
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Boron-rich, cytocompatible block copolymer nanoparticles by polymerization-induced self-assembly†
Lin-Chiang Sherlock
Huang‡
abc,
Dao
Le‡
 abd,
I-Lun
Hsiao
ef,
Susanne
Fritsch-Decker
e,
Christian
Hald
ab,
Su-Ching
Huang
g,
Jen-Kun
Chen
g,
Jih Ru
Hwu
c,
Carsten
Weiss
e,
Min-Hua
Hsu
*h and
Guillaume
Delaittre
*abi
abd,
I-Lun
Hsiao
ef,
Susanne
Fritsch-Decker
e,
Christian
Hald
ab,
Su-Ching
Huang
g,
Jen-Kun
Chen
g,
Jih Ru
Hwu
c,
Carsten
Weiss
e,
Min-Hua
Hsu
*h and
Guillaume
Delaittre
*abi
aInstitute of Biological and Chemical Systems – Functional Molecular Systems (IBCS-FMS), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
bInstitute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology (KIT), Engesserstrasse 15, 76131 Karlsruhe, Germany
cDepartment of Chemistry, National Tsing Hua University, Hsinchu 30013, Taiwan
dPolymer Chemistry Team (ECP), Institut Parisien de Chimie Moléculaire (IPCM), Sorbonne Université and CNRS, UMR 8232, 4 Place Jussieu, 75005 Paris, France
eInstitute of Biological and Chemical Systems – Biological Information Processing (IBCS-BIP), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
fSchool of Food Safety, College of Nutrition, Taipei Medical University, No. 250, Wuxing St., Taipei 11031, Taiwan
gInstitute of Biomedical Engineering and Nanomedicine, National Health Research Institutes, No. 35, Keyan Rd., Zhunan Township, Miaoli County 35053, Taiwan
hDepartment of Chemistry, National Changhua University of Education, Changhua 50007, Taiwan. E-mail: minghuahsu@cc.ncue.edu.tw
iOrganic Functional Molecules, Organic Chemistry, University of Wuppertal, Gaußstrasse 20, 42119 Wuppertal, Germany. E-mail: delaittre@uni-wuppertal.de; Web: http://www.twitter.com/GPRDelaittre
First published on 5th August 2020
Abstract
Core–shell nanoparticles (NPs) with a boron-rich core were synthesized by RAFT-mediated polymerization-induced self-assembly using a new methacrylic boronate ester monomer. Under specific conditions, sub-100 nm spherical NPs could be obtained at high conversions by either emulsion or dispersion RAFT polymerization using poly(oligo(ethylene glycol) methacrylate) (POEGMA) dithiobenozate-based chain transfer agents. Phenylboronic acid surface-functionalized NPs were obtained using a telechelic POEGMA. Primary data on biocompatibility is provided and suggests suitability as boron delivery agent for boron neutron capture therapy.
Among the various potential techniques to fight cancer and eliminate tumor cells, boron neutron capture therapy (BNCT) is an attractive binary treatment based on (i) delivery of non-toxic boron drugs into tumor cells and (ii) irradiation with a thermal neutron beam to trigger nuclear fission of boron-10 and subsequent production of high-energy alpha particles.1 The key challenge for successful BNCT treatment is the specific accumulation of boron – in any form – in tumor cells. Boronophenylalanine (BPA) has been one of the two major compounds used in BNCT clinical trials for decades, yet requires rather large injected amounts to reach sufficient boron concentrations.2 The low tumor uptake and selectivity, and short retention of BPA are due to its non-selective tumor-targeting functionality, negative charge, and low molecular weight.3 Similar issues are encountered with the other major compound used in BNCT, namely sodium borocaptate. To improve the longer circulation of boron agents and therefore facilitate specific tumor accumulation, several types of nanoparticles (NPs) such as liposome, dendrimer, and block copolymer (BCP) NPs have been evaluated as potential delivery system.4 For instance, boronic acid-containing BCP NPs have been designed and synthesized for BNCT.5–10 In these examples, boron compounds were either encapsulated in or covalently linked to nanoparticles supposed to accumulate in neoplastic regions via the enhanced permeability and retention (EPR) effect.11,12 While they may improve specific delivery, macromolecular approaches to boron delivery for BNCT still exhibit shortcomings such as low boron content per unit weight, and/or complicated and time-consuming particle synthesis. In the present contribution, we sought to harness polymerization-induced self-assembly (PISA) to prepare boron-rich nanoparticles because this method precisely addresses such shortcomings.
PISA has become a very popular method over the last decade to design core–shell polymeric NPs with specific shapes and controlled diameters.13–17 Indeed, PISA not only yields colloidal suspensions with homogeneous morphology directly at high concentrations, but also simultaneously gives access to precise macromolecular architecture with high control over functionality,18,19 with excellent potential for a range of applications.20–27 We thus anticipated that the PISA-based synthesis of nanoparticles with a boron-rich core and a biocompatible shell in a time-efficient manner could be possible. Note that, while boron-containing polymers have recently been in vogue,28–30 no example of boron-containing PISA-made nanomaterials can be found. To this end, we designed a new boronate ester-functionalized methacrylate based on a benzyl methacrylate (BzMA) scaffold – a monomer well-known in the PISA realm – and polymerized it in polar media under radical initiation in the presence of a poly(oligo(ethylene glycol) methacrylate) (POEGMA) macromolecular chain transfer agent (macroCTA). POEGMA was selected for its high solubility in a wide range of solvents, as well as its biocompatibility and propensity to provide a stealth character to coated nanomaterials, thereby enabling application of our NPs as nanomedicines, hence in the BNCT field.31–33 Moreover, since the POEGMA macroCTA is synthesized by reversible addition–fragmentation transfer (RAFT) polymerization, α-functionalized POEGMA macroCTAs can be readily obtained, which allows a straightforward post-functionalization on the surface of nanoparticles, e.g., for targeted delivery. Here, phenylboronic acid was introduced as α end group for specific targeting of sialylated epitopes on the membrane of solid tumors.34 The overall design of the present study is displayed in Scheme 1.
 | ||
| Scheme 1 Synthetic route and compounds employed in the current study for boron-rich nanoparticle formation by RAFT polymerization-induced self-assembly (RAFTPISA). ACVA = 4,4′-azobis(4-cyanopentanoic acid). AIBN = azobisisobutyronitrile. | ||
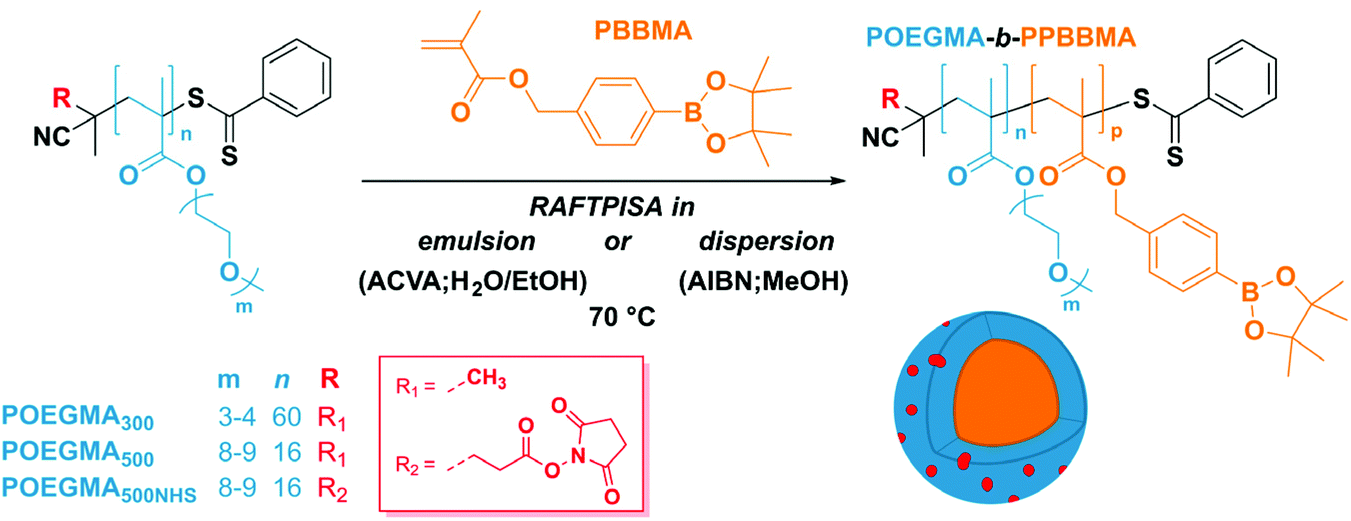
As mentioned above, we designed the boronate ester-containing methacrylate based on BzMA because the latter was successfully employed in multiple RAFT polymerization-induced self-assembly (RAFTPISA) studies.35–39 We chose the ester form of boronic acid to prevent potential issues related to self-dimerization and subsequent uncontrolled crosslinking during synthesis,40,41 as well as to tune solubility with regards to the PISA process.16 4-Pinacolboronylbenzyl methacrylate monomer PBBMA was synthetized in two steps: (i) esterification of commercially available 4-(hydroxymethyl) phenylboronic acid with pinacol and (ii) Steglich esterification with methacrylic acid (see ESI†). Combination of 1H, 13C, and 11B NMR spectroscopy and high-resolution mass spectrometry characterizations confirmed structure and purity of PBBMA, obtained as an oil (Fig. S1–S4†). Before implementing it for the synthesis of amphiphilic block copolymers in dispersed media, the propensity of this monomer to be polymerized by RAFT solution polymerization with a low-molar-mass CTA, namely 2-cyano-2-propyl benzodithioate (CPBD), was first assessed. The polymerization was conducted in N,N-dimethylacetamide (DMAc) at various [PBBMA]/[CPBD] ratios (100, 200, and 400) with a constant [AIBN]/[CPBD] ratio equal to 0.2, at either 0.3 M or 0.6 M of PBBMA (9 and 18 wt%, respectively). As expected, the higher concentration of monomer led to a faster polymerization (Fig. 1A). In most cases, the polymerization fulfilled elementary criteria for a controlled/living polymerization, i.e., linear increase in pseudo-first order plot – at least in the first 8 hours of polymerization, linear increase of number-average molar mass with conversion, and clear shift of size-exclusion chromatography (SEC) traces (Fig. 1 and S5†). However, it was possible to achieve high conversions in a reasonable time frame only at the lowest targeted degree of polymerization (DPth = 100), i.e., at the highest CPBD and AIBN concentrations. Higher DPth systematically involved higher Đ values. Nevertheless, PBBMA showed a sufficient amenability to polymerize via the RAFT process and was subsequently used in emulsion and dispersion polymerizations, without surfactant, in the presence of POEGMA macroCTAs.
 | ||
| Fig. 1 RAFT solution polymerization of PBBMA in DMAc at 70 °C. (A) First-order kinetic plots for various [PBBMA] and DPth. Dashed lines correspond to linear fits. (B) Evolution of number-average molar mass (top) and dispersity (bottom) with PBBMA conversion for [PBBMA] = 0.6 M at various DPth. (C) Size-exclusion chromatograms of polymers obtained at different times for [PBBMA] = 0.6 M and DPth = 100. | ||
A set of macroCTAs was prepared by RAFT solution polymerization of two distinct OEGMA oligomonomers (Mn = 300 or 500 g mol−1) with either CPBD or an N-hydroxysuccinimide ester derivative of CPBD, to introduce α functionality, which should then eventually be presented at the particle surface after RAFTPISA (see Scheme 1). All macroCTAs were obtained with low dispersity (Đ < 1.2) (Table S1†).
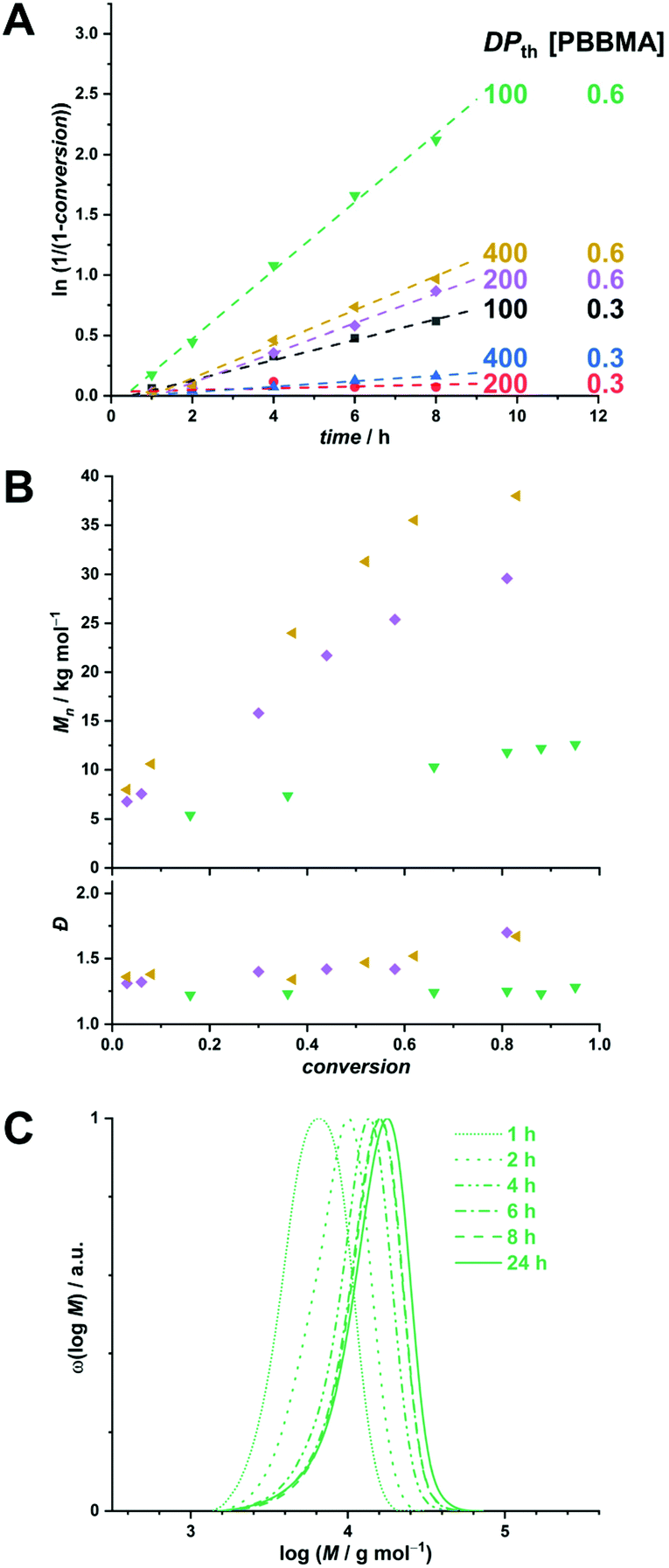
First, we examined the emulsion polymerization of PBBMA in water/EtOH (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vol/vol). All sets of conditions can be found in Table S2.† For this series, POEGMA500 was chosen for its high water solubility (even at high temperatures), which is a vital parameter in PISA. For instance, emulsion RAFTPISA at 70 °C with [PBBMA]/[POEGMA500]/[ACVA] = 100/1/0.2 at 10 wt% solids content (Table S2,† entry 1) led to about 60% conversion after 2 h with a reasonably narrow molar mass distribution (MMD), yet did not significantly proceed further (Fig. S8†). The resulting NPs were stable monodisperse spheres of about 50 nm, as determined by dynamic light scattering (DLS) (Fig. S9†), accompanied by a slight coagulum. Increase in total solids content to 20 wt% allowed higher PBBMA conversions to be reached (94% in 2 h; Table S2,† entry 4), yet with a broadening of the MMD (Fig. 2A). Although the final dispersion contained a homogeneous population of spheres of around 50 nm, as observed by transmission electron microscopy (TEM) (Fig. 2B) and DLS (Fig. S10†), a small amount of coagulum was again present. Increasing [PBBMA]/[POEGMA500] to 200 or 400 at solids contents of 10 or 20 wt% systematically led to early destabilization and low conversions (entries 2, 3, 5, and 6, Table S2†).
:1 vol/vol). All sets of conditions can be found in Table S2.† For this series, POEGMA500 was chosen for its high water solubility (even at high temperatures), which is a vital parameter in PISA. For instance, emulsion RAFTPISA at 70 °C with [PBBMA]/[POEGMA500]/[ACVA] = 100/1/0.2 at 10 wt% solids content (Table S2,† entry 1) led to about 60% conversion after 2 h with a reasonably narrow molar mass distribution (MMD), yet did not significantly proceed further (Fig. S8†). The resulting NPs were stable monodisperse spheres of about 50 nm, as determined by dynamic light scattering (DLS) (Fig. S9†), accompanied by a slight coagulum. Increase in total solids content to 20 wt% allowed higher PBBMA conversions to be reached (94% in 2 h; Table S2,† entry 4), yet with a broadening of the MMD (Fig. 2A). Although the final dispersion contained a homogeneous population of spheres of around 50 nm, as observed by transmission electron microscopy (TEM) (Fig. 2B) and DLS (Fig. S10†), a small amount of coagulum was again present. Increasing [PBBMA]/[POEGMA500] to 200 or 400 at solids contents of 10 or 20 wt% systematically led to early destabilization and low conversions (entries 2, 3, 5, and 6, Table S2†).
 | ||
| Fig. 2 (A) Size-exclusion chromatograms of polymers obtained by PBBMA emulsion RAFTPISA in water/ethanol 3:1 vol/vol at 70 °C with POEGMA500 at DPth = 100, and 20 wt% solids content (entry 4 from Table S2†). (B) Transmission electron micrograph of nanoparticles obtained in the conditions mentioned above, after 2 h. | ||
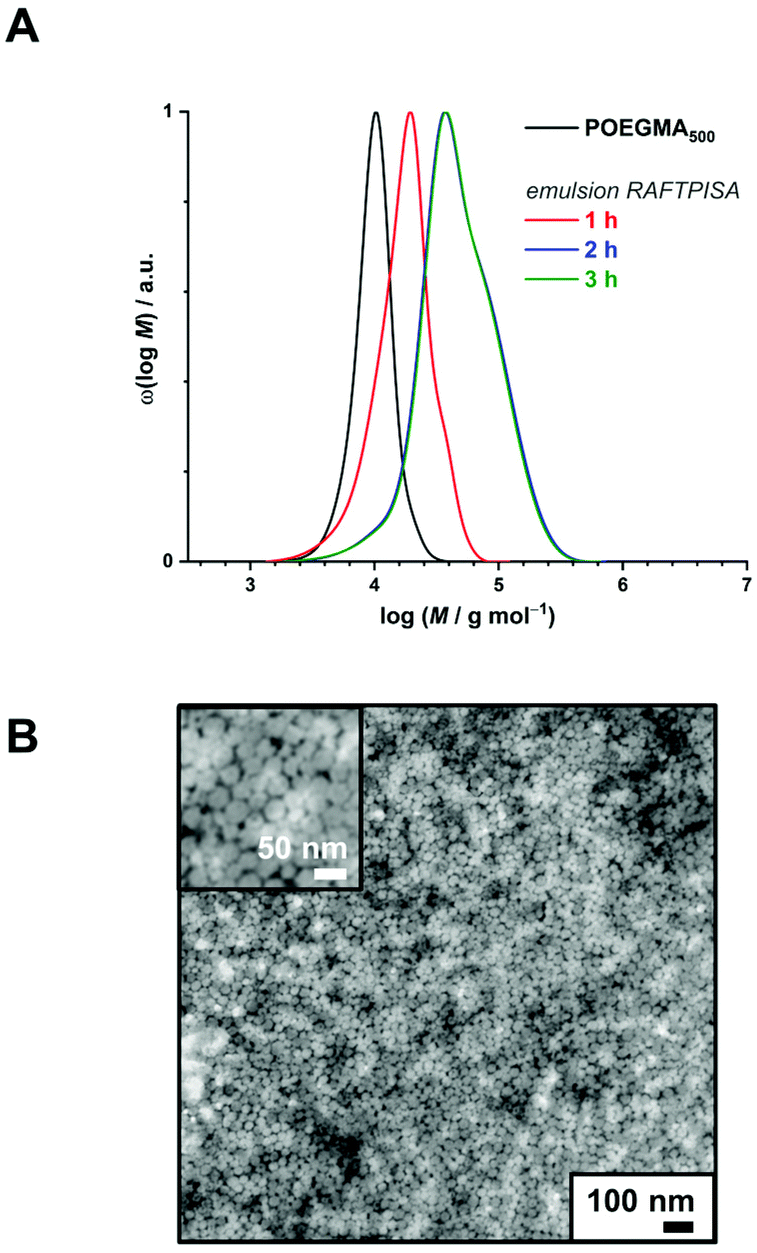
In order to produce a colloidally stable PISA system and achieve easy removal of potentially unreacted monomer, we turned to a dispersion process. Methanol was chosen as polymerization medium because (i) PBBMA was found to be highly soluble in it as opposed to its polymer – which is a requirement for PISA – and (ii) because it is considered a green solvent.42 As the thermoresponsive character of POEGMAs with shorter oligoethylene glycol side chains becomes irrelevant in methanol, we chose to start our investigation on dispersion RAFTPISA with POEGMA300, because this is also a more practical polymer in terms of purification (i.e., separation of residual monomer). A series of experiments was performed by varying DPth (100, 200, and 400) and the total solids content (10, 15, and 30 wt%). With the previously employed [initiator]/[CTA] ratio of 0.2, far-from-complete monomer conversions were obtained after 24 h (Table S3, entries 1–6, as well as Fig. S11†). Increasing [initiator]/[CTA] to 0.5 allowed a nearly full PBBMA conversion for DPth = 100 at 15 and 30 wt%. In those cases, as well as for DPth = 200 at 30 wt%, more than 90% PBBMA was polymerized within 6–8 h and nearly full conversion was eventually reached (Fig. 3A). Molar masses increased with time and conversion, with however a significant apparent amount of remaining macroCTA, and therefore increasing Đ (Fig. 3B). Nevertheless, particle size increased with conversion and retained a homogeneous character (Table S4†). As observed by TEM, pure spherical NPs were obtained with DPth = 100 at 15 wt% (Fig. 3C). Importantly, the hydrodynamic diameters of the NPs remained essentially unchanged when transferred to water with no sign of aggregation (Table S4†). At higher target DPth and/or solids content, PISA syntheses gave more condensed jelly-like solution or gel sedimentation indicating possible formation of worm-like filomicelles/nanofibers (entries 5, 6, and 9, Table S3†). At higher DPth and solids content, i.e., 200–400 and 30 wt%, control was lost both in terms of MMDs (Đ > 2) and colloidal stability (precipitation). When POEGMA300 was replaced with POEGMA500 (Table S5†), kinetics remained similar (Fig. S14†). For DPth = 100 at 15 wt%, a colloidally stable dispersion was obtained, however with conversion-dependent evolution of the dispersion characteristics, following classic observations made in PISA formulations leading to higher-order morphologies: first, fluid dispersions of small nanoparticles; then, gel with large heterogeneous particle size distribution (PSD); finally, viscous dispersions with homogeneous PSD (Fig. S15 and Table S6†). As of now, we have not investigated all these samples departing from classic core–shell spheres in depth, since BNCT has so far typically required spherical nanoparticles. Nevertheless, note that other PISA morphologies, particularly anisotropic structures, could be interesting as well.43
 | ||
| Fig. 3 (A) Conversion vs. time plots for PBBMA dispersion RAFTPISA with POEGMA300 at [AIBN]/[POEGMA300] = 0.5 at various solids contents and DPth. (B) Size-exclusion chromatograms of polymers obtained by PBBMA dispersion RAFTPISA in methanol at 70 °C with POEGMA300 at [AIBN]/[POEGMA300] = 0.5, DPth = 100, and 15 wt% solids content (entry 7, Table S3†). (C) Transmission electron micrograph of nanoparticles obtained after 25 h under conditions corresponding to B (entry 7, Table S3†). | ||
In most dispersion RAFTPISA experiments, the dispersity of the resulting block copolymers was relatively high, with the presence of a low-molar-mass tail in MMDs. While this tail is not uncommon and is typically attributed to a fraction of residual macroCTA, its relatively high intensity was puzzling. We and others did not observe this in the RAFTPISA of BzMA or 2-hydroxypropyl methacrylate with similar macroCTAs.38,44,45 We postulated that some interactions with the SEC stationary phase arising from moieties not present in those previous systems could be responsible for the observed MMD distortions in the present case. In fact, when we incubated PBBMA in methanol at 70 °C, we found that a steady amount of free pinacol (13–14 mol%) was instantaneously formed (Fig. S17†). To investigate this, we added free pinacol to a dispersion RAFTPISA in order to shift the equilibrium between boronate ester on the one hand and boronic acid and pinacol on the other hand towards the former. POEGMA500-mediated RAFTPISA of PBBMA with pinacol (20 mol% with regards to PBBMA) was slightly accelerated (Fig. S18A†), with final nanoparticles very similar in size (see Fig. S19†). More importantly, SEC analysis of final block copolymer evidenced narrower MMDs with a noticeable decrease of the aforementioned tail signal (Fig. S18B†), which is an indirect proof of possibly interference of hydrolyzed boronate ester moieties with a neat RAFTPISA. A possible solution to this issue would be the use of dry methanol.
The motivation of this study being the development of a potential boron delivery agent for BNCT, we sought to preliminarily assess the biocompatibility of the RAFTPISA-made POEGMA-b-PPBBMA NPs. For imaging purposes, fluorescent NPs were prepared by adding a small amount of fluorescein methacrylate (FMA) to a PISA recipe.46 Briefly, nanoparticles NP1 and NP2, without and with surface functional groups, respectively, were obtained from POEGMA500 and POEGMA500NHS, respectively (see ESI†). NHS ester-surface-functionalized NP2 were subsequently reacted with 3-aminophenylboronic acid to produce NP3 with specific targeting agent for sialylated epitopes (Fig. 4A), as mentioned earlier. Successful surface modification to obtain NP3 was confirmed by the appearance of aromatic signals in deuterated polar media, in which the core is invisible (Fig. S20†). The NP dispersions were dialyzed against MeOH, then water, to remove unreacted monomer and switch the dispersant, respectively. Their colloidal characteristics in cell culture media are collated in Table S7.† The fluorescent nature of the NPs was confirmed by fluorescence spectroscopy with λem,max = 515 nm (Fig. S20†).
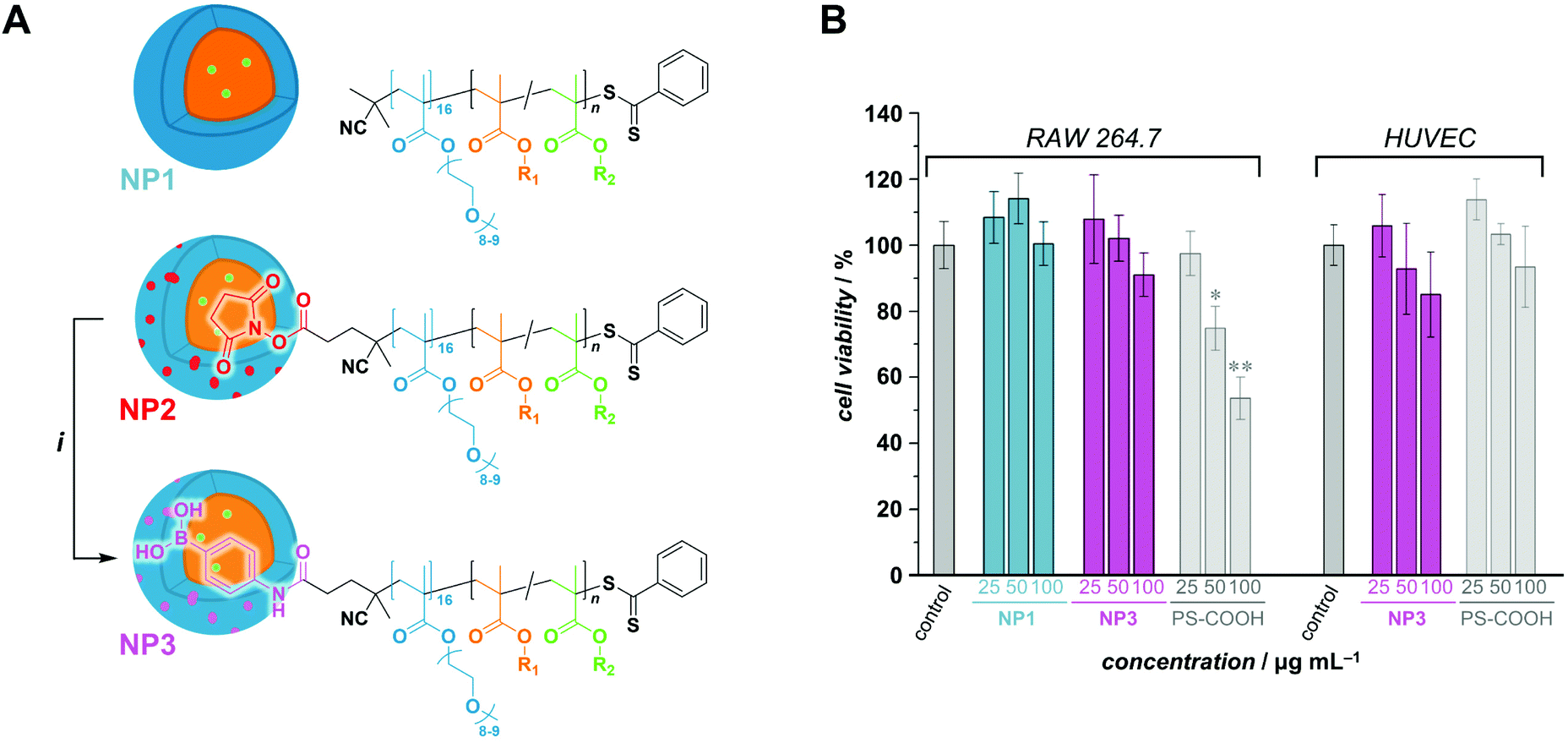 | ||
| Fig. 4 (A) Modification of NHS ester-functionalized POEGMA500NHS-b-PPBBMA nanoparticles. R1 = 4-phenylboronate pinacol ester; R2 = fluorescein. i: 3-aminobenzeneboronic acid, triethylamine, ambient temperature. (B) Cell viability of boron-containing PISA-made nanoparticles NP1 and NP3 in RAW 264.7 cells and HUVECs. Data are representative of 2 independent experiments, each performed in triplicates and shown as means ± SEM. *Significantly different from control group (*p < 0.05, **p < 0.01). | ||
Next, we analyzed interactions of the NPs with scavenger cells. Indeed, clearance by the reticuloendothelial system (RES) typically accounts for the poor delivery of nanomedicines to target tissues.47 Therefore, uptake and cytocompatibility of NP1 and NP3 were tested in RAW 264.7 and HUVEC cells,48 which are widely used as model systems in nanotoxicology and represent macrophages and endothelial cells, respectively, belonging to the RES.45 Fluorescein-labeled, carboxylated polystyrene (PS-COOH) NPs were included as a reference polymeric nanomaterial.49 As seen in Fig. 4B, nanoparticles NP1 and NP3 were non-toxic up to the concentration of 100 μg mL−1 over 24 h of exposure. Note that NP1 could not be assessed with HUVECs because large agglomerates were formed in the corresponding M200/LSGS cell medium (Table S7†) and interfered with image analysis. Compared to PS-COOH NPs, non-functionalized NP1 were taken up at very low amounts in macrophages and adhered to endothelial cells (presumably due to agglomeration). However, 3-aminophenylboronic acid-functionalized NP3, intended for targeting selected cells of interest, did not accumulate in macrophages and endothelial cells (Fig. S21−S23†). Hence, future investigations on clearance of these NPs in vivo as well as their potential to target specific cell types are warranted. The difference in colloidal stability between NP1 and NP3 in these conditions remains to be elucidated. Nevertheless, functional nanoparticles NP3 appear to be promising for BNCT.
While their stealth core–shell architecture and their ease of functionalization with targeting moieties or labels confer decisive advantages over boronophenylalanine (BPA) – the classic BNCT agent – it is particularly interesting to put into perspective the “solubilizing effect” of the present NPs with the maximum solubility of BPA. The latter is indeed soluble at physiological pH only up to about 6 mg mL−1, that is, 0.031 wt% of boron.50 Considering the best working systems here (entries 4 and 7 of Tables S2 and S3,† respectively), boron mass concentrations with one order of magnitude higher are achieved (0.42 and 0.33 wt%, respectively), within non-optimized systems (see calculations in ESI†). At the same time, the boron concentration per mass unit of compound remains in the same range: 5.2 wt% for BPA vs. 2.8 and 2.2 wt%, respectively.
Conclusions
In this communication, we described for the first time the synthesis of amphiphilic diblock copolymer nanoparticles with a boron-rich core by polymerization-induced self-assembly (PISA). A new methacrylic monomer with a pinacol boronate ester was synthesized with good yields from commercially available and cheap reagents in two steps. Its efficient RAFT polymerization was demonstrated for limited degrees of polymerization, both in homogeneous and heterogeneous systems. Using hydrophilic macromolecular transfer agents, PISA could be carried out to obtain sub-100 nm spherical nanoparticles using both emulsion and dispersion polymerization in (hydro)alcoholic media. While they may not be useful for the envisioned application, i.e., boron-neutron capture therapy (BCNT), typical PISA higher-order morphologies such as nanorods/fibers and nano/microvesicles were not investigated, yet could be part of further investigations. Future work should also be directed to refining the optimal block copolymer composition (DP of 1st block vs. DP of 2nd block) to increase the relative boron concentration within the nanoparticles. Finally, encouraged by the preliminary results on biocompatibility reported here, implementation of the new nanoparticles in BNCT, first in vitro, then possibly in vivo, should be performed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L-C. S. H. thanks the Ministry of Science and Technology of the Republic of China (MOST) and the German Academic Exchange Service (DAAD) for his DAAD-MOST Sandwich fellowship. D. L. thanks the Alexander von Humboldt Foundation for his postdoctoral fellowship. J. R. H. and M.-H. H. thank MOST (Grant No. 109-2634-F-007-023) as well. The Macroarc group (ITCP, KIT) and the Levkin lab (IBCS-FMS, KIT) are gratefully acknowledged for access to SEC and DLS instruments, respectively.References
- R. F. Barth, Z. Zhang and T. Liu, Cancer Commun., 2018, 38, 36 CrossRef.
- F. Ali, N. S. Hosmane and Y. Zhu, Molecules, 2020, 25, 828 CrossRef CAS PubMed.
- R. F. Barth, P. Mi and W. Yang, Cancer Commun., 2018, 38, 35 CrossRef PubMed.
- R. L. Moss, Appl. Radiat. Isot., 2014, 88, 2–11 CrossRef CAS PubMed.
- A.-K. Azab, M. Srebnik, V. Doviner and A. Rubinstein, J. Controlled Release, 2005, 106, 14–25 CrossRef CAS PubMed.
- G. Kahraman, O. Beşkardeş, Z. M. O. Rzaev and E. Pişkin, Polymer, 2004, 45, 5813–5828 CrossRef CAS.
- L. Zhang, Y. Lin, J. Wang, W. Yao, W. Wu and X. Jiang, Macromol. Rapid Commun., 2011, 32, 534–539 CrossRef CAS PubMed.
- L.-C. S. Huang, W.-Y. Hsieh, J.-Y. Chen, S.-C. Huang, J.-K. Chen and M.-H. Hsu, Appl. Radiat. Isot., 2014, 88, 89–93 CrossRef PubMed.
- S. Yoneoka, K. C. Park, Y. Nakagawa, M. Ebara and T. Tsukahara, Polymers, 2018, 11, 42 CrossRef PubMed.
- S. Yoneoka, Y. Nakagawa, K. Uto, K. Sakura, T. Tsukahara and M. Ebara, Sci. Technol. Adv. Mater., 2019, 20, 291–304 CrossRef CAS PubMed.
- A. H. Soloway, W. Tjarks, B. A. Barnum, F.-G. Rong, R. F. Barth, I. M. Codogni and J. G. Wilson, Chem. Rev., 1998, 98, 1515–1562 CrossRef CAS PubMed.
- R. F. Barth, Mg. H. Vicente, O. K. Harling, W. S. Kiger, K. J. Riley, P. J. Binns, F. M. Wagner, M. Suzuki, T. Aihara, I. Kato and S. Kawabata, Radiat. Oncol., 2012, 7, 146 CrossRef PubMed.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS PubMed.
- F. D’Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368 CrossRef PubMed.
- D. Le, D. Keller and G. Delaittre, Macromol. Rapid Commun., 2019, 40, 1800551 CrossRef PubMed.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, Macromol. Rapid Commun., 2019, 40, 1800279 CrossRef PubMed.
- X. Zhang, A. F. Cardozo, S. Chen, W. Zhang, C. Julcour, M. Lansalot, J. F. Blanco, F. Gayet, H. Delmas, B. Charleux, E. Manoury, F. D'Agosto and R. Poli, Chem. – Eur. J., 2014, 20, 15505–15517 CrossRef CAS PubMed.
- I. Canton, N. J. Warren, A. Chahal, K. Amps, A. Wood, R. Weightman, E. Wang, H. Moore and S. P. Armes, ACS Cent. Sci., 2016, 2, 65–74 CrossRef CAS PubMed.
- J. Lesage De La Haye, I. Martin-Fabiani, M. Schulz, J. L. Keddie, F. D'Agosto and M. Lansalot, Macromolecules, 2017, 50, 9315–9328 CrossRef CAS.
- D. Keller, A. Beloqui, M. Martínez-Martínez, M. Ferrer and G. Delaittre, Biomacromolecules, 2017, 18, 2777–2788 CrossRef CAS PubMed.
- L. D. Blackman, S. Varlas, M. C. Arno, Z. H. Houston, N. L. Fletcher, K. J. Thurecht, M. Hasan, M. I. Gibson and R. K. O'Reilly, ACS Cent. Sci., 2018, 4, 718–723 CrossRef CAS PubMed.
- F. H. Sobotta, F. Hausig, D. O. Harz, S. Hoeppener, U. S. Schubert and J. C. Brendel, Polym. Chem., 2018, 9, 1593–1602 RSC.
- M. J. Derry, T. Smith, P. S. O'Hora and S. P. Armes, ACS Appl. Mater. Interfaces, 2019, 11, 33364–33369 CrossRef CAS PubMed.
- D. Le, M. Dilger, V. Pertici, S. Diabaté, D. Gigmes, C. Weiss and G. Delaittre, Angew. Chem., Int. Ed., 2019, 58, 4725–4731 CrossRef CAS PubMed.
- F. Cheng and F. Jäkle, Polym. Chem., 2011, 2, 2122–2132 RSC.
- W. L. A. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375–1397 CrossRef CAS PubMed.
- G. Vancoillie and R. Hoogenboom, Polym. Chem., 2016, 7, 5484–5495 RSC.
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- Y. Qi, A. Simakova, N. J. Ganson, X. Li, K. M. Luginbuhl, I. Ozer, W. Liu, M. S. Hershfield, K. Matyjaszewski and A. Chilkoti, Nat. Biomed. Eng., 2016, 1, 2 CrossRef PubMed.
- W. Gao, W. Liu, T. Christensen, M. R. Zalutsky and A. Chilkoti, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16432–16437 CrossRef CAS PubMed.
- S. Deshayes, H. Cabral, T. Ishii, Y. Miura, S. Kobayashi, T. Yamashita, A. Matsumoto, Y. Miyahara, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2013, 135, 15501–15507 CrossRef CAS PubMed.
- E. R. Jones, M. Semsarilar, A. Blanazs and S. P. Armes, Macromolecules, 2012, 45, 5091–5098 CrossRef CAS.
- E. R. Jones, M. Semsarilar, P. Wyman, M. Boerakker and S. P. Armes, Polym. Chem., 2016, 7, 851–859 RSC.
- E. R. Jones, O. O. Mykhaylyk, M. Semsarilar, M. Boerakker, P. Wyman and S. P. Armes, Macromolecules, 2016, 49, 172–181 CrossRef CAS PubMed.
- E. T. Garrett, Y. Pei and A. B. Lowe, Polym. Chem., 2016, 7, 297–301 RSC.
- G. Ng, J. Yeow, J. Xu and C. Boyer, Polym. Chem., 2017, 8, 2841–2851 RSC.
- S. Delpierre, B. Willocq, J. De Winter, P. Dubois, P. Gerbaux and J.-M. Raquez, Chem. – Eur. J., 2017, 23, 6730–6735 CrossRef CAS PubMed.
- R. Tanaka, N. Tonoko, S. Kihara, Y. Nakayama and T. Shiono, Polym. Chem., 2018, 9, 3774–3779 RSC.
- C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927–934 RSC.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed.
- Y. Zhang, Z. Wang, K. Matyjaszewski and J. Pietrasik, Macromol. Rapid Commun., 2019, 40, 1800331 CrossRef PubMed.
- E. Molle, D. Le, T. Norizadeh Abbariki, M. S. Akdemir, M. Takamiya, E. Miceli, O. Kassel and G. Delaittre, ChemPhotoChem, 2019, 3, 1084–1089 CrossRef CAS.
- D. Le, F. Wagner, M. Takamiya, I.-L. Hsiao, G. Gil Alvaradejo, U. Strähle, C. Weiss and G. Delaittre, Chem. Commun., 2019, 55, 3741–3744 RSC.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- I. Hansjosten, J. Rapp, L. Reiner, R. Vatter, S. Fritsch-Decker, R. Peravali, T. Palosaari, E. Joossens, K. Gerloff, P. Macko, M. Whelan, D. Gilliland, I. Ojea-Jimenez, M. P. Monopoli, L. Rocks, D. Garry, K. Dawson, P. J. F. Röttgermann, A. Murschhauser, J. O. Rädler, S. V. Y. Tang, P. Gooden, M. F. A. Belinga-Desaunay, A. O. Khan, S. Briffa, E. Guggenheim, A. Papadiamantis, I. Lynch, E. Valsami-Jones, S. Diabaté and C. Weiss, Arch. Toxicol., 2018, 92, 633–649 CrossRef CAS PubMed.
- O. Lunov, T. Syrovets, C. Loos, J. Beil, M. Delacher, K. Tron, G. U. Nienhaus, A. Musyanovych, V. Mailänder, K. Landfester and T. Simmet, ACS Nano, 2011, 5, 1657–1669 CrossRef CAS PubMed.
- T. R. Lahann, US Pat., US5492900A, 1993 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00710b |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |