
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Glutathione peroxidase mimics based on conformationally-restricted, peri-like, 4,5-disubstituted fluorene dichalcogenides†
Kesar
Jagdev
 a,
Damiano
Tanini
b,
Jack W.
Lownes
a,
Carlotta
Figliola
a,
Louise
Male
a,
Antonella
Capperucci
b and
Richard S.
Grainger
*a
a,
Damiano
Tanini
b,
Jack W.
Lownes
a,
Carlotta
Figliola
a,
Louise
Male
a,
Antonella
Capperucci
b and
Richard S.
Grainger
*a
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK. E-mail: r.s.grainger@bham.ac.uk
bUniversity of Florence, Department of Chemistry “Ugo Shiff”, Via della Lastruccia 13, 1-50019 Sesto Fiorentino, Italy
First published on 30th November 2021
Abstract
Glutathione peroxidase (GPx) regulates cellular peroxide levels through glutathione oxidation. GPx-mimics based on 4,5-disubstituted fluorene diselenides, their oxides, and ditellurides show catalytic activities consistent with conformational restriction about the dichalcogen bond.
Organoselenium compounds play a central role in biological systems and medicinal chemistry.1 The selenocysteine-containing enzyme glutathione peroxidase (GPx) catalyses the reduction of peroxides through oxidation of the endogenous thiol glutathione to glutathione disulfide.2 The build-up of reactive oxygen species such as peroxides is associated with certain disease states, and hence small selenium-containing molecules which can mimic the function of GPx have potential in drug development.1,2 A wide range of GPx mimics containing diverse selenium functionality has been investigated, with the aminoselenide Ebselen 1 reaching phase 3 clinical trials for a variety of diseases associated with oxidative stress (Fig. 1).2,3
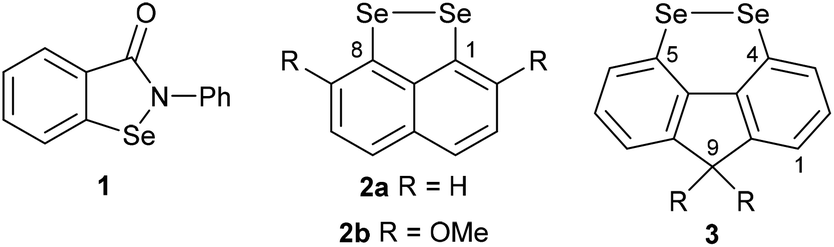 | ||
| Fig. 1 Selenium-containing GPx mimics: Ebselen 1, Back's conformationally-restricted 1,8-peri-substituted naphthalene diselenides 2 and proposed conformationally-restricted 4,5-disubstituted fluorene diselenides 3 in this study. | ||
Diselenides are promising GPx mimics,4–7 with even the simple diphenyl diselenide showing two times greater activity than Ebselen.6 In 2011, Back reported that 1,8-, peri-substituted, naphthalene diselenides 2 show an order of magnitude greater GPx-like activity compared with diphenyl diselenide (Fig. 1).7 Restricting the conformation around the diselenide bond to almost planar, as found in 2, reduces the HOMO–LUMO energy gap and raises the energy of the HOMO compared with conformationally-unrestricted diphenyl diselenide, thereby increases the rate of oxidation of 2 by peroxide in the rate-determining step.
In a search for alternative conformationally-restricted aryl diselenides‡ that show enhanced GPx-like activity and which are amenable to structural variation towards medicinal chemistry applications, we considered the previously unreported 4,5-fluorene diselenides 3 (Fig. 1). As with peri-substituted 1,8-naphthalenes 2, the near planarity of fluorene8 should constrain the geometry of the diselenide bond, and the close proximity of groups in the 4,5-(bay) region should favour dichalcogen bond formation.9 In this paper we report the first investigation into the synthesis and properties of 3, its mono- and trioxides, the corresponding ditelluride and their GPx-like activity.
Fluorene diselenides 3a (R = Me) and 3b (R = Bu) were synthesized from fluorenes 4a and 4b, through quenching the 4,5-dilithiofluorene species, generated using BuLi-TMEDA,9 with elemental selenium (Scheme 1). Diselenide oxidation was investigated, in view of selenium oxides showing potential GPx-like activity, and to compare their behaviour with the analogous 5-membered ring naphthalene bis-selenium species reported by Kice (2a)10 and Back (2b).7 Oxidation with 1.2 equivalents of mCPBA in Et2O gave selenolseleninates 5a and 5b along with recovered starting material. We did not see any evidence of formation of the symmetrical selenenic anhydride in these mono-oxidations, in contrast to the oxidation of 2a, where a mixture of isomeric monoxides is observed.10 Use of a larger excess (3.5 equivalents) of mCPBA resulted in the precipitation of seleninic anhydrides 6 as single stereoisomers in excellent 85–95% yields. These were assigned as the trans, C2-symmetric stereoisomers, rather than the alternative cis, meso structures, on the basis of the equivalent Me groups at C-9 in the 1H and 13C NMR of 6a. Treatment of 6a with KOH in CD3OD formed the dipotassium salt of the bis-seleninic acid 7a, evidenced by 77Se NMR, which upon acidification returned the same stereoisomer 6a in 86% yield, suggesting the trans-isomer is thermodynamic preferred. In contrast, naphthalenes 2 give mixtures of diastereomeric seleninic anhydrides in both selenium oxidation and in base-mediated ring-opening – acidification.7,10 The ditellurides 8a and 8b were also prepared from fluorenes 4a and 4b using tellurium as the quench for the dilithio species.
 | ||
| Scheme 1 Synthesis of 4,5-disubstituted fluorene diselenides 3, selenolseleninates 5, seleninic anhydrides 6 and ditellurides 8, and structures of dipotassium salt of bis-seleninic acid 7a and related biaryl diselenide 9. Reagents and conditions: (i) n-BuLi (4 equiv.), TMEDA (4 equiv.), 60 °C, 4 h, then Se or Te (8 equiv.), THF, −78 °C–rt. (ii) mCPBA (1.2 equiv.), Et2O, 15 min. (iii) mCPBA (3.5 equiv.), Et2O, 15 min. | ||
The X-ray crystal structure of 3a is shown in Fig. 2 and that of 3b (four independent molecules in the unit cell) in the ESI (Fig S1–S5†).11 In contrast to the essentially planar naphthalene diselenides 2a and 2b (C–Se–Se–C dihedral angle 2a: −2.28(13),122b: −1.50(7)13), diselenides 3 are non-planar (C4–Se1–Se2–C5 dihedral angle 3a −41.35(12); 3b average 42.3(20)§) and cause a twist in the fluorene plane (C4–C11–C12–C5 dihedral angle 3a −10.9(4); 3b average 11.3(15)§).14 This C–Se–Se–C dihedral angle is still much smaller than in the conformationally unconstrained diphenyl diselenide (85.4(2), −85.5(3))15 and in the less constrained biaryl diselenide, dibenzo[c,e][1,2]diselenine (9, Scheme 1) (59.0(3), −59.0(4), −57.0(4)).16 The Se–Se bond length in 3b is 2.34416(4) Å, shorter than naphthalene diselenides 2a (2.3639(5) Å) and 2b (2.3552(3) Å), but longer than in diphenyl diselenide (2.3066(7) Å; 2.3073(10) Å) and 9 (mean length 2.323(2) Å).
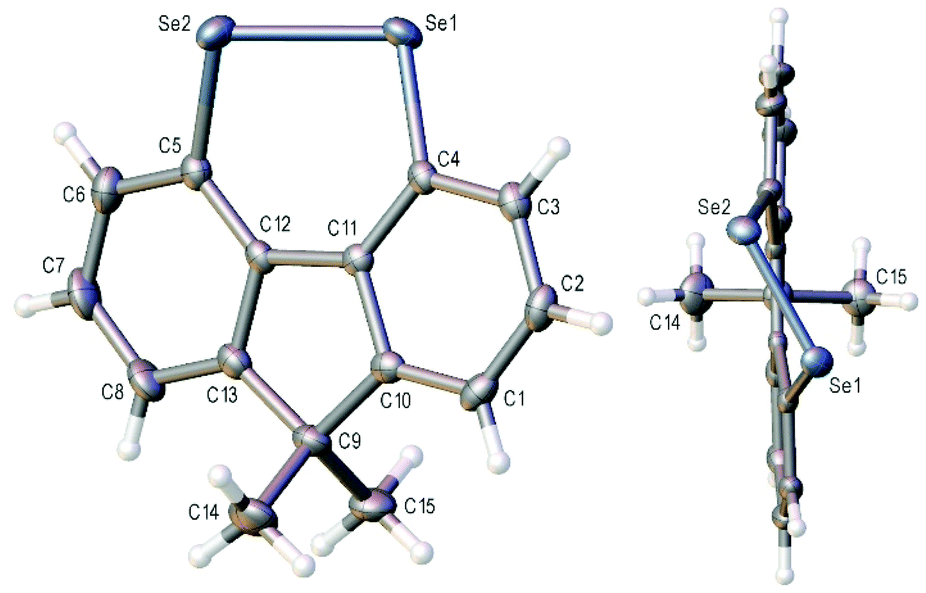 | ||
| Fig. 2 Two views of the crystal structure of diselenide 3a with ellipsoids drawn at the 50% probability level. | ||
The GPx-like catalytic activities of diselenides 3, selenolseleninates 5, seleninic anhydrides 6 and ditellurides 8 were determined using Iwoka's NMR assay,17 which monitors the drop in concentration of dithiotheritol (DTTred) as it is oxidized to the disulfide DTTox over time (Fig. 3). A solvent system of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CD3OD:CDCl3 was used to maintain solubility of all components and hence compare catalytic activity under homogenous conditions, although rates in this solvent system are much slower than in the original report of D2O.18 The times taken for the initial concentration of DTTred to halve (T50), after addition of H2O2 are shown in Table 1. T50 allows catalysts to be compared where there is a rapid initial reaction, as is the case herein for selenolseleninates and seleninic anhydrides, prior to addition of H2O2. Back's naphthalene diselenide 2b, wherein the electron-donating ortho-OMe groups were shown to increase catalytic activity over the non-substituted 2a, was also included, along with a background reaction (no catalyst).
:1 CD3OD:CDCl3 was used to maintain solubility of all components and hence compare catalytic activity under homogenous conditions, although rates in this solvent system are much slower than in the original report of D2O.18 The times taken for the initial concentration of DTTred to halve (T50), after addition of H2O2 are shown in Table 1. T50 allows catalysts to be compared where there is a rapid initial reaction, as is the case herein for selenolseleninates and seleninic anhydrides, prior to addition of H2O2. Back's naphthalene diselenide 2b, wherein the electron-donating ortho-OMe groups were shown to increase catalytic activity over the non-substituted 2a, was also included, along with a background reaction (no catalyst).
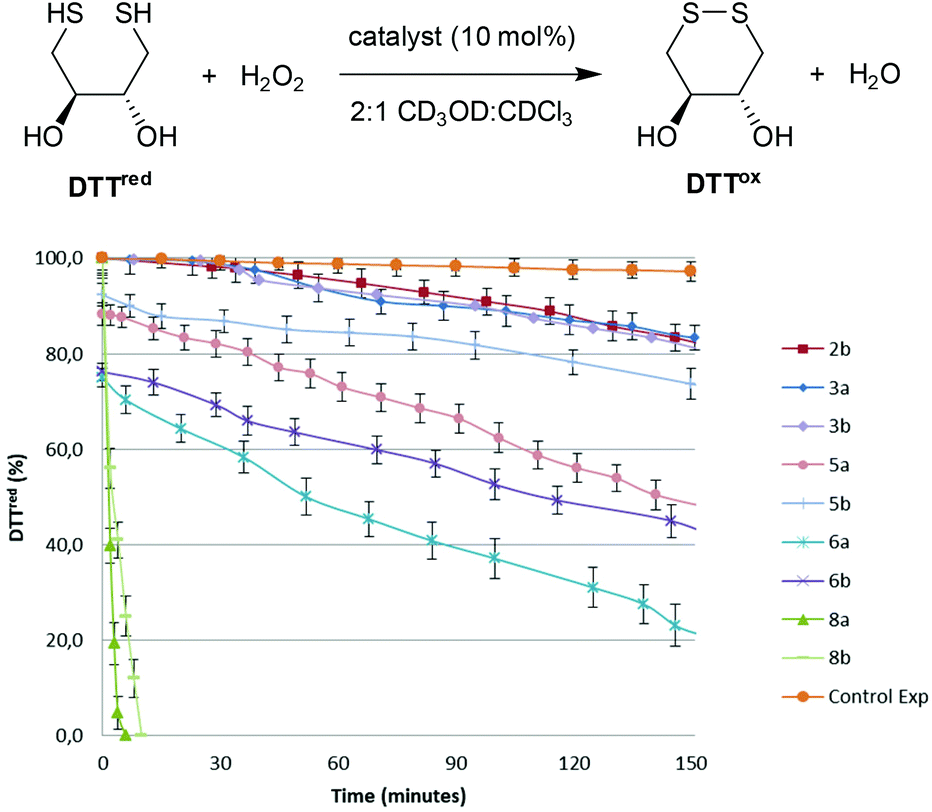 | ||
| Fig. 3 Oxidation of DTTred with H2O2 in the presence of selenium- or tellurium-containing catalysts (10 mol%). Reaction conditions: [DTTred]0 = 0.14 M, [H2O2]0 = 0.14 M, [catalyst] = 0.014 M, 2:1 CD3OD/CDCl3 solution (0.6 mL). Reaction progress monitored by 1H NMR. The mean (±) SD values of three separate experiments are reported. | ||
| Entry | Catalyst | Initial DDTreda (%) |
T
50
(min) |
|---|---|---|---|
| a After addition of 10 mol% catalyst before addition of H2O2. b T 50 is the time required to halve the initial thiol concentration after the addition of H2O2. c Data in parenthesis are the experimental error. | |||
| 1 | 2b | 100 | >300 |
| 2 | 3a | 100 | >300 |
| 3 | 3b | 100 | >300 |
| 4 | 5a | 88 | 141 (±9)c |
| 5 | 5b | 92 | 253 (±17) |
| 6 | 6a | 75 | 52 (±8) |
| 7 | 6b | 75 | 105 (±11) |
| 8 | 8a | 100 | <3 |
| 9 | 8b | 100 | <3 |
All of the selenium- and tellurium-containing compounds 3, 5, 6 and 8 catalyse the oxidation of DTTred to DTTox. Diselenides 3 have comparable activities to the naphthalene diselenide 2b in this assay, despite lacking activating ortho-OMe substituents (Fig. 1 and Table 1, entries 1–3). The selenolseleninates 5 have shorter T50 than the corresponding diselenides 3 (Table 1, entries 4 and 5). Before adding H2O2, approximately 10% of DTTox was detected, pointing to an initial fast reaction that occurs prior to the first NMR reading under these homogenous conditions. A more extensive initial reaction occurs with trioxides 6, with approx. 25% DTTox detected, contributing to the overall shorter T50 (entries 6 and 7). In general, 9,9-dimethyl-substituted fluorenes catalyse the oxidation of DTTred faster than the butyl-substituted systems (compare entries 4 vs. 5, and entries 6 vs. 7). Oxidation using ditellurides 8a and 8b is two orders of magnitude faster that the corresponding diselenides 3a and 3b (entries 8 and 9), with reactions complete within minutes of adding H2O2.19
In order to gain further mechanistic insight into the catalytic cycle, stoichiometric reactions of selenium-containing catalysts were carried out (Scheme 2). Treatment of diselenide 3a with a large excess (10 equiv.) of H2O2 in 2:1 MeOH:CH2Cl2 at room temperature gave slow oxidation to monoxide 5a (Scheme 3, eqn (1)). No higher oxides were detected, and independent treatment of selenolseleninate 5a or seleninic anhydride 6a with H2O2 under these conditions gave no reaction, suggesting 6a is not an intermediate in the catalytic cycle.20
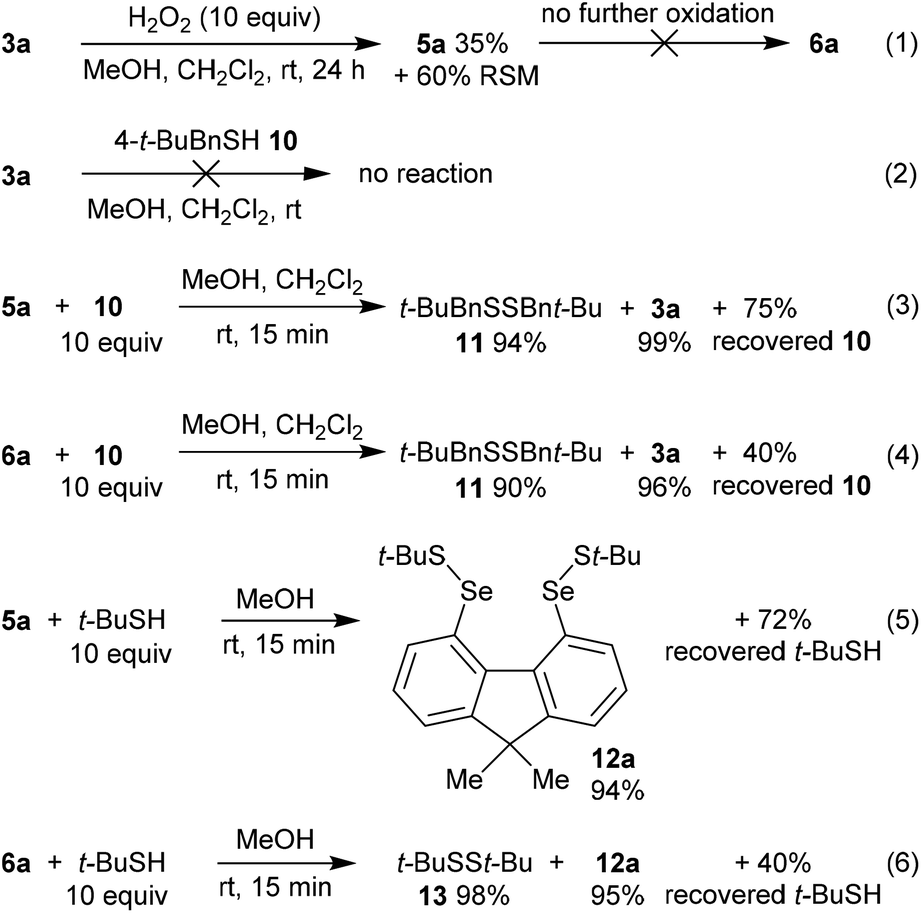 | ||
| Scheme 2 Mechanistic investigations. % yields of recovered thiol are based on theoretical consumption. | ||
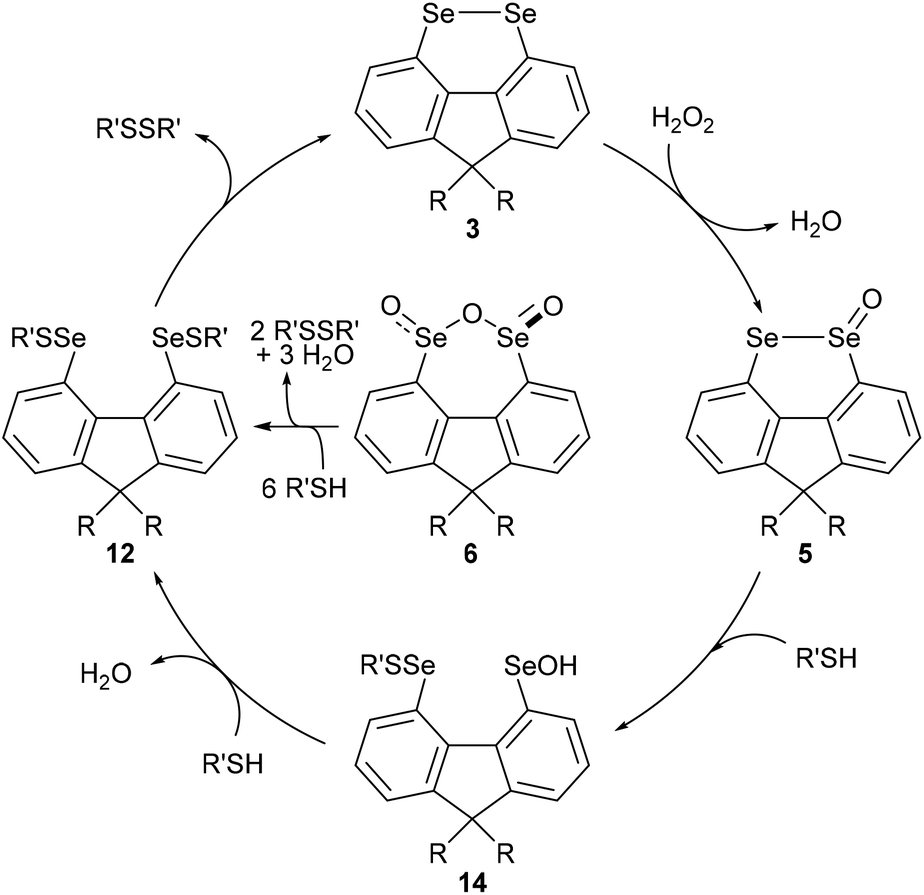 | ||
| Scheme 3 Proposed catalytic cycle for oxidation of thiols to disulfides. | ||
Diselenide 3a does not react with (4-(tert-butyl)phenyl)methanethiol (10)21 in CH2Cl2/MeOH at room temperature (Scheme 2, eqn (2)). However reaction of selenolseleninate 5a with 10 equivalents of 10 gave an essentially instantaneous and quantitative transformation to diselenide 3a and disulfide 11 along with 75% recovered thiol 10, (eqn (3)). Under the same conditions, seleninic anhydride 6a underwent a similarly rapid and high-yielding transformation to 3a and 11 (eqn (4)), where the 90% yield of 11 is based on theoretical consumption of 6 molar equivalents of thiol 10 (stoichiometries in the reactions of 5a and 6a with thiols are shown in the ESI, Scheme S1†).
No intermediate bis-selenium species were observed in the reactions of 5a and 6a with thiol 10. However, reaction of 5a with the bulkier thiol, t-BuSH, gave the bis-selenenyl sulfide 12a (eqn (5)), a potential intermediate in the formation of 3a. Indeed, isolated 12a is slowly transformed over 24 h in solution to diselenide 3a and di-tert-butyl disulfide (13). This rate of this reaction is not changed by addition of 3 equivalents of thiol 10, and no disulfides derived from 10 were formed, only 13. The breakdown of bis-selenenyl sulfide 12a to diselenide 3a and disulfide 13 is thus presumably intramolecular, but given the steric hindrance provided by the t-Bu group, care should be taken in extrapolating these observations to all thiols. Kice reported a similar reaction of t-BuSH with the monoxides of naphthalene diselenide 2a to give isolable 1,8-bis[(tert-butylthio)seleno]naphthalene,10 which led Back to propose bis-selenenyl sulfides as intermediates in the catalytic cycle of 2b.7
The reaction of seleninic anhydride 6a with t-BuSH also gave bis-selenenyl sulfide 12a (eqn (6)), though clearly there are multiple potential intermediates preceding its formation. These intermediates account for the formation of disulfide 13 (98% based on theoretical amount of t-BuSH consumed and consistent with recovery of 4 equivalents of thiol, Scheme S1†), not observed in the reaction of selenolseleninate 5a with t-BuSH.¶
Based on the above observations, a catalytic cycle directly analogous to that proposed by Back for naphthalene diselenides 2 is suggested7 (Scheme 3): this cycle is mechanistically distinct from catalysis by other diselenides, which involve initial Se–Se bond cleavage by reaction with thiols.22 The rate-determining step is the oxidation of diselenide 3 to selenolseleninate 5, which in turn rapidly consumes two equivalents of thiol and forms disulfides via the intermediates 14 (not observed) and 12 (observed as 12a for R = Me, R′ = t-Bu). As noted above, the conversion of 12 to 3 may occur by more than one mechanism and may also be catalysed by thiol: this step is severely slowed in the case of R′ = t-Bu where nucleophilic attack at sulfur is restricted and where an intramolecular mechanism appears most likely. Oxides 5 and 6 initially circumvent the rate-determining oxidation, resulting in overall shorter T50 in the DDT NMR assay. The initial rapid reaction of 5 and 6 with DTTred is evident in Fig. 3. Consumption of 1 equivalent of dithiol DTTred (2 × SH) with the 10 mol% of catalysts 5a and 5b present at the start of the assay should lead to an immediate 10% reduction in the amount of DTTred, which is consistent with the approx. 10% observed initial DTTred (Table 1, entries 4 and 5). Similarly, rapid consumption of 3 equivalents of DDTred (6 × SH) with the starting 10 mol% of catalysts 5a and 5b should give a theoretical 30% reduction in the amount of DDTred, with approx. 25% reduction observed in practice (entries 6 and 7).
Conclusions
In conclusion, readily synthesized, bay-substituted 4,5-fluorene diselenides 3 possess properties analogous to peri-substituted 1,8-naphthalenes 2, including increased GPx-like activity compared with non-conformationally constrained diselenides. Despite a greater twist in the diselenide bond, the catalytic activity of fluorenes 3a and 3b is similar to that of naphthalene 2b in a homogenous DDT redox assay, without the need for additional activation by ortho-OMe groups on the aromatic rings. Moving forward, the fluorene scaffold is particularly amenable to structural variation through incorporation of different functionality at C-9, for example towards water-soluble GPx mimics,18,23 and the close proximity of groups in the 4,5-bay positions may be exploited in other applications based on 1,8-peri-substituted naphthalenes.24,25Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank EPSRC (EP/K039245/1) for funding.Notes and references
- Reviews: (a) C. W. Nogueira, N. V. Barbosa and J. B. T. Rocha, Arch. Toxicol., 2021, 95, 1179–1226 CrossRef CAS PubMed; (b) D. Radomska, R. Czarnomysy, D. Radomski and K. Bielawski, Int. J. Mol. Sci., 2021, 22, 1009 CrossRef CAS PubMed; (c) Z. Chen, H. Lai, L. Hou and T. Chen, Chem. Commun., 2020, 56, 179–196 RSC.
- (a) N. V. Barbosa, C. W. Nogueira, P. A. Nogara, A. F. de Bem, M. Aschner and J. B. T. Rocha, Metallomics, 2017, 9, 1703–1734 CrossRef CAS PubMed; (b) L. Orian and S. Toppo, Free Radicals Biol. Med., 2014, 66, 65–74 CrossRef CAS PubMed.
- Reviews: (a) K. N. Sands, T. A. Tuck and T. G. Back, Chem. – Eur. J., 2018, 24, 9714–9728 CrossRef CAS PubMed; (b) G. Mugesh and H. B. Singh, Chem. Soc. Rev., 2000, 29, 347–357 RSC.
- Reviews: (a) M. D. Tiezza, G. Ribaudo and L. Orian, Curr. Org. Chem., 2019, 23, 1381–1402 CrossRef CAS; (b) M. Álvarez-Pérez, W. Ali, M. A. Marć, J. Handzlik and E. Domínguez-Álvarez, Molecules, 2018, 23, 628 CrossRef PubMed.
- Recent representative examples: (a) K. Arai, Y. Sato, I. Nakajima, M. Saito, M. Sasaki, A. Kanamori and M. Iwaoka, Bioorg. Med. Chem., 2021, 29, 115866 CrossRef CAS PubMed; (b) D. Bhowmick and G. Mugesh, Org. Biomol. Chem., 2015, 13, 9072–9082 RSC; (c) V. P. Singh, J.-f. Poon, R. J. Butcher, X. Lu, G. Mestres, M. K. Ott and L. Engman, J. Org. Chem., 2015, 80, 7385–7395 CrossRef CAS PubMed; (d) P. Prabhu, B. G. Singh, M. Noguchi, P. P. Phadnis, V. K. Jain, M. Iwaoka and K. I. Priyadarsini, Org. Biomol. Chem., 2014, 12, 2404–2412 RSC.
- S. R. Wilson, P. A. Zucker, R.-R. C. Huang and A. Spector, J. Am. Chem. Soc., 1989, 111, 5936–5939 CrossRef CAS.
- D. J. Press and T. G. Back, Org. Lett., 2011, 13, 4104–4107 CrossRef CAS PubMed.
- L. A. Chetkina and V. K. Belsky, Crystallogr. Rep., 2013, 58, 26–48 CrossRef CAS.
- V. B. Bonifácio, J. Morgado and U. Scherf, Synlett, 2010, 1333–1336 CrossRef.
- J. L. Kice, Y.-H. Kang and M. B. Manek, J. Org. Chem., 1988, 53, 2435–2439 CrossRef CAS.
- CCDC 2099301 (3a) and 1470801 (3b)† contain the supplementary crystallographic data for this paper.
- S. M. Aucott, H. L. Milton, S. D. Robertson, A. M. Z. Slawin and J. D. Woollins, Heteroat. Chem., 2004, 15, 530–542 CrossRef CAS.
- C. Figliola, L. Male, P. N. Horton, M. B. Pitak, S. J. Coles, S. L. Horswell and R. S. Grainger, Organometallics, 2014, 33, 4449–4460 CrossRef CAS.
- A. L. Fuller, L. A. S. Scott-Hayward, Y. Li, M. Bühl, A. M. Z. Slawin and J. D. Woollins, J. Am. Chem. Soc., 2010, 132, 5799–5802 CrossRef CAS PubMed.
- We have observed a similar twist in a 4,5-carbazole diselenide: I. A. Pocock, A. M. Alotaibi, K. Jagdev, C. Prior, G. R. Burgess, L. Male and R. S. Grainger, Chem. Commun., 2021, 57, 7252–7255 RSC.
- M. R. Bryce, A. Chesney, A. K. Lay, A. S. Batsanov and J. A. K. Howard, J. Chem. Soc., Perkin Trans. 1, 1996, 2451–2459 RSC.
- K. Kumakura, B. Mishra, K. I. Priyadarsini and M. Iwaoka, Eur. J. Org. Chem., 2010, 440–445 CrossRef.
- The DDT assay has been used in CD3OD: K. Arai, A. Tashiro, Y. Osaka and M. Iwaoka, Molecules, 2017, 22, 354 CrossRef PubMed.
- For higher GPx-like activity of Te derivatives vs. Se derivatives, see (a) Ref. 7; ; (b) D. Tanini, L. Ricci and A. Capperucci, Adv. Synth. Catal., 2020, 362, 1323–1332 CrossRef CAS and references therein.
- For investigation of seleninic anhydrides as GPx mimics see: S.-C. Yu, A. Borchert, H. Kuhn and I. Ivanov, Chem. – Eur. J., 2008, 14, 7066–7071 CrossRef CAS PubMed.
- Chosen as a “non-odourous” alkyl thiol mimic of glutathione: M. Node, K. Kumar, K. Nishide, S.-i. Ohsugi and T. Miyamoto, Tetrahedron Lett., 2001, 42, 9207–9210 CrossRef CAS.
- D. Bhowmick and G. Mugesh, Org. Biomol. Chem., 2015, 13, 10262–10272 RSC.
- N. M. R. McNeil, D. J. Press, D. M. Mayder, P. Garnica, L. M. Doyle and T. G. Back, J. Org. Chem., 2016, 81, 7884–7897 CrossRef CAS PubMed.
- Reviews of peri-substituted dichalcogenides: (a) P. Kilian, F. R. Knight and J. D. Woollins, Coord. Chem. Rev., 2011, 255, 1387–1413 CrossRef CAS; (b) P. Kilian, F. R. Knight and J. D. Woollins, Chem. – Eur. J., 2011, 17, 2302–2328 CrossRef CAS PubMed. For application in other enzyme mimics see: (c) Ref. 13; ; (d) C. Figliola, L. Male, S. L. Horswell and R. S. Grainger, Eur. J. Inorg. Chem., 2015, 3146–3156 CrossRef CAS; (e) S. Mondal, D. Manna, K. Raja and G. Mugesh, ChemBioChem, 2020, 21, 911–923 CrossRef CAS PubMed. For additional applications from our group see: (f) R. S. Grainger, B. Patel and B. M. Kariuki, Angew. Chem., Int. Ed., 2009, 48, 4832–4835 CrossRef CAS PubMed; (g) R. S. Grainger, B. Patel, B. M. Kariuki, L. Male and N. Spencer, J. Am. Chem. Soc., 2011, 133, 5843–5852 CrossRef CAS PubMed; (h) B. Patel, J. Carlisle, S. E. Bottle, G. R. Hanson, B. M. Kariuki, L. Male, J. C. McMurtrie, N. Spencer and R. S. Grainger, Org. Biomol. Chem., 2011, 9, 2336–2344 RSC.
- For increased proton sponge basicity in 4,5-disubstituted fluorene vs. 1,8-naphthalene see: H. A. Staab, T. Saupe and C. Krieger, Angew. Chem., Int. Ed. Engl., 1983, 22, 731–732 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and analytical data, copies of NMR spectra and X-ray crystallography for 3a (CCDC 2099301) and 3b (CCDC 1470801). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ob02153b |
| ‡ Aryl selenides are less toxic than alkyl selenides. See ref. 3a. |
| § For 3b the average value calculated from molecules 1–3 for each parameter is given (see ESI†). |
| ¶ Ph3CSH and 1-adamantylthiol gave the corresponding disulfide and diselenide 3a directly from 6a (see ESI†). |
| This journal is © The Royal Society of Chemistry 2021 |