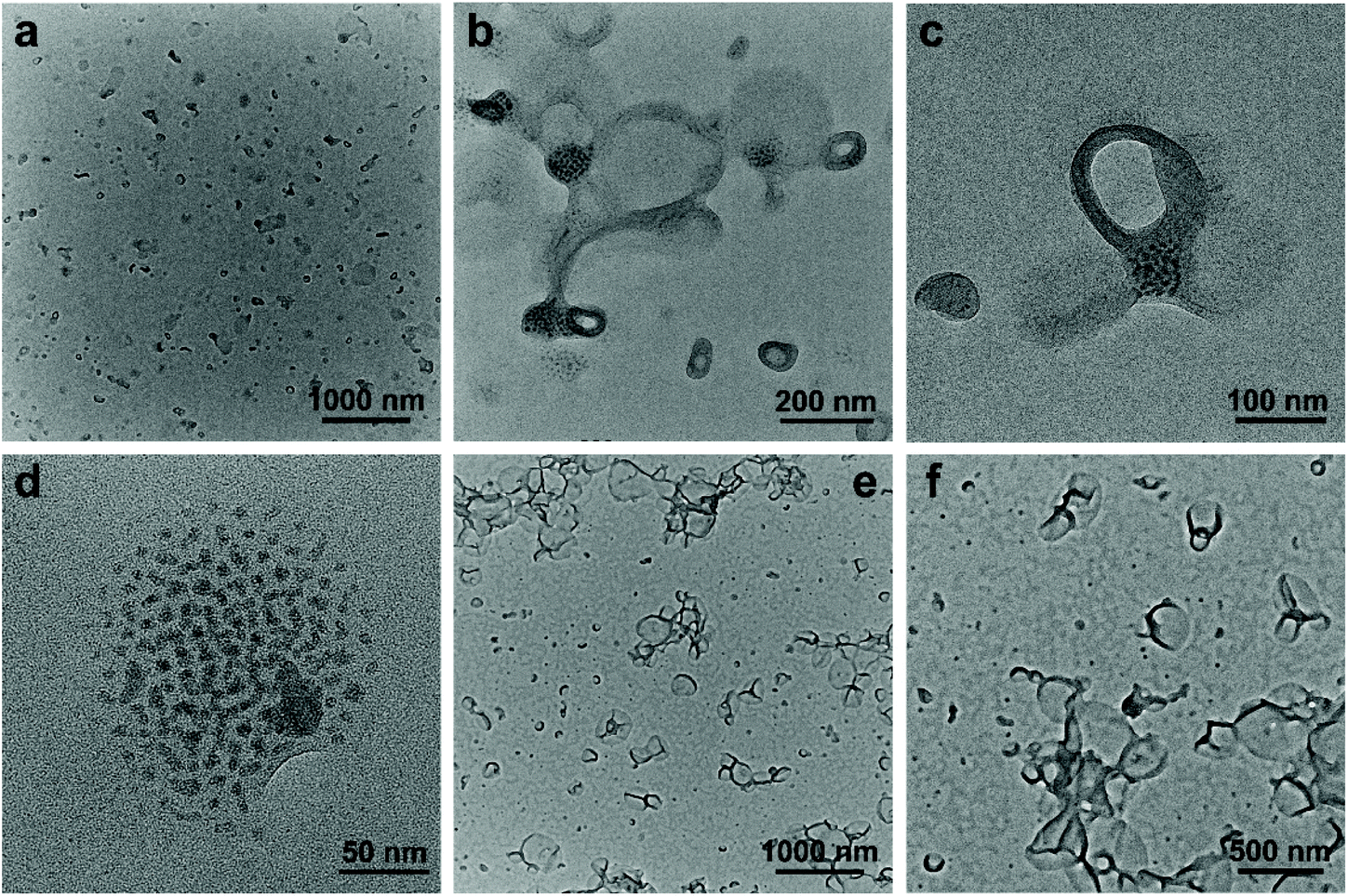 | ||
| Fig. 6 Upon dispersing SEW-4 in the chloroform/methanol mixture solvent with a methanol volume of 10%, typical BF-TEM images show that regular bilayered vesicles coexisted with reverse solid spheres (a–d). When the methanol content was increased to 25 vol%, a pure phase of bilayered vesicles formed (e and f). | ||
Similarly, SEW-5 also formed regular vesicles in the chloroform/methanol mixture solvent with methanol contents of 50 vol% (Fig. S11a and b†) and 90 vol% (Fig. S11c†). However, in the case of SEW-3, the pure phase of vesicular aggregates was formed only at a methanol content of 90 vol% (Fig. S12 and Table S7†). It should be highlighted that all of these regular vesicular aggregates differed completely from the hollow structures formed in the toluene/methanol mixture solvent (Fig. 4 and S9†), but actually belonged to the self-assembly classification of amphiphilic macromolecules in selective solvents.10,12,16–22 Note that the reverse cylindrical, rice-like, or spherical micelles formed by SEW-2–5 in chloroform53 showed an increasing trend of molecular reorganization to create regular vesicles in the chloroform/methanol mixture solvent. This was due to the increasingly loose core–corona interface of the micelle with increasing molecular weight of the PSn arms (n = 26, 39, 57, 81) in chloroform. Meanwhile, the original micellar aggregates coexisted with the unimers of SEW-5, as previously discussed.53
As previously reported,53 the miktoarm star copolymers of SEW-2–5 self-assembled in nonselective solvents such as chloroform, THF, and toluene to create micellelike aggregates with a PSn corona and a [α-SiW12O40]4−/PEG45 core. These micellelike aggregates stemmed from the topological constraints of the chemically different arms of PSn and PEGm in the miktoarm stars, wherein the weak incompatibility was appropriately magnified between the arms. Furthermore, in terms of the solvent quality for both the PS and PEG arms, the difference of the solubility parameters was in the order of chloroform < THF < toluene (Tables S1–S3†). Therefore, the primary micelles were more stable in THF or toluene than in chloroform. This would make it harder for the primary micelles to generate normal micelles in THF/methanol or toluene/methanol mixture solvents than in chloroform/methanol solvents (Fig. 2–7 and S2–S19†).
 | ||
| Fig. 7 Phase diagram of SEW-1–5 shows pathway complexity in the methanol based mixture solvents. | ||
SEW-1 self-assembled to generate nano-sized lamellae in methanol based mixture solvents
As discussed, the self-assembly of SEW-1 in toluene, THF, or chloroform brought about the formation of nano-sized bilayered structures with a PS17 core and [α-SiW12O40]4−/PEG45 corona.53 Upon adding methanol (90 vol%) to toluene, THF, or chloroform solutions, SEW-1 formed nano-sized lamellae with a PS17 core and [α-SiW12O40]4−/PEG45 corona (Fig. S19 and S20†). This was due to the similar bilayered precursors formed by SEW-1 in the nonselective solvents.53Conclusions
In summary, we have demonstrated the hierarchical self-assembly behaviors of POM based miktoarm star copolymers (SEW-1–5, Fig. 7) with pathway complexity in methanol based mixture solvents. The solvent quality for the hierarchical self-assembly is highly dependent on the used solvent combinations and their volume ratios, further leading to tunable intra- and intermolecular interactions between the star copolymers or micelles. Accordingly, a series of nonconventional aggregates are fabricated in a controllable and modular fashion in the cases of SEW-2–5, including bundled fibers, sheetlike assemblies, and hollow spheres. Both the bundles and sheets have micrometric sizes. These self-assembled nanostructures are comparable in appearance to wormlike micelles, bilayered lamellae, and vesicles, respectively, fabricated from amphiphilic block copolymers or miktoarm stars in selective solvents. However, among these complex aggregates, both the cylindrical and spherical micelles originally formed by the miktoarm stars in nonselective solvents (THF, toluene, and chloroform)53 still maintain their core–corona micelle structures in the present methanol based selective solvents. This class of higher-level nanostructures does not resemble the commonly accepted micellelike nano-objects formed from amphiphilic (macro)molecules in selective solvents.1–51,61–64,69,70 The driving forces for such unconventional aggregates are attributed to the intra- and inter-micelle van der Waals attractions occurring under the poor solvent conditions for the PSn arms.The difference is that the miktoarm star of SEW-1 self-assembles to produce nano-sized lamellae with a PS17 core and a [α-SiW12O40]4−/PEG45 corona in the methanol based mixture solvents. This is predictable because of the similar bilayered precursors formed by SEW-1 in the nonselective solvents,53 whereas SEW-3–5 are found to form normal vesicles with a PSn core (n = 39, 57, 81) and a [α-SiW12O40]4−/PEG45 corona in the chloroform/methanol mixture solvent. The reverse cylinders, rices, and spheres of the miktoarm stars originally obtained in chloroform53 undergo molecular reorganization to create normal vesicles in the present selective solvents. These results show that the self-assembly of the miktoarm stars generates micelle-structured intermediates for the final formation of micelle-like aggregates in selective solvents. This mechanism insight shows a sharp contrast to the commonly accepted consideration of single star-shaped copolymers in nonselective solvents.
However, as far as we are concerned, these hierarchical nanostructures and their formation mechanisms cannot be accessed for the regular star copolymers, in which no heavy metal-containing nanoclusters or nanocomposites are used as the cores of the stars, and thus the difference between the core and arms cannot be recognized clearly. In the present study, the POM cluster can be regarded as a TEM-visible probe which allows us to directly access the hierarchical structures of the star copolymers in more detail. Also, a series of intermediate aggregates are clearly observed by using the TEM-visible probe method, providing detailed mechanistic insights into the hierarchical self-assembly processes of the miktoarm star copolymers in solution. The results presented herein not only demonstrate different hierarchical self-assembly behaviors of amphiphilic macromolecules in solution, but also enable the construction of advanced functional materials with unprecedented structural complexity.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We dedicate this paper to Professor Lixin Wu on the occasion of his 60th birthday. This work is supported by the NSFC (21674044 and 21474044), the Fundamental Research Funds for the Central Universities (lzujbky-2020-43), and the Open Project of State Key Laboratory of Supramolecular Structure and Materials of Jilin University (sklssm202003). The project was supported by the Open Research Fund of State Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences (2018–25).Notes and references
- A. H. Gröschel and A. H. E. Müller, Nanoscale, 2015, 7, 11841 RSC.
- W. M. Jacobs and D. Frenkel, J. Am. Chem. Soc., 2016, 138, 2457 CrossRef PubMed.
- B. Li, W. Li, H. Li and L. Wu, Acc. Chem. Res., 2017, 50, 1391 CrossRef PubMed.
- Y. Lu, J. Lin, L. Wang, L. Zhang and C. Cai, Chem. Rev., 2020, 120, 4111 CrossRef PubMed.
- C. Yi, Y. Yang, B. Liu, J. He and Z. Nie, Chem. Soc. Rev., 2020, 49, 465 RSC.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418 CrossRef PubMed.
- Q. Luo, C. Hou, Y. Bai, R. Wang and J. Liu, Chem. Rev., 2016, 116, 13571 CrossRef PubMed.
- C. Gao and G. Chen, Acc. Chem. Res., 2020, 53, 740 CrossRef PubMed.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967 CrossRef PubMed.
- S. Förster and M. Antonietti, Adv. Mater., 1998, 10, 195 CrossRef.
- R. C. Hayward and D. J. Pochan, Macromolecules, 2010, 43, 3577 CrossRef.
- Y. Chen, Macromolecules, 2012, 45, 2619 CrossRef.
- J. Zhang, X.-F. Chen, H.-B. Wei and X.-H. Wan, Chem. Soc. Rev., 2013, 42, 9127 RSC.
- Z. Ge and S. Liu, Chem. Soc. Rev., 2013, 42, 7289 RSC.
- Y. Zhu, B. Yang, S. Chen and J. Du, Prog. Polym. Sci., 2017, 64, 1 CrossRef.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174 CrossRef PubMed.
- L. Zhang and A. Eisenberg, J. Am. Chem. Soc., 1996, 118, 3168 CrossRef.
- Y.-Y. Won, A. K. Brannan, H. T. Davis and F. S. Bates, J. Phys. Chem. B, 2002, 106, 3354 CrossRef.
- P. Bhargava, Y. Tu, J. X. Zheng, H. Xiong, R. P. Quirk and S. Z. D. Cheng, J. Am. Chem. Soc., 2007, 129, 1113 CrossRef PubMed.
- S. J. Byard, C. T. O'Brien, M. J. Derry, M. Williams, O. O. Mykhaylyk, A. Blanazs and S. P. Armes, Chem. Sci., 2020, 11, 396 RSC.
- Y. Han, H. Yu, H. Du and W. Jiang, J. Am. Chem. Soc., 2010, 132, 1144 CrossRef PubMed.
- J. Xiao and J. Du, J. Am. Chem. Soc., 2020, 142, 6569 CrossRef PubMed.
- K. L. Thompson, P. Chambon, R. Verber and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 12450 CrossRef PubMed.
- K. L. Thompson, C. J. Mable, A. Cockram, N. J. Warren, V. J. Cunningham, E. R. Jones, R. Verber and S. P. Armes, Soft Matter, 2014, 10, 8615 RSC.
- Z. Wang, M. C. M. van Oers, F. P. J. T. Rutjes and J. C. M. van Hest, Angew. Chem., Int. Ed., 2012, 51, 10746 CrossRef PubMed.
- Z. Wang, F. P. J. T. Rutjes and J. C. M. van Hest, Chem. Commun., 2014, 50, 14550 RSC.
- L. Cheng, G. Zhang, L. Zhu, D. Chen and M. Jiang, Angew. Chem., Int. Ed., 2008, 47, 10171 CrossRef PubMed.
- J.-H. Kim, W. J. Kwon and B.-H. Sohn, Chem. Commun., 2015, 51, 3324 RSC.
- S. Chae, S. Lee, K. Kim, S. W. Jang and B.-H. Sohn, Chem. Commun., 2016, 52, 6475 RSC.
- C. Yi, Y. Yang and Z. Nie, J. Am. Chem. Soc., 2019, 141, 7917 CrossRef PubMed.
- H. Gao, X. Ma, J. Lin, L. Wang, C. Cai, L. Zhang and X. Tian, Macromolecules, 2019, 52, 7731 CrossRef.
- H. Gao, L. Gao, J. Lin, Y. Lu, L. Wang, C. Cai and X. Tian, Macromolecules, 2020, 53, 3571 CrossRef.
- H. Cui, Z. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647 CrossRef PubMed.
- A. O. Moughton, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2012, 45, 2 CrossRef.
- Z. Zhang, C. Zhou, H. Dong and D. Chen, Angew. Chem., Int. Ed., 2016, 55, 6182 CrossRef PubMed.
- A. H. Gröschel, F. H. Schacher, H. Schmalz, O. V. Borisov, E. B. Zhulina, A. Walther and A. H. E. Müller, Nat. Commun., 2012, 3, 710 CrossRef PubMed.
- A. H. Gröschel, A. Walther, T. I. Löbling, F. H. Schacher, H. Schmalz and A. H. E. Müller, Nature, 2013, 503, 247 CrossRef PubMed.
- T. I. Löbling, O. Borisov, J. S. Haataja, O. Ikkala, A. H. Gröschel and A. H. E. Müller, Nat. Commun., 2016, 7, 12097 CrossRef PubMed.
- T. I. Löbling, O. Ikkala, A. H. Gröschel and A. H. E. Müller, ACS Macro Lett., 2016, 5, 1044 CrossRef.
- J. M. Ren, T. G. McKenzie, Q. Fu, E. H. H. Wong, J. Xu, Z. An, S. Shanmugam, T. P. Davis, C. Boyer and G. G. Qiao, Chem. Rev., 2016, 116, 6743 CrossRef PubMed.
- Y. Zhao, Macromol. Rapid Commun., 2019, 40, 1800571 CrossRef PubMed.
- Y. Zhang, T. Guan, G. Han, T. Guo and W. Zhang, Macromolecules, 2019, 52, 718 CrossRef.
- Y. Zhang, M. Cao, G. Han, T. Guo, T. Ying and W. Zhang, Macromolecules, 2018, 51, 5440 CrossRef.
- Z. Li, E. Kesselman, Y. Talmon, M. A. Hillmyer and T. P. Lodge, Science, 2004, 306, 98 CrossRef PubMed.
- Z. Li, M. A. Hillmyer and T. P. Lodge, Nano Lett., 2006, 6, 1245 CrossRef PubMed.
- F. Xu, D. Wu, Y. Huang, H. Wei, Y. Gao, X. Feng, D. Yan and Y. Mai, ACS Macro Lett., 2017, 6, 426 CrossRef.
- A. Hanisch, A. H. Gröschel, M. Fötsch, M. Drechsler, H. Jinnai, T. M. Ruhland, F. H. Schacher and A. H. E. Müller, ACS Nano, 2013, 7, 4030 CrossRef PubMed.
- D. Voulgaris, C. Tsitsilianist, F. J. Esselink and G. Hadziioannou, Polymer, 1998, 39, 6429 CrossRef.
- D. Voulgaris and C. Tsitsilianist, Macromol. Chem. Phys., 2001, 202, 3284 CrossRef.
- E. R. Zubarev and J. Teng, J. Am. Chem. Soc., 2003, 125, 11840 CrossRef PubMed.
- E. R. Zubarev, J. Xu, A. Sayyad and J. D. Gibson, J. Am. Chem. Soc., 2006, 128, 15098 CrossRef PubMed.
- Q. He, H. Huang, X.-Y. Zheng, J. Xiao, B. Yu, X.-J. Kong and W. Bu, ACS Appl. Mater. Interfaces, 2018, 10, 16947 CrossRef PubMed.
- J. Xiao, Q. He, S. Qiu, H. Li, B. Wang, B. Zhang and W. Bu, Sci. China: Chem., 2020, 63, 792 CrossRef.
- C. M. Hansen, Hansen Solubility Parameters, A Users Handbook, CRC Press, Boca Raton, FL, 2000 Search PubMed.
- Y. Li, J. Q. Pham, K. P. Johnston and P. F. Green, Langmuir, 2007, 23, 9785 CrossRef PubMed.
- S. Okubayashi, Y. Itoh and H. Shosenji, J. Appl. Polym. Sci., 2005, 97, 545 CrossRef.
- J. S. Son and D. S. Ji, Fibers Polym., 2008, 9, 380 CrossRef.
- M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, Oxford, New York, 2003 Search PubMed.
- D. I. Dimitrov, A. Milchev and K. Binder, J. Chem. Phys., 2007, 127, 084905 CrossRef PubMed.
- A. Striolo and S. A. Egorov, J. Chem. Phys., 2007, 126, 014902 CrossRef PubMed.
- Q. Zhang, Y. Liao, L. He and W. Bu, Langmuir, 2013, 29, 4181 CrossRef PubMed.
- Q. Zhang, Y. Liao and W. Bu, Langmuir, 2013, 29, 10630 CrossRef PubMed.
- N. Liu, Q. He and W. Bu, Langmuir, 2015, 31, 2262 CrossRef PubMed.
- N. Liu, Q. He, Y. Wang and W. Bu, Soft Matter, 2017, 13, 4791 RSC.
- M. Niwa, T. Hayashi and N. Higashi, Langmuir, 1990, 6, 263 CrossRef.
- P. M. Saville, I. R. Gentle, J. W. White, J. Penfold and J. R. P. Webster, J. Phys. Chem., 1994, 98, 5935 CrossRef.
- X. Yu, S. Zhong, X. Li, Y. Tu, S. Yang, R. M. Van Horn, C. Ni, D. J. Pochan, R. P. Quirk, C. Wesdemiotis, W.-B. Zhang and S. Z. D. Cheng, J. Am. Chem. Soc., 2010, 132, 16741 CrossRef PubMed.
- K. Holmberg, B. Jönsson, B. Kronberg and B. Lindman, Surfactants and Polymers in Aqueous Solution, 2nd edn, Wiley, Chichester, U.K., 2003 Search PubMed.
- Q. Zhang, L. He, H. Wang, C. Zhang, W. Liu and W. Bu, Chem. Commun., 2012, 48, 7067 RSC.
- Y. Liao, N. Liu, Q. Zhang and W. Bu, Macromolecules, 2014, 47, 7158 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and instruments, packing parameter calculation, dynamic light scattering plots, TEM and SEM images, and additional results and discussion. See DOI: 10.1039/d0py01170c |
| This journal is © The Royal Society of Chemistry 2021 |