
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePredicting the catalytic activity of azolium-based halogen bond donors: an experimentally-verified theoretical study†
Alexandra A.
Sysoeva
 ,
Alexander S.
Novikov
,
Mikhail V.
Il'in
,
Vitalii V.
Suslonov
and
Dmitrii S.
Bolotin
*
,
Alexander S.
Novikov
,
Mikhail V.
Il'in
,
Vitalii V.
Suslonov
and
Dmitrii S.
Bolotin
*
Institute of Chemistry, Saint Petersburg State University, Universitetskaya Nab. 7/9, Saint Petersburg, 199034, Russian Federation. E-mail: d.s.bolotin@spbu.ru
First published on 29th July 2021
Abstract
This report demonstrates the successful application of electrostatic surface potential distribution analysis for evaluating the relative catalytic activity of a series of azolium-based halogen bond donors. A strong correlation (R2 > 0.97) was observed between the positive electrostatic potential of the σ-hole on the halogen atom and the Gibbs free energy of activation of the model reactions (i.e., halogen abstraction and carbonyl activation). The predictive ability of the applied approach was confirmed experimentally. It was also determined that the catalytic activity of azolium-based halogen bond donors was generally governed by the structure of the azolium cycle, whereas the substituents on the heterocycle had a limited impact on the activity. Ultimately, this study highlighted four of the most promising azolium halogen bond donors, which are expected to exhibit high catalytic activity.
Introduction
Over the last decade, substantial research efforts have been focused on investigating new types of organocatalysts owing to their significant advantages over organometallic and coordination catalysts, including low to negligible sensitivity to air and moisture, reduced environmental footprint, and lower toxicity.1,2 In general, organocatalysts can participate in either covalent or noncovalent bonding interactions with a substrate in order to promote a chemical reaction. The former activation mode involves the formation of one or several covalent bonds between the catalyst and a reaction substrate. Typically, heterocyclic carbenes,1,3,4 amines,1,2,5,6 and phosphines7 comprise this type of organocatalyst. The latter activation mode involves noncovalent interactions between the catalyst and a substrate, and this type of reactivity is investigated in the present work.2,8–12An organocatalyst interacting with a substrate via noncovalent interactions typically does so through hydrogen bonding (HB), so the use of HB donors, such as ureas,13–18 squaramides,18–20 and other Brønsted acids,21–23 has led to numerous important advancements in the field of organocatalysis. In addition, noncovalent interactions can be realized via halogen8–10,24–27 or chalcogen27–29 bonding interactions (XB and ChB, respectively), but this type of catalytic activity has not been widely explored. Recent progress in the field of XB-donor catalysis has clearly indicated that cationic iodine(I)- and iodine(III)-containing species have promising potential in terms of organocatalysis owing to their high catalytic activity and remarkable stability under the necessary reaction conditions.
It has been demonstrated that 2-iodoimidazolium10,30–32 and 4-iodo-1,2,3-triazolium10,33–36 salts effectively catalyze an extensive series of organic transformations, including (aza)-Diels–Alder cycloadditions, Michael additions, halide abstractions, olefin reductions, and many other reactions.10 It is worth mentioning that one report also describes the preparation of 5-iodo-1,2,4-triazolium salts and presents their catalytic activity for Michael additions.37 Recently, hypervalent iodine(III) derivatives (i.e., diaryliodonium salts) have been successfully employed for the living cationic polymerization of olefins,38 halide abstractions,39 and carbonyl activations.39–41 Although diaryliodonium salts demonstrate comparable or greater organocatalytic activity than azolium-based iodine(I) derivatives for the studied reactions,39 currently, the latter remains to be the most commonly studied type of XB-donating organocatalyst.
Considering the high catalytic activity and chemical stability of iodine(I)-containing azolium derivatives and the relatively small number of corresponding catalyst types (only three types among 14 possible variations: 2-iodoimidazoilums, 4-iodo-1,2,4-triazoliums, and to a lesser extent, 5-iodo-1,2,4-triazoliums), in this work, we aimed to estimate the potential of all structural types of iodine-containing azolium salts for applications as XB-donating organocatalysts. Thorough quantum-chemical calculations were carried out and verified based on experimental kinetic studies.
Results and discussion
In the first stage of this work, limitations were established regarding the range of substrates to be studied. Although high catalytic activity is a key feature of any catalyst, other characteristics, such as chemical stability and synthetic accessibility also play important roles when selecting an appropriate catalyst. Therefore, iodoazoliums with N–I moieties were excluded from this study because such compounds are subject to homolytic cleavage of this bond, which may induce a series of radical or oxidative transformations. Considering the importance of chemical stability, all oxazolium derivatives were also excluded because the high electrophilicity of these compounds rendered them unstable to reactions with common nucleophiles, including H2O.42–45 Overall, fourteen structural types of iodoazoliums were evaluated in this study while keeping within these limitations (Fig. 1). The pentazoliums were excluded because of the absence of the C atoms in the heterocycle (only N–I bonds could be formed) and its general instability under ordinary conditions.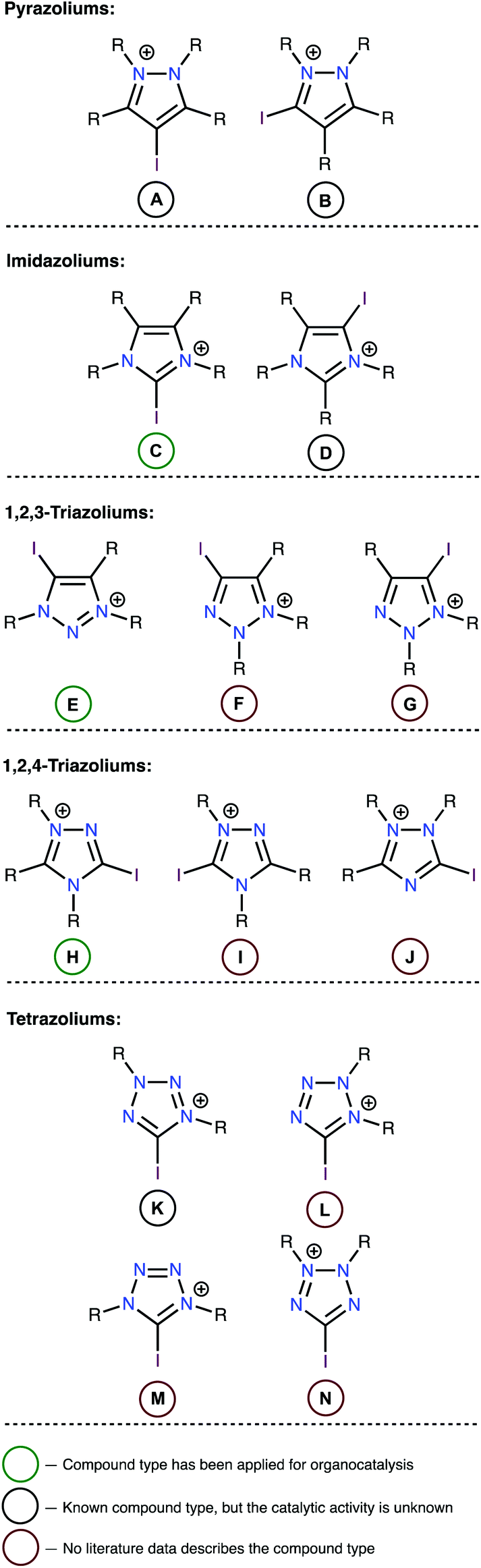 | ||
| Fig. 1 Structures of the studied iodoazoliums. | ||
A review of the relevant literature data indicated that all of iododiazoliums (A–D) are known, but only C has been applied for XB-donating organocatalysis.46–50 The diazoliums A and D served as precursors for the preparation of mesoionic carbene metal complexes by the Huynh51–54 and Bera55–57 groups, respectively. There is only one reported example of a type-B compound (i.e., 1,2-dimethyl-3-iodoindazoium iodide), and its potential applications have not yet been explored. The triazoliums E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33,34,36,58,59 and H37 were successfully utilized as organocatalysts, but no articles describe the preparation and application of compounds F, G, I, or J. The iodine-containing tetrazoliums K–N are also generally unexplored, and only one example of such compounds (i.e., 5-iodo-1,3-diphenyltetrazolium tetrafluoroborate; type K) has been reported.60 However, the catalytic properties of this compound were not studied, although it demonstrated reactivity toward a malonodinitrile anion to give phenylcyanamide (63%) and PhNHN
33,34,36,58,59 and H37 were successfully utilized as organocatalysts, but no articles describe the preparation and application of compounds F, G, I, or J. The iodine-containing tetrazoliums K–N are also generally unexplored, and only one example of such compounds (i.e., 5-iodo-1,3-diphenyltetrazolium tetrafluoroborate; type K) has been reported.60 However, the catalytic properties of this compound were not studied, although it demonstrated reactivity toward a malonodinitrile anion to give phenylcyanamide (63%) and PhNHN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CN)2 (54%).60
C(CN)2 (54%).60
Electrostatic surface potential distribution analysis
The electrostatic potentials on the surfaces of cations (in particular, in halonium cations61) are entirely positive, but anisotropic. The electrostatic surface potential (ESP) distributions62–64 were calculated for all structural types of investigated azoliums A–N, where all R groups were considered as methyl moieties (A*–N*; Fig. 2). The highest potentials (Vs(max)) on the σ-holes of the I atoms fell in the range, 384–541 kJ mol−1, with the cations of the studied azolium types C*, E*, and H* fell within the narrow range of moderate values (441–457 kJ mol−1). With the exception of A*, the quantity of N atoms vicinal to the I atom did not have a clear effect on the Vs(max) of the σ-holes on the I atom because of the overlap of the corresponding values (408–461 vs. 441–541 kJ mol−1 for 1N and 2Ns, respectively). Additionally, Vs(max) did not directly depend on the overall quantity of N atoms in the cycle (2Ns = 384–457; 3Ns = 408–493; 4Ns = 441–541 kJ mol−1). Although it is reasonable to assume that the availability of a lone pair of electrons at the vicinal position to the I atom (as in F*, H*, J*, K*, L*, and N*) may decrease the Vs(max) of the σ-hole, no such relationship was observed. Overall, analysis of the ESP distributions indicated that the Vs(max) of the I atom σ-hole had no appreciable dependence on a series of structural parameters of the azoliums.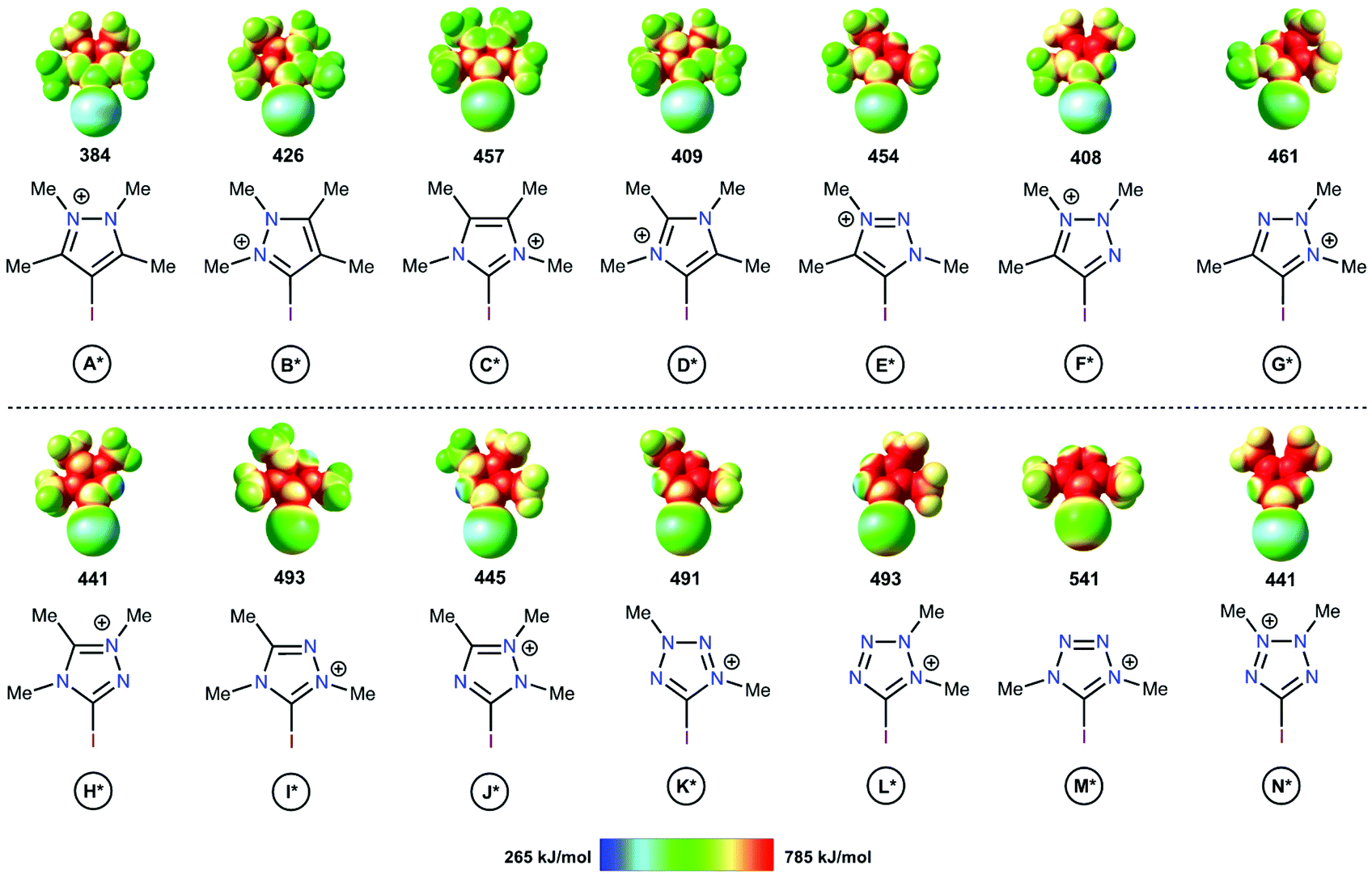 | ||
| Fig. 2 Electrostatic surface potential distributions for the investigated azoliums featuring methyl substituents. The highest potentials on the iodine atom σ-hole are given in kJ mol−1. | ||
Density functional theory calculations
As with other catalysts, the catalytic effect of XB donors originates from decreasing the Gibbs free energy of activation and linear free energy relationships (LFERs) between halogen bonding association constants and calculated electrostatic potentials have been documented previously.65–67 LFERs between catalytic activity and halogen bonding association constants also have been observed.46,68 Therefore, this phenomenon was the basis for the density functional theory (DFT) calculations carried out in this study to determine the activation energies of model reactions involving A*–N*. The hydrolysis of methyl chloride (TS1) and the coupling of ammonia and acetone (TS2) were chosen as simple model reactions (Scheme 1 and Table 1) because XB donors have typically been studied in reactions that either required elimination of a halogen atom or carbonyl activation.10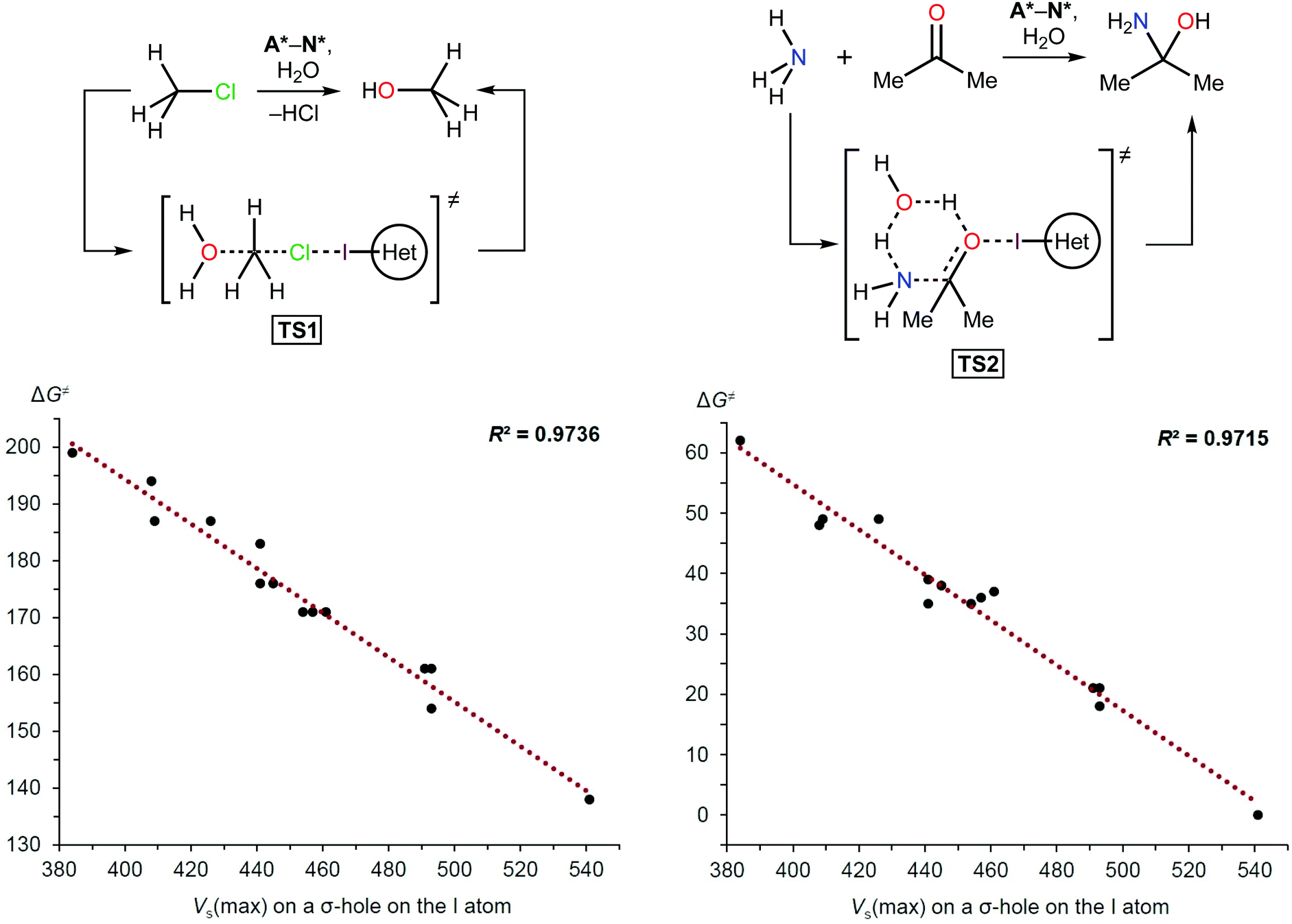 | ||
| Scheme 1 Model reactions (top) and plots showing the correlation between the Gibbs free energy of activation (ΔG≠; kJ mol−1) and Vs(max) on the I atom σ-hole (kJ mol−1). | ||
| Azolium type | V s(max) on the I atom (kJ mol−1) | ΔG≠ (kJ mol−1) | |
|---|---|---|---|
| TS1 | TS2 | ||
| A* | 384 | 199 | 62 |
| F* | 408 | 194 | 48 |
| D* | 409 | 187 | 49 |
| B* | 426 | 187 | 49 |
| N* | 441 | 183 | 39 |
| H* | 441 | 176 | 35 |
| J* | 445 | 176 | 38 |
| E* | 454 | 171 | 35 |
| C* | 457 | 171 | 36 |
| G* | 461 | 171 | 37 |
| K* | 491 | 161 | 21 |
| I* | 493 | 161 | 21 |
| L* | 493 | 154 | 18 |
| M* | 541 | 138 | 0 |
The theoretical data revealed a strong correlation between the Vs(max) of the I atom σ-holes and the activation energies of the model reactions (Scheme 1 and Table 1). This suggested that the Vs(max) value may represent the relative catalytic activity of the studied azoliums. Furthermore, these results highlighted promising structural types for designing and preparing effective organocatalysts, thus also indicating which less active cation types could be disregarded.
Analysis of the Vs(max) values revealed that four azolium compounds (I*, K*, L*, and M*) featured significantly more electropositive I atoms than all other studied cation types; correspondingly, this analysis suggested that pyrazolium A* should be the least catalytically active among all the studied azoliums. To verify this DFT-based conclusion, a series of azoliums with unknown catalytic activities were synthesized, and kinetic measurements were carried out to study their catalytic potential. To achieve adequately representative results, the azolium types that were predicted to potentially exhibit outstanding catalytic activity were selected for these experimental investigations. Considering that the azolium types I, L, and M are yet unknown, and K is represented by only one example in the literature,60 the synthesis and evaluation of these compounds’ reactivities deserve individual in-depth studies. Therefore, pyrazoliums of type-A were chosen as model substrates for experimental verification of the theoretical data because relevant general synthetic strategies for their preparation were available in the literature and they demonstrated suitable stability under ordinary conditions.
Experimental confirmation of the relative catalytic activity of the studied azoliums
The pyrazolium-triflate salts [1–5]OTf (Scheme 2 and Fig. 3) have not been characterized previously, so they were fully characterized in the current study. Furthermore, their catalytic activities were compared with the reactivities of the most abundant azolium-based organocatalysts, i.e., 2-iodoimidazolium and 4-iodo-1-alkyl-1,2,3-triazolium species ([6]OTf and [7]OTf, respectively). The generation of 1-benzoyl-3,5-dimethylpyrazole from acetyl acetone and benzoyl hydrazide (Knorr-type reaction; Scheme 2) was selected as the model reaction for the experimental confirmation of the theoretical data because it involved carbonyl activation and was deemed suitable in our group's previous study regarding the catalytic activity of iodine(III) derivatives.41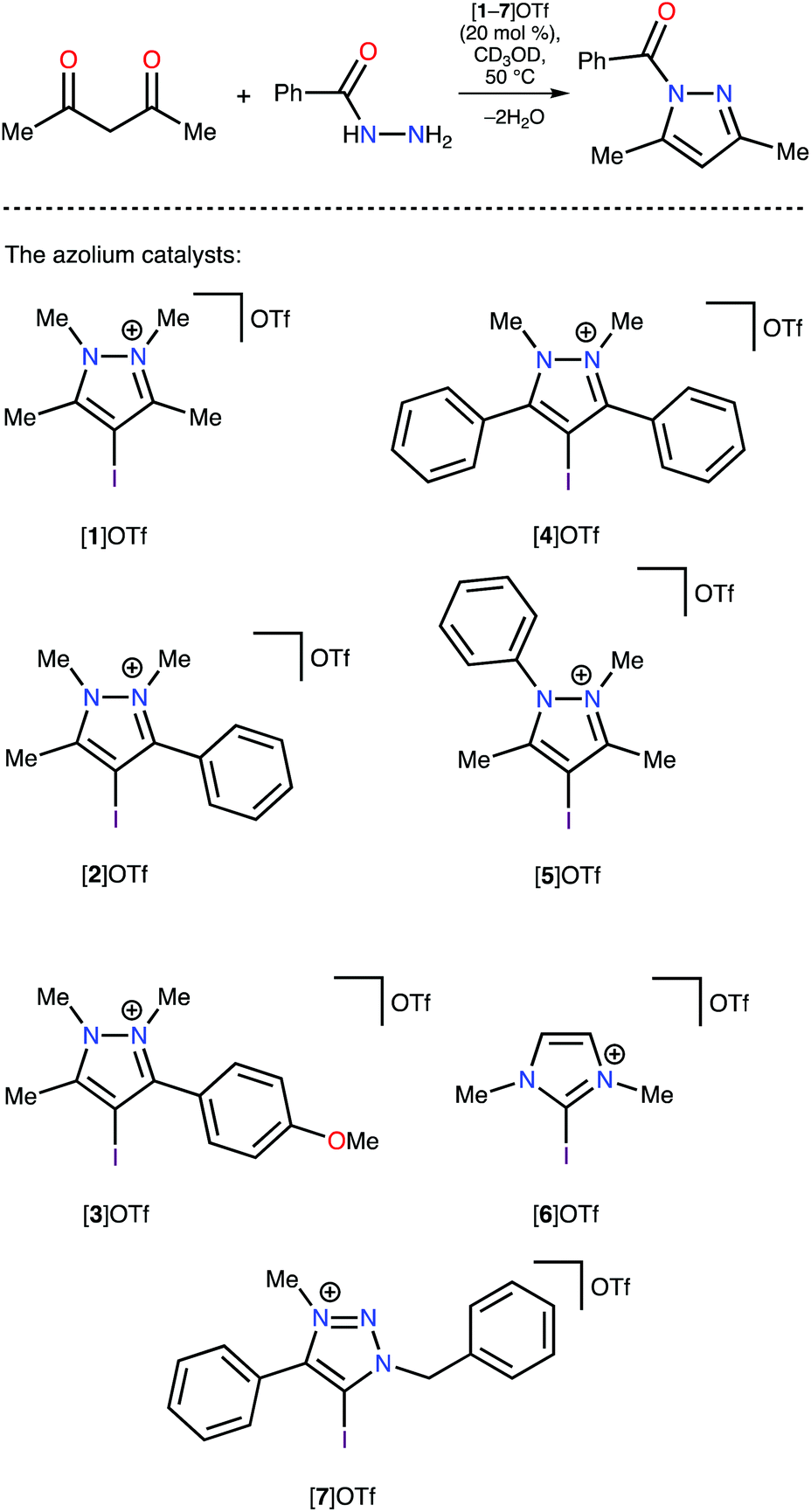 | ||
| Scheme 2 The series of azolium catalysts tested in the Knorr-type reaction. | ||
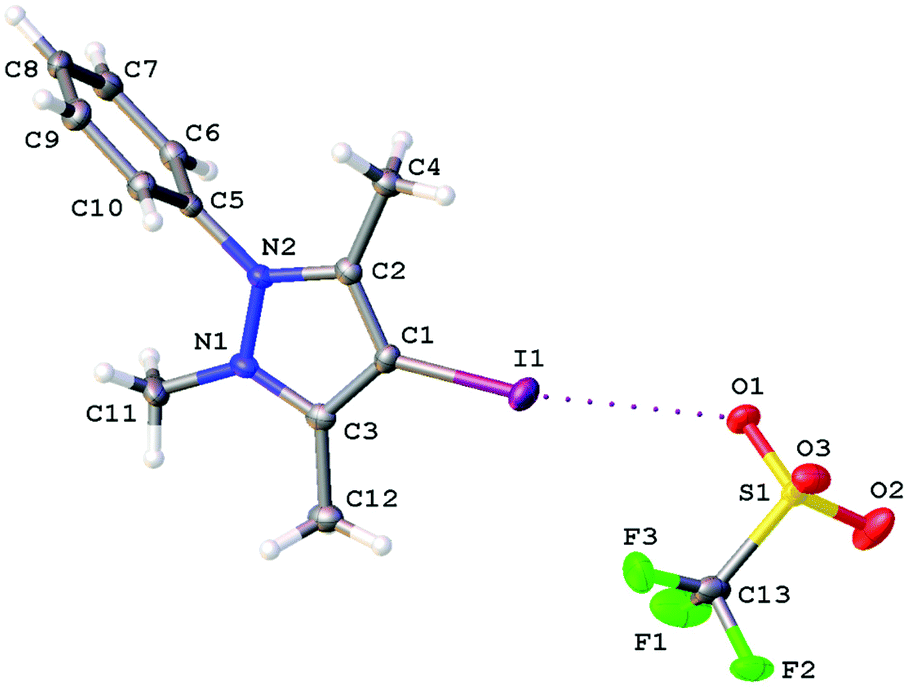 | ||
| Fig. 3 The molecular structure of [5]OTf determined by X-ray diffraction. | ||
Proton nuclear magnetic resonance (1H-NMR) monitoring indicated that the most active species for the test reaction was the triazolium [7]OTf, and the imidazolium [6]OTf was less active (Fig. 4). However, a comparison of the kinetic curves of [6]OTf and [7]OTf within the first 30 min of the reaction indicated similar or even higher catalytic activity of [6]OTf. This observation suggests possible degradation of [6]OTf during the reaction or its inhibition by competitive binding with the product. Huber and co-workers reported similar observations in terms of the kinetics of 2-iodoimidazoliums catalyzing the Mukaiyama reaction.50 The shape of the kinetic curves for [6]OTf and [7]OTf are entirely consistent with the theoretical data, i.e., their nearly identical Vs(max) of the I atom σ-holes (454 and 457 kJ mol−1 for E* and C*, respectively).
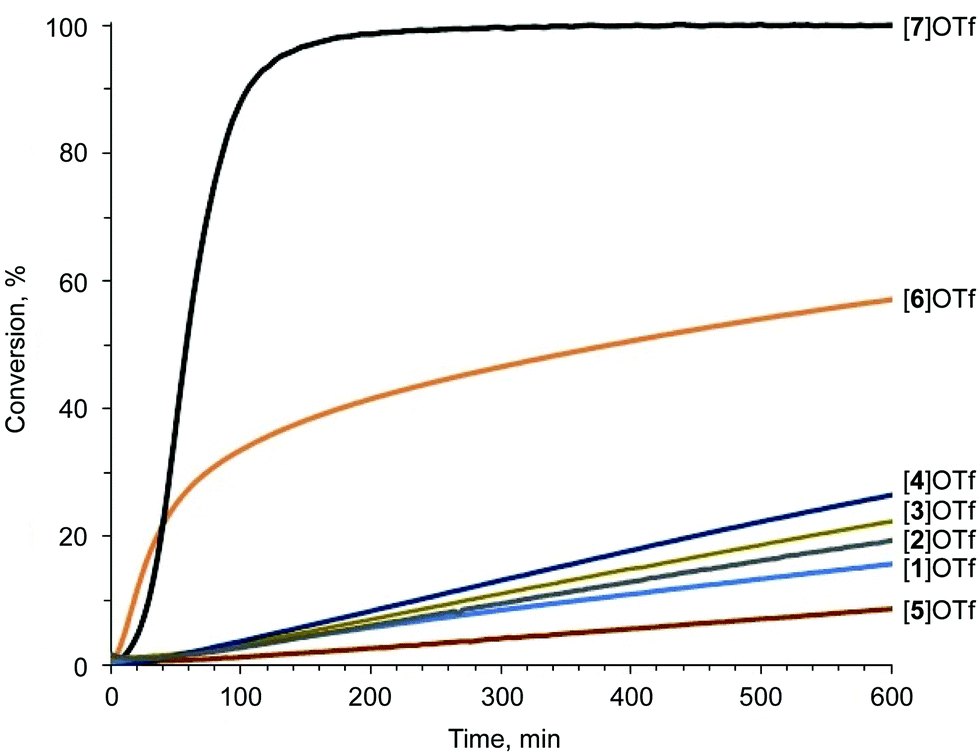 | ||
| Fig. 4 Knorr-type reaction catalytic conversion based on 1H-NMR monitoring. The S-shape of the kinetic curves is caused by the initial accumulation of the intermediate.41 | ||
As expected, following the theoretical calculations, the pyrazoliums [1–5]OTf were significantly less active in the studied reaction, relative to the imidazolium [6]OTf and triazolium [7]OTf compounds, and their catalytic activities did not depend strongly on the nature of substituents on the heterocyclic system. The catalytic activity was enhanced slightly by changing the methyl groups to phenyl moieties, which exhibited weaker electron donating effects at the vicinal position relative to the I atom ([1–4]OTf); however, activity decreased after changing Me to Ph at the N atom ([5]OTf vs. [1]OTf). Therefore, it was concluded that catalytic activity of azolium-based halogen bond donors is generally determined by the structure of the azolium heterocycle, whereas the substituents on the cycle have significantly less influence on the activity. The experimental data obtained fully agree with the DFT calculations, which themselves had a strong correlation with the ESP distribution data. Therefore, ESP distribution analysis could be successfully employed to predict the catalytic activity of azolium-based XB donors. This experimentally-verified theoretical study highlighted the most promising azolium cycles for applications in organocatalysis.
Conclusion
The results of this study support two major conclusions. First, ESP distribution analysis was successfully implemented to evaluate the relative catalytic activity of a series of structurally similar XB donors. This analysis importantly revealed the strong correlation between the positive electrostatic potential of the σ-hole on the halogen atom and the Gibbs free energy of activation of the considered reactions (i.e., halogen abstraction and carbonyl activation). ESP analysis requires significantly less computational time and energy than computing the localization and energies of reaction transition states; therefore, this study demonstrates a relatively facile method for estimating the relative activity of XB donors. Furthermore, the approach for estimating the relative catalytic activities of azolium catalysts presented herein was confirmed experimentally.Second, the ESP analysis considering all possible structural types of iodine-containing azolium-based donors that were stable under ordinary conditions unambiguously indicated that the most promising azolium compounds for organocatalysis applications include one 1,2,4-triazolium and three tetrazolium species (Fig. 5). Neither type has been tested in terms of their organocatalytic activities, nor comprehensively described in terms of other properties.
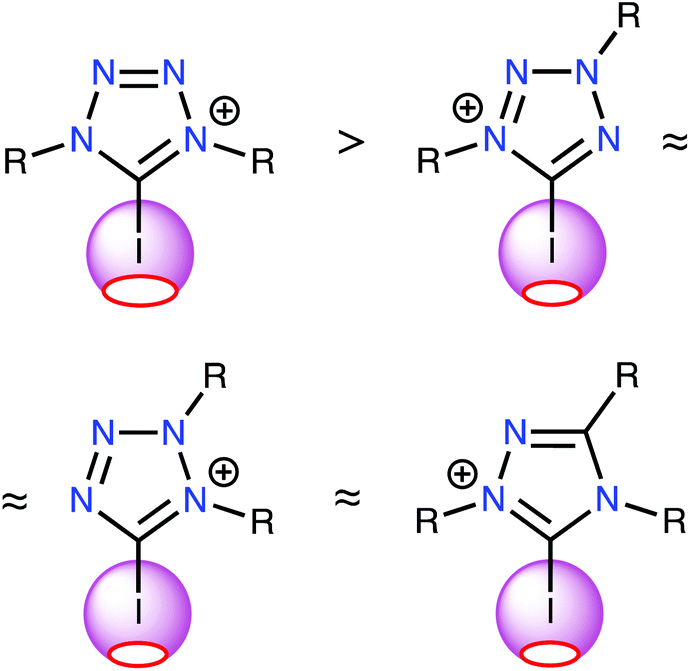 | ||
| Fig. 5 The most promising azolium-based XB-donating organocatalysts and a relative comparison of their Vs(max) values of the σ-hole on the I atom. | ||
These results can also inspire the rational design and development of new XB-donating organocatalysts comprising the most promising azolium features, without requiring blind screening of the activity of such compounds. Further experimental examinations of similar azolium compounds are ongoing in our laboratory.
Experimental section
Materials and instrumentation
All solvents, 1,3-diketones, hydrazine hydrate, phenylhydrazine, PhI(OAc)2, KOH, MeI, iodine, Na2SO4, MgSO4, AgOTf, NaHCO3, Na2S2O3, KI, NaOAc, NH4Cl, 1-methylimidazole, N-iodosuccinimide, phenylacetylene, NaN3, BnBr, and CuI were obtained from commercial sources and used as received. All syntheses were conducted in air. Chromatographic separation was carried out using Macherey-Nagel silica gel 60 M (0.063–0.2 mm). Analytical thin-layer chromatography (TLC) was performed on unmodified Merck ready-to-use plates (TLC silica gel 60 F254) applying UV detection. Melting points (M.p.) were measured on a Stuart SMP30 apparatus in capillaries, and the values reported herein were not corrected. Molar conductivities of ∼1 × 10−3 M solutions in MeOH were measured on a Mettler Toledo FE30 conductometer using an Inlab710 sensor. High resolution electrospray ionization time-of-flight mass-spectra (HRESI-TOF-MS) were obtained on a Bruker maXis spectrometer equipped with an electrospray ionization source. The instrument was operated in positive-ion mode with an m/z range 50–1200. The nebulizer gas flow was 1.0 bar, and the drying gas flow was 4.0 L min−1. For HRESI+-MS, the studied compounds were dissolved in MeOH. Infrared (IR) spectra (4000–400 cm−1) of samples in KBr pellets were recorded on a Shimadzu IR Prestige-21 instrument. 1H- and 13C{1H}-NMR spectra were measured on a Bruker Avance 400 spectrometer in CDCl3, (CD3)2SO, CD3CN, and CD3OD at 25 °C; the residual solvent signal was used as the internal standard. The NMR monitoring kinetic experiments were carried out by measuring the 1H-NMR spectra every 5 min (four scans; repetition time = 4 s) following the initial equilibration period of 5 min on a Bruker Avance III 500 spectrometer in CD3OD at 50 °C; the residual solvent signal was used as the internal standard.X-ray diffraction studies
Crystallographic data for all crystals were obtained using a Rigaku “SuperNova XtaLAB” ([1]I, [5]I) or a Rigaku “Synergy XtaLAB” ([5]OTf) diffractometer, each equipped with a monochromatic micro-focus CuKα X-ray source. All crystals were maintained at 100 K during the data collection. Crystal structures were solved using the ShelXT69 structure solution program and refined using the ShelXL70 structure refinement program incorporated in the Olex271 program package. Empirical absorption corrections were applied using spherical harmonics implemented in the SCALE3 ABSPACK scaling algorithm (Rigaku Oxford Diffraction, 2020). All crystallographic data for this work can be obtained free of charge from the Cambridge Crystallographic Database (CCDC 2085430 for [5]I, 2085431 for [5]OTf, 2085432 for [1]I).
Analytical and spectroscopic data for [1–5]OTf
The compounds [1–7]OTf were characterized by HRESI+-MS, IR, and 1H- and 13C{1H}-NMR spectroscopies. The compounds [6]OTf72 and [7]OTf73 have been characterized previously, and the description of their synthesis and characterization is in the ESI.†The HRESI+ mass-spectra of [1–5]OTf contain peaks corresponding to the quasi-ions [M]+. The IR spectra of [1–5]OTf display one to five weak bands in the range of 3106–2926 cm−1, which were attributed to the C–H stretches. These IR spectra also display one or two medium-to-strong bands in the range of 1648–1457 cm−1, which were attributed to the CN and CC stretches. The strong bands corresponding to the stretching frequencies of the SO and C–F bonds of the triflate anion were observed in the range of 1263–1029 cm−1. In the region of 645–516 cm−1 two medium-to-strong bands attributed to the C–I bonds were detected. The 1H-NMR spectra of [1–5]OTf contain sets of signals from N–CH3 and C–CH3 at 4.24–3.69 and 2.58–2.25 ppm, respectively. The proton resonances from C–C6H5 were detected at 7.72–7.06 ppm, whereas the signals from N–C6H5 were observed at 7.83–7.68 ppm. The 13C{1H}-NMR spectra of [1–5]OTf displayed the following set of signals: 133.12–129.22 (N–C6H5), 131.60–126.05 (C–C6H5), 69.22–65.56 (C–I), 36.39–35.06 (N–CH3), and 14.17–12.95 ppm (C–CH3). Signals from CN carbons in the pyrazolium rings were observed at 150.52–148.05 ppm, whereas the signals from the CF3-group of the triflate anion were detected as a quartet centered around 121.17–120.40 ppm.
Syntheses and characterizations of [1–5]OTf
:hexane = 1:1, v/v). A solution of the 4-iodopyrazole (2.5 mmol) in DMSO (2 mL) was added to a stirred solution of KOH (280 mg, 5 mmol) and MeI (233 μL, 3.75 mmol) in DMSO (2 mL). The resulting solution was stirred overnight at RT, and then H2O (100 mL) was added to the reaction mixture. The product was extracted with EtOAc (3 × 50 mL). The combined organic layers were washed with brine (50 mL), and the organic layer was dried over Na2SO4 and filtered from the inorganics. The solvent was evaporated in vacuo at 50 °C, and the corresponding 1-methyl-4-iodopyrazole was used without additional purification. A solution of MeI (224 μL, 3.6 mmol) in CH3CN (1 mL) was added to a stirred solution of the corresponding 1-methyl-3-iodopyrazole (1.2 mmol) in CH3CN (4 mL). The resulting solution was stirred for 7 d at 85 °C, and the generated precipitate was filtered off, washed with CH3CN (2 × 3 mL) and Et2O (1 × 5 mL), and dried at 50 °C for 2 h in air to give the corresponding 1,2-dimethyl-4-iodopyrazolium iodide, which was used without further purification. A solution of AgOTf (136 mg, 0.53 mmol) in MeOH (2 mL) was added to a stirred solution of the corresponding 1,2-dimethyl-4-iodo-pyrazolium iodide (0.53 mmol) in MeOH (3 mL). The suspension was stirred for 15 min at RT, and the precipitate formed was filtered off, washed with MeOH (15 mL), and the combined organic layers were evaporated in vacuo at 40 °C. The residue was recrystallized from EtOAc (5 mL) to give [1–4]OTf. Yields on the last synthetic step are reported below.
[1]OTf: Yield: 83% (176 mg). M.p.: 159–161 °C. ΛM (CH3OH, 1.09 × 10−3 M): 98.8 Ohm−1 cm−1 mol−1. 1H-NMR (400.13 MHz, CD3CN, ppm): δ = 3.93 (s, 6H, N–CH3), 2.46 (s, 6H, C–CH3). 13C{1H}-NMR (101.61 MHz, CD3CN, ppm): δ = 148.05 (C–CH3), 121.01 (q, 1JCF = 320.7 Hz, CF3), 65.56 (C–I), 35.06 (N–CH3), 12.95 (C–CH3). HRMS (ESI-TOF): m/z calcd for C7H12N2I+: 251.0040; found: 251.0042. IR (KBr, selected bands, cm−1): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3039 (w, C–H), 2926 (w, C–H), 1639 (m, CN), 1553 (m, CC), 1509 (w, CC), 1263 (s, SO or C–F), 1151 (s, SO or C–F), 1032 (s, SO or C–F), 645 (s, C–I), 518 (m–s, C–I).
= 3039 (w, C–H), 2926 (w, C–H), 1639 (m, CN), 1553 (m, CC), 1509 (w, CC), 1263 (s, SO or C–F), 1151 (s, SO or C–F), 1032 (s, SO or C–F), 645 (s, C–I), 518 (m–s, C–I).
[2]OTf. Yield: 64% (157 mg). M.p.: 137–138 °C. ΛM (CH3OH, 9.7 × 10−4 M): 239.2 Ohm−1 cm−1 mol−1. 1H-NMR (400.13 MHz, CDCl3, ppm): δ = 7.66–7.58 (m, 3H, Ph), 7.52–7.49 (m, 2H, Ph), 4.24 (s, 3H, N–CH3), 4.04 (s, 3H, N–CH3), 2.62 (s, 3H, C–CH3). 13C{1H}-NMR (101.61 MHz, CDCl3, ppm): δ = 150.09, 148.71 (C–CH3, C–Ph); 131.60, 129.95, 129.46, 126.05 (Ph); 120.66 (d, 1JCF = 320.7 Hz) 66.25 (C–I); 36.39, 35.99 (N–CH3), 13.89 (C–CH3). HRMS (ESI-TOF): m/z calcd for C12H14N2I+: 313.0196; found: 313.0198. IR (KBr, selected bands, cm−1): = 3056 (w, C–H), 1503 (m, CN), 1461 (m, CC), 1271 (s, C–F), 1260 (s, SO or C–F), 1153 (s, SO or C–F), 1030 (s, C–F), 637 (s, C–I), 517 (m, C–I).
[3]OTf. Yield: 78% (203 mg). M.p.: 148–149 °C. ΛM (CH3OH, 7.3 × 10−4 M): 239.2 Ohm−1 cm−1 mol−1. 1H-NMR (400.13 MHz, CDCl3, ppm): δ = 7.45–7.41 (m, 2H, Ar); 7.11–7.08 (m, 2H, Ar); 4.21 (s, 3H), 4.03 (s, 3H), 3.91 (s, 3H) (N–CH3, O–CH3); 2.60 (s, 3H, C–CH3). 13C{1H}-NMR (101.61 MHz, CDCl3, ppm): δ = 161.98 (Ar); 150.13, 148.54 (CCH3, C–Ar); 131.55, 117.74, 114.92 (Ar); 120.67 (d, 1JCF = 320.5 Hz) 66.41 (C–I); 55.54 (O–CH3); 36.36, 35.97 (N–CH3); 13.90 (C–CH3). HRMS (ESI-TOF): m/z calcd for C13H16ON2I+: 343.0302; found: 343.0308. IR (KBr, selected bands, cm−1): = 3083 (w, C–H), 3036 (w, C–H), 2966 (w, C–H), 2937 (w, C–H), 2839 (w, C–H), 1613 (s, CN), 1573 (m, CC), 1525 (m, CC), 1460 (m, CC), 1263 (s, C–F), 1249 (s, SO or C–F), 1032 (s, SO or C–F), 641 (s, C–I), 517 (s, C–I).
[4]OTf. Yield: 86% (239 mg). M.p.: 175–177 °C. ΛM (CH3OH, 8.8 × 10−4 M): 261.9 Ohm−1 cm−1 mol−1. 1H-NMR (400.13 MHz, CD3OD, ppm): δ = 7.72–7.69 (m, 6H, Ph), 7.65–7.63 (m, 4H, Ph), 4.08 (s, 6H, N–CH3). 13C NMR (101.61 MHz, CD3OD, ppm): δ = 150.52 (C–Ph); 131.39, 129.88, 129.18 and 126.48 (Ph); 120.40 (d, 1JCF = 318.2 Hz); 66.68 (C–I); 35.67 (N–CH3). HRMS (ESI-TOF): m/z calcd for C17H16N2I+: 375.0352; found: 375.0361. IR (KBr, selected bands, cm−1): = 3061 (w, C–H), 2961 (w, C–H), 1608 (m, CN), 1477 (m, CC), 1457 (m, CC), 1276 (s, C–F), 1260 (s, SO or C–F), 1035 (s, SO or C–F), 640 (s, C–I), 517 (m, C–I).
The combined organic layers were washed with brine (50 mL), and the organic layer was dried over Na2SO4 and then filtered. The solvent was evaporated in vacuo at 50 °C to give 3,5-dimethyl-1-phenyl-4-iodopyrazole in 81% yield (1.227 g) as a brown oil, which was used without additional purification. A solution of 3,5-dimethyl-1-phenyl-4-iodopyrazole (988 mg, 3.31 mmol) in MeI (3 mL) was stirred for 3 d at 60 °C, and the precipitate formed was filtered off, washed with Et2O (10 mL), and dried at 50 °C for 2 h in air to give the 1,3,5-trimethyl-2-phenyl-4-iodopyrazolium iodide in 46% yield (672 mg) as a colorless solid. A solution of AgOTf (118 mg, 0.46 mmol) in MeOH (2 mL) was added to a stirred solution of 1,3,5-trimethyl-2-phenyl-4-iodopyrazolium iodide (203 mg, 0.46 mmol) in MeOH (3 mL). The suspension was stirred for 15 min at RT, and the precipitate formed was filtered off, washed with MeOH (15 mL), and the combined organic layers were evaporated in vacuo at 40 °C and recrystallized from EtOAc (5 mL) to give [5]OTf as a colorless solid.
[5]OTf. Yield: 87% (186 mg). M.p.: 163–164 °C. ΛM (CH3OH, 8.2 × 10−4 M): 114.0 Ohm−1 cm−1 mol−1. 1H-NMR (400.13 MHz, (CD3)2SO, ppm): 7.83–7.74 (m, 3H, Ph), 7.71–7.68 (m, 2H, Ph), 3.69 (s, 3H, N–CH3), 2.56 (s, 3H, C–CH3), 2.23 (s, 3H, C–CH3). 13C{1H}-NMR (101.61 MHz, (CD3)2SO, ppm): δ = 149.46, 148.90 (C–CH3); 133.12, 131.95, 131.11, 129.22 (Ph); 121.17 (q, 1JCF = 319.0 Hz, CF3); 69.22 (C–I); 36.36 (N–CH3); 14.17, 13.80 (C–CH3). HRMS (ESI-TOF): m/z calcd for C12H14N2I+: 313.0196; found: 313.0204. IR (KBr, selected bands, cm−1): = 3106 (w, C–H), 3059 (w, C–H), 3019 (w, C–H), 2938 (w, C–H), 1594 (m, CN), 1546 (m, CN), 1504 (s, CC), 1488 (s, CC), 1277 (s, C–F), 1260 (s, SO or C–F), 1224 (s, SO or C–F), 1031 (s, SO or C–F), 648 (s, C–I), 517 (m, C–I).
Computational details
Full geometry optimizations of all model structures were carried out via DFT calculations with the M06-2X functional74 using the Gaussian-09 program package.75 The quasi-relativistic pseudopotential MWB46,76 which described 46 core electrons, and an appropriate contracted basis set were used for the iodine atoms, and standard 6-31G* basis sets were used for all other atoms. This level of theory was successfully applied by us in studies of very similar metal-free objects and processes.41,42 No symmetry restrictions were applied during the geometry optimization procedure. The Hessian matrices were calculated analytically for all optimized model structures (i) to confirm the location of the correct minima or saddle points on the potential energy surface (no imaginary frequencies, or one imaginary frequency, respectively) and (ii) to estimate the thermodynamic parameters (calculated at 25 °C) (see Tables S1 and S2 in the ESI†). The molecular surface electrostatic potential calculations were performed using the Multiwfn program (version 3.7)77 based on the obtained wavefunctions for the optimized equilibrium model structures. The molecular surface electrostatic potentials were visualized in the Chemcraft program (http://www.chemcraftprog.com/). The Cartesian atomic coordinates for all optimized equilibrium model structures are presented in Table S3 in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The theoretical calculations and kinetic experiments in this work were supported by the Russian Science Foundation (grant 20-73-10013), and the synthetic portions were supported by the Russian Foundation for Basic Research (grant 19-03-00044). Physicochemical studies were performed at the Center for Magnetic Resonance, Center for X-ray Diffraction Studies, Chemistry Educational Centre, and Center for Chemical Analysis and Materials Research (all at Saint Petersburg State University).References
- V. Oliveira, M. Cardoso and L. Forezi, Catalysts, 2018, 8, 605 CrossRef.
- Y. Qin, L. Zhu and S. Luo, Chem. Rev., 2017, 117, 9433–9520 CrossRef CAS.
- D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed.
- S. Mondal, S. R. Yetra, S. Mukherjee and A. T. Biju, Acc. Chem. Res., 2019, 52, 425–436 CrossRef CAS.
- T. Chanda and J. C. G. Zhao, Adv. Synth. Catal., 2018, 360, 2–79 CrossRef CAS.
- S. Vellalath and D. Romo, Angew. Chem., Int. Ed., 2016, 55, 13934–13943 CrossRef CAS.
- H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS PubMed.
- R. Tepper and U. S. Schubert, Angew. Chem., Int. Ed., 2018, 57, 6004–6016 CrossRef CAS.
- D. Bulfield and S. M. Huber, Chem. – Eur. J., 2016, 22, 14434–14450 CrossRef CAS PubMed.
- R. L. Sutar and S. M. Huber, ACS Catal., 2019, 9, 9622–9639 CrossRef CAS.
- C. M. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390–2431 CrossRef CAS.
- C. Thomas and B. Bibal, Green Chem., 2014, 16, 1687–1699 RSC.
- M. N. Grayson and K. N. Houk, J. Am. Chem. Soc., 2016, 138, 9041–9044 CrossRef CAS PubMed.
- M. Puripat, R. Ramozzi, M. Hatanaka, W. Parasuk, V. Parasuk and K. Morokuma, J. Org. Chem., 2015, 80, 6959–6967 CrossRef CAS PubMed.
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed.
- X. Fang and C. J. Wang, Chem. Commun., 2015, 51, 1185–1197 RSC.
- O. V. Serdyuk, C. M. Heckel and S. B. Tsogoeva, Org. Biomol. Chem., 2013, 11, 7051–7071 RSC.
- F. E. Held and S. B. Tsogoeva, Catal. Sci. Technol., 2016, 6, 645–667 RSC.
- X. Han, H. B. Zhou and C. Dong, Chem. Rec., 2016, 16, 897–906 CrossRef CAS.
- B. L. Zhao, J. H. Li and D. M. Du, Chem. Rec., 2017, 17, 994–1018 CrossRef CAS PubMed.
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- T. Akiyama and K. Mori, Chem. Rev., 2015, 115, 9277–9306 CrossRef CAS PubMed.
- T. James, M. van Gemmeren and B. List, Chem. Rev., 2015, 115, 9388–9409 CrossRef CAS PubMed.
- S. Schindler and S. M. Huber, in Halogen Bonding II, ed. P. Metrangolo and G. Resnati, Springer, 2015, pp. 167–204 Search PubMed.
- M. Erdelyi, Chem. Soc. Rev., 2012, 41, 3547–3557 RSC.
- D. von der Heiden, A. Vanderkooy and M. Erdélyi, Coord. Chem. Rev., 2020, 407, 213147 CrossRef CAS.
- S. Benz, A. I. Poblador-Bahamonde, N. Low-Ders and S. Matile, Angew. Chem., Int. Ed., 2018, 57, 5408–5412 CrossRef CAS.
- L. Vogel, P. Wonner and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 1880–1891 CrossRef CAS PubMed.
- J. Bamberger, F. Ostler and O. G. Mancheno, ChemCatChem, 2019, 11, 5198–5211 CrossRef CAS PubMed.
- Y. Takeda, D. Hisakuni, C. H. Lin and S. Minakata, Org. Lett., 2015, 17, 318–321 CrossRef CAS PubMed.
- S. H. Jungbauer, S. M. Walter, S. Schindler, L. Rout, F. Kniep and S. M. Huber, Chem. Commun., 2014, 50, 6281–6284 RSC.
- J. P. Gliese, S. H. Jungbauer and S. M. Huber, Chem. Commun., 2017, 53, 12052–12055 RSC.
- M. Kaasik, A. Metsala, S. Kaabel, K. Kriis, I. Jarving and T. Kanger, J. Org. Chem., 2019, 84, 4294–4303 CrossRef CAS.
- A. Dreger, E. Engelage, B. Mallick, P. D. Beer and S. M. Huber, Chem. Commun., 2018, 54, 4013–4016 RSC.
- R. Haraguchi, S. Hoshino, M. Sakai, S. G. Tanazawa, Y. Morita, T. Komatsu and S. I. Fukuzawa, Chem. Commun., 2018, 54, 10320–10323 RSC.
- K. Torita, R. Haraguchi, Y. Morita, S. Kemmochi, T. Komatsu and S. I. Fukuzawa, Chem. Commun., 2020, 56, 9715–9718 RSC.
- R. A. Squitieri, K. P. Fitzpatrick, A. A. Jaworski and K. A. Scheidt, Chem. – Eur. J., 2019, 25, 10069–10073 CrossRef CAS PubMed.
- R. Haraguchi, T. Nishikawa, A. Kanazawa and S. Aoshima, Macromolecules, 2020, 53, 4185–4192 CrossRef CAS.
- F. Heinen, E. Engelage, A. Dreger, R. Weiss and S. M. Huber, Angew. Chem., Int. Ed., 2018, 57, 3830–3833 CrossRef CAS.
- Y. Zhang, J. Han and Z.-J. Liu, RSC Adv., 2015, 5, 25485–25488 RSC.
- S. N. Yunusova, A. S. Novikov, N. S. Soldatova, M. A. Vovk and D. S. Bolotin, RSC Adv., 2021, 11, 4574–4583 RSC.
- M. V. Il'in, L. A. Lesnikova, D. S. Bolotin, A. S. Novikov, V. V. Suslonov and V. Y. Kukushkin, New J. Chem., 2020, 44, 1253–1262 RSC.
- M. V. Il'in and D. S. Bolotin, Chem. Heterocycl. Compd., 2020, 56, 824–828 CrossRef.
- M. V. Il'in, A. A. Sysoeva, D. S. Bolotin, A. S. Novikov, V. V. Suslonov, E. V. Rogacheva, L. A. Kraeva and V. Y. Kukushkin, New J. Chem., 2019, 43, 17358–17366 RSC.
- M. V. Il'in, D. S. Bolotin, V. V. Suslonov and V. Y. Kukushkin, New J. Chem., 2018, 42, 9373–9376 RSC.
- Y. P. Chang, T. Tang, J. R. Jagannathan, N. Hirbawi, S. Sun, J. Brown and A. K. Franz, Org. Lett., 2020, 22, 6647–6652 CrossRef CAS.
- X. Liu and P. H. Toy, Adv. Synth. Catal., 2020, 362, 3437–3441 CrossRef CAS.
- R. L. Sutar, E. Engelage, R. Stoll and S. M. Huber, Angew. Chem., Int. Ed., 2020, 59, 6806–6810 CrossRef CAS PubMed.
- J. Wolf, F. Huber, N. Erochok, F. Heinen, V. Guerin, C. Y. Legault, S. F. Kirsch and S. M. Huber, Angew. Chem., Int. Ed., 2020, 59, 16496–16500 CrossRef CAS PubMed.
- R. L. Sutar, N. Erochok and S. M. Huber, Org. Biomol. Chem., 2021, 19, 770–774 RSC.
- Y. Han and H. V. Huynh, Chem. Commun., 2007, 1089–1091, 10.1039/b615441g.
- Y. Han, L. J. Lee and H. V. Huynh, Chemistry, 2010, 16, 771–773 CrossRef CAS.
- Y. Han, H. V. Huynh and G. K. Tan, Organometallics, 2007, 26, 6581–6585 CrossRef CAS.
- Y. Han and H. V. Huynh, Dalton Trans., 2011, 40, 2141–2147 RSC.
- S. Yadav, I. Dutta, S. Saha, S. Das, S. K. Pati, J. Choudhury and J. K. Bera, Organometallics, 2020, 39, 3212–3223 CrossRef CAS.
- S. Saha, S. Yadav, N. U. D. Reshi, I. Dutta, S. Kunnikuruvan and J. K. Bera, ACS Catal., 2020, 10, 11385–11393 CrossRef CAS.
- A. Tyagi, N. U. D. Reshi, P. Daw and J. K. Bera, Dalton Trans., 2020, 49, 15238–15248 RSC.
- A. Peterson, M. Kaasik, A. Metsala, I. Järving, J. Adamson and T. Kanger, RSC Adv., 2019, 9, 11718–11721 RSC.
- M. Kaasik, S. Kaabel, K. Kriis, I. Järving and T. Kanger, Synthesis, 2019, 2128–2135 CAS.
- S. Araki, Y. Wanibe, F. Uno, A. Morikawa, K. Yamamoto, K. Chiba and Y. Butsugan, Chem. Ber., 1993, 126, 1149–1155 CrossRef CAS.
- G. Cavallo, J. S. Murray, P. Politzer, T. Pilati, M. Ursini and G. Resnati, Int. Union Crystallogr. J., 2017, 4, 411–419 CrossRef CAS.
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS.
- P. Politzer and J. Murray, Crystals, 2019, 9, 165 CrossRef CAS.
- P. Politzer, J. S. Murray, T. Clark and G. Resnati, Phys. Chem. Chem. Phys., 2017, 19, 32166–32178 RSC.
- M. G. Sarwar, B. Dragisic, L. J. Salsberg, C. Gouliaras and M. S. Taylor, J. Am. Chem. Soc., 2010, 132, 1646–1653 CrossRef CAS PubMed.
- E. Dimitrijevic, O. Kvak and M. S. Taylor, Chem. Commun., 2010, 46, 9025–9027 RSC.
- I. Alkorta, G. Sánchez-Sanz and J. Elguero, CrystEngComm, 2013, 15, 3178–3186 RSC.
- N. Momiyama, A. Izumiseki, N. Ohtsuka and T. Suzuki, ChemPlusChem, 2021, 86, 913–919 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- W. He, Y. C. Ge and C. H. Tan, Org. Lett., 2014, 16, 3244–3247 CrossRef CAS PubMed.
- M. Breugst, D. von der Heiden, E. Detmar and R. Kuchta, Synlett, 2017, 1307–1313 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- A. Bergner, M. Dolg, W. Küchle, H. Stoll and H. Preuß, Mol. Phys., 1993, 80, 1431–1441 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2085430–2085432. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ob01158h |
| This journal is © The Royal Society of Chemistry 2021 |