
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacilitating functionalization of benzene-1,3,5-tricarboxamides by switching amide connectivity†
Sandra M. C.
Schoenmakers‡
,
Bart W. L.
van den Bersselaar‡
,
Shikha
Dhiman
,
Lu
Su
 and
Anja R. A.
Palmans
*
and
Anja R. A.
Palmans
*
Laboratory of Macromolecular and Organic Chemistry, Institute for Complex Molecular Systems, Eindhoven University of Technology, Eindhoven, The Netherlands. E-mail: a.palmans@tue.nl
First published on 7th September 2021
Abstract
Synthetic water-compatible supramolecular polymers based on benzene-1,3,5-tricarboxamides (BTAs) have attracted a lot of interest in recent years, as they are uniquely suited to generate functional multicomponent biomaterials. Their morphologies and intrinsic dynamic behaviour mimic fibrous structures found in nature. Moreover, their modularity allows control of the density of functionalities presented on the surface of the fibres when using functionalized BTA monomers. However, such moieties generally comprise a functionality on only one of three side chains, resulting in lengthy synthetic protocols and limited yields. In this work, we avert the need for desymmetrization of the core by starting from commercially available 5-aminoisophthalic acid. This approach eliminates the statistical reactions and reduces the number of synthetic steps. It also leads to the inversion of the connectivity of one of the amides to the benzene core. By combining spectroscopy, light scattering and cryogenic transmission electron microscopy, we confirm that the inversed amide BTAs (iBTAs) form intermolecular hydrogen bonds and assemble into supramolecular polymers, like previously used symmetrical BTAs, albeit with a slight decrease in water solubility. Solubility problems were overcome by incorporating iBTAs into conventional BTA-based supramolecular polymers. These two-component mixtures formed supramolecular fibres with a morphology and dynamic behaviour similar to BTA-homopolymers. Finally, iBTAs were decorated with a fluorescent dye to demonstrate the synthesis of functional monomers, and to visualize their co-assembly with BTAs. Our results show that functionality can be introduced into supramolecular polymers with monomers that slightly differ in their core structure while maintaining the structure and dynamics of the fibres.
Introduction
Nature uses supramolecular interactions to create dynamic and adaptive systems from several structurally diverse building blocks.1–4 Synthetic water-compatible supramolecular polymers have recently emerged as potential biomaterials since their structure and dynamics can mimic the fibrous structures found in nature.5,6 The use of non-covalent interactions allows control of the number of biological recognition motifs presented on the supramolecular structures by mixing in different functional monomers in the desired ratio.7,8 This modular approach has been applied in, for example, supramolecular polymers formed by peptide amphiphiles to create scaffolds for cartilage9 and bone regeneration,10 for targeted therapy for atherosclerosis11 and for the promotion of angiogenesis.12 In addition, supramolecular polymers based on ureido-pyrimidinones (UPys) showed tuneable dynamics by mixing mono- and bivalent variants.13 Both types of monomers were functionalized with charged end groups for the intracellular delivery of siRNA14 and with peptides to make the polymers suitable for cell adhesion.15 Finally, the periphery of various columnar structures formed by discotic molecules were decorated with carbohydrates to trigger cellular uptake16,17 and with biotin to create a fibrous scaffold for protein binding.18,19The self-assembly of discotic benzene-1,3,5-tricarboxamides (BTAs) into supramolecular polymers has been studied extensively in recent years.20 The amides can be connected to the benzene ring via the carbonyl group or via the nitrogen atom, yielding C-centred and N-centred BTAs, respectively. Although both variants self-assemble in organic media, the aggregation and hydrogen bonding was weaker in case of N-centred BTAs.21,22C-Centred BTAs have been modified to be compatible with water by decorating the core amides with a hydrophobic chain of at least eleven carbon atoms to protect the intermolecular hydrogen bonds from interaction with the solvent.23,24 Their water-solubility was ensured by a tetra(ethylene glycol) periphery (BTA-C11-EG4 & BTA-C12-EG4, Chart 1).23,24 The micrometre long supramolecular fibres that formed in water were found to be highly dynamic25,26 and the dynamic exchange of monomers between supramolecular polymers could be tuned by modification of the hydrophilic/hydrophobic balance of the monomers25 or by co-assembling structurally different monomers.27 Recently, these BTA-based supramolecular polymers have been functionalized with benzoxaborole to interact with red blood cells,28 with charged groups for the intracellular delivery of siRNA,29 with carbohydrates for binding to lectins30 and with DNA for protein recruitment.31 These examples illustrate the potential use of BTA-based polymers to access sophisticated biomaterials.
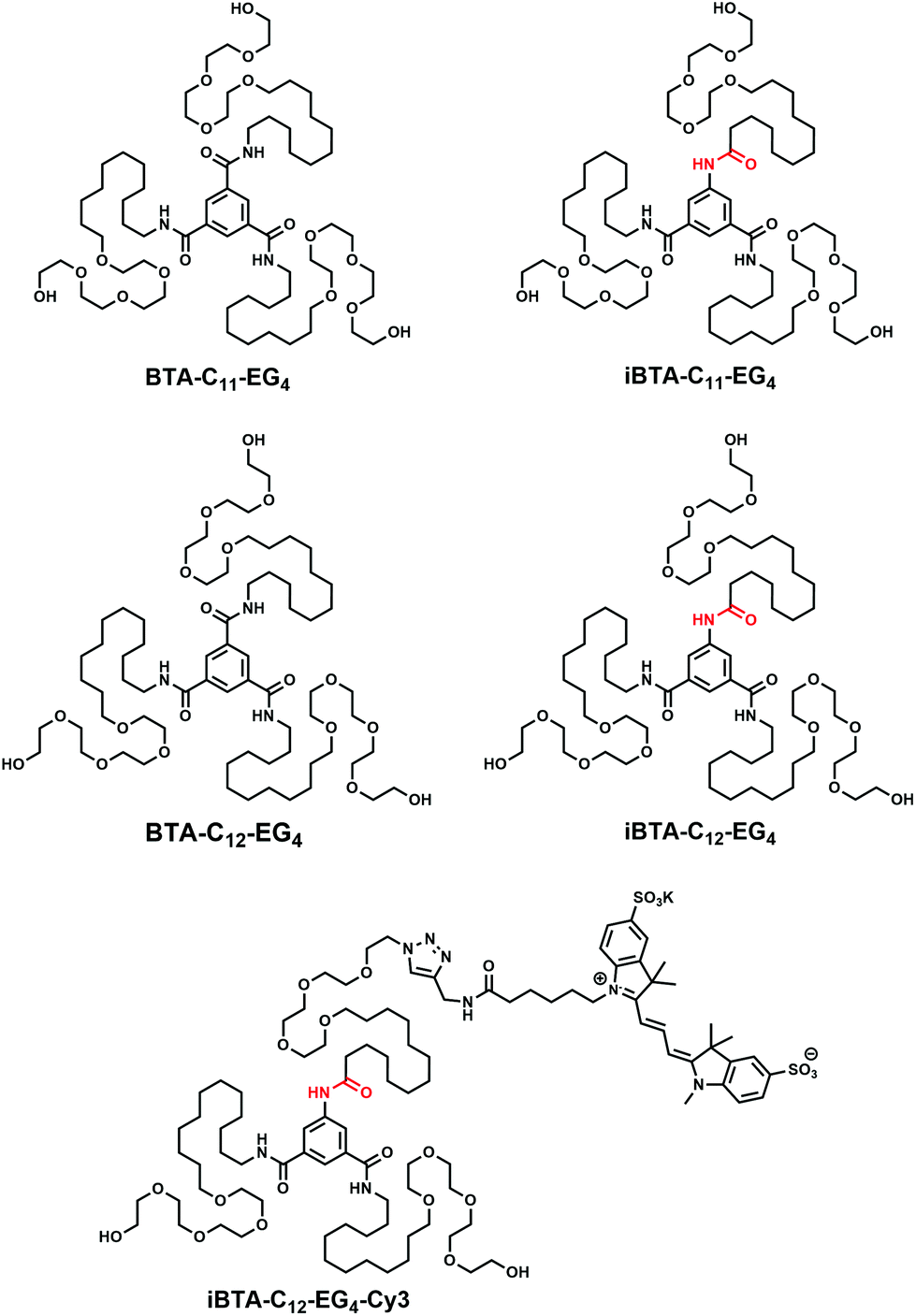 | ||
| Chart 1 Chemical structures of BTA-C11-EG4, iBTA-C11-EG4, BTA-C12-EG4, iBTA-C12-EG4 and iBTA-C12-EG4-Cy3. The inversed amide bonds are indicated in red. | ||
Linear peptide amphiphiles and UPy monomers generally contain only one side that is suitable for functionalization, whereas the discotic molecules comprise a C3-symmetrical core containing three reactive sites for the attachment of functional groups. As it is often favourable to use monovalent monomers to prevent solubility issues and unfavourable steric effects,28,32 this inherently introduces lengthy synthesis protocols for the desymmetrization of the monomers, often affording limited yields. For C-centred BTAs, this involves the statistical deprotection of a trimethyl 1,3,5-benzenecarboxylate core, creating multiple by-products which limits the yield.33 A strategy to circumvent statistical reactions and to introduce one different side arms onto the core is to start from a C2-symmetrical core.34–41 The group of Bouteiller synthesized N-substituted benzene-1-urea-3,5-biscarboxamide (BUBA) monomers with a significantly enhanced yield, starting from a commercially available dimethyl 5-aminoisophthalate core.42 The BUBA molecules showed cooperative self-assembly into long helices in organic media, similar to BTAs. Despite the altered hydrogen-bonding ability of the urea moieties compared to the amides, the BUBA molecules were readily incorporated into the symmetrical BTA-based supramolecular polymers.
Inspired by the work of Bouteiller et al.,42 we here selectively introduce function onto water-compatible BTAs by starting from a C2-symmetrical 5-aminoisophthalic acid core. This affords a BTA core with the connectivity of one of the amides inverted from C-centred to N-centred, which we refer to as iBTA. First, we evaluate if this single inversion of amide connectivity affects the supramolecular self-assembly behaviour by synthesizing two unfunctionalized iBTAs. The aqueous self-assembly of iBTAs with an undecyl (iBTA-C11-EG4, Chart 1) and dodecyl aliphatic chains (iBTA-C12-EG4, Chart 1) is compared to their C-centred counterparts using UV and Fourier transform infrared (FT-IR) spectroscopy, static light scattering (SLS), cryogenic transmission electron microscopy (cryoTEM) and hydrogen/deuterium exchange followed by mass spectrometry (HDX-MS). Furthermore, we explore the incorporation of iBTAs into supramolecular polymers of BTAs. Finally, we report the synthesis of an iBTA functionalized with a cyanine dye (iBTA-C12-E4-Cy3, Chart 1) and visualise its co-assembly with a C-centred BTA using stochastic optical reconstruction microscopy (STORM).43–47 High resolution microscopy images show that the synthetically easily accessible iBTAs can indeed be used for the introduction of function into BTA-based supramolecular polymers.
Results and discussion
Design and synthesis of iBTAs
Non-functional iBTAs (Chart 1) were designed to be atomically identical to their C3-symmetrical counterparts, but differ in the connectivity of one amide bond. The synthesis of iBTAs starts from 5-aminoisophthalic acid or dimethyl 5-aminoisophthalate. For iBTA-C11-EG4 and iBTA-C12-EG4, the side chains consist of an aliphatic spacer (undecyl and dodecyl, respectively) to provide a hydrophobic pocket which protects the hydrogen bonds between the amides of the cores,24 connected to tetra(ethylene glycol) to provide water compatibility.23 In iBTA-C12-EG4-Cy3, the side chain that is attached to the amine is decorated with a fluorescent sulfonated Cyanine3-dye (sulfo-Cy3) that permits STORM imaging. Hereby the possibility to exploit the desymmetrised core for the controlled introduction of function on only one side chain of the monomer is illustrated.The synthesis of the iBTAs requires side chains comprising either an amine or a carboxylic acid group for coupling to the core. Amine-terminated side chains (7a/b, Scheme S1 and section 2 of the ESI†) were synthesized based on optimized literature procedures.23 For the carboxylic acid-terminated chains (11a/b, Scheme S2 and section 3 of the ESI†), a new synthetic procedure was developed. Herein, a carboxylic acid is protected with a tert-butyl group to limit transesterification reactions (Fig. S1, ESI†). A subsequent Williamson ether synthesis with tetra(ethylene glycol) and deprotection of the carboxylic acid yields the desired side chain (11a/b). The desired iBTAs can be obtained via two pathways, which are demonstrated in the synthesis of both iBTA-C11-EG4 and iBTA-C12-EG4. iBTA-C11-EG4 was synthesized by attaching the carboxylic acid terminated side chain (11a) to dimethyl 5-aminobenzene-1,3-dicarboxylate. After hydrolysis of the methyl esters, the diacid (16) was reacted with the amine terminated side chains (7a) (Scheme S3 and section 4 of the ESI†) yielding pure iBTA-C11-EG4 as confirmed by NMR (Fig. S2 and 3, ESI†), FT-IR and LC-MS. In the second pathway for the synthesis of iBTA-C12-EG4 (Scheme 1), the amine terminated side chains (7b) were attached first to 5-aminobenzene-1,3-dicarboxylic acid after activation of the carboxylic acids using thionyl chloride. The resulting intermediate (1) was stable under argon in the fridge. For iBTA-C12-EG4 the synthesis was continued by coupling of the carboxylic acid terminated side chain (11b) to 1 resulted in a benzyl protected iBTA (2) which after hydrogenation yielded iBTA-C12-EG4 in high purity as confirmed by NMR (Fig. S4 and 5, ESI†), FT-IR and LC-MS.
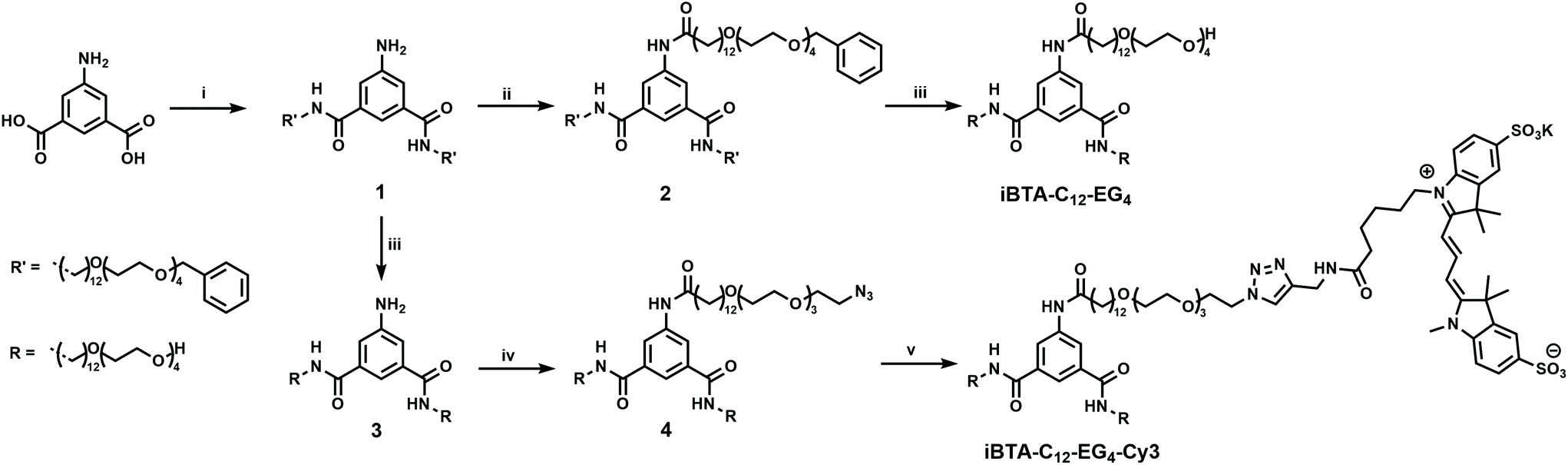 | ||
| Scheme 1 Synthesis of iBTA-C12-EG4 and iBTA-C12-EG4-Cy3. Reagents and conditions: (i) (1) SOCl2, reflux, 2 h. (2) H2N(CH2)12O(CH2CH2O)4OCH2Ph (7b), NEt3, THF, room temperature, 24 h (25%); (ii) (1) HOOC(CH2)12O(CH2CH2O)4CH2Ph (11b), (COCl)2, DMF, DCM, room temperature, 3 h. (2) NEt3, room temperature, 24 h (74%); (iii) H2, 10% Pd/C, MeOH, room temperature, 24 h (iBTA-C12-EG4: 80%; 3: 80%); (iv) HOOC(CH2)12O(CH2CH2O)3CH2CH2N3 (14), DMT-MM, MeOH, room temperature, 24 h (25%); (v) Sulfo-Cy3-alkyne, CuSO4, sodium ascorbate, DMSO, room temperature, 24 h (quant.). | ||
We selected a peripheral azide moiety to introduce a functional group onto the iBTA scaffold, as this allows to directly attach different types of functionalities via copper(I)-catalysed azide–alkyne cycloaddition (CuAAC) or via a Staudinger reduction of the azide to yield a reactive primary amine. The carboxylic acid terminated side chain was therefore equipped with an azide-derivative of tetra(ethylene glycol) that was synthesized according to a literature procedure.48 The azide group in the periphery required a minor adaptation of the synthetic route, since azides are known to form side products in a catalytic hydrogenation due to imine formation.49 Although ammonia can be added to prevent the formation of the intermediate, the catalytic hydrogenation is also effectively hindered by this addition.50 Therefore, 1 was catalytically hydrogenated to 3 prior to the introduction of the azide-decorated side chain (14, Scheme 1). The azide-decorated iBTA-C12-EG4-N3 (4) was obtained in high purity as confirmed by NMR (Fig. S6 and 7, ESI†), FT-IR and LC-MS. Finally, iBTA-C12-EG4-N3 was reacted with an alkyne functionalized sulfo-Cy3-dye in a CuAAC reaction to yield iBTA-C12-EG4-Cy3 as confirmed by LC-MS.
Supramolecular polymerization of iBTA-C11-EG4 and iBTA-C12-EG4 in water
We applied a previously optimised sample preparation protocol to assess the self-assembly of the newly synthesized iBTA monomers into supramolecular polymers.25 Using this protocol, a clear solution was obtained for iBTA-C11-EG4 after overnight equilibration (Fig. S8A, ESI†) but the sample with iBTA-C12-EG4 was turbid, indicating that the material did not completely dissolve. To compensate for the decreased solubility in water of iBTA-C12-EG4, the compound was injected into water from a concentrated stock solution in acetonitrile (ACN), followed by controlled heating and cooling of the sample as described before. A final concentration of 10 vol% of ACN in H2O was required to obtain samples that did not show any scattering (Fig. S8B, ESI†).The self-assembly of the iBTAs in aqueous media was first studied with UV spectroscopy. We previously established that BTA-C11-EG4 and BTA-C12-EG4 form micrometre long supramolecular polymers, with characteristic absorbance bands at 211 and 226 nm (Fig. 1A). In contrast, the UV spectra of iBTAs are red shifted, with iBTA-C11-EG4 showing a maximum (λmax) at 237 nm and iBTA-C12-EG4 at 230 nm. Both spectra also contain a shoulder around 268 nm and a broad band of low intensity between 290–310 nm. The spectra of both iBTA in aqueous media differ significantly compared to those in pure ACN, a solvent in which they are molecularly dissolved (Fig. S8, ESI†). This indicates that iBTAs self-assemble in water. The small differences in λmax between iBTA-C11-EG4 and iBTA-C12-EG4 probably result from the presence of 10% ACN in the latter, which may affect the propensity to form intermolecular hydrogen bonds.51 The broad band between 290–310 nm has previously been observed for symmetrical N-centred BTAs in organic media21 and is connected to the switch in connectivity of one amide bond. The spectra of the iBTAs differ both in shape and intensity compared to their symmetrical BTA counterparts, which suggests that the inversion of one of the amides may affect the packing of the hydrogen bonds.
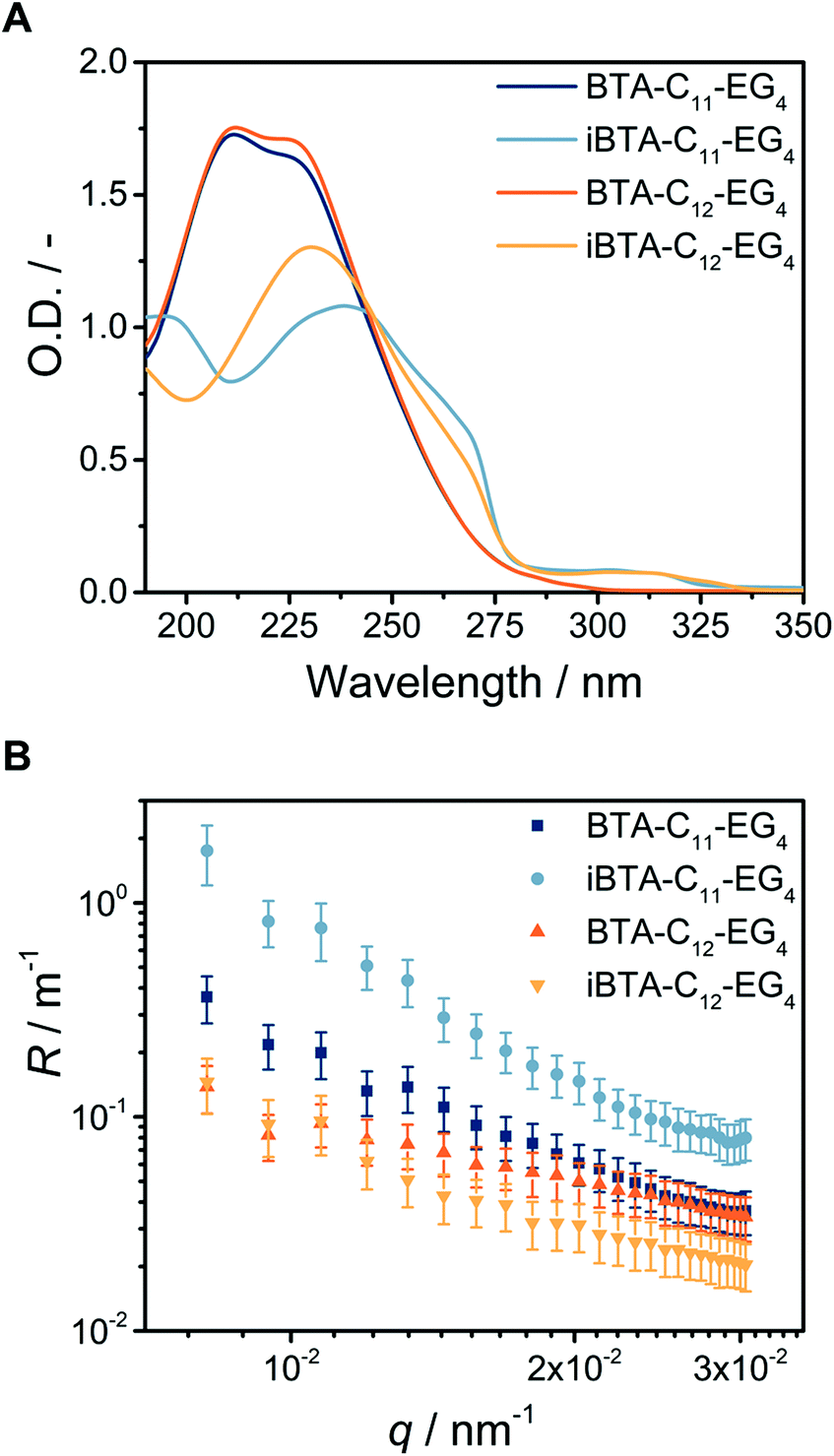 | ||
| Fig. 1 (A) UV spectra of BTA-C11-EG4, iBTA-C11-EG4, BTA-C12-EG4 and iBTA-C12-EG4 in water (c = 500 μM, l = 1 mm, T = 20 °C, 10 vol% ACN was present in the sample of iBTA-C12-EG4). (B) Rayleigh ratio R versus the scattering vector q as calculated from light scattering experiments with BTA-C11-EG4, iBTA-C11-EG4, BTA-C12-EG4 and iBTA-C12-EG4 in water (c = 500 μM, l = 1 cm, λ = 532 nm, T = 20 °C, 10 vol% ACN was present in the sample of iBTA-C12-EG4). | ||
To study the morphologies formed by iBTA-C11-EG4 and iBTA-C12-EG4 in water, SLS and cryoTEM measurements were performed and compared to those of the symmetrical counterparts BTA-C11-EG4 and BTA-C12-EG4. Like BTA-C11-EG4 and BTA-C12-EG4, iBTA-C11-EG4 and iBTA-C12-EG4 show an angular dependence of the Rayleigh ratio R that is typical for the presence of long, elongated structures (Fig. 1B). iBTA-C11-EG4 and iBTA-C12-EG4 show similar slopes, although the Rayleigh ratio was slightly increased for iBTA-C11-EG4 compared to iBTA-C12-EG4. These findings suggest that iBTAs form elongated structures with a similar anisotropy as their symmetrical counterparts, but they differ in size. CryoTEM was used to visualize the morphologies of the iBTAs. The cryoTEM image of iBTA-C11-EG4 shows fibres of several micrometres in length (Fig. 2A and Fig. S9, ESI†), whereas the image of iBTA-C12-EG4 shows fibres of various lengths, mainly below one micrometre (Fig. 2B and Fig. S10, ESI†). There are, however, subtle differences compared to the morphologies formed by symmetrical BTAs. Both C-centred BTAs assemble into micrometre long supramolecular polymers (Fig. S11, ESI†) and for BTA-C12-EG4 a double helix structure was recently revealed with high magnification cryoTEM and image reconstruction,52 whereas such a strand separation is absent in BTA-C11-EG4 (Fig. S11A, ESI†). iBTA-C11-EG4 also lacks the strand separation of a double helix structure (Fig. S9B, ESI†), whereas in the case of iBTA-C12-EG4 only a fraction of the fibres shows strand separation that may indicate the presence of a double helix (Fig. S10B, ESI†). From these results, we conclude that iBTA-C11-EG4 forms fibres of similar morphology as BTA-C11-EG4, and iBTA-C12-EG4 forms fibres that are slightly altered in length and secondary structure compared to BTA-C12-EG4. The decrease in length does not originate from the presence of ACN and is solely a result of the amide bond inversion. Further research with high magnification cryoTEM and image reconstruction should be done to confirm these findings.
 | ||
| Fig. 2 CryoTEM images of (A) iBTA-C11-EG4 and (B) iBTA-C12-EG4 in water (c = 500 μM, 10 vol% ACN was present in the sample of iBTA-C12-EG4) at a magnification of 24000×. | ||
The properties of the supramolecular polymers were further studied with spectroscopic techniques. FT-IR spectroscopy permits investigation of the presence of intermolecular hydrogen bonds in solution. The C3-symmetrical BTAs show an amide I vibration (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch) at 1648 cm−1 in the molecularly dissolved state and a vibration at 1635 cm−1 when intermolecular hydrogen bonds are present in D2O (Fig. S12A and B, ESI†). In the molecularly dissolved state, the amide I vibration of iBTA-C11-EG4 and iBTA-C12-EG4 was positioned at 1642 cm−1 and 1643 cm−1, respectively, with a shoulder around 1670 cm−1. The amide I vibration of both iBTAs was found at 1628 cm−1 after self-assembly in aqueous solution and the shoulder shifted to 1667 cm−1 (Fig. S12C and D, ESI†). This shift of the amide I vibration to lower wavenumbers after self-assembly is indicative for the formation of intermolecular hydrogen bonds. The split of the amide I vibration in case of the iBTAs could arise from the different connectivity of one of the amides in iBTAs. Although the FT-IR spectra of iBTAs are similar, they are shifted towards lower wavenumbers compared to the BTAs, suggesting a different hydrogen bond pattern and/or strength.
O stretch) at 1648 cm−1 in the molecularly dissolved state and a vibration at 1635 cm−1 when intermolecular hydrogen bonds are present in D2O (Fig. S12A and B, ESI†). In the molecularly dissolved state, the amide I vibration of iBTA-C11-EG4 and iBTA-C12-EG4 was positioned at 1642 cm−1 and 1643 cm−1, respectively, with a shoulder around 1670 cm−1. The amide I vibration of both iBTAs was found at 1628 cm−1 after self-assembly in aqueous solution and the shoulder shifted to 1667 cm−1 (Fig. S12C and D, ESI†). This shift of the amide I vibration to lower wavenumbers after self-assembly is indicative for the formation of intermolecular hydrogen bonds. The split of the amide I vibration in case of the iBTAs could arise from the different connectivity of one of the amides in iBTAs. Although the FT-IR spectra of iBTAs are similar, they are shifted towards lower wavenumbers compared to the BTAs, suggesting a different hydrogen bond pattern and/or strength.
In addition, we used the solvatochromic dye Nile Red (NR) to study the presence of hydrophobic regions in the iBTA-based structures. The emission spectra of NR when it is added to iBTA-C11-EG4 show a similar λmax,em (613 nm) compared to that of BTA-C11-EG4 (615 nm, Fig. S13A, ESI†). In contrast, the emission maximum of NR in iBTA-C12-EG4 is red-shifted to 626 nm, compared to that of BTA-C12-EG4 (612 nm, Fig. S13B, ESI†) indicative of a more polar environment, possibly a result of the presence of 10% ACN. We infer from this that a hydrophobic pocket is present in iBTA-C11-EG4, whereas a decisive conclusion cannot be drawn for iBTA-C12-EG4 due to the presence of 10% ACN.
Taken all together, the above results show that iBTAs form a primary supramolecular structure that is similar to that formed by symmetrical BTAs. Long, elongated fibres stabilised by intermolecular hydrogen bonds, which are surrounded by hydrophobic pockets are formed in aqueous media. The morphologies of iBTAs differ in secondary structure from the well-studied BTA-C12-EG4 in the sense that undulations due to a double helix structure are absent in iBTA-C11-EG4 and less pronounced in iBTA-C12-EG4. This may affect the exchange dynamics of monomers between polymers.
To assess the exchange dynamics of iBTAs, we measured hydrogen/deuterium exchange followed by mass spectrometry (HDX-MS). With HDX-MS, the exchange of labile hydrogen atoms to deuterium atoms is followed over time after dilution of an aqueous samples into D2O.25,26 The three outer hydroxyl hydrogen atoms of BTAs and iBTAs are in direct contact with the solvent and will exchange immediately to deuterium atoms. The three amide hydrogen atoms are contained in the hydrophobic pocket and form hydrogen bonds. As a result, their H/D exchange will mainly occur when the monomers move between polymers and are released into the surrounding D2O. HDX-MS is therefore a powerful tool to elucidate the rate of monomer exchange between polymers and permits to evaluate if the change in connectivity of one of the amides affects the exchange dynamics of the supramolecular polymers.
After confirming that the nature of the supramolecular polymers does not change upon dilution (Fig. S14, ESI†), all samples were 100-fold diluted into D2O and the percentage of deuterated analogues was followed over time (Fig. S15 and section 7 of the ESI†). The percentage of fully deuterated molecules can be used to compare the exchange dynamics between the different BTAs and iBTAs (Fig. 3). In all cases, a fast increase of deuteration to BTA6D is observed in the first hour, followed by a more gradual increase. After 48 h, 77% of the BTA-C11-EG4 polymers is fully deuterated but the deuteration takes longer for BTA-C12-EG4 of which 70% of the molecules is fully deuterated after 72 h. 79% of the iBTA-C11-EG4 molecules is completely deuterated after 48 hours and 81% of the iBTA-C12-EG4 molecules is completely deuterated after 72 h. The H/D exchange of both iBTAs follows a similar trend, indicating that it is not greatly influenced by the length of the hydrophobic chains. iBTA-C11-EG4 initially shows a faster exchange than BTA-C11-EG4, but the percentage of fully deuterated molecules is almost the same after 48 h. The percentage of fully deuterated molecules is higher for iBTA-C12-EG4 than for BTA-C12-EG4 over the whole period measured. This could indicate that the inversion of the amide bond increases the exchange dynamics, but it could also be an effect of the small percentage of ACN still present after the preparation of the sample.51 All in all, the exchange dynamics of both iBTAs and BTAs do not show large differences, indicating that the rates of exchange of molecules between polymers is rather similar.
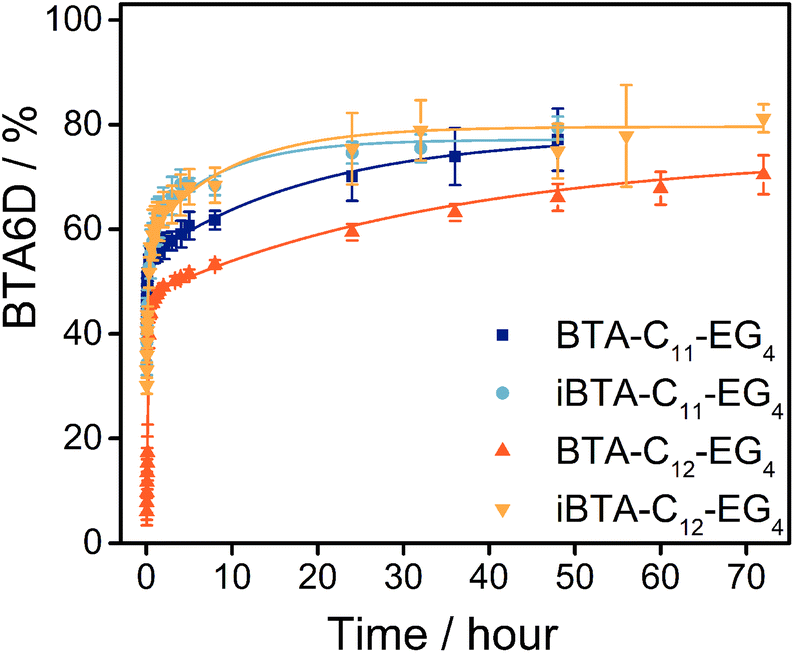 | ||
| Fig. 3 The percentage of fully deuterated BTA-C11-EG4, iBTA-C11-EG4, BTA-C12-EG4 and iBTA-C12-EG4 as a function of time after the 100× dilution of 500 μM aqueous samples into D2O (T = room temperature, 10 vol% ACN was present in the sample of iBTA-C12-EG4). The symbols represent the average and the error bars the standard deviation calculated from three independent measurements. The bi-exponential growth functions were added to guide the eye. | ||
Supramolecular copolymerization of BTA-C12-EG4 and iBTA-C12-EG4
The iBTAs have been designed to incorporate functional monomers into supramolecular polymers of BTAs, which requires the co-assembly of BTAs and iBTAs. To evaluate the ability of the molecules to copolymerize, samples of BTA-C12-EG4 with several percentages of iBTA-C12-EG4 were prepared. BTA-C12-EG4 and iBTA-C12-EG4 were mixed in the solid state before dissolving in water and applying the heating–cooling protocol as described before. ACN was not required to obtain homogeneous samples, since BTA-C12-EG4 helps to solubilize iBTA-C12-EG4 indicating their possible copolymerization. UV spectra of mixtures at a concentration of 500 μM do not show any changes in the absorption bands up to 10% of iBTA-C12-EG4 (Fig. 4A). At higher percentages of iBTA, a change in the absorption spectra is observed but the spectra never overlap with those of self-sorted systems (Fig. S16A, ESI†), suggesting an interaction between BTA-C12-EG4 and iBTA-C12-EG4. The UV spectra indicate that iBTA and BTA form copolymers over a wide range of concentrations (Fig. S16B, ESI†). The formation of supramolecular copolymers of BTA-C12-EG4 and iBTA-C12-EG4 was further corroborated with SLS (Fig. S17, ESI†), which revealed the presence of structures of similar dimensions as BTA-C12-EG4 polymers.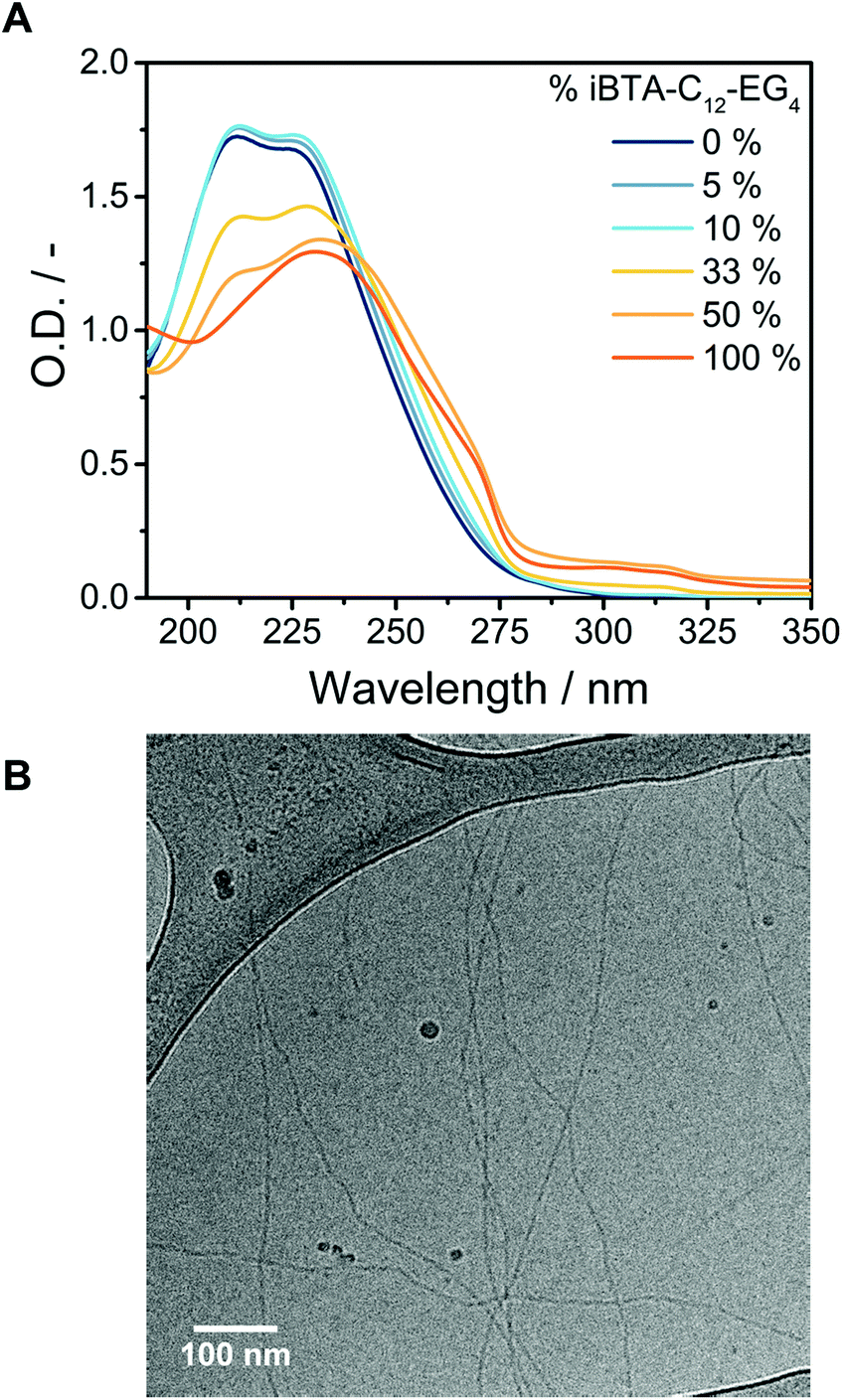 | ||
Fig. 4 (A) UV spectra of mixtures of BTA-C12-EG4 and iBTA-C12-EG4 in different ratios in water (c = 500 μM, l = 1 mm, T = 20 °C). (B) CryoTEM image of the mixture of BTA-C12-EG4 with 10% iBTA-C12-EG4 in water (c = 500 μM) at a magnification of 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×. The dark spherical objects are crystalline ice particles. 000×. The dark spherical objects are crystalline ice particles. | ||
Biomedical applications generally require only a fraction of the monomers within the supramolecular polymers to be functionalized.28,32 We therefore focus on the supramolecular copolymers containing a maximum of 10% of iBTA-C12-EG4. CryoTEM images verified the formation of the supramolecular copolymers upon mixing BTA-C12-EG4 with 10% iBTA-C12-EG4 (Fig. 4B). The copolymers are micrometres long and contain the double helix secondary structure as previously observed for BTA-C12-EG4 (Fig. S18, ESI†).52 HDX-MS experiments of the 10% co-assembly confirmed the interaction between BTA-C12-EG4 and iBTA-C12-EG4 (Fig. S19, ESI†). Additionally, those experiments revealed that the exchange dynamics of the copolymer match with those of the homopolymer of BTA-C12-EG4. From these results, we conclude that the structure and dynamics of BTA-C12-EG4 supramolecular polymers are not altered by the incorporation of a small percentage of iBTA-C12-EG4.
Introducing functionalized iBTAs into BTA-based supramolecular polymers
The aforementioned spectroscopic and microscopic experiments confirmed the insignificant changes in structure and monomeric packing between BTA and iBTA by inversion of one of the amide bonds. Although these experiments suggest copolymerization of BTA and iBTA, STORM was used to obtain visual confirmation for the incorporation of iBTA monomers into BTA-C12-EG4 fibres. Hereto, iBTA was functionalized with the fluorescent dye sulfo-Cy3 (iBTA-C12-EG4-Cy3). Dye-functionalized iBTA forms fibrous structures when mixed with iBTA-C12-EG4 as evidenced by cryoTEM (Fig. S20 and 21, ESI†). Our main interest is, however, to corroborate if iBTA-C12-EG4-Cy3 can introduce its functionality into fibres of BTA-C12-EG4. For this, a sample was prepared of supramolecular copolymers based on BTA-C12-EG4 with 10% iBTA-C12-EG4-Cy3 incorporated. Additionally, 5% BTA-C12-EG4-Cy5 was added for visualization of the polymers of BTA-C12-EG4 to confirm that the dye-labelled iBTAs co-assemble with BTA-C12-EG4 polymers and to rule out self-sorting of the iBTAs (see Fig. 5A for chemical structures relevant to this experiment). To avoid degradation of the dye at higher temperatures, the samples were prepared by few additional steps after the heating–cooling protocol (section 1, ESI†). The sample was equilibrated for several days, diluted into PBS and adsorbed on a coverslip. Dual channel imaging was performed by simultaneous excitation of Cy3 (green) and Cy5 (red). Normal resolution total internal reflection fluorescence (TIRF) microscopy and super resolution STORM imaging reveal micrometre long supramolecular polymers that contain both Cy3- and Cy5-labeled monomers (Fig. 5B), thereby confirming that the functionalized iBTA is present within the supramolecular polymers of BTA-C12-EG4 (see Fig. S22 and 23 for additional images, ESI†).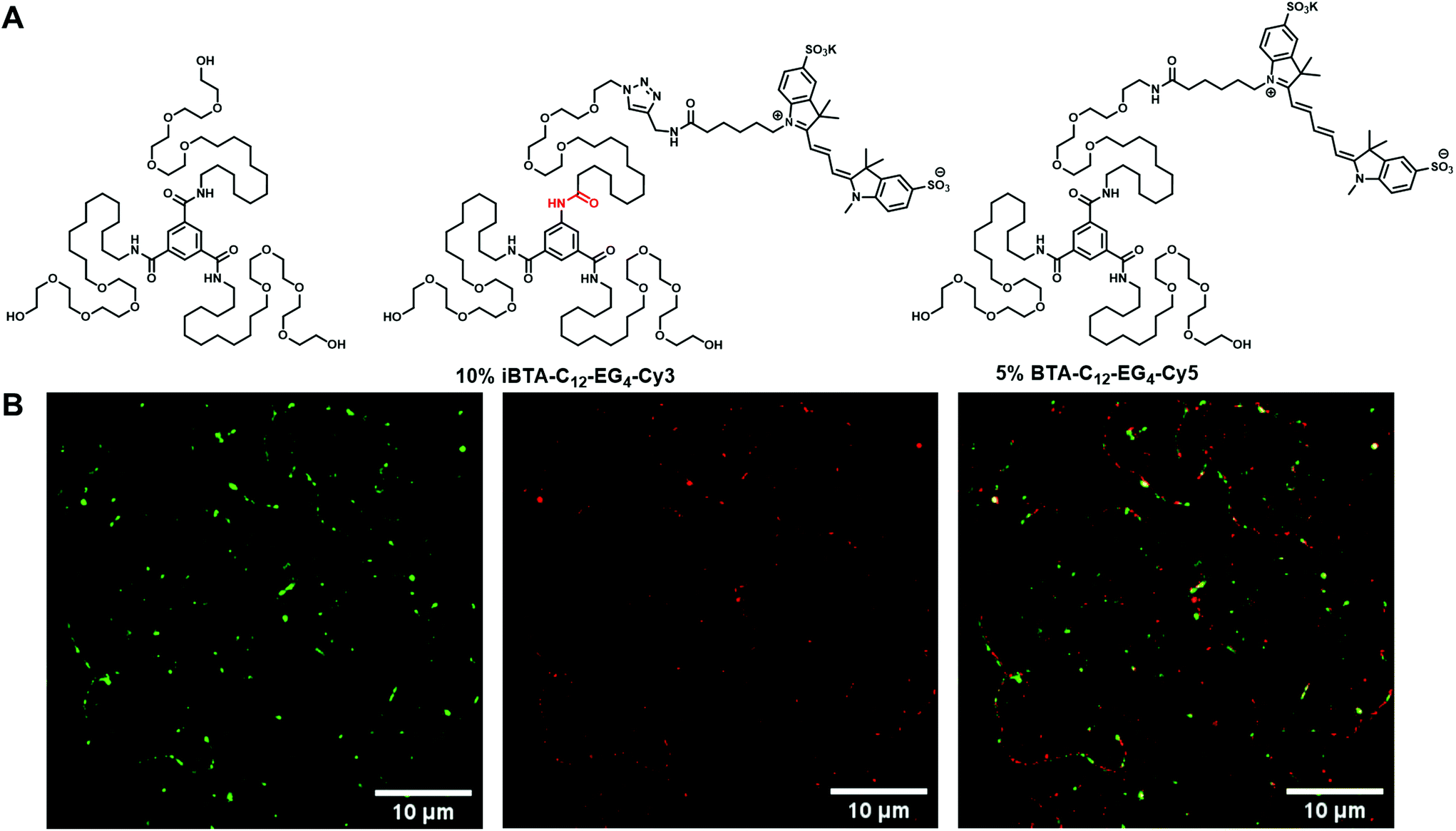 | ||
| Fig. 5 STORM experiment of a BTA polymer functionalized with iBTA. (A) Chemical structures of the components used in this experiment. A polymer of BTA-C12-EG4 was mixed with 10% iBTA-C12-EG4-Cy3 and 5% BTA-C12-EG4-Cy5. (B) STORM image of BTA-C12-EG4 mixed with dye-functionalized iBTA and BTA. The separate Cy3 (λex = 561 nm, green colour) and Cy5 (λex = 647 nm, red colour) channels and the merged image are presented. | ||
Conclusions
In conclusion, we demonstrated the synthesis of iBTAs by starting from a C2-symmetrical core to facilitate access to BTA-based supramolecular polymers that can be easily appended with a tuneable amount of functional groups. Hereto, we changed the terminal group of one of the side chains from an amine to a carboxylic acid, which required the same amount of synthetic steps and thus did not affect the synthetic effort. Furthermore, an azide moiety—acting as a handle for further functionalisation—was introduced in a three step synthesis similar to the procedure for an amine terminated side chain. By using a carboxylic acid terminated side chain in combination with C2-symmetrical 5-aminoisophtlalic acid, the statistical deprotection of trimethyl benzene-1,3,5-tricarboxylate was circumvented. The synthetic effort to obtain the desired core molecule was hereby reduced from 6 steps to 3 steps. Although the overall yield of the novel route is slightly lower than that of the previously reported method, we expect these yields to be improved upon further optimization of the reaction conditions.Since this newly developed synthesis protocol results in the inversion of the connectivity of one of the amide bonds, we first studied if this mutation would hamper the self-assembly of the iBTAs with unfunctional monomers. iBTA-C11-EG4 forms supramolecular polymers of similar morphology and dynamics as its C-centred counterpart. iBTA-C12-EG4, on the other hand, shows a reduced water solubility compared to BTA-C12-EG4, although they are atomically identical. By adding ACN as a cosolvent, the solubility is enhanced while the formation of supramolecular polymers is retained. The difference in solubility between iBTA-C11-EG4 and iBTA-C12-EG4, which differ by one methylene unit only, illustrates once more the importance of the delicate balance between hydrophobic and hydrophilic interactions that should be taken into consideration in the design of new supramolecular building blocks.
Despite their different amide bond orientation, iBTAs are fully compatible with symmetrical BTAs and the molecules were copolymerised in several ratios without the need for a cosolvent. Especially at lower percentages of iBTA, which is desired for the introduction of biological motifs, the copolymers adopt a morphology and exchange dynamics identical to homopolymers of the BTA. Visual proof of the co-assembly was provided by STORM microscopy of a dye-functionalized iBTA-C12-EG4.
Our results exemplify that iBTAs are a versatile platform for the functionalization of BTA-based polymers without altering the properties of the polymers. We envision that the introduction of function via building blocks with a small mismatch in core structure is not restricted to the BTA-based system presented here and this strategy should be applied to other building blocks used in the quest towards multipurpose synthetic biomaterials. Our future work will focus on the interaction of functional iBTAs embedded in BTA-based systems with biological material.
Experimental
Materials and methods
BTA-C11-EG4,24BTA-C12-EG423 and BTA-C12-EG4-Cy545 were synthesized according to literature procedures. All reagents and chemicals used were obtained from commercial sources at the highest purity available and used without further purification. All solvents were of AR quality and purchased from Biosolve. Deuterated compounds were obtained from Cambridge Isotope Laboratories and stored over 4 Å molecular sieves. Dry solvents were obtained using MBraun solvent purification system (MB SPS-800). Water for aqueous samples was purified on an EMD Millipore Milli-Q Integral Water Purification System. Glassware was dried in an oven at 135 °C overnight prior to reactions under dry conditions. Reactions were followed by thin-layer chromatography (TLC) using 60-F254 silica gel plates from Merck and visualized by UV light at 254 nm and/or staining (ninhydrin, bromocresol green, potassium permanganate, iodine chamber). Flash column chromatography was performed on a Grace Reveleris X2 chromatography system using Reveleris Silica Flash cartridges. Reversed phase column chromatography was achieved using a reversed phase KP-C18-HS SNAP column or a Reveleris C18 reversed-phase flash cartridge. All 1H NMR and 13C NMR spectra were recorded on Bruker Ultrashield spectrometers (400 MHz for 1H NMR and 100 MHz for 13C NMR). Proton chemical shifts are reported in ppm (δ) downfield from trimethyl silane (TMS) using the resonance frequency of the deuterated solvent (CDCl3; 7.26 ppm, DMSO-d6; 2.50 ppm) as the internal standard. Peak multiplicities are abbreviated as s: singlet; d: doublet; m: multiplet; t: triplet, p: pentet, dd: double doublet. Carbon chemical shifts are reported in ppm (δ) downfield from TMS using the resonance frequency of the deuterated solvents (CDCl3; 77.16 ± 0.06 ppm; DMSO-d6; 39.52 ± 0.06 ppm) as the internal standard. Infrared spectra were recorded using a PerkinElmer Spectrum Two FT-IR spectrometer equipped with a PerkinElmer Universal ATR Two Accessory. All solid-state spectra were measured at room temperature from 500 cm−1 to 4000 cm−1 and were averaged over 16 scans. Liquid chromatography mass spectroscopy (LC-MS) spectra were acquired using a device consisting of multiple components: Shimadzu SCL-10 A VP system controller with Shimadzu LC-10AD VP liquid chromatography pumps (with an Alltima C18 3 u (50 × 2.1 mm) reversed-phase column and gradients of water), a Shimadzu DGU 20A3 prominence degasser, a Thermo Finnigan surveyor auto sampler, a Thermo Finnigan surveyor PDA detector and a Thermo Scientific LCW Fleet. All samples were dissolved in 1:1 H2O:ACN in ca. 0.1 mg mL−1 concentration. Matrix assisted laser absorption/ionization mass time of flight mass spectroscopy (MALDI-TOF-MS) spectra were obtained on a Bruker Autoflex Speed. α-Cyano-4-hydroxycinnamic acid (CHCA) or trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCBT) were used as matrices. All samples were dissolved in either DCM or CHCl3 with a concentration of 1.0 mg mL−1.
Synthetic procedures
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2Br), 2.16 (t, J = 7.3 Hz, 2H, tBuOOCC2), 1.77 (p, J = 7.0 Hz, 2H, CH2C2CH2Br), 1.48 (m, 2H, tBuOOCCH2C2), 1.39 (s, 9H,
2Br), 2.16 (t, J = 7.3 Hz, 2H, tBuOOCC2), 1.77 (p, J = 7.0 Hz, 2H, CH2C2CH2Br), 1.48 (m, 2H, tBuOOCCH2C2), 1.39 (s, 9H, ![[t with combining low line]](https://www.rsc.org/images/entities/i_char_0074_0332.gif)
![[B with combining low line]](https://www.rsc.org/images/entities/char_0042_0332.gif)
![[u with combining low line]](https://www.rsc.org/images/entities/char_0075_0332.gif) OOC), 1.24 (s, 12H, aliphatic). 13C NMR (100 MHz, CDCl3δ): 173.36, 79.90, 63.12, 35.63, 34.06, 32.84, 29.46, 29.42, 29.40, 29.28, 29.09, 28.76, 28.18, 28.13, 25.11. FT-IR (ATR) ν (cm−1): 3459, 3007, 2975, 2926, 2855, 1729, 1457, 1392, 1366, 1255, 1151, 1043, 952, 919, 848, 755, 723, 645, 563, 462.
2Br), 2.20 (t, J = 7.5 Hz, 2H, tBuOOCC2CH2), 1.85 (p, J = 7.0 Hz, 2H, C2CH2Br), 1.69–1.66 (m, 2H, tBuOOCCH2C2), 1.45–1.36 (m, 11H, OOCCH2, C2CH2CH2Br), 1.36–1.21 (m, 14H, aliphatic). 13C NMR (100 MHz, CDCl3δ): 173.36, 79.89, 60.40, 35.64, 34.06, 32.85, 29.54, 29.50, 29.45, 29.43, 29.29, 29.10, 28.77, 28.18, 28.13, 25.12. FT-IR (ATR) ν (cm−1): 3424, 2975, 2916, 2850, 1718, 1473, 1366, 1218, 1150, 1108, 847, 718.
2O), 3.66–3.64 (m, 14H, O(C2)2O), 3.58–3.56 (m, 2H, O(C2)2O), 3.44 (t, J = 6.8 Hz, 2H, CH2CH2C2O), 2.20 (t, J = 7.5 Hz, 2H, tBuOOCC2), 1.62–1.52 (m, 4H, tBuOOCCH2C2 & CH2C2CH2O), 1.44 (s, 9H, OOCCH2), 1.35–1.20 (m, 14H, aliphatic). 13C NMR (100 MHz, CDCl3δ): 173.37, 128.36, 127.75, 127.59, 79.89, 73.25, 71.56, 70.66, 70.62, 70.05, 69.44, 35.64, 31.89, 29.64, 29.59, 29.56, 29.50, 29.48, 29.32, 29.11, 28.13, 26.10, 25.13, 22.71, 14.13. FT-IR (ATR) ν (cm−1): 2925, 2855, 1730, 1455, 1366, 1250, 1144, 1103, 1043, 946, 848, 736, 698, 464.
OOC), 1.24 (s, 12H, aliphatic). 13C NMR (100 MHz, CDCl3δ): 173.36, 79.90, 63.12, 35.63, 34.06, 32.84, 29.46, 29.42, 29.40, 29.28, 29.09, 28.76, 28.18, 28.13, 25.11. FT-IR (ATR) ν (cm−1): 3459, 3007, 2975, 2926, 2855, 1729, 1457, 1392, 1366, 1255, 1151, 1043, 952, 919, 848, 755, 723, 645, 563, 462.
2Br), 2.20 (t, J = 7.5 Hz, 2H, tBuOOCC2CH2), 1.85 (p, J = 7.0 Hz, 2H, C2CH2Br), 1.69–1.66 (m, 2H, tBuOOCCH2C2), 1.45–1.36 (m, 11H, OOCCH2, C2CH2CH2Br), 1.36–1.21 (m, 14H, aliphatic). 13C NMR (100 MHz, CDCl3δ): 173.36, 79.89, 60.40, 35.64, 34.06, 32.85, 29.54, 29.50, 29.45, 29.43, 29.29, 29.10, 28.77, 28.18, 28.13, 25.12. FT-IR (ATR) ν (cm−1): 3424, 2975, 2916, 2850, 1718, 1473, 1366, 1218, 1150, 1108, 847, 718.
2O), 3.66–3.64 (m, 14H, O(C2)2O), 3.58–3.56 (m, 2H, O(C2)2O), 3.44 (t, J = 6.8 Hz, 2H, CH2CH2C2O), 2.20 (t, J = 7.5 Hz, 2H, tBuOOCC2), 1.62–1.52 (m, 4H, tBuOOCCH2C2 & CH2C2CH2O), 1.44 (s, 9H, OOCCH2), 1.35–1.20 (m, 14H, aliphatic). 13C NMR (100 MHz, CDCl3δ): 173.37, 128.36, 127.75, 127.59, 79.89, 73.25, 71.56, 70.66, 70.62, 70.05, 69.44, 35.64, 31.89, 29.64, 29.59, 29.56, 29.50, 29.48, 29.32, 29.11, 28.13, 26.10, 25.13, 22.71, 14.13. FT-IR (ATR) ν (cm−1): 2925, 2855, 1730, 1455, 1366, 1250, 1144, 1103, 1043, 946, 848, 736, 698, 464.
1H NMR (400 MHz, CDCl3δ): 7.35–7.29 (m, 5H, Ar), 4.57 (s, 2H, ArC2O), 3.66–3.64 (m, 14H, O(C2)2O), 3.58–3.56 (m, 2H, O(C2)2O), 3.44 (t, J = 6.8 Hz, 2H, CH2CH2C2O), 2.20 (t, J = 7.5 Hz, 2H, tBuOOCC2), 1.62–1.52 (m, 4H, tBuOOCCH2C2 & CH2C2CH2O), 1.44 (s, 9H, OOCCH2), 1.35–1.20 (m, 16H, aliphatic). 13C NMR (100 MHz, CDCl3δ): 138.29, 128.35, 127.74, 127.58, 79.88, 77.22, 73.25, 71.56, 70.66, 70.63, 70.06, 69.45, 35.65, 29.65, 29.60, 29.51, 29.32, 29.11, 28.14, 26.10, 25.14. FT-IR (ATR) ν (cm−1): 2922, 2853, 1728, 1455, 1365, 1247, 1148, 1101, 845, 737, 698. LC-MS: m/z calculated for C32H56O7 + Na+: 575.39 [M + Na]+; observed 575.42; m/z calculated for C28H46O6 + NH4+: 496.70 [transesterification/elimination product + NH4]+; observed 496.25 (section 2 of the ESI†).
OOCCH2), 7.31 (m, 5H, Ar), 4.49 (s, 2H, ArC2O), 3.60–3.40 (m, 16H, O(CH2)2O), 3.33 (m, 2H, CH2C2O), 2.18 (t, J = 7.3 Hz, 2H, HOOCC2CH2), 1.47 (m, 4H, HOOCCH2C2 & C2CH2O), 1.33–1.15 (m, 14H, aliphatic). 13C NMR (100 MHz, DMSO-d6δ): 174.95, 138.95, 128.66, 127.93, 127.81, 72.49, 70.77, 70.28, 69.95, 69.60, 34.15, 31.73, 29.68, 29.49, 29.43, 29.37, 29.22, 29.03, 28.85, 26.12, 24.97, 22.58, 14.43. FT-IR (ATR) ν (cm−1): 3584–2440, 2928, 2852, 1733, 1706, 1456, 1353, 1251, 1096, 938, 845, 735, 698. MALDI-TOF-MS: m/z calculated for C27H46O7 + Na+: 505.31 [M + Na]+; observed 505.35; m/z calculated for C27H46O7 + K+: 521.42 [M + K]+; observed 521.32.
OOCCH2), 7.31 (m, 5H, Ar), 4.49 (s, 2H, ArC2O), 3.60–3.40 (m, 16H, O(C2)2O), 3.33 (m, 2H, CH2C2O), 2.18 (t, J = 7.3 Hz, 2H, HOOCCH2C2), 1.47 (m, 4H, HOOCCH2C2 & C2CH2O), 1.33–1.15 (m, 16H, aliphatic). 13C NMR (100 MHz, DMSO-d6δ): 174.95, 138.94, 128.65, 127.92, 127.81, 72.49, 70.76, 70.32, 70.28, 70.24, 69.94, 69.60, 34.12, 31.73, 29.67, 29.49, 29.45, 29.37, 29.33, 29.21, 29.01, 26.11, 24.96. FT-IR (ATR) ν (cm−1): 3719–2365, 2921, 2856, 1737, 1703, 1454, 1349, 1242, 1101, 940, 739, 698. MALDI-TOF-MS: m/z calculated for C28H48O7 + Na+: 519.33 [M + Na]+; observed 519.33; m/z calculated for C28H48O7 + K+: 535.44 [M + K]+; observed 535.30.
2)2O), 3.58 (m, 2H, O(C2)2O), 3.45 (t, J = 6.8 Hz, 2H, CH2CH2C2O), 3.39 (t, J = 5.1 Hz, 2H, N3C2), 2.20 (t, J = 7.5 Hz, 2H, tBuOOCC2CH2), 1.56 (m, 4H, tBuOOCCH2C2 & CH2C2CH2O), 1.44 (s, 9H, OOCH2), 1.21 (m, 16H, aliphatic). 13C NMR (100 MHz, CDCl3δ): 79.88, 77.21, 71.56, 70.71, 70.65, 70.06, 50.71, 35.65, 31.89, 29.65, 29.60, 29.49, 29.32, 29.11, 29.03, 28.13, 26.11, 25.13, 22.70, 14.12. FT-IR (ATR) ν (cm−1): 3455, 2922, 2853, 2104, 1729, 1460, 1367, 1247, 1147, 1102, 936, 851. LC-MS: m/z calculated for C25H49N3O6 + Na+: 510.35 [M + Na]+; observed 510.25; m/z calculated for C21H39N3O5 + Na+: 436.28 [transesterification/elimination product + Na]+; observed 436.17 (section 2 of the ESI†).
2), 3.60–3.40 (m, 16H, O-(C2)2-O), 3.33 (m, 2H, CH2C2O), 2.18 (t, J = 7.3 Hz, 2H, HOOCC2CH2), 1.47 (m, 4H, HOOCCH2C2 & C2CH2O), 1.33–1.15 (m, 16H, aliphatic). 13C NMR (100 MHz, DMSO-d6δ): 174.95, 70.77, 70.31, 70.27, 70.18, 69.95, 69.72, 60.22, 50.47, 34.12, 31.73, 29.67, 29.50, 29.45, 29.38, 29.33, 29.21, 29.02, 28.84, 26.12, 24.96, 22.57, 21.22, 14.55, 14.42. FT-IR (ATR) ν (cm−1): 3650–2450, 2924, 2854, 2098, 1731, 1708, 1454, 1349, 1284, 1243, 1102, 931, 837. MALDI-TOF-MS: m/z calculated for C21H41N3O6 + Na+: 454.29 [M + Na]+; observed 454.27.
COCH2), 8.50 (s, 2H, core next to amide), 8.15 (s, 1H, core between esters), 7.31 (m, 5H, Ar), 4.48 (s, 2H, ArC2O), 3.89 (s, 6H, 2× ArCOOC3), 3.49 (m, 16H, O(C2)2O), 3.34 (m, 2H, CH2C2O), 2.33 (t, J = 7.2 Hz, 2H, NHCOC2CH2), 1.59 (m, 2H, NHCOCH2C2), 1.45 (m, 2H, C2CH2O), 1.27 (m, 14H, aliphatic). 13C NMR (100 MHz, DMSO-d6δ): 172.42, 165.77, 140.69, 138.92, 131.08, 128.65, 127.92, 127.80, 124.22, 123.85, 72.47, 70.75, 70.31, 70.26, 70.22, 69.93, 69.58, 60.23, 52.99, 36.86, 31.73, 29.66, 29.47, 29.43, 29.34, 29.21, 29.03, 28.85, 26.11, 25.37, 22.58, 21.24, 14.56, 14.44. FT-IR (ATR) ν (cm−1): 3322, 2925, 2855, 1725, 1698, 1604, 1553, 1453, 1438, 1342, 1240, 1102, 1003, 949, 906, 873, 794, 759, 721, 698, 542, 464. LC-MS: m/z calculated for C37H55NO10 + H+: 674.39 [M + H]+; observed 674.25; m/z calculated for C37H55NO10 + NH4+: 691.41 [M + NH4]+; observed 691.17.
), 4.57 (s, 2H, ArC2O), 3.69–3.56 (m, 16H, O(C2)2O), 3.45–3.38 (m, 2H, CH2CH2C2O), 2.30–2.27 (t, J = 7.3 Hz, 2H, ArNHCOC2CH2), 1.58–1.47 (m, 2H, CH2C2CH2O), 1.47–1.44 (m, 2H, ArNHCOCH2C2), 1.27 (m, 14H, aliphatic). 13C NMR (100 MHz, DMSO-d6δ): 181.91, 128.66, 127.93, 72.47, 70.76, 70.26, 69.59, 36.86, 33.68, 32.83, 29.66, 29.49, 26.34, 26.12, 23.06. FT-IR (ATR) ν (cm−1): 3500–2500, 2917, 2855, 1625, 1559, 1435, 1358, 1095, 949, 779, 719, 697. MALDI-TOF-MS: m/z calculated for C35H51NO10 + Li+: 652.29 [M + Li]+; observed 652.39; m/z calculated for C35H51NO10 + Na+: 668.34 [M + Na]+; observed 668.37.
COCH2), 8.24 (s, 2H, core protons), 7.98 (s, 1H, core proton between regular amides), 7.34–7.27 (m, 15H, Ar), 6.52 (m, 2H, 2× ArCONCH2), 4.57 (s, 6H, 3× ArC2O), 3.69–3.54 (m, 48H, O(C2)2O), 3.47–3.38 (m, 10H, 3× CH2CH2C2O, 2× CONHC2CH2), 2.43–2.39 (t, J = 7.5 Hz, 2H, ArNHOCC2CH2), 1.73–1.68 (m, 2H, 1.85 ArNHOCCH2C2), 1.60–1.53 (m, 10H, 3× CH2C2CH2O, 2× ArCONHCH2C2), 1.36–1.21 (m, 42H, aliphatic). 13C NMR (100 MHz, CDCl3δ): 128.36, 127.77, 127.61, 77.23, 73.25, 71.55, 70.60, 70.05, 69.42, 64.47, 38.63, 29.60, 29.58, 29.51, 29.43, 29.36, 29.24, 26.94, 26.06, 25.38. FT-IR (ATR) ν (cm−1): 3320, 2926, 2853, 1648, 1536, 1425, 1349, 1096, 734, 697. LC-MS: m/z calculated for C87H141N3O18 + NH4+: 1534.05 [M + NH4]+; observed 1534.50; m/z calculated for C80H135N3O18 + H+: 1426.98 [M-benzyl + H]+; observed 1426.58; m/z calculated for C73H129N3O18 + H+: 1336.94 [M-2benzyl + H]+; observed 1336.58; m/z calculated for C66H123N3O18 + H+: 1246.89 [M-3benzyl + H]+; observed 1246.58; m/z calculated for C87H141N3O18 + 2(NH4+): 776.04 [M + 2NH4]2+; observed 776.33; m/z calculated for C80H135N3O18 + NH4+ + H: 722.51 [M-benzyl + NH4 + H]2+; observed 772.75; m/z calculated for C87H141N3O18 + 2(NH4+) + H+: 517.70 [M + 2NH4 + H]3+; observed 517.75.
COCH2), 8.24 (s, 2H, core protons), 7.98 (s, 1H, core proton between regular amides), 7.12 (m, 2H, 2× ArCONCH2), 3.69–3.62 (m, 48H, O(C2)2O), 3.47–3.38 (m, 10H, 3× CH2CH2C2O, 2× CONHC2CH2), 2.43–2.39 (t, J = 7.5 Hz, ArNHOCC2CH2), 1.73–1.68 (m, 2H, ArNHOCCH2C2), 1.60–1.53 (m, 10H, 3× CH2C2CH2O, 2× ArCONHCH2C2), 1.36–1.21 (m, 42H, aliphatic). 13C NMR (100 MHz, CDCl3δ): 173.00, 166.78, 139.34, 135.45, 120.89, 72.55, 71.54, 70.49, 70.45, 70.37, 70.34, 70.10, 69.82, 61.46, 40.30, 37.34, 29.62, 29.44, 29.40, 29.36, 29.33, 29.19, 26.92, 25.96, 25.47. FT-IR (ATR) ν (cm−1): 3460, 3346, 3074, 2927, 2853, 1729, 1656, 1595, 1546, 1460, 1428, 1346, 1286, 1244, 1106, 943, 882, 837, 707, 697. MALDI-TOF-MS: m/z calculated for C66H123N3O18 + Na+: 1268.87 [M + Na]+; observed 1268.69; m/z calculated for C66H123N3O18 + K+: 1284.98 [M + K]+; observed 1284.85.
CH2), 4.57 (s, 4H, ArC2O), 3.68–3.62 (m, 28H, O(C2)2O), 3.58–3.56 (m, 4H, O(C2)2O), 3.45–3.38 (m, 8H, CH2CH2C2O, ArCONHC2CH2CH2), 1.60–1.54 (m, 8H, CH2C2CH2O, ArCONHCH2C2CH2), 1.38–1.26 (m, 32H, aliphatic). 13C NMR (100 MHz, CDCl3δ): 128.36, 127.76, 127.60, 77.21, 73.26, 71.54, 70.63, 70.06, 29.60, 29.49, 29.44, 29.43, 29.42, 29.40, 29.23, 26.05. FT-IR (ATR) ν (cm−1): 3322, 2924, 2854, 1619, 1593, 1524, 1454, 1348, 1291, 1100, 860, 732, 698. LC-MS: Calculated Mw: m/z calculated for C62H101N3O12 + H+: 1080.75 [M + H]+; observed 1080.58; m/z calculated for C62H101N3O12 + H++Na+: 551.87 [M + H + Na]2+; observed 551.75; m/z calculated for C55H95N3O12 + H+: 990.70 [M-benzyl + H]+; observed 990.58.
1H NMR (400 MHz, CDCl3δ): 8.19 (s, 2H, core protons), 7.93 (s, 1H, core proton), 7.73 (m, 1H, ArNCOCH2), 7.34–7.27 (m, 15H, Ar), 6.39 (m, 2H, 2× ArCONCH2), 4.56 (s, 6H, ArC2O), 3.69–3.62 (m, 42H, O(C2)2O), 3.57–3.55 (m, 6H, O(C2)2O), 3.47–3.38 (m, 10H, 3× CH2CH2C2O, 2× ArCONHC2CH2), 2.43–2.39 (t, J = 7.5 Hz, 2H, ArNHCOC2CH2), 1.73–1.67 (m, 2H, 1.85 ArNHCOCH2C2), 1.60–1.53 (m, 10H, 3× CH2C2CH2O, 2× ArNHCOCH2C2), 1.36–1.21 (m, 48H, aliphatic). 13C NMR (100 MHz, CDCl3δ): 128.36, 127.76, 127.60, 77.22, 73.25, 71.55, 70.66, 70.61, 70.05, 69.44, 64.45, 29.48, 29.23, 26.93, 26.07, 25.37. FT-IR (ATR) ν (cm−1): 3315, 2924, 2854, 1642, 1536, 1451, 1349, 1293, 1247, 1104, 749, 698. LC-MS: m/z calculated for C90H147N3O18 + H+: 1559.08 [M + H]+; observed 1558.83; m/z calculated for C77H137N3O18 + H+: 1393.00 [M-2benzyl + H]+; observed 1378.75; m/z calculated for C90H147N3O18 + 2(Na+): 802.03 [M + 2Na]2+; observed 802.25; m/z calculated for C90H147N3O18 + 3(Na+): 542.35 [M + 3Na]3+; observed 542.75.
COCH2), 8.22 (s, 2H, core protons), 7.95 (s, 1H, core proton between regular amides), 6.61 (m, 2H, 2× ArCONCH2), 3.69–3.62 (m, 48H, O(C2)2O), 3.47–3.38 (m, 10H, 3× CH2CH2C2O, 2× ArCONHC2CH2), 2.43–2.39 (t, J = 7.5 Hz, 2H, 2× ArNHCOC2CH2), 1.73–1.68 (m, 2H, ArNHCOCH2C2), 1.60–1.53 (m, 10H, 3× CH2C2CH2O, 2× ArCONHCH2C2), 1.36–1.21 (m, 48H, aliphatic). 13C NMR (100 MHz, CDCl3δ): 172.60, 166.46, 139.23, 135.60, 120.62, 72.64, 71.57, 71.55, 70.62, 70.60, 70.56, 70.53, 70.27, 70.03, 61.69, 40.30, 37.49, 29.52, 29.46, 29.44, 29.40, 29.37, 29.35, 29.31, 29.22, 26.92, 26.04, 26.00, 25.43. FT-IR (ATR) ν (cm−1): 3608–3068, 2924, 2854, 1637, 1603, 1546, 1444, 1335, 1272, 1118, 935, 884, 721. LC-MS: m/z calculated for C69H129N3O18 + H+: 1288.84 [M + H]+; observed 1288.67; m/z calculated for C69H129N3O18 + 2(H+): 644.98 [M + 2H]2+; observed 944.92; m/z calculated for C69H129N3O18 + 3(H+): 430.32 [M + 3H]3+; observed 430.58.
:EtAc 50:50). Hereafter, the mixture was filtered over Celite and concentrated in vacuo to obtain the product as a white solid. Yield: 18.6 mg, 80%. 1H NMR (400 MHz, CDCl3δ): 7.51 (s, 1H, core), 7.20 (s, 2H, core), 6.71 (s, 2H, 2× ArCONCH2), 3.72–3.57 (m, 32H, O(C2)2O), 3.44–3.36 (m, 8H, CH2CH2C2O, ArCONHC2CH2CH2), 1.58–1.54 (m, 8H, CH2C2CH2O, ArCONHCH2C2CH2), 1.38–1.24 (m, 32H, aliphatic). 13C NMR (100 MHz, DMSO-d6δ): 79.79, 79.46, 79.13, 72.81, 70.80, 70.30, 70.28, 70.25, 69.95, 60.65, 29.69, 29.52, 29.50, 29.37, 29.24, 26.83, 26.13. FT-IR (ATR) ν (cm−1): 3298, 2925, 2855, 1640, 1555, 1462, 1348, 1312, 1250, 1208, 1102, 941, 884. MALDI-TOF-MS: m/z calculated for C48H89N3O12 + Na+: 922.63 [M + Na]+; observed 922.66; m/z calculated for C48H89N3O12 + K+: 938.74 [M + K]+; observed 938.62.
COCH2), 7.95 (s, 1H, core proton between regular amides), 6.59 (s, 2H, 2× ArCONCH2), 3.72–3.57 (m, 48H, O(C2)2O), 3.44–3.37 (m, 12H, 3× CH2CH2C2O, 2× ArCONHC2CH2, OCH2C2N3), 2.41 (t, J = 8.0 Hz, 2H, NHCOC2), 1.72–1.53 (water influence, m, 12H, 1× NHCOCH2C2CH2, 2× CONHCH2C2CH2, 3× C2CH2O(CH2)2O), 1.37–1.16 (m, 48H, aliphatic chain). 13C NMR (100 MHz, CDCl3δ): 172.43, 166.30, 139.11, 135.63, 120.52, 77.34, 77.02, 76.70, 72.62, 71.57, 70.70, 70.64, 70.61, 70.55, 70.52, 70.27, 70.06, 70.03, 70.01, 61.70, 50.69, 40.28, 37.58, 29.61, 29.53, 29.51, 29.49, 29.43, 29.37, 29.26, 29.15, 26.87, 26.06, 26.03, 25.44. FT-IR (ATR) ν (cm−1): 3464, 3305, 2926, 2856, 2102, 1643, 1594, 1539, 1441, 1346, 1278, 1109, 942, 888. LC-MS: m/z calculated for C69H128N6O17 + H+: 1313.94 [M + H]+; observed 1313.58; m/z calculated for C69H128N6O17 + Na+: 1335.92 [M + Na]+; observed 1335.83; m/z calculated for C69H128N6O17 + 2(H+): 657.48 [M + 2H]2+; observed 657.33; m/z calculated for C69H128N6O17 + H+ + Na+: 668.47 [M + H + Na]2+; observed 668.42; m/z calculated for C69H128N6O17 + 2(Na+): 679.46 [M + 2Na]2+; observed 679.50.
Author contributions
S. M. C. S., B. W. L. B., L. S. and A. R. A. P. planned the experiments. S. M. C. S., B. W. L. B. and S. D. carried out the experiments. S. M. C. S., B. W. L. B. and A. R. A. P. wrote the manuscript. All the authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Dutch Ministry of Education, Culture and Science (Gravity program 024.001.035) and the European Research Council (H2020-EU.1.1., SYNMAT project, ID 788618). A. J. H. Spiering and Nicholas Matsumoto are gratefully acknowledged for the synthesis of BTA-C11-EG4 and BTA-C12-EG4. Svenja Herziger and Christoph Böttcher are gratefully acknowledged for their support in the structural characterization of BTA-C11-EG4. We would like to thank E. W. Meijer for fruitful discussions.References
- C. Sanchez, H. Arribart and M. M. G. Guille, Nat. Mater., 2005, 4, 277–288 CrossRef CAS PubMed.
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef CAS PubMed.
- D. A. Fletcher and R. D. Mullins, Nature, 2010, 463, 485–492 CrossRef CAS PubMed.
- D. Lingwood and K. Simons, Science, 2010, 327, 46–50 CrossRef CAS PubMed.
- O. J. G. M. Goor, S. I. S. Hendrikse, P. Y. W. Dankers and E. W. Meijer, Chem. Soc. Rev., 2017, 46, 6621–6637 RSC.
- E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS PubMed.
- P. Y. W. Dankers, M. C. Harmsen, L. A. Brouwer, M. J. A. Van Luyn and E. W. Meijer, Nat. Mater., 2005, 4, 568–574 CrossRef CAS PubMed.
- K. Petkau-Milroy, M. H. Sonntag and L. Brunsveld, Chem. – Eur. J., 2013, 19, 10786–10793 CrossRef CAS PubMed.
- R. N. Shah, N. A. Shah, M. M. Del Rosario Lim, C. Hsieh, G. Nuber and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3293–3298 CrossRef CAS PubMed.
- A. Mata, Y. Geng, K. J. Henrikson, C. Aparicio, S. R. Stock, R. L. Satcher and S. I. Stupp, Biomaterials, 2010, 31, 6004–6012 CrossRef CAS PubMed.
- N. A. Mansukhani, E. B. Peters, M. M. So, M. S. Albaghdadi, Z. Wang, M. R. Karver, T. D. Clemons, J. P. Laux, N. D. Tsihlis, S. I. Stupp and M. R. Kibbe, Macromol. Biosci., 2019, 19, 1900066 CrossRef PubMed.
- R. Mammadov, B. Mammadov, S. Toksoz, B. Aydin, R. Yagci, A. B. Tekinay and M. O. Guler, Biomacromolecules, 2011, 12, 3508–3519 CrossRef CAS PubMed.
- S. I. S. Hendrikse, S. P. W. Wijnands, R. P. M. Lafleur, M. J. Pouderoijen, H. M. Janssen, P. Y. W. Dankers and E. W. Meijer, Chem. Commun., 2017, 53, 2279–2282 RSC.
- M. H. Bakker, R. E. Kieltyka, L. Albertazzi and P. Y. W. Dankers, RSC Adv., 2016, 6, 110600–110603 RSC.
- R. C. van Gaal, B. D. Ippel, S. Spaans, M. I. Komil and P. Y. W. Dankers, J. Polym. Sci., 2021, 1–14 Search PubMed.
- D. Straßburger, N. Stergiou, M. Urschbach, H. Yurugi, D. Spitzer, D. Schollmeyer, E. Schmitt and P. Besenius, ChemBioChem, 2018, 19, 912–916 CrossRef PubMed.
- K. Petkau-Milroy, M. H. Sonntag, A. H. A. M. van Onzen and L. Brunsveld, J. Am. Chem. Soc., 2012, 134, 8086–8089 CrossRef CAS PubMed.
- K. Petkau-Milroy, M. H. Sonntag, A. Colditz and L. Brunsveld, Int. J. Mol. Sci., 2013, 14, 21189–21201 CrossRef PubMed.
- M. K. Müller, K. Petkau and L. Brunsveld, Chem. Commun., 2011, 47, 310–312 RSC.
- S. Cantekin, T. F. A. de Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125 RSC.
- P. J. M. Stals, J. C. Everts, R. De Bruijn, I. A. W. Filot, M. M. J. Smulders, R. Martín-Rapún, E. A. Pidko, T. F. A. De Greef, A. R. A. Palmans and E. W. Meijer, Chem. – Eur. J., 2010, 16, 810–821 CrossRef CAS PubMed.
- J. Van Herrikhuyzen, P. Jonkheijm, A. P. H. J. Schenning and E. W. Meijer, Org. Biomol. Chem., 2006, 4, 1539–1545 RSC.
- C. M. A. Leenders, L. Albertazzi, T. Mes, M. M. E. Koenigs, A. R. A. Palmans and E. W. Meijer, Chem. Commun., 2013, 49, 1963–1965 RSC.
- C. M. A. Leenders, M. B. Baker, I. A. B. Pijpers, R. P. M. Lafleur, L. Albertazzi, A. R. A. Palmans and E. W. Meijer, Soft Matter, 2016, 12, 2887–2893 RSC.
- X. Lou, R. P. M. Lafleur, C. M. A. Leenders, S. M. C. Schoenmakers, N. M. Matsumoto, M. B. Baker, J. L. J. van Dongen, A. R. A. Palmans and E. W. Meijer, Nat. Commun., 2017, 8, 15420 CrossRef PubMed.
- X. Lou, S. M. C. Schoenmakers, J. L. J. van Dongen, M. Garcia-Iglesias, N. M. Casellas, M. Fernández-Castaño Romera, R. P. Sijbesma, E. W. Meijer and A. R. A. Palmans, J. Polym. Sci., 2021, 59, 1151–1161 CrossRef CAS PubMed.
- B. N. S. Thota, X. Lou, D. Bochicchio, T. F. E. Paffen, R. P. M. Lafleur, J. L. J. van Dongen, S. Ehrmann, R. Haag, G. M. Pavan, A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2018, 57, 6843–6847 CrossRef CAS PubMed.
- G. Morgese, B. F. M. Waal, S. Varela-Aramburu, A. R. A. Palmans, L. Albertazzi and E. W. Meijer, Angew. Chem., 2020, 59, 17382–17386 CrossRef.
- M. H. Bakker, C. C. Lee, E. W. Meijer, P. Y. W. Dankers and L. Albertazzi, ACS Nano, 2016, 10, 1845–1852 CrossRef CAS PubMed.
- S. I. S. Hendrikse, L. Su, T. P. Hogervorst, R. P. M. Lafleur, X. Lou, G. A. van der Marel, J. D. C. Codee and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 13877–13886 CrossRef CAS PubMed.
- S. P. W. Wijnands, W. Engelen, R. P. M. Lafleur, E. W. Meijer and M. Merkx, Nat. Commun., 2018, 9, 65 CrossRef PubMed.
- K. Petkau-Milroy and L. Brunsveld, Eur. J. Org. Chem., 2013, 3470–3476 CrossRef CAS.
- J. Roosma, T. Mes, P. Leclère, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2008, 130, 1120–1121 CrossRef CAS PubMed.
- J. J. Van Gorp, J. A. J. M. Vekemans and E. W. Meijer, Mol. Cryst. Liq. Cryst., 2003, 397, 191–205 CrossRef.
- S. Bera, S. K. Maity and D. Haldar, CrystEngComm, 2014, 16, 4834–4841 RSC.
- N. San-José, A. Gómez-Valdemoro, S. Ibeas, F. C. García, F. Serna and J. M. García, Supramol. Chem., 2010, 22, 325–338 CrossRef.
- S. Kumar, S. Bera, S. K. Nandi and D. Haldar, Soft Matter, 2021, 17, 113–119 RSC.
- M. U. Kassack, K. Braun, M. Ganso, H. Ullmann, P. Nickel, B. Böing, G. Müller and G. Lambrecht, Eur. J. Med. Chem., 2004, 39, 345–357 CrossRef CAS PubMed.
- M. Blomenhofer, S. Ganzleben, D. Hanft, H. W. Schmidt, M. Kristiansen, P. Smith, K. Stoll, D. Mäder and K. Hoffmann, Macromolecules, 2005, 38, 3688–3695 CrossRef CAS.
- Y. Zhou, M. Xu, T. Yi, S. Xiao, Z. Zhou, F. Li and C. Huang, Langmuir, 2007, 23, 202–208 CrossRef CAS PubMed.
- F. V. Gruschwitz, M. C. Fu, T. Klein, R. Takahashi, T. Higashihara, S. Hoeppener, I. Nischang, K. Sakurai and J. C. Brendel, Macromolecules, 2020, 53, 7552–7560 CrossRef CAS.
- Y. Li, L. Dubreucq, B. G. Alvarenga, M. Raynal and L. Bouteiller, Chem. – Eur. J., 2019, 25, 10650–10661 CrossRef CAS PubMed.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–795 CrossRef CAS PubMed.
- B. Huang, M. Bates and X. Zhuang, Annu. Rev. Biochem., 2009, 78, 993–1016 CrossRef CAS PubMed.
- L. Albertazzi, D. van der Zwaag, C. M. A. Leenders, R. Fitzner, R. W. van der Hofstad and E. W. Meijer, Science, 2014, 344, 491–495 CrossRef CAS PubMed.
- R. M. P. Da Silva, D. Van Der Zwaag, L. Albertazzi, S. S. Lee, E. W. Meijer and S. I. Stupp, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- S. Pujals, N. Feiner-Gracia, P. Delcanale, I. Voets and L. Albertazzi, Nat. Rev. Chem., 2019, 3, 68–84 CrossRef.
- L. Albertazzi, F. J. Martinez-Veracoechea, C. M. A. Leenders, I. K. Voets, D. Frenkel and E. W. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12203–12208 CrossRef CAS PubMed.
- M. Lange, A. L. Pettersen and K. Undheim, Tetrahedron, 1998, 54, 5745–5752 CrossRef CAS.
- H. Sajiki, Tetrahedron Lett., 1995, 36, 3465–3468 CrossRef CAS.
- R. P. M. Lafleur, X. Lou, G. M. Pavan, A. R. A. Palmans and E. W. Meijer, Chem. Sci., 2018, 9, 6199–6209 RSC.
- R. P. M. Lafleur, S. Herziger, S. M. C. Schoenmakers, A. D. A. Keizer, J. Jahzerah, B. N. S. Thota, L. Su, P. H. H. Bomans, N. A. J. M. Sommerdijk, A. R. A. Palmans, R. Haag, H. Friedrich, C. Böttcher and E. W. Meijer, J. Am. Chem. Soc., 2020, 142, 17644–17652 CrossRef CAS PubMed.
- T. Aiba, M. Sato, D. Umegaki, T. Iwasaki, N. Kambe, K. Fukase and Y. Fujimoto, Org. Biomol. Chem., 2016, 14, 6672–6675 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, synthetic procedures for sidechains and BTA-C11-EG4, supporting spectroscopy and microscopy images, details of the HDX-MS analysis, 1H and 13C NMR spectra of all new compounds. See DOI: 10.1039/d1ob01587g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |