
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functional nanostructures by NiCCo-PISA of helical poly(aryl isocyanide) copolymers†
Sètuhn
Jimaja
 abc,
Yujie
Xie
ab,
Jeffrey C.
Foster
b,
Daniel
Taton
c,
Andrew P.
Dove
*b and
Rachel K.
O'Reilly
*b
abc,
Yujie
Xie
ab,
Jeffrey C.
Foster
b,
Daniel
Taton
c,
Andrew P.
Dove
*b and
Rachel K.
O'Reilly
*b
aDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK
bSchool of Chemistry, University of Birmingham, Edgbaston B15 2TT, UK. E-mail: r.oreilly@bham.ac.uk; a.dove@bham.ac.uk
cLaboratoire de Chimie des Polymères Organiques, Université de Bordeaux/CNRS École Nationale Supérieure de Chimie, de Biologie & de Physique, 33607 Cedex Pessac, France
First published on 23rd July 2020
Abstract
Herein, we present a straightforward and versatile methodology to achieve functional polymeric nano-objects that contain helical cores. Nickel-catalysed coordination polymerisation-induced self-assembly (NiCCo-PISA) of helical poly(aryl isocyanide) amphiphilic diblock copolymers was conducted, affording micelles containing controllable quantities of activated ester groups (i.e. pentafluorophenyl esters) in the core that were subsequently modified using post-polymerisation modification (PPM) with amine nucleophiles. Three amines bearing different functionalities (alcohol, trifluoro and a maleimide dye) were successfully introduced into the nano-object cores as verified via NMR and FT-IR spectroscopy, while the retention of helicity within the resulting diblock copolymers was confirmed by circular dichroism (CD) spectroscopy. Changes in nanostructure morphology following modification were monitored by dynamic light-scattering (DLS), confirming the disassembly of the nano-objects when the core hydrophilicity was increased through the introduction of polar functionalities. These readily-synthesised and modified nanostructures containing helical cores are valuable scaffolds for use in applications such as circularly polarised luminescence, enantioselective chemistry or chiral separation.
Introduction
Post-polymerisation modification (PPM) is a versatile approach that has been widely employed to introduce functionality to polymeric materials.1–4 The PPM approach bypasses the potentially time-consuming steps of synthesising novel functional monomers and precludes difficulties arising from the protection and deprotection of functionalities incompatible with polymerisation processes. Thus, PPM often reduces the number of synthetic steps required for material syntheses and provides precursor materials that can be modified with a wide variety of functional groups.5–15For nanomaterials, reactive functionality provides a convenient and polyvalent means of introducing valuable moieties such as dyes,16–18 catalysts or drugs.19 PPM can also be employed to dynamically modify nanostructure properties, endowing such constructs with the capability to respond to environmental stimuli.20–23 For example, Rifaie-Graham et al. developed a diblock copolymer system that was modified through two PPM steps to incorporate donor–acceptor Stenhouse adduct-derived dyes with distinct excitation wavelengths. The polymersomes assembled from these copolymers exhibited light-responsive permeability-switch behaviour. The polymersomes were loaded with complementary enzymes, one type of polymersome with glucose oxidase and the other with horseradish peroxidase. Treatment of the resulting suspension with purpurogallin led to the formation of a wavelength-selective biocatalysis cascade that oxidised glucose on demand, which could be followed spectrophotometrically by measuring the chemiluminescent oxidation of purpurogallin.24 Wooley and co-workers developed polyphosphoester block copolymers fitted with alkyne25 or alkene26,27 pendant groups that could be readily functionalised using thiol nucleophiles. They demonstrated that PPM of these alkyne-containing phosphodiester scaffolds via thiol–yne chemistry allowed for facile modification of the polymer charge in water. Self-assembly of the modified copolymers yielded micelles with differential surface charge—non-ionic, anionic, cationic and zwitterionic—impacting the cytotoxicity of the particles toward mouse macrophage cells. In this instance, PPM enabled the creation of a library of polymers from a single scaffold.
Pentafluorophenol (PFP) esters are activated electrophiles reactive towards amines and alcohols that can be employed in PPM. For the synthesis of such scaffolds by controlled radical polymerisation, both pentafluorophenyl methacrylate (PFPMA) and pentafluorophenyl acrylate (PFPA) have been exploited as comonomers to incorporate the reactive functionality.28–32 In one example, Meijer and co-workers developed single-chain polymeric nanoparticles (SCNPs) composed of polyacrylate copolymers containing PFPA.33 This system was readily modified by reaction with different amines to affix the SCNPs with precise functionality; in this case, ligands capable of binding copper for the catalysis of azide–alkyne cycloaddition and depropargylation reactions, or Jeffamine for photosensitisation.33 More recently, PFPMA was employed by Couturaud et al. as a core-forming monomer for reversible addition–fragmentation chain transfer (RAFT)-mediated polymerisation-induced self-assembly (PISA) in DMSO.34 The resulting nanostructures were subsequently cross-linked via the activated ester groups with disulphide-containing diamine crosslinkers to achieve redox-responsive nanoparticles that degraded in the presence of reducing agents.
PFP-derived monomers can undergo PISA in DMSO due to their high solubility and tendency to produce polymers that become insoluble upon elongation. Based on our successful implementation of nickel-catalysed coordination-PISA (NiCCo-PISA) of aryl isocyanides in DMSO,35 we hypothesised that a PFP aryl isocyanide could be used as a core-forming monomer either as such or via copolymerisation with menthyl-ester aryl isocyanide (MAIC), while a PEG-ester aryl isocyanide (PAIC) served as the stabilising block.36 Wu and co-workers developed such a PFP aryl isocyanide (FAIC) comonomer to introduce electrophile handles into helical polyisocyanides (PIC).37–39 They demonstrated that FAIC readily polymerised under the same conditions as MAIC. Based on the similar solvophobicity of FAIC and MAIC, their copolymerisation represented an ideal system for the synthesis of helical and functionalisable nanostructures using NiCCo-PISA. Further, we reasoned that the resulting nanostructures would maintain their helicity subsequent to modification with various functional nucleophiles due to the static nature of the PIC helices.
In this study, we explore the synthesis, helicity, and self-assembly behaviour of P(PAIC)-b-P(MAIC-co-FAIC) block copolymer nanostructures synthesised by NiCCo-PISA. Further, we investigate the possibility of their modification with different nucleophiles and the influence of added functionality on the nano-objects’ size, stability and helicity retention. The combination of the simplicity of synthesising helix-containing nanostructures by NiCCo-PISA and the wide range of potential modifications possible by PPM provides a modular platform for the preparation of functional and helical nanomaterials.
Results and discussion
Functionalisable NiCCo-PISA micelles
To develop functionalised particles via NiCCo-PISA, it was necessary to introduce FAIC to the formulation without disrupting the polymerisation or self-assembly processes. In order to investigate the former, MAIC and FAIC were copolymerised in THF, a common solvent for both homopolymers, aiming for a degree of polymerisation (DP) of 30 and using different comonomer feed ratios to achieve P(MAIC-co-FAIC) copolymers containing 0–100 mol% of FAIC units (Fig. 1A). After purification by repeated precipitation in methanol, analysis of the copolymers by SEC indicated no apparent differences in molecular weight distribution between the copolymers, with number average molecular weights (Mn) ranging between 4.4–4.5 kDa and dispersity (ĐM) between 1.13–1.21 (Fig. S5†) across the series.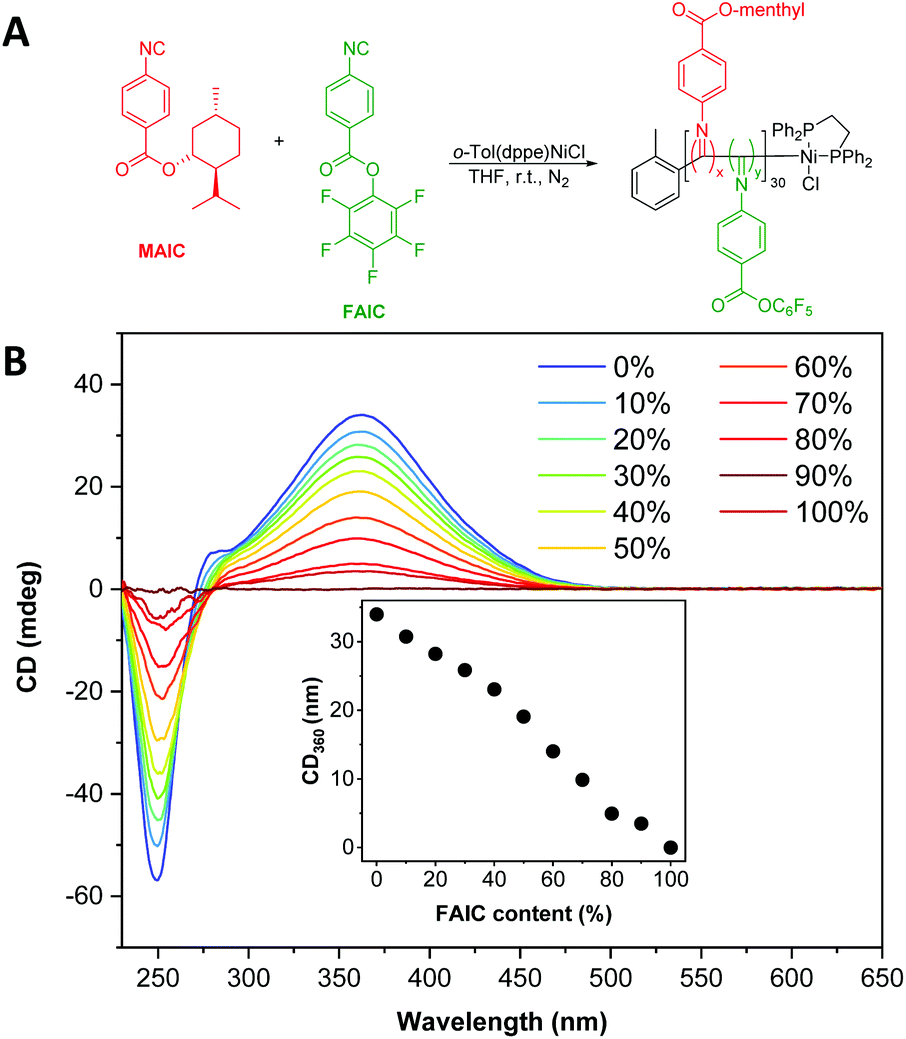 | ||
| Fig. 1 (A) Synthesis scheme of P(MAIC)x-co-P(FAIC)y where x + y = 30. (B) CD spectra (THF, 0.5 mg mL−1) of P(MAIC)x-co-P(FAIC)y copolymers with FAIC content ranging from 0 to 100 mol%. | ||
The successful incorporation of FAIC units into the copolymers was evidenced by 19F NMR spectroscopy (Fig. S6†). In particular, the broad signals at −153, −158 and −162 ppm in the 19F NMR spectra corresponded to the incorporated PFP moieties, proved in agreement with the monomer spectrum. The fluorine signal intensity increased with the quantity of FAIC present in the copolymer with the exception of the P(FAIC)30 homopolymer, likely as a consequence of its poor solubility in chloroform decreasing its signal intensity. Overall, these data demonstrated that copolymerisation of FAIC with MAIC did not appear to negatively influence molecular weight or molecular weight distribution (MWD).
The copolymers exhibited a clear signal at λ = 360 nm, assigned to the n–π* transition of the PIC backbone C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, consistent with previous literature reports.46–48 The helicity of the copolymers, as quantified by the strength of the signal at λ = 360 nm in the CD spectra, was found to vary linearly with the content of MAIC (i.e. the chiral monomer), decreasing until no CD absorption was present for P(FAIC)30 in the absence of chiral information (Fig. 1B).
N, consistent with previous literature reports.46–48 The helicity of the copolymers, as quantified by the strength of the signal at λ = 360 nm in the CD spectra, was found to vary linearly with the content of MAIC (i.e. the chiral monomer), decreasing until no CD absorption was present for P(FAIC)30 in the absence of chiral information (Fig. 1B).
We next sought to understand the impact of including the relatively more hydrophobic FAIC comonomer in our PISA formulation. To this end, NiCCo-PISA was conducted using PAIC to form the stabiliser block with DPP(PAIC) = 20 and DPsolvophobic (i.e. DPP(MAIC) + DPP(FAIC)) of 30 aiming for spherical micelles (Scheme 1). Different FAIC loadings were targeted in a series of experiments: 20, 50 and 100 mol% of the core block. The resulting P(PAIC)20-b-(P(MAIC)y-co-P(FAIC)z)30 diblock copolymers were named based on their FAIC content: D20%, D50% and D100%. A control containing no PFP moieties (D0%) was prepared to verify the stability of MAIC and PAIC units toward nucleophiles. SEC of the purified D0%, D20% and D50% diblock copolymers displayed no detectable differences in MWD, indicating that the polymerisation process was not impacted by the incorporation of FAIC as demonstrated earlier for the P(MAIC)x-co-P(FAIC)y series. In contrast, the D100% copolymer showed at least two distinct populations, hinting that the self-assembly had an impact on the growth of the polymer as seen in our previous study on NiCCo-PISA.
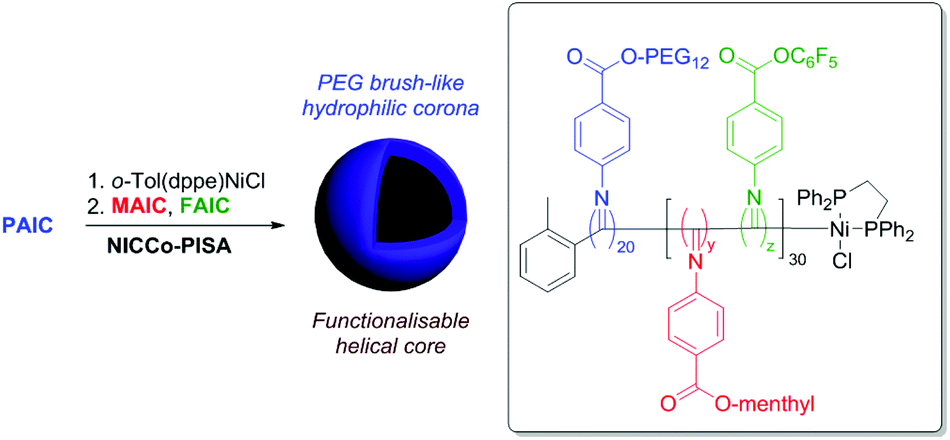 | ||
| Scheme 1 NiCCo-PISA to prepare functional P(PAIC)20-b-(P(MAIC)x-co-P(FAIC)y)30 diblock copolymer nano-objects. | ||
DLS analysis of the diblock copolymer assemblies obtained from NiCCo-PISA was conducted to ensure that the particle size distribution was not affected by the incorporation of FAIC into the copolymer. Indeed, no major change in size was detected for D20% or D50% compared to D0% (Fig. 2). However, for D100%, the size distribution and correlogram indicated that some aggregation was occurring, likely caused by their increased core hydrophobicity.
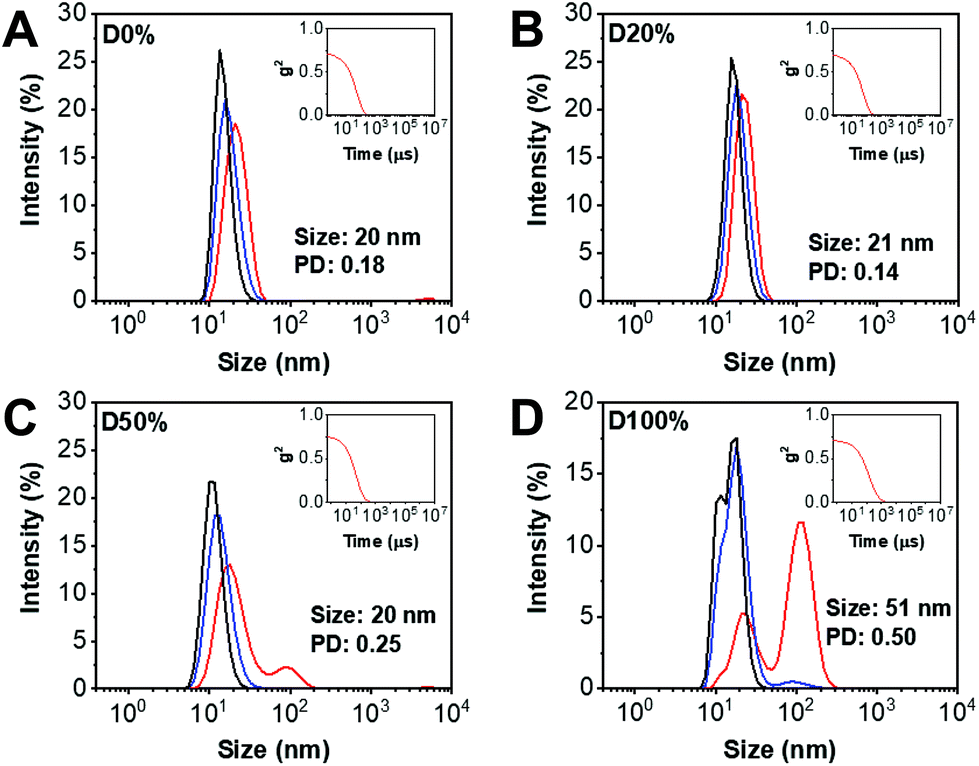 | ||
| Fig. 2 Size distributions of (A) D0%, (B) D20%, (C) D50% and (D) D100% in DMSO obtained by DLS (1 mg mL−1). The intensity (red line), volume (blue line) and number (black line) distributions are displayed. The insets show the correlograms. | ||
The diblock copolymers were also analysed by CD spectroscopy (Fig. 3), showing that helicity was retained during the self-assembly process, as was presented previously for NiCCo-PISA to achieve P(PAIC)20-b-P(MAIC)30. However, a decrease in the CD signal was observed with increasing FAIC content, concordant with the CD analysis conducted on the P(MAIC)x-co-P(FAIC)y series, resulting from the reduced proportion of chiral MAIC units in the core-forming blocks.
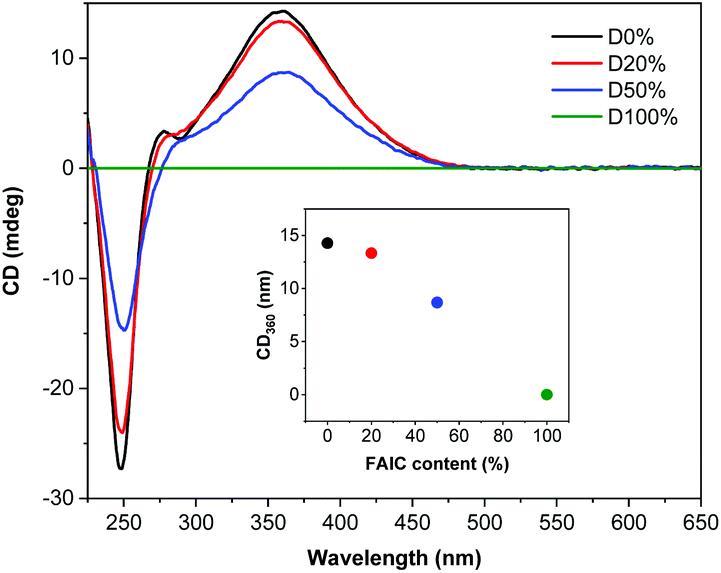 | ||
| Fig. 3 CD (THF, 0.5 mg mL−1) spectra of P(PAIC)20-b-(P(MAIC)y-co-P(FAIC)z)30 with 0, 20, 50 and 100 mol% of FAIC. | ||
Functionalisation with ethanolamine and trifluoroethylamine
PPM of D0%, D20% and D50% using different primary amine nucleophiles was then evaluated (Scheme 2). Nucleophiles were chosen with groups that could be easily detected following PPM. In particular, we supposed that successful incorporation of ethanolamine (EOA) would decorate the copolymer with alcohol functionalities that should be detectable by FT-IR while trifluoroethylamine (TFEA) would introduce a new fluorine handle that could be followed using 19F NMR spectroscopy. In addition, substitution with EOA was expected to drastically alter the polarity of the nanoparticle cores.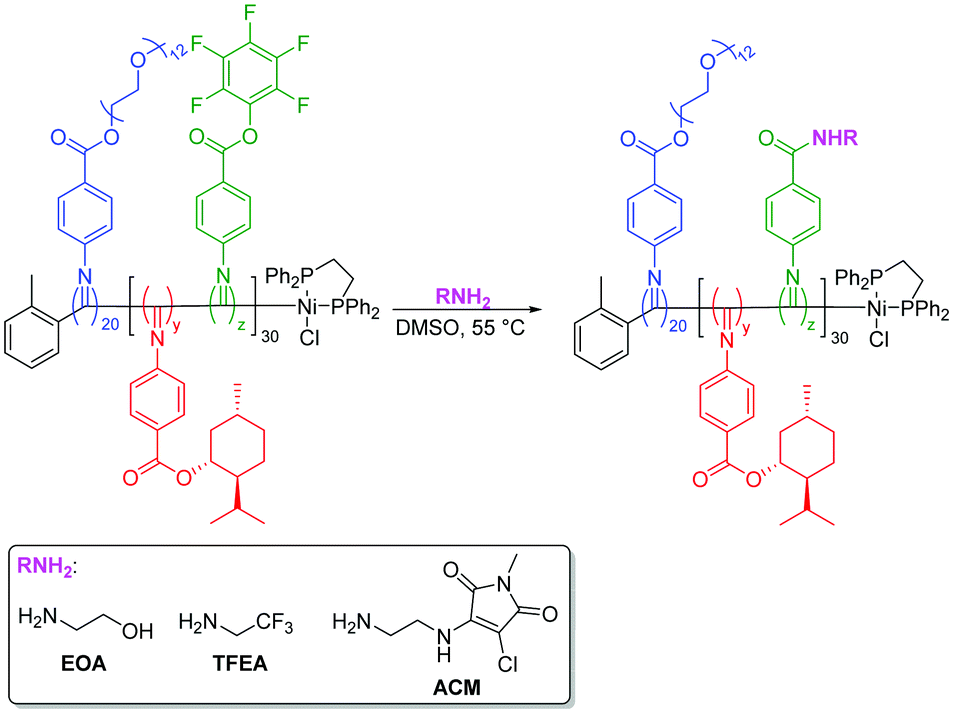 | ||
| Scheme 2 PPM of P(PAIC)20-b-(P(MAIC)y-co-P(FAIC)z)30 with various primary amines: EOA, TFEA, and amine chloride maleimide dye. | ||
D0%, D20% and D50% were treated in situ with EOA and TFEA directly following NiCCo-PISA. These aminolysis reactions were carried out in the absence of base to avoid hydrolysis of the PFP esters.34 An excess of amine was thus employed to ensure quantitative conversion to amide product. The reactions were allowed to stir for 24 h before an aliquot was taken and purified by dialysis to remove unreacted amine and phenolic by-products prior to analysis.
SEC analysis of the copolymers before and after substitution showed no significant change in MWD for D20%+EOA, D20%+TFEA, or D50%+TFEA, while high molecular weight tailing was observed for D50%+EOA (Fig. 4 and Fig. S7†). In the case of D50%+EOA, the broad MWD was attributed to interaction of the installed alcohol and amide functionalities with the SEC columns while in the case of D50%+TFEA, only the amides interacted with the column.
 | ||
| Fig. 4 Normalised SEC RI molecular weight distributions (THF + 2% v/v NEt3, 40 °C, PS standards) of D0%, D20%, and, D50% and before and after treatment with EOA and purification by dialysis. | ||
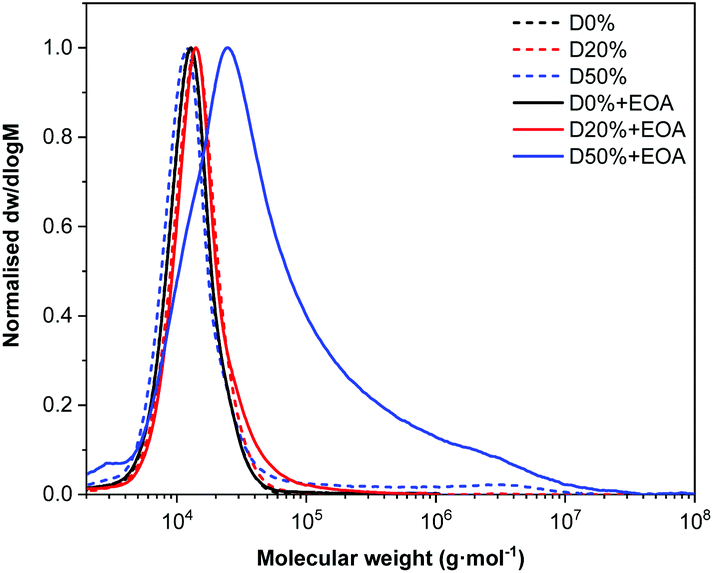
By FT-IR, several bands revealed the success and completion of the substitution reactions. First, a broad band at 3350 cm−1 indicated the presence of O–H (alcohol) and N–H (amide) groups and therefore the successful insertion of EOA into the copolymer (Fig. 5). The signal's strength was directly related to the quantity of the associated functionality; thus a stronger signal was evident for D50%+EOA compared to D20%+EOA. The absence of amide and alcohol signals for D0%+EOA demonstrated the stability of MAIC and PAIC to the reaction conditions. In addition, the absence of four bands in particular indicated the successful substitution of PFP units: 1761 cm−1 (CO) and 1246 cm−1 (C–O) associated with the ester group; 1516 cm−1 (CC) for the PFP aromatic unit and 1037 cm−1 for C–F (Fig. S8†). Their disappearance suggested the aminolysis reaction reached completion. A similar FT-IR spectroscopic analysis was performed for D0%, D20% and D50% after treatment with TFEA (Fig. S9†). It is noteworthy that the unreacted copolymers were purified by dialysis and the absence of COOH signal confirms the stability of the PFP units in aqueous environment.
 | ||
| Fig. 5 FT-IR of D0% (black), D20% (red) and D50% (blue) following substitution reaction with EOA. | ||
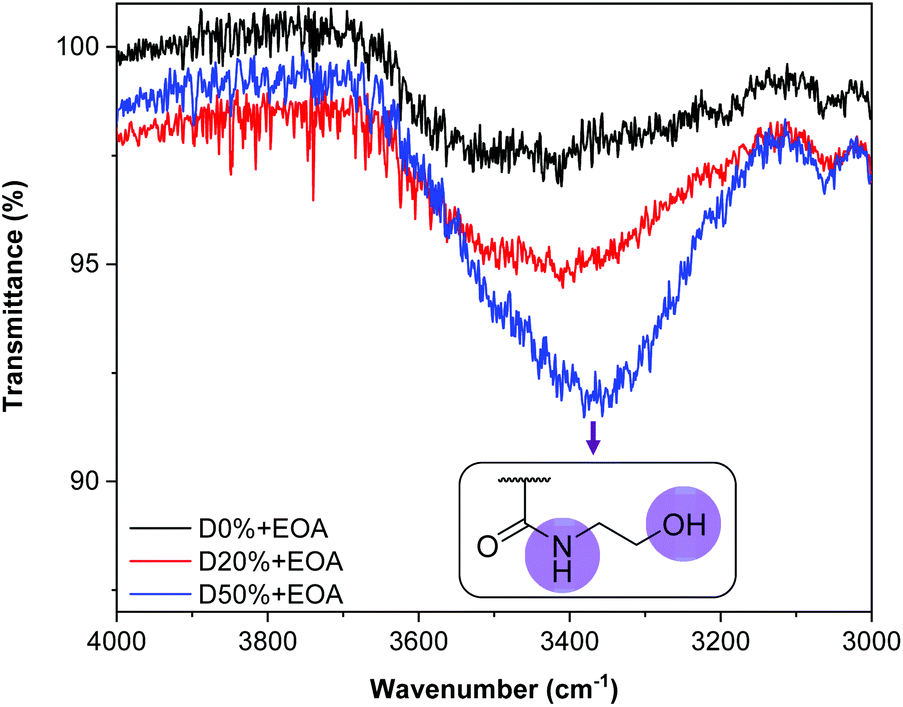
To further verify the consumption of PFP units, 19F NMR spectroscopy of D0%, D20% and D50% before and after reaction with EOA or TFEA was conducted (Fig. 6 and Fig. S10†). The complete disappearance of these signals in the presence of amine suggested quantitative conversion during aminolysis. The absence of signals associated with molecular PFP also showed that the reaction by-product, pentafluorophenol, was successfully removed during the purification step. In the case of the TFEA substituted copolymers, i.e.D20%+TFEA and D50%+TFEA, the appearance of a new singlet at −72 ppm, assigned to the installed CF3 group (Fig. 6), indicated the successful incorporation of TFEA. The broadened signal relative to the spectrum of the pure monomer provided further evidence of the attachment of CF3 to the rigid copolymer chain. All copolymers were also analysed by 1H NMR spectroscopy, where a new signal could be detected for D50%+EOA in concordance with the EOA CH2 protons at 2.65 ppm (Fig. S11 and S12†).
 | ||
| Fig. 6 19F NMR spectra of D20% and D50% in CDCl3 (377 MHz, 298 K) before and after treatment with TFEA and purification by dialysis. The 19F NMR spectrum of TFEA is also provided for comparison. | ||
The nano-object suspensions obtained from NiCCo-PISA and following PPM were also analysed by DLS (Fig. S13, Fig. S14† and Table 1) to verify their stability towards changes in core polarity from substitution of the PFP units. In brief, the size of the assemblies did not change for D0%+EOA and D0%+TFEA, indicating a good stability of the non-reactive nanostructure to the reaction conditions. Similarly, no change was found in the hydrodynamic diameters (Dh) of the D20+EAO, D20%+TFEA and D50%+TFEA nanostructures. However, there was a clear indication of a size change for D50%+EOA, where the Dh after substitution increased from 20 to 34 nm and the size distribution (PD) from 0.25 to 1. This broad distribution of sizes can be explained by the increase in polarity and therefore solubility of the nanostructure's core, with their overall increase in solvophilicity leading to a partial disassembly of the nano-objects.
| Polymer |
D
h![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (nm) a (nm) |
M
n, SECb (kDa) |
Đ
Mb |
CD360c (mdeg) |
|---|---|---|---|---|
| a Particle size measured by DLS with PD in parenthesis. b Determined by SEC (THF + 2% v/v NEt3) using PS standards. c CD (THF, 0.5 mg mL−1) signal at λ = 360 nm. d Molecular weight distribution was outside the calibration range. | ||||
| D0% | 20 (0.18) | 11.2 | 1.24 | 14 |
| D0% + EAO | 20 (0.16) | 11.4 | 1.22 | — |
| D0% + TFEA | 20 (0.17) | 11.4 | 1.25 | — |
| D20% | 21 (0.14) | 12.4 | 1.24 | 13 |
| D20% + EOA | 19 (0.17) | 13.5 | 1.38 | 13 |
| D20% + TFEA | 18 (0.21) | 13.0 | 1.20 | 13 |
| D50% | 20 (0.25) | 10.5 | 1.34 | 9 |
| D50% + EOA | 34 (1.00) | —d | —d | 8 |
| D50% + TFEA | 23 (0.32) | —d | —d | 8 |
CD spectroscopy was employed to determine the change in helicity after aminolysis (Fig. 7). No significant changes were observed for any of the four copolymers when comparing spectra before and after PPM. The signal at λ = 360 nm remained constant at ca. 13 mdeg and 9 mdeg for the D20% and D50% substituted copolymers, respectively, while the signal at 250 nm decreased slightly. This latter absorption band was derived from the PFP aromatic rings in the copolymer scaffold and therefore was expected to decrease subsequent to substitution. These data confirmed the stability of the helical backbone to the reaction conditions and their resilience to the substitution of 20% or 50% of the core units.
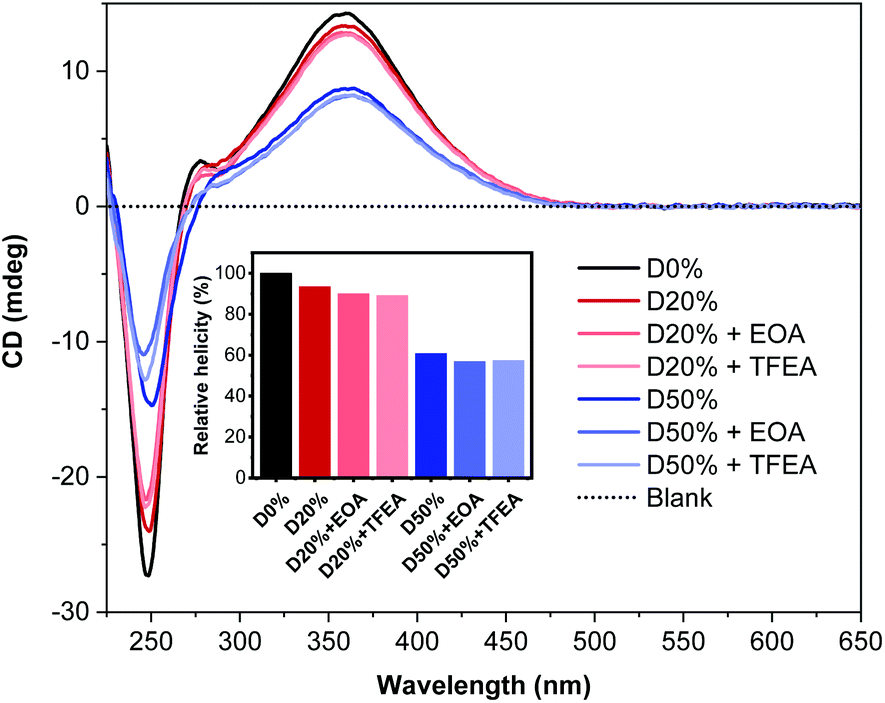 | ||
| Fig. 7 CD (THF, 0.5 mg mL−1) spectra of D0%, D20%, and, D50% before and after treatment with EOA or TFEA and purification by dialysis. The inset shows the helicity (CD at λ = 360 nm) of the copolymers compared to D0%. | ||
Functionalisation with maleimide dye
To further leverage PPM to study changes in the self-assembly behaviour and composition of the diblock copolymer nanostructures, a fluorescent aminochloromaleimide (ACM) dye was synthesised (Scheme 2, see also ESI†) for introduction into the nano-object cores. Substituted maleimides like ACM possess solvent-dependent fluorescence lifetimes and emissions, which have been used extensively in our group for chemico-fluorescent responsive copolymers, fluorescent nanogels and the labelling of proteins, enzymes and polymers.18,40–44 Following PPM, the copolymers were analysed as before.By FT-IR, decreases in the FAIC signals were detectable for both reactions along with an increase of the amide (N–H) signal (Fig. S15†), signifying some, but incomplete, ACM incorporation. Observation of the signal at 1760 cm−1 assigned to the amide function (CO) indicated there were still PFP units in both D20%+ACM and D50%+ACM (Fig. 8A). However, these signals were substantially broader following substitution. We reasoned that the resonance associated with the newly formed amides could also occupy this region, complicating calculation of conversion. Thus, the following spectroscopic analyses are more qualitative than quantitative and were simply used to confirm the presence or absence of dye and to evaluate the dye's environment. Interestingly, PFP units were still detected after the incomplete reactions which demonstrated their stability under the reaction conditions.
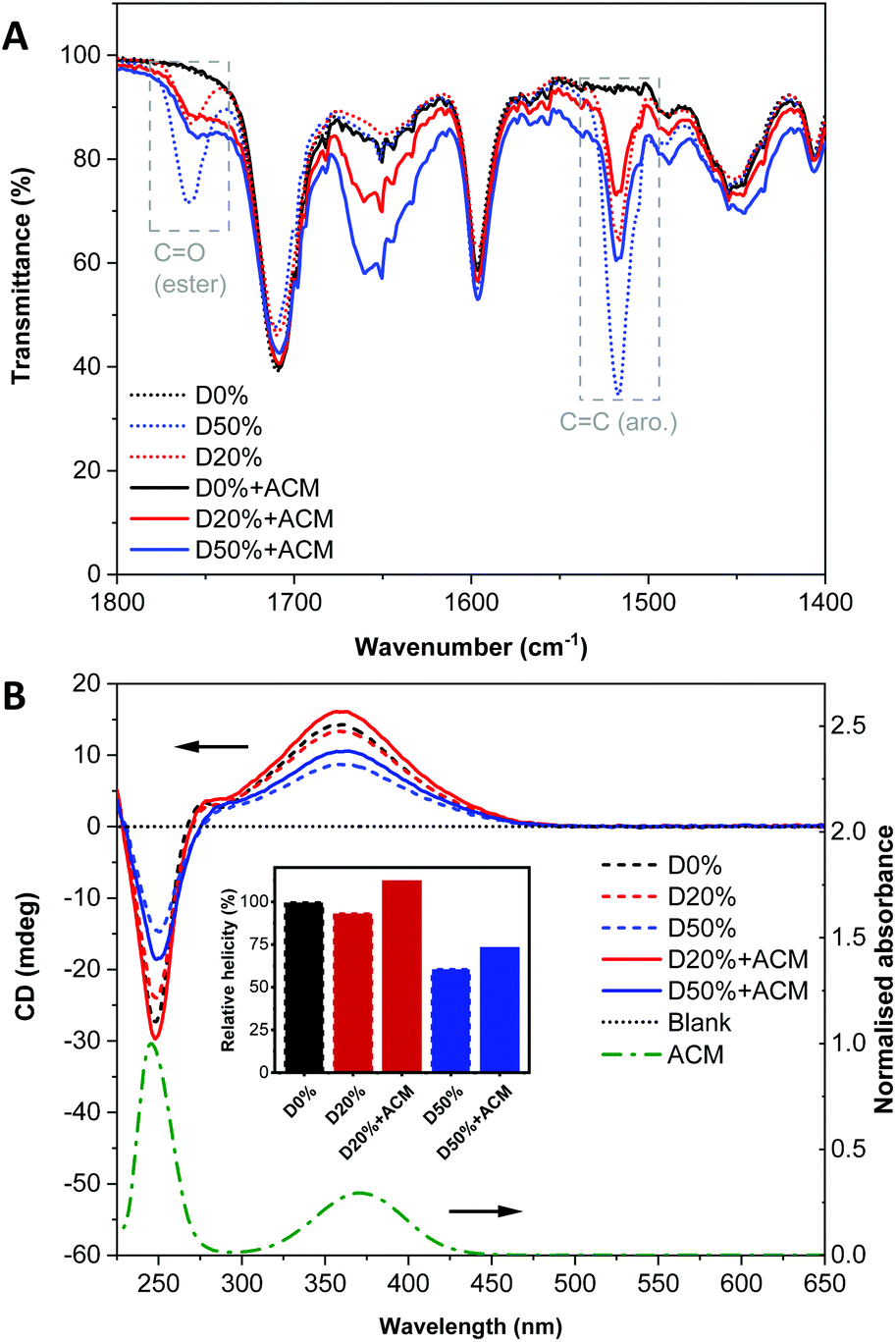 | ||
| Fig. 8 (A) FT-IR and (B) CD (THF, 0.5 mg mL−1) spectra of the D0% (black), D20% (red) and D50% (blue) copolymers before (dotted lines) and after PPM with ACM (solid lines). UV/Vis spectrum (THF, 0.5 mg mL−1) of free ACM. The inset shows the helicity (CD at λ = 360 nm) of the copolymers compared to D0%. | ||
SEC analysis of the dye-substituted copolymer showed MWDs for D0%+ACM and D20%+ACM similar to their parent copolymers, while the D50%+ACM copolymer exhibited clear tailing in this region (Fig. S16†). Here, the ACM-derived secondary amine and/or imide functionalities were assumed to interact with the SEC column in a similar manner to the alcohol functionalities of the D50%+EOA copolymer.
CD spectroscopy showed an increase of the CD signals at λ = 250 nm and 360 nm for both dye-substituted copolymers (Fig. 8B). It was known that the ACM unit is UV-active in these regions as evidenced by its UV/Vis spectrum. It should also be noted that the ACM moiety possesses no inherent chirality. CD spectroscopy measures the absorbance of chiral species; thus, the increase in absorbance at ca. λ = 360 nm in the CD spectra of D20%+ACM and D50%+ACM further corroborated the successful introduction of ACM into the helical environment of the diblock copolymer.
As before, the obtained diblock copolymer nanostructures were analysed by DLS. The D20%+ACM copolymer formed micelles of similar size to the unmodified diblock copolymer (22 vs. 21 nm). The size distributions and correlogram for D50%+ACM indicated a possible partial disassembly of the nanostructures with a Dh of 16 nm and a PD of 0.37 vs. 0.25 for the unmodified copolymer (Fig. S17†). The higher hydrophilicity of the dye compared with the PFP units likely increased the solvability of the core, inducing increased solvation of the micelle unimer constituents.
Fluorescence spectrophotometry was utilised to monitor ACM fluorescence from the modified diblock copolymers. The original polymer, D0%, was not fluorescent in the absence of dye substitution. In contrast, an emission band was detected at λem = 488 nm for both of the dye-substituted polymers when excited at λex = 375 nm in THF, in line with the fluorescence emission of the free ACM. Moreover, a solution containing a mixture of the copolymer D0% and the free dye displayed a signal at the same wavelength, proving that the copolymer itself did not influence λem (Fig. S18†). After transfer of the species to water, λem for nanostructures of D20%+ACM and D50%+ACM shifted to 510 nm (Δλ = 22 nm). By comparison, the redshift in emission of the free dye was more substantial (λem = 577 nm, Δλ = 89 nm). The observed differences in emission in H2O between the polymer-bound and free dye are consistent with previous studies from our group and reflect the environmental dependence of ACM fluorescence (Fig. 9).42,43 Indeed, the similarities in emission behaviour of D50%+ACM in both THF and H2O further confirm the attachment of the ACM moieties to the core block of the diblock copolymer nano-objects. In addition the blue shift in emission between the free dye in water and the dye inside the nanostructure cores implies the polarity of the latter resembles that of THF.
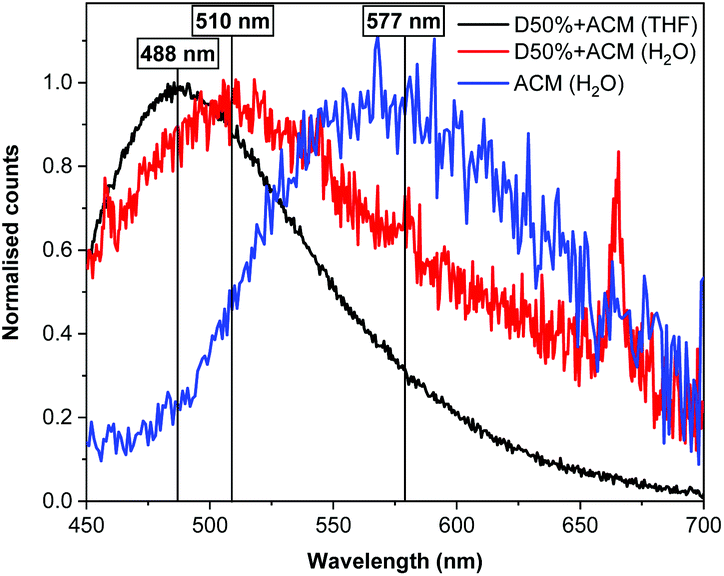 | ||
| Fig. 9 Fluorescence spectra (λex = 375 nm) for D50%+ACM and free ACM in THF or H2O. | ||
Fluorescence lifetime measurements displayed vast discrepancies depending on the solvent and/or attachment of the ACM dyes (Fig. S19A and Table S2†). Both D20%+ACM and D50%+ACM had similar average fluorescence lifetimes (τAv,a) of 6.5 and 7.7 ns, respectively, and exhibited shorter lifetimes than the free dye in THF that had a τAv,a = 14.8 ns, providing further evidence of ACM's attachment to the diblock copolymers. To further investigate the influence of the spatial location of the dye, D20% or D50% were mixed with the appropriate quantity of free ACM in THF, displaying lifetimes of 13.8 and 14.2 ns, respectively, showing no differences between the free and polymer-bound dyes. This reinforced the idea that the observed decrease of lifetime in the initial study occurred as a consequence of the dye being in close proximity to the polymer chains (Fig. S19B†). Fluorescence lifetime measurements of D20%+ACM, D50%+ACM or free dye in water were very short – around 1 ns – indicating the dye was readily quenched regardless of attachment. Maleimide dyes are effectively quenched in protic solvents as a consequence of electron driven proton transfer from the hydrogen bonding between water and the fluorophore (Fig. S19C†).45 Thus, the fact that reduced lifetimes were observed for D20%+ACM and D50%+ACM implied the presence of water in the nanoparticle cores was sufficient in quantity to quench fluorescence.
Conclusions
Helical-core micelles that could easily be functionalised by different nucleophiles were synthesised via copolymerisation of FAIC and MAIC aryl isocyanide monomers by NiCCo-PISA. The resulting diblock copolymers and nanostructures were analysed by complementary techniques, showing the successful introduction of a range of functionalities by PPM including alcohols, fluorinated units and fluorescent dyes. Moreover, the stability of the core's helicity to the PPM conditions was confirmed. The PFP units inside the hydrophobic core were shown to be resistant to hydrolysis under the reaction conditions and during dialysis, indicating that these micelles could be employed for modification under aqueous conditions. When hydrophilic amines were utilised (i.e. EOA and ACM), partial disassembly of the micelles was observed for the D50% diblock copolymer nano-objects, demonstrating the effect of the modification of the core's polarity on the nano-objects’ structural integrity. The introduction of ACM inside the micelle cores confirmed, via solvatochromatism, their covalent attachment within the hydrophobic cores of the nanostructures. Lifetime measurements indicated the presence of water in the core that readily quenched the grafted dyes. These micelles represent versatile helical scaffolds for the preparation of functionalised nanostructures, with potential applications as chiral fluorescent probes, enantiomer-specific stimuli-responsive systems or stereoselective nanoreactors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Union (SUSPOL-EJD 642671), ERC (grant number 615142), EPSRC and the University of Birmingham.Notes and references
- J.-F. Lutz and B. S. Sumerlin, The Role of Click Chemistry in Polymer Synthesis, in Click Chemistry for Biotechnology and Materials Science, ed. J. Lahann, John Wiley & Sons, Ltd., 2009, pp. 69–88 Search PubMed.
- N. K. Boaen and M. A. Hillmyer, Chem. Soc. Rev., 2005, 34, 267–275 RSC.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS.
- K. A. Günay, P. Theato and H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1–28 CrossRef.
- M. B. Larsen, S.-J. Wang and M. A. Hillmyer, J. Am. Chem. Soc., 2018, 140, 11911–11915 CrossRef CAS.
- C. J. Atkins, G. Patias, J. S. Town, A. M. Wemyss, A. M. Eissa, A. Shegiwal and D. M. Haddleton, Polym. Chem., 2019, 10, 646–655 RSC.
- T. Krappitz, P. Feibusch, C. Aroonsirichock, V. P. Hoven and P. Theato, Macromolecules, 2017, 50, 1415–1421 CrossRef CAS.
- P. K. Kuroishi, M. J. Bennison and A. P. Dove, Polym. Chem., 2016, 7, 7108–7115 RSC.
- T. R. Barlow, J. C. Brendel and S. Perrier, Macromolecules, 2016, 49, 6203–6212 CrossRef CAS.
- R. J. Ono, S. Q. Liu, S. Venkataraman, W. Chin, Y. Y. Yang and J. L. Hedrick, Macromolecules, 2014, 47, 7725–7731 CrossRef CAS.
- Y.-C. M. Wu and T. M. Swager, J. Am. Chem. Soc., 2019, 141, 12498–12501 CrossRef CAS.
- N. H. Park, M. Fevre, Z. X. Voo, R. J. Ono, Y. Y. Yang and J. L. Hedrick, ACS Macro Lett., 2016, 5, 1247–1252 CrossRef CAS.
- D. N. Crisan, O. Creese, R. Ball, J. L. Brioso, B. Martyn, J. Montenegro and F. Fernandez-Trillo, Polym. Chem., 2017, 8, 4576–4584 RSC.
- P. R. Sruthi and S. Anas, J. Polym. Sci., 2020, 58, 1039–1061 CrossRef CAS.
- T. Kubo, C. A. Figg, J. L. Swartz, W. L. A. Brooks and B. S. Sumerlin, Macromolecules, 2016, 49, 2077–2084 CrossRef CAS.
- C. Battistella, Y. Yang, J. Chen and H.-A. Klok, ACS Omega, 2018, 3, 9710–9721 CrossRef CAS.
- A. Das and P. Theato, Macromolecules, 2015, 48, 8695–8707 CrossRef CAS.
- M. P. Robin and R. K. O'Reilly, Chem. Sci., 2014, 5, 2717–2723 RSC.
- Y. Zhong, B. J. Zeberl, X. Wang and J. Luo, Acta Biomater., 2018, 73, 21–37 CrossRef CAS.
- N. Busatto, J. L. Keddie and P. J. Roth, Polym. Chem., 2020, 11, 704–711 RSC.
- X. Liu, D. Hu, Z. Jiang, J. Zhuang, Y. Xu, X. Guo and S. Thayumanavan, Macromolecules, 2016, 49, 6186–6192 CrossRef CAS.
- P. Theato, B. S. Sumerlin, R. K. O'Reilly and T. H. Epps III, Chem. Soc. Rev., 2013, 42, 7055–7056 RSC.
- M. S. Rolph, M. Inam and R. K. O'Reilly, Polym. Chem., 2017, 8, 7229–7239 RSC.
- O. Rifaie-Graham, S. Ulrich, N. F. B. Galensowske, S. Balog, M. Chami, D. Rentsch, J. R. Hemmer, J. Read de Alaniz, L. F. Boesel and N. Bruns, J. Am. Chem. Soc., 2018, 140, 8027–8036 CrossRef CAS.
- S. Zhang, J. Zou, F. Zhang, M. Elsabahy, S. E. Felder, J. Zhu, D. J. Pochan and K. L. Wooley, J. Am. Chem. Soc., 2012, 134, 18467–18474 CrossRef CAS.
- Y. H. Lim, G. S. Heo, Y. H. Rezenom, S. Pollack, J. E. Raymond, M. Elsabahy and K. L. Wooley, Macromolecules, 2014, 47, 4634–4644 CrossRef CAS.
- S. Cho, G. S. Heo, S. Khan, J. Huang, D. A. Hunstad, M. Elsabahy and K. L. Wooley, Macromolecules, 2018, 51, 3233–3242 CrossRef CAS.
- F. D. Jochum and P. Theato, Macromolecules, 2009, 42, 5941–5945 CrossRef CAS.
- Q. Zhang, P. Schattling, P. Theato and R. Hoogenboom, Eur. Polym. J., 2015, 62, 435–441 CrossRef CAS.
- H. Gaballa, S. Lin, J. Shang, S. Meier and P. Theato, Polym. Chem., 2018, 9, 3355–3358 RSC.
- M. I. Gibson, E. Fröhlich and H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4332–4345 CrossRef CAS.
- S. Lin, A. Das and P. Theato, Polym. Chem., 2017, 8, 1206–1216 RSC.
- Y. Liu, T. Pauloehrl, S. I. Presolski, L. Albertazzi, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2015, 137, 13096–13105 CrossRef CAS.
- B. Couturaud, P. G. Georgiou, S. Varlas, J. R. Jones, M. C. Arno, J. C. Foster and R. K. O'Reilly, Macromol. Rapid Commun., 2019, 40, 1800460 CrossRef.
- S. Jimaja, S. Varlas, Y. Xie, J. C. Foster, D. Taton, A. P. Dove and R. K. O'Reilly, ACS Macro Lett., 2020, 9, 226–232 CrossRef CAS.
- N. Liu, C.-H. Ma, R.-W. Sun, J. Huang, C. Li and Z.-Q. Wu, Polym. Chem., 2017, 8, 2152–2163 RSC.
- J. Yin, L. Xu, X. Han, L. Zhou, C. Li and Z.-Q. Wu, Polym. Chem., 2017, 8, 545–556 RSC.
- L. Xu, X.-H. Xu, N. Liu, H. Zou and Z.-Q. Wu, Macromolecules, 2018, 51, 7546–7555 CrossRef CAS.
- M. Su, N. Liu, Q. Wang, H. Wang, J. Yin and Z.-Q. Wu, Macromolecules, 2016, 49, 110–119 CrossRef CAS.
- M. P. Robin, M. W. Jones, D. M. Haddleton and R. K. O'Reilly, ACS Macro Lett., 2012, 1, 222–226 CrossRef CAS.
- M. P. Robin, P. Wilson, A. B. Mabire, J. K. Kiviaho, J. E. Raymond, D. M. Haddleton and R. K. O'Reilly, J. Am. Chem. Soc., 2013, 135, 2875–2878 CrossRef CAS.
- A. B. Mabire, M. P. Robin, W.-D. Quan, H. Willcock, V. G. Stavros and R. K. O'Reilly, Chem. Commun., 2015, 51, 9733–9736 RSC.
- Y. Xie, J. T. Husband, M. Torrent-Sucarrat, H. Yang, W. Liu and R. K. O'Reilly, Chem. Commun., 2018, 54, 3339–3342 RSC.
- J. T. Husband, A. C. Hill and R. K. O'Reilly, Polym. Int., 2019, 68, 1247–1254 CrossRef CAS.
- M. Staniforth, W.-D. Quan, T. N. V. Karsili, L. A. Baker, R. K. O'Reilly and V. G. Stavros, J. Phys. Chem. A, 2017, 121, 6357–6365 CrossRef CAS.
- S. Asaoka, A. Joza, S. Minagawa, L. Song, Y. Suzuki and T. Iyoda, ACS Macro Lett., 2013, 2, 906–911 CrossRef CAS.
- F. Takei, K. Yanai, K. Onitsuka and S. Takahashi, Chem. – Eur. J., 2000, 6, 983–993 CrossRef CAS.
- J. Lee, S. Shin and T.-L. Choi, Macromolecules, 2018, 51, 7800–7806 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00791a |
| This journal is © The Royal Society of Chemistry 2021 |