
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of 5,7-diarylindoles via Suzuki–Miyaura coupling in water†
Vijayaragavan
Elumalai
 and
Jørn H.
Hansen
*
and
Jørn H.
Hansen
*
Chemical Synthesis and Analysis Division, Department of Chemistry, UiT The Arctic University of Norway, Hansine Hansens veg 54, 9037 Tromsø, Norway. E-mail: jorn.h.hansen@uit.no
First published on 16th November 2021
Abstract
The synthesis of novel 5,7-diaryl and diheteroaryl indoles has been explored via efficient double Suzuki–Miyaura coupling. The method notably employs a low catalyst loading of Pd(PPh3)4 (1.5 mol%/coupling) and water as the reaction solvent to obtain 5,7-diarylated indoles without using N-protecting groups in up to 91% yield. The approach is also suitable for N-protected and 3-substituted indoles and constitutes an important green and convenient arylation strategy for the benzenoid ring of indoles. The synthesized diarylindoles are fluorescent.
Introduction
Indole is an essential nitrogen-containing heterocycle and a central scaffold in several natural products and other bioactive molecules.1,2 Consequently, a range of synthetic methods and strategies have been described for the functionalization of indoles.3 C2- and C3-functionalization of the pyrrole ring has been thoroughly investigated, and this has been extensively reviewed by Sandtorv.3a The innate high reactivity of the pyrrole ring has left functionalization on the benzenoid ring positions (C4–7) a challenging task.4 Frost and co-workers5 reported in 2017 a detailed review of site-selective functionalization on the benzenoid ring. In addition, a complete review of C4-indole functionalization has been recently assembled by Ackermann and Pilarski in 2018.6 There is clearly a need for selective functionalization methods of the benzenoid ring in order to access sparsely explored parts of indole chemical space.Diarylindoles are important precursors for the synthesis of biologically active compounds, pharmaceuticals and organic materials. 2,3-Diarylindoles have several important biological activities7 and, thus, a range of methodologies have been described for the synthesis of such structures.8 Other substitution patterns than 2,3-diaryl have not been extensively reported. 5,7-Bis(4-methoxyphenyl)indole has been previously described and was generated via a Kumada-coupling using 5,7-dibromoindole as the precursor (Scheme 1).9 This approach has very limited scope, requires strongly basic and otherwise very specific reaction conditions (e.g. ether solvents). Poly(5,7-bis(2-thiophene)-indole), a polymer used in electrochromic devices, is synthesized from 5,7-bis(2-thiophene)-indole (BTI).10a Diarylindoles often exhibit fluorescent properties and are, arguably, useful building blocks in organic materials chemistry.11 The potential for applications of novel diarylindoles both as bioactives and in materials chemistry, combined with the limited number of studies of these compounds, particularly in the 5,7-disubstituted series, became the impetus for developing a practical approach to the synthesis of novel compounds in this area of chemical space.
 | ||
| Scheme 1 Comparison of previous approaches and current work. | ||
Synthetic approaches via transition metal-catalysed reactions are not abundant for double arylation and typically require N-protecting groups.8,9 Several examples of multiple Suzuki–Miyaura couplings on indoles and related heterocycles exist,10b–e however, none in which 5,7-diarylindoles were generated. One approach was found involving a Ru-catalyzed C–H activation (Scheme 1),8c however, the scope was very narrow and this approach required a carboxylic acid directing group in the 6-position and prolonged reaction time at high temperature in order to generate the 5,7-diarylindoles. In this paper, we demonstrate a straightforward, protecting-group free and green approach to 5,7-diarylindoles employing a highly efficient Suzuki–Miyaura coupling in water to access a range of novel 5,7-disubstituted indoles.
Results and discussion
To obtain 5,7-diarylindoles we envisioned starting from commercially available 5,7-dibromoindole (1) which could undergo double cross-coupling with readily available arylboronic acids. Initially, classical conditions for the Suzuki–Miyaura coupling were explored, including palladium tetrakistriphenylphosphine as catalyst, DMF/water as solvent, sodium carbonate as base and tert-butylammonium iodide as an additive (Table 1).12 The studies of coupling 1 with phenylboronic acid revealed that full conversion and excellent yield (95%) could be obtained at 120 °C with 3 equivalents of boronic acid present. Moreover, the reaction was completed efficiently in a microwave reactor after one hour employing only 3 mol% of palladium catalyst, effectively 1.5 mol% per functionalization event. A similar result was found for reaction under thermal heating in a sealed tube, but the microwave reaction was more convenient to conduct. Furthermore, the study also showed that protection of the indole nitrogen is not necessary in this chemistry. The double functionalization worked effectively in different solvent systems such as methanol, methanol/water, acetone/water, THF/water and ethanol/water (full conversion). The reaction in pure methanol without adding TBAI was just as effective as the binary systems, suggesting that this additive was not necessary in the polar solvent system and that binary solvent mixtures were not necessary either to achieve an efficient transformation. These observations led to testing pure water (without TBAI) as the solvent, which yielded essentially full conversion and 83% isolated yield of the 5,7-diphenylindole 3. Another interesting observation from the survey arose from monitoring the formation of mono-coupled products (4 + 5 combined). When using acetonitrile as solvent, it appeared that mono-coupling was the favoured product (4.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio). A similar selectivity was observed in THF and suggested the possibility of a solvent-controlled mono-coupling, a phenomenon that has not been studied systematically to our knowledge. To investigate this, a survey of solvents and catalysts was conducted in which only 1.5 equivalents of boronic acid was employed, and otherwise keeping the reaction conditions the same as before (Table 2). It was found that 4 (5-bromo-7-phenylindole) is the predominant species when the conversion is below ca. 50% in acetonitrile or THF. All three species 3–5 are detectable at higher conversions and only the doubly coupled product 3 is detectable when using water or methanol as solvents. 30% isolated yield of 4 was obtained at 54% conversion of 1 with negligible amounts of 5 and 3 present. Although not a great synthetic result, it is interesting and may be one of few direct routes to this disubstituted indole. In summary, our survey of the reaction variables led to a set of green and mild conditions for double Suzuki–Miyaura couplings of 5,7-dibromoindole 1 including low catalyst loading and water as the reaction solvent, but also demonstrated that a variety of solvent choices are available as long as a protic co-solvent is also added. This feature is useful in cases where the solubility of reaction components needs to be modified.
:1 ratio). A similar selectivity was observed in THF and suggested the possibility of a solvent-controlled mono-coupling, a phenomenon that has not been studied systematically to our knowledge. To investigate this, a survey of solvents and catalysts was conducted in which only 1.5 equivalents of boronic acid was employed, and otherwise keeping the reaction conditions the same as before (Table 2). It was found that 4 (5-bromo-7-phenylindole) is the predominant species when the conversion is below ca. 50% in acetonitrile or THF. All three species 3–5 are detectable at higher conversions and only the doubly coupled product 3 is detectable when using water or methanol as solvents. 30% isolated yield of 4 was obtained at 54% conversion of 1 with negligible amounts of 5 and 3 present. Although not a great synthetic result, it is interesting and may be one of few direct routes to this disubstituted indole. In summary, our survey of the reaction variables led to a set of green and mild conditions for double Suzuki–Miyaura couplings of 5,7-dibromoindole 1 including low catalyst loading and water as the reaction solvent, but also demonstrated that a variety of solvent choices are available as long as a protic co-solvent is also added. This feature is useful in cases where the solubility of reaction components needs to be modified.
|
|
|||||
|---|---|---|---|---|---|
| No. | Solvent | Deviation from conditions | Conversion (GC) | ||
| 1 (%) | 3 , (%) | 4 + 5b (%) | |||
|
a General procedure: In a microwave vial, 5,7-dibromoindole 1 (1.0 mmol), phenylboronic acid 2 (3.0 mmol), Na2CO3 (2.0 mmol) and Pd(PPh3)4 (0.03 mmol, 3 mol%) were added. The vial was sealed, flushed with a stream of dry nitrogen and added the solvent (2.5 mL) or solvent/H2O (4:1 ratio, 2.5 mL) via the septum. The vial was sonicated for 30 s and heated under microwave irradiation for 1 hour at 120 °C.
b Conversion based on GC. nd = not detected.
c Isolated yield.
|
|||||
| 1 | DMF/H2O | TBAI (10 mol%) | >99 | 95 (80)c | nd |
| 2 | DMF | — | 65 | 15 | 50 |
| 3 | DMF/H2O | 150 °C, 10% Pd/S-Phos | >99 | >99 | nd |
| 4 | MeOH/H2O | TBAI (10 mol%) | >99 | >99 | nd |
| 5 | MeOH | — | >99 | >99 | nd |
| 6 | Acetone/H2O | TBAI (10 mol%) | >99 | >99 | nd |
| 7 | Acetone | — | 71 | 10 | 61 |
| 8 | MeCN/H2O | TBAI (10 mol%) | 93 | 46 | 47 |
| 9 | MeCN | — | 81 | 15 | 66 |
| 10 | THF/H2O | TBAI (10 mol%) | >99 | >99 | nd |
| 11 | THF | — | 76 | 15 | 61 |
| 12 | EtOH/H2O | TBAI (10 mol%) | 98 | 98 | nd |
| 13 | H 2 O | — | 96 | 96 (83) | nd |

|
|
||||||
|---|---|---|---|---|---|---|
| No. | Solvent | Pd-Catalyst | Conversion (GC) | |||
| 1 (%) | 4 , (%) | 5 (%) | 3 (%) | |||
|
a General procedure: In a microwave vial, 5,7-dibromoindole 1 (1 mmol), phenylboronic acid 2 (1.5 mmol), Na2CO3 (2 mmol), and Pd-catalyst (0.03 mmol, 3 mol%) were added. The vial was sealed and flushed with stream of dry nitrogen and added the solvent (2.5 ml)/solvent/H2O (4:1 ratio, 2.5 ml) via the septum. The vial was sonicated for 30 s and heated under microwave irradiation for 1.5 hours at 120 °C.
b Conversion based on GC.
c Isolated yield. nd-not detected.
d Ligand S-phos (6 mol%) was used.
e Reaction performed at ambient temperature.
f 2 equivalents of boronic acid was used.
|
||||||
| 1 | MeCN | Pd(PPh 3 ) 4 | 54 | 53(30%) | nd | 1 |
| 2 | MeCN/H2O | Pd(PPh3)4 | 87 | 40 | 23 | 23 |
| 3 | H2O | Pd(PPh3)4 | >99 | nd | nd | 99 |
| 4 | MeOH/H2O | Pd(PPh3)4 | >99 | Traces | Traces | 99 |
| 5 | THF | Pd(PPh3)4 | 49 | 38 | 8 | 2 |
| 6 | MeCN | Pd(PPh3)2CI2 | 51 | 50 | nd | 1 |
| 7 | MeCN | Pd2(dba)3 | Traces | Traces | Traces | nd |
| 8d | MeCN | Pd(PPh3)4 | 41 | 35 | 4 | 1 |
| 9e | MeCN | Pd(PPh3)4 | Traces | Traces | Traces | nd |
| 10f | MeCN | Pd(PPh3)4 | 63 | 41 | 15 | 6 |
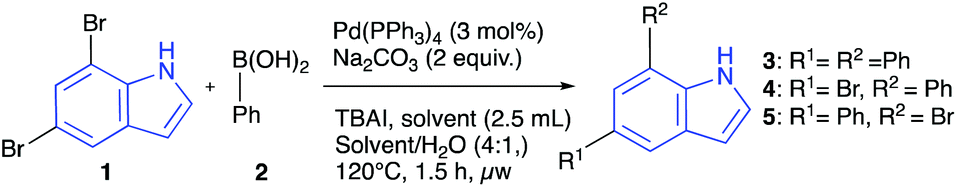
The scope of electron-rich arylboronic acids was studied first (Scheme 2 in blue). These are well-tolerated and afforded moderate to excellent yields in most cases. Some of the reactions were also conducted in DMF/water or EtOH/water for comparison, and these all yielded similar results as in pure water. In the para-substituted series, both ethyl (3c) and methoxy (3d) afforded good 62% and 76% isolated yields of 3c. Boc- and acetyl protected amino groups yielded 46% and 42% yields of 3j and 3k, respectively. It appears that the good π-donating methoxy group is generally increasing the yields, because also meta and ortho-methoxy substituted products 3i and 3h were formed in good yields of 70% and 85% respectively. The latter further shows that the reaction is relatively insensitive to steric clash with the ortho-substituent of the boronic acid. This notion was further challenged with ortho-methyl and ortho-hydroxy substituents affording 3b and 3f in 70% and 82% yields, respectively. The good yield of the latter further supports that good π-donation is an important factor to increase the yield. The introduction of substituents in both ortho-positions led to somewhat diminished yields. ortho-Dimethyl and dimethoxy patterns still afforded products 3l and 3e in 61% and 51% yields, respectively. Attempts to introduce mesityl groups did not result in any practically important product formation of 3g under these reaction conditions.
 | ||
| Scheme 2 Scope of arylboronic acids in the indole double arylation. a Reaction procedure: In a microwave vial, 5,7-dibromoindole 1 (1 mmol), arylboronic acid 2 (3 mmol), Na2CO3 (2 mmol), and Pd(PPh3)4 (0.03 mmol, 3 mol%) were added. The vial was sealed and flushed with stream of dry nitrogen and added H2O (2.5 ml) via the septum. The vial was sonicated for 30 s and heated under microwave irradiation for 1 hour at 120 °C. b Conversion based on GC/isolated yield. c Reaction was performed at 140 °C for 90 min. d In DMF/H2O 4:1. e Mixture (both mono and bis-coupling) of products were observed. f Reaction was performed in EtOH/H2O at 120 °C for 1 hour. | ||
In the series of electron-withdrawing groups, generally lower to moderate yields were observed. The para and ortho-chlorosubstituted products 3n and 3o were formed in 53% and 60% yields, respectively, under the standard reaction conditions. The remaining electron-withdrawing systems 3p–3t were formed in rather low 13–46% yields, thus demonstrating that such groups have a detrimental effect on the yield. Some improvement could be observed when increasing reaction times and temperatures. For example, the conversion was improved from 31% to 91% with an isolated yield of 72% of the para-cyano substituted product 3m, suggesting that high yields are also possible in this series. Overall, the examples demonstrate tolerance towards halides, carboxy-, cyano- and nitro-groups and that ortho, para and meta-substituent patterns can be used in this chemistry.
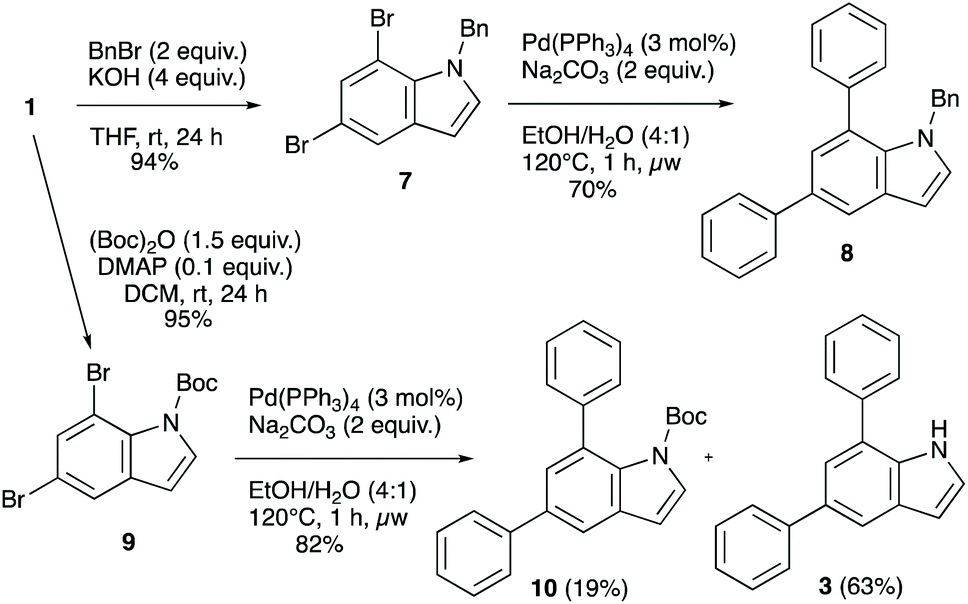
Another aspect of relevance to the synthetic utility of the double coupling approach would be compatibility with some commonly employed protecting groups at nitrogen. The chemistry is nicely compatible with a free N–H, which is a major strength, but protecting groups are sometimes desired in synthesis for various reasons. We therefore employed benzyl- and Boc-protected model indoles in order to demonstrate that these are also tolerated in the arylation chemistry (Scheme 3), and the benzyl-protected 5,7-dibromoindole 7 underwent a clean transformation to the diarylindole 8 in 70% isolated yield. The N-Boc protected system 9 also underwent double arylation with a very good overall yield of 82%, but was mostly deprotected under the reaction conditions (63% free NH product). This lability of the Boc-group under thermal conditions is not unexpected and may allow for simultaneous diarylation/deprotection if the conditions are optimized further. These examples demonstrate that the diarylation chemistry is compatible with N-protecting groups.
 | ||
| Scheme 3 Double arylation with some common N-protecting groups. | ||
Finally, we employed some heterocyclic boronic acids in this chemistry under the standard reaction conditions as well as 3-substituted indole 1a (Scheme 4). The system employing the indolyl boronic acid is particularly interesting, as it constitutes a synthesis of tris-indoles (Scheme 4A). The parent indole 1 afforded the tris-indole 5a in 52% yield. If a 3-formyl substituted indole was employed in the chemistry, the corresponding tris-indole 6a was formed in 77% yield. Employing the 3-formyl substituted indole further afforded 16–72% yields of 6b–d (Scheme 4B). Concerning heteroaromatic boronic acids, 4-pyridyl boronic acid afforded full conversion and 81% isolated yield of 5b (Scheme 4B). Surprisingly, the furanyl boronic acid only produced trace amounts of product, whereas 2-thiophenyl boronic acid afforded the expected product 5d in excellent chemical yield of 91%. In these reactions, some ethanol was required in the solvent mixture to improve the solubility of the boronic acids.
 | ||
| Scheme 4 Tris-indole formation and diarylation with 3-formylindoles and heteroarylboronic acids. a Reaction procedure: In a microwave vial, 5,7-dibromoindole-3-carbaldehyde 1 (1 mmol), arylboronic acid 2 (3 mmol), Na2CO3 (2 mmol) and Pd(PPh3)4 (0.03 mmol, 3 mol%) were added. The vial was sealed and flushed with stream of dry nitrogen and added EtOH/H2O (4:1 ratio, 2.5 ml) via the septum. The vial was sonicated for 30 s and heated under microwave irradiation for 1 hour at 120 °C. b Conversion based on GC/isolated yield. c Only traces of product was observed when H2O (100%) was used as a solvent. d Obtained an unseparable mixture of ca. 10% mono-functionalized indole and 6d. Yield of 6 was estimated from NMR-ratio of the two components in the isolated material. e In DMF/H2O 4:1. f 50% conversion was obtained in water as solvent. | ||
The 5,7-diarylindoles generated in this study are fluorescent. As such, they are of interest for organic materials science and as potential fluorescent probes in molecular imaging. Although the fluorescent properties of these compounds are not central to this study, we decided to visually ascertain to what extent the differences in structure of the introduced aryl substituents influenced the fluorescence. Solutions of five compounds (3n, 5d, 3m, 3b and 5a) in methanol were irradiated with a simple hand-held UV-lamp (365 nm) and a broad spectrum of fluorescence intensities are evident (Fig. 1). Notably, the novel compound 3m appears to be particularly bright, likely since it is a push–pull electronic system. A more detailed study of these properties will be disclosed in the near future.
 | ||
| Fig. 1 Fluorescence of 5,7-diarylindoles dissolved in MeOH and irradiated with a UV-lamp (365 nm). | ||
Conclusions
In conclusion, we have disclosed a practical and green Suzuki–Miyaura coupling approach for the synthesis of 5,7-diarylindoles in moderate to excellent yields. Some selectivity for mono-coupling at the 7-position can be achieved in acetonitrile and THF. The functionalization proceeds readily in about 1 hour with only 1.5 mol% catalyst loading per coupling in water or water/ethanol solvent mixtures under microwave heating conditions. We have also demonstrated its synthetic utility in the synthesis of tris-indoles and that the chemistry is compatible with some common protecting groups at nitrogen and a formyl group in the indole 3-position. This approach should be a preferred method for the synthesis of 5,7-diarylindoles. The 5,7-diarylindoles display interesting fluorescence properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding for this research project by the Research Council of Norway (grant no. 275043 CasCat).Notes and references
- For some examples of natural products containing the indole moiety, see: (a) N.-Y. Ji, X.-M. Li, L.-P. Ding and B.-G. Wang, Helv. Chim. Acta, 2007, 90, 385–391 CrossRef CAS; (b) S. R. Walker, E. J. Carter, B. C. Huff and J. C. Morris, Chem. Rev., 2009, 109, 3080–3098 CrossRef CAS PubMed; (c) R. J. Melander, M. L. Minvielle and C. Melander, Tetrahedron, 2014, 70, 6363–6372 CrossRef CAS PubMed.
- (a) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2000, 103, 893–930 CrossRef PubMed; (b) M. Z. Zhang, Q. Chen and G. F. Yang, Eur. J. Med. Chem., 2015, 89, 421–424 CrossRef CAS; (c) V. Sharma, P. Kumar and D. Pathak, J. Heterocycl. Chem., 2010, 47, 491–502 CAS; (d) S. Biswal, U. Sahoo, S. Sethy, H. K. S. Kumar and M. Banerjee, Asian J. Pharm. Clin. Res., 2012, 5, 1–6 CAS; (e) A. W. Woodward and B. Bartel, Ann. Bot., 2005, 95, 707–735 CrossRef CAS; (f) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS; (g) B. Trost and M. K. Brennan, Synthesis, 2009, 3003–3025 CrossRef CAS; (h) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489–4497 CrossRef CAS; (i) R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef.
- For a review, see: (a) A. H. Sandtorv, Adv. Synth. Catal., 2015, 357, 2403–2435 CrossRef. For key seminal publications, see: (b) X. Wang, B. S. Lane and D. Sames, J. Am. Chem. Soc., 2005, 127, 4996–4997 CrossRef; (c) D. R. Stuart, E. Villemure and K. Fagnou, J. Am. Chem. Soc., 2007, 129, 12072–12073 CrossRef; (d) N. P. Grimster, C. Guantlett, C. R. A. Godfrey and M. J. Gaunt, Angew. Chem., Int. Ed., 2005, 44, 3125–3129 CrossRef PubMed; (e) R. J. Phipps, N. P. Grimster and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 8172–8174 CrossRef PubMed; (f) N. R. Deprez, D. Kalyani, A. Krause and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 4972–4973 CrossRef CAS; (g) N. Lebrasseur and I. Larrosa, J. Am. Chem. Soc., 2008, 130, 2926–2927 CrossRef CAS PubMed; (h) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608–9644 CrossRef.
- (a) A. K. Pitts, F. O'Hara, R. H. Snell and M. J. Gaunt, Angew. Chem., Int. Ed., 2015, 54, 5451 CrossRef; (b) A. D. Yamaguchi, K. M. Chepiga, J. Yamaguchi, K. Itami and H. M. Davies, J. Am. Chem. Soc., 2015, 137, 644 CrossRef; (c) An approach using stoichiometric Pd is reported: J. Liang, W. Hu, P. Ta and Y. Jia, J. Org. Chem., 2013, 78, 5810 CrossRef PubMed; (d) P. Tao, J. Liang and Y. Jia, Eur. J. Org. Chem., 2014, 5735 CrossRef.
- J. A. Leitch, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 5618 CrossRef.
- J. Kalepu, P. Gandeepan, L. Ackermann and L. T. Pilarski, Chem. Sci., 2018, 9, 4203–4216 RSC.
- (a) J. Szmuszkovicz, E. M. Glenn, R. V. Heinzelman, J. B. Hester, Jr. and G. A. Youngdale, J. Med. Chem., 1966, 9, 527–536 CrossRef PubMed; (b) B. F. McAdam, F. Catella-Lawson, I. A. Mardini, S. Kapoor, J. A. Lawson and G. A. FitzGerald, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 272–277 CrossRef PubMed; (c) H. El-Diwani, S. S. Nakkady, O. H. Hishmat, O. A. El-Shabrawy and S. S. Mahmoud, Pharmazie, 1992, 47, 178–181 Search PubMed; (d) H. I. El-Diwani, N. A. M. M. Shmeiss and N. M. Saleh, Pol. J. Chem., 1995, 69, 470–475 Search PubMed; (e) N. A. M. M. Shmeiss, M. M. F. Ismail, H. I. El-Diwani, A. B. Hassan and S. A. Nada, Modell. Meas. Control, C, 1997, 55, 11–36 Search PubMed; (f) R. L. Jones, Y.-M. Qian, H. Wise, H. N. C. Wong, W.-L. Lam, H.-W. Chan, A. P. C. Yim and J. K. S. Ho, J. Cardiovasc. Pharmacol., 1997, 29, 525–535 CrossRef PubMed; (g) O. H. Hishmat, M. Y. Ebeid, S. S. Nakkady, M. M. Fathy and S. S. Mahmoud, Bull. Chim. Farm., 1999, 138, 259–266 CAS.
- (a) Y. Liu and G. W. Gribble, Tetrahedron Lett., 2000, 41, 8717–8721 CrossRef CAS; (b) M. F. Ibad, D. S. Zinad, M. Hussain, A. Ali, A. Villinger and P. Langer, Tetrahedron, 2013, 69, 7492–7504 CrossRef CAS; (c) M. Simonetti, D. M. Cannas, A. Panigrahi, S. Kujawa, M. Kryjewski, P. Xie and I. Larrosa, Chem. – Eur. J., 2017, 23, 549–553 CrossRef CAS PubMed.
- S. Ishikawa and K. Manabe, Tetrahedron, 2011, 67, 10156–10163 CrossRef CAS.
- (a) X. Yang, C. Liu, J. Guo, L. Wang and G. Nie, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2356–2364 CrossRef CAS; (b) J. Pang, S. Yuan, J. Qin, M. Wu, C. T. Lollar, J. Li, N. Huang, B. Li, P. Zhang and H.-C. Zhou, J. Am. Chem. Soc., 2018, 140, 12328–12332 CrossRef CAS PubMed; (c) S. T. Handy, T. Wilson and A. Muth, J. Org. Chem., 2007, 72, 8496–8500 CrossRef CAS PubMed; (d) S. C. Anderson and S. T. Handy, Synthesis, 2010, 2721–2724 CAS; (e) N. Bisballe, M. Hedidi, C. S. Demmer, F. Chevallier, T. Roisnel, V. Dorcet, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis, G. Bentabed-Ababsa, L. Bunch and F. Mongin, Eur. J. Org. Chem., 2018, 3904–3913 CrossRef CAS.
- M. Miyasaka, A. Fukushima, T. Satoh, K. Hirano and M. Miura, Chem. – Eur. J., 2009, 15, 3674–3677 CrossRef CAS PubMed.
- (a) N. Miyaura, T. Yanagi and A. Suzuki, Synth. Commun., 1981, 11, 513–519 CrossRef CAS; (b) P. R. Eastwood, Tetrahedron Lett., 2000, 41, 3705–3708 CrossRef CAS; (c) A. F. Littke, S. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020–4028 CrossRef CAS; (d) R. R. González, L. Liguori, A. Martinez Carrillo and H.-R. Bjørsvik, J. Org. Chem., 2005, 70, 9591–9594 CrossRef; (e) I. Hoffmann, B. Blumenröder, S. O. N. Thumann, S. Dommer and J. Schatz, Green Chem., 2015, 17, 3844 RSC; (f) V. Elumalai, A. H. Sandtorv and H.-R. Bjørsvik, Eur. J. Org. Chem., 2016, 1344–1354 CrossRef; (g) V. Elumalai and H.-R. Bjørsvik, ChemistrySelect, 2017, 2, 9387 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization and NMR-spectra. See DOI: 10.1039/d1ob02058g |
| This journal is © The Royal Society of Chemistry 2021 |