
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The dehydration of N-acetylglucosamine (GlcNAc) to enantiopure dihydroxyethyl acetamidofuran (Di-HAF)†
Cornelis H. M.
van der Loo
 a,
Mark L. G.
Borst
b,
Kees
Pouwer
b and
Adriaan J.
Minnaard
*a
a,
Mark L. G.
Borst
b,
Kees
Pouwer
b and
Adriaan J.
Minnaard
*a
aDepartment of Chemical Biology, Stratingh Institute for Chemistry, University of Groningen, Groningen, The Netherlands. E-mail: A.J.Minnaard@RUG.nl
bSymeres B.V., Kadijk 3, 9747 AT Groningen, The Netherlands
First published on 2nd November 2021
Abstract
The first multi-gram synthesis of enantiopure dihydroxyethyl acetamidofuran (Di-HAF) is reported. Under optimized conditions, GlcNAc dehydrates in pyridine in the presence of phenylboronic acid and triflic acid to afford Di-HAF in 73% yield and 99.3% ee in just 30 minutes. This protocol opens the door for further research on this bio-renewable building block which is now available as a chiral pool synthon. A plausible mechanism of its formation and of the subsequent dehydration of Di-HAF into well-known 3-acetamido-5-acetylfuran (3A5AF) is proposed.
Introduction
Valorisation of biobased raw materials has attracted much attention in recent years.1,2 In the biorefinery concept, biomass is converted into fuels, and building blocks for the chemical industry. Cellulose, nature's most abundant biopolymer, has received prime attention from the scientific community, but chitin, the second most abundant biopolymer, is arguably equally interesting.3,4 The exoskeleton of crustaceans is a major source of chitin and is copiously present in waste streams of shellfish processing plants. Chitin is a polysaccharide with repeating units of N-acetylglucosamine (GlcNAc). The monomer GlcNAc is readily obtained via acidic, enzymatic or thermal depolymerization.5,6 Research has shown that GlcNAc can be dehydrated into the substituted furan derivates 5-(hydroxymethyl)furfural (5-HMF), 3-acetamido-5-acetylfuran (3A5AF) and dihydroxyethyl acetamidofuran (Di-HAF, Scheme 1) in 65%,7 60%8 and 40% yield,9 respectively (Scheme 2).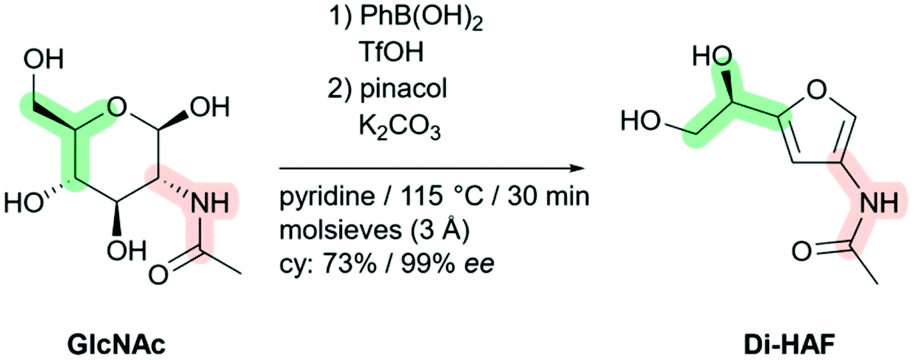 | ||
| Scheme 1 Synthesis of enantiopure dihydroxyethyl acetamidofuran (Di-HAF) by dehydration of N-acetyl glucosamine (GlcNAc). | ||
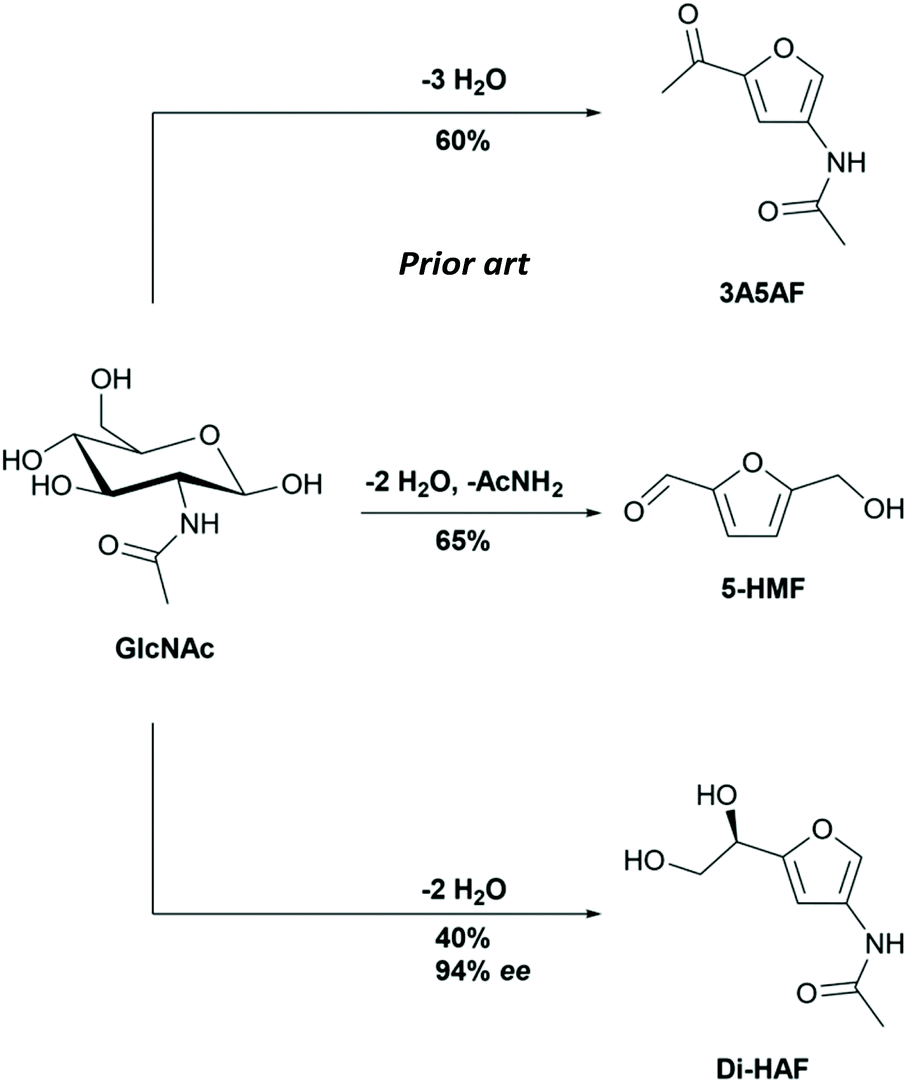 | ||
| Scheme 2 GlcNAc derived substituted furan building blocks. | ||
While the preparation and use of the two furan derivatives 5-HMF and 3A5AF has been studied actively in recent years,8,10–14 the furanyl diol dihydroxyethyl acetamidofuran (Di-HAF) attracted less attention. This bis-dehydration product of GlcNAc was identified by Kuhn and Krüger in 195615 as an intermediate in the Morgan–Elson method for quantitative determination of hexosamines16 and was named chromogen III.15 We believe that the name “chromogen” does not do justice to the potential applications of this compound and therefore prefer to use the more general acronym: Di-HAF.
Unlike the other furans derived from GlcNAc, Di-HAF retains a chiral center, originally at the C5 position of GlcNAc. As chirality plays a crucial role in drug development and virtually all chiral drugs currently are enantiopure,17Di-HAF is potentially valuable building block. The other enantiomer of GlcNAc, and therefore of Di-HAF, is available as well although its synthesis takes several steps.18
In addition to the chiral center, the acetamido group of GlcNAc also remains present and therefore Di-HAF is a so-called Haber-independent nitrogen-containing compound.12 Nitrogen containing compounds are omnipresent in pharmaceuticals, textiles, soaps and surfactants,4 and the vast majority of the nitrogen in these compounds comes from ammonia produced in the Haber–Bosch process.12 This process is notoriously energy inefficient and utilizing a bio-renewable source would be a more sustainable and energy efficient alternative.12
The synthesis of Di-HAF was reported by Kuhn and Kruger shortly after its identification.9GlcNAc was dehydrated in pyridine at reflux for 48 hours. A mixture of mono-dehydrated, bis-dehydrated (Di-HAF), and tris-dehydrated products (3A5AF) was obtained (Scheme 3). This mixture was separated in an elaborate purification procedure comprising chromatography over alumina, crystallization, recrystallization of the mother liquor, chromatographic purification of the mother liquor and another recrystallization. Ultimately, Di-HAF was isolated in 40% yield with an enantiopurity of approximately 94% ee. Probably because of its low yield and tedious purification, it took more than half a century before the group of Osada revisited the synthesis and used continuous flow chemistry to prepare Di-HAF in high temperature water in just 35% yield.19,20 This procedure was run on mg-scale, and the authors did not elaborate upon the obtained optical purity.
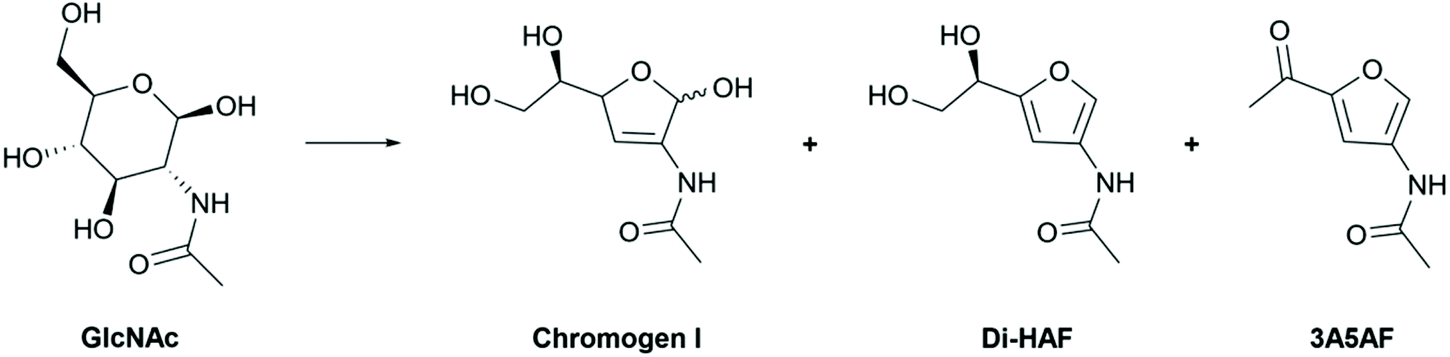 | ||
| Scheme 3 Dehydration products of GlcNAc as identified by Kuhn and Kruger. | ||
Convinced that a straightforward, scalable preparation of Di-HAF will lead to its use as a versatile building block in the fine chemical and pharmaceutical industry, we decided to study the dehydration of GlcNAc to Di-HAF. In the course of this study, we noticed that the proposed mechanisms of the dehydration of GlcNAc had to be scrutinized and corrected.
Two different mechanisms describing the dehydration have been reported in literature (Scheme 4).8,19–21 In solution, GlcNAc is largely present in its pyranose form (S16, ESI†), and the first step of both mechanisms involves ring-chain tautomerism to arrive at 1. From this point two different pathways have been proposed. Kerton et al.8,21 proposed pathway A, based on the identification of various dehydration side products and an NMR study of the intermediates. Nucleophilic attack of the C4 hydroxyl group on the carbonyl group affords the furanose form (2). The extra-annular ketone functionality is assumed to be formed by elimination of the primary alcohol followed by keto–enol tautomerism. Elimination of two molecules of water, in an unspecified order, provides the tris-dehydrated product 3A5AF. Some caveats in the published data were noted, however. The reported 13C NMR and 1H NMR spectra of the reaction intermediates21 show distinct peaks corresponding to Di-HAF,19 but this structure is not included in the mechanism. In addition, boric acid was found to significantly improve the yield of the dehydration,8 a phenomenon that was attributed to a boric acid induced equilibrium shift between the pyranose- and furanose forms. However, elimination of the primary hydroxy group does not directly benefit from such a shift. Arguably, these observations are more in support of the mechanism proposed by Osada et al.19,20 (pathway B). In this proposal, the first step is elimination of the C3 hydroxyl group that occurs in the ring-opened form. Enolization of the aldehyde opens the door for an E1cb-type elimination mechanism to provide α,β-unsaturated aldehyde 6. In principle, the keto–enol equilibrium between 1 and 5 will also result in epimerization to give the C2-epimer of GlcNAc: N-acetylmannosamine (ManAc). To support this part of the mechanism, we dehydrated ManAc under the reported conditions, which indeed afforded Di-HAF (S6, ESI†). This confirms enolization is likely the first step in the mechanism. Aldehyde 6 is the ring-opened form of chromogen I and, after ring closure, the anomeric hydroxyl group eliminates to form oxocarbenium ion 7. Deprotonation leads to aromatization and Di-HAF is obtained.
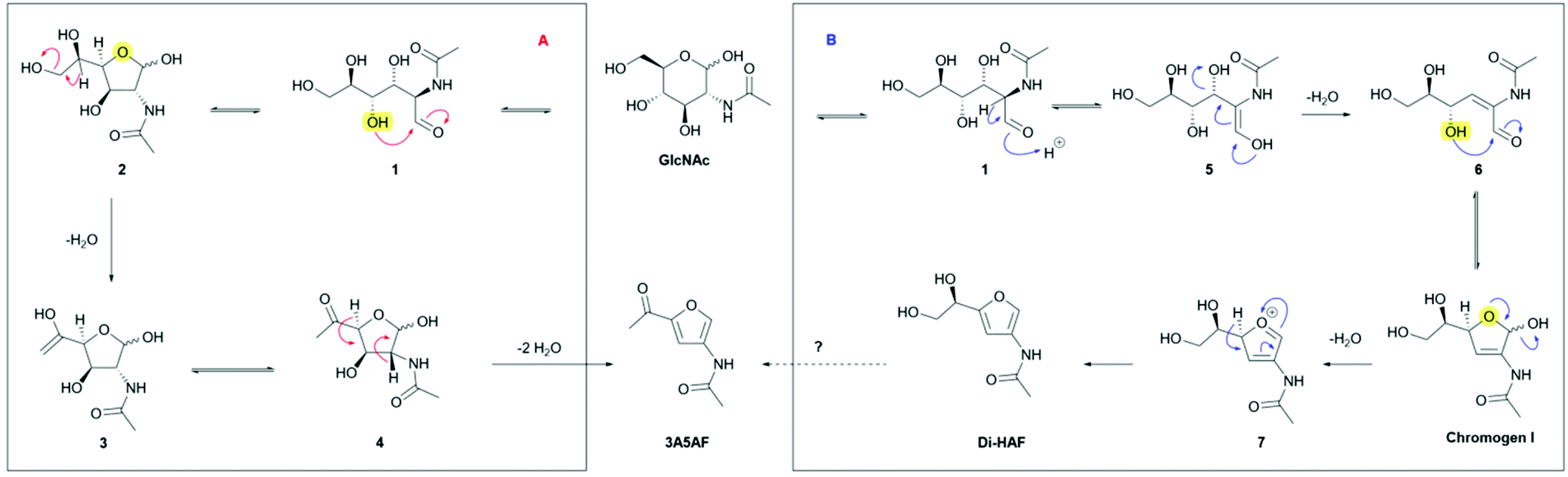 | ||
| Scheme 4 Dehydration mechanisms of GlcNAc according to literature. (A) Proposed mechanism to 3A5AF by Kerton et al. (B) Proposed mechanism to Di-HAF by Osada et al. | ||
Elimination of the primary hydroxy group in Di-HAF would furnish 3A5AF. However, this step has not been experimentally verified and it is not easy to envision why this elimination reaction would take place. Elimination of the “furanylic” hydroxy group seems considerably more likely. This would lead, after tautomerization, to an aldehyde but neither this aldehyde nor its reaction products have been identified.
Here we propose an alternative, detailed mechanism of the dehydration of Di-HAF to 3A5AF and report a scalable method for multi-gram synthesis of enantiopure Di-HAF in high yield and with a straightforward purification (Scheme 1).
Results and discussion
To allow the synthesis of Di-HAF in an acceptable yield, a thorough understanding of the dehydration mechanism of GlcNAc was required. To study this mechanism, we performed H/D-exchange experiments on Di-HAF by stirring the latter in slightly acidic D2O (S8, ESI†). 1H NMR clearly showed H/D-exchange at the 2-position of the furan ring, likely facilitated by electron donation of the neighbouring acetamide. Based on this observation, we propose the mechanism for dehydration of Di-HAF as shown in Scheme 5.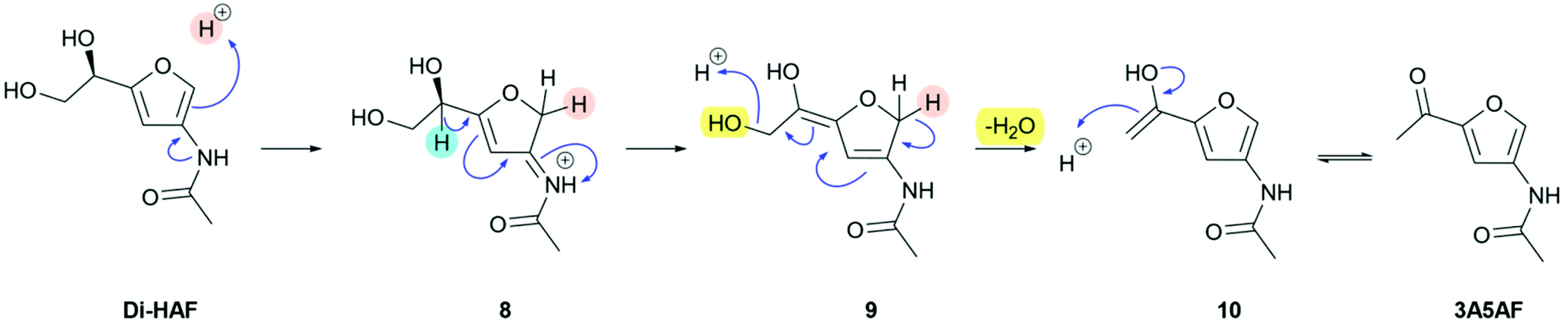 | ||
| Scheme 5 Proposed mechanism of the dehydration of Di-HAF to afford 3A5AF. | ||
Protonation of the furan ring at the 2-position affords intermediate 8. After deprotonation of the furanylic position, diene 9 is obtained. Deprotonation of the dihydrofuran leads to conjugate elimination and expels the third molecule of water. Subsequent keto–enol tautomerization provides 3A5AF. To confirm that Di-HAF is indeed the precursor of 3A5AF, we performed the synthesis of 3A5AF starting from Di-HAF. Under acid catalysis in pyridine, 3A5AF is formed in 64% yield (S7, ESI†). This confirmed that indeed 3A5AF is formed from Di-HAF which means that a high yielding synthesis of Di-HAF is in principle possible, provided that further reaction of Di-HAF to 3A5AF is avoided.
Our strategy for the optimization of the synthesis of Di-HAF was to improve the procedure reported by Kuhn and Kruger9 with the goal to increase yield, strongly simplify isolation and retain the enantiopurity of Di-HAF.
Temperature sensitivity of Di-HAF had been demonstrated by Osada et al.19 We assumed that the prolonged heating required for the reaction in the procedure of Kuhn and Kruger results in the formation of humins, which are undefined polymeric by-products formed during the dehydration of carbohydrates.13,22 Therefore, we decided to investigate the use of a catalyst in order to shorten the reaction time. The benefit of acid catalysis on the dehydration of carbohydrates has been demonstrated in the furfural synthesis from glucose23,24 and the dehydration of GlcNAc in refluxing pyridine with hydrochloric acid proved to be completed within 24 hours. The reaction, however, did not stop at the desired bis-dehydrated stage (Di-HAF) but continued to the tris-dehydrated product (3A5AF). To increase selectivity we used an observation of Kerton et al.8,21,25 who discovered an accelerating effect of boric acid in the synthesis of 3A5AF. It was proposed that via a boric acid complex the pyranose-furanose equilibrium is shifted towards the furanose form, after which the primary alcohol eliminates to form the ketone.8 We showed, however, that the ketone forms in the final step. Based on our understanding of the mechanism, we assume that boric acid forms a cyclic boronate ester with the terminal hydroxyl groups of GlcNAc. This “funnels” the dehydrated intermediate 6 (Scheme 4) into the furanose form and this is followed by the second dehydration step. Therefore, the reaction intermediates of the boric acid mediated dehydration of GlcNAc probably include the boronate ester of chromogen I and the boronate ester of Di-HAF (Scheme 6). Boronate esters from boric acid are known to be relatively labile, so in situ hydrolysis liberates the diol and further conversion to 3A5AF takes place. Hydrolysis is necessary because boron reduces the electron density at the oxygen atom that is part of the boronate ester, which makes protonation less favourable and thereby prevents elimination. Therefore we imagined, that a boronic acid derivate that forms a more stable boronate ester than boric acid, would accelerate the formation of the furan ring even more but prevent the third elimination step to 3A5AF. Hydrolysis of the boronate ester under mild conditions áfter the reaction would then furnish Di-HAF.
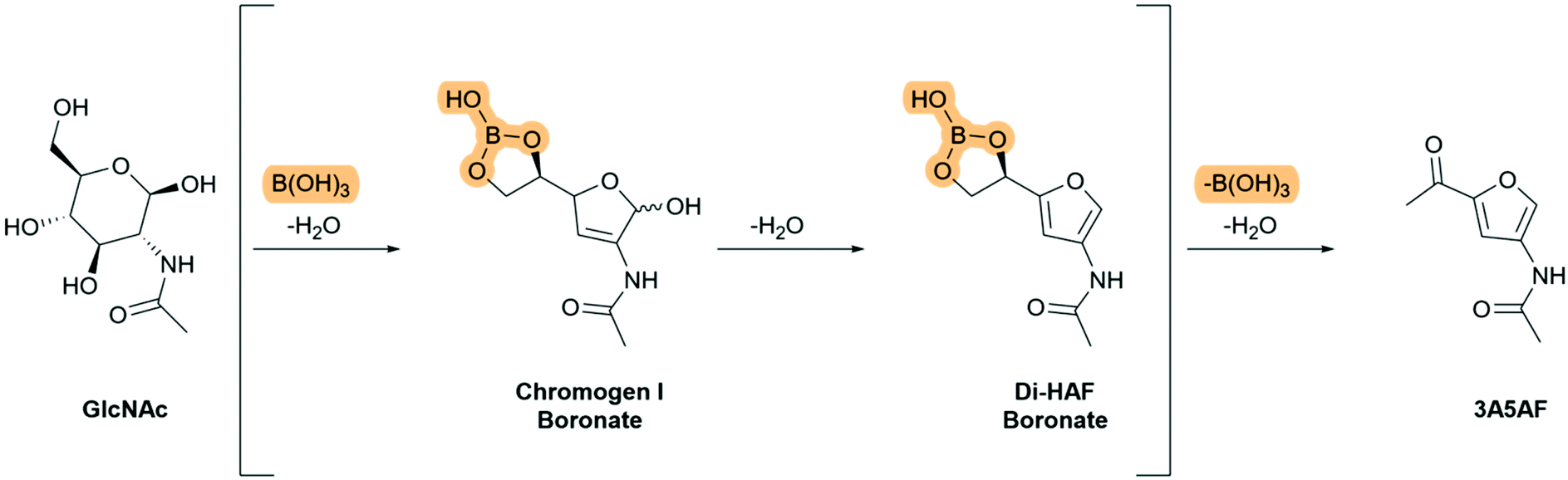 | ||
| Scheme 6 Proposed boronate ester intermediates in the boric acid mediated dehydration of GlcNAc to 3A5AF. | ||
Phenylboronic acid is relatively low-cost, and forms stable esters with carbohydrates.26 Therefore, we screened various combinations of phenylboronic acid and commonly used Brønsted-acids as shown in Scheme 7. To our delight, the formation of ketone 3A5AF was not observed in any of these experiments, indicating that phenylboronic acid effectively suppresses the third elimination reaction. Because of incomplete conversion after 90 minutes, only low yields were found with no acid- or weak acid catalysts (Table 1, entries 1–3). With stronger acids (entries 4–7) the starting material was completely consumed and the phenylboronate ester of Di-HAF (11) was formed in good to excellent yield (73%–95%). We were slightly surprised to find a correlation between acid strength and reaction rate because in pyridine, dissociation of an acid is expected, so the pyridinium ion would be the protonating species in each case. However, as the pKa of the acid decreases, the coordination between the pyridinium ion and the conjugated base becomes weaker.27 The increased concentration of free pyridinium could be responsible for acceleration of the reaction.
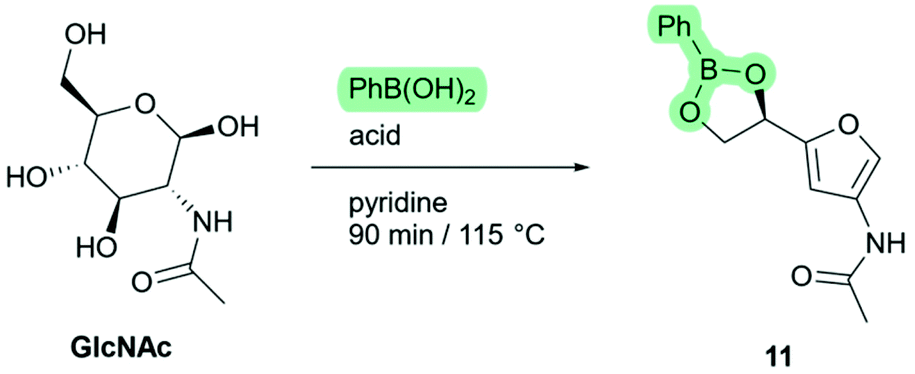 | ||
| Scheme 7 Phenylboronate mediated dehydration of GlcNAc. | ||
We had found earlier that the reaction with hydrochloric acid (in the absence of phenylboronic acid) required 24 hours to go to completion. Phenylboronic acid alone (entry 1) showed only a few percent conversion after 90 minutes. Interestingly, apparently due to a synergistic effect between phenylboronic acid and the acid catalyst, a significant increase in reaction rate and yield was observed. We assume this is because the two additives accelerate different stages of the dehydration mechanism. Phenylboronic acid directs the starting material into the favourable furanose form, and the acid catalyses the actual dehydration steps. This complimentary effect is especially pronounced with the combination of phenylboronic acid and triflic acid which furnished boronate ester 11 in high yield (95%, entry 7). Further investigations of these conditions revealed that the reaction was complete within 30 minutes, corresponding to a 102 fold rate increase relative to the original procedure.
With the optimum reaction conditions identified, we focused on the partial racemization, initially observed by Kuhn and Kruger. Chiral HPLC analysis showed that the new method with phenylboronic acid and triflic acid furnished Di-HAF with 95% ee, similar to that reported by Kuhn and Kruger (94% ee).9 However, racemization increased upon scale-up and on larger scale (50 g, vide infra) the ee dropped to 86%. It turned out that at least two recrystallizations were required to increase the enantiopurity to >99% ee, obviously at the expense of the overall yield. To gain a better understanding of the racemization mechanism involved, a screening was performed. A solution of Di-HAF in pyridine was stirred for 1 hour at 115 °C in the presence of water (10 v/v%) and in dry solvent (molecular sieves, 10 w/v%) with acid (HCl, pTsOH, 1.0 eq.) or base (NaHCO3, 1.0 eq.). Although racemization was in all cases modest, it was more pronounced under wet acidic conditions (HCl, −4.7% ee) while dry acidic conditions also showed a smaller decrease in enantiomeric excess (HCl, −2.1% ee). It is worth mentioning that completely water-free conditions cannot be obtained as under acidic conditions water is formed in situ by dehydration of Di-HAF.
Based on these observations, different racemization pathways were envisioned (Scheme 8). The H/D exchange experiment, carried out earlier, showed that protonation takes place at the 2-position of the furan ring leading to 8 (pathway A). According to the mechanism proposed in Scheme 5, a deprotonation-protonation sequence of the furanylic position leads to racemisation (3-Rac). Subsequent deprotonation affords racemic Di-HAF (Di-HAF-Rac). A second position available for protonation, resulting in overall racemization, is the furanylic hydroxyl group (pathway B). Under wet conditions, the first steps follow the known mechanism of the Piancantelli rearrangement.28 In this case, nucleophilic attack of water at the 2-position of the furan ring (12) and conjugated substitution expels the protonated hydroxyl group (13). Subsequent nucleophilic attack of water on the allylic double bond (“SN2′-type”) results in the formation of Di-HAF-Rac. In addition to these mechanisms, variants of these cannot be excluded. For example, attack of the primary hydroxy group in 12 leading to the epoxide and subsequent ring opening with water also furnishes Di-HAF-Rac.
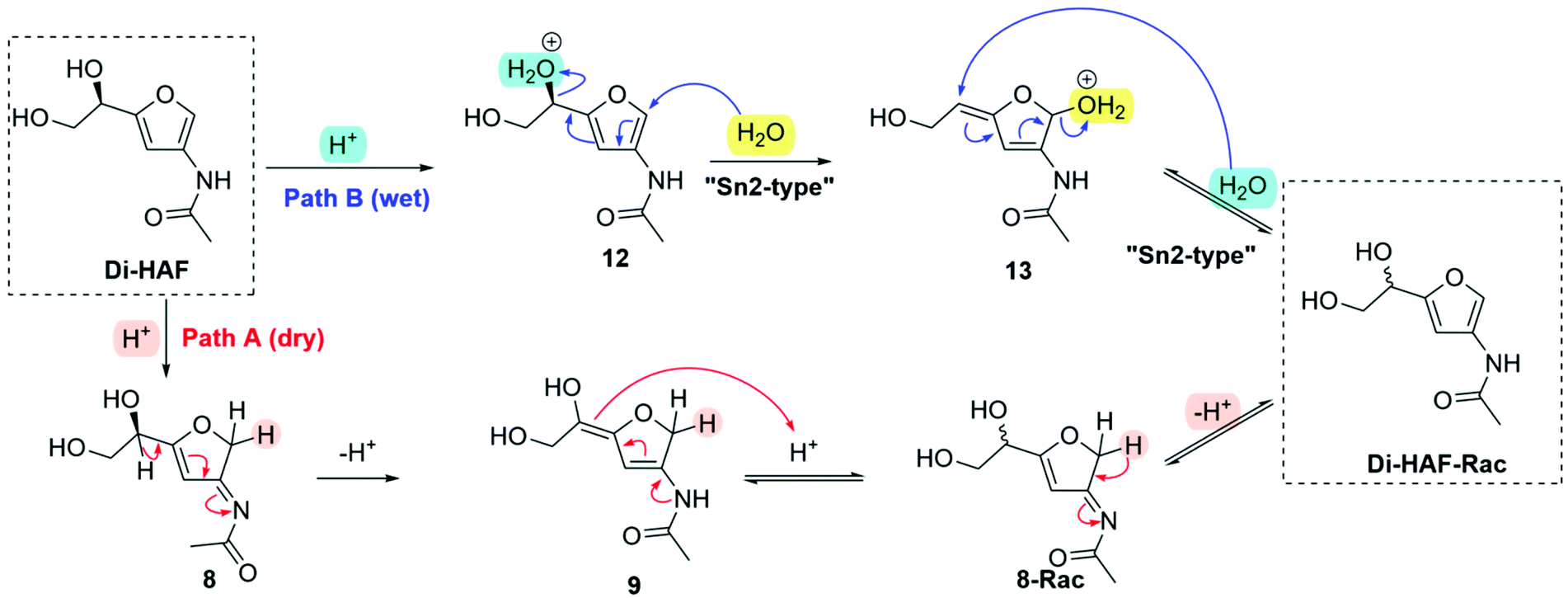 | ||
| Scheme 8 Proposed racemization pathways of Di-HAF. Path A: under dry conditions; Path B: under wet conditions. | ||
The mechanisms deduced from the racemization study revealed that acid catalysis is key in the racemization. Acid, however, also significantly increases the rate and yield of the dehydration of GlcNAc and therefore cannot be easily omitted.
The presence of water proved to be of significance as well. We decided to focus first on the efficient removal of the water that is formed during the reaction. Four equivalents of water are generated; two during the formation of the boronate ester and two during the elimination reactions. The released water was scavenged with powdered molecular sieves (3 Å) and this significantly supressed racemization. The drop in enantiopurity was limited to 1–2% versus 16% observed without water scavenging. We were satisfied with this result as, if required, even higher ee can be reached in a single crystallization. Based on the significant suppression of racemization observed in the presence of powdered molecular sieves, it is likely that water induced racemization is the dominant racemization mechanism, so pathway B of Scheme 8.
With the synthetic problems solved, we set out to address the issue of isolation. The original purification protocol consists of multiple chromatographic purifications and recrystallizations, which hampers larger scale synthesis. The procedure should be time-efficient and allow for multigram scale synthesis. Dehydration of GlcNAc with phenylboronic acid affords the boronate ester of Di-HAF, and to liberate the diol functionality we needed mild conditions to cleave the boronate ester. We found that if the reaction mixture is treated with a small excess of pinacol, Di-HAF is produced via transesterification leading to a readily separated mixture of Di-HAF and phenylboronic acid pinacol ester.
Initially, removal of the solvent (pyridine) under reduced pressure led to noticeable degradation. This was attributed to the limited stability of Di-HAF in the presence of the reaction byproducts. To avoid this, the reaction mixture was treated with both solid potassium carbonate and pinacol. This neutralized acids and liberated the product. Di-HAF was subsequently precipitated by the addition of heptane as anti-solvent and collected by filtration over a patch of silica. The solid residue on the silica was washed with ethyl acetate and subsequently flushed through the silica with a mixture of dichloromethane and methanol (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The solvent could subsequently be removed in vacuo without decomposition of the product. A straightforward crystallization from acetonitrile afforded Di-HAF in 73% yield and excellent enantio- and chemical purity (99.3% ee, >99%). To demonstrate the scalability of the method, the reaction was performed on 50 g scale and afforded to our satisfaction 30.5 g Di-HAF, corresponding to a yield of 73%, with an optical purity of 99.1% ee (S5, ESI†).
:1). The solvent could subsequently be removed in vacuo without decomposition of the product. A straightforward crystallization from acetonitrile afforded Di-HAF in 73% yield and excellent enantio- and chemical purity (99.3% ee, >99%). To demonstrate the scalability of the method, the reaction was performed on 50 g scale and afforded to our satisfaction 30.5 g Di-HAF, corresponding to a yield of 73%, with an optical purity of 99.1% ee (S5, ESI†).
Conclusions
A thorough study of the one-step synthesis of dihydroxyethyl acetamidofuran (Di-HAF) from N-acetylglucosamine resulted in a versatile and scalable procedure. A combination of phenylboronic acid and triflic acid offers a synergistic effect that almost doubled the highest reported yield (40% → 73%). Phenylboronic acid improved chemoselectivity, while triflic acid drastically increased the reaction rate. The reaction time was decreased from days to minutes (48 h → 30 min). A mild work-up procedure, combined with a straightforward crystallization provides Di-HAF in 73% yield and excellent chemical- and optical purity (>99%, 99.3% ee). A preparative reaction on 50 g scale confirms that the process is scalable, effecting neither yield nor purity.In the course of the study it became clear that the reported mechanism of the dehydration of GlcNAc proposed in literature was incomplete. Therefore this mechanism was scrutinized and supplemented with the dehydration of Di-HAF to the tris-dehydrated product 3A5AF. This mechanism offers a plausible explanation for the formation of the ketone functionality that is found in 3A5AF.
The availability of multi-gram quantities of enantio-pure Di-HAF opens the door for further research on this versatile building block as starting material for several reactions. In Scheme 9 a selection of potentially interesting reactions is shown. Furfuryl alcohols are known to readily undergo rearrangements such as the Piancatelli- and Achmatowicz rearrangement. The acid mediated Piancatelli rearrangement produces cyclopentenone derivates (15). Piancatelli products are important motifs in natural- and pharmaceutical products28,29 and have been used for the synthesis of diversity oriented compound libraries.30 The oxidative Achmatowicz rearrangement turns the furan ring back into the six membered pyran ring (16). Many bioactive natural products contain such a unit31 and the value of these structures has also been shown in the work of O'Doherty who converted furfuryl alcohols into unnatural carbohydrates via their Achmatowicz products.32
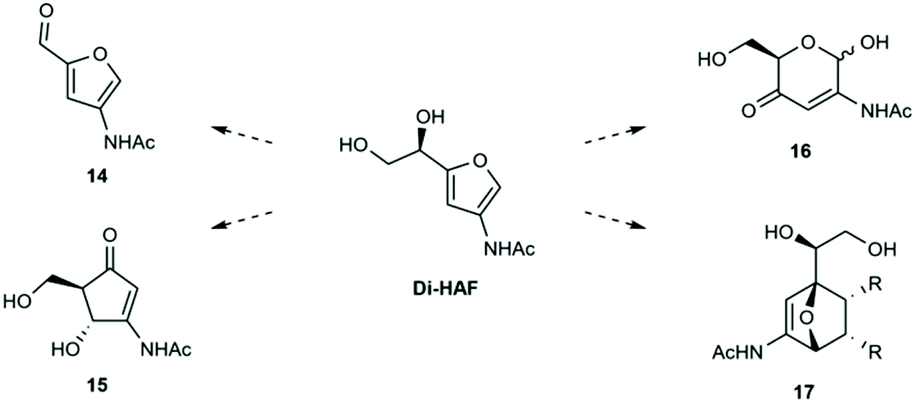 | ||
| Scheme 9 Potentially interesting reactions with Di-HAF. | ||
In addition to rearrangement products, Di-HAF contains handles that allow for further modifications. The vicinal diol-moiety can probably be converted into an aldehyde by periodate treatment (the Malaprade reaction), which affords the 3-acetamide-furfural analogue 14. Furfural, the corresponding dehydration product of xylose, has a plethora of applications in industry, from flavouring agent to lubricant and building block of pharmaceuticals.33 The 3-acetamide analogue of furfural could provide complementary applications. Recently, Bruijnincx et al.34,35 showed that furfural derivatives can be employed in Diels–Alder cyclo-additions with methyl maleimide. Currently, our group is investigating the possibilities of accessing substituted 7-oxa-norbornenes (17) via Diels–Alder cycloadditions with Di-HAF.
Experimental
General procedure for the preparation of (R)-N-(5-(1,2-Dihydroxyethyl)furan-3-yl)acetamide (Di-HAF)
Triflic acid (1 eq.) was added to a suspension of N-acetylglucosamine (GlcNAc) (1 eq.), phenylboronic acid (1.5 eq.) and molecular sieves (3 Å, powdered 5 mass eq.) in pyridine (0.2 M). The flask was flushed with nitrogen and heated to reflux temperature (116 °C). After 30 min the mixture was cooled down to room temperature. Pinacol (2 eq.) and potassium carbonate (2 eq.) were added and stirring was continued for 30 min. The mixture was poured into a stirred suspension of silica (3 mass eq.) in heptane (10 vol.) and the suspension was filtered over a patch of silica (4 cm layer). The residue was washed with heptane (10 vol.), ethyl acetate (10 vol.) and subsequently with dichloromethane-methanol (9:1, 20 vol). The dichloromethane-methanol solution was concentrated in vacuo (45 °C) to afford a brown oil. The crude was crystallized from acetonitrile (4 vol.) to afford Di-HAF as a light brown solid (yield: 73%, >99% ee). 1H NMR (400 MHz, D2O) δ 7.74 (s, 1H), 6.31 (s, 1H), 4.65 (t, J = 6.0 Hz, 1H), 3.80–3.68 (m, 2H), 2.02 (s, 3H). 13C NMR (100 MHz, D2O) δ 171.93, 152.55, 132.64, 124.27, 102.97, 67.36, 63.27, 21.95.
Author contributions
All authors took part in the conceptualization of this study. C. van der Loo carried out the experimental work, C. van der Loo and A. Minnaard wrote the original draft of the manuscript. All authors contributed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The CCC Carbobased program is acknowledged for funding and Dr A. Heeres (Hanze University of Applied Sciences) for valuable discussions. We thank the analytical department of Symeres Groningen for analytical support, in particular chiral HLPC, and R. Sneep (University of Groningen) for HRMS analysis.Notes and references
- X. Chen, H. Yang and N. Yan, Chem. – Eur. J., 2016, 22, 13402–13421 CrossRef CAS PubMed.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- F. M. Kerton, Y. Liu, K. W. Omari and K. Hawboldt, Green Chem., 2013, 15, 860–871 RSC.
- N. Yan and X. Chen, Nature, 2015, 524, 155–157 CrossRef CAS.
- M. Nikahd, J. Mikusek, L. J. Yu, M. L. Coote, M. G. Banwell, C. Ma and M. G. Gardiner, J. Org. Chem., 2020, 85, 4583–4593 CrossRef CAS.
- M. N. V. Ravi Kumar, R. A. A. Muzzarelli, C. Muzzarelli, H. Sashiwa and A. J. Domb, Chem. Rev., 2004, 104, 6017–6084 CrossRef.
- H. Zang, S. Yu, P. Yu, H. Ding, Y. Du, Y. Yang and Y. Zhang, Carbohydr. Res., 2017, 442, 1–8 CrossRef CAS.
- M. W. Drover, K. W. Omari, J. N. Murphy and F. M. Kerton, RSC Adv., 2012, 2, 4642–4644 RSC.
- R. Kuhn and G. Kruger, Chem. Ber., 1957, 90, 264–277 CrossRef CAS.
- K. W. Omari, J. E. Besaw and F. M. Kerton, Green Chem., 2012, 14, 1480–1487 RSC.
- T. T. Pham, A. C. Lindsay, S. W. Kim, L. Persello, X. Chen, N. Yan and J. Sperry, ChemistrySelect, 2019, 4, 10097–10099 CrossRef CAS.
- T. T. Pham, A. C. Lindsay, X. Chen, G. Gözaydin, N. Yan and J. Sperry, Sustainable Chem. Pharm., 2019, 13, 100143 CrossRef.
- D. Padovan, H. Kobayashi and A. Fukuoka, ChemSusChem, 2020, 13, 3594–3598 CrossRef CAS PubMed.
- J. Wang, H. Zang, S. Jiao, K. Wang, Z. Shang, H. Li and J. Lou, Sci. Total Environ., 2020, 710, 136293 CrossRef CAS PubMed.
- R. Kuhn and G. Krüger, Eur. J. Inorg. Chem., 1956, 89, 1473–1486 CAS.
- J. A. Cifonelli and A. Dorfman, J. Biol. Chem., 1958, 231, 11–18 CrossRef CAS.
- R. Jayakumar and R. Vadivel, Org. Med. Chem. Int. J., 2018, 5, 555661 Search PubMed.
- E. Lattová and L. Petruš, Carbohydr. Res., 1992, 235, 289–293 CrossRef.
- M. Osada, K. Kikuta, K. Yoshida, K. Totani, M. Ogata and T. Usui, Green Chem., 2013, 15, 2960–2966 RSC.
- M. Osada, S. Shoji, S. Suenaga and M. Ogata, Fuel Process. Technol., 2019, 195, 106154–106160 CrossRef CAS.
- X. Chen, S. L. Chew, F. M. Kerton and N. Yan, Green Chem., 2014, 16, 2204–2212 RSC.
- C. Zhou, J. Zhao, A. E. A. Yagoub, H. Ma, X. Yu, J. Hu, X. Bao and S. Liu, Egypt. J. Pet., 2017, 26, 477–487 CrossRef.
- R. J. Van Putten, J. C. Van Der Waal, E. De Jong, C. B. Rasrendra, H. J. Heeres and J. G. De Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS.
- R. J. Van Putten, J. N. M. Soetedjo, E. A. Pidko, J. C. Van Der Waal, E. J. M. Hensen, E. De Jong and H. J. Heeres, ChemSusChem, 2013, 6, 1681–1687 CrossRef CAS.
- X. Chen, Y. Liu, F. M. Kerton and N. Yan, RSC Adv., 2015, 5, 20073–20080 RSC.
- S. Manhas and M. S. Taylor, J. Org. Chem., 2017, 82, 11406–11417 CrossRef CAS.
- E. R. Berg, D. D. Green, D. C. Moliva, B. T. Bjerke, M. W. Gealy and D. J. Ulness, J. Phys. Chem. A, 2008, 112, 833–838 CrossRef CAS PubMed.
- C. Verrier, S. Moebs-Sanchez, Y. Queneau and F. Popowycz, Org. Biomol. Chem., 2018, 16, 676–687 RSC.
- C. Piutti and F. Quartieri, Molecules, 2013, 18, 12290–12312 CrossRef CAS.
- T. T. Pham, X. Chen, T. Söhnel, N. Yan and J. Sperry, Green Chem., 2020, 22, 1978–1984 RSC.
- A. K. Ghosh and M. Brindisi, RSC Adv., 2016, 6, 111564–111598 RSC.
- M. F. Cuccarese and G. A. O'Doherty, Asymmetric Synth. II More Methods Appl, 2013, pp. 249–259 Search PubMed.
- M. Kabbour and R. Luque, Furfural as a platform chemical: From production to applications, Elsevier B.V., 2019 Search PubMed.
- R. C. Cioc, M. Lutz, E. A. Pidko, M. Crockatt, J. C. Van Der Waal and P. C. A. Bruijnincx, Green Chem., 2021, 23, 367–373 RSC.
- R. Cioc, T. Smak, M. Crockatt, J. K. Van der Waal and P. C. A. Bruijnincx, Green Chem., 2021, 23, 5503–5510 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob02004h |
| This journal is © The Royal Society of Chemistry 2021 |