
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Automated glycan assembly of peptidoglycan backbone fragments†
Pietro
Dallabernardina
a,
Valentina
Benazzi
ab,
Jon D.
Laman
 c,
Peter H.
Seeberger
ad and
Felix F.
Loeffler
*a
c,
Peter H.
Seeberger
ad and
Felix F.
Loeffler
*a
aDepartment of Biomolecular Systems, Max Planck Institute of Colloids and Interfaces, Am Muehlenberg 1, 14476 Potsdam, Germany. E-mail: felix.loeffler@mpikg.mpg.de
bUniversity of Pavia, Department of Organic Chemistry, V.le Torquato Taramelli, 10, 27100 Pavia, Italy
cDepartment of Pathology & Medical Biology, University Medical Center Groningen, Groningen, The Netherlands
dFreie Universität Berlin, Institute of Chemistry and Biochemistry, Arnimallee 22, 14195 Berlin, Germany
First published on 25th October 2021
Abstract
We report the automated glycan assembly (AGA) of different oligosaccharide fragments of the bacterial peptidoglycan (PGN) backbone. Iterative addition on a solid support of an acetyl glucosamine and a new muramic acid building block is followed by cleavage from the solid support and final deprotection providing 10 oligosaccharides up to six units.
Peptidoglycans (PGN) are an essential component of the cell wall of both Gram-positive and Gram-negative bacteria. PGN serve a structural function, counteracting osmotic pressure and maintaining cell shape, and protect against external threats.1,2 The main PGN structure is made of linear glycan strands cross-linked by short peptide chains (Fig. 1b).3 Several structural modifications at both the glycan part, such as acetylation, and the stem peptide are possible in order to give rise to hundreds of structural variants.4 Nevertheless, the structure of the glycan backbone is generally conserved and it is composed of two alternating amino sugars (Fig. 1a), N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc), linked by a β-1–4 bond. The D-lactoyl group of each MurNAc residue is connected through an amide bond to a stem peptide that can be up to five amino acid residues long.5 Many modifications can occur at the peptide chain,2 in particular at the third amino acid. The majority of Gram-positive bacteria have an L-Lys at position three, while the majority of Gram-negative bacteria have a non-proteinogenic amino acid A2pm (2,6-diaminopimelic acid). The final dipeptide might present some modifications due to acquired resistance to antibiotics.6 Stem peptides from different glycan strands may be cross-linked either directly or by an interpeptide bridge consisting of one to seven amino acid residues.2
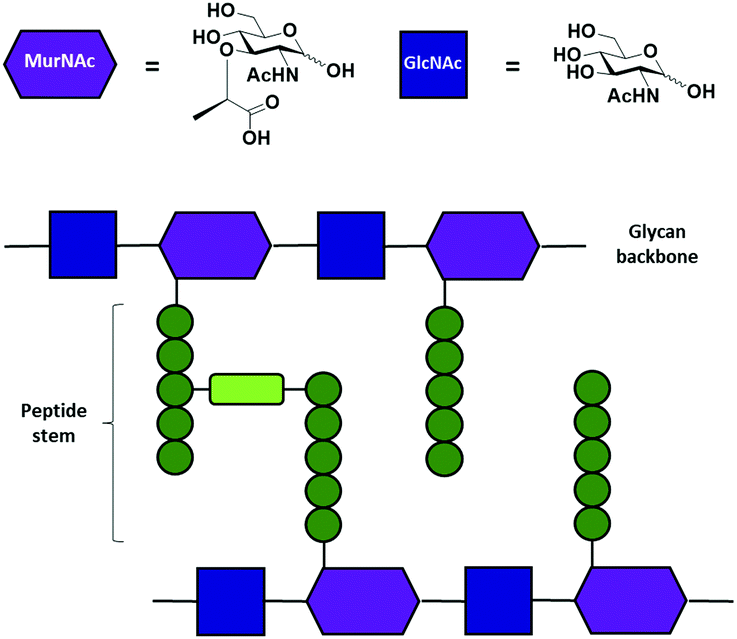 | ||
| Fig. 1 (a) Symbolic representation and structures of the two amino sugars, N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc). (b) Schematic representation of the peptidoglycan structure. The different glycan strands are branched on the MurNAc residues with a sequence of amino acids (dark green circles) called stem peptide. An interpeptide bridge (light green box) may connect the two stem peptides. | ||
In the past two decades, PGNs drew interest not only as a possible target for antibiotics,7 but also for their role as immunostimulatory molecules,8–11 as well as in mouse brain development and behavior.12 It was suggested that PGN fragments have many diverse roles as signalling molecules, including communication between bacteria and bacteria with the host, as well as pathogenesis in animals and plants.5 PGNs have also been correlated with some autoimmune and chronic inflammatory diseases.13–16 In particular, some studies14,17–19 have demonstrated that peptidoglycan fragments are present within phagocytes in the brain of multiple sclerosis patients and that anti-PGN antibodies are present in cerebrospinal fluid during active disease, suggesting a possible link between these molecules and disease. Therefore, the level and identity of PGNs in the blood, urine, and tissues, might serve as a potential marker for progression of autoimmune and inflammatory diseases, and, hence, as a tool in therapy monitoring.13
Given the biological relevance of these molecules, insight into PGN structure is essential to understand the role of PGNs in pathogenesis, discovering the mechanisms involved in their release, and how they are detected by bacterial versus human host cells.5 Even though different proteins with PGN binding activity have been identified, the exact epitopes they recognize remain poorly characterized.20 A major drawback in the characterization of the binding motif is the complexity in the purification of the PGN fragments from biological sources.20 Therefore, to obtain pure compounds it is necessary to resort to chemical synthesis. The chemical synthesis of such oligosaccharide libraries is not trivial and requires an immense synthetic effort.21–24 Automated glycan assembly (AGA)25 has already proven its versatility in the generation of oligosaccharide collections.26,27 Iterative coupling of different building blocks (BB) on a solid support allows for the synthesis of well-defined oligo- and polysaccharide structures in a fast manner.
Here, we report the AGA of ten fragments related to the glycan strands of PGNs using two different monosaccharide BBs. To prepare the compound collection, we designed BBs 1 and 2 (Scheme 1) that can be synthesized starting from an unprotected glucosamine. The amine at C2 was equipped with a trichloroacetyl (TCA) as participating protecting group, to ensure β selectivity during glycosylation. The TCA can be converted into the desired acetyl group upon reduction during the final deprotection.
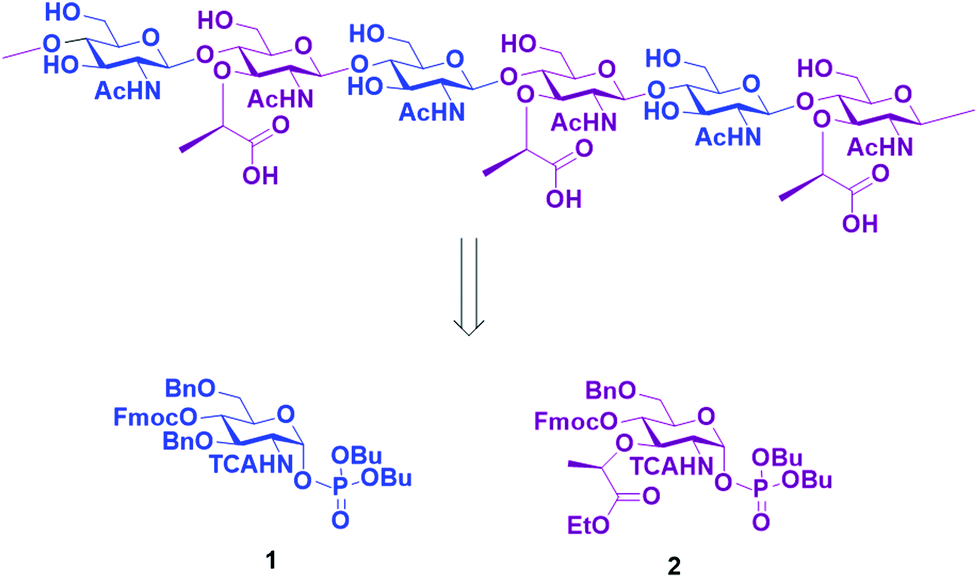 | ||
| Scheme 1 Retrosynthetic approach to obtain the glycan backbone of PGN and the two BB required for the synthesis. | ||
Fluorenylmethoxycarbonyl (Fmoc) was installed at C4 and used as a temporary protecting group for β1–4 chain elongation. A lactic ester was installed in position three of the BB 2 as precursor for the lactic acid moiety of the muramic acid. All other positions were protected with benzyl (Bn) permanent protecting groups. The phosphate leaving group was preferred to other leaving groups due to observed higher coupling efficiency in AGA for glucosamine BBs.28 The two desired BBs 1 and 2 are accessible starting from the unprotected glucosamine with a divergent synthetic pathway from glucosamine thioglycoside 3.29 Glycosyl phosphate building block 1 was prepared in nine steps, following published procedures.28–30
BB 2 was synthesized starting from thiogycoside 329 (Scheme 2) that was treated with NaH and (–)-ethyl (S)-2-trifluromethanesulfonyl propionate, previously prepared in situ from ethyl L-(–)-lactate and triflic anhydride,31 to obtain the monosaccharide 4 with the ethyl propionate at C3. Different solvents were tested. Initially, DMF was used, but only starting material was recovered. In THF the reaction appeared to proceed to completion, but the desired compound was obtained in an inseparable mixture with different side products. Compound 3 proved insoluble in dichloromethane. Therefore, we added some drops of DMF, to completely dissolve the starting material. This mixture gave the best result, affording 4 in 53% yield. The regioselective opening of the benzylidene was attempted using the Broensted acid trifluoroacetic acid and triethylsilane32 as hydride donor. These conditions provided a low yield since a side product originated from the condensation of the free hydroxyl group on position 4 and the ethyl ester. This side product was previously reported when Kinzy et al. used a Broensted acid to hydrolyze the benzylidene acetal.33
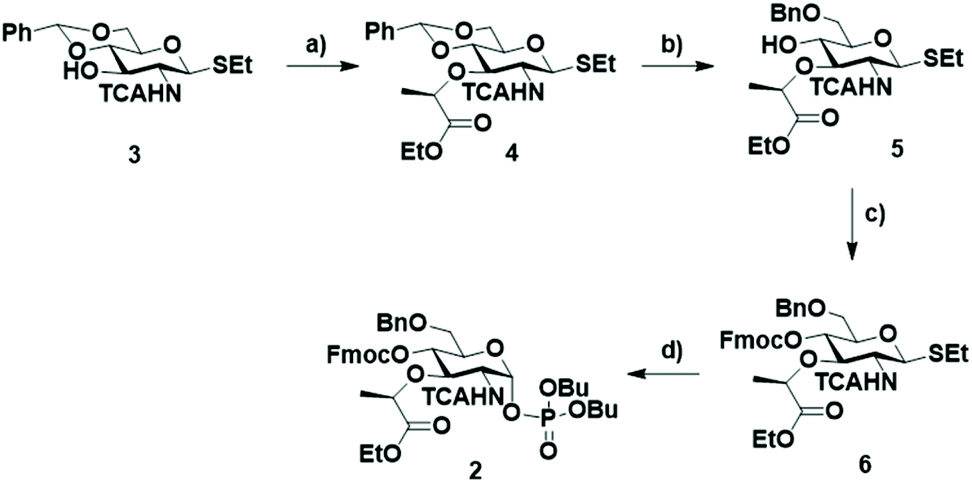 | ||
| Scheme 2 Synthesis of the MurNAc BB 2. Reagents and conditions: (a) (–)-ethyl (S)-2-trifluoromethylsulfonyloxy propionate, NaH, DMF/CH2Cl2, 0 °C → rt, 53%; (b) Me3N·BH3, BF3·OEt2, CH3CN, 0 °C → rt, 65%; (c) FmoCl, pyridine, CH2Cl2, rt, 93%; (d) (BuO)2P(O)OH, NIS, TfOH, 0 °C → rt, 83%. | ||
Therefore, another strategy for the selective benzylidene opening, involving Me3N·BH3 and the Lewis acid BF3·OEt2 in acetonitrile was adopted.34 Under these conditions, the formation of the lactone side product was not observed. Finally, the hydroxyl group on C4 was protected with Fmoc to give 6 and subsequently, the thioglycoside was transformed into the corresponding α-glycosyl phosphate 2.
With BB 1 and 2 in hand, we moved forward with the synthesis of our collection of oligosaccharides using AGA (Scheme 3). Merrifield resin functionalized with a photocleavable linker was used as solid support.35 The BBs were coupled to the solid support using TMSOTf-promoted glycosylation, employing one cycle and five equivalents of BB. Then, an acidic capping solution was used to acetylate the unreacted hydroxyl groups and, thereby, minimizing deletion sequences.36
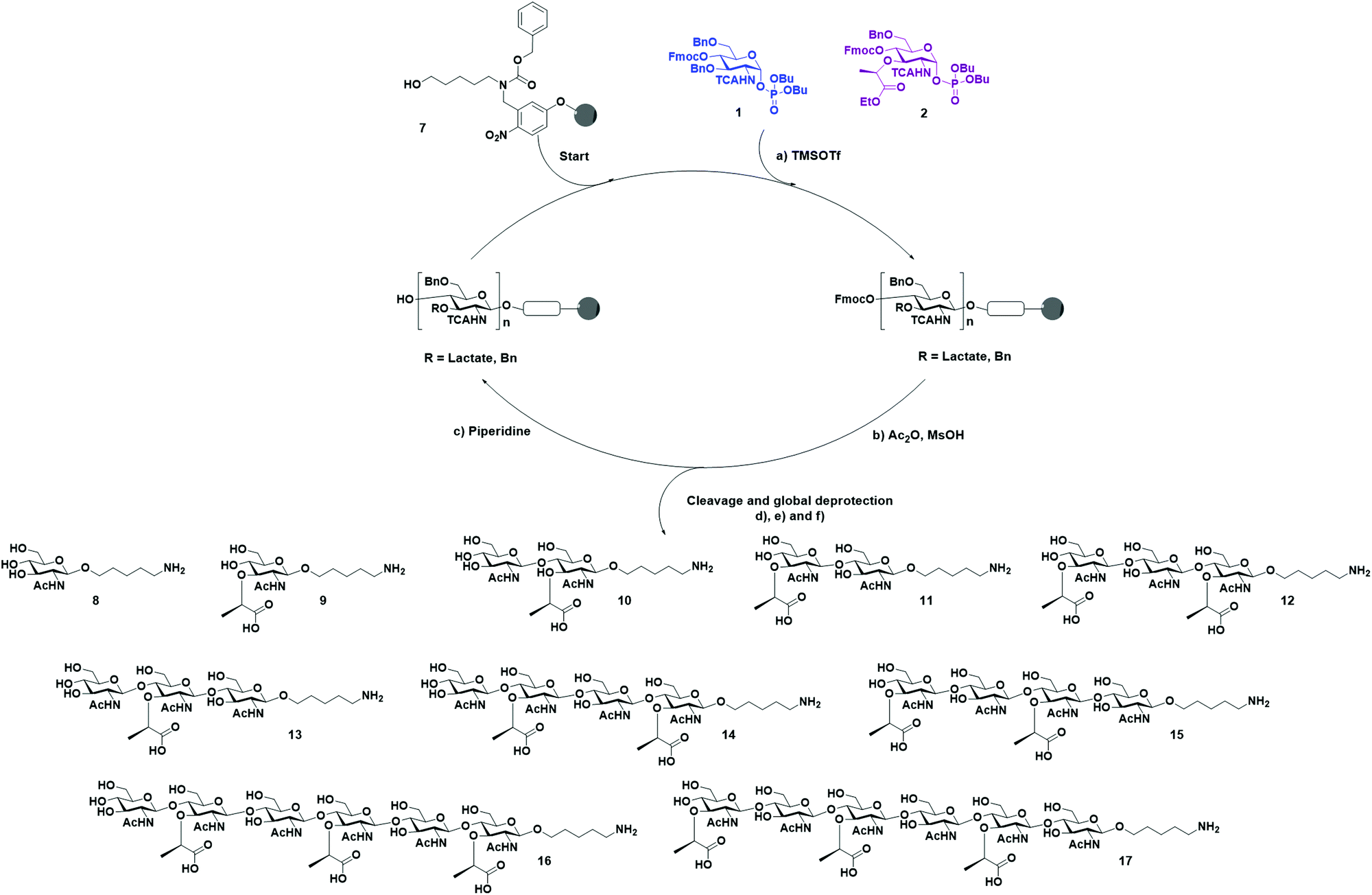 | ||
| Scheme 3 Automated glycan assembly of PGN oligosaccharides. Reagents and conditions: (a) 1 × 5 equiv. BB1, TMSOTf, CH2Cl2, −30 °C (5 min) → −15 °C (35 min); (b) 1 cycle of a 10% acetic anhydride and 2% methanesulfonic acid in CH2Cl2, 25 °C; (c) 3 cycles of 20% piperidine in DMF, 25 °C (5 min) (Module B); (d) CH2Cl2, hν (365 nm); (e) LiOH, THF/1,4-dioxane/H2O, 2–24 h; (f) H2, Pd/C, H2O/HOAc, 16–48 h. 8: 27%; 9: 42%; 10: 20%; 11: 10%; 12: 11%; 13: 9%; 14: 3%; 15: 8%; 16: 3%; 17: 2%. | ||
Subsequently, the Fmoc carbonate was cleaved from the structure, preparing the monosaccharide for the installation of the next unit. This cycle of coupling, capping, and deprotection was repeated until the desired sequence was achieved. Once AGA was completed, the oligosaccharides were cleaved from the resin using UV light (365 nm) in a continuous flow photoreactor.35 After purification of the desired oligosaccharides from the deletion sequences, the basic hydrolysis of the lactoyl esters to carboxylic acids was performed using LiOH in a solution of THF/dioxane/H2O.31 The remaining protecting groups were cleaved by hydrogenolysis with Pd/C and H2. Under these conditions, the TCA group was reduced to acetamide. This last step was more problematic, since after several days of hydrogenolysis, we still observed the presence of multiple chlorine atoms by MALDI. Therefore, for the hexamers were hydrogenated under high pressure to achieve complete conversion in 48 h. The relatively low yield is caused by the inefficiency of the photocleavage from the resin together with the rather low reactivity of the building blocks, which led to the presence of deletion sequences. Finally, ten different PGN glycan backbones of alternating MurNAc and GlcNAc moieties, equipped with an aminolinker were obtained: the two monomers 8 and 9, two disaccharides 10 and 11, two trimers 12 and 13, two tetramers 14 and 15, and two hexamers 16 and 17.
Conclusions
Ten oligosaccharide fragments of the PGN backbone were synthesized. The use of a new MurNAc BB enabled the fast synthesis of a collection of oligosaccharides up to hexamers. These oligosaccharides can be used to create glycan microarrays for the characterization of peptidoglycan directed antibodies. Advancing the idea from recently published approaches, where molecules on microarrays were modified directly on the glass slide after in situ synthesis37 or printing,38 we will follow a similar strategy. We envision to couple different stem peptide variants from solution, forming the amide bond directly with the arrayed oligosaccharides. These arrays will support the identification of epitopes recognized by anti-PGN antibodies, and can potentially help to close current knowledge gaps in autoimmune disorders.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the German Federal Ministry of Education and Research (BMBF, grant no. 13XP5050A), the Max-Planck-Fraunhofer Collaboration Project (Glyco3Display), and the Max Planck Society for financial support. In addition, the authors would like to thank Martina Delbianco for the fruitful discussions and the members of the Department of Biomolecular Systems for the technical support, especially Eva Settels and Olaf Niemeyer. Open Access funding provided by the Max Planck Society.References
- J. V. Höltje, Microbiol. Mol. Biol. Rev., 1998, 62, 181–203 CrossRef PubMed.
- W. Vollmer, D. Blanot and M. A. de Pedro, FEMS Microbiol. Rev., 2008, 32, 149–167 CrossRef CAS PubMed.
- J. H. Rogers, H. R. Perkins and J. B. Ward, Microbial Cell Walls and Membranes, Springer, Netherlands, 1980 Search PubMed.
- P. Schumann, in Methods in Microbiology, 2011, vol. 38, pp. 101–129 Search PubMed.
- O. Irazoki, S. B. Hernandez and F. Cava, Front. Microbiol., 2019, 10, 500 CrossRef PubMed.
- A. K. Yadav, A. Espaillat and F. Cava, Front. Microbiol., 2018, 9, 2064 CrossRef PubMed.
- M. A. Kohanski, D. J. Dwyer and J. J. Collins, Nat. Rev. Microbiol., 2010, 8, 423–435 CrossRef CAS PubMed.
- S. E. Girardin, I. G. Boneca, L. A. M. Carneiro, A. Antignac, M. Jéhanno, J. Viala, K. Tedin, M.-K. Taha, A. Labigne, U. Zähringer, A. J. Coyle, P. S. D. Stefano, J. Bertin, P. J. Sansonetti and D. J. Philpott, Science, 2003, 300, 1584–1587 CrossRef CAS PubMed.
- S. E. Girardin, L. H. Travassos, M. Hervé, D. Blanot, I. G. Boneca, D. J. Philpott, P. J. Sansonetti and D. Mengin-Lecreulx, J. Biol. Chem., 2003, 278, 41702–41708 CrossRef CAS PubMed.
- K. L. Woodhams, J. M. Chan, J. D. Lenz, K. T. Hackett and J. P. Dillard, Infect. Immun., 2013, 81, 3490–3498 CrossRef CAS PubMed.
- M. A. Boudreau, J. F. Fisher and S. Mobashery, Biochemistry, 2012, 51, 2974–2990 CrossRef CAS PubMed.
- A. Gonzalez-Santana and R. D. Heijtz, Trends Mol. Med., 2020, 26, 729–743 CrossRef CAS PubMed.
- Z. Huang, J. Wang, X. Xu, H. Wang, Y. Qiao, W. C. Chu, S. Xu, L. Chai, F. Cottier, N. Pavelka, M. Oosting, L. A. B. Joosten, M. Netea, C. Y. L. Ng, K. P. Leong, P. Kundu, K.-P. Lam, S. Pettersson and Y. Wang, Nat. Microbiol., 2019, 4, 766–773 CrossRef CAS PubMed.
- P. J. Shaw, M. J. Barr, J. R. Lukens, M. A. McGargill, H. Chi, T. W. Mak and T.-D. Kanneganti, Immunity, 2011, 34, 75–84 CrossRef CAS PubMed.
- B. L. Jutras, R. B. Lochhead, Z. A. Kloos, J. Biboy, K. Strle, C. J. Booth, S. K. Govers, J. Gray, P. Schumann, W. Vollmer, L. K. Bockenstedt, A. C. Steere and C. Jacobs-Wagner, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 13498–13507 CrossRef CAS PubMed.
- L. Visser, H. J. d. Heer, L. A. Boven, D. van Riel, M. van Meurs, M.-J. Melief, U. Zähringer, J. van Strijp, B. N. Lambrecht, E. E. Nieuwenhuis and J. D. Laman, J. Immunol., 2005, 174, 808–816 CrossRef CAS PubMed.
- I. A. Schrijver, M. van Meurs, M.-J. Melief, C. W. Ang, D. Buljevac, R. Ravid, M. P. Hazenberg and J. D. Laman, Brain, 2001, 124, 1544–1554 CrossRef CAS PubMed.
- W. G. Branton, J. Q. Lu, M. G. Surette, R. A. Holt, J. Lind, J. D. Laman and C. Power, Sci. Rep., 2016, 6, 37344 CrossRef CAS PubMed.
- J. D. Kriesel, P. Bhetariya, Z.-M. Wang, D. Renner, C. Palmer and K. F. Fischer, Sci. Rep., 2019, 9, 1387 CrossRef PubMed.
- N. Wang, A. Hirata, K. Nokihara, K. Fukase and Y. Fujimoto, Pept. Sci., 2016, 106, 422–429 CrossRef CAS PubMed.
- S. Inamura, K. Fukase and S. Kusumoto, Tetrahedron Lett., 2001, 42, 7613–7616 CrossRef CAS.
- S. Inamura, Y. Fujimoto, A. Kawasaki, Z. Shiokawa, E. Woelk, H. Heine, B. Lindner, N. Inohara, S. Kusumoto and K. Fukase, Org. Biomol. Chem., 2006, 4, 232–242 RSC.
- N. Wang, H. Hasegawa, C.-Y. Huang, K. Fukase and Y. Fujimoto, Chem. – Asian J., 2017, 12, 27–30 CrossRef CAS PubMed.
- M. Lee, D. Hesek, D. A. Dik, J. Fishovitz, E. Lastochkin, B. Boggess, J. F. Fisher and S. Mobashery, Angew. Chem., Int. Ed., 2017, 56, 2735–2739 CrossRef CAS PubMed.
- O. J. Plante, E. R. Palmacci and P. H. Seeberger, Science, 2001, 291, 1523–1527 CrossRef CAS PubMed.
- P. H. Seeberger, Acc. Chem. Res., 2015, 48, 1450–1463 CrossRef CAS PubMed.
- M. Guberman and P. H. Seeberger, J. Am. Chem. Soc., 2019, 141, 5581–5592 CrossRef CAS PubMed.
- T. Tyrikos-Ergas, V. Bordoni, G. Fittolani, M. A. Chaube, A. Grafmüller, P. H. Seeberger and M. Delbianco, Chem. – Eur. J., 2021, 27, 2321–2325 CrossRef CAS PubMed.
- J. Kandasamy, F. Schuhmacher, H. S. Hahm, J. C. Klein and P. H. Seeberger, Chem. Commun., 2014, 50, 1875–1877 RSC.
- L. Kröck, D. Esposito, B. Castagner, C.-C. Wang, P. Bindschändler and P. H. Seeberger, Chem. Sci., 2012, 3, 1617–1622 RSC.
- R. Enugala, M. J. D. Pires and M. M. B. Marques, Carbohydr. Res., 2014, 384, 112–118 CrossRef CAS PubMed.
- M. P. DeNinno, J. B. Etienne and K. C. Duplantier, Tetrahedron Lett., 1995, 5, 669–672 CrossRef.
- W. Kinzy and R. R. Schmidt, Liebigs Ann. Chem., 1987, 407–415 CrossRef CAS.
- M. Oikawa, W.-C. Liu, Y. Nakai, S. Koshida, K. Fukase and S. Kusumoto, Synlett, 1996, 1179–1180 CrossRef CAS.
- S. Eller, M. Collot, J. Yin, H. S. Hahm and P. H. Seeberger, Angew. Chem., Int. Ed., 2013, 52, 5868–5861 CrossRef PubMed.
- Y. Yu, A. Kononov, M. Delbianco and P. H. Seeberger, Chem. – Eur. J., 2018, 24, 6075–6078 CrossRef CAS PubMed.
- M. Mende, A. Tsouka, J. Heidepriem, G. Paris, D. S. Mattes, S. Eickelmann, V. Bordoni, R. Wawrzinek, F. F. Fuchsberger, P. H. Seeberger, C. Rademacher, M. Delbianco, A. Mallagaray and F. F. Loeffler, Chem. – Eur. J., 2020, 26, 9954–9963 CrossRef CAS PubMed.
- C. Ruprecht, M. P. Bartetzko, D. Senf, A. Lakhina, P. J. Smith, M. J. Soto, H. Oh, J.-Y. Yang, D. Chapla, D. V. Silva, M. H. Clausen, M. G. Hahn, K. W. Moremen, B. R. Urbanowicz and F. Pfrengle, Angew. Chem., Int. Ed., 2020, 59, 12494–12498 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob01987b |
| This journal is © The Royal Society of Chemistry 2021 |