
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A reference scale of cucurbit[7]uril binding affinities†
Mohammad A.
Alnajjar
 a,
Werner M.
Nau
*a and
Andreas
Hennig
*ab
a,
Werner M.
Nau
*a and
Andreas
Hennig
*ab
aDepartment of Life Sciences and Chemistry, Jacobs University Bremen, Campus Ring 1, 28759 Bremen, Germany. E-mail: w.nau@jacobs-university.de
bInstitute of Chemistry of New Materials, Universität Osnabrück, Barbarastr. 7, 49080 Osnabrück, Germany. E-mail: andreas.hennig@uni-osnabrueck.de
First published on 3rd August 2021
Abstract
The accurate determination of ultra-high binding affinities in supramolecular host–guest chemistry is a challenging endeavour because direct binding titrations are generally limited to affinities <106 M−1 due to sensitivity constraints of common titration methods. To determine higher affinities, competitive titrations are usually performed, in which one compound with a well established binding affinity serves as a reference. Herein, we propose a reference scale for such competitive titrations with the host cucurbit[7]uril (CB7) comprising binding affinities in the range from 103 to 1015 M−1. The suggested reference compounds are commercially available and will aid in the future determination of CB7 binding affinities for stimuli-responsive host–guest systems.
Introduction
The determination of binding constants is quintessential in supramolecular host–guest chemistry, because it provides a direct measure for analyzing the strength of intermolecular interactions between host and guest.1–3 Commonly, binding constants are determined by titrations, in which a physical property such as the chemical shift in NMR spectroscopy or the absorbance or fluorescence in optical spectroscopy is monitored, while the concentrations of the binding partners (host or guest) are being varied.4–7 The resulting binding titration curves are subsequently fitted to a suitable binding model to obtain the association constant, Ka, and, thereby, the interaction energy, ΔG, via ΔG = R·T![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnKa, where T is the absolute temperature and R the universal gas constant. Alternatively, isothermal titration calorimetry (ITC) is used to determine the thermodynamics of binding processes by measuring the heat evolved or absorbed upon association between guest and host to afford Ka, the complexation stoichiometry, n, and the binding enthalpy, ΔH. The binding entropy, ΔS, can be extracted according to ΔG = ΔH − TΔS.8–11
lnKa, where T is the absolute temperature and R the universal gas constant. Alternatively, isothermal titration calorimetry (ITC) is used to determine the thermodynamics of binding processes by measuring the heat evolved or absorbed upon association between guest and host to afford Ka, the complexation stoichiometry, n, and the binding enthalpy, ΔH. The binding entropy, ΔS, can be extracted according to ΔG = ΔH − TΔS.8–11
The reliability of the results gained from these titrations depends on a number of factors, which have been comprehensively summarized.6 For example, the analysis of more involved binding phenomena, such as the formation of ternary complexes involving different modes of cooperativity, requires a careful experimental planning and a critical data analysis. As another example, Benesi–Hildebrand plots remain popular, although it has often been noted that data linearization by (double)-reciprocal plots introduces large errors, which are not considered by standard least-squares fitting methods.6
However, even with careful consideration of all of these factors, binding constants with high reliability and, ideally, inter-laboratory reproducibility are often challenging to obtain. As an example from our own research, we have performed numerous binding titrations with cucurbit[7]uril (CB7) and acridine orange (AO) and obtained values that ranged from 5.7 × 104 M−1 to 3.1 × 106 M−1.12–15 We could finally trace this back to an undesirable interaction of AO with the walls of quartz glass cuvettes, which could be reduced, but not completely eliminated, in poly(methylmethacrylate) cuvettes.15
With respect to the family of the pumpkin-shaped cucurbit[n]urils (CBs with n = 5–8, 10 and 14),16–19 numerous host–guest binding constants have been reported for hydrocarbons, dyes, drugs, amino acids, and even for selected amino acid residues or sequences in peptides and proteins.20–22 The resulting data compilations are of indisputable scientific merit, but the reported individual values are occasionally difficult to compare due to variations in experimental conditions while in other cases they appear contradictory even if reported for the same conditions. For example, several binding constants have only been measured in buffer solutions with high salt concentrations;23 the associated data points can only be considered as apparent binding constants due to the competitive binding of cations at CB portals.24,25 A particular challenge arises from the very high binding affinities of CBs (Ka > 109 M−1).26,27 These preclude direct host–guest titrations, because the required nanomolar concentrations are typically too low to afford a detectable spectroscopic or calorimetric response.4–7 Access to CB binding constants is further limited by the very slow host–guest exchange rates at ultra-high affinities (Ka > 1012 M−1), which have been noted at several instances, for example for the protonated forms of amino-substituted cyclohexanes, adamantanes, diamantanes, and ferrocenes.26–32
Among the CB homologues, the focus is on the intermediary sized CB7, which is notoriously known for its extremely strong binding (up to Ka = 7.2 × 1017 M−1).27 This ultrahigh affinity has immense potential in biotechnology as well as analytical chemical applications,33–36 for example, CB7-beads can selectively capture proteins labelled with 1-trimethylammoniomethylferrocene from complex heterogenous protein mixtures.37
In an effort to obtain structure–activity relationships and to tune the binding affinities with CB7, Isaacs and co-workers have used competitive titrations with sub-stoichiometric amounts of the host and an excess of two competing guest molecules and established a reference scale of binding affinities in order to determine ultra-high binding affinities by multistep 1H NMR competition experiments.26,28,29 However, their key reference compound to assess binding constants with nanomolar and higher affinity, (3-aminopropyl)[(trimethylsilyl)methyl]amine, is not readily available.23,26–28,38
In order to facilitate the determination of CB7 host–guest binding affinities, we introduce herein a series of reference compounds that allow affinity determinations by competitive titrations in the range from <103 M−1 to >1015 M−1 (Scheme 1). The purpose of our rather analytical-chemical supramolecular study was not to measure new affinities of additional compounds, but rather to provide robust, mutually cross-checked, and reproducible values for already studied compounds that are readily and broadly accessible.
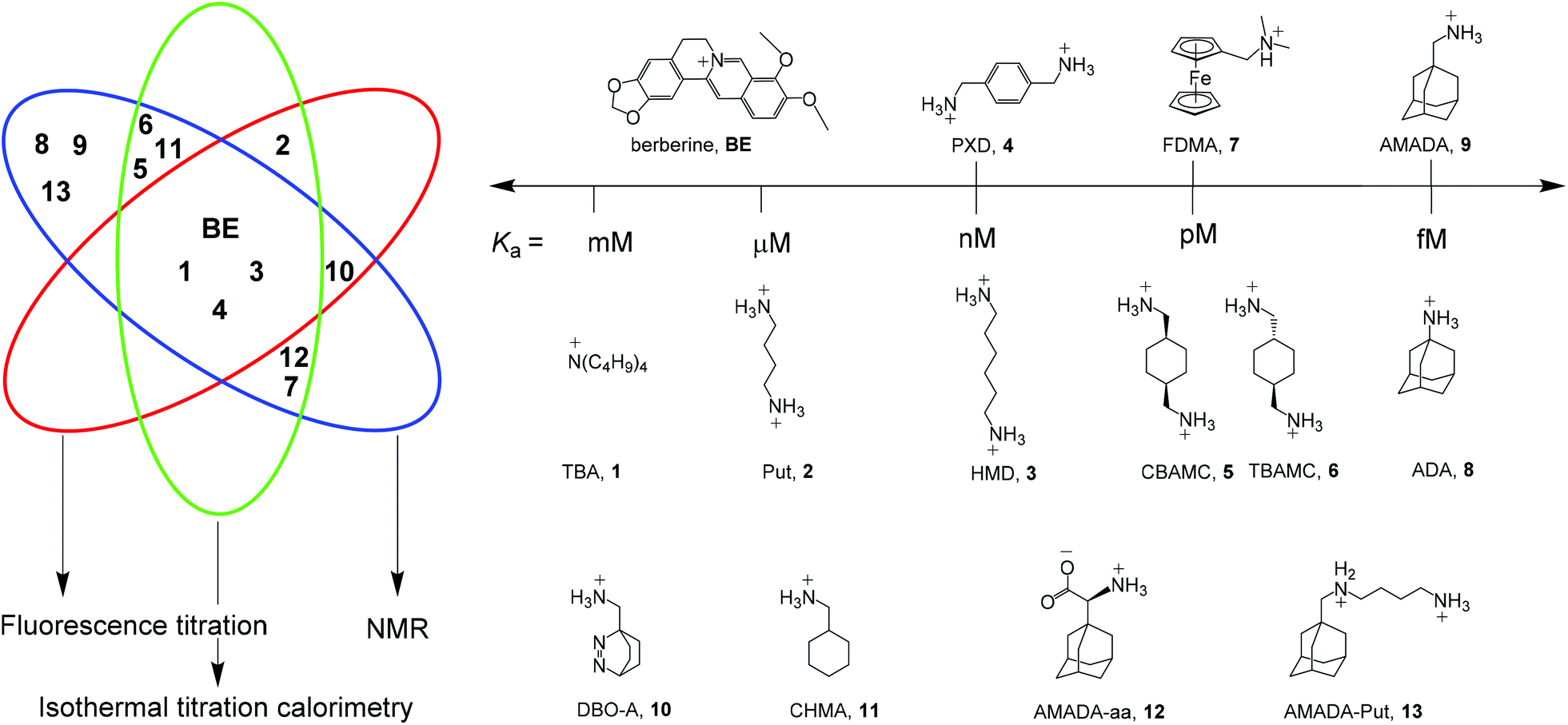 | ||
| Scheme 1 Structures of established reference compounds BE and 1–9 to determine the affinity of guests 10–13. | ||
As a central entry point to this series, we have selected berberine (BE), for which we could confirm a recently reported and very thoroughly determined binding constant.39 Based on direct fluorescence and ITC titrations as well as competitive NMR titrations against putrescine (Put, 2), we propose a reference value of (2.36 ± 0.20) × 107 M−1 for BE. Competitive fluorescence titrations with BE and control experiments by NMR and ITC suggested hexamethylenediamine (HMD, 3) and p-xylylenediamine (PXD, 4) as the desirable reference compounds with nM affinity. For pM affinity, the cis- and trans-isomers of 1,4-bis(aminomethyl)-cyclohexane (CBAMC, 5 and TBAMC, 6) as well as N,N-dimethylaminomethylferrocene (FDMA, 7) were explored. At the upper end, adamantylamine (ADA, 8) could serve as potential reference compound with fM affinity. The proposed binding constants of all reference compounds and their errors are based on repeated measurements with various methods and we discuss herein the advantages and limitations of each reference compound. Finally, we use our reference scale to report the binding affinities of compounds 10–13, which we have previously used in various contexts without accurately determining their binding affinities.15,21,31,40–43
Results and discussion
Reference compound for μM affinity: berberine
The most desirable property of a potential reference compound for binding constant determinations is the possibility to directly measure its affinity by various methods. This suggests to establish a fluorescent dye as a first reference compound, which allows one to combine the results from optical spectroscopy, ITC, and, eventually, NMR spectroscopy. The possibility to jointly use these methods with the same compound is, however, rarely met, because many dyes tend to aggregate at the millimolar concentrations required for NMR. For example, acridine orange, which has been widely used with CB7,12,42,44 shows clear signs of aggregation around 10 μM in water, which renders it unsuitable for binding constant determinations by NMR.45,46 Isaacs and co-workers explored the fluorescent dye 3,6-diaminoacridine (proflavine) as a reference compound,27 which also showed significant peak broadening at concentrations higher than 0.5 mM, even in the presence of CB7.27Based on these considerations, we selected BE as an alternative fluorescent reference compound. The fluorescence of BE is insensitive to pH and complex formation with CB7 is known to enhance the fluorescence intensity about 500-fold, ITC gives a pronounced heat response, 1H NMR spectroscopy shows well-separated peaks of complexed and free BE, a slow exchange on the 1H NMR timescale, and no signs of aggregation up to its solubility limit of ca. 2.5 mM.51 This rather unique combination affords easily detectable signals upon complexation and allows a mutual verification of binding constants measured by fluorescence, ITC, and NMR.
In order to determine the binding constant of BE with minimal uncertainty, we have initially re-evaluated data sets from eight randomly selected fluorescence titrations (n = 8), which were performed by different individuals in our lab during the previous years with different commercial and self-synthesized CB7 batches. Global fitting of this extended data set gave a binding constant of (1.91 ± 0.14) × 107 M−1 (Fig. S1†), whereas measurements from ITC (Ka = (1.20 ± 0.10) × 107 M−1, n = 3, Fig. S2†) suggested a slightly lower binding constant with non-overlapping error ranges.
When we compared our result with the CB7·BE binding constants reported in the literature, we noted that Miskolczy and Biczók found a binding constant of (2.4 ± 0.3) × 107 M−1 in purified water, which had been freshly distilled from a diluted KMnO4 solution.39 They noted that trace impurities in commercial HPLC-quality water could give a lower binding affinity, and we were intrigued whether our Millipore “ultrapure” water could be responsible for the observed deviations in the measured CB7·BE binding affinity. Indeed, when we re-measured the binding constant of BE with CB7 in water distilled from KMnO4, our value (Ka = (2.43 ± 0.39) × 107 M−1, Fig. S3†) was in perfect agreement with the value documented by Miskolczy and Biczók. Moreover, ITC measurements with water distilled from KMnO4 also gave a higher binding constant of (2.26 ± 0.40) × 107 M−1 (Fig. S4†), in very good agreement with the value from fluorescence spectroscopy. The detrimental influence on the apparent binding affinity was also noted with putrescine (Put, 2), which gave Ka = (1.42 ± 0.14) × 106 M−1 with water from our Millipore purification system and (1.85 ± 0.10) × 106 M−1 with water distilled from KMnO4 by ITC (see section 4.4 in ESI†). Combining the results from fluorescence and ITC gave the binding affinity of BE to CB7 and its error as Ka = (2.36 ± 0.20) × 107 M−1 (see section 3.1 in ESI† for error calculation), which we propose as the first reference value of a reference scale of CB7 binding affinities (Table 1 and Fig. 1).23,39,51–54
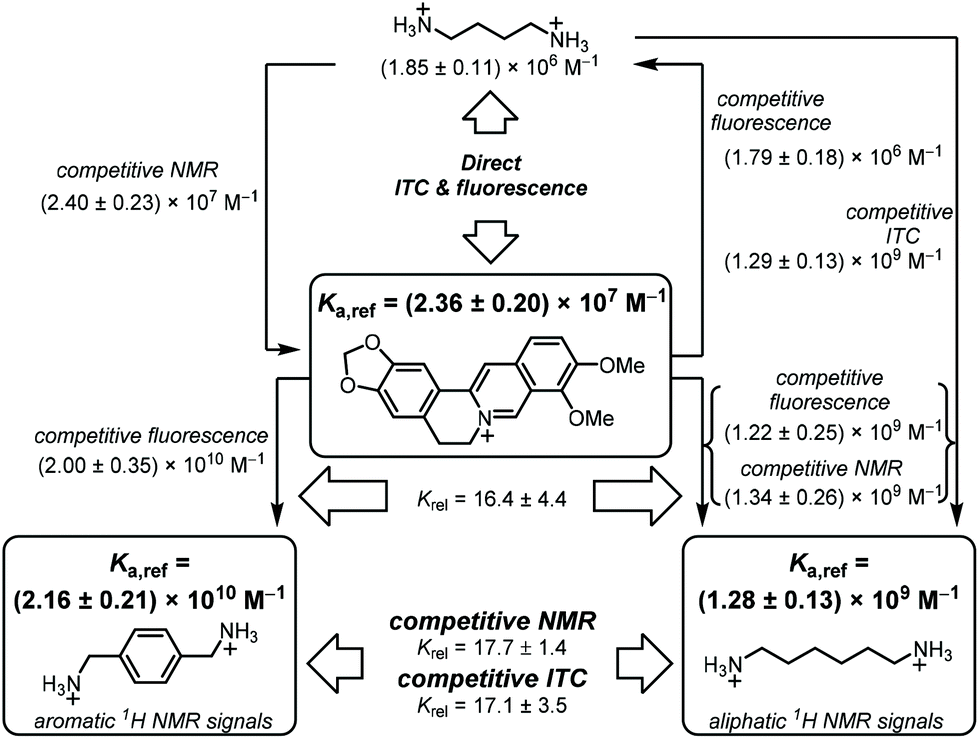 | ||
| Fig. 1 Flow scheme for determination and mutual verification of binding constants in the range of 106 to 1010 M−1. | ||
| Compound | K a (M−1) | Recommended Ka (M−1) | Literature value(s) | Ref. | ||
|---|---|---|---|---|---|---|
| NMRa | ITC | Fluorescenceb | ||||
| a Measured by competitive NMR (unless indicated differently). b Measured by fluorescence displacement with BE (except for BE itself). c Measured by direct NMR titration. d Measured by direct ITC. e Measured by direct fluorescence titration. f Measured by competitive ITC. g Binding constants could not be determined due to the quantitative displacement of BE. h No literature value available. i Binding constants could not be determined due to slow exchange. j Different values reported in main text and ESI.† | ||||||
| TBA, 1 | (4.10 ± 0.52) × 103c |
(4.75 ± 0.56) × 103d |
(4.67 ± 0.52) × 103 | (4.51 ± 0.31) × 103 | 2.8 × 103c |
47 |
| Put, 2 | n.d. | (1.85 ± 0.11) × 106d |
(1.79 ± 0.18) × 106 | (1.82 ± 0.11) × 106 | 7.9 × 105d |
48 |
| BE | (2.40 ± 0.22) × 107 | (2.26 ± 0.39) × 107d |
(2.43 ± 0.39) × 107e |
(2.36 ± 0.20) × 107 | 2.4 × 107d |
39 |
| HMD, 3 | (1.34 ± 0.26) × 109 | (1.29 ± 0.13) × 109f |
(1.22 ± 0.25) × 109 | (1.28 ± 0.13) × 109 | 2.1 × 109f |
29 and 49 |
| 1.4 × 108f |
48 | |||||
| PXD, 4 | (2.27 ± 0.30) × 1010 | (2.20 ± 0.40) × 1010f |
(2.00 ± 0.35) × 1010 | (2.16 ± 0.21) × 1010 | 3.3 × 109f |
48 |
| 1.2 × 1010f |
27 | |||||
| CBAMC, 5 | (6.01 ± 0.67) × 1012 | (5.62 ± 0.68) × 1012f |
n.d.g | (5.81 ± 0.48) × 1012 | n.a.h | n.a.h |
| TBAMC, 6 | (6.57 ± 0.48) × 1012 | (6.62 ± 0.77) × 1012f |
n.d.g | (6.60 ± 0.46) × 1012 | n.a.h | n.a.h |
| FDMA, 7 | (2.40 ± 0.30) × 1012 | (2.38 ± 0.34) × 1012f |
n.d.g | (2.39 ± 0.23) × 1012 | 2.0 × 1012f |
49 |
| 2.4 × 1012f |
50 | |||||
| ADA, 8 | (1.04 ± 0.15) × 1015 | n.d.i | n.d.g | (1.04 ± 0.15) × 1015 | 1.7 × 1014f |
50 |
| 1.1 × 1011f |
48 | |||||
| AMADA, 9 | (5.26 ± 0.61) × 1015 | n.d.i | n.d.g | (5.26 ± 0.61) × 1015 | 7.7 × 1014a |
50j |
| 9 × 1014a |
50j |
|||||
The value of the CB7·BE binding constant was therefore additionally confirmed by NMR spectroscopy. Competitive NMR measurements, in which an excess of a competitor is added to displace a sizeable fraction of the reference compound from a sub-stoichiometric amount of the host,23,28 were considered as the only suitable approach, because a direct determination of the binding constant is prevented by the high affinity of the CB7·BE complex. The latter would require low micromolar concentrations, which are incompatible with standard 1H NMR instruments operating at 400 or 500 MHz.
As potential competitors, the tetrabutylammonium cation (TBA, 1) and Put (2) were considered with binding affinities in the millimolar and micromolar range. TBA (1) was immediately disregarded due to precipitation in the NMR tube containing a mixture of TBA (1), BE, and CB7, which singled out Put (2). As a starting point, the binding constant of Put (2) was determined by a direct ITC titration as (1.85 ± 0.11) × 106 M−1 (n = 1) and by competitive fluorescence titrations as (1.79 ± 0.18) × 106 M−1 (n = 6, see section 4.4 in ESI† for data and 3.2 for error calculation). Combining the values and errors of these titrations led to a reference value for Put (2) of (1.82 ± 0.11) × 106 M−1. In the next step, the relative binding affinity (Krel = Ka,BE/Ka,Put) of Put (2) was determined by competitive NMR. This requires the identification of well-resolved peaks of complexed and free BE or Put (2),52 and we considered the 1H NMR signals of protons 1, 2, 3, and 5 + 11 in their free (without prime) and complexed form (with prime) as suitable (Fig. 2). Integration of the 1H NMR peak areas of the signals gave Krel by eqn (ESI-6) to (ESI-11),† and the average and standard deviation of these four sets of peaks was next calculated from triplicate measurements (see section 3.3.3 in ESI†), which gave a value of Krel = 13.18 ± 0.60; by using the reference Ka value of Put (2), we obtained a Ka value of (2.40 ± 0.22) × 107 M−1 for BE by competitive NMR (eqn (ESI-18)†). This value is in perfect agreement with the proposed reference value for BE from the direct titrations, such that we confidently recommend it as a reference compound with micromolar affinity (see lower part of Fig. 1), which can conversely be used to conduct measurements with higher-affinity binders, those in the nanomolar range.
 | ||
| Fig. 2 1H NMR spectrum of 0.84 mM CB7, 1.16 mM Put (2) and 0.91 mM BE in D2O at pD 7.4. The four signals marked in bold italic were used for integration and determination of Krel. | ||
Reference compounds for nM affinity
In order to establish reference compounds with nanomolar affinity, we decided for HMD (3) and PXD (4), with reported binding constants of 2.1 × 109 M−1 and 1.8 × 109 M−1.28–30 The trimethylsilylated reference compound synthesized by the Isaacs group was elegantly selected for its well-separated NMR peak in the region around 0 ppm,26 whereas HMD (3) and PXD (4) complement each other by covering the aliphatic and aromatic regions of the NMR spectrum. Consequently, the aliphatic HMD (3) can be used to determine binding affinities of aromatic competitors with minimal probability of overlapping NMR peaks and the aromatic PXD (4) can be used for aliphatic competitors (Fig. 1).As a first step, we have determined the binding affinity of HMD (3) by competitive fluorescence titrations with BE (Fig. S6†), which yielded a value of Ka = (1.22 ± 0.25) × 109 M−1 (n = 6, see section 3.2 in ESI† for error calculation), whereas the displacement titration with PXD (4) was more challenging. With typical concentrations of BE and CB7, we noted that the titration curve could not be reliably fitted, because the strong binding of PXD (4) led to quantitative displacement under the standard conditions. To remedy, we increased the concentration of BE from 2 μM to 25 μM to account for the high affinity of PXD (4). To eliminate potential interferences from the inner filter effect at this high concentration, the commonly used 10 × 10 mm cuvette was replaced with a 10 × 4 mm cuvette. When BE was excited along the 4 mm path, a linear dependence of fluorescence intensity on concentration was obtained (Fig. S11†). In this way, competitive fluorescence titrations with PXD (4) (Fig. 3) gave a binding constant of Ka = (2.00 ± 0.35) × 1010 M−1 (n = 6, see section 3.2 in ESI† for error calculation).
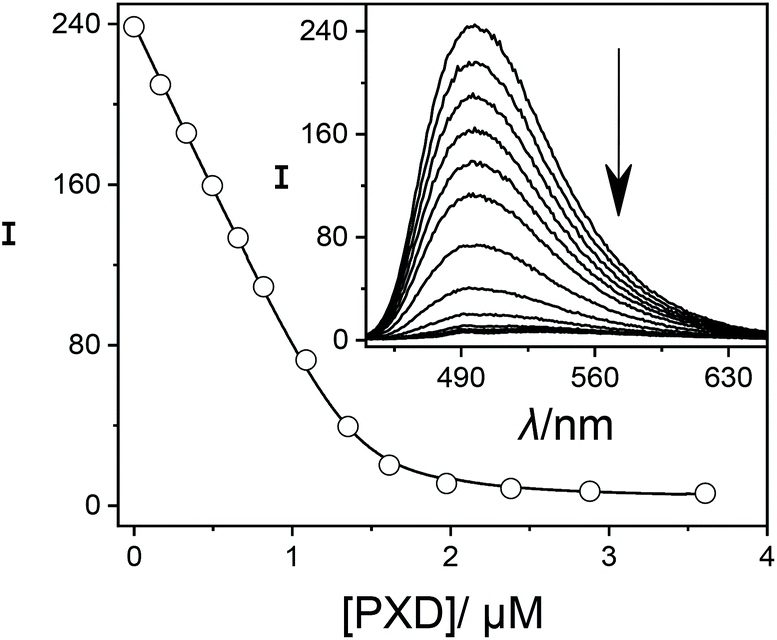 | ||
| Fig. 3 Fluorescence displacement titration (λex = 420 nm, λem = 490 nm) of 25.0 μM BE and 1.4 μM CB7 upon addition of different concentrations of PXD (4). The inset shows the spectral variations. | ||
Our results revealed some discrepancies with the reported literature values.28,29 For example, it had been reported that HMD (3) (Ka = 2.1 × 109 M−1) binds about equally strong to CB7 as PXD (4) (2.1 × 109 M−1versus 1.8 × 109 M−1), whereas we found an order of magnitude higher affinity for PXD (4, Krel = 16.4 ± 4.4, by fluorescence titration). To obtain an independent confirmation, competitive 1H NMR measurements with HMD (3) and PXD (4) were performed next (Fig. 4). The 1H NMR spectra of mixtures of HMD (3), PXD (4), and a limiting quantity of CB7, exhibited clear spectral differentiation with no overlap between the peak positions of free and complexed HMD (3) and PXD (4); only the peaks ascribed to the benzylic protons of uncomplexed PXD (4) overlapped with the CB7 peaks at 4.20 ppm (Fig. 2). The relative binding constant and its standard deviation could accordingly be calculated by using all six combinations of the integrated peak areas of the two PXD (4) peak pairs (the CB7 peak area was subtracted from the integrated peak area of B) and three HMD (3) peak pairs (eqn (ESI-11)†). This gave Krel = Ka,PXD/Ka,HMD = 17.7 ± 1.4, which agrees very well with the Krel value obtained by fluorescence (16.4 ± 4.4) and clearly confirmed that PXD (4) binds more strongly to CB7 than HMD (3).
 | ||
| Fig. 4 1H NMR competition experiment with 1.0 mM CB7, 1.6 mM PXD (4), and 5.7 mM HMD (3) in D2O at pD 7.4. Signals for PXD (4) are assigned with uppercase letters and those for HMD (3) with Arabic numerals. Dashes indicate the respective CB7 complexes. The integrated peak areas are given below the x-axis. | ||
The binding constant of HMD (3) was further confirmed by 1H NMR using BE as a competitive binder (Fig. S9†), which afforded Ka = (1.34 ± 0.26) × 109 M−1, and by ITC by using Put (2) as a competitive binder (Fig. S7,†Ka = (1.29 ± 0.13) × 109 M−1). Both values agree very well with our fluorescence value, such that we suggest to use a reference value of (1.28 ± 0.13) × 109 M−1 for HMD (3) (see eqn (ESI-2) and (ESI-3)†). This value lies within the range of reported literature values for HMD (3) binding to CB7, which varied from 1.4 × 108 M−1 to 2.1 × 109 M−1 (see Fig. 1 and Table 1 for an overview).29,48
The absolute Ka value of PXD (4) cannot be measured against berberine by NMR, because the required concentration of berberine exceeds the solubility limit of BE, but it was further confirmed by competitive ITC against HMD (3) (Fig. S12,†Ka = (2.20 ± 0.40) × 1010 M−1). Combining the results from ITC, NMR, and fluorescence suggests a reference value of (2.16 ± 0.21) × 1010 M−1 for the binding affinity of PXD (4) to CB7 (Fig. 1), which lies above the range of previously reported binding affinities (Table 1).27,49
It is noteworthy that we scrutinized our results in water distilled from KMnO4 and data obtained in initial exploration experiments with Millipore water. A re-evaluation of the data in our Millipore water clearly suggested that the potential trace impurities had only a small, but sizeable influence on direct titrations, whereas the influence in competitive titrations was negligible. For example, when BE displacement titrations with HMD (3) in Millipore water were re-evaluated with the reference Ka value of BE in water distilled from KMnO4, the obtained binding constants of HMD (3) were identical, within error, to the results from titrations in water distilled from KMnO4. Also, for Put (2), direct ITC titrations revealed differences in Millipore water and in water distilled from KMnO4 (see previous section), whereas fluorescence displacement titrations with Put (2) and BE in Millipore water gave the same binding constants as in water distilled from KMnO4, when the reference Ka value of BE in water distilled from KMnO4 was used in the fitting. It is further interesting to note, that the relative binding constants of Put (2) and BE in H2O at pH 7.0 (by fluorescence) and in D2O at pD 7.4 (by NMR) were identical within our rigorously determined error ranges, which excludes any undesirable interferences in our commercial D2O in competitive NMR titrations. From these observations, one may jump to the conclusion that competitive titrations always afford reliable relative binding constants even if the concentrations of known or unknown impurities (such as buffer salts or the unknown impurity in the employed Millipore water) vary. However, this is correct only if the impurity or salt competes with both analytes of interest in the same manner. In reality, this is certainly not always the case, e.g. the presence of cations affects the binding of different hydrocarbons to CB6 to a different degree, likely due to the variable involvement of ternary complexes.55
Reference compounds for pM affinity
With increasing affinity, competitive titrations become more and more time-demanding, because of the slow exchange kinetics associated with high-affinity guests. This prevents the use of reference compounds with Ka > 1012 M−1 in competitive ITC titrations, because equilibration times of hours to days would be required after every injection. As an alternative, competitive ITC titrations have been performed with high millimolar concentrations of the more rapidly exchanging guests PXD (4) or HMD (3) with nanomolar affinity by Kaifer48 and by Kim, Inoue and Gilson.50 This strategy is, however, not possible with competitive 1H NMR titrations, because the determination of ratios of components in a mixture with small peak areas (low concentration of the complex) and large peak areas (high concentration of the free competitor) is often lacking accuracy.23,56 The Isaacs group has therefore utilized 1-adamantyl-pyridinium as a relatively fast exchanging high-affinity reference compound with picomolar affinity for competitive 1H NMR titrations.26,27As potential alternatives, we introduce herein cis- and trans-1,4-bis(aminomethyl)-cyclohexane (CBAMC, 5, and TBAMC, 6) as well as N,N-dimethylaminomethyl-ferrocene (FDMA, 7) as potential reference compounds for CB7 binding affinities in the pM range. As an initial experiment, the relative binding constant of CBAMC (5) and TBAMC (6) were assessed by NMR and ITC against PXD (4) (Table 1, see Fig. S23, S25, S48, and S49†), which indicated that the affinity of both isomers to CB7 is very similar and that the affinity of the trans isomer is only 1.1-fold higher than that of the cis isomer. Interestingly, the cis isomer CBAMC (5) showed, however, more rapid exchange kinetics (Fig. S50†) and was thus further investigated in more detail.
CBAMC (5) shows clearly separated free and bound peaks for the CH2-N methanaminium group (at 2.87 and 2.55 ppm), the C–H group of the cyclohexane ring (at 1.64 and 0.71 ppm), and for four cyclohexane CH2 protons (at 1.85 and 0.15 ppm) (Fig. S24†). The competitive NMR titration of CBAMC (5) against PXD (4) (Fig. S25†) was evaluated as described above by considering all six combinations of the three CBAMC (5) compound peak pairs and two PXD (4) reference peak pairs with eqn (ESI-11).† This gave a Krel value of 278 ± 14, which was converted into a binding constant of (6.01 ± 0.67) × 1012 M−1 by NMR using eqn (ESI-16) and (ESI-18).† We also determined the binding affinity of CBAMC (5) by ITC (n = 2) by titrating CBAMC (5) into a solution containing the PXD·CB7 complex (Fig. S23†). This furbished a binding constant of (5.62 ± 0.68) × 1012 M−1, in good agreement with the NMR value, which leads us to propose a reference value of (5.81 ± 0.48) × 1012 M−1 for CBAMC (5). In addition, FDMA (7) was measured against PXD (4) by NMR and ITC (Fig. S39 and S40†), which yielded binding constants of (2.40 ± 0.30) × 1012 M−1 and (2.38 ± 0.34) × 1012 M−1, respectively; we suggest a reference value of (2.39 ± 0.23) × 1012 M−1 for FDMA (7) (Table 1).
The 1-ferrocenyl-trimethylmethanammonium cation had been used previously within a series of binding constant determinations,28 but based on our results, we consider ferrocene derivatives, such as FDMA (7), to be overall less useful than CBAMC (5). First, we noted that many of the ferrocene peaks overlap with the CB7 peaks in 1H NMR spectroscopy (Fig. S38†), such that only a single peak appeared suitable for integration. Second, FDMA (7) has a limited solubility (<12 mM), which is too low for competitive titrations of guests with femtomolar affinity (see below). And third, we noted that the 1H NMR peaks of a FDMA (7) solution in D2O became significantly broadened after a few days, which may be due to slow decomposition of FDMA (7). The advantage of FDMA (7) compared to CBAMC (5) is its significantly faster exchange rate (see Fig. S32 and S33†), which is, however, still too slow to be useful for competitive ITC. As a consequence, FDMA (7) may be a useful reference compound when high binding affinities need to be rapidly screened, whereas CBAMC (5) will likely provide more accurate affinities. A literature value for CBAMC (5) has, so far, not been reported, but our value for FDMA (7) is gratifyingly in excellent agreement with the value reported by Kim, Inoue, and Gilson (2.4 × 1012 M−1) and in very good agreement with the value by Kaifer (2.0 × 1012 M−1).49,50
Reference compounds for fM affinity
Several guests with femtomolar and higher affinity have been reported for CB7, which include diamantane, adamantane, and bicyclooctane derivatives,27,50 but the extremely slow exchange kinetics in combination with the lack of established reference compounds renders the determination of these ultrahigh affinities often challenging and time-consuming. For example, adamantylamine (ADA, 8) has been very often used as a prototypical ultrahigh-affinity guest, but its reported binding constant varies largely and ranges from 1.1 × 1011 M−1 to 4.2 × 1014 M−1.48,50With the goal to extend our reference scale to compounds with binding affinities in the femtomolar range, we considered ADA (8) as well as aminomethyladamantane (AMADA, 9), also because they are both commercially available. The Krel values of both compounds were determined by 1H NMR competition experiments with CBAMC (5) as the reference. To ensure full relaxation of the host–guest equilibration mixtures, two samples were prepared for each competitor, in which, first, CBAMC (5) was pre-mixed with CB7, and second, the competitor was pre-mixed with CB7 before addition of the other compound. Subsequently, 1H NMR spectra were recorded after varying time periods until both mixtures were fully equilibrated, as indicated by both having reached the same degree of complexation (Fig. S32 and S37†). Integration of three peaks per spectrum gave Krel = 179 ± 14 for ADA (8) and 906 ± 73 for AMADA (9) using eqn (ESI-11) to (ESI-15).† This value was converted into a binding constant of (1.04 ± 0.15) × 1015 M−1 for ADA (8) and (5.26 ± 0.61) × 1015 M−1 for AMADA (9) by NMR using eqn (ESI-16) and (ESI-18).† Notably, our value for ADA (8) is about fivefold higher than the originally reported values in the literature,28,48,50 which underlines the challenge to accurately measure such ultra-high binding affinities.
When comparing the principal suitability of ADA (8) and AMADA (9) as potential reference compounds, it is apparent that the dissociation kinetics of ADA (8) is much faster than that of AMADA (9) (Fig. S32 and S37†). In fact, the chemical exchange of AMADA (9) in 1H NMR competition experiment is so slow that it would require unreasonably long equilibration times in routine binding titrations (Fig. S37†). This is unfortunate, because the peaks of the CH2-N methanaminium protons of AMADA (9) and its CB7 complex are very well separated (at 2.68 and 2.46 ppm for the free complexed form) and can thus be very reliably integrated.
Reference compound for mM affinity
To complete the list of suitable reference compounds also at the lower end of the affinity scale, we included TBA (1), which could serve as a potential reference in the mM range for important biological molecules such as carnitine, trimethyllysine, or amino acids.47,48,57,58 The binding constant of TBA (1) was measured by ITC, fluorescence displacement with BE and by a direct 1H NMR titration (see Table 1 and section 4.5 in ESI†). This provided an average binding constant of (4.51 ± 0.31) × 103 M−1, which we propose as the reference value for this low affinity range.Binding constants of high-affinity guests
After setting up a reference scale of binding affinities, we determined the binding constant of compounds 10–13 (Table 2). (2,3-Diazabicyclo[2.2.2]oct-2-enyl)methylamine (DBO-A, 10) and the putrescine derivative of aminomethyladamantane (AMADA-Put, 13) are guests with a supposedly high binding affinity to CB7. For example, AMADA-Put (13) was previously introduced by us to determine the number of reactive surface functional groups on micro- and nanoparticles15,40,41 and as a ditopic guest in a supramolecular switch,31 and DBO-A (10) may be useful in time-resolved assays with CB7.59,60 However, the binding constants of 10 and 13 have so far not been reported. In addition, we were interested in the binding affinity of an amino acid derivative of AMADA (S)-2-(adamantan-1-yl)-2-aminoacetic acid hydrochloride (AMADA-aa, 12) to CB7, and in the affinity of cyclohexylmethylamine (CHMA, 11). The latter was previously explored by us as an anchor group in the design of reporter dyes for CB7-based sensor systems.43| Compound | Method | K a/M−1 | Literature value |
|---|---|---|---|
| a Measured by competition NMR against PXD (4). b Measured by fluorescence displacement with BE. c Literature value not available. d Measured by competitive ITC against HMD (3). e Measured by competitive ITC in ref. 29. f Measured by competition NMR against CBAMC (5). | |||
| DBO-A, 10 | NMR | (2.18 ± 0.31) × 1010a |
n.a.c |
| Fluorescence | (2.07 ± 0.21) × 1010b |
||
| CHMA, 11 | NMR | (8.80 ± 1.10) × 1010a |
1.3 × 1011e |
| ITC | (8.43 ± 0.43) × 1010d |
||
| AMADA-aa, 12 | NMR | (1.55 ± 0.32) × 1012f |
n.a.c |
| AMADA-Put, 13 | NMR | (2.02 ± 0.39) × 1016f |
n.a.c |
With the now set up reference scale and the respective uncertainty ranges, the binding constant for each compound was measured by competition experiments using the reference values and errors in Table 1. The binding constant of DBO-A (10) was measured by 1H NMR competition with PXD (4) and by fluorescence displacement titration with BE. 1H NMR, using the integrated peak areas of eight peaks (Fig. S28†), gave a Ka value of (2.18 ± 0.31) × 1010 M−1 and global fitting (n = 6) of fluorescence titrations (Fig. S26†) gave a binding constant for DBO-A (10) of (2.07 ± 0.21) × 1010 M−1. Noteworthy, the fluorescence displacement titration was performed as described above for PXD (4) to avoid quantitative binding and the fluorescence of DBO-A (10) itself was undetectable under these conditions due to the low brightness of the azo chromophore. The binding constant of DBO-A (10) is lower than the analogous aliphatic bicyclooctane,50 which is in agreement with a decreased hydrophobicity of DBO derivatives due to the significant dipole moment and high water-solubility resulting from the azo group.61
The Ka value of AMADA-Put (13) was expected to be in the femtomolar range and was accordingly determined against CBAMC (5) as a competitor. AMADA-Put (13) exhibited extremely slow exchange kinetics. Especially, when CBAMC (5) was added to the AMADA-Put (13) complex with CB7, more than six months were required to reach equilibrium. The obtained Krel of AMADA-Put (13) against CBAMC (5) was (3.47 ± 0.59) × 103, which gave a Ka value of (2.02 ± 0.39) × 1016 M−1 (see Table 2 and Fig. S43, 44†). The increased affinity of aminoalkylated adamantane derivatives compared to the unsubstituted amines was previously noted and explained through a primary ammonium looping model.26,62 In agreement with this model, the ratio of the Krel values of 13 and 9 agrees very well with the related aminoalkylated adamantylamines from Isaacs.26
The amino acid AMADA-aa (12) has two ionizable functional groups, and typical pKa values for the α-carboxylic acid (<2.5) and α-amino groups (>8.5) in amino acids suggest that AMADA-aa (12) prevails in its zwitterionic, neutral form at neutral pH. Using PXD (4) as a competitor, the relative binding constant was measured by 1H NMR competition using six integratable peaks, which gave Krel = 72 ± 13 and Ka = (1.55 ± 0.32) × 1012 M−1 (see Table 2 and Fig. S41, 42†). As expected, AMADA-aa (12) has a significantly lower binding affinity than AMADA (9), because the negatively charged carboxylate anion introduces repulsive ion-dipole interactions with the carbonyl groups at the CB7 rim.42,63
Finally, the affinity of CHMA (11) to CB7 was obtained via competitive ITC by injecting CHMA (11) into a solution containing the HMD·CB7 complex, which established Ka = (8.43 ± 0.43) × 1010 M−1 (Table 2 and Fig. S21†). This value is slightly lower than the literature value, which is attributed to the higher Ka value of HMD (3) used in the competition experiment in the literature;29 the relative binding constants of our experiments and the ITC titration in the literature differ only by 10%. To additionally confirm the revised value, the binding constant of CHMA (11) was also measured by 1H NMR competition using PXD (4) as a competitor, which gave Ka = (8.8 ± 1.1) × 1010 M−1 (Fig. S22†).
Conclusions
We have established a series of reference compounds for the determination of CB7 binding affinities by competitive host–guest titrations. The compounds (Scheme 1) have been selected to cover a wide range of affinities (mM–fM), to be amenable to various measurement techniques, and to allow ready and broad access as well as high reproducibility on account of commercial sample availability. The fluorescent dye berberine was established as a central compound with μM affinity, which was measured by fluorescence spectroscopy, ITC, and 1H NMR. Within a series of cross-validated competition experiments, we next established hexamethylenediamine and p-xylylenediamine as references with nM affinity, cis-1,4-bis(aminomethyl)-cyclohexane and N,N-dimethylaminomethylferrocene as references with pM affinity (with the latter showing faster exchange), and we propose adamantylamine as reference compound with fM affinity.To illustrate an immediate application of the reference scale, we determined the binding constants of four compounds of interest, DBO-A (10), AMADA-aa (12), AMADA-Put (13), and CHMA (11), by the corresponding competitive titrations with the respective reference compounds. For example, CHMA (11) was tested as an anchor group in the design of new host-dye reporter pairs with CB743 and AMADA-Put (13) was used to determine the number of reactive surface functional groups15,40,41 and as a ditopic guest in a supramolecular switch.31 The amino acid AMADA-aa (12) could be introduced into peptides and become useful in sensing applications, for example in supramolecular tandem membrane and enzyme assays.20,42,60,64–67
Experimental section
See ESI† for Experimental details.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the DFG (HE 5967/6-1 and NA 686/15-1) for financial support. We also thank the co-workers of the Nau group, who shared their previous BE titrations with us, namely, Dr Suhang He, Dr Mohamed Nilam, and Ms Kedamawit Dessalegn.Notes and references
- F. Biedermann and H.-J. Schneider, Chem. Rev., 2016, 116, 5216–5300 CrossRef CAS PubMed
.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394–418 RSC
-
K. A. Connors, Binding Constants: The Measurement of Molecular Complex Stability, John Wiley & Sons, New York, Chichester, Brisbane, Toronto, Singapore, 1987 Search PubMed
- K. Hirose, J. Inclusion Phenom. Macrocyclic Chem., 2001, 39, 193–209 CrossRef CAS
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC
- I. Jarmoskaite, I. AlSadhan, P. P. Vaidyanathan and D. Herschlag, eLife, 2020, 9, e57264 CrossRef CAS PubMed
- M. M. Pierce, C. S. Raman and B. T. Nall, Methods, 1999, 19, 213–221 CrossRef CAS PubMed
-
L. Mazzei, S. Ciurli and B. Zambelli, in Methods Enzymol, ed. A. L. Feig, Academic Press, 2016, vol. 567, pp. 215–236 Search PubMed
- M. R. Duff Jr., J. Grubbs and E. E. Howell, J. Visualized Exp., 2011, 2796, DOI:10.3791/2796
- S. Leavitt and E. Freire, Curr. Opin. Struct. Biol., 2001, 11, 560–566 CrossRef CAS PubMed
- P. Montes-Navajas, A. Corma and H. Garcia, ChemPhysChem, 2008, 9, 713–720 CrossRef CAS PubMed
- G. Ghale, N. Kuhnert and W. M. Nau, Nat. Prod. Commun., 2012, 7, 343–348 CrossRef CAS PubMed
- M. Shaikh, J. Mohanty, P. K. Singh, W. M. Nau and H. Pal, Photochem. Photobiol. Sci., 2008, 7, 408–414 CrossRef CAS PubMed
- A. Hennig, A. Hoffmann, H. Borcherding, T. Thiele, U. Schedler and U. Resch-Genger, Chem. Commun., 2011, 47, 7842–7844 RSC
- W. A. Freeman, W. L. Mock and N. Y. Shih, J. Am. Chem. Soc., 1981, 103, 7367–7368 CrossRef CAS
- J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee, J.-K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540–541 CrossRef CAS
- A. I. Day, R. J. Blanch, A. P. Arnold, S. Lorenzo, G. R. Lewis and I. Dance, Angew. Chem., Int. Ed., 2002, 41, 275–277 CrossRef CAS PubMed
- A. Day, A. P. Arnold, R. J. Blanch and B. Snushall, J. Org. Chem., 2001, 66, 8094–8100 CrossRef CAS PubMed
- G. Ghale and W. M. Nau, Acc. Chem. Res., 2014, 47, 2150–2159 CrossRef CAS PubMed
- W. M. Nau, G. Ghale, A. Hennig, H. Bakirci and D. M. Bailey, J. Am. Chem. Soc., 2009, 131, 11558–11570 CrossRef CAS PubMed
- F. Biedermann and W. M. Nau, Angew. Chem., Int. Ed., 2014, 53, 5694–5699 CrossRef CAS PubMed
- L. Cao and L. Isaacs, Supramol. Chem., 2014, 26, 251–258 CrossRef CAS
- S. Zhang, L. Grimm, Z. Miskolczy, L. Biczók, F. Biedermann and W. M. Nau, Chem. Commun., 2019, 55, 14131–14134 RSC
- H. Tang, D. Fuentealba, Y. H. Ko, N. Selvapalam, K. Kim and C. Bohne, J. Am. Chem. Soc., 2011, 133, 20623–20633 CrossRef CAS PubMed
- D. Sigwalt, M. Šekutor, L. Cao, P. Y. Zavalij, J. Hostaš, H. Ajani, P. Hobza, K. Mlinarić-Majerski, R. Glaser and L. Isaacs, J. Am. Chem. Soc., 2017, 139, 3249–3258 CrossRef CAS PubMed
- L. Cao, M. Šekutor, P. Y. Zavalij, K. Mlinarić-Majerski, R. Glaser and L. Isaacs, Angew. Chem., Int. Ed., 2014, 53, 988–993 CrossRef CAS PubMed
- S. Liu, C. Ruspic, P. Mukhopadhyay, S. Chakrabarti, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2005, 127, 15959–15967 CrossRef CAS PubMed
- M. V. Rekharsky, T. Mori, C. Yang, Y. H. Ko, N. Selvapalam, H. Kim, D. Sobransingh, A. E. Kaifer, S. Liu, L. Isaacs, W. Chen, S. Moghaddam, M. K. Gilson, K. Kim and Y. Inoue, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20737–20742 CrossRef CAS PubMed
- D. Ma, P. Y. Zavalij and L. Isaacs, J. Org. Chem., 2010, 75, 4786–4795 CrossRef CAS PubMed
- Y.-C. Liu, W. M. Nau and A. Hennig, Chem. Commun., 2019, 55, 14123–14126 RSC
- C. Marquez and W. M. Nau, Angew. Chem., Int. Ed., 2001, 40, 3155–3160 CrossRef CAS PubMed
- A. E. Kaifer, W. Li and S. Yi, Isr. J. Chem., 2011, 51, 496–505 CrossRef CAS
- J. Murray, J. Sim, K. Oh, G. Sung, A. Lee, A. Shrinidhi, A. Thirunarayanan, D. Shetty and K. Kim, Angew. Chem., Int. Ed., 2017, 56, 2395–2398 CrossRef CAS PubMed
- X. Zhou, X. Su, P. Pathak, R. Vik, B. Vinciguerra, L. Isaacs and J. Jayawickramarajah, J. Am. Chem. Soc., 2017, 139, 13916–13921 CrossRef CAS PubMed
- S.-R. Wang, J.-Q. Wang, G.-H. Xu, L. Wei, B.-S. Fu, L.-Y. Wu, Y.-Y. Song, X.-R. Yang, C. Li, S.-M. Liu and X. Zhou, Adv. Sci., 2018, 5, 1800231 CrossRef PubMed
- D.-W. Lee, K. M. Park, M. Banerjee, S. H. Ha, T. Lee, K. Suh, S. Paul, H. Jung, J. Kim, N. Selvapalam, S. H. Ryu and K. Kim, Nat. Chem., 2011, 3, 154–159 CrossRef CAS PubMed
- We classify as “not readily available” all compounds that were not listed as “in stock” by at least two suppliers when this study was conducted.
- Z. Miskolczy and L. Biczók, J. Phys. Chem. B, 2014, 118, 2499–2505 CrossRef CAS PubMed
- A. Hennig, H. Borcherding, C. Jaeger, S. Hatami, C. Würth, A. Hoffmann, K. Hoffmann, T. Thiele, U. Schedler and U. Resch-Genger, J. Am. Chem. Soc., 2012, 134, 8268–8276 CrossRef CAS PubMed
- M. Nilam, M. Ahmed, M. A. Alnajjar and A. Hennig, Analyst, 2019, 144, 579–586 RSC
- A. Hennig, H. Bakirci and W. M. Nau, Nat. Methods, 2007, 4, 629–632 CrossRef CAS PubMed
- M. A. Alnajjar, J. Bartelmeß, R. Hein, P. Ashokkumar, M. Nilam, W. M. Nau, K. Rurack and A. Hennig, Beilstein J. Org. Chem., 2018, 14, 1961–1971 CrossRef CAS PubMed
- B. D. Wagner, N. Stojanovic, G. Leclair and C. K. Jankowski, J. Inclusion Phenom. Macrocyclic Chem., 2003, 45, 275–283 CrossRef CAS
- J. Liu, N. Jiang, J. Ma and X. Du, Eur. J. Org. Chem., 2009, 4899–4899 CrossRef
- M. Sayed, S. Jha and H. Pal, Phys. Chem. Chem. Phys., 2017, 19, 24166–24178 RSC
- A. D. St-Jacques, I. W. Wyman and D. H. Macartney, Chem. Commun., 2008, 40, 4936–4938 RSC
- N. Dong, J. He, T. Li, A. Peralta, M. R. Avei, M. Ma and A. E. Kaifer, J. Org. Chem., 2018, 83, 5467–5473 CrossRef CAS PubMed
- W. S. Jeon, K. Moon, S. H. Park, H. Chun, Y. H. Ko, J. Y. Lee, E. S. Lee, S. Samal, N. Selvapalam, M. V. Rekharsky, V. Sindelar, D. Sobransingh, Y. Inoue, A. E. Kaifer and K. Kim, J. Am. Chem. Soc., 2005, 127, 12984–12989 CrossRef CAS PubMed
- S. Moghaddam, C. Yang, M. Rekharsky, Y. H. Ko, K. Kim, Y. Inoue and M. K. Gilson, J. Am. Chem. Soc., 2011, 133, 3570–3581 CrossRef CAS PubMed
- M. Megyesi, L. Biczók and I. Jablonkai, J. Phys. Chem. C, 2008, 112, 3410–3416 CrossRef CAS
- Z. Miskolczy, L. Biczók, M. Megyesi and I. Jablonkai, J. Phys. Chem. B, 2009, 113, 1645–1651 CrossRef CAS PubMed
- D. Bhowmik, M. Hossain, F. Buzzetti, R. D'Auria, P. Lombardi and G. S. Kumar, J. Phys. Chem. B, 2012, 116, 2314–2324 CrossRef CAS PubMed
- G.-Q. Wang, L. Guo, L.-M. Du and Y.-L. Fu, Microchem. J., 2013, 110, 285–291 CrossRef CAS
- M. Florea and W. M. Nau, Angew. Chem., Int. Ed., 2011, 50, 9338–9342 CrossRef CAS PubMed
- T. D. W. Claridge, S. G. Davies, M. E. C. Polywka, P. M. Roberts, A. J. Russell, E. D. Savory and A. D. Smith, Org. Lett., 2008, 10, 5433–5436 CrossRef CAS PubMed
- Z.-Z. Gao, J.-L. Kan, L.-X. Chen, D. Bai, H.-Y. Wang, Z. Tao and X. Xiao, ACS Omega, 2017, 2, 5633–5640 CrossRef CAS PubMed
- I. W. Wyman and D. H. Macartney, Org. Biomol. Chem., 2010, 8, 253–260 RSC
- C. Marquez, F. Huang and W. M. Nau, IEEE Trans. Nanobiosci., 2004, 3, 39–45 CrossRef PubMed
- A. Hennig and W. M. Nau, Front. Chem., 2020, 8, 806 CrossRef CAS PubMed
- W. M. Nau, M. Florea and K. I. Assaf, Isr. J. Chem., 2011, 51, 559–577 CrossRef CAS
- J. Hostaš, D. Sigwalt, M. Šekutor, H. Ajani, M. Dubecký, J. Řezáč, P. Y. Zavalij, L. Cao, C. Wohlschlager, K. Mlinarić-Majerski, L. Isaacs, R. Glaser and P. Hobza, Chem. – Eur. J., 2016, 22, 17226–17238 CrossRef PubMed
- D. Sobransingh and A. E. Kaifer, Chem. Commun., 2005, 5071–5073 RSC
- S. Peng, A. Barba-Bon, Y.-C. Pan, W. M. Nau, D.-S. Guo and A. Hennig, Angew. Chem., 2017, 129, 15948–15951 CrossRef
- A. Barba-Bon, Y.-C. Pan, F. Biedermann, D.-S. Guo, W. M. Nau and A. Hennig, J. Am. Chem. Soc., 2019, 141, 20137–20145 CrossRef CAS PubMed
- F. Biedermann, G. Ghale, A. Hennig and W. M. Nau, Commun. Biol., 2020, 3, 383 CrossRef CAS PubMed
- M. Nilam, S. Collin, S. Karmacharya, A. Hennig and W. M. Nau, ACS Sens., 2021, 6, 175–182 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, experimental details, as well as fitting equations. See DOI: 10.1039/d1ob01304a |
| This journal is © The Royal Society of Chemistry 2021 |